

Abstract
Aim:
Histone deacetylase (HDAC) is an attractive target for antitumor therapy. Therefore, the development of novel HDAC inhibitors is warranted.
Materials & methods:
A series of HDAC inhibitors based on N-hydroxycinnamamide fragment was designed as the clinically used belinostat analog using amide as the connecting unit. All target compounds were evaluated for their in vitro HDAC inhibitory activities and some selected compounds were tested for their antiproliferative activities.
Conclusion:
Among them, compound 7e showed an IC50 value of 11.5 nM in inhibiting the HDAC in a pan-HDAC assay, being the most active compound of the series.
Keywords: : anticancer activities, drug design, molecular docking, structure–activity relationship, synthesis
In spite of considerable therapeutic advances, cancer remains a significant public health problem. In the past two decades, there has been a major shift from nonspecific cytotoxic agents that damage both tumor and normal cells to molecular targeted drugs, which are more effective and less harmful to normal cells by focusing on molecular changes that are specific to tumor cells [1]. Histone deacetylase (HDAC) inhibitors are a new class of targeted anticancer drugs, which selectively alter the expression of genes [2,3]. The important role of HDAC in the pathogenesis of cancer has been reported by numerous researchers since the end of last century [4,5]. To date, five HDAC inhibitors have been approved by the US FDA or China FDA (Figure 1). Vorinostat (generally known as SAHA) and romidepsin are regarded as first-generation HDAC inhibitors for the treatment of cutaneous T-cell lymphoma and/or peripheral T-cell lymphoma (PTCL), receiving approval in 2006 and 2009, respectively [6,7]. Belinostat and panobinostat were approved as second-generation HDAC inhibitors with an N-hydroxycinnamamide moiety by the FDA in 2014 and 2015, respectively [8,9]. Chidamide has been only approved in China for the treatment of PTCL in 2014 [10]. Despite the fact that most of the HDAC inhibitors have been approved for relatively rare types of cancer (cutaneous T-cell lymphoma and PTCL), the approval of panobinostat to treat multiple myeloma [9] has shown that HDAC inhibitors are also able to achieve high efficacy in more common tumor types. The clinical efficacy of these drugs validates the inhibition of HDAC as a valuable cancer treatment target. Consequently, there continues to be a growing interest in designing and developing novel HDAC inhibitors that may have improved clinical utility.
Figure 1. . Structures of approved histone deacetylase inhibitors.

Sir James Black, winner of the 1988 Nobel Prize in medicine, proposed that, ‘the most fruitful basis for the discovery of a new drug is to start with an old drug [11].’ Among those approved HDAC inhibitors, belinostat was chosen by us as a starting point for the design of a new HDAC inhibitor because of its potency and the feasibility of chemical modification to the molecule. Most HDAC inhibitors share a common structural pharmacophore model which consists of three parts: a cap group plus a connecting unit (CU) (surface recognition moiety), which interacts with residues on the rim of active site, a saturated or unsaturated linker that occupies the narrow channel and a zinc binding group (ZBG) interacting with the zinc ion at the active site (Figure 2) [12]. In the past few years, a large number of papers have been published by altering the cap, linker and zinc binding portions of the HDAC inhibitors. The structure–activity data have indicated that a hydroxamic acid ZBG possesses the highest HDAC binding affinity [13]. Meanwhile, N-hydroxycinnamamide, which serves as both ZBG and the linker group, is recognized as a superior structure in current HDAC inhibitor designs [14–19]. Two approved HDAC inhibitors (belinostat and panobinostat) and several HDAC inhibitors in clinical trials (pracinostat [SB939] and dacinostat [LAQ824]) all contain an N-hydroxycinnamamide bioactive fragment (Figures 1 & Figure 2). On the other hand, crystal structures of HDAC enzymes exhibit multiple grooves on the surface of the protein around the binding site and the surface recognition moiety is in a position suitable to interact with these grooves [20]. Thus, modification of the surface recognition moiety appears to be the most promising strategy to optimize drug–target interactions.
Figure 2. . Pharmacophore model and structures of several histone deacetylase inhibitors under clinical evaluation.
