

Abstract
Background:
Abdominal aortic aneurysm (AAA) is a severe aortic disease with a high mortality rate in the event of rupture. Pharmacological therapy is needed to inhibit AAA expansion and prevent aneurysm rupture. Transcription factor EB (TFEB), a master regulator of autophagy and lysosome biogenesis, is critical to maintain cell homeostasis. In this study, we aim to investigate the role of vascular smooth muscle cell (VSMC) TFEB in the development of AAA and establish TFEB as a novel target to treat AAA.
Methods:
The expression of TFEB was measured in human and mouse aortic aneurysm samples. We used loss/gain-of-function approaches to understand the role of TFEB in VSMC survival and explored the underlying mechanisms through transcriptome and functional studies. Utilizing vascular smooth muscle cell (VSMC)-selective Tfeb knockout mice and employing different mouse AAA models, we determined the role of VSMC TFEB and a TFEB activator on AAA in vivo.
Results:
We found that TFEB is downregulated in both human and mouse aortic aneurysm lesions. TFEB potently inhibits apoptosis in VSMCs and transcriptome analysis revealed that TFEB regulates apoptotic signaling pathways, especially apoptosis inhibitor B-cell lymphoma 2 (BCL2). BCL2 is significantly upregulated by TFEB and is required for TFEB to inhibit VSMC apoptosis. Consistently, we observed that TFEB deficiency increases VSMC apoptosis and promotes AAA formation in different mouse AAA models. Furthermore, we demonstrated that 2-hydroxypropyl-β-cyclodextrin (HPβCD), a clinical agent used to enhance the solubility of drugs, activates TFEB and inhibits AAA formation and progression in mice. Finally, we found that HPβCD inhibits AAA in a VSMC TFEB-dependent manner in mouse models.
Conclusions:
Our study demonstrated that TFEB protects against VSMC apoptosis and AAA. TFEB activation by HPβCD may be a promising therapeutic strategy for the prevention and treatment of AAA.
Keywords: abdominal aortic aneurysm, vascular biology, smooth muscle cell, autophagy, apoptosis
Introduction:
Abdominal aortic aneurysm (AAA) is the most common type of aneurysm (American Heart Association report, 2018). The risk factors for AAA include older age, male sex, family history, hypertension, elevated cholesterol level, obesity, and preexisting atherosclerotic occlusive disease.1 Rupture of an aneurysm is often lethal with over 85% mortality.1 Current clinical intervention for AAA is limited to endovascular repair or surgical repair with only 10% of all AAA patients eligible, leaving no treatment options for the rest of the AAA patients. This is due to existing comorbidities or aneurysm lesions not suitable for surgical repair, even though these patients are still at high risk of AAA rupture.2 To date, an effective pharmacological treatment for AAA is not available. One key feature of AAA is the depletion of vascular smooth muscle cells (VSMCs) in the media layer of the vessel.3 The loss of VSMCs contributes to the weakness of the aortic wall and ensuing rupture.
Transcription factor EB (TFEB) is a member of the microphthalmia transcription factor (MITF)/TFE family. As a master regulator of autophagy and lysosome biogenesis,4 TFEB has beneficial effects on lysosomal storage diseases and neurodegenerative diseases.5 TFEB has become a promising molecular target for the treatment of cardiovascular disease. Endothelial TFEB reduces atherosclerosis6 and promotes postischemic angiogenesis.7 Macrophage TFEB inhibits inflammation and atherosclerosis.8 However, the role of TFEB in VSMC biology and related vascular diseases remains to be explored.
In the present study, we uncovered a protective role of TFEB in AAA using VSMC-selective Tfeb knockout (KO) mice and shed light on the molecular mechanisms by which TFEB controls VSMC survival. In addition, we found that 2-Hydroxypropyl-β-cyclodextrin (HPβCD), an FDA approved cyclodextrin derivative currently used to increase the solubility of drugs and under phase II clinical trial to treat Niemann-Pick Disease Type C1 (NPC1), activates TFEB and inhibits AAA in multiple mouse models. Our findings demonstrate the potential for use of TFEB activators to treat AAA.
Methods:
An expanded Methods section is available in the online-only Data Supplement. The data, methods used in the analysis and study materials that support the finding of this study are available from the corresponding author upon reasonable request.
Human aortic aneurysm samples
Human thoracic aortic aneurysm (TAA) samples were obtained from the Department of Cardiac Surgery at the University of Michigan with Institutional Review Board approval (Hum00077616). Informed consent was obtained from all subjects. Human abdominal aortic aneurysm (AAA) slides and age-matched control samples (Supplemental Table II) are purchased from Origene (Rockville, MD). The medical information of patients with aortic aneurysm is detailed in Supplemental Tables I and II.
Animal Studies
C57BL/6N-Atm1Brd/a Tfebtm1a(EUCOMM)Wtsi/BcmMmucd mice were produced at the Baylor College of Medicine from embryonic stem cells provided by the Wellcome Trust Sanger Institute within the NIH funded Knockout mouse project. The Tfebflox mice contain loxP sites flanking exons 4 and 5 of Tfeb.7 The Myh11-creERT2 mouse was purchased from the Jackson Laboratory (Stock No: 019079, Bar Harbor, ME). Myh11-creERT2/Tfebflox (TfebΔSMC) mice were generated by crossbreeding Tfebflox mice with Myh11-creERT2 mice. The cre transgene was randomly inserted in the Y chromosome in these animals, for which only knock out (KO) male mice can be generated and, accordingly, only male mice were used in this study9. To induce the cre expression, Myh11-creERT2/Tfebflox mice and Tfebflox control mice were injected intraperitoneally with tamoxifen (T5648, Sigma-Aldrich) at a dose of 75mg/kg/day for 5 consecutive days. Two weeks after tamoxifen administration, AAA was induced in the Tfebflox mice and VSMC-Tfeb KO mice in the different models, which are described in detail in the online supplementary files. All animal work was performed in accordance with the University of Michigan Animal Care and Use Committee.
Data availability
RNA-seq, ChIP-seq, and microarray data were deposited into GEO database (GSE137577 and GSE138000).
Statistics
Statistics tests were performed in R (for RNA-seq, ChIP-seq and microarray) or GraphPad Prism version 7.0 (GraphPad Software, San Diego, CA). Unless indicated otherwise, values are presented as mean ± standard error of the mean (SEM). All data were tested for normality and equal variance. If the data passed those tests, Student t-test was used to compare two groups. One-way ANOVA or two-way ANOVA followed by Holm-Sidak post hoc analysis was used for comparisons among >2 groups. If the data did not pass those tests, Mann-Whitney was used to compare two groups, Kruskal-Wallis test followed by a two-stage step-up method of Benjamini, Krieger and Yekutieli were used for >2 groups. P value < 0.05 was considered statistically significant.
Results:
TFEB is reduced in human and mouse aneurysmal lesions.
Through a paired comparison of TFEB expression in the human proximal ascending aortic aneurysm lesions with that in the adjacent non-lesion aorta, we found that TFEB mRNA and protein abundance are reduced in the human thoracic aortic aneurysm (TAA) lesions (Figure 1A–C, patients’ information in Supplemental Table I). We further determined the TFEB protein in AAA lesion by immunostaining (Supplemental Figure I A, patients’ information in Supplemental Table II). The AAA lesion displayed overt degradation of elastic fibers and decreased TFEB expression in the VSMCs in the media layer of the AAA area (SM22 was used as a smooth muscle cell marker) concomitant with massive loss of VSMCs (Supplemental Figure I A). Consistently, pro-inflammatory cytokines (TNFα, IL1β and IFNγ), which elicit and aggravate aortic aneurysm, reduced TFEB protein in human aortic smooth muscle cells (HASMCs) (Figure 1D). TFEB was also downregulated at the mRNA (Figure 1E) and protein levels in the mouse AAA lesions (Figure 1F) when using the proprotein convertase subtilisin/kexin type 9 (PCSK9)/angiotensin II (AngII) model (Supplemental Figure VI A).10 Our analysis of a dataset11 from the Gene Expression Omnibus (GEO) database revealed that TFEB mRNA is also significantly reduced in the intracranial aneurysm (Supplemental Figure I B). Altogether, our data indicate that vascular TFEB is reduced in the aneurysmal lesions.
Figure 1. TFEB is downregulated in human and mouse aortic aneurysm.

(A) Transcription factor EB (TFEB) mRNA and (B) protein were determined by quantitative polymerase chain reaction (qPCR) and Western blot, respectively, in human proximal ascending aortic aneurysm lesions compared with that in the adjacent normal aorta (n=12 for mRNA, n=8 for protein). (C) TFEB was detected by immunofluorescence in human aneurysmal lesions and adjacent normal aorta. Scale bar, 50 μm. (D) Human aortic smooth muscle cells (HASMCs) were treated with tumor necrosis factor α (TNFα; 20 ng/mL), interleukin 1 β (IL1β; 10 ng/mL) or Interferon γ (IFNγ; 50 ng/mL) for 72 hours. TFEB protein was determined by Western blot (n=3). (E-F) In the proprotein convertase subtilisin/kexin type 9 (PCSK9)/ angiotensin II (AngII) model, TFEB mRNA and protein were determined in the aortas of AngII- and saline-infused mice by qPCR (E; n=15 for saline, n=8 for AngII) or Western blot (F; n=6). Data are presented as mean ± SEM. *p < 0.05, **p < 0.01. Paired t-test for A and B, one-way ANOVA followed by Holm-Sidak post hoc analysis for D, and unpaired t-test for E and F.
TFEB inhibits HASMC apoptosis.
