

Abstract
Introduction
Split notochord syndrome is a rare disorder characterized by fistula formation between the gastrointestinal tract and skin on the dorsum. Prenatal diagnosis is difficult and most cases are diagnosed postnatally.
Clinical findings and diagnosis
A 29-year-old woman, gravida 3 para 2, was referred for fetal cystic chest mass on prenatal ultrasound for congenital pulmonary airway malformation (CPAM). Fetal magnetic resonance imaging (MRI) suggested foregut duplication, and this was confirmed on postnatal thoracotomy with mass excision. A spine dysraphism was suspected on prenatal ultrasound, but was not confirmed on fetal MRI at the time of the study. Neonatal MRI noted vertebral abnormalities, confirming split notochord syndrome. Retrospective examination of the fetal MRI images detected a dysraphism and confirmed the prenatal ultrasound findings.
Outcome
At 17 months of life, the child had mild symptoms of neurogenic bowel, but was meeting all milestones without neurodevelopmental delays. We present a mild form of split notochord syndrome.
Conclusion
Split notochord syndrome is difficult to diagnose prenatally and should be considered when a fetal cystic chest mass is found on ultrasound. Detailed vertebrae assessment may improve detection.
Keywords: Fetal notochord syndrome, Pregnancy, Fetal anomaly, Prenatal diagnosis, Vertebral abnormalities
Highlights
-
•
Split notochord syndrome is difficult to diagnose prenatally.
-
•
Split notochord syndrome should be considered when fetal cystic chest mass is present.
-
•
Anatomic survey should include detailed examination of the vertebrae when fetal chest mass is present.
-
•
Prenatal magnetic resonance imaging identified foregut duplication cyst, but not the vertebral anomalies in this case.
-
•
Proper identification of vertebral anomalies is important for more accurate prenatal counseling.
1. Introduction
Split notochord syndrome, a rare pleomorphic disorder, results from persistent communication of the embryonic endoderm and ectoderm layers; it was first mentioned in the medical literature by Rembe in 1887 [[1], [2], [3], [4], [5]]. The endoderm, the innermost of the three germ cell layers, gives rise to the bladder, digestive and respiratory systems [6]. The ectoderm, the outer germ cell layer, gives rise to the nervous system and skin [6]. Persistent communication between these germ cell layers creates a fistula between the gastrointestinal tract, central nervous system, bladder and skin of the dorsum, resulting in a myriad of anomalies, including splitting of the notochord [[1], [2], [3], [4],6]. The notochord, a transient embryonic structure, is the vertebrae precursor and represents the mesodermal layer [6]. It has a role in the folding of the early embryo, vertebral column segmentation and regulating the development of surrounding tissue, specifically the neural plate and endoderm [6]. Split notochord syndrome may also be described as an occult spinal dysraphism [3]. Timing of notochord splitting, size and location are contributors to the variation and severity of associated malformations and clinical features. [4] Mild forms include isolated neuroenteric cysts. Severe cases include dorsal enteric fistula, severe intracranial and central nervous system malformations such as diastematomyelia (longitudinal splitting of the spinal cord), diplomyelia (duplications of the spinal cord), and myelomeningocele (dorsal spinal sac) [3]. The majority of published cases are found in the pediatric and neonatology literature after postnatal diagnosis [1,2,7]. Due to the variability in anomalies, prenatal diagnosis is difficult and only four cases of prenatally diagnosed split notochord are reported in the literature [[1], [2], [3]]. We present a case of a fetal cystic chest mass that postnatally was confirmed to be a foregut duplication cyst and a component of the split notochord syndrome; we highlight the sonographic features that may improve prenatal diagnosis.
2. Case Presentation
A 29-year-old woman, gravida 3 para 2, was referred to a fetal diagnosis center at 21 weeks of gestation for evaluation of a fetal cystic chest mass. The patient's medical history and family history were noncontributory. Ultrasound revealed a cystic mass in the right thorax measuring 1.9 × 2.1 × 2.6 cm. (Fig. 1), presumed to be a right congenital pulmonary airway malformation (CPAM). The spine images were limited but one area of the cervical thoracic spine on ultrasound was suspicious for dysraphism (Fig. 2). Fetal echocardiogram was normal and no other anomalies were identified.
Fig. 1.

