

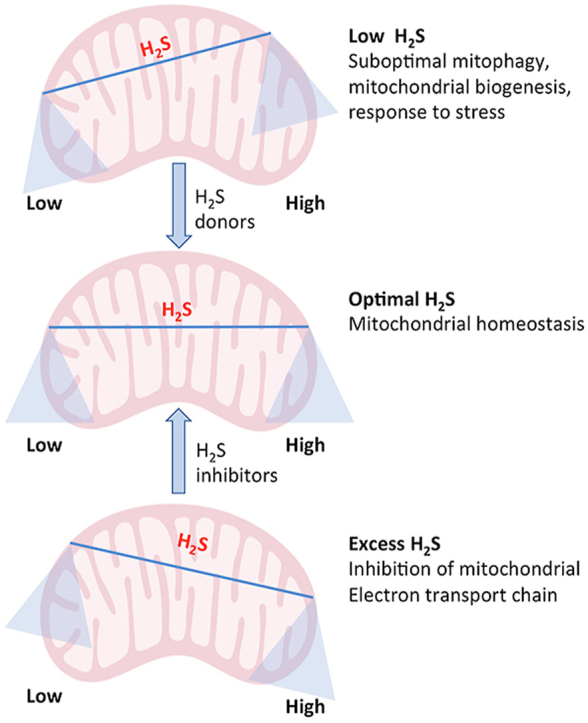
Abstract
Hydrogen sulfide (H2S) was once considered to have only toxic properties, until it was discovered to be an endogenous signaling molecule. The effects of H2S are dose dependent, with lower concentrations being beneficial and higher concentrations, cytotoxic. This scenario is especially true for the effects of H2S on mitochondrial function, where higher concentrations of the gasotransmitter inhibit the electron transport chain, and lower concentrations stimulate bioenergetics in multiple ways. Here we review the role of H2S in mitochondrial function and its effects on cellular physiology.
Keywords: Mitochondria, Hydrogen sulfide, Sulfide oxidation, Bioenergetics, Cell signaling, Sulfhydration/persulfidation
Graphical abstract
Highlights
-
•
Hydrogen sulfide (H2S) plays central roles in mitochondrial homeostasis.
-
•
Both excess H2S and a paucity of H2S have deleterious effects.
-
•
One of the modes by which H2S signals in mitochondria is by sulfhydrating target proteins.
-
•
Administering H2S (where scarcity of H2S occurs) or inhibiting H2S production (in case of excess H2S) may be beneficial.
Abbreviations
- 3-MST
3-mercaptopyruvate sulfur transferase
- AMPK
AMP-activated protein kinase
- CaMKII
Ca2+/calmodulin-dependent protein kinase II
- CAT
cysteine amino transferase
- CBS
cystathionine β-synthase
- CS
citrate synthase
- CSE
cystathionine γ-lyase
- DNMTA
DNA methyltransferase
- DS
Down syndrome
- IDH2
isocitrate dehydrogenase 2
- IRF-1
interferon regulatory factor 1
- LCAD
long chain acyl-CoA dehydrogenase
- mPTP
mitochondrial permeability transition pore
- OXPHOS
oxidative phosphorylation
- PTEN
lipid phosphatase and tensin homolog
- PLP
pyridoxal 5-phosphate
- sAC
soluble adenylyl cyclase
- SPRC
S-propyl-l-cysteine
- SQR
sulfide quinone oxidoreductase
- SDH
succinate dehydrogenase
- TR
thiosulfate reductase
- Trx
thioredoxin
- TrxR
thioredoxin reductase
- TST
thiosulfate sulfurtransferase
- TFAM
mitochondrial transcription factor A
- USP8
ubiquitin specific peptidase 8
1. Introduction
H2S was believed to be a noxious molecule and an environmental hazard until it was discovered to be produced endogenously [1]. H2S is generated in mammals by three enzymes: cystathionine γ-lyase (CSE), cystathionine β-synthase (CBS) and 3-mercaptopyruvate sulfur transferase (3-MST). CSE and CBS are key enzymes in the reverse transsulfuration pathway leading to the transfer of sulfur from homocysteine to cysteine (Fig. 1). CBS condenses serine with homocysteine to form cystathionine, which is then acted on by CSE to produce cysteine. CSE and CBS produce H2S by several different reactions [2]. CSE can use either cysteine or homocysteine in the presence of its cofactor, pyridoxal 5-phosphate (PLP) to produce H2S. CBS does not produce H2S from cysteine alone and prefers a combination of cysteine and homocysteine to produce H2S; CBS also utilizes homocysteine to produce H2S, although when homocysteine levels are high, the enzymatic activity of CBS is inhibited. 3-MST on the other hand produces H2S in conjunction with cysteine amino transferase (CAT) to produce H2S. H2S participates in a wide spectrum of physiological processes in every tissue in the body, functioning as a gaseous signaling molecule or gasotransmitter [[3], [4], [5]]. H2S levels are tightly regulated in cells as either excess or scarcity of the gaseous signaling molecule is detrimental. The mitochondria play a central role in the catabolism of H2S, which regulates its steady state levels.
Fig. 1.

Biosynthetic pathway leading to hydrogen sulfide production. Shown is the transsulfuration pathway, which involves transfer of sulfur from homocysteine to cysteine. Homocysteine is derived from dietary methionine in mammals by the action of methionine-adenosyltransferase (MAT) and methyltransferase (MT) and S-adenosylhomocysteine hydrolase (SAHH). Homocysteine condenses with serine in a condensation reaction catalyzed by cystathionine β-synthase (CBS) to produce cystathionine and water. Cystathionine is acted on by cystathionine γ-lyase (CSE) to generate cysteine. Cysteine is used as a substrate by CSE to generate H2S. CBS prefers a combination of cysteine and homocysteine to produce H2S and cystathionine. Cysteine is utilized by cysteine amino transferase (CAT) to produce 3-mercaptopyruvate, which is the substrate for 3-mercaptopyruvate sulfur transferase (3-MST) to produce hydrogen sulfide (H2S). Homocysteine can also be utilized as a substrate by CSE and CBS to generate H2S.