VSMC depletion is one of the major features in the pathogenesis of aortic aneurysm.3 We found that TFEB knockdown promotes HASMC death induced by either Fas ligand (FasL) or TNFα + cycloheximide (CHX), by measurement of the activity of lactate dehydrogenase (LDH) released into the culture medium. Cell death can be blocked by the pan-caspase inhibitor carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]- fluoromethylketone (zvad), indicating it is an apoptosis-dependent process (Supplemental Figure II A). To further investigate the apoptotic process, we used Western blot to probe for selected apoptosis markers – the cleavage of Poly ADP-ribose polymerase (PARP) and cysteine-aspartic protease, cysteine aspartase 3 (Caspase 3). Adenovirus-mediated TFEB overexpression in HASMCs significantly decreased the cleavage of both PARP and Caspase 3 (Figure 2A), and also reduced Caspase 3 activity (Figure 2B). Consistently, TFEB knockdown in HASMCs significantly increased the cleavage of both PARP and Caspase 3 (Figure 2C) and Caspase 3 activity (Figure 2D). Combined with propidium iodide (PI) staining, Annexin V assay was used to distinguish apoptotic cells (Annexin V+/PI-) from necrotic cells (Annexin V+/PI+). TFEB overexpression significantly reduced apoptotic cells (11.2 ± 0.80%) compared with LacZ control (17.03 ± 0.80%) (Figure 2E), while TFEB knockdown increased apoptotic cells (15.73 ± 0.15%) compared with siRNA-negative control (siCt) group (10.77 ± 0.32%) (Figure 2F). The VSMC apoptosis induced by TFEB deficiency was abolished by zvad, indicating an essential role of the caspase pathway in the process (Figure 2F).
Figure 2. TFEB inhibits apoptosis in human aortic smooth muscle cells (HASMCs).
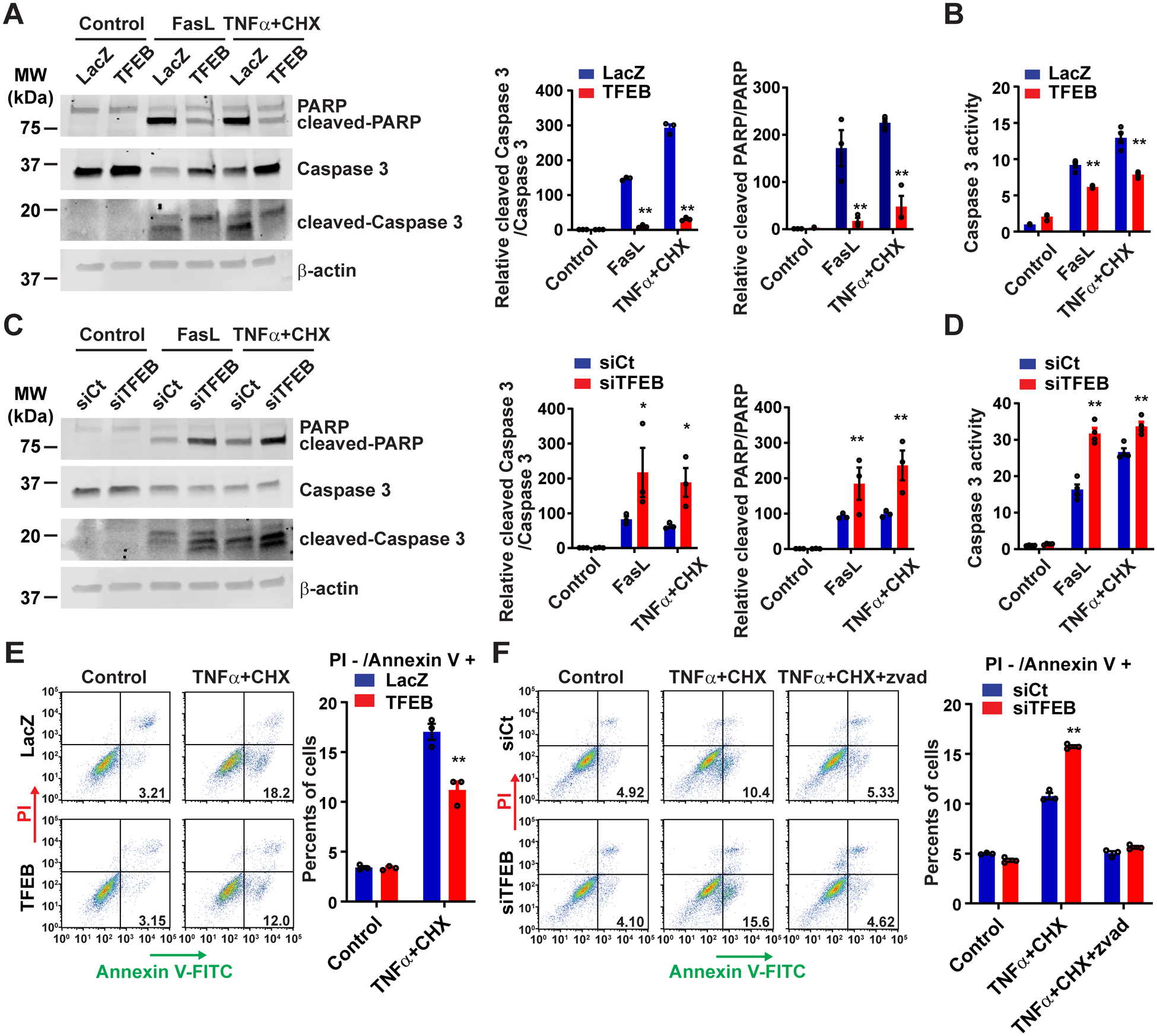
(A-B) HASMCs were infected with adenovirus encoding LacZ (Ad-LacZ) or TFEB (Ad-TFEB) (multiplicity of infection [MOI], 30). After 48 hours, the cells were treated with Fas ligand (FasL; 100 ng/mL) or tumor necrosis factor α (TNFα; 100 ng/mL) + cycloheximide (CHX; 20 μg/mL) for 6 hours. The apoptosis was assessed by Western blot (A) and cysteine-aspartic protease, cysteine aspartase (Caspase 3) activity assay (B). (C-D) HASMCs were transfected with siRNA-negative control (siCt; 30 nM) or siRNA-TFEB (siTFEB; 30 nM). After 72 hours, the cells were treated with FasL or TNFα+CHX for 6 hours. Similarly, the apoptosis was determined by Western blot (C) or Caspase 3 activity assay (D). (E-F) HASMCs were infected with Ad-LacZ or Ad-TFEB (MOI, 30) for 48 hours (E) or transfected with siCt or siTFEB (30 nM) for 72 hours (F), and then the cells were treated with TNFα + CHX for 4 hours before Annexin V/propidium iodide (PI) staining and flow cytometry analysis. Data are presented as mean ± SEM of three independent experiments. *p < 0.05, **p < 0.01 compared with Ad-LacZ or siCt group. Two-way ANOVA followed by Holm-Sidak post hoc analysis for A, B, C, D, E, F. Zvad: carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]- fluoromethylketone
TFEB inhibits HASMC apoptosis through B-cell lymphoma 2 (BCL2).
The apoptotic pathways include the extrinsic pathway (Caspase 8-dependent) and the intrinsic pathway (Caspase 9-dependent). TFEB knockdown significantly increased Caspase 9 cleavage but did not affect Caspase 8 and BH3 interacting domain death agonist (BID, a downstream effector of Caspase 8) (Supplemental Figure II B–C). The activation of the intrinsic pathway is coupled with the loss of mitochondria membrane potential. TFEB knockdown significantly increased the number of Deep Redhigh/TMRMlow cells, indicating that TFEB is critical for maintaining the mitochondria membrane potential (Supplemental Figure II D), which, in turn, is controlled by the BCL2 family proteins during apoptosis.12 Therefore, we determined the expression of the BCL2 family of genes in HASMCs with altered TFEB expression (Supplemental Figure II E–F). In overexpression and knockdown experiments, we found a direct correlation of TFEB with BCL2 expression (Figure 3A–D) but not with Bcl-2-associated X protein (BAX) (Supplemental Figure II E–F). Using chromatin immunoprecipitation sequencing (ChIP-Seq) (Supplemental Figure II G) and ChIP-qPCR (Figure 3E), we demonstrated the binding of TFEB to the human BCL2 promoter. TFEB increased BCL2 promoter-driven luciferase activity and mutation of the putative TFEB binding site diminished the TFEB-dependent effect (Figure 3F). It is noteworthy that pretreatment with BCL2 inhibitors ABT19913 or HA14–114 abolished the anti-apoptotic effect of TFEB in HASMCs, indicating an indispensable role of BCL2 in TFEB-dependent anti-apoptotic effects (Figure 3G).
Figure 3. TFEB inhibits apoptosis through upregulation of B-cell lymphoma 2 (BCL2) in human aortic smooth muscle cells (HASMCs).

(A and B) HASMCs were infected with adenovirus encoding LacZ (Ad-LacZ) or Ad-TFEB (MOI, 30) for 48 hours or (C and D) HASMCs were transfected with siRNA-control (siCt; 30 nM) or siRNA-TFEB (siTFEB; 30 nM) for 72 hours, BCL2 mRNA and protein were determined by quantitative polymerase chain reaction (qPCR) (A, C) or Western blot (B, D). (E) Chromatin immunoprecipitation (ChIP) assay was performed in HASMCs infected with Ad-LacZ or Ad-flag-TFEB (MOI, 30). The binding of TFEB to the BCL2 promoter was determined by qPCR. (F) CV1 cells were transfected with a human BCL2 promoter-driven luciferase vector containing a wild type (WT) or mutated TFEB binding site. After 24 hours, the cells were infected with Ad-LacZ and Ad-TFEB (MOI, 30). The luciferase activity was measured and normalized to Renilla activity. (G) HASMCs were infected with Ad-LacZ and Ad-TFEB (MOI, 30). After 48 hours, the cells were pretreated with ABT199 (200 nM) or HA14–1 (20 μM) for 1 hour followed by treatment with TNFα + CHX for 6 hours. Apoptosis was determined by Western blot. Data are presented as mean ± SEM of three independent experiments. *p < 0.05, **p < 0.01 compared with Ad-LacZ, siCt or IgG group. Unpaired t-test for A, B, C, D and two-way ANOVA followed by Holm-Sidak post hoc analysis for E, F and G.
TFEB inhibits apoptosis independent of autophagy.
