

Abstract
Background
Desmoplastic melanoma (DM) is a rare subtype of spindle cell malignant melanoma characterized by frequent local recurrences and hematogenous spread, but without molecular classification. The aim of the study was to investigate in a DM series the incidence of relevant gene alterations in cancer, the programmed death-ligand 1 (PD-L1) expression status and the association with clinicopathological features and melanoma progression.
Methods
A total of 38 patients were included. Clinical follow-up and the histopathological features of all cases were retrospectively collected. PD-L1 expression by immunohistochemistry (IHC) and BRAF genomic alterations by real-time PCR were determined in 34 samples. Additionally, a molecular analysis by next-generation sequencing was performed in 25 DMs.
Results
Tumors occurred predominantly in men (76%) and in the head and neck region (50%). Most tumors were pure DMs (66%), containing less than 10% of conventional melanoma. Overall, 48% of our cohort harbored TP53 mutations, most of them showing a molecular signature associated with ultraviolet (UV)-oncogenesis, and 29%, BRAF mutations. A positive correlation between TP53 with depth of invasion (P=0.005) and presence of elastosis (P=0.002) was found. High-expression of PD-L1 in tumor cells was observed in 38% of cases and correlated with depth of tumoral infiltration (P=0.003), TP53 (P=0.016), PD-1 (P<0.001) and tumor-infiltrating lymphocytes (TILS) (P<0.001). PD-L1 expression in immune cells correlated with PD-1 (P=0.006), tumoral PD-L1 expression (P=0.029) and TP53 mutation (P=0.002). Survival correlated with depth of invasion (P=0.003), stage of tumors (P=0.015), positive sentinel lymph node (P=0.004), lymph node metastasis (P=0.024) and distant metastasis (P<0.001).
Conclusions
Our results suggest that progressed DMs with deep tumoral infiltration frequently harbor TP53 mutations, PD-L1 expression and present a high inflammatory response, probably related to adaptive immune resistance in this tumor-type.
Keywords: Desmoplastic melanoma (DM), next-generation sequencing (NGS), Oncomine, programmed death-ligand 1 (PD-L1), TP53
Introduction
Desmoplastic melanoma (DM) is an infrequent melanoma subtype, accounting for less than 4% of all cutaneous melanomas, but showing an increasing incidence (1). This neoplasm is histologically recognized by a spindle cell proliferation associated with a collagenous stroma and a characteristic immunophenotype. This immunophenotype lacks specific melanocytic markers expression, such as MART-1, HMB45 or MITF (2). A number of cases are histologically mixed, demonstrating that a second component of common melanoma is recognized in over 10% of the tumor. The development of DM is strongly related to high-ultraviolet (UV) exposure and it mainly affects elderly patients with highly sun-exposed skin (3). Most of the diagnoses are made at advanced stages because they are frequently clinically overlooked, and comparing with typical melanomas, DMs have a higher rate of local recurrences and hematogenous distant metastases, especially to the lung (4,5). Systemic oncological treatments are required for metastatic disease, and driver mutations assessment is usually needed to establish a personalized oncological treatment. Almost 50% of melanomas harbor a driver alteration in BRAFV600E, a molecular target enhancing the MAPK pathway (6). Moving beyond BRAFV600, suppression of immune checkpoints that limit T-cell responses to melanoma have provided targets for cancer immunotherapy. Among the immune checkpoint inhibitors, anti-cytotoxic T-lymphocyte antigen 4 (CTLA-4), programmed death 1 (PD-1) and its ligand 1 (PD-L1) have showed promising therapeutic outcomes and some have been approved for this patient population. PD-L1 expression by immunohistochemistry (IHC) is a biomarker routinely obtained in the clinical care of oncology, generally associated with the response to immunotherapy (7,8); other potential biomarkers are also being studied, such as tumor mutational burden (TMB) (9). Although DMs have a lower incidence of BRAFV600E and NRASQ61K/R mutations than standard melanomas, they are characterized by a high-rate of mutations (high TMB) and UV signature with a higher probability to response using anti-PD-1 drugs (10).
The present study was undertaken to molecularly characterize a cohort of DMs by next-generation sequencing (NGS) using a panel of twenty-two selected genes frequently altered in solid tumors, including actionable driver alterations and tumor suppressor genes, and to explore PD-L1 expression by IHC, which has been considered a marker of immune evasion by neoplastic cells causing tumor proliferation. Our main goal was to investigate in our patient population if TP53 mutations and high-expression of PD-L1 could have a relevant role in tumor aggressiveness and progression, probably related to immune evasion.
We present the following article in accordance with the STROBE reporting checklist (available at http://dx.doi.org/10.21037/atm-20-1846).
Methods
Patients and samples
Patients diagnosed with DM at Hospital Clinic of Barcelona (Barcelona, Spain) from 2007 to 2020 and with available biopsy tissue were included in the study. The study was approved by the Internal Review Board of the Hospital Clinic of Barcelona (HCB/2017/0097) and informed consent was taken from all the patients. The clinical data and patient outcomes were retrieved from the electronic medical records of our institution. This study was conducted in accordance with the principles of the Declaration of Helsinki (as revised in 2013).
