

Abstract
The basic helix-loop-helix transcription factor Bhlhe40 is emerging as a key regulator of immunity during infection, autoimmunity, and inflammatory conditions. We describe roles of Bhlhe40 in the circulating and tissue-resident arms of the immune system, with emphasis on recent work on regulation of cytokine production and proliferation. We explore mechanisms behind these functions in mouse models and human cells, including interactions with other transcription factors, and propose that Bhlhe40 is both a central mediator of inflammation and pathogen control, as well as a crucial regulator for a growing number of tissue-resident leukocyte populations. Finally, we suggest areas for further study that we think will contribute to advances in our understanding of immunity and disease.
Bhlhe40: emerging regulator of immunity
Bhlhe40 (see Box 1) is a member of the basic helix-loop-helix transcription factor (TF) family. Bhlhe40 and its most related family member, Bhlhe41, bind to class B E-box DNA sequences with the consensus motif CACGTG [1, 2]. These proteins oligomerize both as homodimers and heterodimers [1–3]. Many functions for mammalian Bhlhe40 have been identified, including regulating cell cycling, cell death, and differentiation, although these studies were predominantly conducted in cell lines [4]. Bhlhe40/41 are unique from other helix-loop-helix Orange domain-containing transcriptional repressors in that they do not interact with the corepressor Groucho, but instead, repress transcription both in cooperation with histone deacetylases (HDACs) and in an HDAC- independent manner [2, 4]. Bhlhe40 also has a poorly understood potential for transcriptional activation. [5–8]. Bhlhe40 is expressed in both hematopoietic and nonhematopoietic mammalian cell types, with early studies focusing on its role beyond the immune system [4, 9].
Box 1: Nomenclature and available tools.
What’s in a name?
Both Bhlhe40 and Bhlhe41 have had many names over the years. Bhlhe40 was first identified as Stimulated with retinoic acid 13 (Stra13) in P19 embryonal carcinoma cells [82] and Bhlhe41 was initially identified in rat brain and called enhancer of Split and HAiry Related Protein-1 (SHARP-1) [83]. Other names for Bhlhe40 include Deleted in esophageal cancer 1 (Dec1), SHARP-2, Basic helix-loop-helix domain containing, class b, 2 (Bhlhb2), and Clast5, while other names for Bhlhe41 include Dec2 and Bhlhb3. Table I shows the names of mouse strains used to study Bhlhe40 and Bhlhe41. For clarity, here, Bhlhe40 denotes the murine protein and BHLHE40 denotes the human protein.
Table I. Mouse Strains Useful for the Study of Bhlhe40 and Bhlhe41a.
| Mouse strain | Notes | Ref. |
|---|---|---|
| Bhlhe40−/− | Global deletion of Bhlhe40 | [8,10,86] |
| Bhlhe41−/− | Global deletion of Bhlhe41 | [32,87] |
| Cd4-Cre+ Bhlhe40fl/fl | Deletion of Bhlhe40 in T cells | [17,29,60] |
| LysM-Cre+ Bhlhe40fl/fl | Deletion of Bhlhe40 in macrophages, monocytes, and neutrophils | [29, 51] |
| Cd11c-Cre+ Bhlhe40fl/fl | Deletion of Bhlhe40 in DCs and alveolar macrophages | [29] |
| Mrp8-Cre+ Bhlhe40fl/fl | Deletion of Bhlhe40 in neutrophils | [29] |
| Clast5-Tg | Overexpression of Bhlhe40 in B and T cells | [38] |
| Dec1-Tg | Overexpression of Bhlhe40 in T cells | [8] |
| Dec2-Tg | Overexpression of Bhlhe41 in T cells | [88] |
| Bhlhe40GFP | Bhlhe40 BAC transgenic reporter strain | [13] |
| Bhlhe41-Cre-hCD2 | cre-IRES-hCD2 gene expression under control of Bhlhe4l BAC | [39] |
| Bhlhe41Tag | Bhlhe41 with multiple tag epitopes (FLAG and V5) | [39] |
| FLAG-Dec2 | Bhlhe41 with FLAG tag expressed under hCD2 promoter control | [32] |
HHCD2 denotes human CD2.
Bhlhe40−/− mice were initially generated on a mixed genetic background and developed a late onset lymphoproliferative disorder characterized by enlargement of lymphoid organs and immune cell infiltration into peripheral tissues [10]. This phenotype was largely due to a failure to eliminate activated T and B cells. We and others have observed a less severe, partially penetrant form of this phenotype in C57BL/6 Bhlhe40−/− mice, predominantly in older females [8, 11]. Because Bhlhe40 expression is widespread, Bhlhe40 deletion in specific immune cell lineages has been essential to elucidating its functions. Recent work has demonstrated that Bhlhe40 regulates cytokine production in mouse and human CD4+ T cells (see Glossary) as well as regulating proliferation and/or metabolism in mouse myeloid, B, and tissue-resident CD8+ T cells, sometimes in partnership with Bhlhe41. Here, we review these advances and speculate on molecular mechanisms explaining these factors’ functions in specific cell types.
Bhlhe40 in circulating lymphocytes
After activation, circulating B and T cells are licensed for effector functions including proliferation, cytokine secretion, and migration into tissues. CD4+ helper T (TH) cells are specialized to regulate other cells, in part via cytokines. While a substantial body of literature has revealed the transcriptional regulators controlling TH cell subset differentiation, less is known about common regulators of cytokine production across TH cell subsets. Recent work has established a key role for Bhlhe40 in this process.
Bhlhe40 in CD4+ T cells
Autoimmune and alloimmune models.
Experimental autoimmune encephalomyelitis (EAE) is the major animal model of multiple sclerosis (MS). In EAE, mice immunized with myelin antigens develop ascending paralysis and neuroinflammation [12]. EAE induction in Bhlhe40GFP bacterial artificial chromosome reporter mice showed that Bhlhe40 was heterogeneously expressed by CD4+ T cells [13]. Pro-inflammatory cytokine-producing CD4+ T cells were more frequent in the Bhlhe40-positive subset, while Bhlhe40-negative CD4+ T cells were enriched for Foxp3 and IL-10 [13]. These data suggested that Bhlhe40 expression correlated with CD4+ T cell encephalitogenicity. Indeed, Bhlhe40−/− mice were protected from disease in both active and passive EAE models, primarily because Bhlhe40−/− CD4+ T cells were non-pathogenic [11, 13, 14]. In vitro, Bhlhe40 regulated cytokine production in polarized TH cells (see Box 2 for details). In vivo, Bhlhe40−/− mouse CD4+ T cells were impaired in their expression of Csf2 (encoding GM-CSF), a pro-inflammatory cytokine, crucial for T cell-mediated EAE [15, 16]. Additionally, relative to Bhlhe40+/+ cells, Bhlhe40−/− CD4+ T cells produced an increased amount of IL-10 [11, 17] -- known to restrain TH cell effector functions and to protect mice from autoimmunity [18]. IL-10 receptor blockade rendered Bhlhe40−/− mice susceptible to EAE, indicating that excessive IL-10 contributed to EAE resistance in Bhlhe40−/− mice [11]. Nevertheless, hundreds of genes were dysregulated in Bhlhe40−/− TH cells, suggesting that other Bhlhe40-dependent genes contributed to EAE susceptibility [11, 14].
Box 2. Bhlhe40 and in vitro T helper cell polarization.
T cell receptor (TCR) and costimulation together with cytokine signals instruct TH cell differentiation and proliferation. Differentiated TH cells secrete a variety of cytokines acting on other immune cells and non-hematopoietic cells during infection and autoimmunity. In vitro, naïve CD4+ T cells can be differentiated into TH1, TH2, TH17, and induced regulatory T (iTREG) cells with surrogate TCR stimulation and co-stimulation in the presence of polarizing cytokines. Bhlhe40/BHLHE40 is absent in naïve mouse and human T cells but is upregulated after anti-CD3 and anti-CD28 stimulation [11, 14]. In vitro-polarized mouse TH1 and TH17 cells have shown higher Bhlhe40 expression than TH2 cells [11]. Although in vitro expression of classical lineage-specifying transcription factors (TF) and most signature genes of each TH lineage do not require Bhlhe40, this TF is essential for normal cytokine production [11]. IFN-γ secretion is reduced in mouse Bhlhe40−/− and Cd4-Cre+ Bhlhe40fl/fl TH1 cells [11, 17]; this phenotype may be due to Bhlhe40’s proposed role as a co-factor for T-bet, the master regulator of TH1 cells [7], although this interaction has not been observed in TH1 cells [17]. Moreover, mouse Bhlhe40−/− TH1, TH2, and TH17 lineages show reduced GM-CSF but enhanced IL-10 production relative to Bhlhe40+/+ cells [11, 17]. Bhlhe41 is specifically expressed in TH2 cells, and mouse Bhlhe41−/− TH2 cells exhibit impaired proliferation and production of IL-4, IL-5, and IL-13 in vitro [32]. Bhlhe41 shares some overlapping functions with Bhlhe40, and thus, may compensate in Bhlhe40−/− TH2 cell cultures. Upregulated Bhlhe40 was reported in mouse iTREG in an expression microarray analysis [11]. An older study using Bhlhe40−/− and transgenic mice claimed that Bhlhe40 in complex with Runx1 activated the Il2ra locus (encoding CD25) and maintained TREG, suggesting a role for Bhlhe40 in mouse iTREG cells [8].
