

Abstract
Bile acids are synthesized in the liver, stored in the gallbladder, and secreted into the duodenum at meals. Apical sodium-dependent bile acid transporter (ASBT), an ileal Na+-dependent transporter, plays the leading role of bile acid absorption into enterocytes, where bile acids are delivered to basolateral side by ileal bile acid binding protein (IBABP) and then released by organic solute transporter OSTα/β. The absorbed bile acids are delivered to the liver via portal vein. In this process called “enterohepatic recycling”, only 5% of the bile acid pool (~3 g in human) is excreted in feces, indicating the large recycling capacity and high transport efficacy of ASBT-mediated absorption. Therefore, bile acid transporter-mediated oral drug delivery has been regarded as a feasible and potential strategy to improve the oral bioavailability. This review introduces the key factors in enterohepatic recycling, especially the mechanism of bile acid uptake by ASBT, and the development of bile acid-based oral drug delivery for ASBT-targeting, including bile acid-based prodrugs, bile acid/drug electrostatic complexation and bile acid-containing nanocarriers. Furthermore, the specific transport pathways of bile acid in enterocytes are described and the recent finding of lymphatic delivery of bile acid-containing nanocarriers is discussed.
Keywords: oral drug delivery, bile acid, enterohepatic circulation, transporter, ASBT, lymphatic delivery
Graphical Abstract
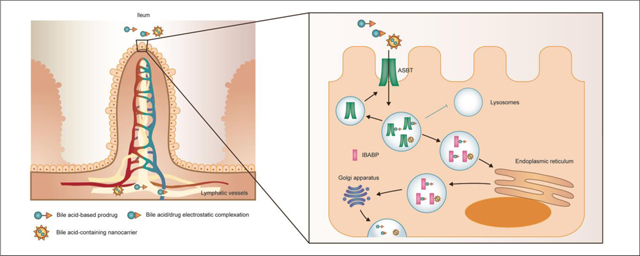
1. Introduction
Oral administration has been regarded as one of the most desirable drug delivery routes due to ease of administration, patient compliance, adjusted dose, and flexibility in the design of dosage forms [1–3]. However, oral bioavailability is largely dominated by physicochemical properties of drugs and biological barriers in the gastrointestinal track (GIT). This often limits the oral administration of a variety of experimental drugs [4].
The first biological barrier in the GIT, the variation of GI lumen pH from harsh acidic environment (pH = 1–2.5) in the stomach to neutral (pH = 6.5–8) in the intestine, may cause pH-induced oxidation and hydrolysis of active pharmaceutical ingredients (API) [5]. Digestive enzymes, especially gastric enzymes, break down biologics into smaller ones. For instance, pepsin, a typical gastric enzyme, cleaves peptide bonds and degrades biopharmaceuticals, resulting in a complex mixture of smaller peptides [6]. It is noted that pancreatic enzymes, despite being digestive, are not considered as a main hinderance for oral drug delivery, since most of them are only abundant in the duodenum, where the drug transit time is short for degradation and the enzyme concentration sharply drops in the jejunum and ileum [4]. Apart from the lumen environment, the mucus layer is regarded as an essential barrier to drug transport to the epithelial layer [7–9]. Mucus is secreted by goblet cells, and its turnover occurs in several minutes to a few hours to aid the elimination of potentially harmful compounds [7]. The viscosity and the turnover rate of the mucus play as both steric and dynamic barriers, requiring the drug delivery materials to navigate through or adhere to the mucus layer [10]. Furthermore, the naked protein core of mucin, electrolytes and poly- or oligosaccharides can form low-affinity interactions with drugs via hydrophobic interactions, electrostatic force, or hydrogen bonding, which might impede absorption [11, 12]. After penetrating the mucus, the next obstacle is the epithelial layer [13]. In theory, hydrophilic drugs can diffuse via the paracellular pathway; however, this pathway is limited by the drug size due to the presence of tight junctions (with the space radius around 8 Å) [14, 15]. For transcellular pathways, the drugs transport via passive partition or specific uptake mechanisms. Although lipophilic drugs are favored in this pathway, the high density of polar head groups in the lipid membrane lowers the drug permeability [4, 16]. After being internalized into the epithelial cells, drugs can be either trapped in membrane-bound subcellular compartments, such as lysosomes, which sequester the drugs from passing though the cell layer or cause the degradation by lysosomal enzymes [17], or expelled back into gastrointestinal (GI) lumen by efflux pumps [18]. The biological factors highly attract research attention to improve oral bioavailability.
According to Lipinski’s Rule of Five, an orally active drug should follow the criteria including: 1) no more than 5 H-bond donors; 2) no more than 10 H-bond receptors; 3) molecular weight less than 500; 4) logP lower than 5; 5) no more than 10 rotatory bonds [19]. This suggests that small molecules with optimal balance in hydrophilicity/hydrophobicity allow appropriate oral bioavailability. Unfortunately, there are more than 40% of drugs with market approval and 90% of molecules in the discovery pipeline are poorly water-soluble [20]. For these insoluble drugs, the oral absorption is hindered by low concentrations and slow dissolution rates [2]. To overcome this problem, there has been a good deal of investigation, especially in the preparation of nanosuspensions, which includes “bottom-up” and “top-down” approaches, emulsions and microemulsions, supercritical fluids, dry-cogrinding, and nanojet technologies [21–26]. These studies make a significant progress in improving the oral drug absorption. However, for those drugs whose absorption is mainly limited by poor permeation or first-pass effects, simply increasing solubility by nanosuspension might not be of value [27]. Compared with improving the solubility, enhancing the permeability is more complicated, and the common methods include permeation enhancers (PE) and transporter/receptor-targeting [28–30].
There are a variety of receptors and transporters expressed in the epithelium of the intestine, which can be targeted for endocytosis so as to increase the drug absorption. Table 1 summarizes selected receptors and transporters considered important for drug absorption [31]. Compared with PEs, this strategy is specific and less toxic by acting on certain sites [32]. Therefore, it has been one of the most popular topics in the oral administration research field.
Table 1.
Selected receptors/transporters used for oral absorption.
| Name | Type | Substrates | References |
|---|---|---|---|
| Neonatal Fc receptor (FcRn) | receptor | IgG, albumin | [39] |
| Folic acid receptor | receptor | Vitamin B9 (folic acid) | [45] |
| Cubulin | receptor | Intrinsic factor-Vitamin B12 | [34] |
| TfR | receptor | Transferrin | [46] |
| αvβ3 | receptor | iRGD, FQS peptide | [47] |
| Oligopeptide transporter (PEPT1) | transporter | β-lactam antibiotics, bestatin, etc. | [48] |
| Organic anion transporter (OAT) | transporter | Bile salts, thyroid hormones, conjugated steroids, etc. | [49] |
| Monocaboxylate transporter (MCT) | transporter | Atorvastatin, ketoprofen, naproxen, etc. | [50, 51] |
| Amino acid transporter ATB0,+ | transporter | Valacyclovir, ganciclovir, etc. | [52] |
| Sodium-dependent glucose transporter (SGLT) | transporter | D-glucose, D-galactose | [31, 53] |
| Sodium-dependent bile acid transporter (ASBT) | transporter | Bile acid, acyclovir, gabapentin | [54] |
Cubilin is one of the commonest targets utilized for enhancing the oral administration by utilizing vitamin B12 modification. B12 is a vital micronutrient for all live cells and can only be acquired through diet [33]. After it is released in the stomach, B12 interacts with haptocorrin (HC) to avoid hydrolysis in acidic conditions (pH < 3). It is transported to the duodenum (pH > 5), where HC is digested and B12 binds to intrinsic factor, the ligand of cubilin [34]. In this way, B12 can be internalized into epithelial cells by receptor-mediated endocytosis. According to the study by Long et al., a vitamin B12-modified amphiphilic sodium alginate derivative (CSAD-VB12) was synthesized and prepared into nanoparticles with insulin encapsulated (CSAD-VB12/INS). Compared with FITC-insulin solution and FITC-CSAD/INS, FITC-CSAD-VB12/INS demonstrated higher uptake efficacy in intestinal villus cells of Type 1 diabetic (T1D) mice. The blood glucose level decreased by 54% with CSAD-VB12/INS in 12 h, significantly higher than 25% of CSAD/INS [35]. Despite numerous studies emphasizing the efficacy of B12 in protein drug absorption, the feasibility of B12-mediated oral administration is still limited due to its efficiency, which reportedly exhibits low absorption capacity (1–2 μg per day in human) [36]. In this case, the dose can hardly meet the therapeutic windows in clinic.
Neonatal Fc receptor (FcRn) is another favored receptor in oral administration by designing IgG-based drug. Knowledge of this receptor dates back to 1958 when Brambell reported the receptor mediating IgG uptake from mother to fetus, and it was identified several years later [37]. In primates, FcRn is expressed throughout adult life in distal epithelial cells, and in most cases, in intracellular vesicles [38, 39]. After IgG is internalized via non-specific pinocytosis, it encounters FcRn in early endosomes, where the acidic environment facilitates the interactions between IgG and FcRn [39]. The FcRn-IgG complex is then transported via endosomes (but avoiding lysosomes) to extracellular spaces, where the pH rises to neutral and the FcRn-IgG are disassociated, with IgG transcytosed and FcRn recycled [39]. This mechanism is expected to promote the transcytosis of IgG or albumin-modified materials. For instance, Pridgen et al prepared IgG-conjugated PLA-PEG nanoparticles and found the absorption efficacy was 11.5-fold compared with unconjugated particles [40]. There are still issues remaining in this strategy. One main question is that fetuses mainly take maternal IgG, while neonates already produce their own IgG within a few days or weeks of life [41, 42], leaving a doubt as to whether the FcRn can still play the role as human grow up. In addition, human FcRn lacks binding affinity to IgG of many species, which might limit its preclinical to clinical transformation [43]. Besides, once Fc fragment is conjugated with a drug, there is a risk of overactivation of the immune system [44].
Overall, most receptor/transporter-mediated strategies are limited by their specificity, potential toxicity, absorption capacity or other particular issues. To overcome this drawback, more research is focusing on the bile acid and its intestinal transporters due to its safety, high capacity and efficacy.
2. Bile acid and bile acid transporter
Bile acids are important regulatory molecules derived from cholesterol in the hepatocytes [55, 56]. In the liver, bile is formed at canaliculae and then modified in bile ducts with electrolytes and water. The bile acids secreted from hepatocytes enter the bile and are stored in the gallbladder, transport to the duodenum during sight, smell, or ingestion of food. In the ileum, most of the bile acids are absorbed by enterocytes, enter the portal vein, and are transported to the liver, completing the “enterohepatic recycling” process (Figure 1)[57]. Most physiological roles of bile acids are played in this process, of which better understanding helps to explain the feasibility of application of bile acids in oral drug delivery.
Figure 1.
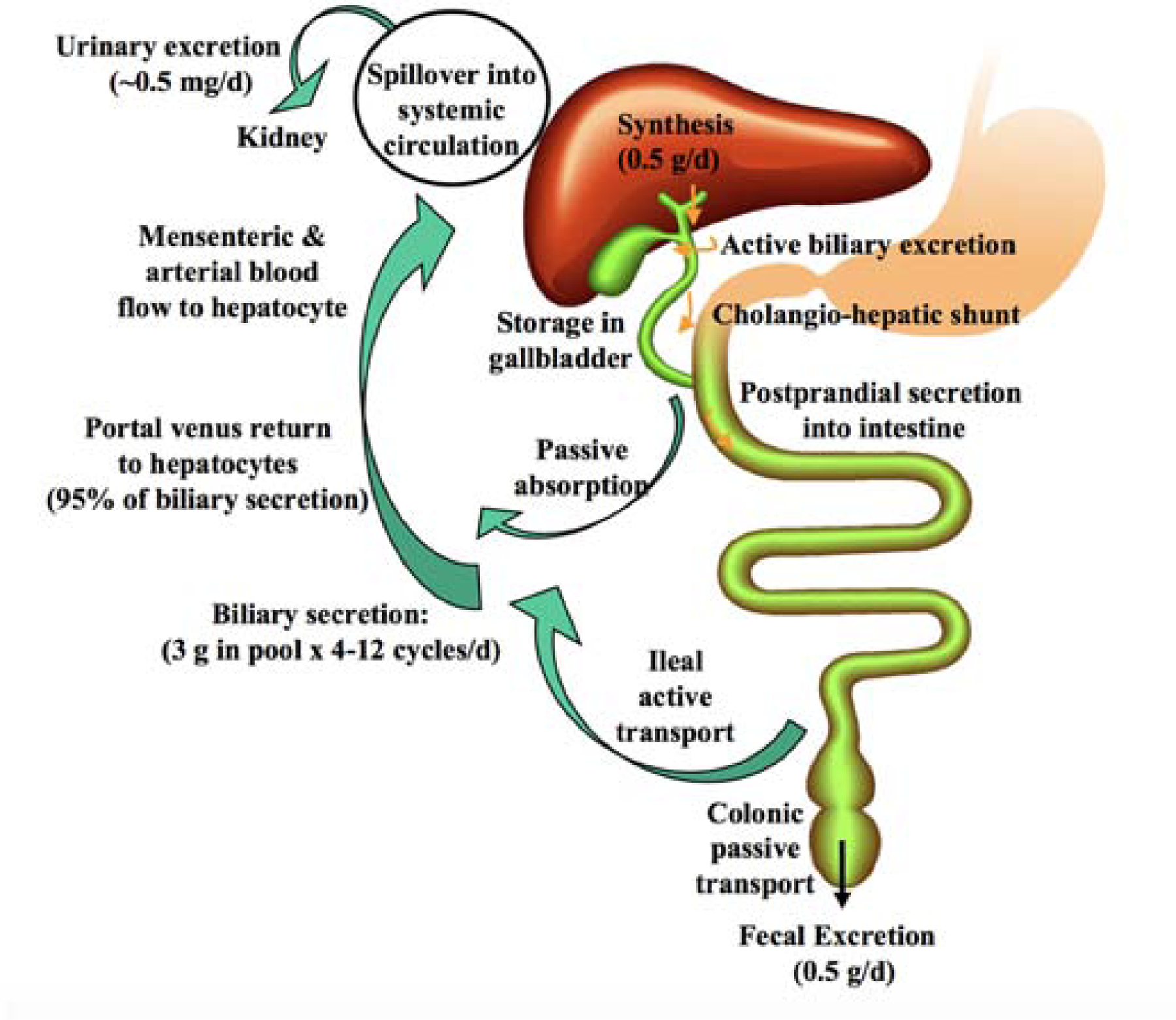
Enterohepatic recycling of bile acids. Bile acids are synthesized in the liver and stored in the gallbladder. At meals, bile acids are excreted to the duodenum for fat emulsification. In small intestine, most bile acids are reabsorbed in the ileum by apical sodium-dependent bile salt transporter (ASBT) and transported back to the liver via portal blood circulation. In this process, about 95% of bile acids are recycled to the liver, and only 5% (~0.5 g) is excreted, which is replenished by the newly synthesized bile acid in the liver. Reproduced, with permission, from reference [58].
2.1. Bile acid physiochemical properties and biological functions
As shown in Figure 2, bile acids in higher vertebrates mainly consist of C24 steroids nucleus, and some are in a conjugated state with glycine or taurine [59, 60]. The steroids nucleus is composed of three six-member rings and a five-member ring. The structure endows the bile acids with the amphiphilic characteristics: the hydroxyl groups are directed to the hydrophilic face (α-face, concave side) and increase its solubility together with the carboxylic side chain; the hydrophobic groups (C18 and C19), on the contrary, are oriented to the hydrophobic face (β-face, convex side) [59]. Consequently, bile acids demonstrate high surface activity: the hydrophobic face of the bile acid monomer desires to eliminate its contact with water, inducing the formation of micelles with the hydrophilic side forming the external surface. Critical micellar concentration (CMC) of the bile acids is around 1.4 – 70 mM, differed by the types of bile acids [61].
Figure 2.
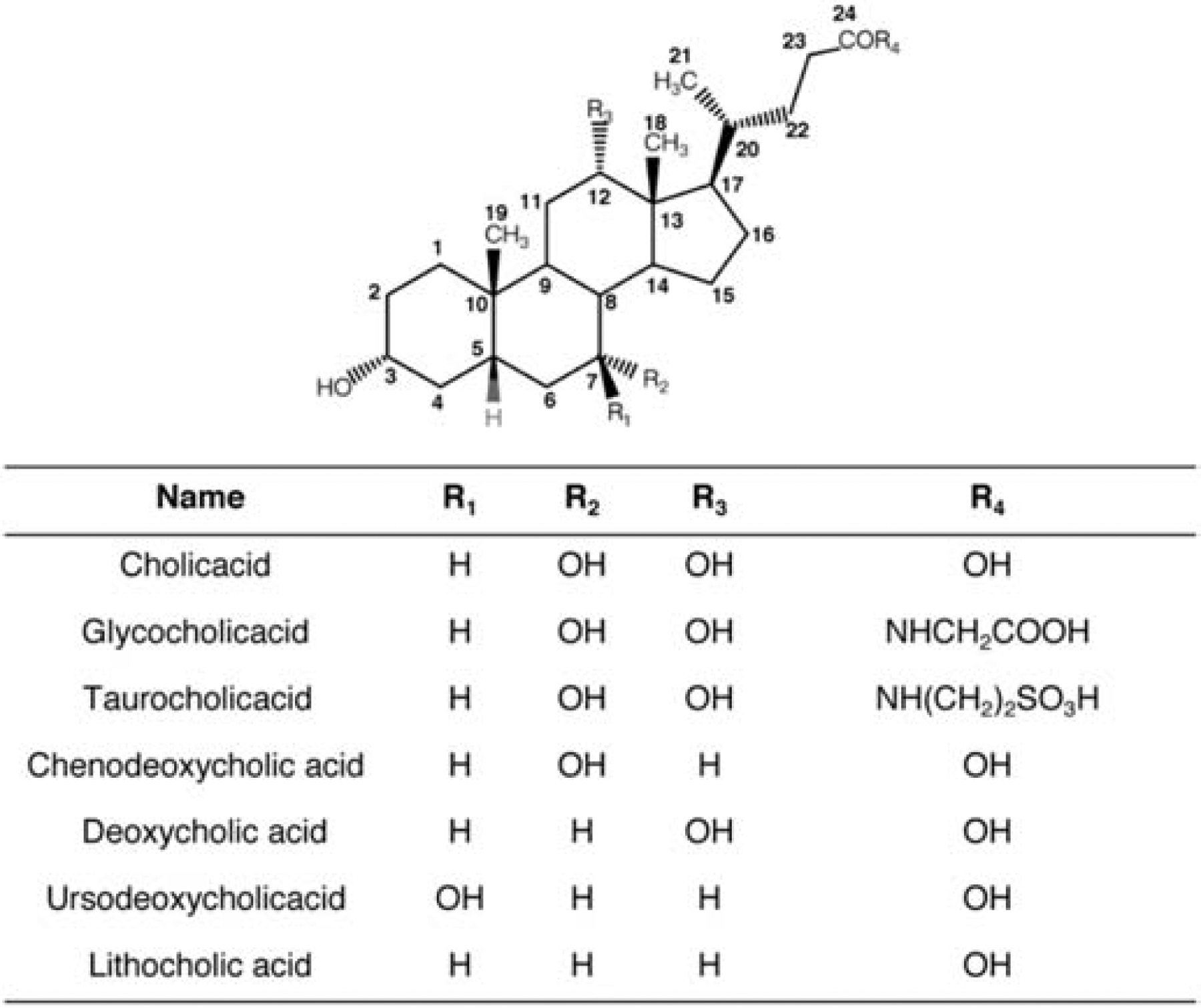
Common bile acid chemical structure and classification.
