

Abstract
We recently reported that N-adamantyl-4-methylthiazol-2-amine (KHG26693) attenuates glutamate-induced oxidative stress and inflammation in the brain. In this study, we investigated KHG 26693 as a therapeutic agent against glutamate-induced autophagic death of cortical neurons. Treatment with KHG26693 alone did not affect the viability of cultured cortical neurons but was protective against glutamate-induced cytotoxicity in a concentration-dependent manner. KHG26693 attenuated the glutamate-induced increase in protein levels of LC3, beclin-1, and p62. Whereas glutamate decreased the phosphorylation of PI3K, Akt, and mTOR, these levels were restored by treatment with KHG26693. These results suggest that KHG26693 inhibits glutamate-induced autophagy by regulating PI3K/Akt/mTOR signaling. Finally, KHG26693 treatment also attenuated glutamate-induced increases in reactive oxygen species, glutathione, glutathione peroxidase, and superoxide dismutase levels in cortical neurons, indicating that KHG26693 also protects cortical neurons against glutamate-induced autophagy by regulating the reactive oxygen species scavenging system.
Keywords: Autophagy, Cortical neurons, Glutamate, N-adamantyl-4-methylthiazol-2-amine, Neurotoxicity
INTRODUCTION
Excess glutamate and activation of glutamate receptors result in excitotoxic neuronal injury and may contribute to neurological disorders, including neurodegenerative diseases (1-3). However, the precise mechanisms of glutamate-induced neuro-toxicity are not fully understood yet. Mitochondrial dysfunction and oxidative stress are two major events in glutamate-induced toxicity. Glutamate-induced ROS (reactive oxygen species) production results in oxidative stress, subsequently altering mitochondrial dynamics (4). The ROS-induced oxidative modification of macromolecules contributes to the pathology of neurological diseases (5). The high consumption of oxygen, high concentration of iron, and low levels of moderate protective antioxidant systems in the brain make this organ particularly vulnerable to oxidative stress (6). As such, antioxidants that scavenge free radicals may be useful for treating oxidative stress-related toxicity in the brain (7). Glutamate-induced neuronal damage may include mechanisms involving autophagy (8, 9). Under normal conditions, autophagic activity is prosurvival, preventing excessive ROS production and induction of inflammatory responses (10). However, dysregulation of autophagy may critically affect immune system function and subsequently induce autophagic cell death (11, 12). Autophagic cell death is implicated in glutamate-induced cytotoxicity, and measures to inhibit autophagy attenuate glutamate-induced neuronal death (13, 14). Glutamate induces an imbalance of mitochondrial dynamics and activates autophagy markers such as beclin-1 and LC3-II (15), but the cross-regulation of glutamate neurotoxicity and autophagy is not clear. Thus, therapeutic strategies against glutamate-induced autophagic cell death have not been intensively investigated.
To develop protective agents against glutamate neurotoxicity, we synthesized a thiazole amine derivative, N-adamantyl-4-methylthiazol-2-amine (KHG26693) (16), and found that it protects cortical cells against Aβ-induced neuronal injury (17) and inhibits lipopolysaccharide-induced brain inflammation (11, 18). We recently found that KHG26693 also attenuates glutamate-induced oxidative stress and inflammation in the brain (19). In the present study, we further investigated the mechanism by which KHG26693 protects neurons against glutamate-induced toxicity and autophagic cell death.
RESULTS AND DISCUSSION
KHG26693 attenuates glutamate-induced death of cortical neurons
Although autophagy is generally neuroprotective, recent studies reports that over-stimulated autophagy may play a role in glutamate-induced neuronal cell death and the regulation of autophagy pathway in glutamate-induced neurotoxicity may be a useful strategy to modulate neuronal cell death (8, 9, 13). It also reported that suppression of autophagy might be beneficial in neurodegenerative damage (20). To our knowledge, however, the physiological role of autophagy in glutamate-induced neuronal cell death are rarely understood so far. In this study, therefore, we investigated the role of autophagy in glutamate-induced cortical neuronal injury. We also examined the effect of autophagy inhibition on the viability of glutamate-induced neuronal cells together with the effects of KHG26603 on this injury.