The hematoxylin-eosin stained slides from the primary DM were reviewed to assess elastosis (elastic tissue degeneration in dermis caused by UV), ulceration, mitoses number, perineural invasion, vascular invasion and tumor-infiltrating lymphocytes (TILS). In one case, the primary melanoma was not available and elastosis and TILS were assessed from the tumoral recurrence. All the cases were reviewed by three pathologists (LA, CF, PC).
Inflammatory component assessment
After reviewing the hematoxylin-eosin stained slides from primary DMs, the lymphoid infiltrate was scored in three categories: 0, absence of inflammatory component; 1, mild presence of lymphoid cells, mainly inside the tumor; 2, marked inflammatory component with lymphoid cells and aggregates within the tumoral parenchyma and at the tumor-stroma interface. Tumors with brisk lymphoid inflammatory component (score 2) were considered positive.
IHC
Four-micron sections were cut from whole-tumor section and immunostaining with PD-L1 22C3 clone pharmDx (RTU, lot number 11086384) and with anti-PD-1 mouse monoclonal antibody (NAT105 clone, Ventana; RTU, lot number V001526) were performed using an automated staining platform (Autostainer Link 48; Dako, Glostrup, Denmark and BenchMark ULTRA platform; Roche Diagnostics, Switzerland, respectively) according to manufacturer instructions. Positivity for PD-L1 was scored and reported by using a three cut-point system based on tumor proportion score (TPS) in cell membrane (convincing partial or complete linear membrane staining, independently of intensity): 0, TPS <5%; 1, TPS ≥5–25%; 2 (high-positivity), TPS ≥25% (11). Additionally, PD-L1 expression (membrane and/or cytoplasmic at any intensity) in mononuclear immune cells (lymphoid cells and macrophages) adjacent to the tumor was assessed and classified in two categories: 0, IC-PD-L1 <5%; 1, IC-PD-L1 ≥5%. In addition, the combined PD-L1 score (CPS), defined as the number of PD-L1-positive cells (tumor cells, lymphocytes, macrophages) divided by the total number of viable tumors cells multiplied by 100, was scored in two levels: 0, CPS <10; 1, CPS ≥10 (12). The immunohistochemical staining results for PD-1 were evaluated on the basis of percentage positivity. All immunohistochemical studies were evaluated by two pathologists (LA and CF). For controversial cases, a consensus was reached over a double-headed microscope.
DNA isolation
For DNA purification from formalin-fixed paraffin-embedded (FFPE) samples, the QIAampDNA FFPE Kit (Qiagen, Hilden, Germany) was employed, following the manufacturer’s instructions. Concentrations of the purified DNAs were determined using the Qubit ds DNA HS assay (Life Technologies Gaithersburg, USA). Samples with ≥2.5 ng DNA/mL were diluted to this concentration.
NGS sample preparation, sequencing run and data processing
Purified DNA (10 ng) was used as a template to generate libraries using the Ion Torrent OncomineTM Solid Tumor DNA Panel kit (OST; Thermo Fisher Scientific, Massachusetts, USA) according to manufacturer’s instructions. The OST panel analyzes the mutational status of 22 genes (EGFR, ALK, ERBB2, ERBB4, FGFR1, FGFR2, FGFR3, MET, DDR2, KRAS, PIK3CA, BRAF, AKT1, PTEN, NRAS, MAP2K1, STK11, NOTCH1, CTNNB1, SMAD4, FBXW7 and TP53). Purified libraries were pooled at 20 pM and following ion spheres the pooled libraries were sequenced using Ion Personal Genome Machine (Thermo Fisher Scientific) platform (13). Ion Reporter Server (Thermo Fisher Scientific) was used to align the read data to the human reference genome nineteenth version sequence, to call sequence variants, and to generate an interactive report for visualization of the sequencing results.
Real-time PCR for BRAF mutation test
The Idylla (Biocartis, Mechelen, Belgium) is a single-use cartridge-based test, fully automated real-time PCR (RT-PCR), and designed for detection of BRAFV600E, E2, D, K, R and M mutation. The test does not require any sample procedure or DNA isolation; all reagents required for the allele-specific RT-PCR test are included in the cartridge. FFPE tissue sections per sample (specimen requirements) were processed according to manufacturer’s instructions.
Statistical analysis
Based on all the valid data obtained, we performed a descriptive analysis of all the variables of interest. The results of quantitative variables are given as median or mean values and range. Qualitative variables are given as absolute and relative frequencies (percentages). Data were analyzed with the SPSS statistical package (version 23.0; SPSS Inc., Chicago, IL, USA). The test used for comparison of qualitative variables was Fisher’s exact test. Differences in quantitative variables were analyzed with Student’s t-test. All statistical tests were two-sided, and significance was considered with a α-risk of 0.05.
Overall survival (OS) was defined as the time from the DM diagnosis to the date of death or to the last date of follow-up. Progression-free survival (PFS) was defined as the time from the DM diagnosis to local recurrence or metastasis. Survival data were analyzed by the Kaplan-Meier method, and survival curves were compared using the log-rank test. Cases with missing data for analyzes were omitted and the remaining data were analyzed.