TFEB has emerged as a master regulator of lysosomal biogenesis and autophagy. To determine whether autophagy mediates the anti-apoptotic effect of TFEB, we used autophagy inhibitors - Bafilomycin A1 (BFA) or 3-Methyladenine (3MA) to block autophagy and used TNFα + CHX, Fas ligand or oxidized low-density lipoprotein (oxLDL) to induce apoptosis in VSMCs. Both Fas ligand and oxLDL promote VSMC apoptosis and AAA formation15, 16. Under these different pro-apoptotic conditions, autophagy inhibitors (3MA and BFA) cannot abolish the anti-apoptotic effect of TFEB in HASMCs (Supplemental Figure III). Autophagy related 7 (ATG7) gene encodes an E1-like enzyme in the ubiquitin-like conjugation systems, which is essential for the autophagosome biogenesis. Knockdown of Atg7 cannot abolish the anti-apoptotic effect of TFEB either (Supplemental Figure IV A). Furthermore, we used siRNA against Beclin 1, which is required for autophagic cell death17, to block autophagy in HASMCs. Our results indicate that Beclin 1 knockdown cannot attenuate the anti-apoptotic effect of TFEB in HASMCs either (Supplemental Figure IV B–C). Collectively, neither pharmacological inhibition (3MA and BFA) nor genetic inactivation of autophagy (siAtg7 and siBeclin1) can attenuate the TFEB-dependent inhibitory effects on VSMC apoptosis in vitro.
Generation of VSMC selective Tfeb KO mice.
To determine the role of TFEB in VSMC biology in vivo, we generated VSMC selective Tfeb KO (TfebΔSMC) mice by crossbreeding Tfeb floxed (Tfebflox) mice with Myh11-cre/ERT2 mice (Supplemental Figure V A). The Tfeb knockout efficiency was determined by both qPCR and Western blot (Supplemental Figure V B–D). Consistent with the in vitro data, Tfeb knockout in vivo also significantly reduced BCL2 mRNA and protein abundance without altering BAX expression in the aorta (Supplemental Figure V B–D). The protein abundance of microphthalmia family members, specifically MITF and TFE3, in the aorta from Tfebflox and TfebΔSMC mice was not significantly changed under basal conditions (Supplemental Figure V E–F). Autophagy related 5 (ATG5) and 7 (ATG7) are critical for autophagy and the deficiency of Atg5 18 or Atg7 19 in VSMCs leads to AAA formation in mice. However, VSMC-Tfeb KO did not influence ATG5 or ATG7 protein abundance in the mouse aorta (Supplemental Figure V G–H).
VSMC-Tfeb KO aggravates aneurysm development and VSMC apoptosis in the PCSK9/AngII model.
We determined whether TFEB is crucial to maintain VSMC homeostasis in vivo using TfebΔSMC mice and multiple AAA murine models. Hyperlipidemia plus Angiotensin II (AngII) infusion is the most commonly used murine AAA model. AAV encoding PCSK9 D377Y (a gain-of-function mutation) was used to induce hyperlipidemia and combined with AngII infusion to induce AAA10 (Supplemental Figure VI B). Tfeb KO in VSMCs significantly increased both the maximal diameter of the abdominal aorta (1.51mm) and the incidence of the AAA (64%) compared with that in Tfebflox mice (maximal diameter of 1.182 mm and AAA incidence of 40.9%) after AngII infusion (Figure 4A–C, Supplemental Figure VII A). Blood pressure and plasma lipid profiles were comparable between Tfebflox mice and TfebΔSMC mice (Supplemental Figure VII B–C). Macrophage infiltration is critical for the development of AAA. We determined the macrophage infiltration in the mouse PCSK9/AngII model by immunostaining for Mac2, a macrophage marker. However, we did not observe a significant difference in the number of macrophages in the aorta between TfebΔSMC mice and control Tfebflox mice (Supplementary Figure VII D). VSMC-Tfeb KO increased elastin disruption and degradation (Figure 4D–E). Matrix metallopeptidase 2 (MMP2) and matrix metallopeptidase 9 (MMP9) cleave elastin, a major vascular extracellular matrix, contributes to the pathogenesis of aortic aneurysm. We determined the expression of MMP2/9 in the mouse aorta by Western blot and MMP2/9 activity was assessed by zymography. We found that VSMC-Tfeb KO increases MMP9 expression and activity, but it has no significant effects on MMP2 (Supplementary Figure VII E–F). VSMC-Tfeb KO promotes VSMC apoptosis assessed by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining in the aortic wall (Figure 4F–G). TFEB is a master regulator of autophagy in various cell types and tissues.20 We assessed autophagy in the mouse aorta by Western blot. TfebΔSMC mice showed impaired autophagy with decreased LC3 turnover (LC3-II/LC3-I) in the aorta (Supplementary Figure VII G).
Figure 4. Vascular smooth muscle cell (VSMC)-Tfeb KO aggravates abdominal aortic aneurysm (AAA) formation in the proprotein convertase subtilisin/kexin type 9 (PCSK9)/ angiotensin II (AngII) and β-aminopropionitrile (BAPN)/AngII models.
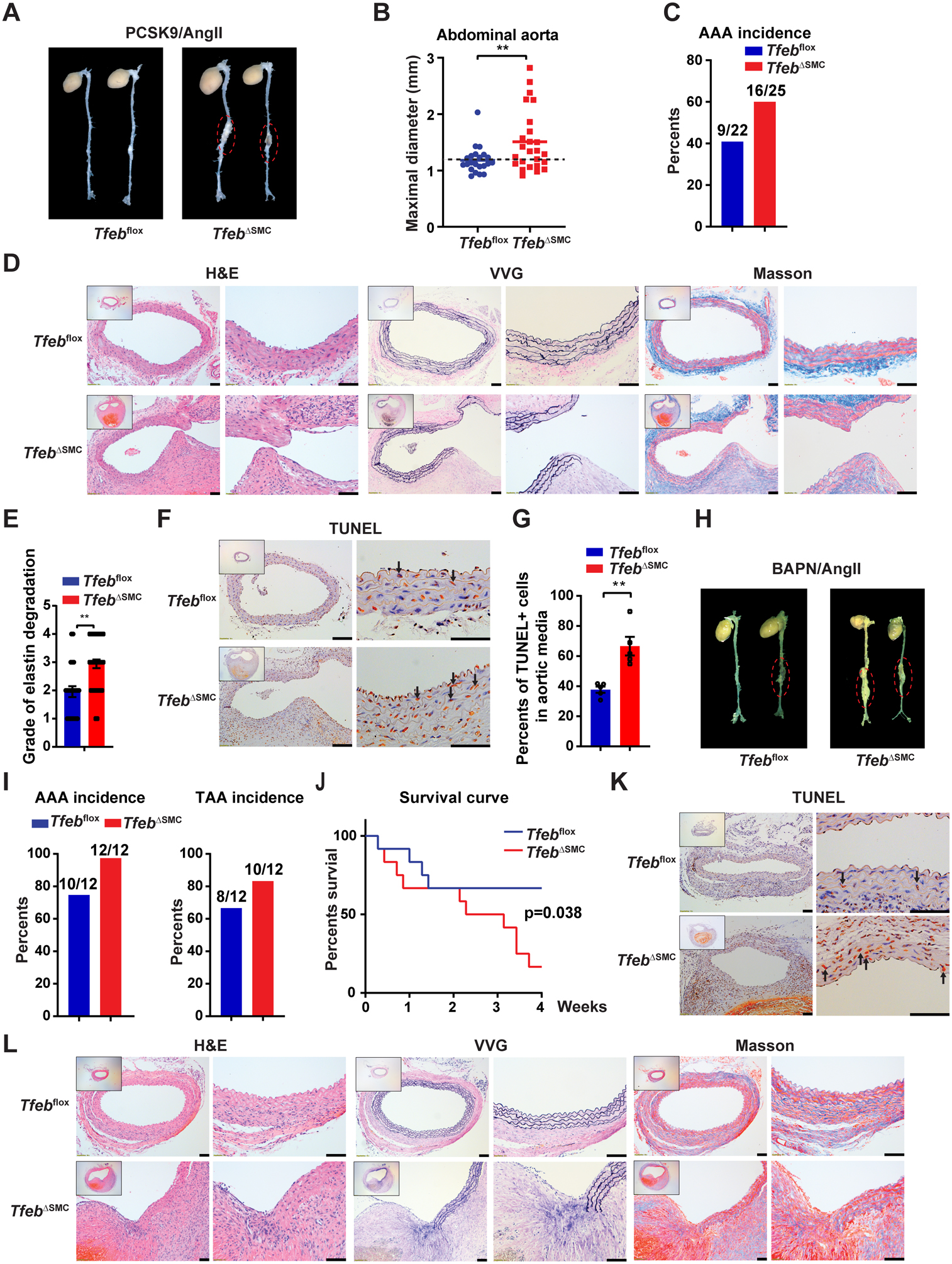
(A-G) In the PCSK9/AngII model, Tfebflox and TfebΔSMC mice were injected (i.p.) with adeno-associated virus (AAV)-PCSK9 D337Y and fed a western diet. After 2 weeks, the mice were infused with AngII (1,000 ng/kg/min) for another 4 weeks. (A) Representative images of AAA. (B) Maximal diameter of the abdominal aorta. (C) AAA incidence. (D) Representative hematoxylin and eosin (H&E), Verhoeff-Van Gieson (VVG) and Masson’s trichrome staining of the mouse aorta. (E) Grade of elastin degradation in the aortic wall. (F-G) Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining of the aortic wall. Normal cell nuclei are blue; apoptotic cell nuclei pare brown. Arrows point to apoptotic cells. The percentage of TUNEL positive nuclei in the aortic media was determined. In (A-E), n=22 for Tfebflox mice and n=25 for TfebΔSMC mice. (H-L) In the BAPN/AngII model, Tfebflox and TfebΔSMC mice were infused with both AngII (1,000 ng/kg/min, 4 weeks) and BAPN (150 mg/kg/min, during the first 2 weeks). (H) Representative images of AAA. (I) AAA and TAA incidence. (J) Survival curve. (K) TUNEL staining of the aortic wall. (L) Representative H&E, VVG and Masson’s trichrome staining of the mouse aorta. In (H-L), n=12 for each genotype. Low-magnification images in D, F, K and L show the entire vascular wall at the site of analysis. Data are presented as mean ± SEM. **p < 0.01. Mann-Whitney test for B, unpaired t-test for E and G, and Mantel-Cox method for J. Scale bar, 50 μm.