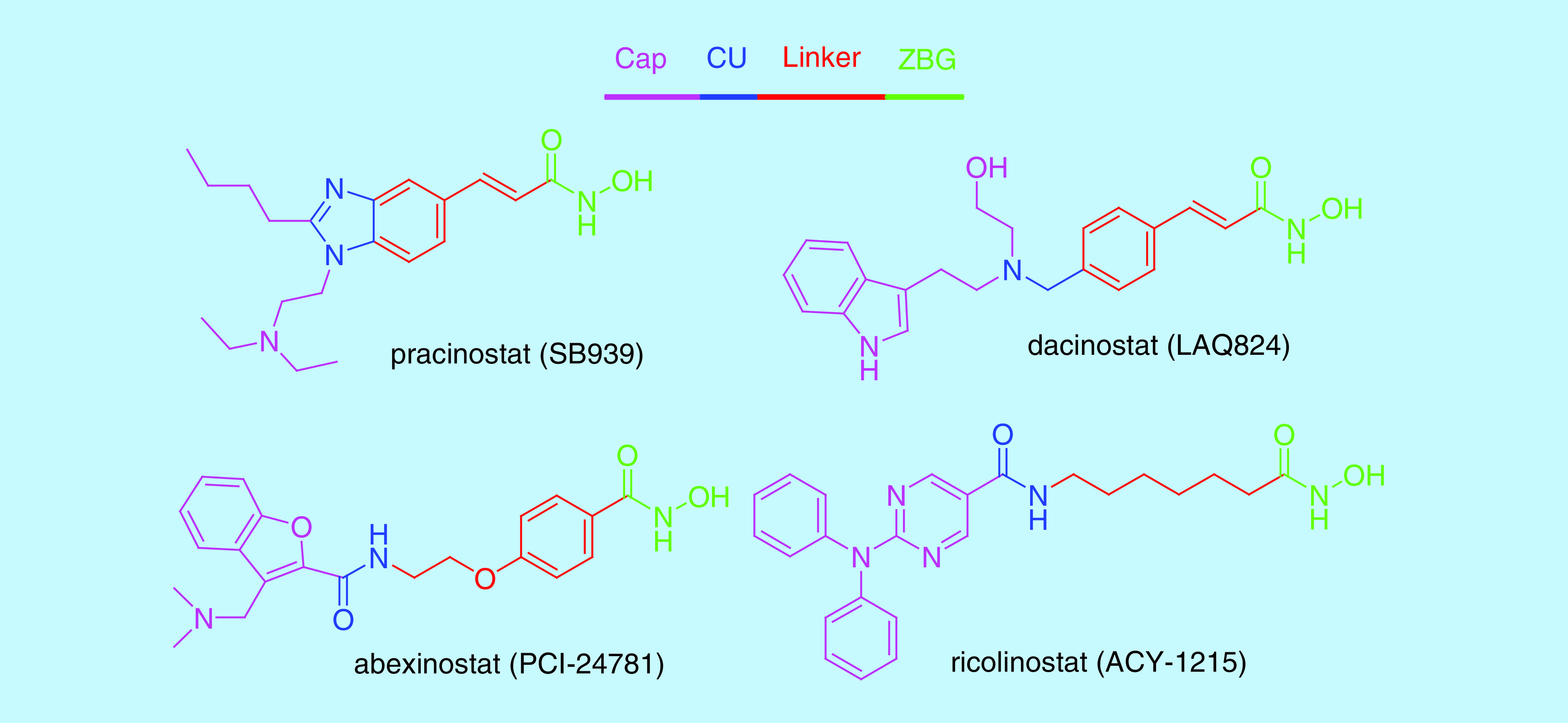
Having decided to maintain an N-hydroxycinnamamide as the ZBG and linker groups, our design ideas focused on the CU which connects the cap group and linker. The CU of belinostat is a sulfonamide, which has a torsion angle between 45° and 90° [21,22]. Although this drug had been known for several years, no structure–activity relationship (SAR) studies had been reported in the scientific literature on the substitution of this belinostat moiety with other CU groups. Thus, whether there is any difference between sulfonamide and other similar groups on HDAC inhibition remains to be clarified. We propose to replace the sulfonamide moiety with an amide moiety which has a strong preference for planarity, with a secondary amide possessing a preferred C–N–C(=O)–C torsion angle of 180°, in anticipation of an altered preference for the position of the surface recognition moiety relative to the rest of the molecule and thus differences in the SAR of the amide series. On the other hand, amide moiety is an active fragment in the surface recognition moiety of many HDAC inhibitors including SAHA, chidamide and intracellular active form of romidepsin (Figure 1) [23]. Other synthetic HDAC inhibitors in clinical trials such as abexinostat (PCI-24781), ricolinostat (ACY-1215) and entinostat (MS275) containing an amide moiety, especially in the surface recognition moiety (Figure 2), have shown promising in vitro and in vivo potency. More importantly, the sulfonamide moiety in belinostat is not present in other HDAC inhibitors, which might be potentially responsible for its off-target side effects.