As shown in Fig. 1A, glutamate treatment for 12 h at 37°C reduced the viability of cultured cortical neurons in a concentration-dependent manner. We have used 5 mM glutamate to perform the subsequent experiments because it was moderate concentration for glutamate-induced cell injury. KHG26693 significantly attenuated glutamate-induced cell death at different concentrations (5-50 µM) compared with glutamate-treated group (P < 0.01) (Fig. 1B). We have selected 20 µM KHG 26693 to perform the subsequent experiments because this concentration was sufficient to attenuate glutamate-induced cell injury. KHG26693 alone showed no effects on the cell viability of cortical neurons under the doses up to 50 µM (Fig. 1B).
Fig. 1.
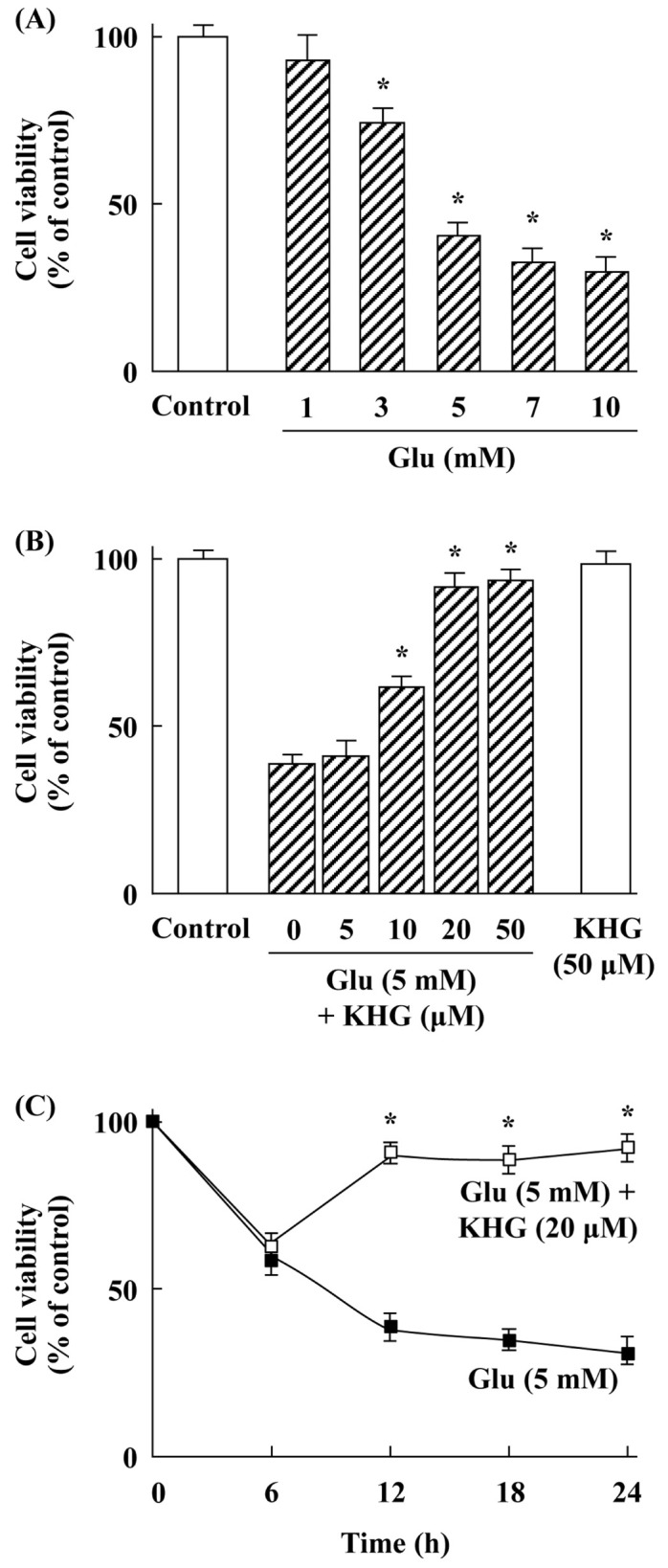
Effects of KHG26693 on glutamate-induced cortical neuron injury. Cell viability was determined by MTT assay. (A) Glutamate treatment for 12 h at 37°C decreased cell viability in a concentration-dependent manner (1, 3, 5, 7, and 10 mM). (B) Treatment of KHG26693 at different concentrations (5-50 M) for 12 h at 37°C ameliorated glutamate (5 mM)-induced neuron injury. (C) Time courses of KHG26693 (20 µM) effects on glutamate (5 mM)-induced toxicity. Data are presented as means ± S.D (n = 3). *Indicates statistical significance between glutamate group and glutamate group treated with KHG26693 (P < 0.01) (n = 3).