Results
Clinicopathological data at diagnosis
Thirty-eight patients diagnosed with DM were included in the patient cohort. Tumors occurred predominantly in males, with a male-to-female ratio over 3:1, and a median age at diagnosis of 65 years (range, 20–82 years). Most tumors were located in the head and neck region (50%, 19/38), followed by the trunk (32%, 12/38) and the extremities (18%, 7/38). The majority of tumors were diagnosed as pure DM (25/38, 66%), composed of an amelanotic spindle, atypical cell proliferation (Figure 1). Among pure DMs, nine cases showed an in situ and/or conventional melanoma component accounting for less than 10%. Thirteen tumors were diagnosed as mixed DMs (34%), of which non-desmoplastic components were observed in more than 10% of tumor cells content. All tumors arose de novo without an associated nevus.
Figure 1.

An example of a pure DM showing a high-inflammatory response and PD-L1 expression (score TPS 2; score IC-PD-L1 1). (A) The tumor is composed of a dermal proliferation of spindle, atypical melanocytic cells displayed in a desmoplastic fibrotic stroma. Tumor cells are admixed with a brisk lymphocytic component (TILS score 2) (H&E 40×). (B) Protein S-100 immunohistochemistry highlights the neoplastic cells infiltrating the dermis. The neoplastic cells are strongly and diffusely positive, showing the characteristic immunophenotype of DM (S-100 40×). (C) Expression of PD-L1 by melanoma cells admixed with immune cells, which also show positivity for PD-L1 (PD-L1 100×). (D) Expression of PD-L1 by melanoma cells, showing a membrane and cytoplasmic positivity (PD-L1 200×). DM, desmoplastic melanoma; H&E, hematoxylin and eosin; IC, immune cells; PD-L1, programmed death ligand 1; TILS, tumor-infiltrating lymphocytes; TPS, tumor proportion score.
The deepest tumoral infiltration (Breslow index) ranged from 0.4 to 8.3 mm (median 3). Ulceration was observed in eight tumors (22%); perineural invasion in 19 (50%) and vascular invasion in two cases. An index mitotic of ≥1 mitosis per mm2 was shown in 23 tumors (61%). The presence of elastosis, indicative of chronic UV exposure was shown in 21 DMs (55%): 18 tumors from the head and neck, two from the trunk (on the sternum and shoulder), and one from the arm.
At diagnosis, stages of 37 patients were recorded, while the complete clinical information of one patient was missed. Eleven patients were in stage I (30%); 20, in stage II (54%); 5, in stage III (13%), and one in stage IV (3%). Sentinel lymph node removal was performed in 34 patients and histopathological study showed melanoma metastasis in 6 cases (18%).
PD-L1 expression is associated with TILS
PD-L1 and PD-1 IHC were performed on 35 different DM specimens, but only 34 have met the criteria for adequacy (more than 100 viable neoplastic cells, Figure 2). Expression of PD-L1 was observed either on tumor cells and/or immune cells and was assessed for TPS, IC-PD-L1, and CPS (see methods, Figure 3). Negative or scattered occasional tumor stained cells (TPS 0) was observed in 15 (44%) out of the 34 evaluable samples, whereas 19 tumors (56%) expressed PD-L1. Six (18%) were classified in TPS 1 and thirteen (38%) in TPS 2. PD-L1-expression in immune cells adjacent to the tumor was ≥5% (IC-PD-L1 score 1) in 24 cases (71%); and negative in 10 cases (29%). The PD-L1CPS assessment was classified below 10 (score 0) in 18 cases (53%) and ≥10 (score 1) in 16 cases (47%). PD-1 expression (≥1) was observed in 32 (94%) out of 34 samples (Figure 3D).
Figure 2.

Flow chart diagram of DM samples. DM, desmoplastic melanoma; IHC, immunohistochemistry; PD-1, programmed death 1; PD-L1, programmed death ligand 1; NA, not assessable; NGS, next-generation sequencing; OST, Oncomine solid tumor DNA kit; RT-PCR, real-time PCR.
Figure 3.

Expression of PD-L1. (A) PD-L1 positive case on tumor and immune cells (PD-L1 22C3 clone); (B) association plot of TILS and PD-L1 expression in tumor cells (TPS score); (C) association plot of TILS and PD-L1 expression in immune cells (IC score); (D) bar chart showing the PD-L1 and PD-1 expression in DM patients. DM, desmoplastic melanoma; IC, immune cells; PD-L1, programmed death ligand 1; TILS, tumor-infiltrating lymphocytes; TPS, tumor proportion score.
Moreover, TILS evaluation was scored in all cases (N=38) and an inflammatory response was totally absent (score 0) in three cases (8%); mild (score 1) in 14 cases (37%) and marked (score 2) in 21 tumor samples (55%). TILS correlated with high-expression of PD-L1 in tumor cells (TPS 2) (P<0.001), PD-L1 in immune cells (IC-PD-L1) (P=0.013) (Table 1), and CPS (P<0.001). TPS showed a statistical association with IC-PD-L1 (P=0.029) and PD-1 (P<0.001).