On the basis of above analyses, in the present study, we describe the design and synthesis of new N-hydroxycinnamamide-based HDAC inhibitors by using N-phenylamide as the surface recognition moiety and N-hydroxycinnamamide as the linker and ZBG groups to mimic the structure of belinostat to retain the biological activity. The most active compound from this work showed strong HDAC inhibitory potency in the low nanomolar range and had effects superior to those of known inhibitors such as SAHA and belinostat.
Materials & methods
Chemistry
All chemicals were obtained from commercial suppliers and were used without purification. Reaction progress was monitored by thin-layer chromatography on silica gel plates. Chromatographic purification was performed on silica gel columns (Qingdao Haiyang Chemical Co. Ltd, Qingdao, Shandong, China, 200–300 mesh size). The yields reported were not optimized. Melting points were measured on an X-4 melting-point apparatus and were uncorrected. 1H NMR and 13C NMR spectra were recorded at 400 and 100 MHz in DMSO-d6 with chemical shift (δ) given in p.p.m. relative to tetramethylsilane (TMS) as internal standard. Multiplicities were indicated, s (singlet), d (doublet), t (triplet), m (multiplet), dd (doublet of doublets), etc; coupling constant (J) was given in Hertz (Hz). High resolution mass spectra (HRMS) were recorded using electrospray ionization (ESI) and time-of-flight mass analysis.
Synthesis of compounds
The general methods for the synthesis of target compounds 7 and 12 are outlined in Figures 3 & Figure 4. Commercially available 3-bromobenzoic acid 1 underwent Heck reaction with methyl acrylate 2 under basic conditions catalyzed by palladium(II) acetate to afford methyl cinnamate 3. Treatment of 3 with thionyl chloride gave acyl chloride 4, which was directly used in the next step without purification. The next step was the condensation of acyl chloride 4 with different amine 5a-p in dichloromethane to obtain intermediates 6a-p, which reacted with hydroxylamine in the presence of sodium hydroxide as a base to obtain the hydroxamic acid functions, affording the final compounds 7a-p.
Figure 3. . Synthesis of belinostat analogs 7a-p.

Reagents and conditions: (A) Pd(OAc)2, P(o-Tol)3, Et3N, DMF, 110°C; (B) SOCl2, DMF(cat), toluene, reflux; (C) RNH2 5a-p, Et3., DCM, 0°C to rt; (D) NH2OH, NaOH, MeOH:DCM = 2:1.
Figure 4. . Synthesis of compounds 12q-s.

Reagents and conditions: (A) H2SO4, MeOH, reflux; (B) RPhBr 10q-s, K2CO3, DMF, reflux; (C) NH2OH, NaOH, MeOH:DCM = 2:1.
Compounds 12q-s were prepared using commercially available 3-(3-hydroxyphenyl)acrylic acid 8 as the starting material. In the first step, 8 was reacted with methanol in the presence of sulfuric acid at reflux to give ester 9. Then, the appropriately substituted aryl bromides 10q-s were reacted with methyl 3-(3-hydroxyphenyl)acrylate 9 in the presence of potassium carbonate in N,N-dimethylformamide to afford intermediates 11q-s, which were then converted to hydroxamic acids 12q-s by the same method described above.
Synthesis of (E)-3-(2-(methoxycarbonyl)vinyl)benzoic acid (3)
A solution of 3-bromobenzoic acid 1 (3.0 g, 15 mmol), methyl acrylate 2 (6.45 g, 75 mmol), palladium acetate (67 mg, 0.3 mmol), tri-o-tolylphosphine (92 mg, 0.3 mmol) and triethylamine (202 mg, 2 mmol) in dimethylformamide (50 ml) was heated at 110°C under nitrogen for 5 h. After the reaction mixture had been cooled, it was acidified with dilute hydrochloric acid, and the resulting solid was filtered and recrystallized from acetone to give the product 3 (2.15 g) as a white solid in 70% yield.
General synthetic procedure for compounds 6a-p
The acid 3 (1.03 g, 5 mmol), thionyl chloride (1.1 ml, 15 mmol) and dimethylformamide (one drop) were mixed in toluene and stirred at 90°C for 1 h to form the acid chloride 4. Then, the excess thionyl chloride and toluene were distilled off under reduced pressure, and the residue was used in the next step without further purification. The amine 5a-q (5 mmol) was dissolved in dichloromethane (20 ml) and cooled to 0°C. Triethylamine (7 mmol) was added to the amine solution followed by acyl chloride 4 in dichloromethane drop-wise. The solution was then allowed to warm to room temperature and was stirred for 1–2 h (monitored by thin-layer chromatography). Upon completion, 3N hydrochloric acid solution was added to the reaction mixture to adjust pH to 2. The organic layer was separated, and the aqueous layer was extracted with dichloromethane (3 × 10 ml). The combined organic layer was dried over anhydrous magnesium sulfate, filtered and concentrated in vacuo. The residue was purified by column chromatography (petroleum ether/ethyl acetate, 3/1) to afford the amide 6.