Glutamate at 5 mM concentration caused a cortical neuronal cell injury as the time prolonged (Fig. 1C). However, treatment with 20 µM KHG26693 significantly attenuated glutamate-induced neuronal cell death (P < 0.01) (Fig. 1C). Taken together, these results suggest that KHG26693 efficiently protect cortical neurons from glutamate-induced cytotoxicity in cultured cortical neuronal cells.
KHG26693 attenuates glutamate-induced autophagy in cortical neurons
Autophagy is modulated in a cooperative process by several autophagy marker proteins such as LC3 (microtubule-associated protein 1 light chain 3), p62, and beclin 1. Recent studies reported that glutamate-induced cortical neuronal cell injury is related with protein expression of LC3, p62, and beclin-1 (13, 21). To determine the involvement of neuronal autophagy in glutamate-induced cortical neuronal cell injury, we examined the expression levels of LC3, p62, and beclin-1 at 12 h after 5 mM glutamate treatment in the presence or absence of 20 µM KHG26693. Glutamate treatment dramatically enhanced the protein expressions of LC3 and beclin-1 compared with the control group as shown in Fig. 2A-C, suggesting that the autophagy activation was increased during the process of glutamate-induced neuronal cell damage. These results are consistent with previous studies reported by other groups, confirming that glutamate may enhance autophagy in cortical neurons (13, 21). However, KHG26693 significantly suppressed the glutamate-induced protein levels of LC3 and beclin-1 close to the control group (Fig. 2A-C), suggesting that down-regulation of autophagy activation may play a critical role in the work of KHG26693.
Fig. 2.

Effects of KHG26693 on glutamate-induced changes in protein levels of LC3, beclin-1, and p62 in cortical neurons. (A) Equal amounts of crude extracts were immunoblotted using primary antibodies against each protein. (B-D) Bar graphs showing quantification of protein levels calculated using densitometry; the ratio of protein intensity to β-actin intensity was assessed. Data are presented as means ± S.D (n = 3). *Indicates statistical significance between glutamate group and glutamate group treated with KHG26693 (P < 0.01) (n = 3).
In contrast to the enhanced expression of LC3, the protein level of p62 was markedly decreased by glutamate treatment (Fig. 2A and 2D), suggesting the possibility for the degradation of this LC3 binding protein by autophagy as reported before by other investigators (22). Once again, KHG26693 efficiently attenuated the protein level of p62 (Fig. 2A and 2D), indicating that glutamate enhanced autophagy activation in cultured cortical neurons and the neuronal injury was rescued via suppression of autophagy by KHG26693. Therefore, it is possible that the attenuation of autophagy activity was coincided with neuroprotective function of KHG26693. Taken together, the results in Fig. 1 and Fig. 2 suggest that KHG26693 may play an important role to protect cortical neurons from the glutamate-induced neuronal cell injury via suppression of autophagy.
Effects of KHG26693 on glutamate-induced PI3K/Akt/mTOR signaling
Mechanisms for the regulation of autophagy is very complicated and involves various and distinct signaling process. Previous studies suggested that PI3K/Akt/mTOR may be important signaling pathways against autophagy activation in neuronal cells (21, 23, 24). It also has been reported that activation of the PI3K/Akt/mTOR signaling pathway promotes necrosis through the suppression of autophagy (25) and caffeine induces apoptosis by enhancement of autophagy via PI3K/Akt/mTOR inhibition (26). Actually, PI3K/Akt plays a crucial role in the proliferation of adult hippocampal neural progenitor cells (27). Thus, this signaling pathway represents a potential target for controlling glutamate-induced autophagy in neuronal diseases (21, 28).
In these experiments, we examined whether KHG26693 acts through the PI3K/Akt/mTOR signaling pathways to suppress glutamate-induced neuronal injury. Whereas the phosphorylation of PI3K, Akt, and mTOR was reduced in cells treated with glutamate, phosphorylation status was maintained in cells also treated with KHG26693 (Fig. 3A-D). These results suggest that KHG26693 inhibits autophagy by preventing glutamate-induced suppression of PI3K/Akt/mTOR signaling.
Fig. 3.