Prenatal ultrasound demonstrated a right-sided chest mass measuring 1.9 × 2.1 × 2.6 cm with leftward shift of the heart due to mass effect.
Fig. 2.
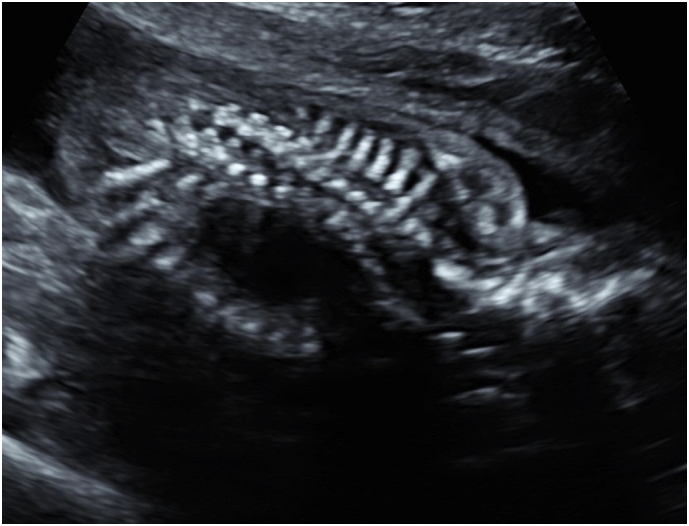
Sonographic image of fetal spine at 21 weeks of gestation demonstrating spine dysraphism.
The chest mass enlarged throughout gestation. The borders became thickened similar to fetal bowel bringing the diagnosis into question (Fig. 3). Fetal MRI was obtained at 30 2/7 weeks and showed a 4.7 × 3.3 × 4.8 cm coffee-bean-shaped cystic structure centered in the right posterior chest without true septations in close proximity to the distal esophagus and the right main stem bronchus consistent with probable foregut duplication system. There were no vertebral or spine anomalies reported on fetal MRI.
Fig. 3.
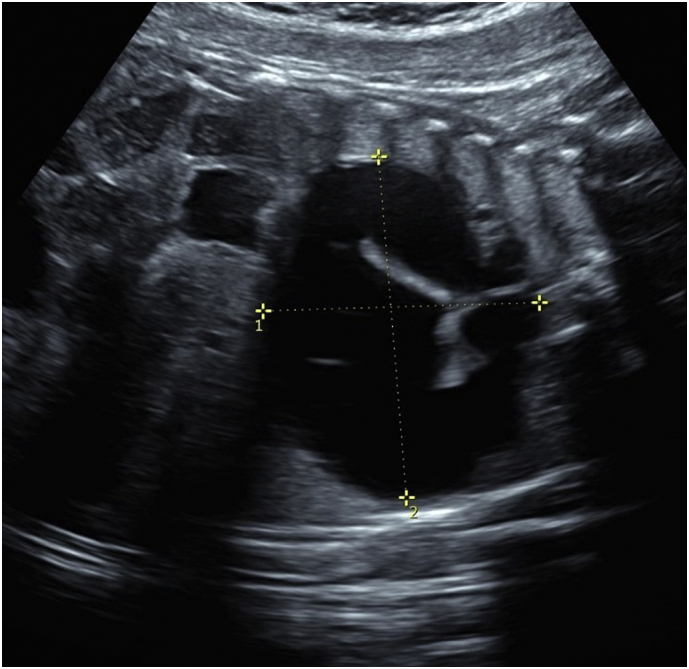
Repeat ultrasound at 37 weeks of gestation showed the persistent mass measuring 6.14 × 4.2 cm without fetal hydrops or polyhydramnios.
Neonatal MRI of the chest and spine showed a large posterior thoracic cyst measuring 6.6 × 4.2 × 5.7 cm suggestive of a foregut duplication cyst, with associated vertebral body anomalies (Fig. 4) involving the lower cervical and upper thoracic vertebrae. A small spinal syrinx was seen, with concern for spinal cord tethering.
Fig. 4.

T2 Weighted image on neonatal MRI demonstrating a vertebral body defect (arrow).
Thoracotomy with intact mass excision was performed with drainage of thick, non-purulent mucus. Intraoperative examination revealed that the mass was separate from the esophagus. Pathologic assessment confirmed foregut duplication cyst. Cytogenetic microarray was normal.