Table 1. Relation of clinicopathological characteristics with TP53 mutations and PD-L1 expression.
| Clinicopathological characteristics | N (%) or median (range) |
TP53 mutation (P) |
TPS PD-L1 high-expression (P) |
IC PD-L1 (P) |
|---|---|---|---|---|
| Sex | ||||
| Male | 29 (76%) | 0.035* | 0.961 | 0.566 |
| Female | 9 (24%) | |||
| Age | ||||
| Median (range) | 65 [20–82] | 0.793 | 0.340 | 0.360 |
| Site | ||||
| Head & neck | 19 (50%) | 0.123 | 0.429 | 0.329 |
| Extremities & trunk | 19 (50%) | |||
| UV exposure | ||||
| Elastosis | 22 (58%) | 0.002* | 0.152 | 0.362 |
| Absence of elastosis | 16 (42%) | |||
| Histological subtype | ||||
| Pure | 25 (66%) | 0.217 | 0.455 | 0.524 |
| Mixed | 13 (34%) | |||
| Depth of invasion | ||||
| Median (range), mm | 3 [0.4–8.3] | 0.005* | 0.003* | 0.410 |
| Mitoses per mm2 | ||||
| <1 | 15 (39%) | 0.051 | 0.075 | 0.283 |
| ≥1 | 23 (61%) | |||
| Ulceration | ||||
| Absent | 30 (79%) | 0.329 | 0.357 | 0.208 |
| Present | 8 (21%) | |||
| TILS | ||||
| Absent & mild | 17 (45%) | 0.044* | <0.001* | 0.013* |
| Marked | 21 (55%) | |||
| PD-1 | ||||
| Median positivity (range) | 5 [0–40] | 0.004* | <0.001* | 0.006* |
| Perineural invasion | ||||
| Absent | 19 (50%) | 0.675 | 0.290 | 0.678 |
| Present | 19 (50%) | |||
| Vascular invasion | ||||
| Absent | 36 (95%) | 0.763 | 0.679 | 0.532 |
| Present | 2 (5%) | |||
| Sentinel lymph node (34 cases) | ||||
| Negative | 28 (82%) | 0.369 | 0.756 | 0.711 |
| Positive | 6 (18%) | |||
| AJCC stage (37 cases) | ||||
| I | 11 (30%) | 0.031* | 0.191 | 0.489 |
| II | 20 (54%) | |||
| III | 5 (13%) | |||
| IV | 1 (3%) |
*, significant P values. AJCC, American Joint Committee of Cancer; IC, immune cells; PD-1, programmed death 1; PD-L1, programmed death ligand 1; TILS, tumor-infiltrating lymphocytes; TPS, tumor proportion score.
TP53 mutations, but not BRAF mutations, are associated with elastosis and tumoral infiltration
First, the BRAF testing by RT-PCR was prioritized because is a biomarker that determines the course of therapy. Four samples were unappropriated for testing (Figure 2) because low tumor tissue percentage was available for molecular analysis. BRAF alterations were identified in six DM tumor samples (18%). Among them, five carried a BRAFV600 (V600E/V600E2/V600D) alteration and one a BRAF V600 (V600K/V600R/V600M) mutation.
Subsequently, twenty-five of the 38 (66%) DMs were considered appropriate for NGS analysis (Figure 2). Four (16%) samples were unappropriated for NGS assay because a low DNA quality was obtained. A molecular informative result was acquired in seventeen specimens and a genetic alteration was recorded in 52% of them (11/21), corresponding to the twenty-two identified alterations. Molecular alterations (non-synonymous mutations) detected by NGS are shown in Table 2. The genes with the highest mutational prevalence were TP53 (52%, 11/21), BRAF (19%, 4/21), ERBB4 (14%, 3/21) and MET (10%, 2/21). Other genes identified at lower incidence (<3%) were: KRAS, STK11, SMAD4 and NRAS. Co-occurrence of oncogenic mutations was detected in 38% (8/21) of the samples. One patient presented three concurrent alterations and two patients carried four different mutations. On the other hand, two patients (12%) showed a unique TP53 G199R mutation and another one a single BRAFD594N mutation. Both single mutations occurred in patients with pure DM histology and with a low depth of tumoral infiltration.
Table 2. Hot-spot mutations identified by next generation sequencing with the Oncomine solid tumor panel.