The main function of bile acids in the gastrointestinal tract is aiding the digestion and absorption of dietary fat. The amphiphilic bile acids emulsify the lipids in small intestine, enhance the activity of pancreatic lipase at the lipid-water interface and incorporate with the digested fats into micelles, which can then transport through the hydrophilic mucus layer and reach the brush border of enterocytes [62]. About 200–600 mg of bile acids are synthesized by the liver per day (80–90 kg body weight), about 5% of the total bile acid pool (3 g) [63]. This plays an important role in metabolism by transforming insoluble cholesterol to small soluble molecules that are easily secreted, accounting for 50% elimination of cholesterol [64]. Biosynthesis of bile acids is regulated by CYP7A1 activity in feedback manner, which responses to the activation of bile acid nuclear receptor Farnesoid X receptor (FXR). Bile acids also interact with gut microbiota, which is closely related to some diseases like obesity, type diabetes and inflammatory bowel disease [65].
2.2. Bile acid secretion from liver
2.2.1. Basolateral (sinusoidal) bile acid uptake into hepatocytes
The bile acids secreted from hepatocytes are from two sources: newly synthesized and recycled. For recycled bile acids, it requires uptake by hepatocytes from sinusoidal blood to start another cycle. The uptake of bile acids into hepatocytes is efficient (about 75–90%) but not simple [66]. Up to now, researchers have clarified two main pathways: Na+-dependent process and Na+-independent process. In Na+ dependent process, the uptake of conjugated bile acids is driven by an inward Na+ gradient under Na+/K+-ATPase and intracellular negative electrical gradient, with the assistance by the transporter called sodium-dependent taurocholic co-transporting polypeptide (NTCP) [67]. The Na+-independent process, on the contrary, is favored for unconjugated bile salts utilizing another transporter family, organic anion transporting polypeptides (OTAP).
2.2.2. Bile acid intracellular trafficking and secretion from hepatocytes into bile
Following the uptake into the hepatocytes, bile acids are rapidly transferred for canalicular secretion within seconds [68]. Usually, bile acids bind to the cytosolic proteins including 3α-hydroxysteroid dehydrogenase, glutathione S-transferase or liver fatty acid binding protein (L-FABP) and are transported by diffusion whereas the lipophilic bile acid are associated with membrane-bound compartments [68, 69]. After reaching the canalicular membrane, the newly synthesized or recycled bile acids are secreted by some transporters in ATP-dependent manner, which is the rate limiting step of the whole transcytosis through the hepatocytes [70]. Bile salts export pump (BSEP, ABCB11) is the most specific transporter in this process, which is exclusively expressed on the canalicular membrane and has high affinity with conjugated monovalent bile acids, whereas it shows little affinity to the unconjugated bile acids [71]. Another transporter participating in bile acid efflux is multidrug resistance-associated protein 2 (MDR2), which is favored for divalent substrate transport like sulfated or glucuronic bile acids, but has no capacity for monovalent ones [64, 72]. Some other proteins like MDR1, MDR3 and MDR4, have low expression level in the canalicular side and work by pumping the bile acid back to the sinusoidal blood when the apical efflux does not efficiently relieve the high-load bile acid in hepatocytes [72]. After secretion into the bile, most bile acids are stored in gallbladder, and a small amount is reabsorbed in cholangiocytes and recycled to hepatocytes [71].
2.3. Bile acid uptake and lipid absorption
2.3.1. Lipid absorption and chylomicron
Lipophilic component absorption is the most important function of bile acids, among which fat and insoluble vitamins are typical examples. Upon ingestion of meal, bile acids are ejected from the gallbladder into duodenum and emulsify dietary lipids to enhance their absorption [73]. The hydrophobic portion of bile acids intercalates into the lipids, specifically, the predominant dietary lipids including triacylglycerols, phospholipids, and cholesterol esters, and it assists to break the large lipid droplets into small ones, which significantly increases their surface area and accessibility to pancreatic lipases [74]. With the hydrolysis by lipases, the lipids are transformed into fatty acids as their common hydrolysis products as well as respective products of monoacylglycerols, lysophospholipids, and free cholesterol [74]. After disassociation with lipase, the fatty acid, glycerol and cholesterol remains associated with bile acids in mixed micelles and hence increase their permeation across the mucus layer and uptake by enterocytes [75]. Generally, the lipids are absorbed in the upper part of the small intestine via two mechanisms: diffusion when the lipid concentration in lumen is higher than that in cytoplasm, or transporter-mediated pathway in an opposite case [76]. The main important fat/cholesterol transporters include cluster determinant 36 (CD36), fatty acid transport protein (FATP), and Niemann-Pick C1-like 1 (NPC1L1). [76].
After internalization, the lipid droplets bind to fatty acid binding proteins (FABP) at the inner side of cell membrane and then transport to endoplasmic reticulum (ER), where the hydrolyzed products are esterified back into triacylglycerols, phospholipids, and cholesterol esters [74, 76]. During the transport in ER and Golgi apparatus, the lipids are packaged together into spherical lipoproteins called chylomicrons, with cholesterol esters and triacylglycerols in the core and phospholipids and cholesterol on the surface [77]. Chylomicrons are then extruded from Golgi apparatus in the exocytic vesicles and released from basolateral membrane. For other lipophilic components like vitamin E, they have similar transport pathway to the lipids with the aid from bile acids [75].
2.3.2. Lymphatic delivery
As Figure 3 shows, after exocytosis from enterocytes, chylomicrons are mainly transported to lacteal and then enter the systemic circulation via lymphatic vessels and the thoracic duct [78]. The claim of lymphatic transport for lipid into blood stream can be backdated to 1662 by physician G. Aselli [79]. It is controversial whether chylomicrons are transported into lacteals by transcytosis or paracellular route, and the transport is further promoted by the pumping activities from resided muscles around the lacteals and the driving force of intrinsic contractility of the collecting lymphatics [80].
Figure 3.
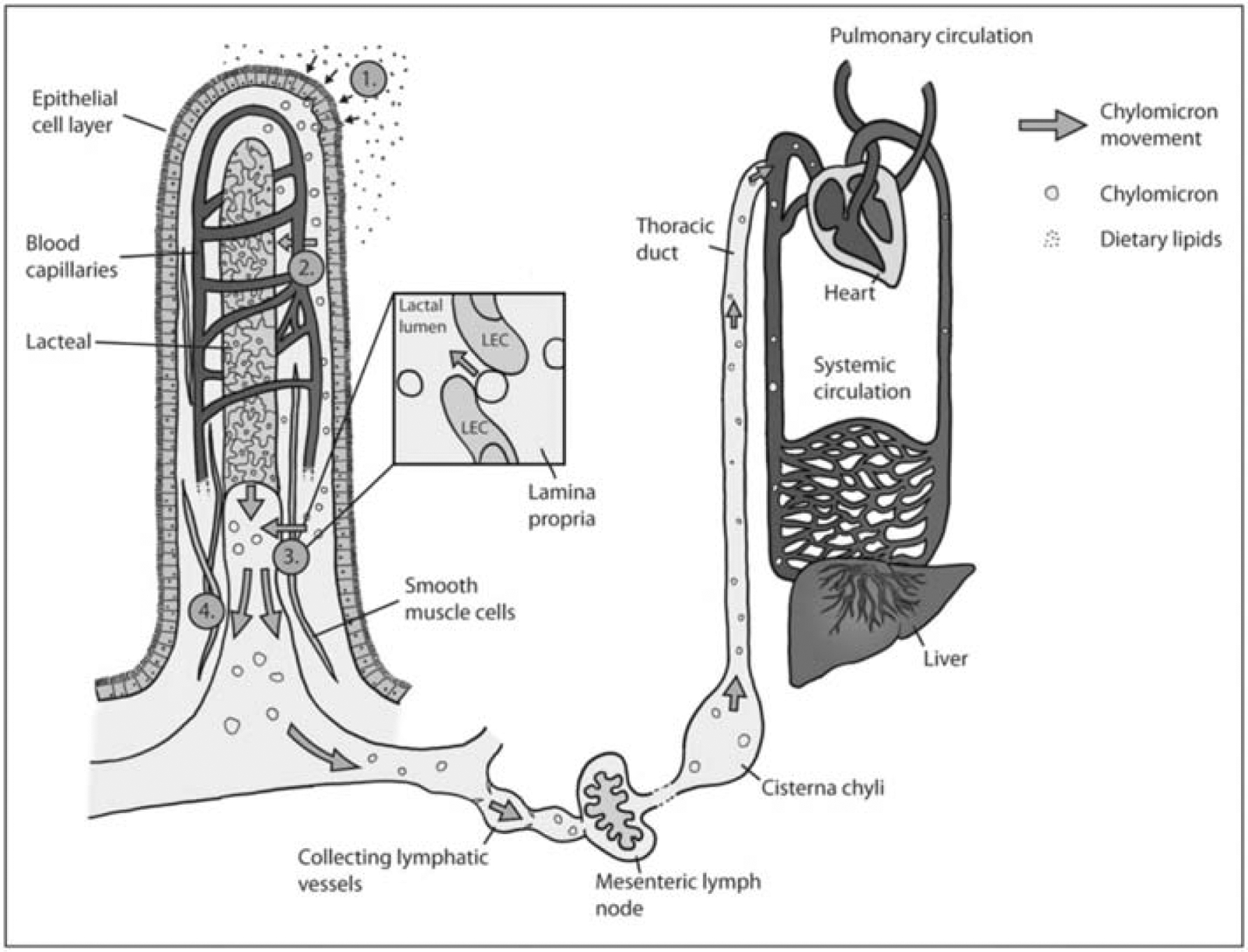
Stages of dietary lipid absorption from intestinal lumen into villi, transporting though the enterocytes and collected by lacteals into systemic circulation. Reproduced, with permission, from reference [79].
Most drugs enter portal veins after transcytosis through enterocytes due to the higher transport rate compared with intestinal lymph, while lacteals are mainly utilized for taking up the particles that are highly lipophilic (LogP > 5) or too large to enter blood streams [81]. The lymphatic delivery is attracting extensive research interests because of several specific advantages: 1) the drugs transported via lymphatic pathway directly enter systemic circulation without going through first-pass effects by the liver [82]; 2) the route works as an alternative pathway with the pharmacokinetics characteristics including double peaks and “flip-flop” behaviors, which avoids the rapid distribution of drugs into organs and tissues and reduces toxicity [83, 84]; 3) this pathway can be utilized by targeting to M cells, a special immune cells distributed in follicle-associated epithelium (FAE) among the epithelial cells [81]. The normal function of M cells is to rapidly uptake, presenting antigens and microorganism to immune systems to initiate the immune response [85]. Without microvilli and mucus on the apical surface, M cells demonstrate higher accessibility to cargos in lumen, and then facilitate the transcytosis with less enzyme degradation by reducing lysosomal navigation [81, 86]. Despite M cells encompassing less than 1% of intestinal cells, many studies have been focusing on M cell-targeting due to its outstanding transport activity, such as toll-like receptor-4 (TLR-4), α5β1 integrin, glycoprotein 2, cellular prion protein, etc. [86] The chylomicron pathway is a potential way for utilizing the amphiphilic property of bile acids to enhance the uptake of drugs, which will be further discussed in later sections.
2.3.3. Uptake of bile acids
Although unconjugated bile acids with high pKa can be transported into enterocytes by passive diffusion, it only accounts for a small faction [87]. In enterohepatic circulation, about 95% of the bile acids are reabsorbed and returned to the liver for recycling, which is attributed to Na+-dependent Apical Sodium Dependent Bile acid Transporter (ASBT, sometimes written as IBAT), with only 5% excreted in feces [63]. Differing from the lipid/bile acid mixed micelles, the uptake of free bile acids mainly occurred in the distal ileum by ASBT. ASBT consists of 348 amino acids, exhibiting 35% identity and 63% amino acid sequence similarity with NTCP [88]. As a symporter, ASBT exploits the Na+ gradient to pump bile acids across the apical membrane, with the stoichiometry of two Na+ for one bile acid molecule [64].
Several mechanisms of ASBT transport have been suggested. One was based on the study of ASBT from N. meningitidis (ASBTNM), simply indicating that the conformation of ASBTNM was regulated by Na+ binding, which drove the movement of panel domain and the transformation between outward and inward facing [89]. Another one, based on the first model, set up a three-dimensional model with Yersinia frederiksenii (ASBTYf), which revealed that a conserved “crossover” region, where the two helices crossed, altered the accessibility of bile acids with the rotation of substrate-binding domain [90]. However, a new study casted the suspect to these models and stimulated the binding process, illustrating that the bile acid binding was prior to the Na+ for lower energy state and the Na+ binding was in order by “knock-on” model, while the releasing of bile acids was caused by the electrostatic repulsion due to the occupation of third Na+ binding sites [91]. These studies promote the investigation of specific dynamics of ASBT and remain a potential area for future exploration.
ASBT follows endocytosis process by multivalency interactions with bile acids and enters the subcellular organelles, and then returns back to the membrane. Al-Hilal and coworkers conjugated tetrameric deoxycholic acids to low molecular weight heparin to synthesize LHe-tetraD and tested its endocytosis and intracellular behaviors [92]. It was found that ASBT was colocalized with LHe-tetraD in MDCK-ASBT cells; after shuttling from membrane to cytoplasm, ASBT is recycled to plasma membrane by removal of LHe-tetraD. It was speculated that recycling endosomes were involved in this process due to the observation of colocalization between ASBT and recycling endosome marker protein rab11 in SK-BR-3 cells, a cell line with high ASBT expression. Our research group further explored the ASBT behavior following the treatment of glycocholic acid-conjugated polystyrene nanoparticles (G40/CPN, where 40 represented degree of substitution) in SK-BR-3 cells [93]. As Figure 4 shows, G40/CPN remained bound to ASBT at 3 h after treatment, while ASBT was separated from the G40/CPN and dispersed in cytoplasm after 6 h and recycled to the cell membrane surface. These results suggested the co-internalization role of ASBT for bile acids uptake, yet the specific mechanism of separation of bile acid-ASBT complex still needs further exploration.
Figure 4.
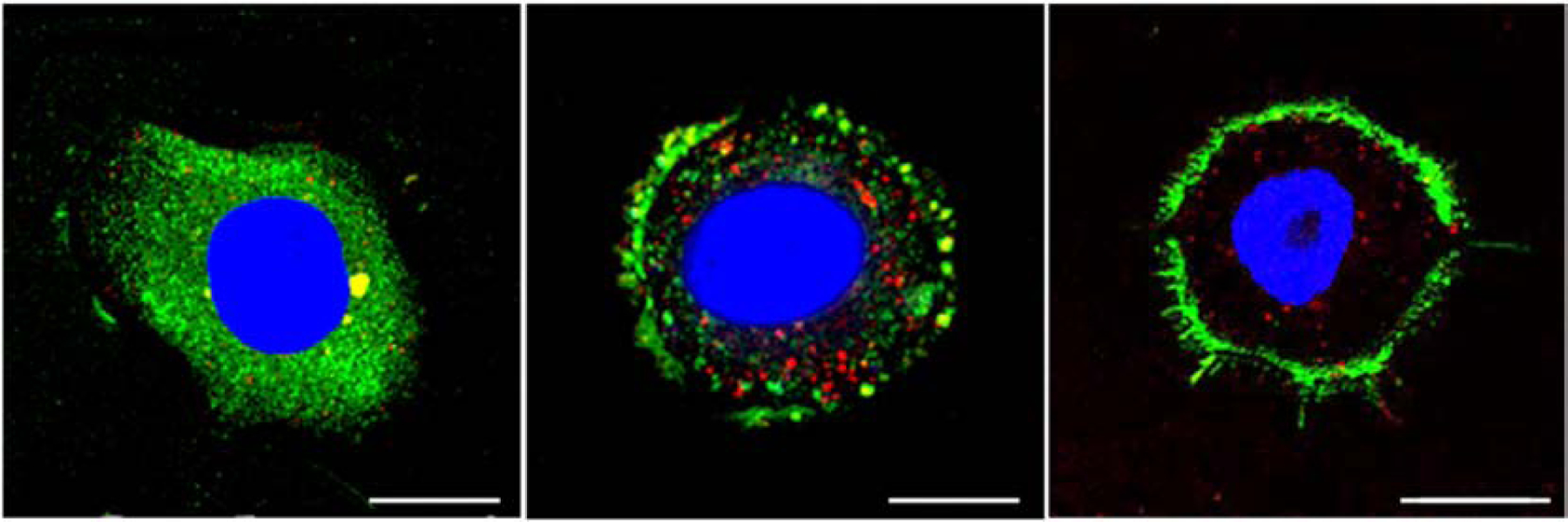
Distribution of ASBT after incubation with G40/CPN in SK-BR-3 cells for 3, 6 and 24 h. Green: ASBT, Red: G40/CPN, Blue: nuclei. Reproduced, with permission, from reference [93].
2.4. Intracellular delivery and exocytosis of bile acids in enterocytes
After bile acids are taken up into intestinal epithelial cells, ileal bile acid binding protein (IBABP) binds them and shuttles them to the basolateral membrane [94]. IBABP is a 14 kD cytosolic protein that shares the similar structure with FABP, including a β-barrel and a helix-turn helix motif over the binding cavity [95]. Each IBABP presents two bile acid binding sites, one interacting with 3-OH of bile acids via hydrogen bond, another with 7-OH or 12-OH [96]. In Al-Hilal’s research, colocalization between LHe-tetraD and IBABP was observed in SK-BR-3 cells, and it was confirmed that LHe-tetraD induced the upregulation of IBABP in 1 h [92]. However, the specific mechanism of IBABP-bile acid delivery mechanism is not fully elucidated.
Once reaching the basolateral membrane, bile acid is exported mainly by the heteromeric organic solute transporter OSTα/β [97]. OSTα domain consists of 340 amino acids with 7 transmembrane domains and β domain of 128 amino acids with single transmembrane domains [98]. OSTα/β is distributed in many tissues, including intestine, kidney, and liver adrenal glands, and it demonstrates a wide substrate spectrum [99]. This transporter is independent of Na+ and not sensitive to pH, K+, Cl−, and ATP depletion, which indicates the bile acids efflux mechanism is facilitated diffusion [100]. After exocytosis, bile acids are transported to the liver and taken up by hepatocytes for another round of enterohepatic circulation.
2.5. Bile acid transporter
As introduced before, bile acid transporters are key factors to facilitate the transcytosis in the liver and intestine in enterohepatic circulation. The main bile acid transporters are summarized in Table 2. It should be noted that some transporters are expressed in various organs. MDR3 is expressed in basolateral membrane of both hepatocytes and enterocytes [68], while evidence is lacking whether this transporter assists efflux of bile acids in enterocytes and we only list its liver distribution. And some transporters, like OTAP, are a large family of proteins with multiple functions, [101] and we mainly focus on the function relevant to bile acid transport and do not distinguish their specific subfamily.
Table 2.