1.1. Hydrogen sulfide and mitochondrial bioenergetics
The mitochondria are the powerhouses of cells and the sites of aerobic respiration, generating ATP via oxidative phosphorylation (OXPHOS), accounting for about 80% of the energy requirements, the remaining 20% being met by glycolysis [6]. The mitochondrial OXPHOS system is composed of five multiprotein complexes (designated complex I–V) [6]. Electrons are transferred from NADH, an intermediate of the Krebs cycle, to NADH coenzyme Q reductase (complex I), which relays them to ubiquinone or coenzyme Q. Coenzyme Q also receives electrons from succinate dehydrogenase (SDH; complex II) and passes them to complex III (cytochrome bc1), which transfers them to cytochrome c, which relays them to complex IV (cytochrome c oxidase) that in turn uses these electrons to reduce molecular oxygen to water (Fig. 2A). The biological effects of H2S follow a bell shaped or biphasic dose-response curve. In the case of mitochondrial function, lower doses of H2S are beneficial, whereas higher doses are inhibitory. One of the earliest reported effects of H2S on mitochondrial function involved an induction of a state of suspended animation, which involved the inhibition of cytochrome c oxidase (Complex IV) of the electron transport chain in the mitochondria [[7], [8], [9], [10]]. H2S binds the copper center of cytochrome c oxidase to inhibit its activity [10,11]. The toxic effects of H2S that led to its classification as an environmental toxin or pollutant was primarily attributed to this property [12]. Another example of excess H2S production is Down syndrome (DS), which is caused by the trisomy of chromosome 21, which causes aberrant expression of genes on the chromosome, causing mental retardation along with vascular and metabolic abnormalities. As CBS is localized on chromosome 21, excess production of H2S was proposed to mediate these abnormalities in DS [13]. In postmortem samples of brains from DS patients, overexpression of CBS was observed which fits in with theory [14]. Similarly, overexpression of 3-MST was also observed in DS fibroblasts, which could contribute to excess H2S and mitochondrial dysfunction [15]. Inhibiting H2S production in fibroblasts derived from DS patients restored mitochondrial bioenergetics [16]. Excess H2S production was also reported in amyotrophic lateral sclerosis (ALS), a disease affecting motor neurons of the brain and spinal cord, leading to paralysis [17,18].
Fig. 2.

Effects of H2S on mitochondria. A) The mitochondrial electron transport chain (ETC) and H2S. The ETC comprises five complexes, designated I through V. Complex I and complex II also donate electrons to CoQ, by oxidation of NADH and succinate, respectively. The electrons are further relayed to complex III and then to complex IV (cytochrome c oxidase) via cytochrome c (cyt c). Cytochrome c oxidase transfers electrons to oxygen (which is the terminal electron acceptor and is reduced to water), while pumping protons across the membrane. The proton motive force is utilized by the F0F1 ATP synthase complex (often referred to as complex V) to catalyze the formation of ATP from ADP. H2S also donates electrons to the ETC to stimulate mitochondrial energetics. The donation of electrons by H2S occurs at the level of coenzyme Q (CoQ) through sulfide quinone oxidoreductase (SQR), forming sulfite (SO32−), sulfate (SO42−) and thiosulfate (S2O32−, denoted as SSO32−) in the process. B) The H2S oxidation pathway. SQR oxidizes H2S using SO32− or glutathione (GSH) as electron acceptors, converting them to S2O32− and glutathione persulfide (GSSH). Thiosulfate reductase (TR) converts S2O32− to GSSH using GSH. Persulfide dioxygenase (ETHE1) in the mitochondrial matrix oxidizes GSSH to SO32−. Sulfite is further oxidized to SO42− by sulfite oxidase (SO). Thiosulfate sulfur transferase (TST), a rhodanese, converts SO32− to S2O32−. Thus, oxidation of H2S in the mitochondria yields SO42− and S2O32−.
H2S was first linked to oxidative phosphorylation in 1986, when it was discovered that Solemya reidi, a gutless clam found in sulfide-rich habitats, oxidized H2S in its tissue mitochondria [19]. Two decades later, H2S was shown to be the first inorganic donor for energy production by mitochondria at low micromolar concentrations [20]. The donation of electrons occurs at the level of coenzyme Q by the action of sulfide quinone oxidoreductase (SQR) on H2S. Coenzyme Q also receives electrons from complex I by oxidation of NADH and Complex II by oxidation of succinate [21]. In addition to these parallel pathways, other oxidation reactions that donate electrons to coenzyme Q are FADH2 generated during fatty acid oxidation, or the oxidation of L-α glycerophosphate in muscle.
1.2. Mitochondrial localization of H2S enzymes
At low concentrations, H2S has beneficial effects on mitochondrial function. Several reports indicate that the biosynthetic enzymes for H2S may be present within the mitochondria. While 3-MST is present in both the cytoplasm and the mitochondria, CSE and CBS, are predominantly cytosolic, but they do translocate to the mitochondria as well. In vascular smooth muscle cells, calcium influx triggers mitochondrial translocation of CSE, a process dependent on translocase of the outer membrane 20 (Tom20) to generate H2S in the mitochondria [22]. The existence of CSE in the mitochondrial compartment was also suggested by earlier studies which report an increase in cystathionine content in rat liver mitochondria treated with propargylglycine, an inhibitor of CSE [23]. CBS, too is associated with mitochondria, and has been reported to be associated with the outer mitochondrial membrane in colon cancer cells, and stimulates mitochondrial bioenergetics [24]. Thus, all the three H2S enzymes modulate mitochondrial function and energetics. Mitochondrial homeostasis is intimately linked to almost all aspects of cellular physiology, several of which are regulated by H2S, hence it is not surprising that effects of the gasotransmitter on mitochondria are pivotal in the cellular functioning of the heart. The effects of H2S on mitochondrial functioning are discussed below.