In the T cell transfer mouse model of colitis, naïve CD4+ T cells are transferred into immunocompromised mice, causing intestinal inflammation and weight loss resembling human colitis [19]. Transferred CD4+ T cells secrete inflammatory cytokines including interferon-γ (IFN-γ) [20], while IL-10 can play a protective role [21]. A study using this model suggested that Bhlhe40 could regulate colitis by controlling the balance between cytokines. Specifically, Cd4-Cre+ Bhlhe40fl/fl naïve CD4+ T cells transferred into Rag1−/− recipient mice failed to induce weight loss [17]. Compared with control Bhlhe40fl/fl CD4+ T cells, Cd4-Cre+ Bhlhe40fl/fl CD4+ T cells showed low IFN-γ, but high IL-10 expression. Thus, Bhlhe40 expression might directly influence colitis by regulating these cytokines, although further robust experiments are needed to show this conclusively.
In contrast to the autoimmune models of EAE and colitis, an alloimmune mouse model of graft-versus-host disease (GvHD) can be induced by transferring donor splenocytes to MHC-mismatched recipient mice [21]. Donor allogeneic CD4+ T cells secrete IFN-γ and GM-CSF -- the latter being an essential driver of inflammation in this model [22–24]. When Bhlhe40−/− donor CD4+ T cells were tested in this model, they showed reduced IFN-γ and GM-CSF expression and caused less weight loss and mortality after transfer, relative to control CD4+ T cells [24]. Notably, similar numbers of IL-10-producing Bhlhe40+/+ and Bhlhe40−/− CD4+ T cells were observed, suggesting that Bhlhe40 could promote GvHD primarily through upregulation of GM-CSF (and perhaps IFN-γ) [24].
Infection models.
Bhlhe40-mediated inflammatory responses can be beneficial in host defense against pathogens. TH1 responses and the balance of IFN-γ and IL-10 are crucial for protective immunity in animal models of toxoplasmosis (caused by Toxoplasma gondii; a protozoan) and tuberculosis (caused by Mycobacterium tuberculosis; a bacterium) [25–28]. A study showed that Cd4-Cre+ Bhlhe40fl/fl mice were highly susceptible to T. gondii and M. tuberculosis infections [17, 29]. Compared with Bhlhe40fl/fl CD4+ T cells, Cd4- Cre+ Bhlhe40fl/fl CD4+ T cells harvested from T. gondii-infected mice secreted less IFN-γ, but more IL-10, after antigen stimulation in vitro [17]. Moreover, in M. tuberculosis-infected Bhlhe40−/− mice, decreased Ifng and increased Il10 transcripts were observed in lung homogenates when compared with infected Bhlhe40+/+ mice [29]. In addition, disrupting IL-10 signaling, by either administering an IL-10R blocking antibody or by generating Bhlhe40−/− Il10−/− mice, rescued the lethal phenotypes resulting from Bhlhe40 deficiency in both disease models; this suggested that these phenotypes were driven by excessive IL-10 production [17, 29]. Of note, Bhlhe40 may also be required in dendritic cells (DC), as Cd11c-Cre+ Bhlhe40fl/fl mice were more susceptible to lethal M. tuberculosis infection than Bhlhe40fl/fl mice [29]. This could be related to Bhlhe40-mediated regulation of Il10 in DCs[29].
Recent work suggests that Bhlhe40 also regulates type 2 immunity. Using Cd4-Cre+ Bhlhe40fl/fl mice, Bhlhe40 was found to be crucial for CD4+ T cell-mediated control of the intestinal helminth Heligmosomoides polygyrus bakeri (H. polygyrus) [30]. Bhlhe40-sufficient mice are able to control helminth infection in part by generating type 2 granulomas, at the expense of damage to their intestinal architecture [30]. Bhlhe40−/− and Cd4-Cre+ Bhlhe40fl/fl mice are unable to generate a protective immune response and granulomas, resulting in impaired helminth control but protection from excessive tissue damage, suggesting that Bhlhe40 mediates immunopathology in this model [30]. Moreover, gene expression analysis of lamina propria CD4+ T cells indicated that the presence of Bhlhe40 was required for normal expression of many helminth-elicited genes [30]. This suggested that, in vivo, the TH2 cell program and related cytokine production was Bhlhe40-dependent, consistent with a CRISPR-Cas9 screen which assessed regulators of in vitro-derived TH2 cells and showed that Bhlhe40 was required to regulate Irf4 [31]. The related TF Bhlhe41 is also required in TH2 cells. Previous work described defects in IL-4, IL-5, and IL-13 production from Bhlhe41−/− mouse TH2 cells in vitro and in vivo after ovalbumin treatment (allergy/asthma model) or after treatment with Schistosoma mansoni eggs (a parasite eliciting potent type 2 immunity) (see Box 2 for further discussion of Bhlhe40/Bhlhe41 in vitro) [32].
In addition, during secondary H. polygyrus infection, Bhlhe40 was required for normal CD4+ T cell production of IL-5 and GM-CSF [30]. While GM-CSF had not been connected to helminth infection, exploring the effects of Bhlhe40 deficiency revealed a combinatorial role for IL-5 and GM-CSF in anti-helminth responses [30]. Csf2rb−/− mice, lacking the common beta chain cytokine receptor component (and therefore, responsiveness to IL-5 and GM-CSF), or combined antibody blockade of IL-5 and GM-CSF, demonstrated impaired recall responses to H. polygyrus [30]. This redundancy could have unexpected implications for atopic diseases. Taken together, we argue that Bhlhe40 can be a double-edged sword, acting to promote inflammatory responses (Figure 1), but sometimes at the cost of increased tissue damage. Investigation into these activities merits further attention.
Figure 1. Bhlhe40 regulates CD4+ T cell cytokines and shapes disease outcomes in mouse models.
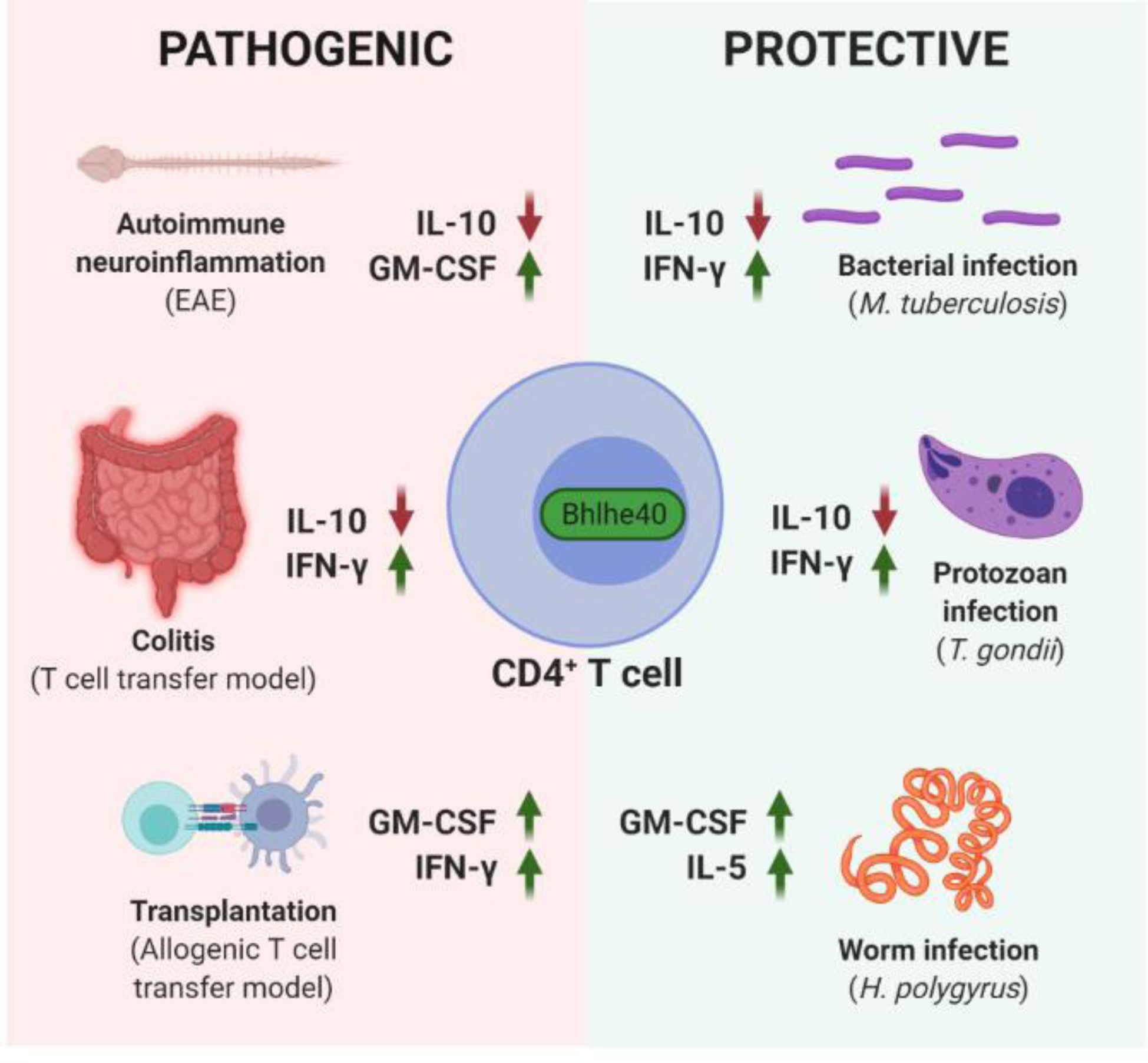
Bhlhe40 is detrimental in murine autoimmune and alloimmune models, mainly because Bhlhe40 drives CD4+ T cell-mediated inflammation by negatively regulating the antiinflammatory cytokine IL-10 and/or positively regulating the proinflammatory cytokines GM-CSF and IFN-γ (Left, “PATHOGENIC”) [11, 14, 17, 24]. However, such Bhlhe40-mediated inflammatory responses can be beneficial in host defense against Mycobacterium tuberculosis (M. tuberculosis), Toxoplasma gondii (T. gondii), and Heligmosomoides polygyrus bakeri (H. polygyrus), in some cases at the cost of increased tissue damage (not shown) (Right, “PROTECTIVE”) [17, 29, 30]. This figure was created using BioRender (https://biorender.com/).