Bile acid transporters
| Name | Distribution | Function | Note | References |
|---|---|---|---|---|
| NTCP | Liver, basolateral | Uptake of bile acids into hepatocytes | Na+-dependent | [67] |
| OATP | Liver, basolateral | Uptake of bile acids into hepatocytes | Na+-independent | [102] |
| FABP | Liver, cytosolic | Shuttle bile acids from basolateral to apical side | For hydrophilic bile acids | [68] |
| MDR1/3/4 | Liver, basolateral | Efflux of bile acids back to blood | Relieve liver load | [72] |
| BSEP | Liver, apical | Efflux of bile acids from hepatocytes | High specificity | [67, 71] |
| MDR2 | Liver, apical | Efflux of bile acids from hepatocytes | Divalent substrates | [64] |
| ASBT | Ileum, apical | Uptake of bile acids into enterocytes | Na+-independent, | [58] |
| IBABP | Ileum, cytosolic | Shuttle bile acids from apical to basolateral side | Unclear mechanism | [94] |
| OSTα/β | Ileum, basolateral | Efflux of bile acids from enterocytes | Facilitated diffusion | [100] |
For oral drug administration, ASBT is a potential target to enhance the absorption of BCS class III/IV drugs due to its high recovery of bile acids in high capacity. But it should be mentioned that some factors can regulate more than one transporter in enterocytes, but in different direction. For example, FXR has been reported to downregulate ASBT expression, similar to NTCP [103], while it induces the expression of IBABP and OSTα/β [104, 105]. In this case, it might be possible that knock-down of FXR to enhance ASBT expression would not promote the absorption bile acid-conjugated drugs, which reminds us to consider the whole transport system when designing a delivery system.
3. Bile acid transporter-mediated oral drug delivery strategies
The use of bile acid transport pathway for drug delivery can be traced back to a patent application in 1948. Bile acids were conjugated with p-aminobenzene sulphonamide to yield N-acrylsulphonamides to treat germ and viral infections attacking the liver [106]. Lack and his colleagues first examined the importance of solute structure to ileum bile acid transporter (known as ASBT later) during 1966 to 1979 [107–109]. In 1994, Dawson and his coworkers first cloned ASBT from hamsters [110]. These studies built up a base for drug delivery using ileal bile acid transporters. In the last several decades, bile acids and ASBT have been investigated for oral administration in various forms, of which the main types are bile acid-conjugated prodrugs, bile acid/drug electrostatic complexation, and bile acid-modified nanocarriers.
3.1. Bile acid-conjugated prodrug strategy
Prodrug refers to inactive derivatives of drug molecules which undergo in vivo enzymatic or biochemical transformation to release active pharmaceutical ingredients (API) [28]. Bile acids can be conjugated to drugs to improve absorption by facilitating ASBT-mediated transport. The bile acid chemical structure provides various sites for prodrug synthesis. Drugs are conjugated directly or using spacers to the positions of C3, C7, C12, C24 by esterification, amidation, reduction, or anhydridization [111].
Kramer, Wess and their coworkers are considered as pioneers in designing bile acid-conjugated prodrugs. Since 1980s, they have developed numerous bile acid-drug conjugates to enhance their delivery. Despite a large portion of their research focusing on the transporters in hepatocytes, they made great achievements in promoting drug absorption by ASBT as well. For example, they modified cholic acid at C3 and C24 to synthesize 3β-(ω-aminoalkoxy)-7α, l2α-dihydroxy-5β-cholan-24-oic acid, conjugated it with peptides of different chain lengths, and tested their absorption after ileum perfusion in rats [112, 113]. It was found that [3H] taurocholate uptake was significantly reduced when incubated with hybrid peptides containing more than four amino acid residues. One of the bile acid-conjugated peptides, named as S3744, were found in bile with Tmax around 12–16 min, while its parent peptide S1037 and the t-butylester S4404 were not absorbed. In addition, it was further confirmed that taurocholate in turn inhibited the uptake of S3744, indicating the competitive effect on the ileum Na+-dependent transporter. This result provides a stepping stone for utilizing oral bile acid prodrugs. However, the technologies and the limited known knowledge might have impeded deeper delving into this area at that time (published in 1994), and more advancement was required for later study. First of all, the in vivo metabolic mechanism of S3744 was still unknown. Although one metabolite was found in bile by thin layer chromatography, the exact structure was unclear. Doubts were cast whether the conjugate could be transformed into parent peptide structure, not to mention the activity revived. Besides this, the specific identity of ASBT was not affirmed by then, hindering the study of specific interaction between the conjugate and the transporter. In the study, the essential proteins were found by photoaffinity, and the pathway was confirmed simply by inhibiting taurocholate uptake or inhibition by taurocholate, lacking molecular biological evidence. In spite of these limitations, the early study of Kramer and Wess opened the gate of oral administration of bile acid-based prodrugs and inspired successors in this field.
Tolle-Sander and coworkers conjugated valacyclovir (valine-modified acyclovir) to chenodeoxycholate at C24 and found acyclovir valylchenodeoxycholate demonstrated more than 10-fold uptake in hASBT-COS cells (202 ± 9 vs 12.9 ± 0.3 pmol/min) compared with acyclovir solution [114]. The existence of Na+ in media led to the uptake of the prodrug increasing to 1.4-fold, with no effect on acyclovir, suggesting the internalization mechanism was changed from passive diffusion to hASBT-mediated transport by chenodeoxycholate modification. Besides, the unconjugated acyclovir recovery was 48.0 ± 5.6% in SD rats’ urine after oral administration of the prodrug, 1.98-fold to that of acyclovir, indicating this prodrug could be highly absorbed and transformed into the active form in vivo. Compared with the early stage study, this research applied ASBT transfection in a cell model and confirmed the in vivo recovery of parent drug, while the drug/transporter interaction/affinity was still limited by taurocholate inhibition.
There have been a variety of other research exploiting the bile acid-conjugated prodrug strategy, which revealed key factors influencing cellular uptake of bile acid-conjugated prodrugs including linkers, charges, and modification sites. For instance, Bhat et al. developed series of C2-C3 annulated glycodeoxycholic acid with steroidal pyrazoles and analyzed their uptake in ASBT-transfected Xenopus laevis oocytes. It was found the uptake was inhibited by taurocholate, affected by β-hydroxyethyl linker, reaching as high as 59%. [115]. Polli’s research team analyzed the optimal structural requirements for uptake using aminopyridine conjugates of chenodeoxycholic acid by 3D-QSAR in hASBT-MDCK cells [116]. It was found that monoanionic conjugates exhibited higher affinity than neutral or chloro-substitute monoanionic conjugates by measuring inhibition by taurocholate. The conclusion moved forward by their further research: gabapentin, a zwitterionic drug with low oral absorption at therapeutic doses, was modified with chenodeoxycholic acid to a series of derivatives [117]. It was confirmed that a single negative charge was preferred for substrate translocation via ASBT in hASBT-MDCK cells, while dianionic compounds showed low affinity and could not be taken as substrate. Balakrishnan et al. examined the uptake of native bile acids in hASBT-MDCK model and proved the necessity of C24-conjugation for transporter-mediated uptake [118]. Due to the intrinsic anti-HIV activity in vitro, cholic acid was linked with a series of amino acids, amino acid analogs, or peptides at C24 and tested as both potential ASBT substrate and HIV-1 protease inhibitor [119]. Choly-D-Asp-β-benzyl ester and cholyl-L-lys-ε-tBOC showed inhibition of taurocholic acid uptake, while only choly-D-Asp-β-benzyl ester showed modest HIV-1 protease inhibitory activity with IC50 = 125 μM. These results depict some clues in guidance of bile acid-conjugated prodrug design and consolidate the relationship between ASBT and bile acid. However, the study is still restricted to medicinal chemistry and mostly focused on cells. The lack of systematic pharmacokinetics and pharmacodynamics (PK/PD) study might prevent applications of these prodrugs in future clinical study. The result seems premature in a degree owing to the absence of tools like imaging and protein expression (except ASBT transfection).
Currently, more imaging technologies, in vivo efficacy and PK analysis have been utilized in bile acid-conjugated prodrugs. To improve oral bioavailability of cytarabine, Sun’s group tested four different cholic acids for conjugation: cholic acid (CA), chenodeoxycholic acid (CDCA), hyodeoxycholic acid (HDCA), and ursodeoxycholic acid (UDCA) conjugated by amide bond at C24 of bile acids [120]. After incubation with HepG2 cells for 48 h, all the conjugates exhibited higher proliferative inhibition effect (over 60%) than cytarabine. The in vitro stability test demonstrated UDCA-cytarabine had the highest prodrug remaining in plasma of rats as well as the highest cytarabine release rate in 2 h, indicating its better metabolic stability. The in vivo PK test showed that UDCA-cytarabine prolonged T1/2 to 15.6 h from 4.0 h of cytarabine, with relative bioavailability 209 ± 45.6%. As a typical example of utilizing bile acid-ASBT interaction to enhance drug in vivo absorption in the last decades, this study covered the drug design, screening, in vitro efficacy test, and PK test, which enriched and improved the development of bile acid-conjugated prodrugs in preclinical study.
Another example is deoxycholic acid-camptothecin (CPT) conjugate (G2) to enhance the oral antitumor efficacy. In Caco-2 3 -D spheroids, 200 μM deoxycholic acid preincubation significantly inhibited the uptake of G2 by 65 %, while no effect was observed on CPT. Although the 3-D cell model is a widely used tool for in vitro penetration study, 3-D cell spheroids are mainly used to mimic solid tumor features such as structure, drug resistance, and growth kinetics [121]. As the enterocyte layer is a monolayer, it is questionable whether 3-D spheroids are suitable as oral absorption models.
With molecular biology technologies, Byun’s team traced ASBT and bile acid-conjugated heparin behavior simultaneously [92]. Apart from the confocal colocalization test of ASBT with LHe-tetraD and endosomes, which is described a bit in 2.3.3 Section, immunoprecipitation-immunoblot analysis of ASBT showed this membrane transporter was distributed in both membrane and plasma after incubation with LHe-tetraD, which was consistent with ASBT recycling. The immunohistochemistry showed the translocation of ASBT from apical to sub-apical mucosa in rat ileum with LHe-tetraD administration in 1 h and gradually recovered in 24 h; meanwhile, the IBABP expression simultaneously increased with the internalization of ASBT (Figure 5). To our knowledge, this was the first time that the bile acid-conjugated prodrug and bile acid transporters were analyzed in subcellular aspects, pushing the bile acid transporter mediated transport into more specific mechanism study.
Figure 5.
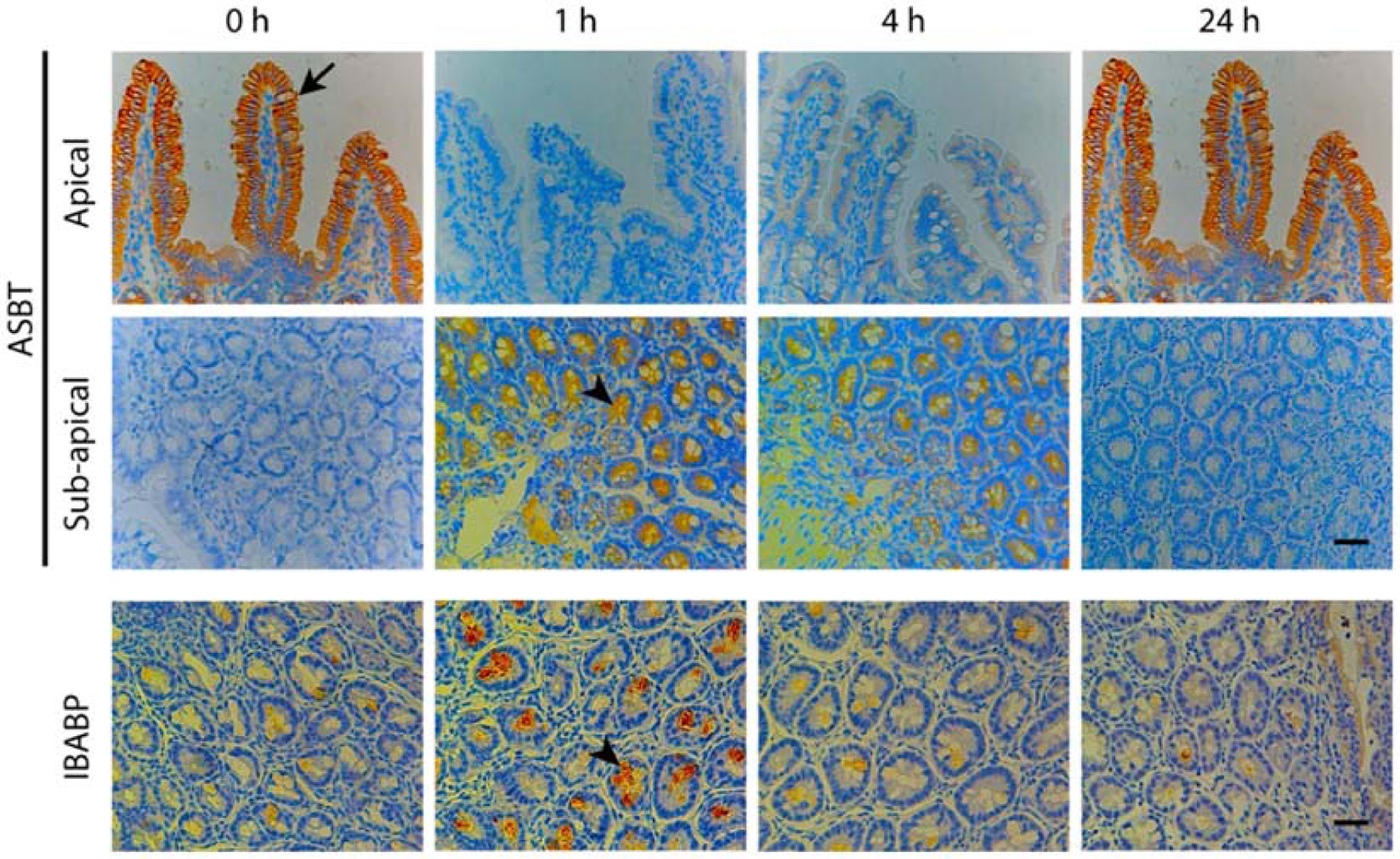
The immunohistochemistry analysis of ASBT distribution in rats after LHe-tetraD treatment. ASBT was internalized from apical membrane to sub-apical compartment in rat ileum after treatment with LHe-tetraD in 1 h, and then gradually recycled to the apical membrane in 24 h. With LHe-tetraD absorption and ASBT uptake, IBABP expression simultaneously increased in 1 h and then decreased. Reproduced, with permission, from reference [92].
3.2. Bile acid/drug electrostatic complexation strategy
Bile acids are anionic at physiological pH and interact with basic (cationic) molecules in the intestine, which might impact their permeation through epithelial cells [122]. Though there has been a long period when ion-pairing with bile acids have been investigated, most studies focused on adjusting the physiochemical properties of drugs rather than focusing on the transporter-targeting. Common ideas are adjusting the polarity of the parent drug by bile acid-drug interaction, or simply using bile acids as an amphiphilic enhancer, which might impact the permeation through the enterocytes.
To improve the permeation of a cationic BCS class III substance, trospium chloride (TC), ionic complexation was prepared between TC and glycochenodeoxycholate (GCDC) or taurodeoxycholate (TDOC), and the permeation was tested in Caco-2 monolayers and rat intestinal excised segments [123]. Both TDOC and GCDC enhanced the permeation through the monolayers and intestinal segments, though GCDC was less effective than TDOC. The permeation enhancement was attributed to the enhancement of passive diffusion. Despite the lack of specific mechanistic study of bile acid-induced permeation, it was interesting to note that oil addition led to reduced absorption. The authors argued that bile acids were consumed by emulsification and thus additional bile acids secreted into intestine in fed condition would not affect existing ion-pairing.
Although native anionic bile acids were primarily used for cation pairing, bile acids could be modified with cationic groups. The resulting cationic bile acids form complexes with anionic drugs. Lee et al. developed an orally available ceftriaxone (CRO) formulation [124]. Cationic cholyethyendiamine (CEA) synthesized from cholic acid and ethylenediamine was ion-paired with anionic CRO at a CRO:CEA = 2:1. After oral administration into rats at dosage 50 mg/kg, the bioavailability of CRO/CEA reached 55.7 ± 18.7% of i.v. injection in 8 h, significantly higher than that of CRO alone (4.3 ± 1.3%). The authors attributed the discrepancy to the increasing lipophilicity after ionic association. Although it was a reasonable and important factor, more aspects should be taken into consideration. One factor is the charge/potential of the complexation, which was less negative compared with CRO. The anionic charge of CRO could impede absorption across the negatively charged cell membrane, thus charge shielding would be favored for enhanced uptake. Another factor was the alternation of transport pathway. The complex was expected to use enterohepatic circulation to aid uptake, which might not be explained by simply increasing lipophilicity. Thus, the absorption was dominated by one or more factors, which requires more investigation.
Another example was the complexation between anionic low molecular weight heparin (LMWH) and cationic deoxycholylethylamine (DCEA) at different ratios for oral administration [125]. Compared with LMWH, LMWH/DCEA demonstrated 25-fold oral bioavailability in rats at 6 h (3.08% vs 0.12%). It was found that at a complex ratio of 1:5, LMWH/DCEA exhibited higher absorption enhancing effect than that of 1:3 and 1:10, owing to the loose structure of 1:3 complex and higher molecular weight of 1:10. With FITC labeling, the LMWH/DECA was observed with the highest intensity in jejunum at 30 min after oral administration, while the strongest was in the ileum was at 60 min, with the fluorescence in the jejunum faded. Despite that increasing lipophilicity was described as the possible explanation by authors, it seems multiple pathways were involved. The rapid absorption in jejunum might be owing to either enhanced diffusion or paracellular transport, or both, while the subsequent dominated uptake in ileum was likely related to the interactions with ASBT, with lower speed but continuous pump-in work.
Electrostatic complexation has also been applied for peptides. Gaowa et al. synthesized cationic EGFR2R-lytic hybrid peptide and mixed with taurodeoxycholate (TDCA) and citric acid at a molar ratio of 1:2:2 to prepare a complex [126]. The zeta potential of peptide, TDCA, and complex were 7.19 ± 1.78 mV, −7.76 ± 5.41 mV and −4.56 ± 0.52 mV, respectively. After incubating with Caco-2 monolayers, the complex significantly reduced the TEER values, indicating the permeation enhancing effect by opening tight junctions. After oral administration in Balb/c mice, the complex demonstrated higher antitumor efficacy than the bulk peptide with tumor volume 318 ± 185 mm3 vs 495 ± 148 mm3 (Figure 6). This is a typical strategy to enhance bile acid/peptide, which requires specific molar ratio of each component and the pH-adjusting substance to maintain the structure and activity of the complex. Although the reduction of TEER indicated that passive diffusion was involved in transport, it should be noted that free TDCA was not removed from the system after preparation of the system. It was inconclusive whether the complex itself could open tight junction and transport via paracellular pathway, or whether it was dependent on the free bile acid effect.
Figure 6.