1.3. The catabolism of H2S in mitochondria
In mammals, H2S is produced by both their own cells and by the intestinal flora [25]. As anaerobic metabolism by resident microbiota in the colon produce significant levels of H2S, the cells of the intestine must defend themselves, by either utilizing or detoxifying excess sulfide. H2S is oxidized to thiosulfate and sulfate in the mitochondria and its rate varies in different tissues [26]. The sulfide oxidation pathway is highly active in cells of the colon. SQR, part of the sulfide oxidation unit (SOU) catalyzes the first step in mitochondrial sulfide oxidation (Fig. 2B) [21,27]. Colon epithelial cells are exposed to high H2S levels and thus these cells harbor an efficient mitochondrial H2S oxidation pathway. In the human colon, the sulfide oxidation pathway enzymes exhibit an apical localization aligned with the host-microbiome interface [28]. H2S is oxidized by SQR, which forms a persulfide and releases two electrons, which are transferred by flavin adenine dinucleotide to CoQ, which then relays them to the ETC. The persulfide is then transferred to an acceptor such as GSH to form GSH persulfide (GSSH) or sulfite (SO32−), which is oxidized to sulfate, as shown in Fig. 2B. GSSH is then oxidized by persulfide dioxygenase (ETHE or SDO (sulfur dioxygenase) to sulfite. Sulfite can be oxidized to sulfate by sulfite oxidase (SO) or reduced by thiosulfate sulfurtransferase (TST/rhodanese) to form thiosulfate (SSO3) by the addition of a persulfide. The sulfane sulfur from thiosulfate can be acted on by another sulfurtransferase called thiosulfate reductase (TR) to form GSSH and SO32− [27]. Thus, in short, steady state levels of H2S are kept in control by the opposing actions of H2S biosynthetic enzymes and H2S degrading enzymes. Suboptimal SQR activity has also been suggested to be a cause of mitochondrial dysfunction in Leigh's disease, where inactivating mutations in SQR can cause an increase in H2S levels, which can then inhibit complex IV [29].
1.4. H2S and second messenger signaling
Similar to NO and CO, H2S influences second messenger signaling involving cyclic nucleotides. H2S is an endogenous inhibitor of phosphodiesterases (PDEs) which degrade cGMP and cAMP to mediate vasorelaxation [30]. Subsequent studies revealed that H2S not only inhibits PDEs, but also stimulates activation of soluble guanylyl cyclases, which synthesize cGMP from GTP, by altering the redox state (reduction of Fe3+ to Fe2+), facilitating its activation by NO [31]. ATP production in mitochondria is regulated by mechanisms involving protein kinase A (PKA), which phosphorylates mitochondrial proteins, including subunits of cytochrome c oxidase. PKA is activated by mitochondrial soluble adenylyl cyclase (sAC) in response to metabolically generated carbon dioxide [32]. The mitochondria too possess PDEs that regulate cyclic nucleotide levels, such as PDE2a, which degrades mitochondrial cAMP and NaHS was reported to inhibit its activity and elevate mitochondrial cAMP levels to augment mitochondrial respiration [32,33]. It should be noted that the first report of sulfide oxidation linked to ATP synthesis in any organism not specifically adapted to a sulfide-rich environment was by Yong and Searcy [34] who showed that chicken liver mitochondria consumed O2 at an accelerated rate when supplied with low concentrations of H2S and that sulfide oxidation was coupled to ATP synthesis.
1.5. H2S and NAD+ metabolism
NAD+ is a cofactor required for several enzymes involved in maintenance of mitochondrial function [35]. In addition to its essential role in the mitochondrial ETC at complex I, as a hydride acceptor to form NADH, which furnishes electrons to the ETC, NAD+ is consumed by enzymes such as the sirtuins and poly ADP ribosyl polymerases (PARPs) to regulate various aspects of cellular physiology such as mitochondrial biogenesis and DNA repair. Decrease in sirtuin activity and NAD+ levels have been linked to aging [36,37]. H2S has been reported to increase NAD+ levels in the vascular endothelium and H2S itself, associated sulfhydration and NAD+ are decreased during aging [38,39]. The sirtuins, SIRT1 and SIRT3 are sulfhydrated, which enhances their activity [40,41]. Accordingly boosting NAD+ levels may improve overall health and lifespan [37,42].
1.6. H2S and oxygen sensing
H2S plays important roles in maintenance of bioenergetics during hypoxia. H2S produced during normoxic conditions is oxidized in the mitochondria, while during hypoxia; this degradation is decreased, leading to an increase in its levels. Oxygen-sensitive H2S metabolism occurs in the mitochondria, which may balance energy requirements [43]. More recently, using a mitochondria‐targeted mass spectrometry probe MitoA, it was shown that hypoxia increases mitochondrial H2S in cardiomyocytes, suggesting a role for the gasotransmitter in oxygen sensing [44]. In addition to short-term oxygen sensing during acute hypoxia, H2S is also involved in long-term oxygen sensing or chronic hypoxia. Decreasing oxygen from 21% to 1% progressively increased H2S production in HEK 293 cells, which was concentrated in the mitochondria [45]. Interestingly, concentration of cysteine, the substrate for generation of H2S, was reported to be about three-fold higher in the mitochondria [22]. The same study also reported mitochondrial translocation of CSE during hypoxia. In addition, during hypoxia, mitochondrial CBS pools are no longer targeted for degradation by the Lon protease due to deoxygenation of its heme group, leading to a six-fold increase in the CBS [21]. Another mechanism involves regulation of protein kinase G (PKG) on oxygen sensing by the carotid body. Under normoxia, PKG, which is stimulated by CO produced by heme oxygenase, phosphorylates CSE at Ser377 inhibiting its activity. During hypoxia, heme oxygenase, whose activity is oxygen dependent, is inactive, leading to decreased phosphorylation of CSE by PKG [46]. Similarly, an interplay of CO and H2S production during hypoxia was also reported in cerebral microvasculature [47]. Whether mitochondria are involved in the process, remains to be determined.