VSMC-Tfeb KO promotes aneurysm rupture and VSMC apoptosis in the β-aminopropionitrile (BAPN)/AngII model.
Unlike PCSK9/AngII model, BAPN/AngII (Supplemental Figure VI C) is known to induce both thoracic aortic aneurysm (TAA) and AAA in mice21. In this model, VSMC-Tfeb KO increased the incidence of both TAA and AAA (Figure 4H–I). Due to the high incidence of aneurysm and rupture in this model, we were able to observe that VSMC-Tfeb KO significantly reduced the survival rate (16.67%) compared with that in Tfebflox mice (66.67%) (Figure 4J). Consistently, Tfeb KO also increased apoptosis (Figure 4K) and elastin degradation (Figure 4L) in the aorta.
VSMC-Tfeb KO induces AAA and VSMC apoptosis upon infusion of AngII alone.
Aortic aneurysms rarely occur in wild type mice (on a chow diet) infused with AngII alone.19 In this condition, with AngII- infusion only (Supplemental Figure VI D), VSMC-Tfeb KO significantly increased the maximal diameter of the abdominal aorta (Figure 5A–B) with a higher incidence of AAA (53.3%) and rupture rate (16.7%) compared with those in Tfebflox mice (incidence of 9.1%, rupture rate of 0%, respectively) (Figure 5C and Supplemental Figure VIII A). Interestingly, the increased aneurysm incidence and maximal diameter of the abdominal aorta can be attenuated by treatment with the apoptosis inhibitor Quinoline-Val-Asp-Difluorophenoxymethylketone (Q-VD-OPh) in vivo (Figure 5A–C). There was no significant difference in plasma lipid profiles (Supplemental Figure VIII B) and systolic blood pressure (Supplemental Figure VIII C) among these three groups. Histologic analysis indicated that VSMC-Tfeb KO increases elastin disruption and degradation in the aortic wall (Figure 5D–E) and TUNEL staining showed an increase of apoptotic cells in the aortic wall of TfebΔSMC mice (Figure 5F). The elastin disruption and apoptosis in vivo can also be rescued by Q-VD-OPh treatment (Figure 5D–F), emphasizing that apoptosis is a key feature mediating the adverse effects of VSMC-Tfeb deficiency in AAA formation.
Figure 5. Vascular smooth muscle cell (VSMC)-Tfeb KO promotes abdominal aortic aneurysm (AAA) in angiotensin II (AngII) infusion-induced hypertension model.
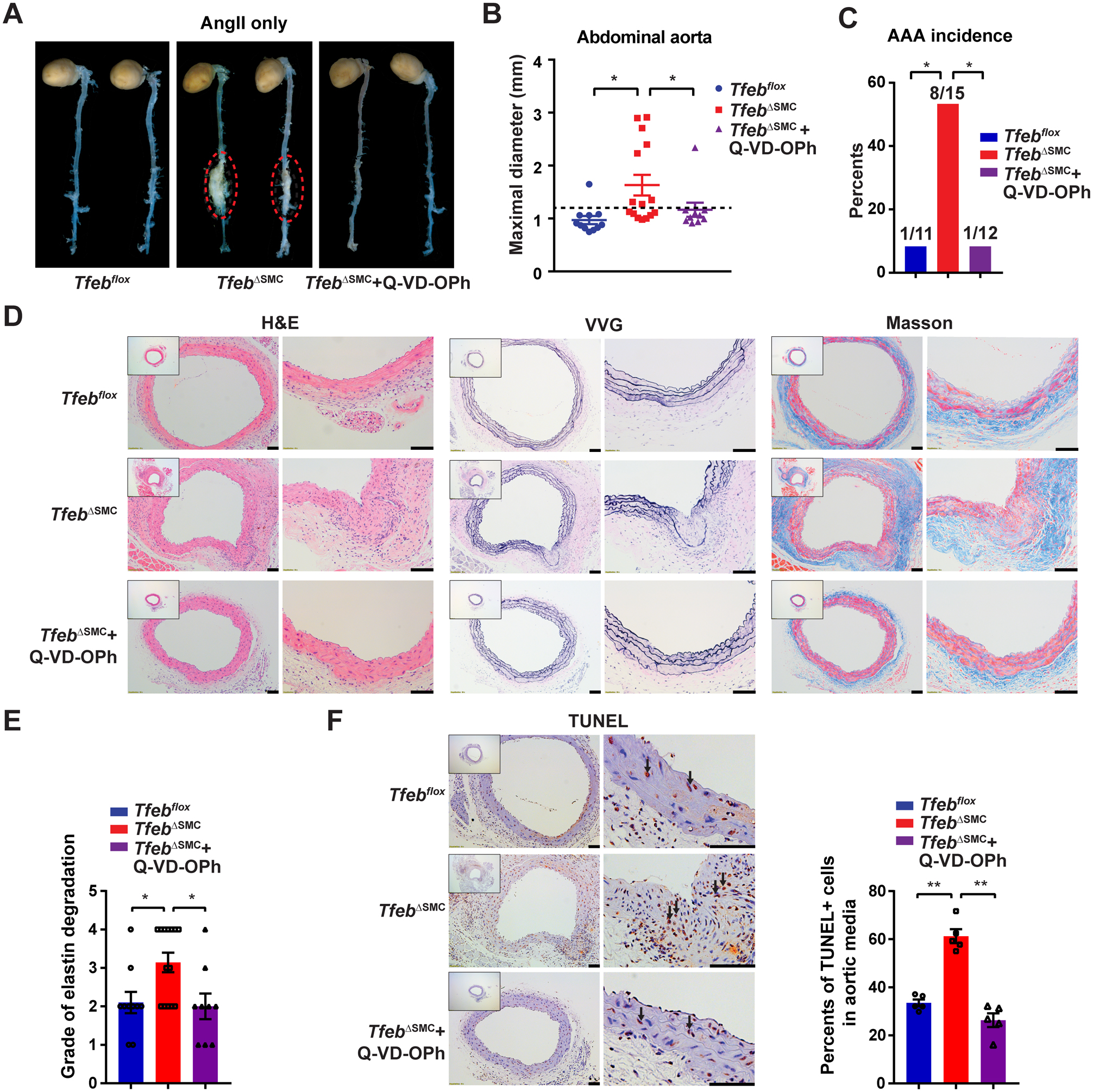
WT (Tfebflox) mice and VSMC-Tfeb KO (TfebΔSMC) mice were infused with AngII (1,000 ng/kg/min) subcutaneously for 8 weeks to induce AAA, and simultaneously administered vehicle control (DMSO) or apoptosis inhibitor Q-VD-OPh (20 mg/kg daily, i.p.) starting one day before AngII infusion. (A) Representative images of the mouse AAA. (B) Maximal diameter of the abdominal aorta. (C) AAA incidence. (D) Representative H&E, VVG and Masson’s trichrome staining of the mouse aorta. (E) Grade of the elastin degradation in the aortic wall. (F) The apoptotic cells were assessed by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining in the aortic wall. Normal cell nuclei are blue; apoptotic cell nuclei are brown. Arrows point to apoptotic cells. The percentage of TUNEL positive nuclei in the aortic media was determined. Low-magnification images in D and F show the entire vascular wall at the site of analysis. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01. Kruskal-Wallis test followed by a two-stage step-up method of Benjamini, Krieger and Yekutieli for B, Fisher’s exact test for C, one-way ANOVA followed by Holm-Sidak post hoc analysis for E and F. (A-E) n=11, 15, 12 for the Tfebflox, TfebΔSMC and TfebΔSMC + Q-VD-OPh group, respectively. Scale bar, 50 μm.
2-hydroxypropyl-β-cyclodextrin (HPβCD) inhibits VSMC apoptosis through activation of TFEB.
TFEB activators induce TFEB nuclear translocation and enhance TFEB transcriptional activity. We determined whether TFEB activators such as HPβCD,22 alexidine dihydrochloride (AD),23 curcumin,24 and naringenin25 could activate TFEB in VSMCs. Although all of them significantly increased TFEB nuclear translocation in HASMCs at the concentrations published previously,22–25 (Supplemental Figure IX A–B), only HPβCD potently inhibits VSMC apoptosis (Supplemental Figure IX C). We further measured apoptosis in VSMCs treated with HPβCD at different doses (6, 8, 10 mg/mL) and found that HPβCD inhibits apoptosis in a dose-dependent manner (Figure 6A). TFEB knockdown abolished the anti-apoptotic effect of HPβCD, indicating an essential role of TFEB in the HPβCD-dependent inhibition of apoptosis (Figure 6B). BCL2 is a direct target of TFEB in VSMCs (Figure 3). HPβCD significantly increased BCL2 at both mRNA and protein levels, and TFEB knockdown abolished the HPβCD effect on BCL2 expression in VSMCs (Figure 6C–D).
Figure 6. 2-hydroxypropyl-beta-cyclodextrin (HPβCD) activates TFEB by reducing intracellular cholesterol.