MRI when the child was 12 months old demonstrated a prominent ventral vertebral defect at T1 and T2 with dorsal body dysplasia at the same level and increased syrinx size. Neural tissue tethering bands extended anteriorly attaching to the posterior vertebral column and appeared to pass directly through the defect. Surgical interventions were not performed due to lack of neurologic deficits.
When the child was 17 months old, MRI of the spine showed a defect in the ventral portion of the spine at the cervicothoracic junction with herniation of soft tissue and decreased size of the focal cervical syrinx. Findings were consistent with split notochord syndrome. Clinically, the child had mild symptoms of neurogenic bowel, and was otherwise asymptomatic. The management plan included neurosurgery follow-up appointments every 6 months and referral to the orthopaedic spine clinic for vertebral defect treatment recommendations. At the age of 17 months, the child remained without concern for a genetic syndrome or dysmorphic features, was meeting milestones and was without neurodevelopmental delays.
3. Discussion
Although CPAM is the most common etiology of fetal chest cystic lesions, the differential should include bronchogenic cyst, pericardial cyst, diaphragmatic hernia, teratoma, esophageal duplication and neurenteric cyst [8]. Close examination for vertebral anomalies should be performed as split notochord syndrome is a consideration if a fetal chest cystic mass is identified. The prenatal spine images in our case suggested a cervico-thoracic dysraphism and were identifiable on retrospective review of the magnetic resonance images after postnatal diagnosis was made. Inclusion of split notochord syndrome on the prenatal differential diagnosis as well as additional views and use of 3-dimenstional ultrasound may have improved proper characterization prenatally. Discussing the possibility of split notochord syndrome and the spine ultrasound images with the radiologist may have also improved prenatal detection. Complications with fetal chest masses include mediastinal shift, altered lung development and pulmonary hypoplasia, polyhydramnios, hydrops and fetal death [3,8]. Serial ultrasound to assess mass growth, mediastinal changes and hydrops development is an important element of pregnancy management. [8] A strength of this report is that serial ultrasound scans were performed to follow the mass size, monitor amniotic fluid volume and assess for development of fetal hydrops.
Almog et al. reported the first two cases of prenatally detected and diagnosed split notochord syndrome in 2001 [2]. Since that time, two additional cases have been reported in the literature, with our case being the fifth [[1], [2], [3]]. Reported neonatal outcomes ranged from normal neurologic function to severe neurodevelopmental delays [[1], [2], [3]]. Neurogenic bowel was present in our case as well as the one reported by Agangi [1]. Herniation of the gastrointestinal organs is associated with more significant gastrointestinal symptoms; Almog reports a case in which the child required duodenal lavage feeds due to herniation of the stomach and Agangi reports a case with bowel herniation and associated moderate constipation and bowel dysfunction even after defect repair [1,2]. Table 1 is a summary of the prenatally diagnosed cases in the literature. Our report concerns a mild case of split notorchord syndrome with minimal neurologic effects.
Table 1.
Summary of Prenatally Diagnosed Split Notochord Syndrome in the Literature.
| Author | Maternal Information | Gestational Age (weeks) | Prenatal Ultrasound Findings and genetic tetsing | Prenatal MRI | Management | Postnatal Findings | Outcome |
|---|---|---|---|---|---|---|---|
| Almog (2001)2 | 27 year old G1P0 | 39 | Severe polydydramnios. Bedside ultrasound in Operating Room just prior to delivery with cystic structure in fetal chest.No tetsing | n/a | Proceeded with Delivery | Respiratory Distress within first few hours of life. Chest Xray: bowel loops in herniated right hemithorax. Spine Xray: severe scoliosis, hemivertibrae at C5, T5 and T7, Split vertebrae T 1–4, 6, 8–12. Surgical findings: herniated bowel loops and pancreas within sack in right hemithorax. Sack released and returned to abdomen with closeure of diaphragmatic defect. Severe kyphoscoliosis present. | No neurologic defecits in the Neonate |