| Case No. | DM type | Depth of invasion (mm) | Gene | Chr | Exon | Mutation: amino acid change |
|---|---|---|---|---|---|---|
| 2 | Pure | 4 | TP53 | 17 | 8 | c.880G>T; p.(Glu294Ter) |
| 2 | Pure | 4 | MET | 7 | 14 | c.2968G>A; p.(Asp990Asn) |
| 6 | Pure | 3 | TP53 | 17 | 7 | c.724T>C; p.(Cys242Arg) |
| 6 | Pure | 3 | BRAF | 7 | 11 | c.1406G>C; p.(Gly469Ala) |
| 10 | Pure | 3 | TP53 | 17 | 6 | c.595G>A; p.(Gly199Arg) |
| 17 | Mixed | 6 | TP53 | 17 | 8 | c.858A>C; p.(Glu286Asp) |
| 17 | Mixed | 6 | TP53 | 17 | 4 | c.293C>T;p.(Pro98Leu) |
| 17 | Mixed | 6 | KRAS | 12 | 2 | c.35G>A; p.(Gly12Asp) |
| 17 | Mixed | 6 | STK11 | 19 | 6 | c.839C>T; p.(Pro280Leu) |
| 20 | Pure | 0.5 | BRAF | 7 | 15 | c.1780G>A; p.(Asp594Asn) |
| 25 | Pure | 6.1 | TP53 | 17 | 8 | c.809T>C; p.(Phe270Ser) |
| 25 | Pure | 6.1 | SMAD4 | 18 | 11 | c.1333C>T; p.(Arg445Ter) |
| 26 | Pure | 5.8 | TP53 | 17 | 8 | c.817C>T: p.(Arg273Cys) |
| 26 | Pure | 5.8 | BRAF | 7 | 15 | c.1742A>G; p.(Asn581Ser) |
| 33 | Pure | 8.3 | TP53 | 17 | 5 | c.533C>T; p.(His179Tyr) |
| 33 | Pure | 8.3 | ERBB4 | 2 | 7 | c.844A>G; p.(Lys282Glu |
| 33 | Pure | 8.3 | ERBB4 | 2 | 6 | c.697G>A; p.(Glu233Lys) |
| 34 | Mixed | 5.8 | TP53 | 17 | 8 | c.833C>T; p.(Pro278Leu) |
| 34 | Mixed | 5.8 | NRAS | 1 | 4 | c.419C>T; p.(Pro140Leu) |
| 34 | Mixed | 5.8 | ERBB4 | 2 | 23 | c.2793T>A; p.(Asp931Glu) |
| 34 | Mixed | 5.8 | MET | 7 | 16 | c.3369T>G; p.(Ile1123Met) |
| 35 | Pure | 7.5 | TP53 | 17 | 8 | c.836G>A; p.(Gly279Glu) |
| 37 | Pure | 6.8 | TP53 | 17 | 6 | c.653T>G; p.(Val218Gly) |
| 37 | Pure | 6.8 | BRAF | 7 | 11 | c.1397G>A; p.(Gly466Glu) |
Chr, chromosome; DM, desmoplastic melanoma.
NGS identified four additional BRAF alterations increasing the BRAF prevalence of our cohort to 29% (10/34). The mutations detected were BRAF G469A, D594N, N581S and G466E. Correlation of BRAF mutation status identified either by RT-PCR or NGS was performed with clinicopathological parameters and no statistical significance was found with any parameter.
Focusing on TP53 mutations, the greatest alteration identified in our cohort of patients was a result of a transition amino acid change (73%, 8/11), in which C-T transition was the substitution most observed (75%, 6/8) (Table 2). Interestingly, seven out of eleven (64%) TP53 mutations were related to UV exposure (C>T, T>C and G>T) (10,14,15). Finally, a correlation between TP53 mutations and clinicopathological features were studied. Significant association was found with sex (P=0.035), presence of elastosis (chronic UV exposure, P=0.002), depth of tumoral infiltration (P=0.005), and staging (P=0.031) (Table 1 and Figure 4).
Figure 4.

Association plots between (A) tumoral infiltration and TP53 mutation, (B) tumoral infiltration and PD-L1 TPS expression, (C) PD-L1 TPS expression and TP53 mutation, and (D) PD-L1 IC expression and TP53 mutation. IC, immune cells; PD-L1, programmed death ligand 1; TILS, tumor-infiltrating lymphocytes; TPS, tumor proportion score.
Correlation between PD-L1 expression with depth of invasion and TP53 mutations
The results obtained so far indicate the relevance of the assessment of PD-L1 status as well as TP53 mutation testing in DM. Correlation between all the clinicopathological and histopathological characteristics studied with PD-L1 TPS high-expression and TP53 alterations are shown in Table 1.
PD-L1 TPS and CPS, but not IC-PD-L1, were found associated with depth of tumor infiltration (P=0.003 and P=0.017 respectively). Moreover, a statistically significant correlation was found between the depth of tumoral infiltration with elastosis (P=0.005) and TILS (P=0.012).
Regarding TP53 alterations, there was a significant correlation with PD-L1 TPS (P=0.016), IC-PD-L1 (P=0.002), and TILS (P=0.044). All TP53 mutated tumors showed PD-L1 expression in inflammatory cells, and nine out of ten TP53 mutated cases (90%) showed PD-L1 TPS expression. The TP53 mutations identified in our cohort of patients and related to PD-L1 expression were: E294*, C242R, G199R, E286D, P98L, F270S, R273C, H179Y, P278L, G279E and V218G. A cross-over proposal interaction between the identified associations is depicted in Figure 5.
Figure 5.
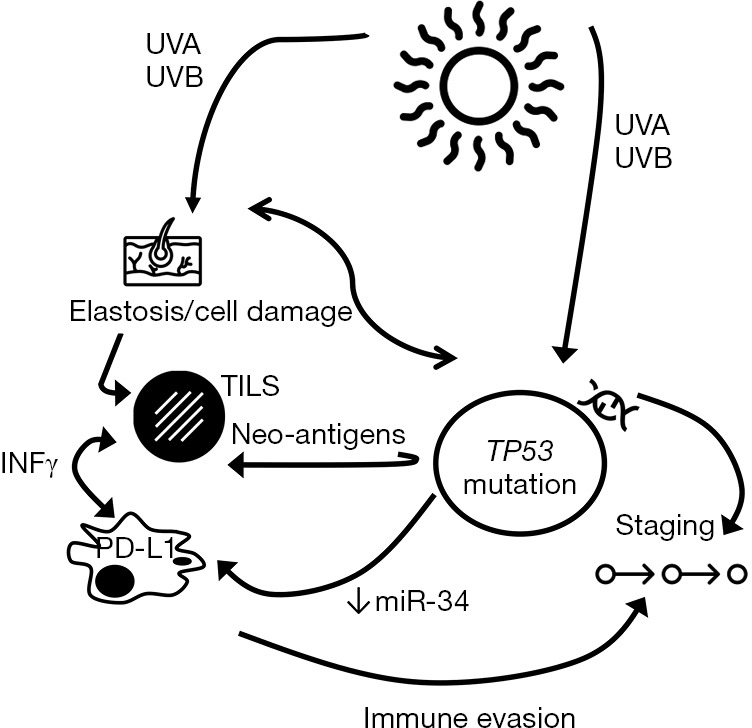
A proposal of the interaction between elastosis, TP53 mutations, TILS, PD-L1 expression and tumor progression. IFNγ, interferon gamma; PD-L1, programmed death ligand 1; TILS, tumor-infiltrating lymphocytes; UV, ultraviolet.