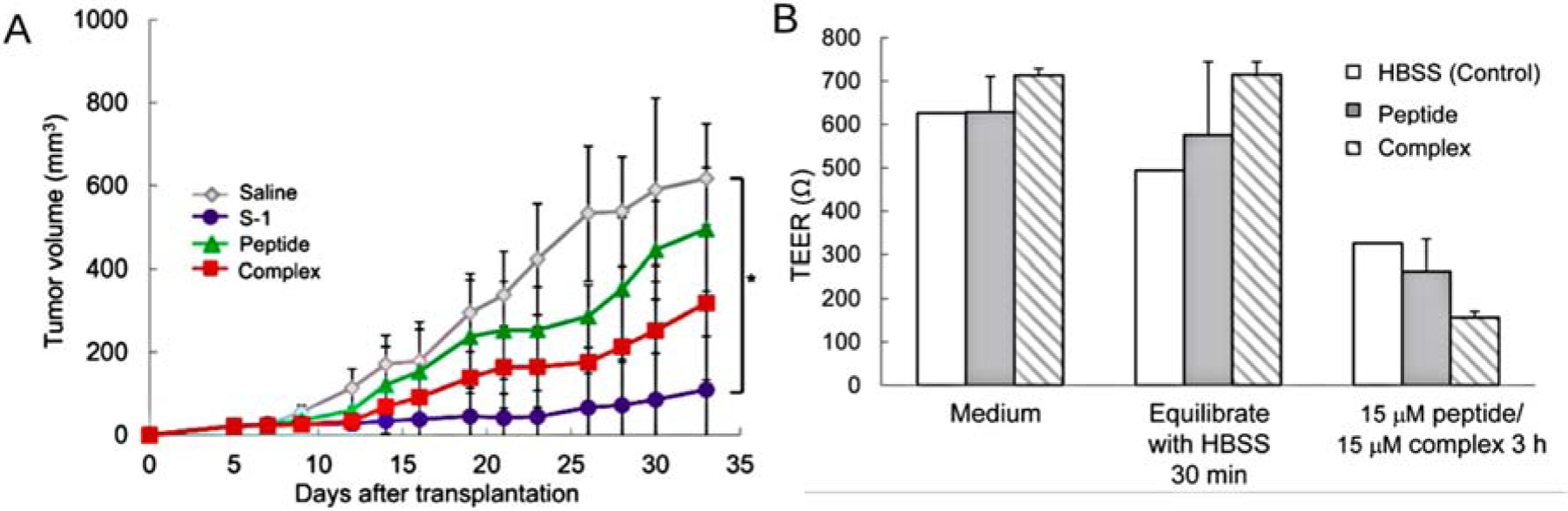
Effect of EGFR2R-lytic hybrid peptide-TDCA-citric acid complex. (A) Measurement of antitumor effect of complex by monitoring tumor size in mice. The complex demonstrated higher tumor inhibition than cationic peptide and S-1 (5-FU oral formulation, positive control). (B) Tight junction opening effect of the complex in Caco-2 cell monolayers. Reproduced, with permission, from reference [126].
Alam et al. reported a method to improve the oral delivery of taurocholate-conjugated low molecular weight heparin derivative (LHT7), where LHT7 was conjugated with a tetrameric deoxycholic acid as LHTD4 and then complexed with deoxycholylethylamine (DCK) [127]. LHTD4 increased the apparent permeability coefficient (Papp) with (74.0 ± 27.2) × 10−7 cm/s, significantly higher than that of LHT7 with (2.6 ± 1.7) × 10−7 cm/s in Caco-2 monolayer. Ion-pairing into LHTD4/DCK further improved the Papp into (135.0 ± 27.3) × 10−7 cm/s. The availability of LHTD4/DCK was 34.8%, while only 3.8% for LHT7/DCK and 14.8% for LHTD4 after oral administration, indicating both chemical conjugation and ion-pairing cooperated to promote the absorption. The authors claimed LHTD4/DCK was transported via ASBT in the endocytosis process by observing that LHTD4/DCK demonstrated higher uptake in MDCK-ASBT cells than MDCK cells, and they hypothesized the conjugation and ion-pairing might increase the local concentration gradient via interaction with ASBT. This was supported by the high distribution of LHTD4 in ileum of treated mice after 6 h. As one of the very few studies clarifying the ASBT role in bile acid/drug electrostatic complex, this study broadened the sight about how bile acid transporter and bile acid-contained drugs interacted. However, it was observed that DCK was disassociated from LHTD4 in intestine and mainly distributed in jejunum. More information would be helpful to explain how free DCK assisted the ASBT-mediated uptake and why it did not behave as a competitive inhibitor.
At present, electrostatic complexation of bile acids and drugs is mainly used in i.v. injection rather than oral administration [128]. One reason might be its lower stability than conjugated prodrug in the harsh GI environment. The amide bond is generally regarded as highly stable towards various reaction conditions such as acidity, basicity, and high temperature [129]. On the contrary, the ionic interaction is greatly weakened in aqueous solution and impaired under high ionic strength. The loose complex structure faces the risk of disassociation in the intestine, which derails the original intention of increasing hydrophobicity and ASBT-targeting. As bile acids themselves can be directly used as permeation enhancers for oral drug administration, for example, for insulin [130], octreotide [131], and gliclazide [132], it is difficult to assert that the improved absorption is owing to the formation of complex or permeation enhancement by free bile acid, if the stability of the complex in GI tract is not clear. This can also be caused by the remaining of free bile acid after complexation, as bile acids are usually excessive in complex preparation and coexisted with the complex. Besides this, varying pH condition in GI tract might affect the equilibrium of ion-pairing and disassociation. These uncertainties might account to the scarcity of bile acid/drug complex transport mechanism study, as multiple pathways might be involved and hard to distinguish in a simple model.
3.3. Bile acid-containing nanocarrier strategy
Nanocarriers have been extensively applied in drug delivery due to their promising properties: high surface area to volume, improved pharmacokinetics and biodistribution, decreased toxicity, increasing drug solubility and stability, controlled release and targeted delivery [133]. In oral drug delivery, nanocarriers help to protect the drug from harsh conditions in GI tract, increase the intestinal absorption, target the specific sites and guarantee a controlled release [134]. Despite arising later than the previous two strategies, bile acid-modified nanocarriers have been ascendant in bile acid transporter-mediated oral drug delivery application and achieve desired diagnosis or therapeutic efficacy by loading various agents and targeting to ASBT.
3.3.1. Liposome
Liposomes mimic cell membranes and has been studied for oral drug delivery since late 1970s due to their high biocompatibility, safety, and large entrapment capacity [135]. However, their oral administration still faces great challenge, as their components are phospholipid and cholesterol in most cases and thus susceptible to GI conditions; lipid digestion sets liposomes at risk of degradation before absorption through the enterocytes [136]. In addition, the permeation through the enterocytes is poor owing to the relatively large size [135]. Some methods are currently used either alone or together to overcome these obstacles, such as using coating materials, changing the liposome components, preparing proliposomes, and specific targeting [137, 138], among which ASBT-targeting became remarkable in recent years.
To improve the intestinal absorption of salmon calcitonin (sCT), Song et al. developed proliposomes containing sCT and taurodeoxycholate (TDC proliposomes) and tested their absorption behavior using Caco-2 and a rat model. Compared with sCT, the transcytosis was increased by 10.8-fold with 3.55-fold decrease of TEER. The duodenum administration of TDC proliposomes resulted in 7.1-fold to that of sCT (0.49% vs 0.069%). As an early study of using bile acid modification to improve intestinal permeation, it illustrated the efficacy of bile acids compared with some other surfactants with relatively low toxicity. Nevertheless, simply ion-pairing to prepare proliposome/bile acid complex might not ensure GI stability, and the transport mechanism was only explained as increasing paracellular pathway, which was at least incomplete in Caco-2 models since proliposomes were able to undergo transcytosis. It set an example for oral liposome delivery with the assistance of bile acids and more adjustments have been applied to conquer these shortcomings.
The polymer-coating method, already used for keeping liposomes stable for several decades, becomes more and more popular in oral administration with the higher risk of liposome degradation in GI tract [139, 140]. Besides, the polymer coating enables the surface modification on the polymers, which might endow liposomes new properties like site-targeting [141]. Wu and coworkers designed a deoxycholic acid and chitosan (CS) conjugate-modified liposomes (DC-LIPs) to enhance the oral absorption of insulin [142]. The deoxycholic acid were conjugated to the positive chitosan chain, and this modified polysaccharide were coated to liposome surface via electrostatic force. The CS-LIPs and DC-LIPs showed significantly higher stability in digestive fluids with slower insulin degradation, while no comparison of coated liposomes with the uncoated. With FITC-insulin loaded, DC-LIPs demonstrated higher permeation in intestine of rats compared with CS-LIPs. The uptake mechanism of DC-LIPs was speculated as ASBT-mediated transport as the uptake was inhibited by preincubation with taurocholate with no effect on CS-LIPs. After oral administration in rats, the blood glucose level was stably reduced to about 45%, 60% and 0% with 50 IU/kg DC-LIPs, CS-LIPs, and free insulin, respectively, with AUC 270.7 ± 26.9, 153.4 ± 27 and 18.89 ± 4.1 μIU·h/mL. On the contrary, the subcutaneous injection of 5 IU/kg free insulin achieved rapid blood glucose level reduction to around 25% in 1 h and recovered to 80% in 5 h with AUC12 h 168.1 ± 23.2 μIU·h/mL. It is noticeable that the colocalization between DC-LIPs and lysosomes gradually decreased after 4 h, suggesting lysosome escape might occur. With promising results, this study offered a feasible way to orally deliver proteins/peptides with stability retained using ASBT-mediated pathway.
Our research group also grafted glycocholic acid to chondroitin sulfate (CS) and coated on exendin-4 loaded liposomes (EL-CSG) by electrostatic complexation (Figure 7) [143]. EL-CSG exhibited only slight size change in digestive fluid, and reduction of exendin 4 (Ex-4) release from 88% to 62% and 51% with CS and CSG coating respectively compared with uncoated liposomes (EL). Oral gavage of EL-CSG showed higher bioavailability (19.5%) than that of EL-CS (4.1%) compared with s.c. injection of Ex-4 solution in normal rats. After glucose administration in type 2 diabetes mellitus rats, the glucose level soared to 238%, 213% and 194% with PBS, 300 μg/kg EL-CS and 20 μg/kg Ex-4 (s.c. injection) oral pretreatment, while 300 μg/kg EL-CSG demonstrated 171% only. The enhanced absorption and efficacy illustrated the effect of ASBT-mediated transport. Interestingly, EL-CSG exhibited similar fat metabolism behavior to free s.c. injection of Ex-4 that both blood cholesterol and LDL level decreased, indicating the ASBT pathway did not alter the cholesterol homeostasis. As ASBT-mediated liposome uptake was in ileum, the endocytosis of liposomes was independent to absorption of dietary lipids, which are processed mainly in the duodenum. In another words, the coating might change the property of liposomes and thus affected their interaction with the fat transporters.
Figure 7.
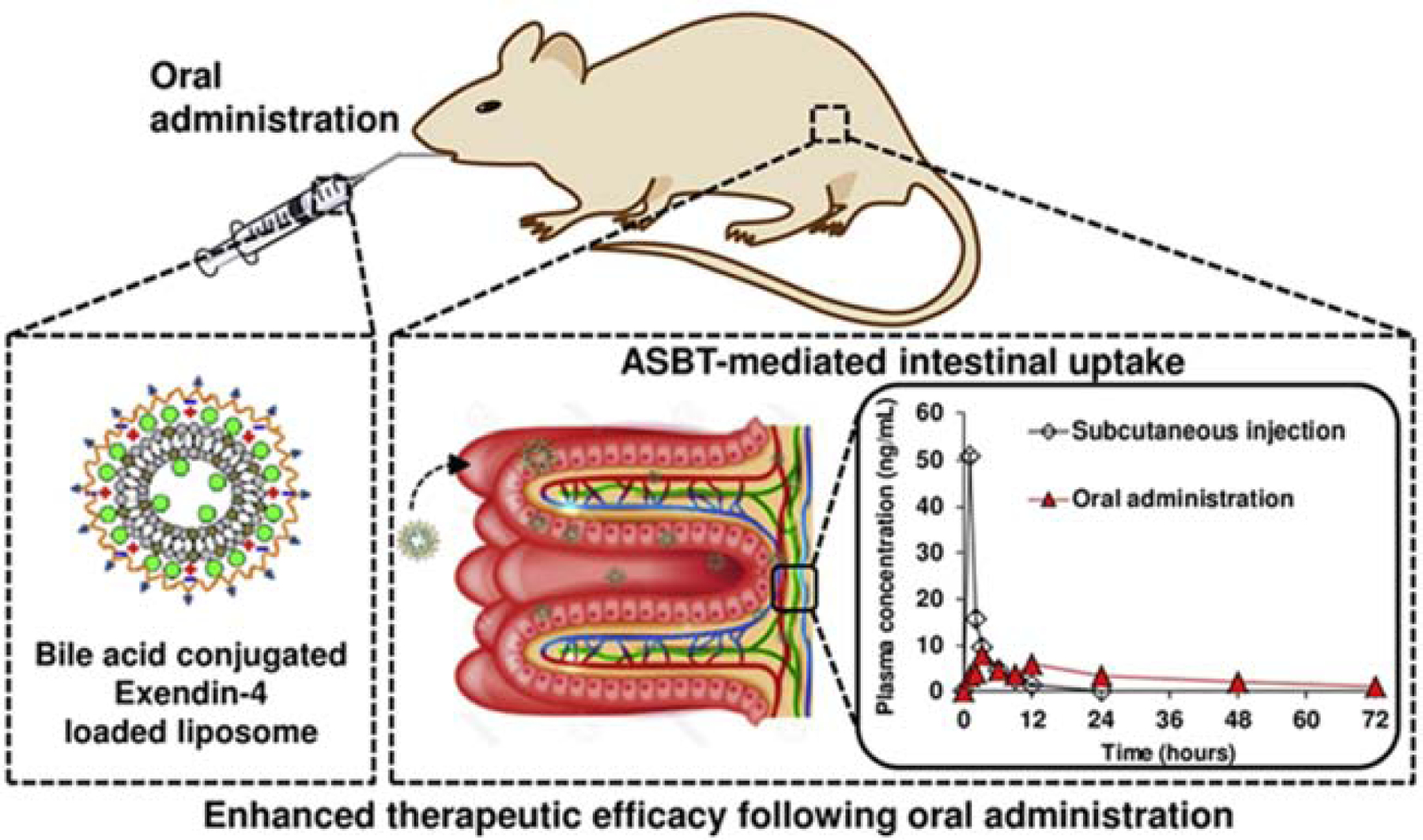
EL-CSG design and its efficacy in oral absorption enhancement. Glycocholic acid was conjugated to CS and then coated on Ex-4 loaded liposomes. After oral administration, EL-CSG was mainly absorbed by ASBT-mediated intestinal uptake in small intestine, with 19.5% oral bioavailability compared with s.c. injection. Reproduced, with permission, from reference [143].
3.3.2. Bilosome
Bilosomes are bilayer vesicular systems composed of bile salts and nonionic surfactant (and charge inducer), which was first described by Conacher et al. for oral delivery of peptide and protein antigens [144]. The main difference where bilosomes are distinguished from liposomes is that bile acids are directly anchored in the bilayer. Compared with traditional liposomes, bilosomes have higher stability in GI fluid and do not undergo oxidative degradation [145]. Although the bilosomes were originally designed for oral immunization, their application has covered the delivery of many APIs nowadays.
Because of its high GI stability, bilosomes are favored for oral administration of many biopharmaceuticals, among which vaccines/antigens are most typical. Haemagglutinin antigen (HA) was entrapped within bilosomes composing of 1-monopalmitoyl glycerol (150 μmol), cholesterol, dicetyl phosphate and deoxycholic acid for influenza treatment [146]. It was found that IgG1 titre was induced and equivalent to s.c. injection in mice, while no IgG2a production was observed, indicating the activation of specific but systematic response. Vyas’s group investigated hepatitis B surface antigen (HBsAg)-loaded bilosomes which were stable and with more than 90% HBsAg remaining in bilosomes after incubation in digestive fluid in 2 h [147]. After intragastric administration in mice, significantly higher sIgA mucosal response was observed compared with alum adsorbed HBsAg formulation. It was found that the bilosomes loading FITC-BSA were highly distributed in gut-associated lymphoid tissues (GALT), while almost no unentrapped FITC-BSA was observed, indicating bilosomes might alter the transport pathway into lymphatic routes. This GALT-targeting might explain the higher immune response with bilosomes. Bilosomes were also used for oral delivery of diphtheria toxoid (DTx) with 88% and 92% DTx retained in the vesicles in simulated gastric fluid and intestinal fluid, respectively [148]. The DTx-loaded bilosomes showed higher sIgA induction in nasal, vaginal, intestinal, and saliva secretions after oral administration in mice compared with alum-adsorbed DTx, and rhodamine-labeled bilosomes demonstrated higher GALT distribution than unloaded DTx. These results illustrate that bilosomes are able to maintain the activity as well as enhance the efficacy compared with conventional vaccines for oral delivery.
Some other APIs can also be administrated by bilosomes. Chen and coworkers prepared fenofibrate-loaded bilosomes with soybean phosphotidylcholine (SPC) and sodium deoxycholate (SDC) and liposomes with SPC and cholesterol (CL) [149]. After oral administration in beagle dogs, the Tmax of the SPC/SDC bilosomes and SPC/CL liposomes were 0.79 and 0.74 h, indicating faster absorption than micronized fenofibrate (Tmax = 1.14 h). Besides, SPC/SDC bilosomes showed bioavailability 1.57-fold higher than SPC/CL liposomes, and 5.13-fold to micronized fenofibrate.
Currently, bilosomes are mainly used in oral delivery to protect biopharmaceuticals from digestive enzymes and harsh pH conditions in GI tract. The evidence of interaction with bile acid transporters, however, is still lacking. The special structure of bilosomes might be an obstacle for utilization of ASBT pathway: when intercalated in the bilayer, the hydrophobic parts of bile acids are oriented to midside and have little opportunity to be exposed to ASBT, especially for those with multilamellar structures. It was once assumed that the enhanced absorption of bilosomes might be led by rapid formation of mixed micelles from vesicles or uptake by M cells into lymphatic systems [149]. Nevertheless, it seems that bilosomes might be used for ASBT targeting with further surface decoration with bile acids, and no further coating is required due to its high GI stability. Further intestinal distribution and formulation-transporter interaction study might provide additional information in the future.
3.3.3. Self-assembled nanoparticle (SaNP)
The amphiphilic property of bile acids promotes the formation of nanoparticles for drug delivery with appropriate modification. Generally, bile acids are usually conjugated with synthetic or biological polymers to form SaNPs for drug delivery, such as poly(lactic-co-glycolic acid) (PLGA), chondroitin sulfate, chitosan, polyethylene glycol (PEG), poly-ε-caprolactone (PCL), and poly(vinyl caprolactam)-poly(vinyl acetate)-poly(ethylene glycol) (Soluplus). Such amphiphilic structure will self-assemble into core-shell nanostructure in water with intra-/intermolecular association between the hydrophobic segments [150]. For instance, self-assembled hybrid nanoparticles (SHNPs) were developed with sodium taurocholate (STC), and Soluplus and Felodipine (FLDP) were loaded as APIs [151]. At STC/FLDP = 1:9, the SHNP showed 40.8 ± 2.7 nm diameter and remained stable with acetate buffer and PBS, with a little decrease in 0.1M HCl. Only 28.1% FLDP was released in 8 h in 0.05% SDS solution. The bioavailability of SHNPs was 1.6-fold to that of STC-free nanoparticles, and the integrity of SHNPS in ileum was confirmed by water-quenching probes. The in situ permeation study showed no discrepancy of Papp among duodenum, jejunum, ileum, and colon, while the ASBT inhibitor Fluvastatin sodium could lead to reduction of Papp up to 58%, and similar permeability was observed in dead intestine lacking of active transporter compared with normal intestine with Fluvastatin sodium, indicating the transport was mediated by ASBT as well as passive diffusion.
Khatun et al. developed taurocholic acid linked heparin-docetaxel nanoparticle (HDTA) as an oral ASBT-targeting system, where docetaxel was used as the active component, and heparin was both a permeation enhancer and an anti-angiogenesis material (Figure 8)[152]. Taurocholic acid linked heparin (HTA) was completely dissolved in the water due to the hydrophilicity, while HDTA formed into 100–120 diameter nanoparticles. After oral administration in SKH1 mice, both HTA and HDTA were observed in epithelial tissues by TEM, and HDTA showed higher AUC value than HTA (338 vs 257 μg·min/mL) at 5 mg/kg dosage. In KB tumor bearing mice, HDTA-rhodamine B was accumulated only in tumor after 24 h, and tumor volume decreased to 54% of control and 63% of the docetaxel group after administration with HDTA4 in 24 days. The tumor volume of HDTA in MDA-MB231 tumor-bearing mice showed 5-fold reduction to the control, while the antitumor efficacy of HTA was 50% less than that of HDTA.