| 2 | 44 year old G6P4 referred for polyhydramnios and suspected congenital diaphragmatic hernia | 28 | Polyhydramnios. Absent stomach below diaphragm, cystic structure in right posterior mediastinum that filled and emptied. Severe Scoliosis and vertebral distortion of thoracic vertebrae with multiple vertebral malformations including hemivertebrae, missing vertebrae and central vertebral body narrowing. 46 XY by amniocenetsis | Counseled and patient desired to continue pregnancy. Preterm prelabor rupture of membranes at 31 weeks with delivery | Significant scoliosis and lordosis of spine at birth with intact skin on the back. Chest Xray: centrally-located heart, normal lungs and stomach filled with barium in right posterior mediastinum. Multiple spine and rib anomalies on Xray. CT scan diagnosed Split Notochord Syndrome. | Feeds through duodenal lavage tube, sepsis, apnea, bradycardia, failure to thrive. Surgical repair Age 4 months with release of stomach.!2 months old: severe developmental delays, normal neurologic examination, severe kyphoscoliosis | |
| Agani 3 (2005)1 | 29 year old G2P0, Obese referred for suspected neural tube defect | 25 | Amniotic fluid: not reported. Small, round hypoechoic cystic mass involving lower sacrum with an oblongated perineal structure with bowel-like contents. No cerebellar or ventricular anomalies. Transvaginal ultrasound confirmed spinal mass with bowel and fluid filled mass originating from lateral gluteal region. Unchanged over 2 additional ultrasounds. Declines tetsing | n/a | Term Cesarean Delivery | Intestinal loop origninating in lateral perineum with serosa on external surface, dorsal enteric fistula opening at base of loop, skin-covered mass in sacral area. Left thigh mildly hypoplastic. Right undescended testis. Abdominal and Spine Xray: rectum ended blindly 3 cm from perineal surface, sacral and coccygeal splitting. Colostomy and partial resection of protruding colon segment. Histology confirmed intestinal origin of mass. MRI and CT: normal brain, complete cleft L4 with tethered cord and lipomyelomeningocele. Multidisciplinary meeting and priority for gastrointestinal repair. Age 2 months: posterior sagital anorectoplasty with identification of rectourethral fistula and loop excision. Colostomy closed Age 6 months. | Normal anus function. Moderate constipation. Neurogenic bladder. Age 3 years: neurosurgical excision of lipomyelomeningocele without improvement in bowel and bladder function |
| Kimya(2007)3 | 35 year old G1P0 | 21 | Amniotic fluid: normal. 25 × 11 mm posterior thoracic hypoechoic bilobar cystic lesion extending towards right lung base, cleaved and deformed adjacent vertebrae | n/a | Perinatal Ethics Committee meeting, offered elective termination, Pregnancy terminated | Autopsy: 3×2×1.5 cm seromucinous multiloculated cyst in posterior mediastinum between heart and posterior chest wall, communicated with epidural space through midline defect Vertebrae 5–7. No evidence of posterior dysraphism in neural tube. Normal abdominal and thoracic organs.Histologic report on the cyst: multiloculated cyst with smooth muscle wall, absent glial tissue | Termination |
| Our Case (2020) | 29 year old G3P2 referred for fetal cystic chest mass concerning for congenital pulmonary airway malformation(CP AM) | 21 | Amniotic Fluid: normal.Cystic chest mass in right thorax measuring 1.9 × 2.1 × 2.6 cm. Limited vertebral views, one area concerning for dysrahism. Normal Fetal Echocardiogram. No other anomalies. Gradual increase in mass size on serial exam. Declined etsting | At 30 weeks, 4.7 × 3.3 × 4.8 cm cystic mass in right posterior chest without septation, near distal esophagus and right mainstem bronchus consistent with probable foregut duplication | Uncomplicated repeat cesarean delivery 38 weeks. | MRI day of life 5 with large, posterior thoracic cyst measuring 6.6 × 4.2 × 5.7 cm suggesting foregut duplication cyst and associated vertebral anomalies in lower cervical and upper thoracic spine, small spinal syrinx with spinal cord tethering constitent with split notochord syndrome. Thoracotomy with mass excision on day of life 15 with drainage of non-purulent mucous. Mass noted to be separate from esophagus. Pathologic evaluation confirmed foregut duplication cyst. Postnatal cytogenic microarray: normal. Discharged home post-operative day 7. | Follow up MRI 12 months old: prominent vertebral defect T1 and T2 with dorsal body dysplasia, anterior tethering and increased syrinx size. No surgical intervention recommended. 17 Months old: MRI defect in ventral portion of spine at cervicothoracic junction, decreased syrinx size.Mild neurogenic bowel, but otherwise normal neurologic examination, meeting milestones and thriving. |
This case is unique in that it is the first in which MRI was used prenatally; MRI was helpful in determining the foregut duplication cyst, but unfortunately did not detect the vertebral anomalies until it was repeated postnatally. Our case is consistent with current literature that most cases are diagnosed in the neonatal and childhood periods [1,2,7].