Effects of KHG26693 on PI3K/Akt/mTOR signaling in glutamate-treated cortical neurons. (A) Equal amounts of crude extracts were immunoblotted using primary antibodies against each protein. (B-D) Bar graphs showing quantification of protein levels calculated using densitometry; the ratios of p-PI3K/PI3K, p-Akt/Akt, and p-mTOR/mTOR are expressed. Data are presented as means ± S.D (n = 3). *Indicates statistical significance between glutamate group and glutamate group treated with KHG26693 (P < 0.01) (n = 3).
KHG26693 mitigates glutamate-induced changes in ROS, GSH, GSH-Px, and SOD levels in cortical neurons
ROS are produced during many normal cellular processes interacting with oxygen. It has been reported that excess production of ROS is one of the critical pathological factors in neurodegenerative diseases and glutamate toxicity is closely related with accelerated production of ROS (29). As shown in Fig. 4A, exposure of cortical neurons to glutamate caused an increase in ROS level up to 3.4-fold, while treatment with KHG26693 significantly suppressed glutamate-induced increases in ROS. Therefore, KHG26693 may protects cortical neurons against glutamate-induced autophagy via suppression of excessive accumulation of ROS. These results are consistent with our current findings that KHG26693 attenuated beta-amyloid-induced ROS production in cultured cortical neurons, further supporting the antioxidant properties of KHG26693 (17).
Fig. 4.

Effects of KHG26693 on ROS (A), GSH (B), GSH-Px (C), and SOD (D) in glutamate-treated cortical neurons. Data are presented as means ± S.D (n = 3). *Indicates statistical significance between glutamate group and glutamate group treated with KHG 26693 (P < 0.01) (n = 3).
Reduced glutathione (GSH) is a cellular antioxidant that detoxifies ROS, and GSH depletion leaves neurons vulnerable to damage from oxidative stress (30). As shown in Fig. 4B, exposure of cortical neurons to glutamate resulted in GSH depletion in company with ROS production compared to control group. However, KHG26693 efficiently attenuated glutamate-induced depletion of GSH (Fig. 4B), suggesting that the protective effect of KHG26693 against glutamate-induced neuronal cell injury may be also related with recovery from GSH depletion.
In addition to GSH, antioxidant enzymes including GSH-Px (glutathione peroxidase) and SOD play an important roles in maintaining the redox homeostasis within cells against ROS production. Therefore, antioxidant enzymes represent a useful therapeutic strategy against cellular oxidative damage. In the present study, we examined the activity change of GSH-Px and SOD in the glutamate treated cortical neurons. ROS scavenging enzymes were down-regulated by glutamate treatment (Fig. 4C and 4D). On the other hand, cells in the group treated with KHG26693 showed significantly higher activities of GSH-Px and SOD than those in the glutamate group (Fig. 4C and 4D). Taken together, our results showed that glutamate reduced GSH level as well as activities of SOD and GSH-Px (Fig. 4). However, KHG26693 treatment attenuated glutamate-induced decrease in SOD, GSH-Px, and GSH level (Fig. 4) through a possible mechanism involving inhibition of ROS production (Fig. 4). These results suggest that KHG26693 may act as a ROS-scavenging antioxidant to protect cortical neurons against glutamate-induced autophagy.
In summary, KHG26693 effectively protects cortical neurons against glutamate-induced toxicity via the suppression of autophagy and ROS production. This neuroprotective effect coincides with the modulation of PI3K/Akt/mTOR signaling. Our results also provide useful insights for strategies aimed at modulating glutamate-induced neuronal cell injury, although the precise mechanisms by which KHG26693 achieves the observed effects should be investigated further.
MATERIALS AND METHODS
Materials
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and dimethyl sulfoxide (DMSO) were purchased from Sigma Chemical Co. (St. Louis, MO). RPMI medium, Dulbecco’s minimum essential medium (DMEM), fetal bovine serum, horse serum, and Fura-2 AM were obtained from Invitrogen (Carlsbad, CA). Anti-LC3, anti-p62, anti-beclin-1, anti-PI3K, anti-phosphorylated (p)-PI3K, anti-Akt, anti-p-Akt, anti-mTOR, anti-p-mTOR, and β-actin were purchased from Cell Signaling Technology (Beverly, MA). N-Adamantyl-4-methylthiazol-2-amine (KHG26693) was synthesized and characterized as previously described (16).