BHLHE40 in human CD4+ T cells.
BHLHE40 is evolutionarily conserved [4, 33]. Similar to findings in murine CD4+ TH cells [13, 14], overexpression and knockout experiments with human circulating CD4+ T cells demonstrated that BHLHE40 expression positively correlated with expression of GM-CSF and other pro-inflammatory cytokines, including IL-17A, IL-22, and TNF [34]. In colorectal cancer patients, BHLHE40 was highly expressed by TH1-like tumor-infiltrating CD4+ T cells, a population enriched in microsatellite-unstable, checkpoint blockade-sensitive tumors, compared to microsatellite-stable, checkpoint-insensitive tumors [35]. Furthermore, anti-CD40 ligand (CD40L) agonist therapy in the MC38 mouse colon cancer model stimulated conventional type 1 DCs to elicit a similar Bhlhe40-expressing TH1-like population in tumors, as identified by single cell RNA-sequencing [36]. These studies support the notion that BHLHE40 is functionally conserved between mice and humans.
Bhlhe40/BHLHE40 in mouse and human B cells
Data regarding expression of Bhlhe40 in B cells have been conflicting, with reports indicating that Bhlhe40 expression is repressed [37–39] or induced upon B cell activation [40]. Variables ranging from B cell type (marginal zone versus follicular B cells in lymphoid organs), or stimulus, might explain to a certain extent these discordant findings. BHLHE40 has been implicated as a regulator of human germinal center B cells based on gene expression analyses of activated human B cells [41, 42], but its function remains untested. Much of the literature on Bhlhe40 and B cells concerns proliferation. Overexpression of Bhlhe40 in a mouse B cell line resulted in less G0/G1-to-S-phase transition relative to controls [37]. In addition, transgenic mice overexpressing Bhlhe40 (Clast5-Tg) exhibited reduced splenic and bone marrow (BM) B cell numbers with impaired proliferative responses to B cell receptor (BCR) crosslinking compared with non-transgenic mice [38]. This study also suggested that Bhlhe40 may be repressed early in B cell development, perhaps to allow proliferation in response to IL-7. Recently, Ikaros was identified as an important regulator of anergic B cells, as Cd23-Cre+ Ikarosfl/fl mice (exhibiting loss of Ikaros in splenic immature B cells) developed an autoimmune syndrome [43]. Of note, Ikaros was proposed to activate expression of Bhlhe40 in anergic B cells, suggesting that this regulation and Bhlhe40 itself may be important for maintenance of anergy [43]. It would therefore be informative to test whether Cd23-Cre+ Bhlhe40fl/fl mice might phenocopy the autoimmunity seen in Cd23-Cre+ Ikarosfl/fl mice. One could speculate that the B cell expansion seen in Bhlhe40−/−mice on a non-C57BL/6 background might be caused by loss of anergy, but this phenotype is confounded by potential B cell-extrinsic effects, namely on T cells [10]. Therefore, Bhlhe40’s function in B2 B cells remains poorly understood, especially concerning its regulation of proliferation and anergy. Further investigation is warranted to understand its functional role in a context- and species-dependent manner.
Of note, B cells are pathogenic in hepatitis C virus-associated mixed cryoglobulinemia (HCV-MC) -- a disease driven by potentially HCV cross-reactive VH1–69+ B cells [44]. In patients presenting with HCV-MC, VH1–69+ B cells have been reported to proliferate less and to express higher amounts of BHLHE40 than B cells from healthy controls; however, whether BHLHE40 can actually regulate the proliferation of these cells remains unclear [45, 46]. Nevertheless, these findings might be clinically relevant as HCV-MC patients are at an elevated risk of developing non-Hodgkin’s lymphoma [47]. Thus, it could be informative to investigate whether B cells from such patients have altered BHLHE40 expression which might contribute to proliferative expansion of malignant clones.
Bhlhe40 in tissue-resident leukocytes
A crucial feature of resident leukocytes is their capacity to locally proliferate for self-renewal and population expansion during an immune response [48, 49]. The transcriptional control of this process is not well understood. Until recently, it remained unclear whether tissue-specific TFs govern this process.
Macrophages
Bhlhe40 is expressed in several mouse myeloid cell subsets, including certain tissue-resident macrophage populations (notably alveolar macrophages (AMs) and peritoneal macrophages), granulocytes, and DCs [13].Natural mutations in E-box motifs bound by Bhlhe40 have correlated with altered binding of the macrophage-lineage specifying transcription factor PU.1 in large peritoneal macrophages (LPMs) [50]. Bhlhe40−/− or LysM-Cre+ Bhlhe40fl/fl conditional knockout mice showed reduced peritoneal macrophage populations in the steady state compared to Bhlhe40+/+ or LysM Cre− Bhlhe40fl/fl mice [51]. Together with models of IL-4 cytokine complex (IL-4c) treatment or helminth infection in mixed BM chimeras and LysM-Cre+ Bhlhe40fl/fl mice, these data indicated that Bhlhe40 was cell-intrinsically required for normal proliferation of tissue-resident LPMs and large pleural macrophages in homeostasis and during type 2 immunity, while dispensable in other macrophage populations [51–53]. A high proportion of Bhlhe40-deficient LPMs expressed Ki67, a marker of proliferating cells, suggesting that cell cycling was dysregulated relative to Bhlhe40-sufficient LPMs [51]. In this study, proliferation of Bhlhe40-deficient LPMs (determined by pHH3 staining and BrdU incorporation) was severely affected during type 2 immunity, despite normal acquisition of alternative activation markers (CD301 and RELMα staining) [51]. Furthermore, ChIP-Seq and gene expression microarray analysis indicated that one function of Bhlhe40 was to transcriptionally repress the TF c-Maf, which antagonizes macrophage proliferation, to allow normal cell cycling [48, 51, 54]. Additionally, thioglycolate-elicited macrophages responding to IL-4c (a mouse model of alternative activation of monocyte-derived macrophages [55, 56]), also exhibited Bhlhe40-dependent cell cycling (BrdU incorporation), suggesting that the peritoneal environment could induce a Bhlhe40-dependent proliferative program in both monocyte-derived and tissue-resident macrophages in these models [51].
A recent report demonstrated that Bhlhe40−/− Bhlhe41−/− mice (but not mice singly deficient for either factor) exhibited pronounced cell-intrinsic defects in AMs, including impaired proliferation (EdU incorporation) and overaccumulation of intracellular lipid stores (Oil Red O and BODIPY staining) [57]. Moreover, in contrast to Bhlhe40-deficient
LPMs, which largely maintained their tissue-specific gene expression program, Bhlhe40/41-deficient AMs exhibited reduced expression of AM-specific genes and increased expression of genes characteristic of other macrophage lineages [57]. Another study reported that Bhlhe41 was expressed at steady state in murine microglia [40]. Furthermore, Bhlhe40 was upregulated in microglia in a mouse model of Alzheimer’s disease induced by overexpression of mutated human APP-PS1 [58], though the functional importance and possible redundancy of these factors in microglia remains unknown. Taken together, these data suggest that Bhlhe40 plays important roles in homeostatic and inflammatory macrophages and is a tissue-specific regulator of macrophage proliferation in mice.
Lymphocytes
B1a cells.
Bhlhe41 is also a key regulator of splenic and peritoneal B1a cells in partnership with Bhlhe40 [39]. Bhlhe40−/− Bhlhe41−/− (and often Bhlhe41−/−) B1a cells exhibit profound defects, including an altered BCR repertoire, impaired signaling, and an increased proportion of Ki67+ cells relative to Bhlhe40+/+ Bhlhe41+/+ B1a cells [39]. In contrast to Bhlhe40-deficient LPMs -- not exhibiting an increase in cell cycle progression beyond G1 phase as compared to Bhlhe40-sufficient LPMs [51] -- a greater proportion of Bhlhe40/41-deficient B1a cells successfully complete cell cycling compared to Bhlhe40/41-sufficient B1a cells [39]. Despite their increased proliferation, Bhlhe40/41-deficient B1a cells are impaired in self-renewal due to increased cell death relative to Bhlhe40/41-sufficient B1a cells, resulting in fewer B1a cells in Bhlhe40−/− Bhlhe41−/− mice [39].
Tissue-resident memory CD8+ T cells.
Transcriptomes of tissue-resident memory CD8+ T cells (TRM) have identified Bhlhe40 as part of the core signature of mouse TRM [59]. Bhlhe40 is required by lung TRM to support survival, normal amounts of effector molecules (including cytokines, chemokines, and granzyme B), and normal mitochondrial function and acetyl coenzyme A (AcCoA) concentration [60]. It is unclear whether Bhlhe40 regulates mitochondrial metabolism in other immune cells. However, lipid accumulation or vacuolation of Bhlhe40/41- (or Bhlhe40-) deficient macrophages has been observed in mice [51, 57]. This is relevant as fatty acids are metabolized to AcCoA within mitochondria; thus, these data might suggest a role for Bhlhe40 in mitochondrial function in other hematopoietic lineages. In an influenza virus infection model, antigen-specific lung CD8+ TRM were reduced in number in Bhlhe40−/− and Cd4-Cre+ Bhlhe40fl/fl mice relative to Bhlhe40+/+ and Cd4-Cre− Bhlhe40fl/fl mice, respectively [60]. This correlated with increased apoptosis in antigen-specific Bhlhe40-deficient lung CD8+ TRM relative to Bhlhe40-sufficient lung CD8+ TRM [60], but whether or not a defect in proliferation also contributed to reduced cell numbers is unclear (see Box 3). Furthermore, Bhlhe40−/− and Cd4-Cre+ Bhlhe40fl/fl mice showed impaired TRM-dependent protective immunity upon influenza virus reinfection, demonstrating the physiologic importance of Bhlhe40 in lung TRM [60].