1.7. Role of sulfhydration in mitochondrial function
Apart from the effects described above, H2S acts on mitochondrial proteins via a posttranslational modification designated as sulfhydration or persulfidation, wherein the –SH groups of cysteine residues are modified to persulfide or SSH groups [[48], [49], [50], [51]]. We have proposed previously that sulfhydration in addition to regulating signaling pathways, it protects against irreversible oxidation of cysteine residues [49]. This was subsequently demonstrated in the case of the lipid phosphatase and tensin homolog (PTEN), a tumor suppressor, and global protection of proteins during not only aging and neurodegeneration, but also during maintenance of physiological signaling [39,52,53]. This is especially relevant in the case of the mitochondria, as the organelle is constantly exposed to free radicals generated during oxidative phosphorylation. Several mitochondrial proteins have been reported to be sulfhydrated (Table 1). S-sulfhydration of the α subunit (ATP5A1) of ATP synthase (F0F1 ATP synthase/complex V) at Cys244 and 294 was reported to increase its activity in HepG2 and HEK293 cell lysates. Sulfhydration of ATP5A1 was upregulated in response to burn injury and decreased in mice lacking CSE implicating a role for CSE-derived H2S in the process [54]. Sulfhydration also exerts protective roles in mitochondrial function in the cardiovascular system. The Ca2+/calmodulin-dependent protein kinase II (CaMKII) is associated with heart failure and in the induction of myocardial mitochondrial injury. CaMKII is sulfhydrated at Cys6 in response to treatment with S-propyl-l-cysteine (SPRC), in a CSE-dependent manner, which decreases its activity in an isoprenaline-induced heart failure model [55]. The protective effect of H2S involved decreased oxidative stress, mitochondrial swelling, mitochondrial permeability transition pore (mPTP) opening and apoptosis. Clearance of damaged mitochondria by mitophagy plays a central role in mitochondrial homeostasis and disruption of this process impacts almost all physiological processes, ranging from cardiovascular functions to neuronal homeostasis. Parkin, an E3-ubiquitin ligase is a key protein involved in clearance of misfolded proteins and dysfunctional mitochondria and mutations in the gene encoding parkin, park2, are linked to autosomal recessive Parkinson's disease [56,57]. Sulfhydration of parkin enhances its E3-ubiquitin ligase activity and promotes clearance of aggregated proteins and facilitates mitophagy [58] (Fig. 3). The recruitment of parkin to damaged mitochondria requires the action of the deubiquitynating enzyme, ubiquitin specific peptidase 8 (USP8), which removes ubiquitin chains from parkin itself to facilitate recruitment of parkin to the mitochondria [59]. USP8 was also reported to be sulfhydrated in response to treatment with H2S donors, which facilitated its interaction with parkin and enhanced its mitochondrial docking in a mouse model of diabetic cardiomyopathy [60]. Other modes of sulfhydration mediated by cysteinyl-tRNA synthetase (CARS) was also suggested to play a role in mitochondrial bioenergetics [61,62]. More recently, an alternate mode of sulfhydration involving mitochondrial cytochrome c was discovered [63]. Reduction of ferric cytochrome c to its ferrous form was associated with increased sulfhydration in vitro. Silencing cytochrome c resulted in decreased sulfhydration of mitochondrial proteins. Consistent with these findings, cytochrome c released during apoptosis correlated with sulfhydration of procaspase 9 and loss of its activity. Levels of sulfhydration is also modulated endogenously by the thioredoxin/thioredoxin reductase (Trx/TrxR) system, and several studies have demonstrated the role of this system in signaling cascades ranging from endoplasmic reticulum stress (ER) to apoptosis [[64], [65], [66], [67]]. Thus, the mitochondrial thioredoxin system (TrxR2/Trx2) may also function to regulate sulfhydration, which may play key roles in cellular function via mitochondrial homeostasis. The relative contributions of the different modes of sulfhydration in mitochondrial function during normal conditions and during stress is an area that warrants further investigation.
Table 1.
Sulfhydration of proteins involved in mitochondrial function and its effects.
| Protein sulfhydrated | Effect on function | Reference |
|---|---|---|
| ATP synthase (F0F1 ATP synthase/complex V) | Stimulates enzyme activity and ATP generation | [54] |
| DJ-1 | Sulfhydration prevents the irreversible oxidation of DJ-1. DJ-1 plays critical roles in maintenance of redox balance in mitochondria. | [39] |
| Interferon Regulatory factor 1 (IRF-1) | Increases binding of IRF-1 at the Dnmta promoter and suppresses its expression, which leads to demethylation of Tfam promoter leading to increased expression TFAM and mitochondrial biogenesis. | [73] |
| Lactate dehydrogenase A (LDHA) | Stimulates LDH activity and increases conversion of lactate to pyruvate, generating NAD+ in the process, and stimulates mitochondrial bioenergetics. | [85] |
| Protein phosphatase 2A (PP2A) | Inhibits PP2A, a negative regulator of AMP kinase (AMPK) which leads to its activation. | [70] |
| p66Shc | Prevents PKCβII-mediated phosphorylation of Ser 36 of p66Shc and its translocation to the mitochondria, thereby preventing oxidative stress in the mitochondria. | [86] |
| Parkin | Activates the E3-ubiquitin ligase activity of parkin, which increases degradation of misfolded proteins. | [58] |
| Peroxisome proliferator-activated receptor-γ coactivator-related protein (PPRC) | Stimulates mitochondrial biogenesis in mouse hepatocytes. | [71] |
| Peroxisome proliferator-activated receptor gamma coactivator- 1α (PGC-1α) | Stimulates mitochondrial biogenesis in mouse hepatocytes. | [71] |
| Sirtuin 1 (SIRT1) | Increases its deacetylase activity and lowers its ubiquitination and reduced its degradation. | [40] |
| Sirtuin 3 (SIRT3) | Increases its deacetylase activity and protects mitochondria against cisplatin-induced kidney injury. Also protects against paraquat mediated liver injury. | [41,87] |
| Ubiquitin specific peptidase 8 (USP8)_ | Increases association of parkin with USP8, which is a deubiquitination enzyme (DUB), which promotes association of parkin to damaged mitochondria to augment mitophagy. | [60] |
Fig. 3.

Role of sulfhydration in mitophagy. The E3 ubiquitin ligase parkin is sulfhydrated which increases the ubiquitination and enhances mitophagy, the clearance of damaged mitochondria. Sulfhydration also facilitates mitophagy by activating the deubiquitinase, ubiquitin specific peptidase 8 (USP8), which removes ubiquitin groups from parkin and promoting its recruitment to damaged mitochondria. Icons of mitochondria generated from BioRender
1.8. H2S and mitochondrial biogenesis
Besides its effects on mitochondrial bioenergetics, H2S stimulates mitochondrial biogenesis. Administration of NaHS in a rat model of cardiac arrest and cardiopulmonary resuscitation preserved mitochondrial function and promoted mitochondrial biogenesis in the brain [68]. Similarly, in a model of ischemia reperfusion, genetic and pharmacologic increases in H2S levels increased mitochondrial biogenesis in the heart [69]. Mice deficient in CSE had decreased cardiac mitochondrial content as compared to their wild-type controls. By contrast, mice overexpressing CSE and mice administered the orally active H2S-donor, SG-1002, displayed enhanced cardiac mitochondrial content. In this system, H2S increased mitochondrial biogenesis by sulfhydrating and inhibiting protein phosphatase 2A (PP2A), which negatively regulates it in an AMP-activated protein kinase (AMPK)-dependent manner [70]. Hepatocytes derived from CSE-/- mice also displayed lower levels of mitochondrial transcription factors and coactivators as compared to wild type [71]. H2S donors increased the expression of the peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α), a key player in mitochondrial biogenesis and also caused its sulfhydration [72]. One of the master regulators for mitochondrial DNA replication is mitochondrial transcription factor A (TFAM). Expression of TFAM is negatively regulated by methylation of its promoter by the DNA methyltransferase, DNMTA. Expression of DNMTA is in turn, repressed by interferon regulatory factor 1 (IRF-1). Sulfhydration of IRF-1 enhances its binding to the DNMTA promoter and represses its expression, thereby preventing methylation of the TFAM promoter to increase its expression and thus, mitochondrial copy number [73]. CSE−/- mice exhibit reduced TFAM expression and mitochondrial copy number, confirming the role of CSE in maintenance of mitochondrial DNA copy number. Thus, H2S donors may alleviate mitochondrial dysfunction caused by inadequate mitochondrial biogenesis.