Association between survival and depth of invasion
Finally, we have retrospectively collected the clinical information and patient outcomes of the 38 patients included in our cohort. All tumors were treated with surgical wide excision; in six patients adjuvant therapy was added: interferon (three patients), radiotherapy (two patients) and pembrolizumab (one patient). There were no survival differences between patients with or without adjuvant therapy. The follow-up of patients showed tumoral recurrences in eight cases (21%) after a median time of 25 months; lymph node metastases in eleven (29%) after a median time of nine months; and distant metastases to the lung, liver and/or skull in 14 (37%) after a median time of 25 months. PFS did not correlate with clinicopathological characteristics; PD-L1 TPS (P=0.231), IC-PD-L1 (P=0.068), PD-L1 CPS (P=0.231), TP53 mutation (P=0.193), or BRAF mutation (P=0.384).
At the time of analysis, 73% of patients were alive. The median OS was 50 months (range, 17–224 months). OS correlated with depth of invasion (P=0.003), stage of tumors (P=0.015), positive sentinel lymph node (P=0.004), lymph node metastasis (P=0.024) and distant metastasis (P<0.001) (Figure 6). No statistical correlations were found between OS and other clinicopathological characteristics, PD-L1TPS (P=0.236), IC-PD-L1 (P=0.514), PD-L1 CPS (0.395), TP53 mutation (P=0.086) or BRAF mutation (P=0.098) (Figure 6).
Figure 6.

Overall survival (OS) plots (A) by stage of tumors, (B) by PD-L1 TPS expression, (C) by PD-L1 expression in immune cells, (D) by PD-L1 CPS expression, (E) by TP53 mutation and (F) by BRAF mutation. PD-L1, programmed death ligand 1; CPS, combined PD-L1 score; TPS, tumor proportion score.
Discussion
Alterations of DNA repair and other signaling pathways play essential roles in the development of the immune system and tumorigenesis (16). Activation of MAPK pathway through oncogene mutations, such as BRAF, NRAS, NF1 or KIT are essential drivers of early stage of development (14). DMs are characterized by harboring activating alterations of the MAPK pathway, such as NF1 loss-of-function mutations, high TMB, but lower frequency of BRAFV600E and NRASQ61K/R mutations compared to other melanoma types (3,17-19). In our series, we have found that over half of DMs harbor TP53 mutations related to UV oncogenesis and, as expected, 29% show BRAFV600 mutations. Molecular alterations were identified by NGS, and those BRAF mutations discovered by NGS, but not by RT-PCR, were due to the fact that the method does not cover the loci altered. NGS enables the testing of multiple genes in one assay with high sensitivity, and currently there are a variety of European Conformity In-Vitro Diagnostic kits, such as the OST DNA kit that we are using for our routine diagnosis for screening of hot spot mutations in solid tumors. In fact, some of these detected mutations in our series are candidates for targeted therapies, such as BRAF and NRAS.
High TMB has been associated with a signature for UV-induced DNA damage and our series comprise a high number of patients with a DM arisen in skin with elastosis. In correlation with these clinicopathological characteristics, a large number of the mutations we have identified can be associated with an indirect DNA damage after UV-radiation. Although the UV-radiation signature is predominantly caused by C>T substitutions at TpC dinucleotides due to repair of 6,4-photoproducts (10,12,14,20,21), other mutations involving T>C and T>A substitutions, potentially caused by indirect DNA damage after UV-radiation characteristic of cyclobutane pyrimidine dimers, have been recently described (15).
It is known that cutaneous melanomas acquire new molecular events while they progress, and different melanoma subtypes can follow different molecular pathways (21). The acquisition of TP53 mutation has been found in metastatic cancer (20) and also in melanomas in advanced stages of progression (21). In addition, the inactivation of TP53 gene has been observed before in high-risk melanomas with high depth of tumoral infiltration, along with the expression of genes promoting cell proliferation (22). Furthermore, TP53-mutated tumors acquire oncogenic functions, including the ability to promote invasion, proliferation and survival. Nevertheless, this is the first study in which TP53 mutations are shown in a series of DMs in advanced stage of tumor infiltration.