Figure 8.
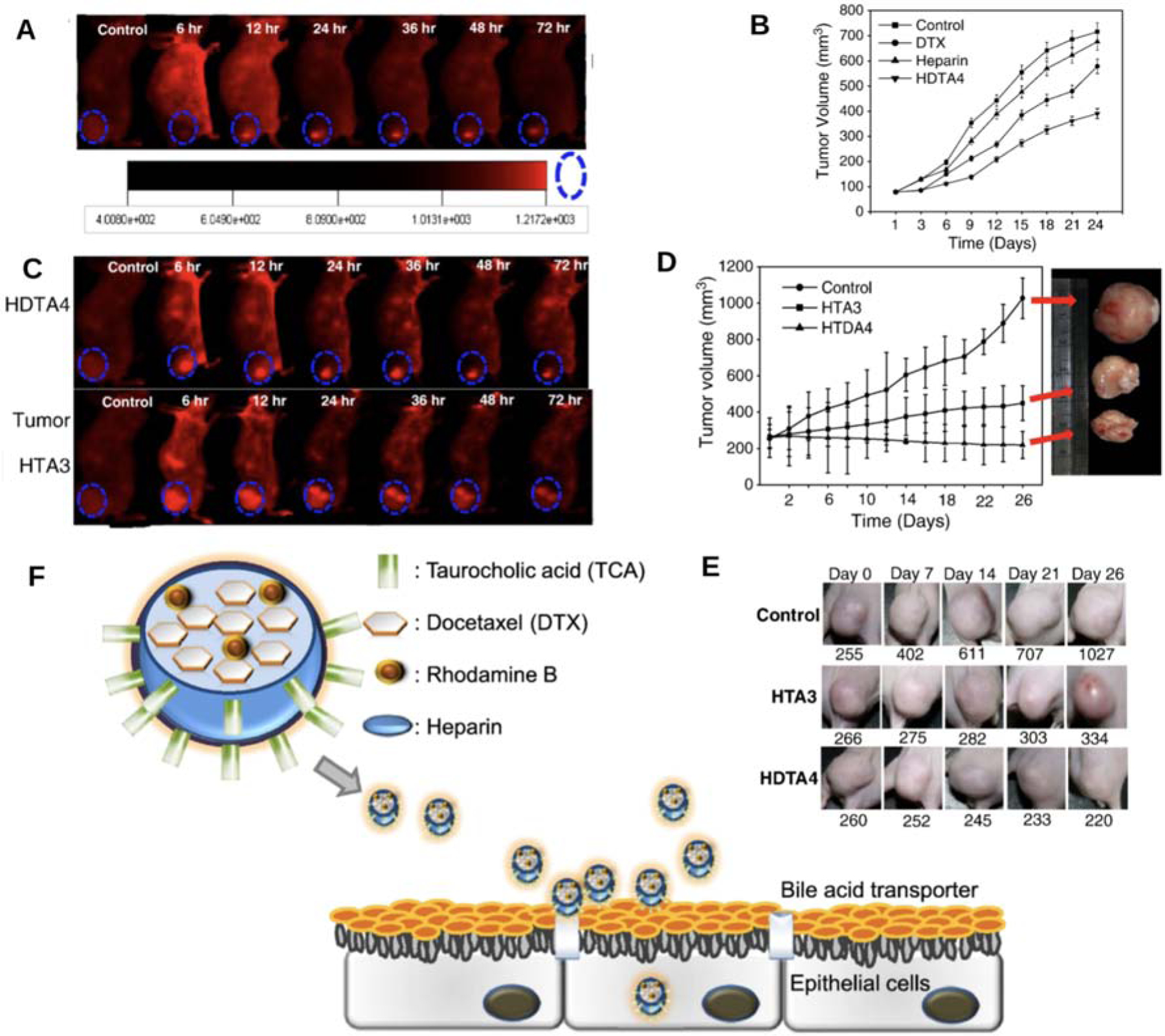
(A–B) Tumor accumulation and antitumor activity of HDTA in KB tumor bearing mice. (C–D) Tumor accumulation and antitumor activity of HDTA4 and HTA3 in MDA-MB231 tumor bearing nude mice. (E) Tumor volume in in MDA-MB231 tumor bearing nude mice after HDTA4 and HTA3 treatment. (F) Schematic illustration of HDTA for oral delivery of DTX using bile acid transporter. Reproduced, with permission, from reference [152].
The same research group also reported low molecular weight heparin (LMWH)-conjugated deoxycholate (DOCA)/(LHD) for loading quantum dots (QDs) [153]. As an imaging agent, the QDs-LHD demonstrated about 200 nm size at pH from 5–9 and only slight size increase in serum with minimal QDs release in 5 days. After oral administration in SKH1 mice, the fluorescence was mainly accumulated in jejunum and ileum, with 62–90% fluorescence intensity in these two sections, and both fluorescence and TEM showed ileum has the highest QDs distribution, indicating the absorption mechanism of QDs-LHD was likely ASBT-mediated.
Chaturvedi and coworkers designed deoxycholic acid (DOCA) conjugated PEGylated polyhydroxybutyrate (PHB) co-polymeric nanoparticles for insulin oral delivery [154]. The amphiphilic DOCA-PEG-PHB preferred assembled into nanoparticles in aqueous solution, and this system was further fabricated into granules and coated with Eudragit S-100. After oral administration in rats, the coated granule showed slow but extended blood glucose level reduction in 12 h, while s.c. injection of insulin produced a sharp decrease of blood glucose level but gradually recovered to the original condition in 12 h. The relative bioavailability of the coated granule was 11.6% of s.c. injection, and the blood insulin level kept being upregulated in 24 h, revealing the controlled release property of this granule. With rhodamine loaded, the PHB-PEG nanoparticles demonstrated diffusive nature in HCT-116 human colon cancer cells, whereas DOCA-PEG-PHB nanoparticles adhered to the cell surface and then internalized by endocytosis to much greater extent. The difference between the two systems implied the DOCA-PEG-PHB nanoparticles were likely to be taken via bile acid transporter.
Apart from conjugated to polymers for nanoparticle formation, bile salts can also be mixed with lipids and polymers for self-assembly. To develop a stable formulation for oral delivery of vitamin K, Nostrum’s research group prepared mixed SaNPs composed of glycocholic acid, egg phosphatidylcholine (EPC), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000] (DSPE-PEG2000) [155]. Compared with commercial product (Konakion® MM), the mixed SaNPs demonstrated higher stability in acidic pH conditions at DSPE-PEG/EPC = 50/50 (mol/mol), with size slightly changed from 10 to 12 nm and 90% vitamin K retained solubilized. Another example is the sodium cholate/phospholipid-mixed SaNPs for enhancement of poorly soluble drug silybin [156]. Compared with silybin-N-methylglucamine, silybin-loaded mixed SaNPs showed slow release of silybin in pH = 1.2 or 7.4 solutions, and 252% relative bioavailability after oral administration in dogs.
Although the amphiphilic structure of bile acids and their polymer conjugates is favored for formation of SaNPs, it should be mentioned that in most cases the hydrophobic groups of bile acids are buried inside the nanoparticles, which might impede interaction with ASBT. It has been reported that deoxycholic acid-heparin could be completely dissolved with addition of DMSO to expose the bile acids, and the solubilized form showed higher oral availability than SaNPs [157]. This suggested that formation of SaNPs indeed shielded the bile acids and hindered the ASBT-mediated transport. Therefore, bile acid-hydrophilic material SaNPs is still not solid enough in ASBT-mediated transport and needs further explanation, though some evidence has been found.
3.3.4. Other bile acid-containing nanocarriers
Other bile acid-containing nanocarriers are also used for oral drug delivery, though not as commonly as those mentioned above. For example, rhodamine B-loaded PLGA nanoparticles were protected by deoxycholate acid (DCA) emulsion to enhance the oral bioavailability [158]. With the existence of DCA, release of rhodamine B was significantly retarded in varied pH conditions. After oral administration in C3H mice, rhodamine B-loaded PLGA nanoparticles showed similar pharmacokinetics behavior with free rhodamine B, while the bioavailability boosted to 1.81-fold with addition of DCA. As PLGA was biodegradable in stomach, its degradation eliminated the difference between nanoparticles and free probes, and the enhancement by DCA illustrated the protection effect for PLGA-nanoparticles. This study provided a new angle of bile acid utilization for oral drug delivery, and it might be further applied with more explanation including the interaction mechanism between DCA and PLGA and the possible enhancement or transporter-targeting in intestine.
3.4. Current status of ASBT-targeted drug delivery
Discovery of ASBT creates new opportunity for enhancing oral bioavailability of absorption-limited drugs by utilizing the enterohepatic circulation of bile acids. In the last several decades, various strategies were developed including bile acid-conjugated prodrugs, electrostatic complexation between bile acids and drugs, and using bile acid nanocarriers, which have made great achievements in enterocytes uptake and oral bioavailability. Some of the strategies are summarized in Table 3. These studies prove the potential of utilizing bile acids in clinics to relieve the dilemma of absorption of BCS III/IV class drugs. Among the numerous bile acid-based studies, in contrast, the illustration of ASBT-targeted strategies is still shallow. One reason is the blurry functions of bile acids in GI tract. As stated above, bile acids can play as permeation enhancers, emulsifiers, ASBT substrates, or a combination of these. Tangible evidence is required to verify the ASBT participation, whether exclusive or synergistic, which requires systematic study from drug delivery to molecular biology studies.
Table 3.
Some strategies of bile acid-based oral drug administration
| Strategies | APIs | Bile acids (or derivatives) | Efficacy | References |
|---|---|---|---|---|
| Prodrug | S1037 | 3-Diazirine-Derivatives of bile acid | Increasing uptake, inhibited by taurocholate | [113] |
| Prodrug | acyclovir | chenodeoxycholate | 198% drug recovery | [114] |
| Prodrug | steroidal pyrazoles | glycodeoxycholic acid | Higher uptake with monoanionic form | [115] |
| Prodrug | gabapentin | chenodeoxycholic acid | Higher uptake with single charge | [117] |
| Prodrug | Asp-β-benzyl ester | cholic acid | Inhibition of taurocholate uptake | [119] |
| Prodrug | cytarabine | ursodeoxycholic acid | Relative bioavailability 209% | [120] |
| Prodrug | camptothecin | deoxycholic acid | 65.48% uptake inhibition by taurocholate in cell spheroids | [121] |
| Prodrug | LWMH | deoxycholic acid | ASBT-colocalization | [92] |
| Electrostatic | trospium chloride | Glycochenodeoxycholate, taurodeoxycholate | Enhanced permeation in Caco-2 | [123] |
| Electrostatic | ceftriaxone | cholyethyendiaine | Relative bioavailability 1300% | [124] |
| Electrostatic | LWMH | deoxycholylethylamine | Relative bioavailability 2500% | [125] |
| Electrostatic | EGFR2R-lytic hybrid peptide | taurodeoxycholate | 64% tumor volume | [126] |
| Prodrug/Electrostatic | LHT7 | tetrameric deoxycholic acid/deoxycholylethylamine | Relative bioavailability 916% | [127] |
| Electrostatic | ibandronate | Nα-deoxycholyl-L-lysyl-methylester | Relative bioavailability 430% | [164] |
| Liposomes | salmon calcitonin | taurodeoxycholate | Relative bioavailability 710% | [165] |
| Liposomes | insulin | deoxycholic acid | Relative bioavailability 143% | [142] |
| Liposomes | Exendin-4 | glycocholic acid | Bioavailability 19.5% to s.c. injection | [143] |
| Bilosomes | haemagglutinin antigen | deoxycholic acid | Higher sIgA titer | [146] |
| Bilosomes | hepatitis B surface antigen | sodium deoxycholate | Higher sIgA titer | [147] |
| Bilosomes | diphtheria toxoid | sodium deoxycholate | Higher sIgA titer | [148] |
| Bilosomes | fenofibrate | sodium deoxycholate | Relative bioavailability 513% | [149] |
| SaNPs | felodipine | sodium taurocholate | Relative bioavailability 160% | [151] |
| SaNPs | vitamin K | glycocholic acid | Higher drug stability/solubility | [155] |
| SaNPs | heparin-docetaxel | taurocholic acid | Relative bioavailability 132% | [152] |
| SaNPs | quantum Dots | deoxycholic acid | Jejunum/ileum accumulation | [153] |
| SaNPs | silybin | sodium cholate | Relative bioavailability 252% | [156] |
| Nanoparticles | rhodamine B | deoxycholic acid | Relative bioavailability 181% | [158] |
Another reason is the interaction mechanism between ASBT and bile acid-based drugs, which might be altered from the way of free bile acids pumping. Although some computational studies depicted the interaction model of ASBT and its substrates [159, 160], it is doubtful whether the small size of ASBT cavity (6 Å × 12 Å × 14 Å) allows the permeation once bile acids are conjugated to drug molecules or formed nanocarriers [161]. On one hand, Byun’s group designed self-assembled nanocomplex with deoxycholic acid (DOCA) conjugated with low molecular weight heparin (LMWH) or protamine, and it was observed that the nanocomplex bound to the Caco-2 cell clusters and then caused cell membrane collapse, which suggested the receptor-mediated uptake mode rather than transporter-mediated [162]. On the other hand, they confirmed the existence of ASBT in the endosomes by gold-conjugated antibody [161], and some other colocalization and taurocholate inhibition experiments substantiate the participation of ASBT in the uptake process. The limited understanding of ASBT in the bile acid-based drug uptake process might affect the design of drug delivery system.
In addition, the interactions between ASBT and bile acids can be blocked by the hidden binding site of bile acids. It has been reported that C3 does not specifically interact with ASBT and is preferred for ASBT-mediated uptake, while C24 modification might lead to slow uptake rate, though the affinity is retained [163]. However, C24 modification is still a very important strategy due to its high chemical accessibility. The C3 modification also assumes risks as hydroxyl group of C3 is essential for IBABP binding as we described before. In some bile acid-containing nanocarriers, long polymer chains increase the steric repulsion and stabilize them from aggregation. Interaction between bile acids and ASBT, nevertheless, can also be disturbed if the bile acids are buried in these polymer chains. These obstacles either weaken the efficacy of ASBT-bile acid interaction or result in the complicated functions of bile acids in intestine, requiring further exploration on formulation optimization and mechanism clarification.
4. Transport pathways of bile acid-based drugs
Although bile acid-based oral drug delivery has been studied for a few decades, its transport pathway research is still in its infancy stage. The permeation pathways through the enterocytes are classified into passive diffusion, transcytosis, and paracellular transport. The main routes of bile acids uptake via ASBT is transcytosis, which includes endocytosis, intracellular transport, and exocytosis pathway.
4.1. Endocytosis of drug in epithelial cells
Endocytosis of the bile acid-based drugs in epithelial cells is the prerequisite and the first step of oral drug absorption [13]. Generally, the drugs can be internalized by multiple pathways in enterocytes, including clathrin-mediated endocytosis (CME), caveolin-dependent endocytosis (CDE), macropinocytosis, and clathrin/caveolin-independent endocytosis (CIE) [166]. CME is based on the progressive and sequential assembly of clathrin-coated vesicles, which contain various transmembrane receptors and corresponding ligands [167]. The heterotetrametric adaptor protein AP2 stabilizes the nascent clathrin-coated vesicles, driving their maturation [168]. Caveolae are cave-like membrane structures located in glycolipids that are rich in cholesterols and sphingolipids, and the attachment of caveolin scaffolding to the plasma membrane enables the expansion of caveolae vesicles and activates CDE [167]. Macropinocytosis relies on the self-protrusion and back fusion of plasma membrane, which is dependent on the activity of Na+/H+ exchanger [169]. Some other CIE routes, mediated by small GTPases including Flotillin, Arf6 or RhoA are not as common as those mentioned above, but are confirmed relevant to the uptake of nanocarriers recently [170].
Up to now, there are only a very few studies clarifying the uptake mechanism of bile acid-based formulations. As previously stated in this review, colocalization between ASBT and bile acid-modified molecules or nanoparticles has been observed [92, 93, 162]. However, it is not clear how the ASBT/bile acid/drug complexes enter the cells, whether with a known pathway or an entirely new mechanism. It was reported that ASBT was associated with lipid raft, and depletion of cholesterol with Methyl-β-Cyclodextrins (MβCD) could reduce the ASBT activity [171]. This discovery implies that ASBT-mediated pathway might be relevant to CDE or at least lipid raft-dependent endocytosis.
Li prepared silybin-loaded nanoliposomes modified with cholic acid (CA-LPs-silybin) and investigated the uptake mechanism on Caco-2 cells [172]. CA-LPs-silybin demonstrated about 2-fold Papp to that of liposomes without cholic acid though the Caco-2 monolayer, suggesting that the ASBT-mediated transport enhanced the permeability. Some endocytic inhibitors were applied to test the CA-LPs-silybin transcytosis through Caco-2 monolayer. It was found that multiple inhibitors could block the transcytosis: chlorpromazine (CME inhibitor), MβCD and nystatin (CDE inhibitors), NaN3 and 4°C temperature (ATP inhibitors), genistein (tyrosine protein kinase inhibitor, relevant to CDE), and cholic acid (ASBT saturation effect), indicating CA-LPs-silybin uptake proceeded via various routes. Besides, the effectiveness of tight junction disrupter did not affect the Papp, which revealed that the liposomes transported by transcytosis rather than paracellular pathway.
Yin’s group synthesized amphiphilic sulfhydryl modified N-deoxycholic acid-N,O-hydroxyethyl chitosan to prepare paclitaxel-loaded micelles (PTX-TGA-DHC). In Caco-2 cells, chlorpromazine and nystatin showed inhibition effect on PTX-TGA-DHC uptake, while amiloride, a macropinocytosis inhibitor, did not disturb the internalization. Therefore, PTX-TGA-DHC uptake was through CME and CIE, but not micropinocytosis. Although the authors confirmed P-gp inhibition by sulfhydryl modification and tight junction opening by chitosan, it is a pity that there was no test whether deoxycholic acid induced the uptake and whether ASBT was involved [173].
Pangeni and coworkers prepared a complex with pemetrexed (PMX) and a bile acid derivative Nα-deoxycholyl-L-lysyl-methylester (DCK) by ion-pairing and mixed it with dispersing agents into oral powder formulation (PMX/DCK-OP) [174]. The endocytosis mechanism was tested in Caco-2 monolayers with various inhibitors. It was found that the transcytosis was significantly inhibited by Act D (ASBT-mediated transport inhibitor), genistein, and amiloride, while chlorpromazine and MβCD did not, indicating the uptake of PMX/DCK-OP was ASBT-mediated, and micropinocytosis was involved in the pathway, but CME not. As for CDE, genistein showed significant uptake inhibition effect but MβCD not. This might be owing to their different action modes, suggesting the uptake was regulated by tyrosine protein kinase while cholesterol had no effect. This result might be a new clue to inspect the conclusion of ASBT-lipid raft association.
4.2. Intracellular transport and exocytosis
After endocytosis, the ASBT/bile acid/drug complexes are delivered by endosomes and separated, followed by the recycling of ASBT as described in Section 2.3.3. The intracellular bile acid-based drugs are hypothesized to be transported by binding to IBABP. Colocalization between LHe-tetraD and IBABP was observed by Al-Hilal’s research as described in Section 2.4. Similarly, Gan’s group also confirmed the colocalization between IBABP and deoxycholic acid-modified nanoparticles (DNPs) in Caco-2 cells [175].