Box 3. The role of Bhlhe40 in T cell proliferation.
A still-unresolved question is the role that Bhlhe40 plays in the proliferation of CD4+ and CD8+ T cells. An early study showed impaired in vitro proliferation of mouse Bhlhe40−/− CD4+ T cells under limited anti-CD3/anti-CD28 antibody stimulation which could be restored upon IL-2 addition [10]. Lymph node cells from Bhlhe40−/− mice immunized with keyhole limpet hemocyanin (KLH) and complete Freund’s adjuvant were less proliferative in vitro after KLH restimulation than cells from Bhlhe40+/+ mice [10]. Others found that without added IL-2, mouse Bhlhe40−/− CD4+ T cells exhibited reduced proliferation in vitro compared to Bhlhe40+/+ CD4+ T cells [14]. In contrast, we and others showed that mouse Bhlhe40-deficient TH-neutral (with or without IL-2), TH1, TH2, and TH17 cells proliferated normally in vitro [17, 30]. Also, encephalitogenic Bhlhe40-expressing CD4+ T cells (assessed via GFP expression in Bhlhe40GFP reporter mice) in the mouse EAE model exhibited a greater frequency of Ki67 positivity relative to GFP-non-expressing CD4+ T cells, suggesting that Bhlhe40 might correlate with cell cycling in vivo, yet awaiting further investigation [13]. Bhlhe41 might also play a role in TH2 cell cycling in light of impaired Bhlhe41−/− TH2 cell proliferation in vitro, introducing additional complexity to this issue [32]. One group recently demonstrated that Bhlhe40 was essential for mouse TRM and tumor-infiltrating lymphocyte accumulation in influenza virus infection and transplantable tumor models, respectively [60]. This was at least in part due to a survival defect of Bhlhe40-deficient cells as demonstrated by increased apoptosis [60], but there might also be a role for Bhlhe40 in CD8+ T cell proliferation. Taken together, these data indicate that the effects of Bhlhe40 on T cell proliferation are complex and may be context-dependent. In light of the importance of the tissue environment in eliciting Bhlhe40-dependent proliferation in other lineages [51, 57], carefully controlled in vivo studies are needed to address the biological importance of Bhlhe40 and Bhlhe41 in T cell proliferation.
Bhlhe40 also plays an important role in tumor-infiltrating CD8+ T cells (TILs). In murine transplantable tumor models (B16-OVA, MC-38, and LLC lines), Bhlhe40−/− mice displayed increased tumor growth relative to Bhlhe40+/+ mice and Bhlhe40−/− TILs displayed decreased production of multiple effector molecules compared to Bhlhe40+/+ TILs [60]. Additionally, PD-1 signaling cell-intrinsically repressed Bhlhe40 expression in TILs in the B16-OVA model, and checkpoint blockade therapy with anti-PD-L1 antibody in this model was ineffective in controlling tumor growth in Bhlhe40−/− and Cd4-Cre+ Bhlhe40fl/fl mice relative to Bhlhe40+/+ and Cd4-Cre− Bhlhe40fl/fl mice [60].
Invariant NKT cells and innate lymphoid cells.
Whether Bhlhe40 is required in other tissue-resident lymphocytes is of considerable interest. In a mouse B16 melanoma model, IFN-γ production from Bhlhe40−/− iNKT cells was impaired, and these cells provided less protection against lung metastases compared to Bhlhe40+/+ iNKT cells [7]. Of note, Bhlhe40 expression appears to be widespread across multiple mouse innate lymphoid cell (ILC) populations. For instance, transcriptional profiling of mouse ILCs in multiple tissues has identified a signature of small intestinal lamina propria ILCs which includes Bhlhe40 [61]. Moreover, single cell RNA-sequencing has revealed Bhlhe40/BHLHE40 expression in both murine and human splenic NK cells [62]. Also, BHLHE40 DNA demethylation has been proposed as a putative biomarker of NK cell activation [63]. However, a functional role for Bhlhe40 in NK cells or other ILCs remains to be investigated. We speculate that Bhlhe40 has a function in ILCs akin to that in mouse TH cells, where it contributes to the regulation of cytokine production [11, 14]. Figure 2 summarizes Bhlhe40/41’s known roles in tissue-resident leukocytes, but in our opinion, the full extent of Bhlhe40’s (and Bhlhe41’s) role in tissue residency remains to be revealed, and careful assessment of individual and shared roles of Bhlhe40 and Bhlhe41 is required.
Figure 2. Roles of Bhlhe40 and Bhlhe41 in murine tissue-resident immune cells.
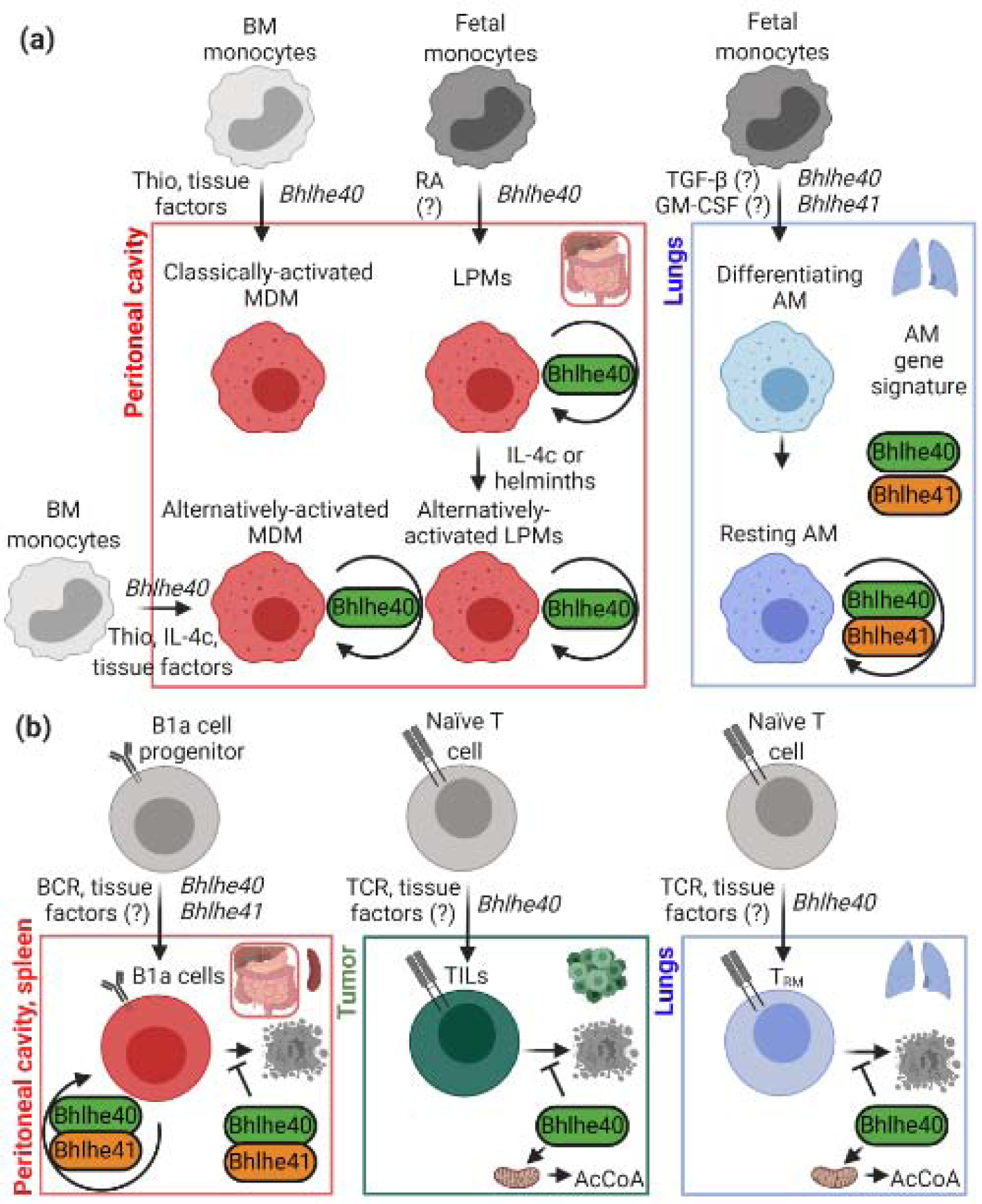
Bhlhe40 plays essential roles in tissue-resident macrophages (a) and lymphocytes (b), cooperating with Bhlhe41 in B1a cells and alveolar macrophages (AM) [39, 51, 57, 60]. In many cases, unknown tissue factors are thought to be important for Bhlhe40 expression, including retinoic acid [50]. These transcription factors regulate the proliferation of several resident leukocyte populations. In macrophages, this involves repression of c-Maf and MafB, but further mechanisms are thought to be involved [51, 57]. Recent work in TRM suggests that Bhlhe40 is also an important regulator of mitochondrial metabolism [60], though it is unclear whether Bhlhe40-deficient TRM are also deficient in proliferation. BM, bone marrow; Thio, thioglycolate; AM, alveolar macrophages; MDM, monocyte-derived macrophages; LPMs, large peritoneal macrophages; IL-4c, IL-4 cytokine complex; TILs, tumor-infiltrating lymphocytes; AcCoA, acetyl coenzyme A; RA, retinoic acid. This figure was created using BioRender (https://biorender.com/).