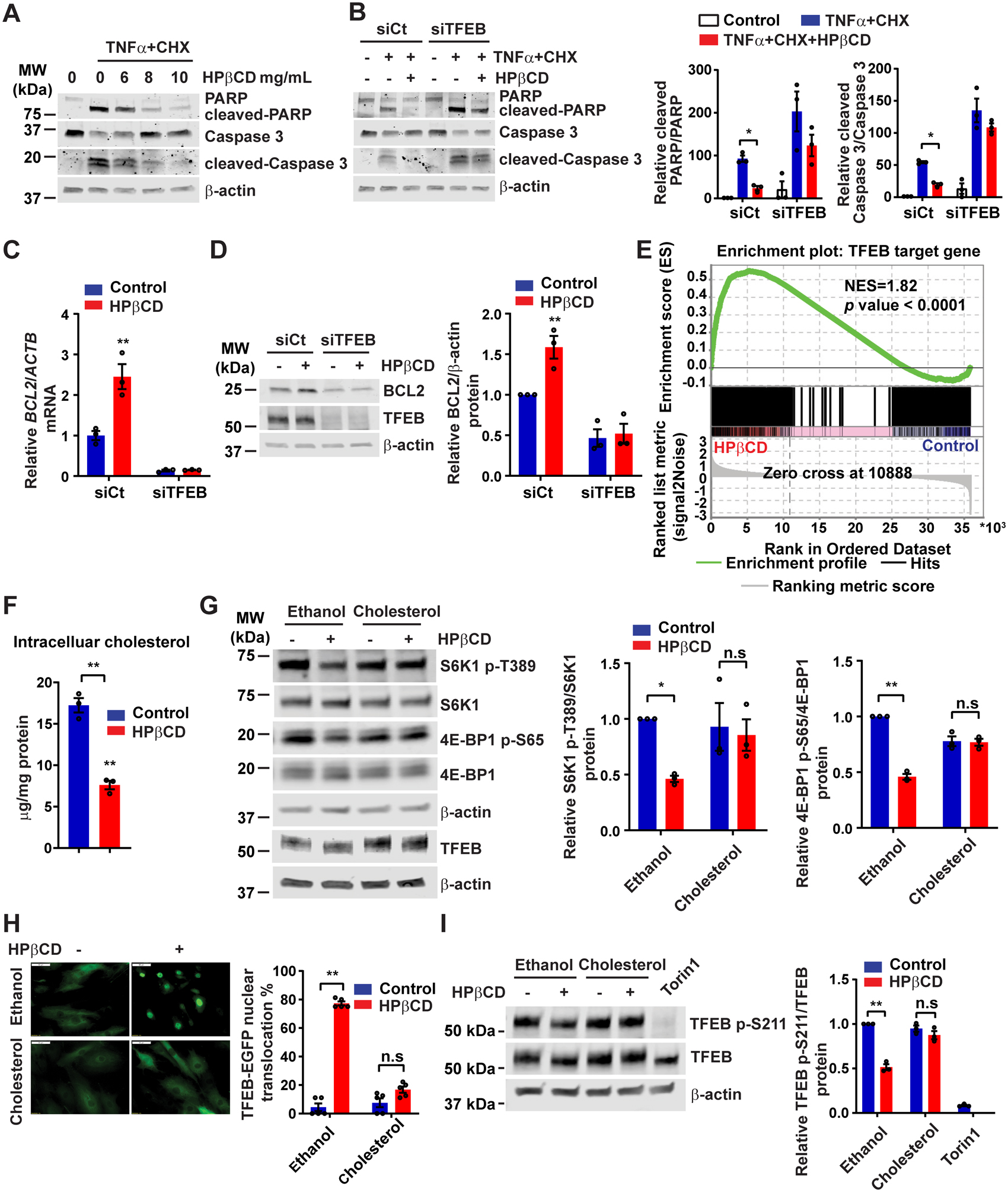
(A) Human aortic smooth muscle cells (HASMCs) were treated with HPβCD at the indicated concentration for 24 hours and then treated with tumor necrosis factor α (TNFα; 100 ng/mL) + cycloheximide (CHX; 20 μg/mL) for 6 hours. The cleavage of poly ADP-ribose polymerase (PARP) and Caspase 3 was determined by Western blot. (B) HASMCs were transfected with siCt or siTFEB (30 nM). After 48 hours, the cells were treated with HPβCD (10 mg/mL) for 24 hours and subsequently treated with TNFα + CHX as in (A). (C-D) HASMCs were transfected with siCt or siTFEB (30 nM). After 48 hours, the cells were treated with HPβCD (10 mg/mL) for 24 hours. B-cell lymphoma 2 (BCL2) expression was determined by quantitative polymerase chain reaction (qPCR) (C) and Western blot (D). (E) HASMCs were treated with HPβCD (10 mg/mL) for 24 hours and RNA was extracted for RNA-sequencing. Gene Set Enrichment Analysis (GSEA) was performed to determine the enrichment of TFEB target genes in the HPβCD-treated group. (F) Intracellular cholesterol was measured in the HASMCs treated with HPβCD (10 mg/mL) for 24 hours. (G) HASMCs were treated with HPβCD (10 mg/mL) in the presence or absence of cholesterol (20 μg/mL) for 6 hours. Mechanistic target of rapamycin complex 1 (mTORC1) activity was determined by Western blot. (H) HASMCs were infected with adenovirus encoding TFEB-enhanced green fluorescent protein (Ad-TFEB-EGFP; MOI, 20), and 24 hours later, the cells were treated with HPβCD (10 mg/mL) in the presence or absence of cholesterol (20 μg/mL) for 6 hours. The TFEB nuclear translocation was analyzed by fluorescent microscopy. (I) HASMCs were infected with Ad-TFEB (MOI, 10), and 48 hours later, the cells were treated with HPβCD (10 mg/mL) in the presence or absence of cholesterol (20 μg/mL) for 6 hours. The phosphorylation of TFEB on Ser211 was determined by Western blot. Data are presented as mean ± SEM of three independent experiments. *p < 0.05, **p < 0.01; n.s: not significant. Two-way ANOVA followed by Holm-Sidak post hoc analysis for B, C, D, G, H, I and unpaired t-test for F. Scale bar, 50 μm.
To gain a more comprehensive understanding of the role of HPβCD in VSMCs, we performed RNA-sequencing of HASMCs treated with HPβCD. We first identified TFEB target genes by merging the differentially expressed genes regulated by TFEB overexpression (by microarray analysis) (Supplemental Table III) and the genes harboring TFEB binding peaks around the transcription start site (TSS) (by ChIP-Seq analysis) (Supplemental Table IV). In total, 740 genes were identified as TFEB target genes in HASMCs (Supplemental Table V). Subsequently, we performed gene set enrichment analysis (GSEA) on the RNA-seq results of HPβCD-treated HASMCs (Supplemental Table VI) with the established TFEB target gene dataset (Supplemental Figure X A). GSEA uncovered that TFEB target genes are highly enriched in the HPβCD treated group with a normalized enrichment score (NES) 1.82 and P value < 0.0001 (Figure 6E). To gain insight into the potential role of TFEB in VSMCs, we performed pathway enrichment analysis of TFEB target genes (740 genes) (Supplemental Table VII). The cellular component analysis suggests that the TFEB target genes most likely belong to lysosome and vacuole-associated genes (Supplemental Figure X B). We also revealed that the top biological pathways with the lowest P value include transmembrane transport (P = 0.0001), ion transport (P = 0.0002) and lipid metabolic process (P = 0.0005) (Supplemental Figure X B). Among the pathways regulated by TFEB in VSMCs, the genes related to lysosome and negative regulator of apoptosis are displayed in the heatmap (Supplemental Figure X C). Besides BCL2, HPβCD also upregulated autophagic TFEB target genes such as alpha-galactosidase A (GLA) and Sequestosome 1 (SQSTM1) as well as the marker for autophagic flux (LC3II/LC3I ratio) in a TFEB-dependent manner (Supplemental Figure X D–E). These data indicate that, as a TFEB activator, HPβCD inhibits apoptosis and shares common downstream genes and pathways with TFEB in VSMCs.
HPβCD activates TFEB by depleting intracellular cholesterol.
Subcellular localization of TFEB is mainly controlled by the status of its phosphorylation at multiple sites such as Ser142 and Ser211.26 Phosphorylated TFEB is retained in the cytoplasm, whereas dephosphorylated TFEB translocates into the nucleus and regulates target gene expression. HPβCD can remove intracellular cholesterol by enhancing cholesterol transport.27 Reduced intracellular cholesterol inhibits mechanistic target of rapamycin complex 1 (mTORC1) activity,28 whereby HPβCD could induce TFEB nuclear translocation. To address this hypothesis, we measured intracellular cholesterol and mTORC1 activity in the HPβCD-treated VSMCs. Our data indicate that HPβCD reduces intracellular cholesterol (Figure 6F) and decreases the phosphorylation of ribosomal protein S6 kinase beta-1 (S6K1) and eIF4E-binding proteins (4E-BP1), two downstream effectors of mTORC1 (Figure 6G). Conversely, addition of cholesterol can attenuate the ability of HPβCD to inhibit mTORC1 activity (Figure 6G) and to induce TFEB nuclear translocation (Figure 6H). We also observed that HPβCD increased the mobility of TFEB in the gel, suggesting an alteration of post-translational modifications in TFEB protein (Figure 6G). Consistently, HPβCD reduced TFEB phosphorylation at Ser211 (Figure 6I), a site required for inhibition of TFEB nuclear translocation. Torin1, a mTORC1 inhibitor, was used as a positive control.
HPβCD inhibits AAA formation and progression.