It should be noted that colocalization between bile acid and IBABP did not tell the specific intracellular stations of bile acid based-drugs. In general, series of endosomes, endoplasmic reticulum (ER), Golgi apparatus, and lysosomes are the most possible docks of endocytic cargoes. Most receptor/transporter-mediated nanocarriers are transported to lysosomes after endocytosis with the acidification of endosomes. Entrapment in lysosomes prevents the transcytosis to the basolateral side of enterocytes; meanwhile, the abundant enzymes can cause the metabolism and degradation of nanocarriers and drugs [176, 177]. Therefore, lysosomal escaping has become an essential topic in formulation design. To study the intracellular destination of bile-based drugs, bile acid-based oral delivery systems and the lysosomes were measured by colocalization or using endosomal acidification inhibitor chloroquine in many studies, and almost all of them confirmed the lysosomal escape [92, 93, 175, 178, 179], suggesting the potential of increasing drug stability and permeation in bile acid-based delivery. The specific mechanism of lysosomal escaping, was not clear.
ER-Golgi apparatus-cell membrane is an essential pathway for cargo secretion, although there is some retrieval delivery from Golgi apparatus to ER [180]. Our research group discovered that glycocholic acid-conjugated polystyrene nanoparticles (G40/CPN) colocalized with ER and Golgi apparatus after uptake into SK-BR-3 cells, indicating both ER and Golgi were involved in the intracellular delivery of G40/CPN [93]. In the PMX/DCK-OP intestinal transport study (see Section 4.1), the ER-Golgi apparatus transport was investigated using the inhibitor brefeldin, which showed Papp was reduced to 43.2% [174]. These results illustrated the transcytosis of those cargoes transporting through ASBT was regulated by both ER and Golgi apparatus.
Although some research claimed the relationship between common transport routes and bile acid-based drugs, there are still many questions to answer. It is still not clear how ASBT interacts with large drug molecules/nanoparticles and is internalized, and at which stage ASBT and bile acids are separated for IBABP binding. To our knowledge, there is no specific study on OSTα/β and bile acid-based drugs. All of these blank fields still require further exploration. In addition, it is worth noting that the mechanism could be varied by different cell line models and formulations, and not all tested cell lines expressing ASBT are from the ileum and may not reflect what exactly happens in the enterocytes in vivo. With proper selection of models and involvement of modern molecular biological technologies, it is expected to find the connections within the numerous studies in the future.
5. New route: intestinal lymphatic transport of bile acid-based drugs
As described in Section 2.3, lymphatic delivery is favored owing to the avoidance of hepatic first-pass effect and slower elimination. In most cases, drugs were delivered to portal veins after transport through the enterocytes, while the lymphatic vessels are mainly utilized for antigens and chylomicron transport. Although the fat absorption needs emulsification and co-absorption with bile acids, the fat digestion and uptake is mainly in the duodenum, differing from ASBT-mediated transport which occurs in the ileum [181]. Therefore, the lymphatic delivery was not considered as the pathway for bile acid-based drug via ASBT endocytosis, until some new discoveries were reported recently.
Zhang and coworkers tested the effect of sodium cholate (SC) on intestinal absorption of candesartan cilexetil loaded lipid nanocarriers (CLN) [182]. Addition of SC enhanced the absorption of candesartan by 1.79-fold. After treatment with cycloheximide, an inhibitor which blocked lymphatic transport pathway by inhibiting chylomicron secretion [183], the AUC(0−t) of CLN and CLN+SC decreased by 20% and 46%, respectively, and showed no significant difference, indicating that blocking the lymphatic route could eliminate the permeation enhancement of SC, and that increasing absorption was due to the SC-induced lymphatic transport. Another example is the development of paclitaxel-loaded bilosomes with sodium deoxycholate (BS-Lips) [184]. Compared with liposomes, BS-Lips exhibited 2.5-fold greater AUC, and cycloheximide treatment led to 20% and 60% reduction in bioavailability of Lip and BS-Lip, respectively. Both of the studies showed that lymphatic delivery was pivotal to the absorption of these bile acid-containing formulation. However, it was still not clear whether the lymphatic transport was relevant to ASBT, as it was unlikely to activate ASBT-mediated pathway for drug delivery simply by co-administration of SC or using bilosomes.
Our research group developed insulin-loaded partially uncapped liposome (IPUL-CST) with chondroitin sulfate-taurocholate acid conjugates [185]. Compared with liposomes without taurocholate, larger uptake in Caco-2 of IPUL-CST and higher insulin distribution in ileum of Balb/c mice demonstrated the ASBT-mediated endocytosis. After oral gavage in rats, the insulin concentration in lymphatic vessels gradually increased in 6 h, and IPUL-CST was observed in lymphatic vessels by TEM imaging, suggesting the relationship between ASBT-mediated endocytosis and lymphatic transport. Similarly, we also found glycocholic acid-conjugated polystyrene nanoparticles (G40/CPN) could be transported to lymphatic systems after oral administration in rats [93]. G40/CPN were observed in mesenteric lymph node and cisterna chyli, and the concentration reached the peak at 3 h after gavage. With the treatment of cycloheximide, the oral bioavailability of G40/CPN significantly decreased to 13%. It was concluded that the G40/CPN shared the chylomicron pathway in ileum and transported via lymphatic vessels, which might give new understanding in transport behavior in distal small intestine. Docetaxel-loaded cationic solid lipid nanoparticles (DSLN-CSG) was also designed with glycocholic acid modification to improve the oral absorption of docetaxel (DTX) [186]. After oral administration in rats, DTX concentration in lymph was comparable with that in plasma, while absence of glycocholic acid resulted in lower concentration in both plasma and lymph.
The latest discovery on lymphatic delivery that shares the pathway of chylomicrons in ileum might explain the high efficacy of bile acid-based drug to a degree. The higher uptake by ASBT-mediated pathway and less hepatic degradation lymphatic delivery greatly improve the permeation and stability of drugs. Despite these exciting achievements, it is still unknown the specific connection between ASBT-mediated uptake and mesenteric lymphatic targeting. In lymphatic targeting study, addition of sodium cholate to carvedilol-loaded niosomes increased the AUC by 40%, and cycloheximide showed 1.8-fold decrease in AUC of sodium cholate-containing niosomes [187]. However, the replacement of sodium cholate by dicetyl phosphate led to the almost same result. This implies that chylomicron pathway is not exclusively triggered by bile acids. Furthermore, it is still early to conclude that bile acids necessarily result in lymphatic delivery. Despite these suspects, it is certain that at least lymphatic delivery is involved in some bile acid-based formulations, and it might direct the new destination of optimizing these oral drug delivery systems.
6. Conclusion and Prospect
The high capacity and specificity in reabsorption of bile acids via ASBT into enterocytes inspires the utilization of bile acid-based drug delivery strategies for improving the oral bioavailability of drugs. Bile acids have widely been used in prodrug design, electrostatic complexation and nanocarriers for permeation enhancement. Although some studies did not confirm the involvement of bile acid transporter, or they implied some other pathways like paracellular route or passive diffusion, more recent studies showed the great efficacy of ASBT-mediated uptake. Mechanism studies on endocytosis, intracellular transport and exocytosis revealed some common ground of various bile acid-based drugs, including the involvement of ER and Golgi complexes, and lysosome escape. New discovery of lymphatic delivery following ASBT-mediated uptake indicates its potential of avoiding first-pass effect and rapid elimination.
More prospective nanocarriers are expected to be applied in bile acid transporter-based oral drug delivery in the future. One of them is mesoporous silica nanoparticles (MSN), which is a promising novel drug vehicle due to its large volume, uniformly mesoporous structure, surface functionality and biocompatibility [188]. With the homogenous pore size, MSN allows fine control of drug load and release kinetics [189]. Zhang et al. reported that MSN improved the bioavailability of telmisartan (TEL) after oral administration: compared with commercialized product Micardis, TEL-loaded MSN showed relatively bioavailability 154.4 ± 28.4% [190]. There have also been efforts to actively modifying MSN. For instance, Porta and coworkers developed folic acid-modified MSN for targeting delivery in U2O cells, a cell line with high folic acid receptor expression. With anticancer drug camptothecin loaded, MSN demonstrated higher cytotoxicity than free camptothecin solution in DMSO (93% vs 50% cell death) [191]. Combining their oral controllable delivery advantage and ligand-modification availability, MSN might be a promising candidate as an ASBT-targeted nanocarrier.
Another potential nanocarrier is nanocrystal, which might be applied in permeation enhancement of BCS IV drugs. Nanocrystals consist essentially of pure API nanoparticles and a small amount of surfactants, which demonstrated high drug load, improved solubility and bioadhension to the intestinal wall [27]. With these superiorities, nanocrystals or amorphous drug granules have been widely studied in oral drug delivery of BCS IV drugs. In the last decades, targeting ligand modification has gradually been a new direction for nanocrystals. Park’s group modified docetaxel nanocrystals (DTX-NCs) with apo-Transferrin (Tf) to improve the uptake in A549 cells. It is shown that Tf-DTX-NCs triggered higher cytotoxicity than DTX-NCs and DTX pure drugs at all tested concentrations [192]. It is interesting that sorafenib nanogranules have been reported to transport through the Caco-2 monolayer via chylomicron routes, indicating the potential of this formulation in lymphatic delivery [193]. Due to their developed manufacturing technologies, it is expected that bile acid-modified nanocrystals might make a difference in improving oral absorption.
Development of ASBT-targeted drug delivery systems has been validated for no more than two decades, before which bile acids have been widely applied in pharmaceutical research. As a novel strategy, ASBT-mediated oral drug delivery demonstrates promising efficacy as well as some unexplored fields. The balance between transporter targeting and enhancer property of bile acids hampered the exclusivity of ASBT-mediated pathway for the uptake of bile acid-based drugs, and the transport mechanisms varied between formulations and cell models, which impeded the understanding of the relationship between the bile acid transporters and cell components that are responsible for cargo delivery. The limited understanding of connection between ASBT and the following lymphatic delivery requires more exploration to control the navigation of drugs. Furthermore, many bile acid-based formulation technologies are still limited in lab scale, whose manufacturing is complicated or expensive. At present, there is no clearly intestinal bile acid transporter targeted drug in clinical research, currently falling behind chemical permeation enhancers like sodium salcaprozate and N-(5-chlorosalicyloyl)-8-aminocaprylic acid [194, 195]. More investigation in mechanistic clarification and formulation optimization is expected to raise the bile acid-targeting oral drug delivery into a new epoch.
Acknowledgements
This work was partially supported by the national Institute of Health (NIH DK114015).
The authors would like to thank Nithya Bala Subrahmanyam from Department of Pharmaceutics and Pharmaceutical Chemistry, University of Utah, for her kindly proofread and editing to improve this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
YHB is the founder of Ileo Science, Inc and FD declares no conflict of interest.
References
- [1].Sharma M, Sharma R, Jain DK, Nanotechnology Based Approaches for Enhancing Oral Bioavailability of Poorly Water Soluble Antihypertensive Drugs, Scientifica (Cairo), 2016 (2016) 8525679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ganta S, Deshpande D, Korde A, Amiji M, A review of multifunctional nanoemulsion systems to overcome oral and CNS drug delivery barriers, Mol Membr Biol, 27 (2010) 260–273. [DOI] [PubMed] [Google Scholar]
- [3].Sastry SV, Nyshadham JR, Fix JA, Recent technological advances in oral drug delivery - a review, Pharm Sci Technolo Today, 3 (2000) 138–145. [DOI] [PubMed] [Google Scholar]
- [4].Homayun B, Lin X, Choi HJ, Challenges and Recent Progress in Oral Drug Delivery Systems for Biopharmaceuticals, Pharmaceutics, 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wang XQ, Zhang Q, pH-sensitive polymeric nanoparticles to improve oral bioavailability of peptide/protein drugs and poorly water-soluble drugs, Eur J Pharm Biopharm, 82 (2012) 219–229. [DOI] [PubMed] [Google Scholar]
- [6].Ahn J, Cao MJ, Yu YQ, Engen JR, Accessing the reproducibility and specificity of pepsin and other aspartic proteases, Biochim Biophys Acta, 1834 (2013) 1222–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Boegh M, Nielsen HM, Mucus as a Barrier to Drug Delivery - Understanding and Mimicking the Barrier Properties, Basic Clin Pharmacol, 116 (2015) 179–186. [DOI] [PubMed] [Google Scholar]
- [8].Shan W, Zhu X, Liu M, Li L, Zhong J, Sun W, Zhang Z, Huang Y, Overcoming the diffusion barrier of mucus and absorption barrier of epithelium by self-assembled nanoparticles for oral delivery of insulin, ACS Nano, 9 (2015) 2345–2356. [DOI] [PubMed] [Google Scholar]
- [9].Li HP, Guissi NE, Su ZG, Ping QN, Sun MJ, Effects of surface hydrophilic properties of PEG-based mucus-penetrating nanostructured lipid carriers on oral drug delivery, Rsc Adv, 6 (2016) 84164–84176. [Google Scholar]
- [10].Brown TD, Whitehead KA, Mitragotri S, Materials for oral delivery of proteins and peptides, Nat Rev Mater, 5 (2020) 127–148. [Google Scholar]
- [11].Butnarasu C, Barbero N, Pacheco D, Petrini P, Visentin S, Mucin binding to therapeutic molecules: The case of antimicrobial agents used in cystic fibrosis, Int J Pharmaceut, 564 (2019) 136–144. [DOI] [PubMed] [Google Scholar]
- [12].Collado-Gonzalez M, Espinosa YG, Goycoolea FM, Interaction Between Chitosan and Mucin: Fundamentals and Applications, Biomimetics, 4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Reinholz J, Landfester K, Mailander V, The challenges of oral drug delivery via nanocarriers, Drug Deliv, 25 (2018) 1694–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Linnankoski J, Makela J, Palmgren J, Mauriala T, Vedin C, Ungell AL, Lazorova L, Artursson P, Urtti A, Yliperttula M, Paracellular porosity and pore size of the human intestinal epithelium in tissue and cell culture models, J Pharm Sci, 99 (2010) 2166–2175. [DOI] [PubMed] [Google Scholar]
- [15].Menard S, Cerf-Bensussan N, Heyman M, Multiple facets of intestinal permeability and epithelial handling of dietary antigens, Mucosal Immunol, 3 (2010) 247–259. [DOI] [PubMed] [Google Scholar]
- [16].Li J, Wang XL, Zhang T, Wang CL, Huang ZJ, Luo X, Deng YH, A review on phospholipids and their main applications in drug delivery systems, Asian J Pharm Sci, 10 (2015) 81–98. [Google Scholar]
- [17].Kaufmann AM, Krise JP, Lysosomal sequestration of amine-containing drugs: analysis and therapeutic implications, J Pharm Sci, 96 (2007) 729–746. [DOI] [PubMed] [Google Scholar]
- [18].Amin ML, P-glycoprotein Inhibition for Optimal Drug Delivery, Drug Target Insights, 7 (2013) 27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lipinski CA, Lombardo F, Dominy BW, Feeney PJ, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv Drug Deliv Rev, 46 (2001) 3–26. [DOI] [PubMed] [Google Scholar]
- [20].Kalepu S, Nekkanti V, Insoluble drug delivery strategies: review of recent advances and business prospects, Acta Pharm Sin B, 5 (2015) 442–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Leuner C, Dressman J, Improving drug solubility for oral delivery using solid dispersions, European Journal of Pharmaceutics and Biopharmaceutics, 50 (2000) 47–60. [DOI] [PubMed] [Google Scholar]
- [22].Patel VR, Agrawal YK, Nanosuspension: An approach to enhance solubility of drugs, J Adv Pharm Technol Res, 2 (2011) 81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Narayan R, Pednekar A, Bhuyan D, Gowda C, Koteshwara KB, Nayak UY, A top-down technique to improve the solubility and bioavailability of aceclofenac: in vitro and in vivo studies, Int J Nanomedicine, 12 (2017) 4921–4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].de Waard H, Frijlink HW, Hinrichs WL, Bottom-up preparation techniques for nanocrystals of lipophilic drugs, Pharm Res, 28 (2011) 1220–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Peng C, Svirskis D, Lee SJ, Oey I, Kwak HS, Chen G, Bunt C, Wen J, Design of microemulsion system suitable for the oral delivery of poorly aqueous soluble beta-carotene, Pharm Dev Technol, 23 (2018) 682–688. [DOI] [PubMed] [Google Scholar]
- [26].Chakravarty P, Famili A, Nagapudi K, Al-Sayah MA, Using Supercritical Fluid Technology as a Green Alternative During the Preparation of Drug Delivery Systems, Pharmaceutics, 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gao L, Liu G, Ma J, Wang X, Zhou L, Li X, Wang F, Application of drug nanocrystal technologies on oral drug delivery of poorly soluble drugs, Pharm Res, 30 (2013) 307–324. [DOI] [PubMed] [Google Scholar]
- [28].Dave VS, Gupta D, Yu M, Nguyen P, Varghese Gupta S, Current and evolving approaches for improving the oral permeability of BCS Class III or analogous molecules, Drug Dev Ind Pharm, 43 (2017) 177–189. [DOI] [PubMed] [Google Scholar]