Primary cultures of cortical neurons and drug treatments
Primary rat cortical neurons were prepared from the brains of embryonic day 16/17 Sprague-Dawley rats as previously described with a slight modification (17, 31). Briefly, dissected cortical tissues were incubated in 1 ml Accutase (Millipore, Bedford, MA) for 10 min at 37°C and then filtered through nylon mesh to obtain a single-cell suspension. Cells (5 × 105 cells/ml) were plated on poly-D-lysine- and laminin-coated multiwell plates and cultured in serum-free DMEM supplemented with 2% B-27 and 0.5 mM glutamine at 37°C in a humidified 5% CO2 incubator. Two days later, the cells were treated with 5 µM cytosine-β-D-arabinofuranoside for 24 h to eliminate nonneuronal cells. The medium was replaced with DMEM supplemented as described above. The cells were cultured for 7 days at 37°C with 5% CO2 to yield purified cortical neurons.
KHG26693 was freshly prepared as a stock solution (10 mM) in DMSO and then diluted to the desired final concentrations in treatment medium as described elsewhere (17). Equivalent amounts of DMSO were used for controls and glutamate-treated cells for all experiments.
Cell viability
Cultured cortical neurons were treated with glutamate at different concentrations for up to 24 h, and then KHG26693 at various concentrations was added to the culture medium as described in the figure legends. Cell viability was determined by MTT reduction assay, as described previously (17). The cultured cells were treated with MTT solution (final concentration, 1 mg/ml) for 4 h, and the formazan formed in the intact cells was dissolved in MTT lysis buffer. The absorbance at 595 nm was recorded with a microplate reader. Cell viability was expressed as a percentage of the control.
Western blotting
Total protein extracts were separated by 10% SDS-polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes, and probed with LC3, p62, beclin-1, p-PI3K, PI3K, p-Akt, Akt, p-mTOR, and mTOR antibodies. Proteins were detected by enhanced chemiluminescence according to the manufacturer’s instructions (Amersham, Buckinghamshire, UK) and analysed using a Molecular Imager ChemiDoc XRS system (Bio-Rad, Hercules, CA) as described elsewhere (17). β-Actin was used to confirm equal protein loading.
Measurement of ROS and GSH level
ROS levels were measured using the oxidant-sensitive probe DCF-DA (2',7'-dichlorofluorescin diacetate) as described previously (11). Fluorescence intensity at 538 nm with excitation at 485 nm was measured with a SpectraMax GEMINI XS spectrophotometer (Molecular Devices, Sunnyvale, CA) according to the manufacturer’s protocol.
GSH levels were measured as described elsewhere with a slight modification (32). Briefly, 15 µl protein-free extracts was mixed with 100 µl 5',5'-dithio-bis(2-nitrobenzoic acid) (6 mM), 875 µl NADPH (0.3 mM), and 10 µl GSH reductase (10 U/ml), and the absorbance change at 412 nm was measured with a spectrophotometer.
Measurement of GSH-Px and SOD enzyme activities
GSH-Px activity was determined by a coupled assay with glutathione reductase as described elsewhere (33). Cell extracts were added to the working solution containing 10 mM NADPH, 84 mM GSH, glutathione reductase, and 15 mM tert-butyl hydroperoxide in a 96-well plate, and absorbance at 340 nm was measured for 3 min at 25°C.
To measure SOD activity, cell extracts were mixed with 10 µM cytochrome c, 50 µM xanthine, and sufficient xanthine oxidase to produce a reduction rate of cytochrome c as described elsewhere (17, 34). The assay was performed at 25°C in 50 mM potassium phosphate buffer (pH 7.8) and 0.1 mM ethylenediaminetetraacetic acid (EDTA).
Statistical analysis
Statistical comparisons were performed by a single-factor ANOVAs followed by Tukey’s post hoc tests. Data from three independent experiments were analyzed and are represented as means ± standard deviations (SDs). Statistical significance was defined as a P-value of <0.01.
ACKNOWLEDGEMENTS
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2018R1D1A3A03000692), by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (2018R1A2B6001743), and by a Student Research Grant from the University of Ulsan College of Medicine, Seoul, Korea.
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicting interests.