1.9. H2S and energy metabolism
Exogenous H2S switches substrate utilization from fatty acid oxidation to glucose in cardiomyocytes of the obese db/db mice by upregulating the expression and activity of the deacetylase, SIRT3, to cause a decrease in acetylation of fatty acid β-oxidation enzyme long chain acyl-CoA dehydrogenase (LCAD) and the acetylation of glucose oxidation enzymes pyruvate dehydrogenase (PDH), isocitrate dehydrogenase (IDH2), and citrate synthase (CS), which decreased LCAD activity and increased the activities of the glucose oxidation enzymes [74]. The H2S biosynthetic enzymes also participate in the maintenance of endothelial bioenergetics. 3-MST plays key roles in the energy metabolism of endothelial cells and its silencing reduced mitochondrial respiration and mitochondrial ATP production, and increased glucose uptake as well as fatty acid β-oxidation. 3-MST silencing resulted in increases in metabolites of the oxidative branch of pentose phosphate pathway (PPP) such as 6‐phosphogluconate, and sedoheptulose‐7‐phosphate, but decreased metabolites of the non-oxidative arm of the PPP, such as ribose 1‐phosphate, reflecting decreased nucleotide synthesis [75]. 3-MST was proposed to act as a regulator of the complex process that has been defined as the “angiogenic/metabolic switch” by which endothelial cells, switch from a quiescent state to a migratory and proliferative state during angiogenesis [76]. Thus, H2S generating enzymes regulate various aspects of energy metabolism to maintain mitochondrial homeostasis.
1.10. Mitochondria-targeted H2S donors
Several diseases are associated with impaired mitochondrial function and H2S donors can be beneficial in cases where there is a paucity of the gaseous signaling molecule. Several mitochondria-targeted H2S donors, which include AP39 and AP123, anethole dithiolethione and hydroxythiobenzamide respectively were developed, which improved mitochondrial functions in several cell types [77,78]. For instance, AP39 improved mitochondrial function in renal epithelial cells, endothelial cells and trophoblasts undergoing oxidative stress [77,79,80]. AP39 was also reported to support mitochondrial bioenergetics in the APP/PS1 primary neurons derived from mouse model of Alzheimer's disease and delay disease progression [81]. AP39 was also harnessed for organ preservation. AP39 protected cardiomyocytes from ischemia-reperfusion injury during cardiac transplantation and had a similar effect on renal grafts as well [82,83]. Thus, these donors hold great promise in the treatment of diseases involving suboptimal mitochondrial function.
2. Conclusions and future perspectives
It is becoming increasingly clear that H2S is both a poison, a fuel and a signaling molecule depending on the context. The deleterious effects of H2S on Complex IV of the mitochondria can be harnessed in a clinical setting under controlled conditions to induce a state of hypometabolism, which may improve surgical outcomes. Similarly, in colon carcinomas, where excess H2S is produced by CBS and utilized as a fuel by cancerous cells, inhibition of CBS may be beneficial. Interestingly, in the context of neurodegenerative diseases such as ALS and DS, excess H2S compromises mitochondrial function and inhibition of H2S production may be beneficial. A point to be noted is that the sulfide oxidation pathway is highly active in cancer cells and in the colon, while it is almost non-functional in neurons, once again adding an additional layer of distinction between cancer and neurodegeneration at the molecular level [21,84]. Past reports of the actions of H2S focused on the toxic effects of the gaseous molecule and the studies were conducted using higher doses of H2S donors. The apparent discrepancy in the effects of H2S stemmed largely from its biphasic effects and this is especially relevant in the context of mitochondrial function. Thus, use of optimal doses of H2S donors or its inhibitors as well as timing, duration and routes of delivery should be carefully considered while targeting diseases involving dysregulated H2S signaling.
Acknowledgements
KK: Supported in part by the National Institutes of Health [R24 DA018055; R01GM123508] and the Professional Staff Congress-City University of New York (PSC-CUNY) [TRADB-49-271]. This work was supported in part by US Public Health Service Grant DA044123 (to S.H.S), the American Heart Association (AHA)-Allen Initiative in Brain Health and Cognitive Impairment (to S.H.S and associates) and the Solve ME/CFS Initiative (SMCI) to BDP.
Contributor Information
Bindu D. Paul, Email: bpaul8@jhmi.edu.
Solomon H. Snyder, Email: ssnyder@jhmi.edu.
Khosrow Kashfi, Email: kashfi@med.cuny.edu.