- [29].Wen H, Jung H, Li X, Drug Delivery Approaches in Addressing Clinical Pharmacology-Related Issues: Opportunities and Challenges, AAPS J, 17 (2015) 1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Shaji J, Patole V, Protein and Peptide drug delivery: oral approaches, Indian J Pharm Sci, 70 (2008) 269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Yang C, Tirucherai GS, Mitra AK, Prodrug based optimal drug delivery via membrane transporter/receptor, Expert Opin Biol Ther, 1 (2001) 159–175. [DOI] [PubMed] [Google Scholar]
- [32].Muheem A, Shakeel F, Jahangir MA, Anwar M, Mallick N, Jain GK, Warsi MH, Ahmad FJ, A review on the strategies for oral delivery of proteins and peptides and their clinical perspectives, Saudi Pharmaceutical Journal, 24 (2016) 413–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Obeid R, Heil SG, Verhoeven MMA, van den Heuvel E, de Groot L, Eussen S, Vitamin B12 Intake From Animal Foods, Biomarkers, and Health Aspects, Front Nutr, 6 (2019) 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Clardy SM, Allis DG, Fairchild TJ, Doyle RP, Vitamin B12 in drug delivery: breaking through the barriers to a B12 bioconjugate pharmaceutical, Expert Opin Drug Deliv, 8 (2011) 127–140. [DOI] [PubMed] [Google Scholar]
- [35].Long L, Lai M, Mao X, Luo J, Yuan X, Zhang LM, Ke Z, Yang L, Deng DY, Investigation Of Vitamin B12-Modified Amphiphilic Sodium Alginate Derivatives For Enhancing The Oral Delivery Efficacy Of Peptide Drugs, Int J Nanomedicine, 14 (2019) 7743–7758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Petrus AK, Fairchild TJ, Doyle RP, Traveling the vitamin B12 pathway: oral delivery of protein and peptide drugs, Angew Chem Int Ed Engl, 48 (2009) 1022–1028. [DOI] [PubMed] [Google Scholar]
- [37].Cervenak J, Doleschall M, Bender B, Mayer B, Schneider Z, Doleschall Z, Zhao Y, Bosze Z, Hammarstrom L, Oster W, Kacskovics I, NFkappaB induces overexpression of bovine FcRn: a novel mechanism that further contributes to the enhanced immune response in genetically modified animals carrying extra copies of FcRn, MAbs, 5 (2013) 860–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hornby PJ, Cooper PR, Kliwinski C, Ragwan E, Mabus JR, Harman B, Thompson S, Kauffman AL, Yan Z, Tam SH, Dorai H, Powers GD, Giles-Komar J, Human and non-human primate intestinal FcRn expression and immunoglobulin G transcytosis, Pharm Res, 31 (2014) 908–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sockolosky JT, Szoka FC, The neonatal Fc receptor, FcRn, as a target for drug delivery and therapy, Adv Drug Deliver Rev, 91 (2015) 109–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Pridgen EM, Alexis F, Kuo TT, Levy-Nissenbaum E, Karnik R, Blumberg RS, Langer R, Farokhzad OC, Transepithelial Transport of Fc-Targeted Nanoparticles by the Neonatal Fc Receptor for Oral Delivery, Sci Transl Med, 5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Shen CJ, Xu HB, Liu D, Veazey RS, Wang XL, Development of serum antibodies during early infancy in rhesus macaques: Implications for humoral immune responses to vaccination at birth, Vaccine, 32 (2014) 5337–5342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lozano NA, Lozano A, Marini V, Saranz RJ, Blumberg RS, Baker K, Agresta MF, Ponzio MF, Expression of FcRn receptor in placental tissue and its relationship with IgG levels in term and preterm newborns, Am J Reprod Immunol, 80 (2018) e12972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ober RJ, Radu CG, Ghetie V, Ward ES, Differences in promiscuity for antibody-FcRn interactions across species: implications for therapeutic antibodies, Int Immunol, 13 (2001) 1551–1559. [DOI] [PubMed] [Google Scholar]
- [44].Seijsing J, Yu S, Frejd FY, Hoiden-Guthenberg I, Graslund T, In vivo depletion of serum IgG by an affibody molecule binding the neonatal Fc receptor, Sci Rep, 8 (2018) 5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Roger E, Kalscheuer S, Kirtane A, Guru BR, Grill AE, Whittum-Hudson J, Panyam J, Folic acid functionalized nanoparticles for enhanced oral drug delivery, Mol Pharm, 9 (2012) 2103–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Du W, Fan Y, Zheng N, He B, Yuan L, Zhang H, Wang X, Wang J, Zhang X, Zhang Q, Transferrin receptor specific nanocarriers conjugated with functional 7peptide for oral drug delivery, Biomaterials, 34 (2013) 794–806. [DOI] [PubMed] [Google Scholar]
- [47].Liu C, Shan W, Liu M, Zhu X, Xu J, Xu Y, Huang Y, A novel ligand conjugated nanoparticles for oral insulin delivery, Drug Deliv, 23 (2016) 2015–2025. [DOI] [PubMed] [Google Scholar]
- [48].Sai Y, Tamai I, Sumikawa H, Hayashi K, Nakanishi T, Amano O, Numata M, Iseki S, Tsuji A, Immunolocalization and pharmacological relevance of oligopeptide transporter PepT1 in intestinal absorption of beta-lactam antibiotics, FEBS Lett, 392 (1996) 25–29. [DOI] [PubMed] [Google Scholar]
- [49].Kalliokoski A, Niemi M, Impact of OATP transporters on pharmacokinetics, Br J Pharmacol, 158 (2009) 693–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Choi JS, Jin MJ, Han HK, Role of monocarboxylic acid transporters in the cellular uptake of NSAIDs, J Pharm Pharmacol, 57 (2005) 1185–1189. [DOI] [PubMed] [Google Scholar]
- [51].Choi JS, Jin MJ, Han HK, Intestinal absorption characteristics of ketoprofen in rats, Biopharm Drug Dispos, 27 (2006) 17–21. [DOI] [PubMed] [Google Scholar]
- [52].Ganapathy ME, Ganapathy V, Amino Acid Transporter ATB0,+ as a delivery system for drugs and prodrugs, Curr Drug Targets Immune Endocr Metabol Disord, 5 (2005) 357–364. [DOI] [PubMed] [Google Scholar]
- [53].Mizuma T, Ohta K, Hayashi M, Awazu S, Intestinal active absorption of sugar-conjugated compounds by glucose transport system: implication of improvement of poorly absorbable drugs, Biochem Pharmacol, 43 (1992) 2037–2039. [DOI] [PubMed] [Google Scholar]
- [54].Dahan A, Khamis M, Agbaria R, Karaman R, Targeted prodrugs in oral drug delivery: the modern molecular biopharmaceutical approach, Expert Opin Drug Deliv, 9 (2012) 1001–1013. [DOI] [PubMed] [Google Scholar]
- [55].Zwicker BL, Agellon LB, Transport and biological activities of bile acids, Int J Biochem Cell Biol, 45 (2013) 1389–1398. [DOI] [PubMed] [Google Scholar]
- [56].Zhou K, Wang J, Xie G, Zhou Y, Yan W, Pan W, Che Y, Zhang T, Wong L, Kwee S, Xiao Y, Wen J, Cai W, Jia W, Distinct Plasma Bile Acid Profiles of Biliary Atresia and Neonatal Hepatitis Syndrome, J Proteome Res, 14 (2015) 4844–4850. [DOI] [PubMed] [Google Scholar]
- [57].Roberts MS, Magnusson BM, Burczynski FJ, Weiss M, Enterohepatic circulation: physiological, pharmacokinetic and clinical implications, Clin Pharmacokinet, 41 (2002) 751–790. [DOI] [PubMed] [Google Scholar]
- [58].Chiang JYL, Ferrell JM, Bile Acid Metabolism in Liver Pathobiology, Gene Expr, 18 (2018) 71–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Monte MJ, Marin JJ, Antelo A, Vazquez-Tato J, Bile acids: chemistry, physiology, and pathophysiology, World J Gastroenterol, 15 (2009) 804–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Chen I, Cassaro S, Physiology, Bile Acids, in: StatPearls, Treasure Island (FL), 2020. [PubMed] [Google Scholar]
- [61].Coello A, Meijide F, Nunez ER, Tato JV, Aggregation behavior of bile salts in aqueous solution, J Pharm Sci, 85 (1996) 9–15. [DOI] [PubMed] [Google Scholar]
- [62].Ko CW, Qu J, Black DD, Tso P, Regulation of intestinal lipid metabolism: current concepts and relevance to disease, Nat Rev Gastroenterol Hepatol, 17 (2020) 169–183. [DOI] [PubMed] [Google Scholar]
- [63].Chiang JYL, Bile Acid Metabolism and Signaling, Comprehensive Physiology, 3 (2013) 1191–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Alrefai WA, Gill RK, Bile acid transporters: structure, function, regulation and pathophysiological implications, Pharm Res, 24 (2007) 1803–1823. [DOI] [PubMed] [Google Scholar]
- [65].Jin LH, Fang ZP, Fan MJ, Huang WD, Bile-ology: from bench to bedside, J Zhejiang Univ Sci B, 20 (2019) 414–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Meier PJ, Molecular mechanisms of hepatic bile salt transport from sinusoidal blood into bile, Am J Physiol-Gastr L, 269 (1995) G801–G812. [DOI] [PubMed] [Google Scholar]
- [67].Stieger B, The role of the sodium-taurocholate cotransporting polypeptide (NTCP) and of the bile salt export pump (BSEP) in physiology and pathophysiology of bile formation, Handb Exp Pharmacol, (2011) 205–259. [DOI] [PubMed] [Google Scholar]
- [68].Meier PJ, Stieger B, Bile salt transporters, Annu Rev Physiol, 64 (2002) 635–661. [DOI] [PubMed] [Google Scholar]
- [69].Boyer JL, Bile formation and secretion, Compr Physiol, 3 (2013) 1035–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Song X, Kaimal R, Yan B, Deng R, Liver receptor homolog 1 transcriptionally regulates human bile salt export pump expression, J Lipid Res, 49 (2008) 973–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Kosters A, Karpen SJ, Bile acid transporters in health and disease, Xenobiotica, 38 (2008) 1043–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Marin JJ, Macias RI, Briz O, Banales JM, Monte MJ, Bile Acids in Physiology, Pathology and Pharmacology, Curr Drug Metab, 17 (2015) 4–29. [DOI] [PubMed] [Google Scholar]
- [73].Birru WA, Warren DB, Ibrahim A, Williams HD, Benameur H, Porter CJH, Chalmers DK, Pouton CW, Digestion of Phospholipids after Secretion of Bile into the Duodenum Changes the Phase Behavior of Bile Components, Mol Pharmaceut, 11 (2014) 2825–2834. [DOI] [PubMed] [Google Scholar]
- [74].Hussain MM, Intestinal lipid absorption and lipoprotein formation, Current Opinion in Lipidology, 25 (2014) 200–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Iqbal J, Hussain MM, Intestinal lipid absorption, Am J Physiol-Endoc M, 296 (2009) E1183–E1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Ko CW, Qu J, Black DD, Tso P, Regulation of intestinal lipid metabolism: current concepts and relevance to disease, Nat Rev Gastro Hepat, 17 (2020) 169–183. [DOI] [PubMed] [Google Scholar]
- [77].Tso P, Balint JA, Formation and transport of chylomicrons by enterocytes to the lymphatics, Am J Physiol, 250 (1986) G715–726. [DOI] [PubMed] [Google Scholar]
- [78].Yanez JA, Wang SW, Knemeyer IW, Wirth MA, Alton KB, Intestinal lymphatic transport for drug delivery, Adv Drug Deliv Rev, 63 (2011) 923–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Hokkanen K, Tirronen A, Yla-Herttuala S, Intestinal lymphatic vessels and their role in chylomicron absorption and lipid homeostasis, Current Opinion in Lipidology, 30 (2019) 370–376. [DOI] [PubMed] [Google Scholar]
- [80].Dixon JB, Mechanisms of chylomicron uptake into lacteals, Ann Ny Acad Sci, 1207 (2010) E52–E57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Managuli RS, Raut SY, Reddy MS, Mutalik S, Targeting the intestinal lymphatic system: a versatile path for enhanced oral bioavailability of drugs, Expert Opin Drug Del, 15 (2018) 787–804. [DOI] [PubMed] [Google Scholar]
- [82].Yanez JA, Wang SWJ, Knemeyer IW, Wirth MA, Alton KB, Intestinal lymphatic transport for drug delivery, Adv Drug Deliver Rev, 63 (2011) 923–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Brocks DR, Davies NM, Lymphatic Drug Absorption via the Enterocytes: Pharmacokinetic Simulation, Modeling, and Considerations for Optimal Drug Development, J Pharm Pharm Sci, 21 (2018) 254s–270s. [DOI] [PubMed] [Google Scholar]
- [84].Zhang XY, Lu WY, Recent advances in lymphatic targeted drug delivery system for tumor metastasis, Cancer Biol Med, 11 (2014) 247–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Gebert A, Rothkotter HJ, Pabst R, M cells in Peyer’s patches of the intestine, Int Rev Cytol, 167 (1996) 91–159. [DOI] [PubMed] [Google Scholar]
- [86].Miller H, Zhang J, Kuolee R, Patel GB, Chen W, Intestinal M cells: the fallible sentinels?, World J Gastroenterol, 13 (2007) 1477–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Alrefai WA, Gill RK, Bile acid transporters: Structure, function, regulation and pathophysiological implications, Pharm Res-Dordr, 24 (2007) 1803–1823. [DOI] [PubMed] [Google Scholar]
- [88].Balakrishnan A, Polli JE, Apical sodium dependent bile acid transporter (ASBT, SLC10A2): a potential prodrug target, Mol Pharm, 3 (2006) 223–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Hu NJ, Iwata S, Cameron AD, Drew D, Crystal structure of a bacterial homologue of the bile acid sodium symporter ASBT, Nature, 478 (2011) 408–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Zhou XM, Levin EJ, Pan YP, McCoy JG, Sharma R, Kloss B, Bruni R, Quick M, Zhou M, Structural basis of the alternating-access mechanism in a bile acid transporter, Nature, 505 (2014) 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Alhadeff R, Ganoth A, Arkin IT, Mechanistic studies of the apical sodium-dependent bile acid transporter, Proteins-Structure Function and Bioinformatics, 83 (2015) 1107–1117. [DOI] [PubMed] [Google Scholar]
- [92].Al-Hilal TA, Chung SW, Alam F, Park J, Lee KE, Jeon H, Kim K, Kwon IC, Kim IS, Kim SY, Byun Y, Functional transformations of bile acid transporters induced by high-affinity macromolecules, Sci Rep-Uk, 4 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Kim KS, Suzuki K, Cho H, Youn YS, Bae YH, Oral Nanoparticles Exhibit Specific High-Efficiency Intestinal Uptake and Lymphatic Transport, Acs Nano, 12 (2018) 8893–8900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Nakahara M, Furuya N, Takagaki K, Sugaya T, Hirota K, Fukamizu A, Kanda T, Fujii H, Sato R, Ileal bile acid-binding protein, functionally associated with the farnesoid X receptor or the ileal bile acid transporter, regulates bile acid activity in the small intestine, Journal of Biological Chemistry, 280 (2005) 42283–42289. [DOI] [PubMed] [Google Scholar]
- [95].D’Onofrio M, Zanzoni S, Munari F, Monaco HL, Assfalg M, Capaldi S, The long variant of human ileal bile acid-binding protein associated with colorectal cancer exhibits sub-cellular localization and lipid binding behaviour distinct from those of the common isoform, Biochim Biophys Acta Gen Subj, 1861 (2017) 2315–2324. [DOI] [PubMed] [Google Scholar]
- [96].Fang CM, Filipp FV, Smith JW, Unusual binding of ursodeoxycholic acid to ileal bile acid binding protein: role in activation of FXR alpha, Journal of Lipid Research, 53 (2012) 664–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Dawson PA, Hubbert M, Haywood J, Craddock AL, Zerangue N, Christian WV, Ballatori N, The heteromeric organic solute transporter alpha-beta, Ostalpha-Ostbeta, is an ileal basolateral bile acid transporter, J Biol Chem, 280 (2005) 6960–6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Boyer JL, OSTalpha-OSTbeta Guards the Ileal Enterocyte From the Accumulation of Toxic Levels of Bile Acids, Cell Mol Gastroenterol Hepatol, 5 (2018) 649–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Ballatori N, Christian WV, Wheeler SG, Hammond CL, The heteromeric organic solute transporter, OSTalpha-OSTbeta/SLC51: a transporter for steroid-derived molecules, Mol Aspects Med, 34 (2013) 683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Soroka CJ, Ballatori N, Boyer JL, Organic solute transporter, OSTalpha-OSTbeta: its role in bile acid transport and cholestasis, Semin Liver Dis, 30 (2010) 178–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Wolkoff AW, Hepatocellular sinusoidal membrane organic anion transport and transporters, Semin Liver Dis, 16 (1996) 121–127. [DOI] [PubMed] [Google Scholar]
- [102].Slijepcevic D, Abbing RLPR, Katafuchi T, Blank A, Donkers JM, van Hoppe S, de Waart DR, Tolenaars D, van der Meer JHM, Wildenberg M, Beuers U, Elferink RPJO, Schinkel AH, van de Graaf SFJ, Hepatic Uptake of Conjugated Bile Acids Is Mediated by Both Sodium Taurocholate Cotransporting Polypeptide and Organic Anion Transporting Polypeptides and Modulated by Intestinal Sensing of Plasma Bile Acid Levels in Mice, Hepatology, 66 (2017) 1631–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Modica S, Moschetta A, Nuclear bile acid receptor FXR as pharmacological target: Are we there yet?, Febs Letters, 580 (2006) 5492–5499. [DOI] [PubMed] [Google Scholar]
- [104].Matsubara T, Li F, Gonzalez FJ, FXR signaling in the enterohepatic system, Molecular and Cellular Endocrinology, 368 (2013) 17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Campana G, Pasini P, Roda A, Spampinato S, Regulation of ileal bile acid-binding protein expression in Caco-2 cells by ursodeoxycholic acid: role of the farnesoid X receptor, Biochem Pharmacol, 69 (2005) 1755–1763. [DOI] [PubMed] [Google Scholar]
- [106].Kramer W, Wess G, Bile acid transport systems as pharmaceutical targets, European Journal of Clinical Investigation, 26 (1996) 715–732. [DOI] [PubMed] [Google Scholar]
- [107].Lack L, Walker JT, Singletary GD, Ileal Bile Salt Transport - in-Vivo Studies of Effect of Substrate Ionization on Activity, American Journal of Physiology, 219 (1970) 487–+. [DOI] [PubMed] [Google Scholar]
- [108].Lack L, Properties and Biological Significance of the Ileal Bile-Salt Transport-System, Environ Health Persp, 33 (1979) 79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Lack L, Weiner IM, Intestinal Bile Salt Transport - Structure-Activity Relationships and Other Properties, American Journal of Physiology, 210 (1966) 1142–&. [DOI] [PubMed] [Google Scholar]
- [110].Shneider BL, Dawson PA, Christie DM, Hardikar W, Wong MH, Suchy FJ, Cloning and Molecular Characterization of the Ontogeny of a Rat Ileal Sodium-Dependent Bile-Acid Transporter, Journal of Clinical Investigation, 95 (1995) 745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Sievanen E, Exploitation of bile acid transport systems in prodrug design, Molecules, 12 (2007) 1859–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Kramer W, Schneider S, 3-Diazirine-Derivatives of Bile-Salts for Photoaffinity-Labeling, Journal of Lipid Research, 30 (1989) 1281–1288. [PubMed] [Google Scholar]
- [113].Kramer W, Wess G, Neckermann G, Schubert G, Fink J, Girbig F, Gutjahr U, Kowalewski S, Baringhaus KH, Boger G, Enhsen A, Falk E, Friedrich M, Glombik H, Hoffmann A, Pittius C, Urmann M, Intestinal-Absorption of Peptides by Coupling to Bile-Acids, Journal of Biological Chemistry, 269 (1994) 10621–10627. [PubMed] [Google Scholar]
- [114].Tolle-Sander S, Lentz KA, Maeda DY, Coop A, Polli JE, Increased acyclovir oral bioavailability via a bile acid conjugate, Mol Pharmaceut, 1 (2004) 40–48. [DOI] [PubMed] [Google Scholar]