Prognosis is dependent on the severity of associated anomalies. Pregnancy termination has been described for cases with severe malformations [3]. In our case, hydrops did not develop, there were no life-limiting anomalies, and the neonate had no significant respiratory or neurologic complications. It differs from prior cases due to the mild disease effects and excellent prognosis, with normal child development and meeting of milestones.
This case highlights the importance of split notochord syndrome as part of the differential and detailed examination of the vertebrae when a fetal cystic chest mass is identified on prenatal ultrasound. Proper identification of an associated vertebral anomaly may lead to improved detection and diagnosis of split notochord syndrome, which is important for more accurate prenatal counseling.
Acknowledgments
Contributors
Abigail M. Ramseyer contributed to case/literature review, manuscript, table, and final approval.
Nafisa Dajani contributed to case/literature review, manuscript, table, figures, and final approval.
Conflict of Interest
The authors declare that they have no conflict of interest regarding the publication of this case report.
Funding
No funding from an external source supported the publication of this case report.
Patient Consent
Informed written consent was obtained prior to submission of this case report.
Provenance and Peer Review
This case report was peer reviewed.
Acknowledgements
The authors would like to thank Donna Eastham, BA for her help in editing and submitting this manuscript.
References
- 1.Agangi A., Paladini D., Bagolan P., Maruotti G.M., Martinelli P. Split notochord syndrome variant: prenatal findings and neonatal management. Prenat. Diagn. 2005;25(1):23–27. doi: 10.1002/pd.1076. Epub 2005/01/22. (15662698) [DOI] [PubMed] [Google Scholar]
- 2.Almog B., Leibovitch L., Achiron R. Split notochord syndrome - prenatal ultrasonographic diagnosis. Prenat. Diagn. 2001;21(13):1159–1162. doi: 10.1002/pd.196. Epub 2002/01/12. (11787043) [DOI] [PubMed] [Google Scholar]
- 3.Kimya Y., Ozyurek E., Yalcinkaya U., Cengiz C., Alyamac Akpynar F. Prenatal diagnosis of the rarely observed split notochord syndrome. Ultrasound Obstet. Gynecol. 2007;29(6):712–713. doi: 10.1002/uog.4003. Epub 2007/05/10. (17487944) [DOI] [PubMed] [Google Scholar]
- 4.Coskun Y., Akman I., Demir M.K., Yapicier O., Somuncu S. A case of split notochord syndrome: Presenting with respiratory failure in the neonatal period. Intractable Rare Dis Res. 2016;5(2):121–123. doi: 10.5582/irdr.2016.01010. Epub 2016/05/20. PubMed PMID: 27195197; PubMed Central PMCID: PMCPMC4869579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singh S, Bothara VP, Rawat JD, Chaubey D, Kumar P, Singh G. A Rare Case of Split Notochord Syndrome. J Neonatal Surg. 2017;6(1):23. Epub 2017/01/14. doi: 10.21699/jns.v6i1.487. PubMed PMID: 28083509; PubMed Central PMCID: PMCPMC5224757. [DOI] [PMC free article] [PubMed]
- 6.Hill M. Notochord 2020. https://embryology.med.unsw.edu.au/embryology/index.php/Notochord cited 2020 23 JUL 2020 Available from.
- 7.Akgur F.M., Ozdemir T., Olguner M., Erbayraktar S., Ozer E., Aktug T. A case of split notochord syndrome: presence of dorsal enteric diverticulum adjacent to the dorsal enteric fistula. J. Pediatr. Surg. 1998;33(8):1317–1319. doi: 10.1016/s0022-3468(98)90179-8. Epub 1998/08/29. (9722015) [DOI] [PubMed] [Google Scholar]
- 8.Çay A., Aydoğdu I., Mirapoglu S.L., Topra K.H. Prenatal diagnosis of mediastinal neurenteric cyst: a case report and review of the literature. J Med Ultrason. 2001;45(4):633–639. doi: 10.1007/s10396-018-0869-y. Epub 2018/02/23. PubMed PMID: 29468491. [DOI] [PubMed] [Google Scholar]