General synthetic procedure for compounds 7a-q
To a solution of compound 6 (2.5 mmol) in methanol (40 ml) and dichloromethane (20 ml) at 0°C was added a solution of hydroxylamine (75 mmol) in the presence of sodium hydroxide (25 mmol). The solution was then allowed to warm to room temperature and was stirred overnight, concentrated and neutralized by adding 3 N hydrochloric acid solution until pH was 7. The resulting solid was filtered and recrystallized from acetone to give the product 7.
(E)-3-(3-(hydroxyamino)-3-oxoprop-1-en-1-yl)-N-phenylbenzamide (7a)
White solid, 0.42 g (yield 60%), mp 176–177°C. 1H NMR (400 MHz, DMSO-d6) δ 10.86 (s, 1H), 10.35 (s, 1H), 9.13 (s, 1H), 8.14 (s, 1H), 7.94 (d, J = 7.7 Hz, 1H), 7.78 (t, J = 6.1 Hz, 3H), 7.64–7.50 (m, 2H), 7.37 (t, J = 7.9 Hz, 2H), 7.12 (t, J = 7.4 Hz, 1H), 6.61 (d, J = 15.8 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 165.6, 163.0, 139.5, 138.0, 136.1, 135.5, 131.1, 129.5, 129.1, 129.0, 126.8, 124.2, 120.8, 120.8. HRMS (ESI): m/z calcd for C16H13N2O3 [M-H]-: 281.0926; found: 281.0911.
(E)-3-(3-(hydroxyamino)-3-oxoprop-1-en-1-yl)-N-(o-tolyl)benzamide (7b)
White solid, 0.65 g (yield 87%), mp 143–144°C. 1H NMR (400 MHz, DMSO-d6) δ 10.02 (s, 1H), 8.18 (s, 1H), 7.93 (d, J = 7.6 Hz, 1H), 7.73 (d, J = 7.6 Hz, 1H), 7.54 (t, J = 7.7 Hz, 1H), 7.44 (d, J = 15.8 Hz, 1H), 7.34 (d, J = 7.5 Hz, 1H), 7.28 (d, J = 7.2 Hz, 1H), 7.20 (d, J = 6.9 Hz, 2H), 6.61 (d, J = 15.8 Hz, 1H), 2.25 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 165.4, 163.1, 136.8, 135.9, 135.6, 134.2, 131.0, 130.8, 129.5, 128.6, 127.1, 126.5, 126.5, 126.5, 121.7, 18.4. HRMS (ESI): m/z calcd for C17H15N2O3 [M-H]-: 295.1083; found: 295.1087.
(E)-3-(3-(hydroxyamino)-3-oxoprop-1-en-1-yl)-N-(m-tolyl)benzamide (7c)
White solid, 0.59 g (yield 80%), mp 160–161°C. 1H NMR (400 MHz, DMSO-d6) δ 10.26 (s, 1H), 8.12 (s, 1H), 7.98–7.85 (m, 1H), 7.77 (d, J = 7.7 Hz, 1H), 7.62–7.49 (m, 4H), 7.24 (t, J = 7.8 Hz, 1H), 6.94 (d, J = 7.5 Hz, 1H), 6.60 (d, J = 15.8 Hz, 1H), 2.31 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 165.6, 162.9, 139.4, 138.3, 138.1, 136.1, 135.4, 132.7, 131.1, 129.6, 129.0, 126.8, 125.0, 121.4, 120.6, 118.0, 21.7. HRMS (ESI): m/z calcd for C17H15N2O3 [M-H]-: 295.1083; found: 295.1090.
(E)-3-(3-(hydroxyamino)-3-oxoprop-1-en-1-yl)-N-(p-tolyl)benzamide (7d)
White solid, 0.54 g (yield 73%), mp 183–184°C. 1H NMR (400 MHz, DMSO-d6) δ 10.83 (s, 1H), 10.25 (s, 1H), 9.12 (s, 1H), 8.12 (s, 1H), 7.92 (d, J = 6.8 Hz, 1H), 7.76 (d, J = 6.6 Hz, 1H), 7.66 (d, J = 7.7 Hz, 2H), 7.54 (d, J = 15.4 Hz, 2H), 7.17 (d, J = 7.7 Hz, 2H), 6.59 (d, J = 15.8 Hz, 1H), 2.28 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 165.4, 162.9, 138.0, 137.0, 136.1, 135.4, 133.2, 131.1, 129.5, 129.5, 129.5, 128.9, 126.7, 120.8, 120.8, 120.7, 21.0. HRMS (ESI): m/z calcd for C17H15N2O3 [M-H]-: 295.1083; found: 295.1084.