Mechanisms of Bhlhe40 activity
Upstream regulation of Bhlhe40.
Different stimuli likely induce Bhlhe40 expression in different cell types (Key Figure, Figure 3). In non-hematopoietic cells, Bhlhe40 can be induced during many physiologic responses, including hypoxia [64, 65] and upon circadian clock oscillation in rodents [66, 67], but these specific inducers have not been investigated in immune cells. Bhlhe40 was first identified as a retinoic acid (RA)-inducible gene [4], and RA maintained Bhlhe40 expression ex vivo in cultured LPMs [50]. As RA supports LPM differentiation [68], it is likely that LPM expression of Bhlhe40 is RA-dependent. However, whether expression of Bhlhe40 in peritoneal B1a cells is likewise RA-elicited is unclear. In AMs, one possible regulator may be GM-CSF, as it is crucial for differentiation of this population [69], and induces Bhlhe40 in cultured murine BM cells [29]. Alternatively, TGF-β might stimulate Bhlhe40 expression in mouse AMs [57], but this remains to be further assessed.
Key Figure, Figure 3. Regulation and functions of Bhlhe40 in mouse hematopoietic cells.
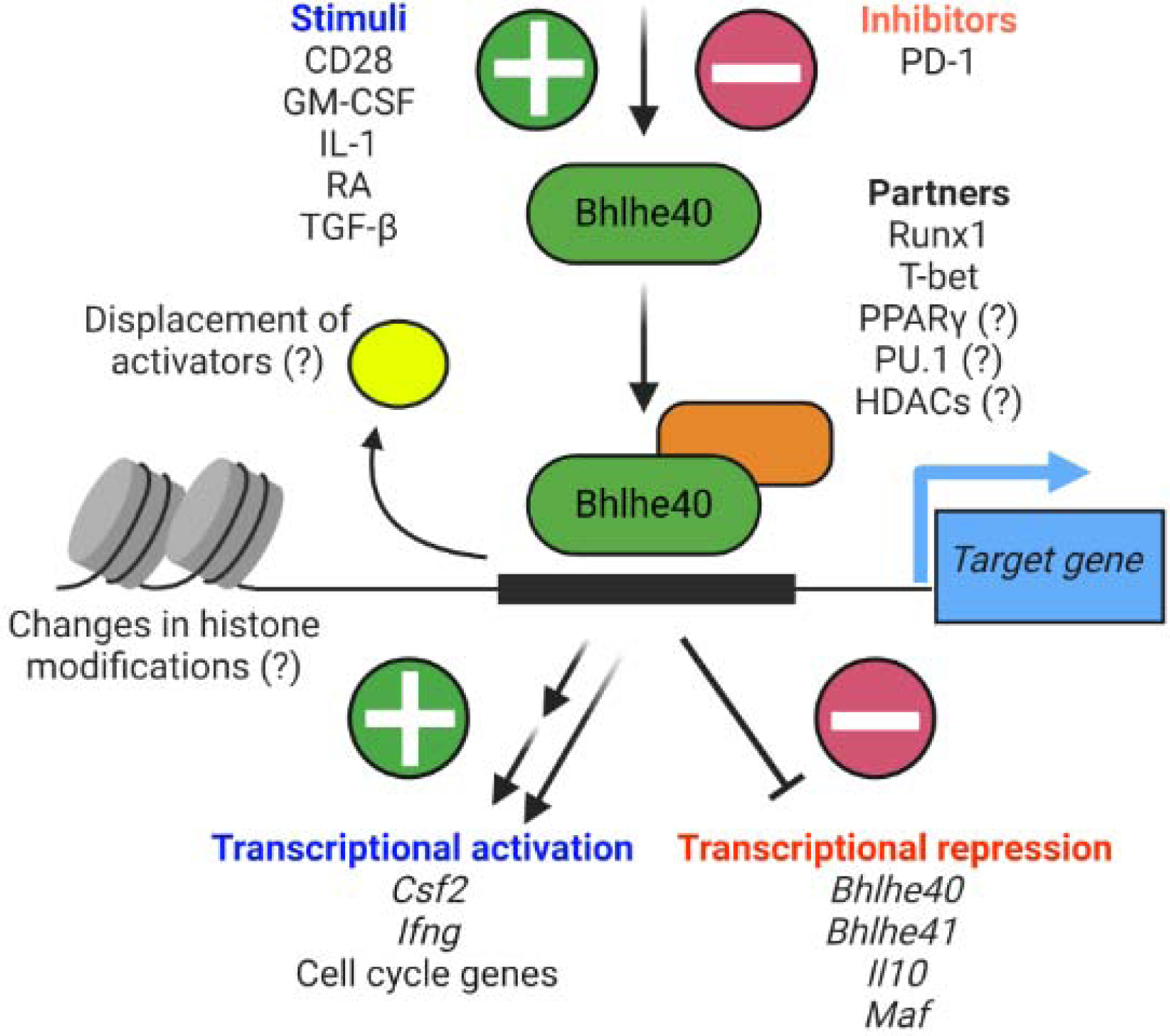
Multiple stimuli induce Bhlhe40 expression in immune cells, including IL-1, retinoic acid (RA), and GM-CSF. IL-1 is partially responsible for Bhlhe40 expression in TH17 cells [13, 70], retinoic acid (RA) can sustain Bhlhe40 expression by in vitro cultured LPMs [50], and GM-CSF can elicit Bhlhe40 expression from bone marrow monocytes in vitro [29]. However, the factors responsible for Bhlhe40 expression in immune cells in vivo are generally poorly understood. The activity of Bhlhe40 is likely dependent on its cell-specific binding partners. Bhlhe40 interacts with HDACs in cell lines and this may be important for its function in vivo [74]. Bhlhe40 is primarily a transcriptional repressor (represented by the single bar-headed line connecting Bhlhe40 to downstream targets), though some targets may be indirectly repressed. A potential mechanism of repression might be that Bhlhe40 displaces transcriptional activators from binding target loci. Bhlhe40 also possesses activating function at some loci. Therefore, while many transcripts that are reduced in the absence of Bhlhe40 are likely indirectly regulated (represented by the two staggered arrows), there are likely some loci directly activated by Bhlhe40 (represented by the single arrow). Human gene targets, including CSF2, MIR146A, and ZC3H12D are not depicted [34]. This figure was created using BioRender (https://biorender.com/).
Reports demonstrate that in mouse CD4+ and CD8+ T cells, Bhlhe40 is partially induced by T cell receptor (TCR) stimulation, but robust expression seems to require CD28-dependent costimulation and cytokine signaling [13, 14, 60]. After T cell activation, IL-1β can enhance Bhlhe40 transcript and protein expression in murine TH17 cells [13, 70, 71]. Following EAE induction, myeloid cells in murine draining lymph nodes can secrete IL-1β, which acts on polarized CD4+ T cells to ensure full Bhlhe40 expression [13, 72, 73]. Other cytokines also likely modulate Bhlhe40 expression. For instance, in a GvHD model, IL-1 receptor (IL-1R) deficiency (in Il1r1−/− mice) or treatment with the IL-1R antagonist Anakinra did not change Bhlhe40 mRNA expression in donor mouse CD4+ T cells, possibly because these IFN-γ-producing cells lacked IL-1R [24]. Regulation of Bhlhe40 differs between diseases, and different factors might stimulate Bhlhe40 expression during type 1, 2, and 3 immunity. It remains to be investigated which signaling pathways (e.g. RAR/RXR, JAK/STAT, SMADs, MAPK, PI3K/Akt) can actually regulate Bhlhe40, and if regulation via these signaling pathways varies between different immune cells.
Mechanisms of regulation by Bhlhe40
Activation vs. repression.
Bhlhe40 can function as both a transcriptional activator and repressor (Key Figure, Figure 3) [5–7, 74]. In murine immune cells, its role as a direct transcriptional activator is poorly understood and may be independent of its binding to E-box motifs; instead, it may depend on interactions with other TF partners at their DNA-binding motifs (possibly T-bet, Runx1, or PU.1) [7, 8, 50]. Thus, studies are needed to establish the basis of Bhlhe40-mediated transcriptional activation in immune cells.
As a transcriptional repressor in cell lines, Bhlhe40 sometimes outcompetes transcriptional activators at target genes [2, 74]. In immune cells, one possible example of this is at the +6 kb region of the Il10 locus, where Bhlhe40 has bound in mouse TH1 cells, GM-CSF-cultured BM cells, and LPMs, as demonstrated by ChIP-Seq [29, 51]. In other work, this enhancer region bound transcriptional activators AP-1, IRF4, and c-Maf in mouse CD4+ T cells [75–78], and we speculate that Bhlhe40 might function to disrupt their binding. Moreover, whether BHLHE40 transcriptionally represses the IL10 locus in human T cells remains unclear [34].