We chose HPβCD to further explore the therapeutic potential of TFEB activation in AAA (Supplemental Figure VI E). In the PSCK9/AngII AAA model, we increased the dose of AngII from 1,000 ng/kg/min to 1,500 ng/kg/min to obtain higher AAA incidence.29 After 4 weeks, HPβCD significantly reduced the maximal diameter of the abdominal aorta and AAA (Figure 7A–C). HPβCD also reduced the degradation of elastin (Figure 7D) and apoptosis (Figure 7E) in the aortic wall. HPβCD treatment did not significantly change body weight, plasma lipid profiles or systolic blood pressure (Supplemental Figure XI A–C). Consistent with the in vitro results showing that HPβCD increased LC3-II/LC3-I ratio in HASMCs (Supplemental Figure X E), administration of HPβCD also enhances autophagy in the aorta in the PCSK9/AngII model (Supplementary Fig XI D). Clearly, it would be of higher clinical relevance to reverse or halt the progression of aortic aneurysm. To address this important issue, we treated the mice with HPβCD after the AAA had been established, starting at week 5 in the PCSK9/AngII model (Supplemental Figure VI F). The diameter of the suprarenal aorta (where most of the aneurysms occur in this model) was monitored by ultrasound imaging30 before and after HPβCD treatment (Supplemental Figure XII A). By analysis of the changes in the maximal diameter of the suprarenal aorta, we found that HPβCD significantly inhibited AAA progression (Supplemental Figure XII B, Figure 7F). To determine whether HPβCD exerts its protective effects against AAA formation mainly through TFEB, we treated Tfebflox mice and TfebΔSMC mice with HPβCD in the PCSK9/AngII AAA model (Supplemental Figure VI G). HPβCD treatment significantly reduced the maximal diameter of the abdominal aorta, AAA incidence and apoptosis in the aortic wall in Tfebflox mice but not in TfebΔSMC mice (Figure 7G–J), indicating that HPβCD inhibited AAA in a TFEB-dependent manner. Taken together, these data demonstrated that HPβCD activates TFEB in VSMCs to inhibit apoptosis and AAA.
Figure 7. 2-hydroxypropyl-beta-cyclodextrin (HPβCD) inhibits abdominal aortic aneurysm (AAA) formation in a vascular smooth muscle cell (VSMC) TFEB-dependent manner.

(A-E) In the proprotein convertase subtilisin/kexin type 9 (PCSK9)/angiotensin II (AngII) model, after inducing hyperlipidemia by AAV-PCSK9 D337Y and western diet, C57BL/6J mice were infused with AngII (1,500 ng/kg/min) for another 4 weeks. Saline (vehicle control) or HPβCD (2 g/kg) were administered (i.p.) twice a week, starting one day before AngII infusion. (A) Representative images of abdominal aortic aneurysm (AAA). (B) Maximal diameter of the abdominal aorta. (C) AAA incidence. (D) Representative H&E, VVG and Masson’s trichrome staining of the mouse aortas. (E) Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining of the aortic wall. In (A-D) n=10 for each group. Low-magnification images in D and E show the entire vascular wall of normal aorta or AAA. (F) To study the aortic aneurysm progression, AAA was first induced in C57BL/6J mice by the method of PCSK9/AngII (1,500 ng/kg/min). The mice were then randomly divided into 2 groups: saline and HPβCD (2 g/kg, i.p. twice a week). The aorta was assessed by ultrasound at week 4 and week 8 after AngII infusion. The change in the internal diameter of the suprarenal aorta was calculated as [diameter (week 8)] – [diameter (week 4)]. In (F), n=9 for each group. (G-J) In PCSK9/AngII model, Tfebflox mice and TfebΔSMC were treated with either saline or HPβCD (2 g/kg, i.p.) starting one day before AngII infusion. (G) Representative images of AAA. (H) Maximal diameter of the abdominal aorta. (I) AAA incidence. (J) TUNEL staining of the aortic wall. In (G-I), n=19 for Tfebflox + Saline, n=12 for Tfebflox + HPβCD, n=16 for TfebΔSMC + Saline and n=13 for TfebΔSMC + HPβCD. (K) Proposed model for TFEB as a therapeutic target in AAA. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01; n.s, not significant. Mann-Whitney test for B, unpaired t-test for E and F, Kruskal-Wallis test followed by two-stage step-up method of Benjamini, Krieger and Yekutieli for H, and two-way ANOVA followed by Holm-Sidak post hoc analysis for J. Scale bar, 50 μm.
Discussion:
AAA is a severe aortic disease with a high risk of rupture. There is an urgent need to discover pharmacologic agents and approaches to prevent, inhibit or even reverse the progression of aortic aneurysm. In this study, we demonstrated that VSMC-Tfeb deficiency aggravates AAA formation in vivo and that HPβCD, a compound approved by FDA for use in humans, limits aneurysm enlargement via activation of TFEB, indicating that pharmacological modulation of TFEB with HPβCD may be a feasible approach for the treatment of AAA (Figure 7K).
Multiple murine AAA models have been developed for the study of human AAA pathogenesis. In the present study, we used three AAA models to demonstrate the inhibitory effect of TFEB on AAA. Injection of AAV encoding PCSK9-D377Y (a gain-of-function mutation) and Western diet feeding can induce hyperlipidemia.10 AAA induced by AngII infusion plus hyperlipidemia recapitulates the major features of human AAA.31 BAPN, a potent lysyl oxidase inhibitor, disrupts the crosslinking of procollagens and tropoelastin and destroys the integrity of the aortic wall.32 Infusion of BAPN together with AngII induces high incidence and rupture of aortic aneurysms.33 The AngII-only infusion model is commonly used as a mouse hypertension model,34 in which the incidence of AAA is very low in wild type mice without additional stimuli such as hyperlipidemia or BAPN.35 In this model, only a few genes such as Atg7 and Timp3 have been reported to induce AAA.19, 36 We found that VSMC-Tfeb KO results in a high incidence (> 50%) of AAA in this model, highlighting the indispensable protective effects of TFEB in VSMC biology and AAA.
VSMCs undergo phenotypic switch during the development of AAA in mice37. We determined the TFEB expression in the SM22 positive cells and found that TFEB protein is reduced in the VSMCs. However, whether TFEB expression in other cell types like macrophages and fibroblasts contributes to the overall reduced levels of TFEB in the AAA lesion remains to be investigated. Of note, TFEB has protective effects on the vasculature at large, including on endothelial cells and in other vascular diseases like atherosclerosis.6
VSMC apoptosis is limited in the healthy aorta but becomes prominent during the development of aortic aneurysm in humans.38 Inhibition of VSMC apoptosis remains a promising strategy to halt aneurysm development.39 In our study, inhibition of apoptosis by Q-VD-OPh significantly reduced the Tfeb KO-induced AAA and VSMC apoptosis in the AngII-only infusion model (Figure 5), and TFEB activation by HPβCD inhibits AAA and VSMC apoptosis in vivo (Figure 7). We revealed a critical protective effect of TFEB on VSMC survival and uncovered a novel TFEB-dependent pathway for preventing AAA. The BCL2 family includes critical mediators in the intrinsic apoptotic pathway. In human aortic aneurysm lesions, BCL2 expression is decreased and negatively correlated with VSMC apoptosis.40 In this study, we uncovered that the upregulation of BCL2 is required for the TFEB-mediated anti-apoptotic effect in VSMCs.
TFEB is a master regulator of lysosomal biogenesis and autophagy. Autophagy either inhibits or activates apoptosis dependent on the cell type and stress intensity.41 Recent studies suggest that defective autophagy increases VSMC death in vivo.37, 42 We found that VSMC-Tfeb KO impairs autophagy in the aorta (Supplementary Figure VII G). Thus, the VSMC apoptosis induced by VSMC-Tfeb KO may result from BCL2-dependent apoptotic pathways and defective autophagy in vivo. Noteworthy, we found that administration of apoptosis inhibitor (Q-VD-OPh) in vivo can rescue the VSMC-Tfeb KO-induced VSMC apoptosis and AAA formation, indicating that inhibition of apoptosis is at least one of the critical mechanisms mediating the effects of TFEB.
Of note, besides autophagy and lysosome genes, TFEB also regulates numerous target genes in human aortic VSMCs as shown in Supplemental Table V. Through pathway enrichment analysis of TFEB target genes, we uncovered, specifically, that TFEB target genes are overrepresented within the membrane transport and lipid metabolism category (Supplemental Figure X B). Transportation and exchange of biological molecules and waste products are necessary to maintain cellular homeostasis. LDL uptake and lipid accumulation in VSMCs has pivotal roles in atherosclerosis formation. TFEB may have diverse effects on VSMCs beyond autophagy and apoptosis and modulate VSMC dysfunction and vascular disorders. The bioinformatics analysis of TFEB target genes provided new insights on the functional study of TFEB in VSMCs. Furthermore, TFEB is expressed not only in VSMCs but also in endothelial cells and macrophages. Normal endothelial cells and macrophages are necessary to protect against AAA formation. In either endothelial cells or macrophages, TFEB may influence AAA as well, which warrants future investigation. However, we found that, in the context of our models, HPβCD can inhibit AAA formation and VSMC apoptosis in a VSMC TFEB-dependent manner (Figure 7). Our studies shed new light on the comprehensive understanding of the TFEB-dependent transcriptional regulation of genes and the diverse biological functions of TFEB in the vascular system.
TFEB can be phosphorylated by intracellular kinases, such as extracellular signal-regulated kinase (ERK)26 and mTORC1,26 whereby TFEB is retained in the cytoplasm. Once TFEB is dephosphorylated by calcineurin,43 it will translocate to the nucleus. HPβCD is an FDA-approved agent to increase the solubility and delivery of lipophilic drugs. Our data suggest that HPβCD activates TFEB by, at least in part, reducing mTORC1 activity in HASMCs. HPβCD decreased intracellular cholesterol (Figure 6F), which could subsequently, inactivated mTORC1 in VSMCs.28 Having a hydrophilic outer surface and a lipophilic central cavity, HPβCD forms inclusion complexes with many lipophilic molecules. Pre-incubation of HPβCD with cholesterol before adding to the cultured VSMCs may have impaired the capability of HPβCD to enter cells, and subsequently attenuated the ability of HPβCD to reduce intracellular cholesterol. Pharmacokinetic analysis using fluorescein isothiocyanate (FITC)-HPβCD indicated that HPβCD undergoes rapid compartmentalization (calculated by the volume of distribution) within the first minute after intravenous administration and is quickly endocytosed by tissues and cells.44 Endocytosed cyclodextrin can reduce the intracellular cholesterol storage by acting from inside endosome/lysosome-like storage organelles rather than by removing cholesterol from the plasma membrane.45 In this manner, cyclodextrin continues to be effective in reducing intracellular cholesterol for several days after extracellular cyclodextrin has been removed.45
Although some TFEB activators can induce TFEB nuclear translocation, they did not inhibit VSMC apoptosis as TFEB overexpression did (Supplemental Figure IX C). The discrepancy may result from the complicated downstream signaling pathways, beyond TFEB, elicited by those drugs. On the other hand, HPβCD potently inhibits VSMC apoptosis in dose- and TFEB-dependent manners. Niemann-Pick Type C disease (NPC) is a recessive lysosomal storage disorder characterized by the accumulation of unesterified cholesterol in lysosome. Animal studies suggest that HPβCD protects against NPC disease46 and atherosclerosis progression47. HPβCD also promotes the clearance of ceroid lipopigment in fibroblasts22 and suppresses M2 polarization in the tumor microenvironment48. Because of its effectiveness and safety, Intravenous Trappsol Cyclo (HPβCD) is in phase II clinical trial to treat NPC1 disease (ClinicalTrials.gov Identifier: NCT01747135, NCT02912793). The in vivo results showed that HPβCD inhibits AAA formation through activation of VSMC TFEB (Figure 7H). Our findings here underscore the potential use of HPβCD for pharmacological activation of TFEB in VSMCs for the treatment of aortic aneurysms.