The tumor microenvironment is crucial for tumor surviving and progression, and a classification based on the inflammatory response and PD-L1 status has been proposed for cancer (23). The majority of melanomas are included in two groups described: (I) tumors showing marked inflammatory response along with PD-L1 TPS expression, or (II) tumors having absence of both inflammatory response and PD-L1 expression. Based on our results, although we have to take them with caution due to the small number of cases, most DMs could be included in the first group, in which evasion of the immune host response is the responsible agent for tumor growth and resistance to therapy. Inflammatory cytokines, especially interferon-gamma (IFNg) released from TILS, are able to up-regulate the expression of PD-L1 in several cell-types, including immune and tumor cells (24). Tumoral cells expressing PD-L1 can suppress local effector T-cell function as an adaptive immune-resistance mechanism. In our series, most DMs showing a high-PDL1 expression have been associated with a marked lymphoid response and depth of invasion, according with the role of PD-L1 on an adaptive response induced by the TILS. These results are similar to previous reports in which lymphoid cell immunophenotyping has demonstrated a high number of CD8 cytotoxic lymphocytes (11,25). Our results have shown a relationship between elastosis, TP53 mutation, high-inflammatory response, PD-L1 expression and depth of tumoral infiltration (Table 1 and Figure 4). These results also suggest that UV-exposure is the responsible agent of cell damage, evidenced through elastosis assessment, and by TP53 and other mutations. Furthermore, the degree of mutagenesis has been associated with the degree of immunogenicity in melanoma (25). In consequence, the immune evasion through high-inflammatory response and PD-L1 expression could be crucial for tumor progression and deep infiltration (Figure 5), which is the most consistent parameter of aggressiveness in melanoma, in agreement with our results. In fact, supporting this hypothesis we have found a significant correlation of both TP53 mutation and PD-L1 expression with the depth of invasion. TP53 tumor suppressor gene is also involved in immune response by regulating inflammatory cytokines and modulating the activation of T-cells and NK cells (26). In this context, Cortez et al. (27) have reported a strong relationship between TP53 and PD-L1 expression through miR-34 and have observed that tumors harboring TP53 mutations presented lower levels of miR-34 and high-PD-L1 expression, which contributes to immune evasion. In the same line, Kraft and colleagues have observed a significant correlation between PD-L1 and p53 overexpression by IHC in a large series of 66 DMs (11). In our study, we have observed a statistical relation between TP53 mutations and PD-L1 IHC expression in tumor cells (P=0.016) and immune cells adjacent to tumor (IC-PD-L1) (P=0.002). These results support the association between both biomarkers. Furthermore, Kraft et al. found a positive relation between PD-L1 TPS expression with the aggressiveness of tumors and follow-up of patients (11) that we were not able to find in our study, probably due to the small number of cases included in our series.
In summary, we demonstrated that NGS enables the testing of multiples genes and, along with PD-L1 expression by IHC, could establish relevant biomarkers to tailor a personalized treatment. In addition, our results identify DMs as a neoplasm related to strong UV-damage and, therefore, these tumors may harbor a high-mutation burden that contributes, as we have observed, to high-PD-L1 expression, immune evasion and tumor progression. This study furthers the understanding of molecular biology and the evolving changes associated with DM progression, which could be considered a good candidate for immunotherapy due to this dynamic pre-existing adaptive immune response. Further analysis on a larger patient cohort is necessary to confirm our results.
Supplementary
The article’s supplementary files as
Acknowledgments
The authors acknowledge Maria Lopez-Alos for her assistance on statistical procedures.
Funding: None.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. This study was conducted in accordance with the principles of the Declaration of Helsinki (as revised in 2013) and was approved by the Internal Review Board of the Hospital Clinic of Barcelona (HCB/2017/0097). All patient data were anonymous. Written informed consents were obtained from the patients.
Footnotes
Reporting Checklist: The authors have completed the STROBE reporting checklist. Available at http://dx.doi.org/10.21037/atm-20-1846
Data sharing statement: Available at http://dx.doi.org/10.21037/atm-20-1846
Peer Review File: Available at http://dx.doi.org/10.21037/atm-20-1846
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/atm-20-1846). Dr. AA reports personal fees and other from BMS, NOVARTIS, PIERRE FABRE, MSD, ROCHE, MERCK, AMGEM, and SANOFI, outside the submitted work. Dr. SP reports grants, personal fees and non-financial support from Almirall, grants and personal fees from Amgen, grants, personal fees and non-financial support from Isdin, non-financial support from Lilly, personal fees from BMS, grants from Novartis, grants, personal fees and non-financial support from Sanofi, grants from Sunpharma, grants, personal fees and non-financial support from La Roche Posay, grants and personal fees from Leo Pharma, grants from Melagenics, grants from Castle, non-financial support from Abbie, personal fees and non-financial support from Pfizer, grants and personal fees from Ojerpharma, personal fees and non-financial support from Roche, outside the submitted work. Dr. CT reports grants and personal fees from Pfizer, grants and personal fees from Novartis, personal fees from Takeda, personal fees from MSD, personal fees from BMS, personal fees from Roche, personal fees from Diaceutics, outside the submitted work. The other authors have no conflicts of interest to declare.