In other instances of repression, Bhlhe40 recruits HDAC1 to silence gene transcription in cell lines [74]. While Bhlhe40 and HDAC1 have not been shown to interact in immune cells, two Bhlhe40-dependent histone acetylation patterns recently emerged. Specifically, genes repressed by Bhlhe40/41 (e.g. Maf, Mafb) exhibit increased H3 acetylation in Bhlhe40−/− Bhlhe41−/− mouse AMs, consistent with HDAC-dependent repression [57]. Conversely, genes activated by Bhlhe40 (e.g. Ifng) exhibit decreased H3 acetylation in Bhlhe40−/− mouse iNKT and CD8+ T cells [7, 60]. In mouse TRM, Bhlhe40 can indirectly promote global histone acetylation (flow cytometric analysis of acetylated H3) by maintaining AcCoA amounts [60] -- an essential substrate for histone acetyl transferases. We speculate that in instances when Bhlhe40 partners with transcriptional activators, Bhlhe40’s interactions with HDACs might be prohibited, eliminating its repressive function.
Select targets of Bhlhe40.
Bhlhe40 and Bhlhe41 bind to their own promoter regions and auto- and cross-repression are seen in murine non-hematopoietic cell lines [2, 29, 51, 74]. Bhlhe40 ChIP-Seq in LPMs has indicated that Bhlhe40 binds multiple proximal sites in the Bhlhe40 and Bhlhe41 loci, and transcriptional data have shown that Bhlhe40−/− mouse LPMs exhibit higher Bhlhe41 expression than Bhlhe40+/+ LPMs [51]. Whether loss of auto- or cross-repression would significantly affect immunity is unknown. Maf has been identified as another target gene that is repressed by Bhlhe40 in TH1 cells and LPMs, as well as by Bhlhe40/41 in mouse AMs [17, 51, 57]. Of note, Bhlhe40 may also be repressed by c-Maf in mouse CD4+ T cells, though the mechanism by which this occurs has not been established [75, 78]. This is a potential instance of complex cross-regulation of Bhlhe40 with another TF.
In multiple studies, GM-CSF production by mouse CD4+ T cells is promoted by Bhlhe40 [11, 13, 14, 30], and candidate binding sites for Bhlhe40 have been identified in the mouse Csf2 locus [11, 30]. In human CD4+ T cells, BHLHE40 supports GM-CSF production, as evidenced by both CRISPR-guided deletion and lentiviral-driven overexpression [34]. However there is no evidence for direct interactions between BHLHE40 and the human CSF2 gene, as tested via BHLHE40 ChIP-Seq analysis in Jurkat T cells [34]; this suggests that BHLHE40 might promote GM-CSF expression indirectly. Two novel immunoregulatory targets are suppressed by BHLHE40 in human CD4+ T cells: miR-146a and ZC3H12D [34]. miR-146a prevents NF-kB activation [79] -- a pathway that induces CSF2-- and the RNase ZC3H12D degrades the mRNAs of several pro-inflammatory cytokines, including GM-CSF [34, 80, 81]. Specifically, ChIP-Seq and luciferase assays have revealed that BHLHE40 can regulate miR-146a indirectly, but can repress ZC3H12D gene expression directly in human CD4+ T cells [34]. Collectively, BHLHE40 appears to function as a direct and indirect transcriptional regulator in mammals (Figure 3). We expect that novel target genes of BHLHE40/41 may be identified when these factors are studied in additional immune cell types.
Cell cycling.
While Bhlhe40 has been linked to proliferation in some cancers and B cells, it has been recently shown to regulate proliferation of several mouse tissue-resident hematopoietic lineages as described above (LPMs, AMs, and B1a cells) [4, 37, 39, 51, 57]. In a seeming contradiction, older studies have linked Bhlhe40 with suppressed cell cycling, while others have linked it to promoting proliferation, particularly in the context of cancer, as recently reviewed [4, 82]. In light of the more recent data in mouse tissue-resident leukocytes [39, 51, 57], we think that these findings might be reconciled if one assumes that Bhlhe40 can enforce normal proliferation in multiple cellular lineages, perhaps inhibiting proliferation and/or preserving proliferative capacity depending on the cell type [4]. Presumably, when Bhlhe40 expression is lost, some populations of cells may inappropriately enter the cell cycle and fail to efficiently complete mitosis, or may undergo cell death [39, 51, 57].
Key downstream targets of Bhlhe40 and Bhlhe41 in cycling immune cells remain unknown. In murine macrophages, beyond Bhlhe40’s (and perhaps Bhlhe41’s) role in repressing Maf, Bhlhe40/41 has also repressed Mafb by as-yet unknown mechanisms [51, 57]. Bhlhe40/41 are suggested to repress E2f genes and support normal expression of common beta chain cytokine receptor components in mouse B1a cells [39]. Moreover, Bhlhe40 can bind loci encoding cell cycle genes whose expression is decreased in Bhlhe40−/− LPMs relative to Bhlhe40+/+ LPMs in mice [51], suggesting possible direct activation. Aside from these targets, identifying other genes controlled by Bhlhe40 might reveal novel regulators of tissue-resident leukocyte proliferation.
Cell identity.
Bhlhe40/41 are variably required for proper expression of lineage identity-related genes in different murine immune cell populations [30, 51, 57], and are likely dependent on their binding partners in specific cell types. In mouse AMs, analysis of binding motifs and gene expression data have suggested that Bhlhe40 possibly cooperates with peroxisome proliferator activated receptor γ (PPARγ) [57]. In addition, given that lineage-specific gene expression programs of H. polygyrus-elicited Bhlhe40−/− mouse CD4+ T cells and homeostatic Bhlhe40−/− Bhlhe41−/− mouse AMs are particularly disrupted, we suspect that in both cases, these factors have possible connections to PPARγ, warranting further investigation [30, 57, 83]. We speculate that there may be crosstalk between Bhlhe40/41 and PPARγ that is required in some lineages for transcriptional activation and cellular identity. Thus, this may be a fruitful area for future study.
Concluding remarks
Bhlhe40 is now established as an essential regulator of mouse and human CD4+ T cells, in addition to a growing number of tissue-resident leukocyte populations; it contributes to controlling cytokine secretion, cell cycling, and metabolism. Bhlhe40 tilts the balance towards inflammation over disease tolerance, suggesting therapeutic potential for stimulating or inhibiting Bhlhe40 in the appropriate setting. Key areas for future research (see Outstanding Questions) include determining the factors and signaling pathways stimulating Bhlhe40 expression in different contexts. Why does Bhlhe40 alone regulate some resident populations while Bhlhe40 and Bhlhe41 jointly control others? Is this division of labor functionally important? What other key partners does Bhlhe40 interact with in immune cells? Bhlhe40 is expressed in multiple other hematopoietic lineages, including ILCs, granulocytes, and DCs, suggesting further interesting biology. Given the rapid pace of research showing Bhlhe40’s importance in anti-tumor, anti-pathogen, autoimmune, and alloimmune responses, we are hopeful that BHLHE40 might be leveraged either as a putative biomarker, or as a candidate target for therapy in a variety of human diseases.
Outstanding Questions.
There are conflicting reports on whether Bhlhe40 regulates T cell proliferation in vitro. Does Bhlhe40 regulate T cell proliferation in vivo?
Does Bhlhe40 predominantly regulate cytokine production by direct transcriptional control or indirectly through other regulators such as c-Maf? Uncovering novel targets of Bhlhe40 could lead to discovery of molecules with unappreciated roles in controlling cytokine production.
Is Bhlhe40 an important regulator of non-hematopoietic cell cytokine production? Bhlhe40 has traditionally had roles in cell proliferation and differentiation in non hematopoietic cells, but its role as a cytokine regulator in these cells is unknown.
Are the roles for Bhlhe40 in CD4+ T helper cell subsets mirrored by key roles in ILCs? As ILCs are tissue-resident sentinels of immunity, understanding the transcriptional pathways that regulate their cytokine production could have important implications for pathogen control early in infection.
In light of Bhlhe40’s role in mitochondrial metabolism in TRM, is Bhlhe40 an important regulator of immunometabolism in other tissue-resident leukocytes?
While Bhlhe40 is a key regulator of specific tissue-resident macrophages in homeostasis, its expression can be induced in monocytes recruited during a type 2 immune response. Is Bhlhe40 important to monocyte-derived, alternatively-activated macrophages during pathological conditions such as fibrosis?
Highlights.
Bhlhe40 is a key regulator of cytokine production by human and mouse T cells.
Bhlhe40 can drive inflammation and enables typical immune responses in autoimmunity, transplantation, cancer, and infection.
Bhlhe40 is a tissue-specific regulator of murine tissue-resident macrophage proliferation.
Bhlhe40 and Bhlhe41 are required in mouse B1a cells for normal proliferation, survival, and BCR repertoire.
Bhlhe40 is essential for mitochondrial metabolism of mouse tissue-resident memory CD8+ T cells and tumor-infiltrating lymphocytes.
Bhlhe40 contributes to controlling the balance between inflammation and disease tolerance, promoting highly inflammatory responses.
Acknowledgements
The authors thank Tara Bradstreet and Elizabeth Schwarzkopf, without whom none of our studies on Bhlhe40 would have been possible and our collaborators, who provided essential scientific, material, and intellectual support. M.E.C. is supported by the National Science Foundation Graduate Research Fellowship program (DGE-1745038). B.T.E. is supported by National Institute of Allergy and Infectious Disease (NIAID) (AI113118 and AI132653).
Glossary
- Active and passive EAE models
EAE induced by immunization with myelin antigen (active model) or adoptive transfer of myelin-specific CD4+ T cells (passive model). Allogeneic: T cell response in the context of transplantation enabled by non-self MHC of either donor (by host T cells) or host (by donor T cells).
- Alternative activation
a broad term for the transcriptional and proteomic changes that occur in macrophages and monocytes during a type 2 immune response.
- Alveolar macrophages (AMs)
lung-resident; responsible for clearing debris and surfactants.