(E)-3-(3-(hydroxyamino)-3-oxoprop-1-en-1-yl)-N-(2-methoxyphenyl)benzamide (7e)
White solid, 0.61 g (yield 78%), mp 125–127°C. 1H NMR (400 MHz, DMSO-d6) δ 9.59 (s, 1H), 8.17 (s, 1H), 7.94 (d, J = 7.7 Hz, 1H), 7.83–7.66 (m, 2H), 7.62–7.41 (m, 2H), 7.21 (td, J = 7.8, 1.8 Hz, 1H), 7.12–7.08 (m, 1H), 6.99 (td, J = 7.6, 1.4 Hz, 1H), 6.63 (d, J = 15.8 Hz, 1H), 3.84 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 165.2, 162.9, 152.2, 137.8, 135.6, 135.6, 131.1, 129.6, 128.7, 127.1, 126.7, 126.5, 125.2, 120.9, 120.7, 111.9, 56.2. HRMS (ESI): m/z calcd for C17H15N2O4 [M-H]-: 311.1032; found: 311.1010.
(E)-3-(3-(hydroxyamino)-3-oxoprop-1-en-1-yl)-N-(3-methoxyphenyl)benzamide (7f)
White solid, 0.62 g (yield 80%), mp 142–144°C. 1H NMR (400 MHz, DMSO-d6) δ 10.33 (s, 1H), 8.13 (s, 1H), 7.90 (d, J = 7.7 Hz, 1H), 7.74 (d, J = 7.6 Hz, 1H), 7.55 (t, J = 7.7 Hz, 1H), 7.50–7.42 (m, 2H), 7.39 (dd, J = 7.9, 1.7 Hz, 1H), 7.26 (t, J = 8.1 Hz, 1H), 6.70 (dd, J = 8.2, 2.5 Hz, 1H), 6.61 (d, J = 15.8 Hz, 1H), 3.76 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 165.7, 163.1, 159.9, 140.8, 136.7, 136.0, 135.8, 131.0, 129.9, 129.5, 128.7, 126.5, 121.8, 113.0, 109.7, 106.5, 55.5. HRMS (ESI): m/z calcd for C17H15N2O4 [M-H]-: 311.1032; found: 311.1030.
(E)-3-(3-(hydroxyamino)-3-oxoprop-1-en-1-yl)-N-(4-methoxyphenyl)benzamide (7g)
White solid, 0.49 g (yield 63%), mp 172–174°C. 1H NMR (400 MHz, DMSO-d6) δ 10.25 (s, 1H), 8.14 (s, 1H), 7.93 (d, J = 7.6 Hz, 1H), 7.78–7.68 (m, 3H), 7.62–7.45 (m, 2H), 6.93 (d, J = 8.8 Hz, 2H), 6.62 (d, J = 15.8 Hz, 1H), 3.74 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 165.2, 162.9, 156.1, 137.7, 136.1, 135.5, 132.6, 130.9, 129.5, 128.8, 126.6, 122.4, 122.4, 120.9, 114.2, 114.2, 55.6. HRMS (ESI): m/z calcd for C17H15N2O4 [M-H]-: 311.1032; found: 311.1035.
(E)-N-(2-aminophenyl)-3-(3-(hydroxyamino)-3-oxoprop-1-en-1-yl)benzamide (7h)
White solid, 0.47 g (yield 63%), mp 165–167°C. 1H NMR (400 MHz, DMSO-d6) δ 9.77 (s, 1H), 8.19 (s, 1H), 7.97 (d, J = 7.8 Hz, 1H), 7.76 (d, J = 7.7 Hz, 1H), 7.61–7.49 (m, 2H), 7.18 (d, J = 7.8 Hz, 1H), 6.99 (t, J = 7.6 Hz, 1H), 6.80 (d, J = 8.0 Hz, 1H), 6.67–6.55 (m, 2H), 4.94 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 165.5, 163.0, 143.7, 138.1, 135.7, 135.4, 131.0, 129.5, 129.1, 127.2, 127.1, 126.9, 123.5, 120.6, 116.7, 116.5. HRMS (ESI): m/z calcd for C16H14N3O3 [M-H]-: 296.1035; found: 296.1028.