References
- 1.Beauchamp R.O., Jr., Bus J.S., Popp J.A., Boreiko C.J., Andjelkovich D.A. A critical review of the literature on hydrogen sulfide toxicity. Crit. Rev. Toxicol. 1984;13:25–97. doi: 10.3109/10408448409029321. [DOI] [PubMed] [Google Scholar]
- 2.Olson K.R. H(2)S and polysulfide metabolism: conventional and unconventional pathways. Biochem. Pharmacol. 2018;149:77–90. doi: 10.1016/j.bcp.2017.12.010. [DOI] [PubMed] [Google Scholar]
- 3.Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol. Rev. 2012;92:791–896. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- 4.Paul B.D., Snyder S.H. Gasotransmitter hydrogen sulfide signaling in neuronal health and disease. Biochem. Pharmacol. 2018;149:101–109. doi: 10.1016/j.bcp.2017.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paul B.D., Snyder S.H. Modes of physiologic H2S signaling in the brain and peripheral tissues. Antioxidants Redox Signal. 2015;22:411–423. doi: 10.1089/ars.2014.5917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Papa S. Mitochondrial oxidative phosphorylation changes in the life span. Molecular aspects and physiopathological implications. Biochim. Biophys. Acta. 1996;1276:87–105. doi: 10.1016/0005-2728(96)00077-1. [DOI] [PubMed] [Google Scholar]
- 7.Blackstone E., Morrison M., Roth M.B. H2S induces a suspended animation-like state in mice. Science. 2005;308:518. doi: 10.1126/science.1108581. [DOI] [PubMed] [Google Scholar]
- 8.Hill B.C. Interactions of sulphide and other ligands with cytochrome c oxidase. An electron-paramagnetic-resonance study. Biochem. J. 1984;224:591–600. doi: 10.1042/bj2240591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petersen L.C. The effect of inhibitors on the oxygen kinetics of cytochrome c oxidase. Biochim. Biophys. Acta. 1977;460:299–307. doi: 10.1016/0005-2728(77)90216-x. [DOI] [PubMed] [Google Scholar]
- 10.Nicholls P., Kim J.K. Sulphide as an inhibitor and electron donor for the cytochrome c oxidase system. Can. J. Biochem. 1982;60:613–623. doi: 10.1139/o82-076. [DOI] [PubMed] [Google Scholar]
- 11.Modis K. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part II. Pathophysiological and therapeutic aspects. Br. J. Pharmacol. 2014;171:2123–2146. doi: 10.1111/bph.12368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Szabo C., Papapetropoulos A. International union of basic and clinical pharmacology. CII: pharmacological modulation of H2S levels: H2S donors and H2S biosynthesis inhibitors. Pharmacol. Rev. 2017;69:497–564. doi: 10.1124/pr.117.014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kamoun P. Mental retardation in Down syndrome: a hydrogen sulfide hypothesis. Med. Hypotheses. 2001;57:389–392. doi: 10.1054/mehy.2001.1377. [DOI] [PubMed] [Google Scholar]
- 14.Ichinohe A. Cystathionine beta-synthase is enriched in the brains of Down's patients. Biochem. Biophys. Res. Commun. 2005;338:1547–1550. doi: 10.1016/j.bbrc.2005.10.118. [DOI] [PubMed] [Google Scholar]
- 15.Panagaki T., Randi E.B., Szabo C. Role of 3-mercaptopyruvate sulfurtransferase in the regulation of proliferation and cellular bioenergetics in human Down syndrome fibroblasts. Biomolecules. 2020;10 doi: 10.3390/biom10040653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Panagaki T., Randi E.B., Augsburger F., Szabo C. Overproduction of H2S, generated by CBS, inhibits mitochondrial Complex IV and suppresses oxidative phosphorylation in Down syndrome. Proc. Natl. Acad. Sci. U. S. A. 2019;116:18769–18771. doi: 10.1073/pnas.1911895116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davoli A. Evidence of hydrogen sulfide involvement in amyotrophic lateral sclerosis. Ann. Neurol. 2015;77:697–709. doi: 10.1002/ana.24372. [DOI] [PubMed] [Google Scholar]
- 18.Spalloni A. Impact of pharmacological inhibition of hydrogen sulphide production in the SOD1G93A-ALS mouse model. Int. J. Mol. Sci. 2019;20 doi: 10.3390/ijms20102550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Powell M.A., Somero G.N. Hydrogen sulfide oxidation is coupled to oxidative phosphorylation in mitochondria of Solemya reidi. Science. 1986;233:563–566. doi: 10.1126/science.233.4763.563. [DOI] [PubMed] [Google Scholar]
- 20.Goubern M., Andriamihaja M., Nubel T., Blachier F., Bouillaud F. Sulfide, the first inorganic substrate for human cells. Faseb. J. 2007;21:1699–1706. doi: 10.1096/fj.06-7407com. [DOI] [PubMed] [Google Scholar]
- 21.Abou-Hamdan A. Oxidation of H2S in mammalian cells and mitochondria. Methods Enzymol. 2015;554:201–228. doi: 10.1016/bs.mie.2014.11.042. [DOI] [PubMed] [Google Scholar]
- 22.Fu M. Hydrogen sulfide (H2S) metabolism in mitochondria and its regulatory role in energy production. Proc. Natl. Acad. Sci. U. S. A. 2012;109:2943–2948. doi: 10.1073/pnas.1115634109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ohta J. Increase in cystathionine content in rat liver mitochondria after D,L-propargylglycine administration. Amino Acids. 1995;9:111–122. doi: 10.1007/BF00805832. [DOI] [PubMed] [Google Scholar]
- 24.Szabo C. Tumor-derived hydrogen sulfide, produced by cystathionine-beta-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc. Natl. Acad. Sci. U. S. A. 2013;110:12474–12479. doi: 10.1073/pnas.1306241110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gibson G.R., Cummings J.H., Macfarlane G.T. Competition for hydrogen between sulphate-reducing bacteria and methanogenic bacteria from the human large intestine. J. Appl. Bacteriol. 1988;65:241–247. doi: 10.1111/j.1365-2672.1988.tb01891.x. [DOI] [PubMed] [Google Scholar]
- 26.Bartholomew T.C., Powell G.M., Dodgson K.S., Curtis C.G. Oxidation of sodium sulphide by rat liver, lungs and kidney. Biochem. Pharmacol. 1980;29:2431–2437. doi: 10.1016/0006-2952(80)90346-9. [DOI] [PubMed] [Google Scholar]
- 27.Hildebrandt T.M., Grieshaber M.K. Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J. 2008;275:3352–3361. doi: 10.1111/j.1742-4658.2008.06482.x. [DOI] [PubMed] [Google Scholar]
- 28.Libiad M. Hydrogen sulfide perturbs mitochondrial bioenergetics and triggers metabolic reprogramming in colon cells. J. Biol. Chem. 2019;294:12077–12090. doi: 10.1074/jbc.RA119.009442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Friederich M.W. Pathogenic variants in SQOR encoding sulfide:quinone oxidoreductase are a potentially treatable cause of Leigh disease. J. Inherit. Metab. Dis. 2020 doi: 10.1002/jimd.12232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bucci M. Hydrogen sulfide is an endogenous inhibitor of phosphodiesterase activity. Arterioscler. Thromb. Vasc. Biol. 2010;30:1998–2004. doi: 10.1161/ATVBAHA.110.209783. [DOI] [PubMed] [Google Scholar]