- Anergic
quiescent, hyporesponsive state of T and B cells generally induced by exposure to cognate antigen in the absence of costimulation as a mechanism of tolerance.
- Atopic diseases
allergic conditions such as allergic rhinitis, asthma, and eczema (atopic dermatitis) characterized by inappropriate immune responses to typically innocuous antigens.
- Bacterial artificial chromosome (BAC)
used to stably insert large portions of DNA into the genome, such as fluorescent reporter constructs.
- B1a B cells
one of two subtypes of innate-like, antibody-producing B1 lymphocytes (T cell-independent).
- B2 B cells
adaptive, antibody-producing lymphocytes (T cell-dependent).
- CD4+ T cells
helper T cells regulating the activity of other immune cells. Divided into several subsets including T helper (TH) type 1, TH2, TH17, and regulatory T cells (TREG).
- CD8+ T cells
cytotoxic T cells capable of killing target cells and producing cytokines.
- Cell cycle
process of cell division. Resting (G0 phase) cells proliferate by sequentially passing through G1, S, G2, and M phases.
- Checkpoint blockade
use of biologic agents to block inhibitory immunoreceptors.
- ChIP-Seq
method to determine transcription factor binding or chromatin modifications throughout the genome.
- Colitis
inflammation of the colon.
- Dendritic cells
professional antigen presenting cells encompassing several subsets, including cDC1, cDC2, and plasmacytoid DC.
- Experimental autoimmune encephalomyelitis (EAE)
animal model of multiple sclerosis; results in immune cell infiltration into the central nervous system, demyelination, and ascending paralysis.
- Granulocytes
immune cells characterized by prominent cytoplasmic granules (neutrophils, eosinophils, and basophils).
- Granuloma
structure formed by immune cells to encase pathogens/antigens during certain infections.
- Graft-versus-host disease
allogeneic reaction occurring post-transplant when donor (graft) lymphocytes attack recipient (host) tissue.
- Helminths
parasitic worms which establish chronic infections.
- Innate lymphoid cells
lack rearranged antigen-specific receptors capable of rapid cytokine production; include ILC1 (TH1-like), ILC2 (TH2-like), and ILC3 (TH17-like) subsets, as well as ILC1-like NK cells and lymphoid tissue inducer cells.
- Monocytes
circulating myeloid cells generated from primitive and definitive hematopoiesis. Fetal liver monocytes give rise to most tissue-resident macrophage populations. Adult bone marrow monocytes are recruited to peripheral tissues in homeostasis and differentiate into macrophages and so-called monocyte-derived dendritic cells during inflammation.
- Peritoneal macrophages
embryonically-derived large serous cavity and monocyte-derived small serous cavity macrophages found in the peritoneum.
- Thioglycolate broth
media preparation eliciting robust recruitment of monocyte-derived macrophages to the peritoneum.
- Tissue-resident memory T cells
frontline mediators of protective memory responses stably occupying tissue sites without exchange with the circulation.
- Type 2 immunity
characterized by TH2 cell responses and cytokines including IL-4, IL-5, and IL-13 observed in helminth infections and allergy.
- Vacuolation
the appearance of numerous membrane-enclosed fluid-filled vesicles within cells.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.St-Pierre B et al. (2002) Stra13 homodimers repress transcription through class B E-box elements. J. Biol. Chem 277, 46544–46551 [DOI] [PubMed] [Google Scholar]
- 2.Azmi S et al. (2003) mSharp-1/DEC2, a basic helix-loop-helix protein functions as a transcriptional repressor of E box activity and Stra13 expression. J. Biol. Chem 278, 20098–20109 [DOI] [PubMed] [Google Scholar]
- 3.Sato F et al. (2004) Functional analysis of the basic helix-loop-helix transcription factor DEC1 in circadian regulation. Interaction with BMAL1. Eur. J. Biochem 271, 4409–4419 [DOI] [PubMed] [Google Scholar]
- 4.Ow JR et al. (2014) Stra13 and Sharp-1, the non-grouchy regulators of development and disease. Curr. Top. Dev. Biol 110, 317–338 [DOI] [PubMed] [Google Scholar]
- 5.Qian Y et al. (2014) DEC1 coordinates with HDAC8 to differentially regulate TAp73 and ΔNp73 expression. PLoS One 9, e84015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Y et al. (2006) The expression of antiapoptotic protein survivin is transcriptionally upregulated by DEC1 primarily through multiple sp1 binding sites in the proximal promoter. Oncogene 25, 3296–3306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kanda M et al. (2016) Transcriptional regulator Bhlhe40 works as a cofactor of T-bet in the regulation of IFN-γ production in iNKT cells. Proc. Natl Acad. Sci. U. S. A 113, E3394–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miyazaki K et al. (2010) The role of the basic helix-loop-helix transcription factor Dec1 in the regulatory T cells. J. Immunol 185, 7330–7339 [DOI] [PubMed] [Google Scholar]
- 9.Kato Y et al. (2014) DEC1/STRA13/SHARP2 and DEC2/SHARP1 coordinate physiological processes, including circadian rhythms in response to environmental stimuli. Curr. Top. Dev. Biol 110, 339–372 [DOI] [PubMed] [Google Scholar]
- 10.Sun H et al. (2001) Defective T cell activation and autoimmune disorder in Stra13-deficient mice. Nat. Immunol 2, 1040–1047 [DOI] [PubMed] [Google Scholar]
- 11.Lin CC et al. (2014) Bhlhe40 controls cytokine production by T cells and is essential for pathogenicity in autoimmune neuroinflammation. Nat. Commun 5, 3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mendel I et al. (1995) A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2b mice: fine specificity and T cell receptor V beta expression of encephalitogenic T cells. Eur. J. Immunol 25, 1951–1959 [DOI] [PubMed] [Google Scholar]
- 13.Lin CC et al. (2016) IL-1-induced Bhlhe40 identifies pathogenic T helper cells in a model of autoimmune neuroinflammation. J. Exp. Med 213, 251–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martínez-Llordella M et al. (2013) CD28-inducible transcription factor DEC1 is required for efficient autoreactive CD4+ T cell response. J. Exp. Med 210, 1603–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Codarri L et al. (2011) RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat. Immunol 12, 560–567 [DOI] [PubMed] [Google Scholar]
- 16.El-Behi M et al. (2011) The encephalitogenicity of TH17 cells is dependent on IL-1 and IL-23-induced production of the cytokine GM-CSF. Nat. Immunol 12, 568–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu F et al. (2018) The transcription factor Bhlhe40 is a switch of inflammatory versus antiinflammatory Th1 cell fate determination. J. Exp. Med 215, 1813–1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bettelli E et al. (2003) IL-10, a key effector regulatory cytokine in experimental autoimmune encephalomyelitis. J. Autoimmun 20, 265–267 [DOI] [PubMed] [Google Scholar]
- 19.Powrie F et al. (1994) Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity 1, 553–562 [DOI] [PubMed] [Google Scholar]
- 20.Eri R et al. (2012) T cell transfer model of colitis: a great tool to assess the contribution of T cells in chronic intestinal inflammation. Methods Mol. Biol 844, 261–275 [DOI] [PubMed] [Google Scholar]
- 21.Shlomchik WD (2007) Graft-versus-host disease. Nat. Rev. Immunol 7, 340–352 [DOI] [PubMed] [Google Scholar]
- 22.Ullrich E et al. (2018) BATF-dependent IL-7RhiGM-CSF+ T cells control intestinal graft-versus-host disease. J. Clin. Invest 128, 916–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tugues S et al. (2018) Graft-versus-host disease, but not graft-versus-leukemia immunity, is mediated by GM-CSF-licensed myeloid cells. Sci. Transl. Med 10, eaat8410. [DOI] [PubMed] [Google Scholar]
- 24.Piper C et al. (2020) Pathogenic Bhlhe40+ GM-CSF+ CD4+ T cells promote indirect alloantigen presentation in the GI tract during GVHD. Blood 135, 568–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hunter CA et al. (1994) Production of gamma interferon by natural killer cells from Toxoplasma gondii-infected SCID mice: regulation by interleukin-10, interleukin-12, and tumor necrosis factor alpha. Infect. Immun 62, 2818–2824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cooper AM et al. (1993) Disseminated tuberculosis in interferon gamma gene-disrupted mice. J. Exp. Med 178, 2243–2247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flynn JL et al. (1993) An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J. Exp. Med 178, 2249–2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moreira-Teixeira L et al. (2017) T Cell-Derived IL-10 Impairs Host Resistance to Mycobacterium tuberculosis Infection. J. Immunol 199, 613–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huynh JP et al. (2018) Bhlhe40 is an essential repressor of IL-10 during Mycobacterium tuberculosis infection. J. Exp. Med 215, 1823–1838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jarjour NN et al. (2020) BHLHE40 Promotes TH2 Cell-Mediated Antihelminth Immunity and Reveals Cooperative CSF2RB Family Cytokines. J. Immunol 204, 923–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henriksson J et al. (2019) Genome-wide CRISPR Screens in T Helper Cells Reveal Pervasive Crosstalk between Activation and Differentiation. Cell 176, 882–896.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang XO et al. (2009) Requirement for the basic helix-loop-helix transcription factor Dec2 in initial TH2 lineage commitment. Nat. Immunol 10, 1260–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Teramoto M et al. (2001) Gene structure and chromosomal location of a human bHLH transcriptional factor DEC1 × Stra13 × SHARP-2/BHLHB2. J. Biochem 129, 391–396 [DOI] [PubMed] [Google Scholar]
- 34.Emming S et al. (2020) A molecular network regulating the proinflammatory phenotype of human memory T lymphocytes. Nat. Immunol 21, 388–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang L et al. (2018) Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 564, 268–272 [DOI] [PubMed] [Google Scholar]