(E)-N-(4-(dimethylamino)phenyl)-3-(3-(hydroxyamino)-3-oxoprop-1-en-1-yl)benzamide (7i)
White solid, 0.58 g (yield 71%), mp 175–177°C. 1H NMR (400 MHz, DMSO-d6) δ 10.85 (s, 1H), 10.10 (s, 1H), 9.12 (s, 1H), 8.12 (s, 1H), 7.98–7.85 (m, 1H), 7.73 (s, 1H), 7.60–7.52 (m, 4H), 6.79 (d, J = 6.1 Hz, 2H), 6.60 (d, J = 15.6 Hz, 1H), 2.89 (s, 6H). 13C NMR (100 MHz, DMSO-d6) δ 164.8, 163.0, 147.8, 138.2, 136.3, 135.4, 130.8, 129.5, 129.3, 128.9, 126.6, 122.3, 122.3, 120.6, 113.0, 113.0, 41.0, 41.0. HRMS (ESI): m/z calcd for C18H18N3O3 [M-H]-: 324.1348; found: 324.1351.
(E)-N-(2-bromophenyl)-3-(3-(hydroxyamino)-3-oxoprop-1-en-1-yl)benzamide (7j)
White solid, 0.62 g (yield 69%), mp 107–109°C. 1H NMR (400 MHz, DMSO-d6) δ 10.17 (s, 1H), 8.20 (s, 1H), 7.97 (d, J = 7.8 Hz, 1H), 7.79 (d, J = 7.7 Hz, 1H), 7.73 (d, J = 7.9 Hz, 1H), 7.64–7.51 (m, 3H), 7.48-7.40 (m, 1H), 7.24 (td, J = 7.7, 1.7 Hz, 1H), 6.61 (d, J = 15.8 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 165.5, 162.9, 137.9, 136.9, 135.6, 135.1, 133.2, 131.6, 129.7, 129.4, 128.9, 128.6, 128.6, 126.6, 121.1, 120.8. HRMS (ESI): m/z calcd for C16H12BrN2O3 [M-H]-: 359.0031; found: 359.0017.
(E)-N-(3-bromophenyl)-3-(3-(hydroxyamino)-3-oxoprop-1-en-1-yl)benzamide (7k)
White solid, 0.64 g (yield 71%), mp 160–162°C. 1H NMR (400 MHz, DMSO-d6) δ 10.49 (s, 1H), 8.14 (s, 1H), 8.11 (s, 1H), 7.93 (d, J = 7.5 Hz, 1H), 7.77 (d, J = 5.4 Hz, 2H), 7.63–7.50 (m, 2H), 7.37–7.27 (m, 2H), 6.61 (d, J = 15.8 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 165.9, 162.8, 141.2, 137.8, 135.6, 131.4, 131.1, 129.6, 129.0, 126.8, 123.0, 121.9, 120.9, 119.4. HRMS (ESI): m/z calcd for C16H12BrN2O3 [M-H]-: 359.0031; found: 359.0021.
(E)-N-(4-bromophenyl)-3-(3-(hydroxyamino)-3-oxoprop-1-en-1-yl)benzamide (7l)
White solid, 0.59 g (yield 65%), mp 200–202°C. 1H NMR (400 MHz, DMSO-d6) δ 10.83 (s, 1H), 10.45 (s, 1H), 9.12 (s, 1H), 8.12 (s, 1H), 7.92 (d, J = 7.6 Hz, 1H), 7.77 (d, J = 8.7 Hz, 3H), 7.63–7.46 (m, 4H), 6.59 (d, J = 15.8 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 165.8, 163.0, 138.9, 138.0, 135.8, 135.5, 131.9, 131.9, 131.3, 129.6, 129.0, 126.8, 122.7, 122.7, 120.7, 115.9. HRMS (ESI): m/z calcd for C16H12BrN2O3 [M-H]-: 359.0031; found: 359.0034.
(E)-N-(2-fluorophenyl)-3-(3-(hydroxyamino)-3-oxoprop-1-en-1-yl)benzamide (7m)
White solid, 0.63 g (yield 84%), mp 165–167°C. 1H NMR (400 MHz, DMSO-d6) δ 10.30 (s, 1H), 8.19 (s, 1H), 7.97 (d, J = 7.9 Hz, 1H), 7.78 (d, J = 8.0 Hz, 1H), 7.67–7.46 (m, 4H), 7.40-7.19 (m, 4H), 6.65 (d, J = 15.8 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 164.2 (J = 264.4 Hz), 156.3 (J = 245.6 Hz), 137.8, 135.6, 135.0, 131.4, 129.6, 129.1, 127.7, 127.5 (J = 7.6 Hz), 126.9, 126.1, 126.0, 124.8 (J = 3.5 Hz), 120.9, 116.3 (J = 19.7 Hz). HRMS (ESI): m/z calcd for C16H12FN2O3 [M-H]-: 299.0832; found: 299.0819.