References
- 1.Feng Z, Wu X, Chen V, et al. Incidence and survival of desmoplastic melanoma in the United States, 1992-2007. J Cutan Pathol 2011;38:616-24. 10.1111/j.1600-0560.2011.01704.x [DOI] [PubMed] [Google Scholar]
- 2.Weissinger SE, Keil P, Silvers DN, et al. A diagnostic algorithm to distinguish desmoplastic from spindle cell melanoma. Mod Pathol 2014;27:524-34. 10.1038/modpathol.2013.162 [DOI] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas Network . Genomic Classification of Cutaneous Melanoma. Cell 2015;161:1681-96. 10.1016/j.cell.2015.05.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jaroszewski DE, Pockaj BA, DiCaudo DJ, et al. The clinical behavior of desmoplastic melanoma. Am J Surg 2001;182:590-5. 10.1016/S0002-9610(01)00819-4 [DOI] [PubMed] [Google Scholar]
- 5.Murali R, Shaw HM, Lai K, et al. Prognostic factors in cutaneous desmoplastic melanoma: a study of 252 patients. Cancer 2010;116:4130-8. 10.1002/cncr.25148 [DOI] [PubMed] [Google Scholar]
- 6.Ascierto PA, Kirkwood JM, Grob JJ, et al. The role of BRAF V600 mutation in melanoma. J Transl Med 2012;10:85. 10.1186/1479-5876-10-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karachaliou N, Pilotto S, Teixidó C, et al. Melanoma: oncogenic drivers and the immune system. Ann Transl Med 2015;3:265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teixidó C, González-Cao M, Karachaliou N, et al. Predictive factors for immunotherapy in melanoma. Ann Transl Med 2015;3:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan TA, Yarchoan M, Jaffee E, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol 2019;30:44-56. 10.1093/annonc/mdy495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eroglu Z, Zaretsky JM, Hu-Lieskovan S, et al. High response rate to PD-1 blockade in desmoplastic melanomas. Nature 2018;553:347-50. 10.1038/nature25187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kraft S, Fernandez-Figueras MT, Richarz NA, et al. PDL1 expression in desmoplastic melanoma is associated with tumor aggressiveness and progression. J Am Acad Dermatol 2017;77:534-42. 10.1016/j.jaad.2017.05.007 [DOI] [PubMed] [Google Scholar]
- 12.Bellmunt J, de Wit R, Vaughn DJ, et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N Engl J Med 2017;376:1015-26. 10.1056/NEJMoa1613683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castillo P, Marginet M, Jares P, et al. Implementation of an NGS panel for clinical practice in paraffin-embedded tissue samples from locally advanced and metastatic melanoma patients. Explor Target Antitumor Ther 2020;1:101-8. 10.37349/etat.2020.00006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell 2012;150:251-63. 10.1016/j.cell.2012.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hayward NK, Wilmott JS, Waddell N, et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017;545:175-80. 10.1038/nature22071 [DOI] [PubMed] [Google Scholar]
- 16.Rivera-Munoz P, Malivert L, Derdouch S, et al. DNA repair and the immune system: From V(D)J recombination to aging lymphocytes. Eur J Immunol 2007;37 Suppl 1:S71-S82. 10.1002/eji.200737396 [DOI] [PubMed] [Google Scholar]
- 17.Jahn SW, Kashofer K, Halbwedl I, et al. Mutational dichotomy in desmoplastic malignant melanoma corroborated by multigene panel analysis. Mod Pathol 2015;28:895-903. 10.1038/modpathol.2015.39 [DOI] [PubMed] [Google Scholar]
- 18.Shain AH, Garrido M, Botton T, et al. Exome sequencing of desmoplastic melanoma identifies recurrent NFKBIE promoter mutations and diverse activating mutations in the MAPK pathway. Nat Genet 2015;47:1194-9. 10.1038/ng.3382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wiesner T, Kiuru M, Scott SN, et al. NF1 Mutations Are Common in Desmoplastic Melanoma. Am J Surg Pathol 2015;39:1357-62. 10.1097/PAS.0000000000000451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goswami RS, Patel KP, Singh RR, et al. Hotspot mutation panel testing reveals clonal evolution in a study of 265 paired primary and metastatic tumors. Clin Cancer Res 2015;21:2644-51. 10.1158/1078-0432.CCR-14-2391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shain AH, Yeh I, Kovalyshyn I, et al. The Genetic Evolution of Melanoma from Precursor Lesions. N Engl J Med 2015;373:1926-36. 10.1056/NEJMoa1502583 [DOI] [PubMed] [Google Scholar]
- 22.Badal B, Solovyov A, Di Cecilia S, et al. Transcriptional dissection of melanoma identifies a high-risk subtype underlying TP53 family genes and epigenome deregulation. JCI Insight 2017;2:e92102. 10.1172/jci.insight.92102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taube JM, Anders RA, Young GD, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med 2012;4:127ra37. 10.1126/scitranslmed.3003689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012;12:252-64. 10.1038/nrc3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frydenlund N, Leone D, Yang S, et al. Tumoral PD-L1 expression in desmoplastic melanoma is associated with depth of invasion, tumor-infiltrating CD8 cytotoxic lymphocytes and the mixed cytomorphological variant. Mod Pathol 2017;30:357-69. 10.1038/modpathol.2016.210 [DOI] [PubMed] [Google Scholar]
- 26.Shatz M, Menendez D, Resnick MA. The human TLR innate immune gene family is differentially influenced by DNA stress and p53 status in cancer cells. Cancer Res 2012;72:3948-57. 10.1158/0008-5472.CAN-11-4134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cortez MA, Ivan C, Valdecanas D, et al. PDL1 Regulation by p53 via miR-34. J Natl Cancer Inst 2015;108:djv303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as