In conclusion, we demonstrated a protective effect of TFEB against VSMC apoptosis and AAA. Based on our findings, we revealed molecular mechanisms underlying the TFEB-dependent inhibition of AAA and demonstrated that the TFEB activator HPβCD could be used as a new therapeutic approach for AAA.
Supplementary Material
Clinical perspective.
What is new?
TFEB expression is reduced in human aneurysms.
Vascular smooth muscle cell (VSMC)-selective Tfeb knockout promotes abdominal aortic aneurysm (AAA) development via induction of VSMC apoptosis in mice.
The TFEB activator 2-hydroxypropyl-beta-cyclodextrin (HPβCD) inhibits AAA formation in a TFEB-dependent manner.
What are the clinical implications?
We identified that HPβCD, a compound rendered safe and currently under clinical trial for Niemann-Pick type C1 disease, could prevent AAA formation and progression by activating TFEB.
TFEB could serve as a novel therapeutic target for the prevention and non-surgical treatment of AAA.
Acknowledgments:
Author Contributions: H. Lu, J. Sun, W. Liang and Y. Fan performed experiments and analyzed results; H. Lu and Y. Fan wrote the article; Z. Chang, O. Rom, Y. Zhao, G. Zhao, W. Xiong, H. Wang, T. Zhu, Y. Guo, L. Chang, M.T. Garcia-Barrio, J. Zhang provided the technical support and contributed to the discussion of the project and article. M.T. Garcia-Barrio did critical editing; Y. Fan and Y.E. Chen designed research and discussed results.
Sources of Funding: This study is supported by National Institutes of Health grants R01HL138094 and R01HL145176 (Y. Fan), R01HL068878, R01HL137214, and R01HL134569 (Y.E. Chen), R01HL138139 (J. Zhang), and American Heart Association grants 17PRE33400179 (H. Lu) and 18PRE34000005 (W. Liang).
Nonstandard Abbreviations and Acronyms
- AAA
Abdominal aortic aneurysm
- AngII
Angiotensin II
- BAPN
β-aminopropionitrile
- BAX
Bcl-2-associated X protein
- BCL2
B-cell lymphoma 2
- BFA
Bafilomycin A1
- Caspase
Cysteine-aspartic protease, cysteine aspartase
- ChIP
Chromatin immunoprecipitation
- CHX
Cycloheximide
- FasL
Fas ligand
- GEO
Gene Expression Omnibus
- GO
Gene ontology
- HASMC
Human aortic smooth muscle cell
- H&E
Hematoxylin and eosin
- HPβCD
2-hydroxypropyl-beta-cyclodextrin
- IL1β
Interleukin 1 β
- IFNγ
Interferon γ
- LC3
Microtubule-associated protein 1A/1B-light chain 3
- 3MA
3-Methyladenine
- MITF
Microphthalmia transcription factor
- MMP
Matrix metallopeptidase
- MOI
multiplicity of infection
- mTORC1
Mechanistic target of rapamycin complex 1
- NES
Normalized enrichment score
- NPC1
Niemann-Pick disease Type C1
- oxLDL
Oxidized low-density lipoprotein
- PARP
Poly ADP-ribose polymerase
- PCSK9
Proprotein convertase subtilisin/kexin type 9
- Q-VD-OPh
Quinoline-Val-Asp-Difluorophenoxymethylketone
- TAA
Thoracic aortic aneurysm
- TFEB
Transcription factor EB
- TMRM
Tetramethylrhodamine
- TNFα
Tumor necrosis factor α
- TUNEL
Terminal deoxynucleotidyl transferase dUTP nick end labeling
- VSMC
Vascular smooth muscle cell
- VVG
Verhoeff-Van Gieson
- Zvad
Carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]- fluoromethylketone
Footnotes
Disclosures: Authors declare no competing interests.
Supplemental Materials
Supplemental Methods
References:
- 1.Kent KC. Clinical practice. Abdominal aortic aneurysms. New Eng J Med. 2014;371:2101–2108. doi: 10.1056/NEJMcp1401430 [DOI] [PubMed] [Google Scholar]
- 2.Chaikof EL, Dalman RL, Eskandari MK, Jackson BM, Lee WA, Mansour MA, Mastracci TM, Mell M, Murad MH, Nguyen LL, et al. The Society for Vascular Surgery practice guidelines on the care of patients with an abdominal aortic aneurysm. J Vasc Surg. 2018;67:2–77 e2. doi: 10.1016/j.jvs.2017.10.044 [DOI] [PubMed] [Google Scholar]
- 3.Rowe VL, Stevens SL, Reddick TT, Freeman MB, Donnell R, Carroll RC and Goldman MH. Vascular smooth muscle cell apoptosis in aneurysmal, occlusive, and normal human aortas. J Vasc Surg. 2000;31:567–576. doi: 10.1067/mva.2000.102847 [DOI] [PubMed] [Google Scholar]
- 4.Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. doi: 10.1126/science.1204592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Decressac M and Bjorklund A. TFEB: Pathogenic role and therapeutic target in Parkinson disease. Autophagy. 2013;9:1244–1246. doi: 10.4161/auto.25044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu H, Fan Y, Qiao C, Liang W, Hu W, Zhu T, Zhang J and Chen YE. TFEB inhibits endothelial cell inflammation and reduces atherosclerosis. Sci Signal. 2017;10: eaah4214. doi: 10.1126/scisignal.aah4214 [DOI] [PubMed] [Google Scholar]
- 7.Fan Y, Lu H, Liang W, Garcia-Barrio MT, Guo Y, Zhang J, Zhu T, Hao Y, Zhang J and Chen YE. Endothelial TFEB (Transcription Factor EB) Positively Regulates Postischemic Angiogenesis. Circ Res. 2018;122:945–957. doi: 10.1161/circresaha.118.312672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evans TD, Jeong SJ, Zhang X, Sergin I and Razani B. TFEB and trehalose drive the macrophage autophagy-lysosome system to protect against atherosclerosis. Autophagy. 2018;14:724–726. doi: 10.1080/15548627.2018.1434373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wirth A, Benyó Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horváth B, Maser-Gluth C, Greiner E, et al. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med. 2008;14:64–68. doi: 10.1038/nm1666 [DOI] [PubMed] [Google Scholar]
- 10.Lu H, Howatt DA, Balakrishnan A, Graham MJ, Mullick AE and Daugherty A. Hypercholesterolemia Induced by a PCSK9 Gain-of-Function Mutation Augments Angiotensin II-Induced Abdominal Aortic Aneurysms in C57BL/6 Mice-Brief Report. Arterioscler thromb vasc biol. 2016;36:1753–1757. doi: 10.1161/atvbaha.116.307613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakaoka H, Tajima A, Yoneyama T, Hosomichi K, Kasuya H, Mizutani T and Inoue I. Gene expression profiling reveals distinct molecular signatures associated with the rupture of intracranial aneurysm. Stroke. 2014;45:2239–2245. doi: 10.1161/strokeaha.114.005851 [DOI] [PubMed] [Google Scholar]
- 12.Kale J, Osterlund EJ and Andrews DW. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ. 2017;25:65. doi: 10.1038/cdd.2017.186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vandenberg CJ and Cory S. ABT-199, a new Bcl-2–specific BH3 mimetic, has in vivo efficacy against aggressive Myc-driven mouse lymphomas without provoking thrombocytopenia. Blood. 2013;121:2285–2288. doi: 10.1182/blood-2013-01-475855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skommer J, Wlodkowic D, Mättö M, Eray M and Pelkonen J. HA14-1, a small molecule Bcl-2 antagonist, induces apoptosis and modulates action of selected anticancer drugs in follicular lymphoma B cells. Leuk Res. 2006;30:322–331. doi: 10.1016/j.leukres.2005.08.022 [DOI] [PubMed] [Google Scholar]
- 15.Liu Z, Fitzgerald M, Meisinger T, Batra R, Suh M, Greene H, Penrice AJ, Sun L, Baxter BT and Xiong W. CD95-ligand contributes to abdominal aortic aneurysm progression by modulating inflammation. Cardiovasc Res. 2018;115:807–818. doi: 10.1093/cvr/cvy264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hsieh C-C, Yen M-H, Yen C-H and Lau Y-TJCr. Oxidized low density lipoprotein induces apoptosis via generation of reactive oxygen species in vascular smooth muscle cells. 2001;49:135–145. [DOI] [PubMed] [Google Scholar]
- 17.Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH and Lenardo MJ. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–1502. doi: 10.1126/science.1096645 [DOI] [PubMed] [Google Scholar]
- 18.Clément M, Chappell J, Raffort J, Lareyre F, Vandestienne M, Taylor Annabel L, Finigan A, Harrison J, Bennett Martin R, Bruneval P, et al. Vascular Smooth Muscle Cell Plasticity and Autophagy in Dissecting Aortic Aneurysms. Arterioscler thromb vasc biol. 2019;39:1149–1159. doi: 10.1161/ATVBAHA.118.311727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramadan A, Singh KK, Quan A, Plant PJ, Al-Omran M, Teoh H and Verma S. Loss of vascular smooth muscle cell autophagy exacerbates angiotensin II-associated aortic remodeling. J Vasc Surg. 2018;68:859–871. doi: 10.1016/j.jvs.2017.08.086 [DOI] [PubMed] [Google Scholar]