(E)-N-(4-chlorophenyl)-3-(3-(hydroxyamino)-3-oxoprop-1-en-1-yl)benzamide (7n)
White solid, 0.45 g (yield 57%), mp 191–193°C. 1H NMR (400 MHz, DMSO-d6) δ 10.83 (s, 1H), 10.46 (s, 1H), 9.11 (s, 1H), 8.12 (s, 1H), 7.92 (d, J = 6.7 Hz, 1H), 7.80 (dd, J = 17.0, 7.7 Hz, 3H), 7.56 (t, J = 12.5 Hz, 2H), 7.43 (d, J = 8.3 Hz, 2H), 6.60 (d, J = 15.7 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 165.7, 162.9, 138.5, 138.0, 135.8, 135.5, 131.3, 129.6, 129.0, 129.0, 129.0, 127.8, 126.8, 122.3, 122.3, 120.8. HRMS (ESI): m/z calcd for C16H12ClN2O3 [M-H]-: 315.0536; found: 315.0546.
(E)-3-(3-(hydroxyamino)-3-oxoprop-1-en-1-yl)-N-(pyridin-2-yl)benzamide (7o)
White solid, 0.43 g (yield 60%), mp 164-165°C. 1H NMR (400 MHz, DMSO-d6) δ 10.93 (s, 1H), 8.41 (d, J = 4.5 Hz, 1H), 8.28–8.16 (m, 2H), 7.99 (d, J = 7.5 Hz, 1H), 7.86 (t, J = 7.8 Hz, 1H), 7.77 (d, J = 7.3 Hz, 1H), 7.60-7.45 (m, 2H), 7.19 (t, J = 5.9 Hz, 1H), 6.64 (d, J = 15.7 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 166.1, 163.2, 152.6, 148.4, 138.6, 135.9, 135.1, 131.4, 129.5, 128.9, 126.8, 121.9, 120.4, 115.2. HRMS (ESI): m/z calcd for C15H12N3O3 [M-H]-: 282.0879; found: 282.0868.
(E)-3-(3-(hydroxyamino)-3-oxoprop-1-en-1-yl)-N-(thiazol-2-yl)benzamide (7p)
White solid, 0.49 g (yield 68%), mp 196–198°C. 1H NMR (400 MHz, DMSO-d6) δ 8.32 (s, 1H), 8.06 (d, J = 7.7 Hz, 1H), 7.71 (d, J = 7.6 Hz, 1H), 7.62-7.39 (m, 3H), 7.13 (s, 1H), 6.61 (d, J = 15.8 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 166.1, 163.0, 162.3, 138.0, 137.8, 135.5, 135.4, 131.4, 129.7, 129.4, 127.0, 120.6, 113.2. HRMS (ESI): m/z calcd for C13H10N3O3S [M-H]-: 288.0443; found: 288.0414.
Synthesis of methyl coumarate 9
Coumaric acid 8 (1.64g, 10 mmol) was suspended in methanol (20 ml), and two drops of concentrated sulfuric acid were added. The mixture was refluxed overnight. The solvent was removed in vacuo, and the residue was purified by column chromatography (petroleum ether/ethyl acetate, 4/1) to afford the methyl coumarate 9 (1.65 g) as a white solid in 93% yield [24].
General synthetic procedure for compounds 11q-s
To a solution of ester 9 (0.89 g, 5 mmol) in dimethylformamide (25 ml) was added 4-substituted bromobenzene 10 (5 mmol) and potassium carbonate (2.07 g, 15 mmol). The mixture was refluxed overnight. The reaction mixture was quenched with water, and extracted with ethyl acetate (3 × 10 ml). The solvent was removed in vacuo, and the residue was purified by column chromatography (dichloromethane) to afford the compounds 11q-s.
General synthetic procedure for compounds 12q-s
Compounds 12q-s were prepared from the corresponding ester 11q-s as described in the synthesis of 7a-p.