- [115].Bhat L, Jandeleit B, Dias TM, Moors TL, Gallop MA, Synthesis and biological evaluation of novel steroidal pyrazoles as substrates for bile acid transporters, Bioorganic & Medicinal Chemistry Letters, 15 (2005) 85–87. [DOI] [PubMed] [Google Scholar]
- [116].Zheng X, Pan Y, Acharya C, Swaan PW, Polli JE, Structural requirements of the ASBT by 3D-QSAR analysis using aminopyridine conjugates of chenodeoxycholic acid, Bioconjug Chem, 21 (2010) 2038–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Rais R, Fletcher S, Polli JE, Synthesis and In Vitro Evaluation of Gabapentin Prodrugs That Target the Human Apical Sodium-Dependent Bile Acid Transporter (hASBT), J Pharm Sci-Us, 100 (2011) 1184–1195. [DOI] [PubMed] [Google Scholar]
- [118].Balakrishnan A, Wring SA, Polli JE, Interaction of native bile acids with human apical sodium-dependent bile acid transporter (hASBT): Influence of steroidal hydroxylation pattern and C-24 conjugation, Pharm Res-Dordr, 23 (2006) 1451–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Kagedahl M, Swaan PW, Redemann CT, Tang M, Craik CS, Szoka FC, Oie S, Use of the intestinal bile acid transporter for the uptake of cholic acid conjugates with HIV-1 protease inhibitory activity, Pharm Res-Dordr, 14 (1997) 176–180. [DOI] [PubMed] [Google Scholar]
- [120].Zhang D, Li DP, Shang L, He ZG, Sun J, Transporter-targeted cholic acid-cytarabine conjugates for improved oral absorption, Int J Pharmaceut, 511 (2016) 161–169. [DOI] [PubMed] [Google Scholar]
- [121].Costa EC, Moreira AF, de Melo-Diogo D, Gaspar VM, Carvalho MP, Correia IJ, 3D tumor spheroids: an overview on the tools and techniques used for their analysis, Biotechnology Advances, 34 (2016) 1427–1441. [DOI] [PubMed] [Google Scholar]
- [122].Pavlovic N, Golocorbin-Kon S, Danic M, Stanimirov B, Al-Salami H, Stankov K, Mikov M, Bile Acids and Their Derivatives as Potential Modifiers of Drug Release and Pharmacokinetic Profiles, Frontiers in Pharmacology, 9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Heinen CA, Reuss S, Amidon GL, Langguth P, Ion Pairing with Bile Salts Modulates Intestinal Permeability and Contributes to Food-Drug Interaction of BCS Class III Compound Trospium Chloride, Mol Pharmaceut, 10 (2013) 3989–3996. [DOI] [PubMed] [Google Scholar]
- [124].Lee S, Kim SK, Lee DY, Chae SY, Byun Y, Pharmacokinetics of a new, orally available ceftriaxone formulation in physical complexation with a cationic analogue of bile acid in rats, Antimicrob Agents Chemother, 50 (2006) 1869–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Lee DY, Lee J, Lee S, Kim SK, Byun Y, Liphophilic complexation of heparin based on bile acid for oral delivery, Journal of Controlled Release, 123 (2007) 39–45. [DOI] [PubMed] [Google Scholar]
- [126].Gaowa A, Horibe T, Kohno M, Kawakami K, Bile Acid as an Effective Absorption Enhancer for Oral Delivery of Epidermal Growth Factor Receptor-Targeted Hybrid Peptide, J Pharm Sci, 107 (2018) 1322–1329. [DOI] [PubMed] [Google Scholar]
- [127].Alam F, Al-Hilal TA, Chung SW, Seo D, Mahmud F, Kim HS, Kim SY, Byun Y, Oral delivery of a potent anti-angiogenic heparin conjugate by chemical conjugation and physical complexation using deoxycholic acid, Biomaterials, 35 (2014) 6543–6552. [DOI] [PubMed] [Google Scholar]
- [128].Nurunnabi M, Khatun Z, Revuri V, Nafiujjaman M, Cha S, Cho S, Huh KM, Lee YK, Design and strategies for bile acid mediated therapy and imaging, Rsc Adv, 6 (2016) 73986–74002. [Google Scholar]
- [129].Mahesh S, Tang KC, Raj M, Amide Bond Activation of Biological Molecules, Molecules, 23 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Jalali A, Moghimipour E, Akhgari A, Enhancing Effect of Bile Salts on Gastrointestinal Absorption of Insulin, Trop J Pharm Res, 13 (2014) 1797–1802. [Google Scholar]
- [131].Michael S, Thole M, Dillmann R, Fahr A, Drewe J, Fricker G, Improvement of intestinal peptide absorption by a synthetic bile acid derivative, cholylsarcosine, Eur J Pharm Sci, 10 (2000) 133–140. [DOI] [PubMed] [Google Scholar]
- [132].Al-Salami H, Butt G, Tucker I, Mikov M, Influence of the semisynthetic bile acid MKC on the ileal permeation of gliclazide in vitro in healthy and diabetic rats treated with probiotics, Methods Find Exp Clin Pharmacol, 30 (2008) 107–113. [DOI] [PubMed] [Google Scholar]
- [133].Din FU, Aman W, Ullah I, Qureshi OS, Mustapha O, Shafique S, Zeb A, Effective use of nanocarriers as drug delivery systems for the treatment of selected tumors, Int J Nanomed, 12 (2017) 7291–7309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Reinholz J, Landfester K, Mailander V, The challenges of oral drug delivery via nanocarriers, Drug Delivery, 25 (2018) 1694–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].He HS, Lu Y, Qi JP, Zhu QG, Chen ZJ, Wu W, Adapting liposomes for oral drug delivery, Acta Pharmaceutica Sinica B, 9 (2019) 36–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Wu W, Lu Y, Qi JP, Oral delivery of liposomes, Therapeutic Delivery, 6 (2015) 1239–1241. [DOI] [PubMed] [Google Scholar]
- [137].Hua SS, Orally administered liposomal formulations for colon targeted drug delivery, Frontiers in Pharmacology, 5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Daeihamed M, Dadashzadeh S, Haeri A, Akhlaghi MF, Potential of Liposomes for Enhancement of Oral Drug Absorption, Current Drug Delivery, 14 (2017) 289–303. [DOI] [PubMed] [Google Scholar]
- [139].Sehgal S, Rogers JA, Polymer-coated liposomes: improved liposome stability and release of cytosine arabinoside (Ara-C), J Microencapsul, 12 (1995) 37–47. [DOI] [PubMed] [Google Scholar]
- [140].Dong C, Rogers JA, Polymer-Coated Liposomes - Stability and Release of Asa from Carboxymethyl Chitin-Coated Liposomes, Journal of Controlled Release, 17 (1991) 217–224. [Google Scholar]
- [141].Watanabe K, Kaneko M, Maitani Y, Functional coating of liposomes using a folate-polymer conjugate to target folate receptors, Int J Nanomed, 7 (2012) 3679–3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Wu S, Bin W, Tu B, Li X, Wang W, Liao S, Sun C, A Delivery System for Oral Administration of Proteins/Peptides Through Bile Acid Transport Channels, J Pharm Sci, 108 (2019) 2143–2152. [DOI] [PubMed] [Google Scholar]
- [143].Suzuki K, Kim KS, Bae YH, Long-term oral administration of Exendin-4 to control type 2 diabetes in a rat model, J Control Release, 294 (2019) 259–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Conacher M, Alexander J, Brewer JM, Oral immunisation with peptide and protein antigens by formulation in lipid vesicles incorporating bile salts (bilosomes), Vaccine, 19 (2001) 2965–2974. [DOI] [PubMed] [Google Scholar]
- [145].Rajput T, Chauhan MK, Bilosome: A Bile Salt Based Novel Carrier System Gaining Interest in Pharmaceutical Research, Journal of Drug Delivery and Therapeutics, 7 (2017). [Google Scholar]
- [146].Mann JF, Ferro VA, Mullen AB, Tetley L, Mullen M, Carter KC, Alexander J, Stimson WH, Optimisation of a lipid based oral delivery system containing A/Panama influenza haemagglutinin, Vaccine, 22 (2004) 2425–2429. [DOI] [PubMed] [Google Scholar]
- [147].Shukla A, Khatri K, Gupta PN, Goyal AK, Mehta A, Vyas SP, Oral immunization against hepatitis B using bile salt stabilized vesicles (bilosomes), Journal of Pharmacy and Pharmaceutical Sciences, 11 (2008) 58–66. [DOI] [PubMed] [Google Scholar]
- [148].Shukla A, Singh B, Katare OP, Significant systemic and mucosal immune response induced on oral delivery of diphtheria toxoid using nano-bilosomes, Brit J Pharmacol, 164 (2011) 820–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Chen YP, Lu Y, Chen JM, Lai J, Sun J, Hu FQ, Wu W, Enhanced bioavailability of the poorly water-soluble drug fenofibrate by using liposomes containing a bile salt, Int J Pharmaceut, 376 (2009) 153–160. [DOI] [PubMed] [Google Scholar]
- [150].Elnaggar YS, Multifaceted applications of bile salts in pharmacy: an emphasis on nanomedicine, Int J Nanomedicine, 10 (2015) 3955–3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Zhang S, Cui D, Xu J, Wang J, Wei Q, Xiong S, Bile acid transporter mediated STC/Soluplus self-assembled hybrid nanoparticles for enhancing the oral drug bioavailability, Int J Pharm, 579 (2020) 119120. [DOI] [PubMed] [Google Scholar]
- [152].Khatun Z, Nurunnabi M, Reeck GR, Cho KJ, Lee YK, Oral delivery of taurocholic acid linked heparin-docetaxel conjugates for cancer therapy, J Control Release, 170 (2013) 74–82. [DOI] [PubMed] [Google Scholar]
- [153].Khatun Z, Nurunnabi M, Cho KJ, Lee YK, Imaging of the GI tract by QDs loaded heparin-deoxycholic acid (DOCA) nanoparticles, Carbohydr Polym, 90 (2012) 1461–1468. [DOI] [PubMed] [Google Scholar]
- [154].Chaturvedi K, Ganguly K, Kulkarni AR, Rudzinski WE, Krauss L, Nadagouda MN, Aminabhavi TM, Oral insulin delivery using deoxycholic acid conjugated PEGylated polyhydroxybutyrate co-polymeric nanoparticles, Nanomedicine, 10 (2015) 1569–1583. [DOI] [PubMed] [Google Scholar]
- [155].Sun F, Jaspers TC, van Hasselt PM, Hennink WE, van Nostrum CF, A Mixed Micelle Formulation for Oral Delivery of Vitamin K, Pharm Res, 33 (2016) 2168–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Yu JN, Zhu Y, Wang L, Peng M, Tong SS, Cao X, Qiu H, Xu XM, Enhancement of oral bioavailability of the poorly water-soluble drug silybin by sodium cholate/phospholipid-mixed micelles, Acta Pharmacol Sin, 31 (2010) 759–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Kim SK, Huh J, Kim SY, Byun Y, Lee DY, Moon HT, Physicochemical conjugation with deoxycholic acid and dimethylsulfoxide for heparin oral delivery, Bioconjug Chem, 22 (2011) 1451–1458. [DOI] [PubMed] [Google Scholar]
- [158].Samstein RM, Perica K, Balderrama F, Look M, Fahmy TM, The use of deoxycholic acid to enhance the oral bioavailability of biodegradable nanoparticles, Biomaterials, 29 (2008) 703–708. [DOI] [PubMed] [Google Scholar]
- [159].Zheng XW, Ekins S, Raufman JP, Polli JE, Computational Models for Drug Inhibition of the Human Apical Sodium-Dependent Bile Acid Transporter, Mol Pharmaceut, 6 (2009) 1591–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Sabit H, Mallajosyula SS, MacKerell AD, Swaan PW, Transmembrane Domain II of the Human Bile Acid Transporter SLC10A2 Coordinates Sodium Translocation, Journal of Biological Chemistry, 288 (2013) 32394–32404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Hu NJ, Iwata S, Cameron AD, Drew D, Crystal structure of a bacterial homologue of the bile acid sodium symporter ASBT, Nature, 478 (2011) 408–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Park J, Choi JU, Kim K, Byun Y, Bile acid transporter mediated endocytosis of oral bile acid conjugated nanocomplex, Biomaterials, 147 (2017) 145–154. [DOI] [PubMed] [Google Scholar]
- [163].Kramer W, Transporters, Trojan horses and therapeutics: suitability of bile acid and peptide transporters for drug delivery, Biological Chemistry, 392 (2011) 77–94. [DOI] [PubMed] [Google Scholar]
- [164].Park JW, Hwang SR, Jeon OC, Moon HT, Byun Y, Enhanced oral absorption of ibandronate via complex formation with bile acid derivative, J Pharm Sci-Us, 102 (2013) 341–346. [DOI] [PubMed] [Google Scholar]
- [165].Song KH, Chung SJ, Shim CK, Enhanced intestinal absorption of salmon calcitonin (sCT) from proliposomes containing bile salts, J Control Release, 106 (2005) 298–308. [DOI] [PubMed] [Google Scholar]
- [166].Deng F, Zhang H, Wang X, Zhang Y, Hu H, Song S, Dai W, He B, Zheng Y, Wang X, Zhang Q, Transmembrane Pathways and Mechanisms of Rod-like Paclitaxel Nanocrystals through MDCK Polarized Monolayer, ACS Appl Mater Interfaces, 9 (2017) 5803–5816. [DOI] [PubMed] [Google Scholar]
- [167].Mulcahy LA, Pink RC, Carter DR, Routes and mechanisms of extracellular vesicle uptake, J Extracell Vesicles, 3 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Kadlecova Z, Spielman SJ, Loerke D, Mohanakrishnan A, Reed DK, Schmid SL, Regulation of clathrin-mediated endocytosis by hierarchical allosteric activation of AP2, J Cell Biol, 216 (2017) 167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Swanson JA, Shaping cups into phagosomes and macropinosomes, Nat Rev Mol Cell Bio, 9 (2008) 639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Song SY, Cong WS, Zhou SR, Shi YJ, Dai WB, Zhang H, Wang XQ, He B, Zhang Q, Small GTPases: Structure, biological function and its interaction with nanoparticles, Asian J Pharm Sci, 14 (2019) 30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Annaba F, Sarwar Z, Kumar P, Saksena S, Turner JR, Dudeja PK, Gill RK, Alrefai WA, Modulation of ileal bile acid transporter (ASBT) activity by depletion of plasma membrane cholesterol: association with lipid rafts, Am J Physiol-Gastr L, 294 (2008) G489–G497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Li Y, Zhu CY, Enhanced hepatic-targeted delivery via oral administration using nanoliposomes functionalized with a novel DSPE-PEG-cholic acid conjugate, Rsc Adv, 6 (2016) 28110–28120. [Google Scholar]
- [173].Yu YF, Huo MR, Fu Y, Xu W, Cai H, Yao LL, Chen QY, Mu Y, Zhou JP, Yin TJ, N-Deoxycholic acid-N,O-hydroxyethyl Chitosan with a Sulfhydryl Modification To Enhance the Oral Absorptive Efficiency of Paclitaxel, Mol Pharmaceut, 14 (2017) 4539–4550. [DOI] [PubMed] [Google Scholar]
- [174].Pangeni R, Jha SK, Maharjan R, Choi JU, Chang KY, Choi YK, Byun Y, Park JW, Intestinal transport mechanism and in vivo anticancer efficacy of a solid oral formulation incorporating an ion-pairing complex of pemetrexed with deoxycholic acid derivative, Int J Nanomed, 14 (2019) 6339–6356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Fan W, Xia D, Zhu Q, Li X, He S, Zhu C, Guo S, Hovgaard L, Yang M, Gan Y, Functional nanoparticles exploit the bile acid pathway to overcome multiple barriers of the intestinal epithelium for oral insulin delivery, Biomaterials, 151 (2018) 13–23. [DOI] [PubMed] [Google Scholar]
- [176].Cui Y, Shan W, Zhou R, Liu M, Wu L, Guo Q, Zheng YX, Wu JW, Huang Y, The combination of endolysosomal escape and basolateral stimulation to overcome the difficulties of “easy uptake hard transcytosis” of ligand-modified nanoparticles in oral drug delivery, Nanoscale, 10 (2018) 1494–1507. [DOI] [PubMed] [Google Scholar]
- [177].Hu YB, Dammer EB, Ren RJ, Wang G, The endosomal-lysosomal system: from acidification and cargo sorting to neurodegeneration, Transl Neurodegener, 4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Wu SW, Bin W, Tu BY, Li XF, Wang W, Liao SL, Sun CS, A Delivery System for Oral Administration of Proteins/Peptides Through Bile Acid Transport Channels, J Pharm Sci-Us, 108 (2019) 2143–2152. [DOI] [PubMed] [Google Scholar]
- [179].He W, Yang K, Fan LF, Lv YQ, Jin Z, Zhu SM, Qin C, Wang Y, Yin LF, Denatured globular protein and bile salt-coated nanoparticles for poorly water-soluble drugs: Penetration across the intestinal epithelial barrier into the circulation system and enhanced oral bioavailability, Int J Pharmaceut, 495 (2015) 9–18. [DOI] [PubMed] [Google Scholar]
- [180].Kienzle C, von Blume J, Secretory cargo sorting at the trans-Golgi network, Trends Cell Biol, 24 (2014) 584–593. [DOI] [PubMed] [Google Scholar]
- [181].Westergaard H, Dietschy JM, The mechanism whereby bile acid micelles increase the rate of fatty acid and cholesterol uptake into the intestinal mucosal cell, J Clin Invest, 58 (1976) 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Zhang Z, Gao F, Jiang S, Chen L, Liu Z, Yu H, Li Y, Bile salts enhance the intestinal absorption of lipophilic drug loaded lipid nanocarriers: mechanism and effect in rats, Int J Pharm, 452 (2013) 374–381. [DOI] [PubMed] [Google Scholar]
- [183].Wang T, Luo Y, Biological fate of ingested lipid-based nanoparticles: current understanding and future directions, Nanoscale, 11 (2019) 11048–11063. [DOI] [PubMed] [Google Scholar]
- [184].Zhang B, Xue A, Zhang C, Yu J, Chen W, Sun D, Bile salt liposomes for enhanced lymphatic transport and oral bioavailability of paclitaxel, Pharmazie, 71 (2016) 320–326. [PubMed] [Google Scholar]
- [185].Kim KS, Kwag DS, Hwang HS, Lee ES, Bae YH, Immense Insulin Intestinal Uptake and Lymphatic Transport Using Bile Acid Conjugated Partially Uncapped Liposome, Mol Pharm, 15 (2018) 4756–4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Kim KS, Youn YS, Bae YH, Immune-triggered cancer treatment by intestinal lymphatic delivery of docetaxel-loaded nanoparticle, J Control Release, 311–312 (2019) 85–95. [DOI] [PubMed] [Google Scholar]
- [187].Arzani G, Haeri A, Daeihamed M, Bakhtiari-Kaboutaraki H, Dadashzadeh S, Niosomal carriers enhance oral bioavailability of carvedilol: effects of bile salt-enriched vesicles and carrier surface charge, Int J Nanomedicine, 10 (2015) 4797–4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Bharti C, Nagaich U, Pal AK, Gulati N, Mesoporous silica nanoparticles in target drug delivery system: A review, Int J Pharm Investig, 5 (2015) 124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Vallet-Regi M, Balas F, Arcos D, Mesoporous materials for drug delivery, Angew Chem Int Edit, 46 (2007) 7548–7558. [DOI] [PubMed] [Google Scholar]
- [190].Zhang YZ, Wang JC, Bai XY, Jiang TY, Zhang Q, Wang SL, Mesoporous Silica Nanoparticles for Increasing the Oral Bioavailability and Permeation of Poorly Water Soluble Drugs, Mol Pharmaceut, 9 (2012) 505–513. [DOI] [PubMed] [Google Scholar]
- [191].Porta F, Lamers GEM, Morrhayim J, Chatzopoulou A, Schaaf M, den Dulk H, Backendorf C, Zink JI, Kros A, Folic Acid-Modified Mesoporous Silica Nanoparticles for Cellular and Nuclear Targeted Drug Delivery, Adv Healthc Mater, 2 (2013) 281–286. [DOI] [PubMed] [Google Scholar]
- [192].Choi JS, Park JS, Development of docetaxel nanocrystals surface modified with transferrin for tumor targeting, Drug Des Dev Ther, 11 (2017) 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Yin YJ, Deng HL, Wu K, He B, Dai WB, Zhang H, Fu JJ, Le Y, Wang XQ, Zhang Q, A multiaspect study on transcytosis mechanism of sorafenib nanogranules engineered by high-gravity antisolvent precipitation, Journal of Controlled Release, 323 (2020) 600–612. [DOI] [PubMed] [Google Scholar]
- [194].Aungst BJ, Absorption Enhancers: Applications and Advances, Aaps J, 14 (2012) 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Maher S, Brayden DJ, Casettari L, Illum L, Application of Permeation Enhancers in Oral Delivery of Macromolecules: An Update, Pharmaceutics, 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]