- 20.Napolitano G and Ballabio A. TFEB at a glance. J Cell Sci. 2016;129:2475–2481. doi: 10.1242/jcs.146365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanematsu Y, Kanematsu M, Kurihara C, Tsou T-L, Nuki Y, Liang EI, Makino H and Hashimoto T. Pharmacologically induced thoracic and abdominal aortic aneurysms in mice. Hypertension. 2010;55:1267–1274. doi: 10.1161/HYPERTENSIONAHA.109.140558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song W, Wang F, Lotfi P, Sardiello M and Segatori L. 2-Hydroxypropyl-beta-cyclodextrin promotes transcription factor EB-mediated activation of autophagy: implications for therapy. J biol chem. 2014;289:10211–10222. doi: 10.1074/jbc.M113.506246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang C, Niederstrasser H, Douglas PM, Lin R, Jaramillo J, Li Y, Oswald NW, Zhou A, McMillan EA, Mendiratta S, et al. Small-molecule TFEB pathway agonists that ameliorate metabolic syndrome in mice and extend C. elegans lifespan. Nat Commun. 2017;8:2270. doi: 10.1038/s41467-017-02332-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang J, Wang J, Xu J, Lu Y, Jiang J, Wang L, Shen HM and Xia D. Curcumin targets the TFEB-lysosome pathway for induction of autophagy. Oncotarget. 2016;7:75659–75671. doi: 10.18632/oncotarget.12318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin L, Zeng W, Zhang F, Zhang C and Liang W. Naringenin Ameliorates Acute Inflammation by Regulating Intracellular Cytokine Degradation. J Immunol. 2017;199:3466–3477. doi: 10.4049/jimmunol.1602016 [DOI] [PubMed] [Google Scholar]
- 26.Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31:1095–1108. doi: 10.1038/emboj.2012.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pontikis CC, Davidson CD, Walkley SU, Platt FM and Begley DJ. Cyclodextrin alleviates neuronal storage of cholesterol in Niemann-Pick C disease without evidence of detectable blood-brain barrier permeability. J Inherit Metab Dis. 2013;36:491–498. doi: 10.1007/s10545-012-9583-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Castellano BM, Thelen AM, Moldavski O, Feltes M, van der Welle RE, Mydock-McGrane L, Jiang X, van Eijkeren RJ, Davis OB, Louie SM, et al. Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science. 2017;355:1306–1311. doi: 10.1126/science.aag1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prins PA, Hill MF, Airey D, Nwosu S, Perati PR, Tavori H, F. Linton M, Kon V, Fazio S and Sampson UK. Angiotensin-Induced Abdominal Aortic Aneurysms in Hypercholesterolemic Mice: Role of Serum Cholesterol and Temporal Effects of Exposure. PLOS ONE. 2014;9:e84517. doi: 10.1371/journal.pone.0084517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Au - Sawada H, Au - Chen JZ, Au - Wright BC, Au - Moorleghen JJ, Au - Lu HS and Au - Daugherty A. Ultrasound Imaging of the Thoracic and Abdominal Aorta in Mice to Determine Aneurysm Dimensions. JoVE. 2019:e59013. doi: 10.3791/59013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Daugherty A, Manning MW and Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest. 2000;105:1605–1612. doi: 10.1172/jci7818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wagenseil JE and Mecham RP. New insights into elastic fiber assembly. Birth defects res C Embryo today. 2007;81:229–240. doi: 10.1002/bdrc.20111 [DOI] [PubMed] [Google Scholar]
- 33.Anzai A, Shimoda M, Endo J, Kohno T, Katsumata Y, Matsuhashi T, Yamamoto T, Ito K, Yan X, Shirakawa K, et al. Adventitial CXCL1/G-CSF expression in response to acute aortic dissection triggers local neutrophil recruitment and activation leading to aortic rupture. Circ res. 2015;116:612–623. doi: 10.1161/circresaha.116.304918 [DOI] [PubMed] [Google Scholar]
- 34.Lerman Lilach O, Kurtz Theodore W, Touyz Rhian M, Ellison David H, Chade Alejandro R, Crowley Steven D, Mattson David L, Mullins John J, Osborn J, Eirin A, et al. Animal Models of Hypertension: A Scientific Statement From the American Heart Association. Hypertension. 2019;73:e87–e120. doi: 10.1161/HYP.0000000000000090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deng Gary G, Martin-McNulty B, Sukovich Drew A, Freay A, Halks-Miller M, Thinnes T, Loskutoff David J, Carmeliet P, Dole William P and Wang Y-X. Urokinase-Type Plasminogen Activator Plays a Critical Role in Angiotensin II–Induced Abdominal Aortic Aneurysm. Circ res. 2003;92:510–517. doi: 10.1161/01.RES.0000061571.49375.E1 [DOI] [PubMed] [Google Scholar]
- 36.Basu R, Fan D, Kandalam V, Lee J, Das SK, Wang X, Baldwin TA, Oudit GY and Kassiri Z. Loss of Timp3 gene leads to abdominal aortic aneurysm formation in response to angiotensin II. J biol chem. 2012;287:44083–44096. doi: 10.1074/jbc.M112.425652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clement M, Chappell J, Raffort J, Lareyre F, Vandestienne M, Taylor AL, Finigan A, Harrison J, Bennett MR, Bruneval P, et al. Vascular Smooth Muscle Cell Plasticity and Autophagy in Dissecting Aortic Aneurysms. Arterioscler Thromb Vasc Biol. 2019;39:1149–1159. doi: 10.1161/ATVBAHA.118.311727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Henderson EL, Geng Y-J, Sukhova GK, Whittemore AD, Knox J and Libby PJC. Death of smooth muscle cells and expression of mediators of apoptosis by T lymphocytes in human abdominal aortic aneurysms. Circulation. 1999;99:96–104. [DOI] [PubMed] [Google Scholar]
- 39.Yamanouchi D, Morgan S, Kato K, Lengfeld J, Zhang F, Liu BJ. Effects of caspase inhibitor on angiotensin II-induced abdominal aortic aneurysm in apolipoprotein e–deficient mice. Aterioscler Throm Vasc Biol. 2010;30:702–707. [DOI] [PubMed] [Google Scholar]
- 40.Durdu S, Deniz GC, Balci D, Zaim C, Dogan A, Can A, Akcali KC and Akar AR. Apoptotic vascular smooth muscle cell depletion via BCL2 family of proteins in human ascending aortic aneurysm and dissection. Cardiovasc therapeut. 2012;30:308–316. doi: 10.1111/1755-5922.12007 [DOI] [PubMed] [Google Scholar]
- 41.Mariño G, Niso-Santano M, Baehrecke EH and Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81. doi: 10.1038/nrm3735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Osonoi Y, Mita T, Azuma K, Nakajima K, Masuyama A, Goto H, Nishida Y, Miyatsuka T, Fujitani Y, Koike M, et al. Defective autophagy in vascular smooth muscle cells enhances cell death and atherosclerosis. Autophagy. 2018;14:1991–2006. doi: 10.1080/15548627.2018.1501132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso C, Forrester A, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015;17:288–299. doi: 10.1038/ncb3114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Varadi J, Hermenean A, Gesztelyi R, Jeney V, Balogh E, Majoros L, Malanga M, Fenyvesi E, Szente L, Bacskay I, et al. Pharmacokinetic Properties of Fluorescently Labelled Hydroxypropyl-Beta-Cyclodextrin. Biomolecules. 2019;9:509. doi: 10.3390/biom9100509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rosenbaum AI, Zhang G, Warren JD and Maxfield FR. Endocytosis of beta-cyclodextrins is responsible for cholesterol reduction in Niemann-Pick type C mutant cells. Proc Natl Acad Sci U S A. 2010;107:5477–482. doi: 10.1073/pnas.0914309107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tanaka Y, Yamada Y, Ishitsuka Y, Matsuo M, Shiraishi K, Wada K, Uchio Y, Kondo Y, Takeo T, Nakagata N, et al. Efficacy of 2-Hydroxypropyl-beta-cyclodextrin in Niemann-Pick Disease Type C Model Mice and Its Pharmacokinetic Analysis in a Patient with the Disease. Bio pharm bull. 2015;38:844–851. doi: 10.1248/bpb.b14-00726 [DOI] [PubMed] [Google Scholar]
- 47.Zimmer S, Grebe A, Bakke SS, Bode N, Halvorsen B, Ulas T, Skjelland M, De Nardo D, Labzin LI, Kerksiek A, et al. Cyclodextrin promotes atherosclerosis regression via macrophage reprogramming. Sci transl med. 2016;8:333ra50. doi: 10.1126/scitranslmed.aad6100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fang L, Hodge J, Saaoud F, Wang J, Iwanowycz S, Wang Y, Hui Y, Evans TD, Razani B and Fan D. Transcriptional factor EB regulates macrophage polarization in the tumor microenvironment. Oncoimmunology. 2017;6:e1312042. doi: 10.1080/2162402x.2017.1312042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chang L, Villacorta L, Zhang J, Garcia-Barrio MT, Yang K, Hamblin M, Whitesall SE, D’Alecy LG and Chen YE. Vascular smooth muscle cell-selective peroxisome proliferator-activated receptor-gamma deletion leads to hypotension. Circulation. 2009;119:2161–2169. doi: 10.1161/circulationaha.108.815803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li T, Yu B, Liu Z, Li J, Ma M, Wang Y, Zhu M, Yin H, Wang X, Fu Y, et al. Homocysteine directly interacts and activates the angiotensin II type I receptor to aggravate vascular injury. Nat Commun. 2018;9:11. doi: 10.1038/s41467-017-02401-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq, ChIP-seq, and microarray data were deposited into GEO database (GSE137577 and GSE138000).