REFERENCES
- 1.Blandini F, Greenamyre JT, Nappi G. The role of glutamate in the pathophysiology of Parkinson's disease. Funct Neurol. 1996;11:3–15. [PubMed] [Google Scholar]
- 2.Olney JW. Excitotoxicity, apoptosis and neuropsychiatric disorders. Curr Opin Pharmacol. 2003;3:101–109. doi: 10.1016/S1471489202000024. [DOI] [PubMed] [Google Scholar]
- 3.Kalia LV, Kalia SK, Salter MW. NMDA receptors in clinical neurology: excitatory times ahead. Lancet Neurol. 2008;7:742–755. doi: 10.1016/S1474-4422(08)70165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patel M, Day BJ, Crapo JD, et al. Requirement for superoxide in excitotoxic cell death. Neuron. 1996;16:345–355. doi: 10.1016/S0896-6273(00)80052-5. [DOI] [PubMed] [Google Scholar]
- 5.Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97:1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- 6.Margaill I, Plotkine M, Lerouet D. Antioxidant strategies in the treatment of stroke. Free Radic Biol Med. 2005;39:429–443. doi: 10.1016/j.freeradbiomed.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 7.Havsteen BH. The biochemistry and medical significance of the flavonoids. Pharmacol Ther. 2002;96:67–202. doi: 10.1016/S0163-7258(02)00298-X. [DOI] [PubMed] [Google Scholar]
- 8.Bigford GE, Alonso OF, Dietrich D, et al. A novel protein complex in membrane rafts linking the NR2B glutamate receptor and autophagy is disrupted following traumatic brain injury. J Neurotrauma. 2009;26:703–720. doi: 10.1089/neu.2008.0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kulbe JR, Levy JMM, Coultrap SJ, et al. Excitotoxic glutamate insults block autophagic flux in hippocampal neurons. Brain Res. 2016;1542:12–19. doi: 10.1016/j.brainres.2013.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, Li YB, Yin JJ, et al. Autophagy regulates inflammation following oxidative injury in diabetes. Autophagy. 2013;9:272–277. doi: 10.4161/auto.23628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim EA, Han AR, Choi J, et al. Anti-inflammatory mechanisms of N-adamantyl-4-methylthiazol-2-amine in lipopolysaccharide-stimulated BV-2 microglial cells. Int Immunopharmacol. 2014;22:73–83. doi: 10.1016/j.intimp.2014.06.022. [DOI] [PubMed] [Google Scholar]
- 12.Chen ZF, Li YB, Han JY, et al. The double-edged effect of autophagy in pancreatic beta cells and diabetes. Autophagy. 2011;7:12–16. doi: 10.4161/auto.7.1.13607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim H, Choi J, Ryu J, et al. Activation of autophagy during glutamate-induced HT22 cell death. Biochem Biophys Res Commun. 2009;388:339–344. doi: 10.1016/j.bbrc.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 14.Ma YM, Ibeanu G, Wang LY, et al. Selenium suppresses glutamate‑induced cell death and prevents mitochondrial morphological dynamic alterations in hippocampal HT22 neuronal cells. BMC Neurosci. 2017;18:15. doi: 10.1186/s12868-017-0337-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumari S, Mehta SL, Li PA. Glutamate Induces Mitochondrial Dynamic Imbalance and Autophagy Activation: Preventive Effects of Selenium. PLoS One. 2012;7:e39382. doi: 10.1371/journal.pone.0039382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang SJ, Lee WJ, Kim EA, et al. Effects of N-adamantyl-4-methylthiazol-2-amine on hyperglycemia, hyperlipidemia and oxidative stress in streptozotocin-induced diabetic rats. Eur J Pharmacol. 2014;736:26–34. doi: 10.1016/j.ejphar.2014.04.031. [DOI] [PubMed] [Google Scholar]
- 17.Cho CH, Kim EA, Kim J, et al. N-Adamantyl-4-methylthiazol-2-amine suppresses amyloid β-induced neuronal oxidative damage in cortical neurons. Free Rad Res. 2016;50:678–690. doi: 10.3109/10715762.2016.1167277. [DOI] [PubMed] [Google Scholar]