- 36.Zhang L et al. (2020) Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell 181, 442–459.e29 [DOI] [PubMed] [Google Scholar]
- 37.Seimiya M et al. (2002) Clast5/Stra13 is a negative regulator of B lymphocyte activation. Biochem. Biophys. Res. Commun 292, 121–127 [DOI] [PubMed] [Google Scholar]
- 38.Seimiya M et al. (2004) Impaired lymphocyte development and function in Clast5/Stra13/DEC1-transgenic mice. Eur. J. Immunol 34, 1322–1332 [DOI] [PubMed] [Google Scholar]
- 39.Kreslavsky T et al. (2017) Essential role for the transcription factor Bhlhe41 in regulating the development, self-renewal and BCR repertoire of B-1a cells. Nat. Immunol 18, 442–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kin NW et al. (2008) DNA microarray gene expression profile of marginal zone versus follicular B cells and idiotype positive marginal zone B cells before and after immunization with Streptococcus pneumoniae. J. Immunol 180, 6663–6674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lefebvre C et al. (2010) A human B-cell interactome identifies MYB and FOXM1 as master regulators of proliferation in germinal centers. Mol. Syst. Biol 6, 377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Y et al. (2016) Global gene regulation during activation of immunoglobulin class switching in human B cells. Sci. Rep 6, 37988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schwickert TA et al. (2019) Ikaros prevents autoimmunity by controlling anergy and Toll-like receptor signaling in B cells. Nat. Immunol 20, 1517–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carbonari M et al. (2005) Hepatitis C virus drives the unconstrained monoclonal expansion of VH1–69-expressing memory B cells in type II cryoglobulinemia: a model of infection-driven lymphomagenesis. J. Immunol 174, 6532–6539 [DOI] [PubMed] [Google Scholar]
- 45.Visentini M et al. (2012) Clonal B cells of HCV-associated mixed cryoglobulinemia patients contain exhausted marginal zone-like and CD21low cells overexpressing Stra13. Eur. J. Immunol 42, 1468–1476 [DOI] [PubMed] [Google Scholar]
- 46.Camponeschi A et al. (2018) DEC1/STRA13 is a key negative regulator of activation-induced proliferation of human B cells highly expressed in anergic cells. Immunol. Lett 198, 7–11 [DOI] [PubMed] [Google Scholar]
- 47.Monti G et al. (2005) Incidence and characteristics of non-Hodgkin lymphomas in a multicenter case file of patients with hepatitis C virus-related symptomatic mixed cryoglobulinemias. Arch. Intern. Med 165, 101–105 [DOI] [PubMed] [Google Scholar]
- 48.Sieweke MH and Allen JE (2013) Beyond stem cells: self-renewal of differentiated macrophages. Science 342, 1242974. [DOI] [PubMed] [Google Scholar]
- 49.Beura LK et al. (2018) Intravital mucosal imaging of CD8+ resident memory T cells shows tissue-autonomous recall responses that amplify secondary memory. Nat. Immunol 19, 173–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gosselin D et al. (2014) Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 159, 1327–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jarjour NN et al. (2019) Bhlhe40 mediates tissue-specific control of macrophage proliferation in homeostasis and type 2 immunity. Nat. Immunol 20, 687–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jenkins SJ et al. (2011) Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science 332, 1284–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ruckerl D and Allen JE (2014) Macrophage proliferation, provenance, and plasticity in macroparasite infection. Immunol. Rev 262, 113–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Soucie EL et al. (2016) Lineage-specific enhancers activate self-renewal genes in macrophages and embryonic stem cells. Science 351, aad5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gundra UM et al. (2014) Alternatively activated macrophages derived from monocytes and tissue macrophages are phenotypically and functionally distinct. Blood 123, e110–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gundra UM et al. (2017) Vitamin A mediates conversion of monocyte-derived macrophages into tissue-resident macrophages during alternative activation. Nat. Immunol 18, 642–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rauschmeier R et al. (2019) Bhlhe40 and Bhlhe41 Transcription Factors Regulate Alveolar Macrophage Self-Renewal and Identity. EMBO J. 38, e101233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Krasemann S et al. (2017) The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 47, 566–581.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Milner JJ et al. (2017) Runx3 programs CD8+ T cell residency in non-lymphoid tissues and tumours. Nature 552, 253–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li C et al. (2019) The Transcription Factor Bhlhe40 Programs Mitochondrial Regulation of Resident CD8+ T Cell Fitness and Functionality. Immunity 51, 491–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robinette ML et al. (2015) Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat. Immunol 16, 306–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Crinier A et al. (2018) High-Dimensional Single-Cell Analysis Identifies Organ-Specific Signatures and Conserved NK Cell Subsets in Humans and Mice. Immunity 49, 971–986.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wiencke JK et al. (2016) The DNA methylation profile of activated human natural killer cells. Epigenetics 11, 363–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miyazaki K et al. (2002) Identification of functional hypoxia response elements in the promoter region of the DEC1 and DEC2 genes. J. Biol. Chem 277, 47014–47021 [DOI] [PubMed] [Google Scholar]
- 65.Yun Z et al. (2002) Inhibition of PPAR gamma 2 gene expression by the HIF-1-regulated gene DEC1/Stra13: a mechanism for regulation of adipogenesis by hypoxia. Dev. Cell 2, 331–341 [DOI] [PubMed] [Google Scholar]
- 66.Honma S et al. (2002) Dec1 and Dec2 are regulators of the mammalian molecular clock. Nature 419, 841–844 [DOI] [PubMed] [Google Scholar]
- 67.Butler MP et al. (2004) Dec1 and Dec2 expression is disrupted in the suprachiasmatic nuclei of Clock mutant mice. J. Biol. Rhythms 19, 126–134 [DOI] [PubMed] [Google Scholar]
- 68.Okabe Y and Medzhitov R (2014) Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell 157, 832–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shibata Y et al. (2001) GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity 15, 557–567 [DOI] [PubMed] [Google Scholar]
- 70.Lee HG et al. (2019) Pathogenic function of bystander-activated memory-like CD4+ T cells in autoimmune encephalomyelitis. Nat. Commun 10, 709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lin CC and Edelson BT (2017) New Insights into the Role of IL-1β in Experimental Autoimmune Encephalomyelitis and Multiple Sclerosis. J. Immunol 198, 4553–4560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ronchi F et al. (2016) Experimental priming of encephalitogenic Th1/Th17 cells requires pertussis toxin-driven IL-1β production by myeloid cells. Nat. Commun 7, 11541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mufazalov IA et al. (2017) IL-1 signaling is critical for expansion but not generation of autoreactive GM-CSF+ Th17 cells. EMBO J. 36, 102–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sun H and Taneja R (2000) Stra13 expression is associated with growth arrest and represses transcription through histone deacetylase (HDAC)-dependent and HDAC-independent mechanisms. Proc. Natl. Acad. Sci. U. S. A 97, 4058–4063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gabryšová L et al. (2018) c-Maf controls immune responses by regulating disease-specific gene networks and repressing IL-2 in CD4+ T cells. Nat. Immunol 19, 497–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jones EA and Flavell RA (2005) Distal enhancer elements transcribe intergenic RNA in the IL-10 family gene cluster. J. Immunol 175, 7437–7446 [DOI] [PubMed] [Google Scholar]
- 77.Ahyi AN et al. (2009) IFN regulatory factor 4 regulates the expression of a subset of Th2 cytokines. J. Immunol 183, 1598–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gabryšová L and O’Garra A (2018) Regulating the regulator: Bhlhe40 directly keeps IL-10 in check. J. Exp. Med 215, 1767–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Taganov KD et al. (2006) NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. U. S. A 103, 12481–12486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Minagawa K et al. (2014) Posttranscriptional modulation of cytokine production in T cells for the regulation of excessive inflammation by TFL. J. Immunol 192, 1512–1524 [DOI] [PubMed] [Google Scholar]
- 81.Wawro M et al. (2017) Intact NYN/PIN-Like Domain is Crucial for the Degradation of Inflammation-Related Transcripts by ZC3H12D. J. Cell. Biochem 118, 487–498 [DOI] [PubMed] [Google Scholar]
- 82.Kiss Z et al. (2020) Non-circadian aspects of BHLHE40 cellular function in cancer. Genes Cancer 11, 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen T et al. (2017) PPAR-γ promotes type 2 immune responses in allergy and nematode infection. Sci. Immunol 2, eaal5196. [DOI] [PubMed] [Google Scholar]
- 84.Boudjelal M et al. (1997) Overexpression of Stra13, a novel retinoic acid-inducible gene of the basic helix-loop-helix family, inhibits mesodermal and promotes neuronal differentiation of P19 cells. Genes Dev 11, 2052–2065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rossner MJ et al. (1997) SHARPs: mammalian enhancer-of-split- and hairy-related proteins coupled to neuronal stimulation. Mol. Cell. Neurosci 9, 460–475 [DOI] [PubMed] [Google Scholar]
- 86.Jiang X et al. (2008) BHLHB2 controls Bdnf promoter 4 activity and neuronal excitability. J. Neurosci 28, 1118–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rossner MJ, et al. Disturbed clockwork resetting in Sharp-1 and Sharp-2 single and double mutant mice. PLoS One, 3 (2008), Article e2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu Z et al. (2009) Dec2 promotes Th2 cell differentiation by enhancing IL-2R signaling. J. Immunol 183, 6320–6329 [DOI] [PubMed] [Google Scholar]