- 31.Zhou Z. Regulation of soluble guanylyl cyclase redox state by hydrogen sulfide. Pharmacol. Res. 2016;111:556–562. doi: 10.1016/j.phrs.2016.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Acin-Perez R. Cyclic AMP produced inside mitochondria regulates oxidative phosphorylation. Cell Metabol. 2009;9:265–276. doi: 10.1016/j.cmet.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Modis K., Panopoulos P., Coletta C., Papapetropoulos A., Szabo C. Hydrogen sulfide-mediated stimulation of mitochondrial electron transport involves inhibition of the mitochondrial phosphodiesterase 2A, elevation of cAMP and activation of protein kinase A. Biochem. Pharmacol. 2013;86:1311–1319. doi: 10.1016/j.bcp.2013.08.064. [DOI] [PubMed] [Google Scholar]
- 34.Yong R., Searcy D.G. Sulfide oxidation coupled to ATP synthesis in chicken liver mitochondria. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2001;129:129–137. doi: 10.1016/s1096-4959(01)00309-8. [DOI] [PubMed] [Google Scholar]
- 35.Canto C., Menzies K.J., Auwerx J. NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metabol. 2015;22:31–53. doi: 10.1016/j.cmet.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Imai S., Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014;24:464–471. doi: 10.1016/j.tcb.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bonkowski M.S., Sinclair D.A. Slowing ageing by design: the rise of NAD(+) and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol. 2016;17:679–690. doi: 10.1038/nrm.2016.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Das A. Impairment of an endothelial NAD(+)-H2S signaling network is a reversible cause of vascular aging. Cell. 2019;176:944–945. doi: 10.1016/j.cell.2019.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zivanovic J. Selective persulfide detection reveals evolutionarily conserved antiaging effects of S-sulfhydration. Cell Metabol. 2019;30:1152–1170 e1113. doi: 10.1016/j.cmet.2019.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Du C. Sulfhydrated sirtuin-1 increasing its deacetylation activity is an essential epigenetics mechanism of anti-atherogenesis by hydrogen sulfide. Antioxidants Redox Signal. 2019;30:184–197. doi: 10.1089/ars.2017.7195. [DOI] [PubMed] [Google Scholar]
- 41.Yuan Y. S-sulfhydration of SIRT3 by hydrogen sulfide attenuates mitochondrial dysfunction in cisplatin-induced acute kidney injury. Antioxidants Redox Signal. 2019;31:1302–1319. doi: 10.1089/ars.2019.7728. [DOI] [PubMed] [Google Scholar]
- 42.Fang E.F. NAD(+) in aging: molecular mechanisms and translational implications. Trends Mol. Med. 2017;23:899–916. doi: 10.1016/j.molmed.2017.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Olson K.R. Hydrogen sulfide as an oxygen sensor. Antioxidants Redox Signal. 2015;22:377–397. doi: 10.1089/ars.2014.5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arndt S. Assessment of H2S in vivo using the newly developed mitochondria-targeted mass spectrometry probe MitoA. J. Biol. Chem. 2017;292:7761–7773. doi: 10.1074/jbc.M117.784678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Olson K.R. Extended hypoxia-mediated H2 S production provides for long-term oxygen sensing. Acta Physiol. 2020;228 doi: 10.1111/apha.13368. [DOI] [PubMed] [Google Scholar]
- 46.Yuan G. Protein kinase G-regulated production of H2S governs oxygen sensing. Sci. Signal. 2015;8:ra37. doi: 10.1126/scisignal.2005846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morikawa T. Hypoxic regulation of the cerebral microcirculation is mediated by a carbon monoxide-sensitive hydrogen sulfide pathway. Proc. Natl. Acad. Sci. U. S. A. 2012;109:1293–1298. doi: 10.1073/pnas.1119658109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mustafa A.K. H2S signals through protein S-sulfhydration. Sci. Signal. 2009;2:ra72. doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paul B.D., Snyder S.H. H(2)S signalling through protein sulfhydration and beyond. Nat. Rev. Mol. Cell Biol. 2012;13:499–507. doi: 10.1038/nrm3391. [DOI] [PubMed] [Google Scholar]
- 50.Paul B.D., Snyder S.H. H2S: a novel gasotransmitter that signals by sulfhydration. Trends Biochem. Sci. 2015;40:687–700. doi: 10.1016/j.tibs.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Filipovic M.R., Zivanovic J., Alvarez B., Banerjee R. Chemical biology of H2S signaling through persulfidation. Chem. Rev. 2018;118:1253–1337. doi: 10.1021/acs.chemrev.7b00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Greiner R. Polysulfides link H2S to protein thiol oxidation. Antioxidants Redox Signal. 2013;19:1749–1765. doi: 10.1089/ars.2012.5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Doka E. Control of protein function through oxidation and reduction of persulfidated states. Sci Adv. 2020;6 doi: 10.1126/sciadv.aax8358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Modis K. S-Sulfhydration of ATP synthase by hydrogen sulfide stimulates mitochondrial bioenergetics. Pharmacol. Res. 2016;113:116–124. doi: 10.1016/j.phrs.2016.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu D. Amelioration of mitochondrial dysfunction in heart failure through S-sulfhydration of Ca(2+)/calmodulin-dependent protein kinase II. Redox Biol. 2018;19:250–262. doi: 10.1016/j.redox.2018.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ge P., Dawson V.L., Dawson T.M. PINK1 and Parkin mitochondrial quality control: a source of regional vulnerability in Parkinson's disease. Mol. Neurodegener. 2020;15:20. doi: 10.1186/s13024-020-00367-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shimura H. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- 58.Vandiver M.S. Sulfhydration mediates neuroprotective actions of parkin. Nat. Commun. 2013;4:1626. doi: 10.1038/ncomms2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Durcan T.M. USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from parkin. EMBO J. 2014;33:2473–2491. doi: 10.15252/embj.201489729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sun Y. Exogenous H2S promoted USP8 sulfhydration to regulate mitophagy in the hearts of db/db mice. Aging Dis. 2020;11:269–285. doi: 10.14336/AD.2019.0524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fujii S., Sawa T., Motohashi H., Akaike T. Persulfide synthases that are functionally coupled with translation mediate sulfur respiration in mammalian cells. Br. J. Pharmacol. 2019;176:607–615. doi: 10.1111/bph.14356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Akaike T. Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat. Commun. 2017;8:1177. doi: 10.1038/s41467-017-01311-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vitvitsky V. Cytochrome c reduction by H2S potentiates sulfide signaling. ACS Chem. Biol. 2018;13:2300–2307. doi: 10.1021/acschembio.8b00463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Krishnan N., Fu C., Pappin D.J., Tonks N.K. H2S-Induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci. Signal. 2011;4:ra86. doi: 10.1126/scisignal.2002329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wedmann R. Improved tag-switch method reveals that thioredoxin acts as depersulfidase and controls the intracellular levels of protein persulfidation. Chem. Sci. 2016;7:3414–3426. doi: 10.1039/c5sc04818d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Doka E. A novel persulfide detection method reveals protein persulfide- and polysulfide-reducing functions of thioredoxin and glutathione systems. Sci Adv. 2016;2 doi: 10.1126/sciadv.1500968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Braunstein I. Opposing effects of polysulfides and thioredoxin on apoptosis through caspase persulfidation. J. Biol. Chem. 2020;295:3590–3600. doi: 10.1074/jbc.RA119.012357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pan H. Protective and biogenesis effects of sodium hydrosulfide on brain mitochondria after cardiac arrest and resuscitation. Eur. J. Pharmacol. 2014;741:74–82. doi: 10.1016/j.ejphar.2014.07.037. [DOI] [PubMed] [Google Scholar]
- 69.Calvert J.W. Genetic and pharmacologic hydrogen sulfide therapy attenuates ischemia-induced heart failure in mice. Circulation. 2010;122:11–19. doi: 10.1161/CIRCULATIONAHA.109.920991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shimizu Y. Hydrogen sulfide regulates cardiac mitochondrial biogenesis via the activation of AMPK. J. Mol. Cell. Cardiol. 2018;116:29–40. doi: 10.1016/j.yjmcc.2018.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Untereiner A.A. Stimulatory effect of CSE-generated H2S on hepatic mitochondrial biogenesis and the underlying mechanisms. Nitric Oxide. 2016;58:67–76. doi: 10.1016/j.niox.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 72.Untereiner A.A., Wang R., Ju Y., Wu L. Decreased gluconeogenesis in the absence of cystathionine gamma-lyase and the underlying mechanisms. Antioxidants Redox Signal. 2016;24:129–140. doi: 10.1089/ars.2015.6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li S., Yang G. Hydrogen sulfide maintains mitochondrial DNA replication via demethylation of TFAM. Antioxidants Redox Signal. 2015;23:630–642. doi: 10.1089/ars.2014.6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sun Y. Exogenous H2S switches cardiac energy substrate metabolism by regulating SIRT3 expression in db/db mice. J. Mol. Med. (Berl.) 2018;96:281–299. doi: 10.1007/s00109-017-1616-3. [DOI] [PubMed] [Google Scholar]
- 75.Abdollahi Govar A. 3-Mercaptopyruvate sulfurtransferase supports endothelial cell angiogenesis and bioenergetics. Br. J. Pharmacol. 2020;177:866–883. doi: 10.1111/bph.14574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Eelen G., de Zeeuw P., Simons M., Carmeliet P. Endothelial cell metabolism in normal and diseased vasculature. Circ. Res. 2015;116:1231–1244. doi: 10.1161/CIRCRESAHA.116.302855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Szczesny B. AP39, a novel mitochondria-targeted hydrogen sulfide donor, stimulates cellular bioenergetics, exerts cytoprotective effects and protects against the loss of mitochondrial DNA integrity in oxidatively stressed endothelial cells in vitro. Nitric Oxide. 2014;41:120–130. doi: 10.1016/j.niox.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gero D. The novel mitochondria-targeted hydrogen sulfide (H2S) donors AP123 and AP39 protect against hyperglycemic injury in microvascular endothelial cells in vitro. Pharmacol. Res. 2016;113:186–198. doi: 10.1016/j.phrs.2016.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ahmad A., Szabo C. Both the H2S biosynthesis inhibitor aminooxyacetic acid and the mitochondrially targeted H2S donor AP39 exert protective effects in a mouse model of burn injury. Pharmacol. Res. 2016;113:348–355. doi: 10.1016/j.phrs.2016.09.013. [DOI] [PubMed] [Google Scholar]
- 80.Covarrubias A.E. AP39, a modulator of mitochondrial bioenergetics, reduces antiangiogenic response and oxidative stress in hypoxia-exposed trophoblasts: relevance for preeclampsia pathogenesis. Am. J. Pathol. 2019;189:104–114. doi: 10.1016/j.ajpath.2018.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhao F.L. AP39, a mitochondria-targeted hydrogen sulfide donor, supports cellular bioenergetics and protects against alzheimer's disease by preserving mitochondrial function in APP/PS1 mice and neurons. Oxid Med Cell Longev. 2016;2016:8360738. doi: 10.1155/2016/8360738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhu C. Supplementing preservation solution with mitochondria-targeted H2 S donor AP39 protects cardiac grafts from prolonged cold ischemia-reperfusion injury in heart transplantation. Am. J. Transplant. 2019;19:3139–3148. doi: 10.1111/ajt.15539. [DOI] [PubMed] [Google Scholar]
- 83.Lobb I. Hydrogen sulfide protects renal grafts against prolonged cold ischemia-reperfusion injury via specific mitochondrial actions. Am. J. Transplant. 2017;17:341–352. doi: 10.1111/ajt.14080. [DOI] [PubMed] [Google Scholar]
- 84.Tabares-Seisdedos R., Rubenstein J.L. Inverse cancer comorbidity: a serendipitous opportunity to gain insight into CNS disorders. Nat. Rev. Neurosci. 2013;14:293–304. doi: 10.1038/nrn3464. [DOI] [PubMed] [Google Scholar]
- 85.Untereiner A.A., Olah G., Modis K., Hellmich M.R., Szabo C. H2S-induced S-sulfhydration of lactate dehydrogenase a (LDHA) stimulates cellular bioenergetics in HCT116 colon cancer cells. Biochem. Pharmacol. 2017;136:86–98. doi: 10.1016/j.bcp.2017.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xie Z.Z. Sulfhydration of p66Shc at cysteine59 mediates the antioxidant effect of hydrogen sulfide. Antioxidants Redox Signal. 2014;21:2531–2542. doi: 10.1089/ars.2013.5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liu Z., Wang X., Li L., Wei G., Zhao M. Hydrogen sulfide protects against paraquat-induced acute liver injury in rats by regulating oxidative stress, mitochondrial function, and inflammation. Oxid Med Cell Longev. 2020;2020:6325378. doi: 10.1155/2020/6325378. [DOI] [PMC free article] [PubMed] [Google Scholar]