- 18.Cho CH, Kim J, Ahn JY, et al. N-Adamantyl-4-methylthiazol-2-amine suppresses lipopolysaccharide-induced brain inflammation by regulating NF-κB signaling in mice. J Neuroimmunol. 2015;289:98–104. doi: 10.1016/j.jneuroim.2015.10.016. [DOI] [PubMed] [Google Scholar]
- 19.Yang SJ, Kim EA, Chang MJ, et al. N-Adamantyl-4-methylthiazol-2-amine attenuates glutamate-induced oxidative stress and inflammation in the brain. Neurotox Res. 2017;32:107–120. doi: 10.1007/s12640-017-9717-x. [DOI] [PubMed] [Google Scholar]
- 20.Xing S, Zhang Y, Li J, et al. Beclin 1 knockdown inhibits autophagic activation and prevents the secondary neurodegenerative damage in the ipsilateral thalamus following focal cerebral infarction. Autophagy. 2012;8:63–76. doi: 10.4161/auto.8.1.18217. [DOI] [PubMed] [Google Scholar]
- 21.Yin WY, Ye Q, Huang HJ, et al. Salidroside protects cortical neurons against glutamate-induced cytotoxicity by inhibiting autophagy. Mol Cell Biochem. 2016;419:53–64. doi: 10.1007/s11010-016-2749-3. [DOI] [PubMed] [Google Scholar]
- 22.Ichimura Y, Kominami E, Tanaka K, et al. Selective turnover of p62/A170/SQSTM1 by autophagy. Autophagy. 2008;4:1063–1066. doi: 10.4161/auto.6826. [DOI] [PubMed] [Google Scholar]
- 23.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2012;22:124–131. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Wang W, Li D, et al. IGF-1 alleviates NMDA-induced excitotoxicity in cultured hippocampal neurons against autophagy via the NR2B/PI3K-AKT-mTOR pathway. J Cell Physiol. 2014;229:1618–1629. doi: 10.1002/jcp.24607. [DOI] [PubMed] [Google Scholar]
- 25.Wu YT, Tan HL, Huang Q, et al. Activation of the PI3K-Akt-mTOR signaling pathway promotes necrotic cell death via suppression of autophagy. Autophagy. 2009;5:824–834. doi: 10.4161/auto.9099. [DOI] [PubMed] [Google Scholar]
- 26.Saiki S, Sasazawa Y, Imamichi Y, et al. Caffeine induces apoptosis by enhancement of autophagy via PI3K/Akt/mTOR/p70S6K inhibition. Autophagy. 2011;7:176–187. doi: 10.4161/auto.7.2.14074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peltier J, O'Neill A, Schaffer DV. PI3K/Akt and CREB regulate adult neural hippocampal progenitor proliferation and differentiation. Dev Neurobiol. 2007;67:1348–1361. doi: 10.1002/dneu.20506. [DOI] [PubMed] [Google Scholar]
- 28.Harris H, Rubinsztein DC. Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol. 2012;8:108–117. doi: 10.1038/nrneurol.2011.200. [DOI] [PubMed] [Google Scholar]
- 29.Qureshi GA, Baig S, Sarwar M, et al. Neurotoxicity, oxidative stress and cerebrovascular disorders. Neurotoxicol. 2004;25:121–138. doi: 10.1016/S0161-813X(03)00093-7. [DOI] [PubMed] [Google Scholar]
- 30.Ma S, Liu H, Jiao H, et al. Neuroprotective effect of ginkgolide K on glutamate-induced cytotoxicity in PC 12 cells via inhibition of ROS generation and Ca2+ influx. Neurotoxicol. 2012;33:59–69. doi: 10.1016/j.neuro.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 31.Nie BM, Jiang XY, Cai JX, et al. Panaxydol and panaxynol protect cultured cortical neurons against Abeta25-35-induced toxicity. Neuropharmacology. 2008;54:845–853. doi: 10.1016/j.neuropharm.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 32.Lombardi G, Varsaldi F, Miglio G, et al. Cabergoline prevents necrotic neuronal death in an in vitro model of oxidative stress. Eur J Pharmacol. 2002;457:95–99. doi: 10.1016/S0014-2999(02)02683-3. [DOI] [PubMed] [Google Scholar]
- 33.Rattanajarasroj S, Unchern S. Comparable attenuation of Abeta(25-35)-induced neurotoxicity by quercitrin and 17 beta-estradiol in cultured rat hippocampal neurons. Neurochem Res. 2010;35:1196–1205. doi: 10.1007/s11064-010-0175-6. [DOI] [PubMed] [Google Scholar]
- 34.Han A, Yang JW, Na JH, et al. Protective effects of N,4,5-trimethylthiazol-2-amine hydrochloride on hypoxia-induced β-amyloid production in SH-SY5Y cells. BMB Rep. 2019;52:439–444. doi: 10.5483/BMBRep.2019.52.7.231. [DOI] [PMC free article] [PubMed] [Google Scholar]