(E)-3-(3-(4-aminophenoxy)phenyl)-N-hydroxyacrylamide (12q)
Pale yellow solid, 0.41 g (yield 60%), mp 148–150°C. 1H NMR (400 MHz, DMSO-d6) δ 10.79 (s, 1H), 9.09 (s, 1H), 7.39 (d, J = 15.8 Hz, 1H), 7.33 (t, J = 7.9 Hz, 1H), 7.20 (d, J = 7.6 Hz, 1H), 7.04 (s, 1H), 6.86 (d, J = 8.1 Hz, 1H), 6.79 (d, J = 8.5 Hz, 2H), 6.61 (d, J = 8.5 Hz, 2H), 6.37 (d, J = 15.8 Hz, 1H), 5.04 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 163.1, 159.8, 146.0, 145.7, 138.5, 136.8, 130.7, 122.0, 121.4, 121.4, 119.9, 118.1, 115.5, 115.5, 115.0. HRMS (ESI): m/z calcd for C15H13N2O3 [M-H]-: 269.0926; found: 269.0901.
(E)-N-hydroxy-3-(3-(4-(methylamino)phenoxy)phenyl)acrylamide (12r)
Pale yellow solid, 0.47 g (yield 66%), mp 95–97°C. 1H NMR (400 MHz, DMSO-d6) δ 7.43-7.27 (m, 2H), 7.18 (d, J = 6.9 Hz, 1H), 7.03 (s, 1H), 6.85 (t, J = 8.6 Hz, 3H), 6.57 (d, J = 8.4 Hz, 2H), 6.38 (d, J = 15.6 Hz, 1H), 5.62 (s, 1H), 2.67 (d, J = 3.0 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 162.9, 159.9, 147.5, 145.5, 137.7, 137.1, 130.7, 121.8, 121.5, 121.5, 120.4, 117.9, 115.0, 113.0, 113.0, 30.5. HRMS (ESI): m/z calcd for C16H15N2O3 [M-H]-: 283.1083; found: 283.1095.
(E)-3-(3-(4-(dimethylamino)phenoxy)phenyl)-N-hydroxyacrylamide (12s)
Pale yellow solid, 0.40 g (yield 54%), mp 134–136°C. 1H NMR (400 MHz, DMSO-d6) δ 10.75 (s, 1H), 9.06 (s, 1H), 7.40 (d, J = 15.8 Hz, 1H), 7.34 (t, J = 7.9 Hz, 1H), 7.22 (d, J = 7.5 Hz, 1H), 7.07 (s, 1H), 6.95 (d, J = 9.0 Hz, 2H), 6.91-6.86 (m, 1H), 6.77 (d, J = 9.1 Hz, 2H), 6.39 (d, J = 15.8 Hz, 1H), 2.88 (s, 6H). 13C NMR (100 MHz, DMSO-d6) δ 163.0, 159.6, 148.1, 146.3, 138.3, 137.0, 130.8, 122.1, 121.2, 121.2, 120.1, 118.2, 115.4, 114.1, 114.1, 41.0, 41.0. HRMS (ESI): m/z calcd for C17H17N2O3 [M-H]-: 297.1239; found: 297.1212.
Pan-HDAC inhibition assay
The HDAC activity assay kit (Fluorometric) ab156064 (Abcam, Cambridge, UK) was used to measure the HDAC inhibition activity of compound 7. In duplicate, ddH2O, HDAC Assay Buffer and Fluoro-Substrate Peptide or Fluoro-Deacetylated Peptide were added to microtiter plate wells. Then, compound 7, SAHA, belinostat or DMSO (control) in which compound was dissolved was added to the assay wells and mixed. Next, Developer was added to each well of the microtiter plate and mixed well. Reactions were initiated by adding 5 μl of HDAC to each well and mixed thoroughly at room temperature (RT) and incubated for 20 min at RT. A total of 20 μl of Stop Solution and 5 μl of Developer were added to each well of the microtiter plate and mixed thoroughly. The reaction mixture was incubated for 20 min at RT and the fluorescence intensity was read for 30–60 min at 1–2-min intervals using a microplate fluorescence reader at Ex/Em = 355 nm/460 nm. The average fluorescence of each data point based on the duplicates was determined. The efficacy of inhibition of compound 7 on the HDAC activity was calculated as follows: inhibition effect = [compound 7 Assay – DMSO Control Assay]. The IC50 was defined as the compound concentration required to inhibit HDAC activity by 50%. The rate of reaction was measured and calculated while the reaction velocity remained constant.
Molecular docking
To study the interactions between HDAC and compounds 7e (most active) and 7b (least active), molecular docking studies were performed using the Glide program of Schrödinger Suite 2017-1 [25]. The X-ray structure of HDAC2 in complex with SAHA (PDB ID: 4LXZ.pdb) was used for the present study, and the active site was gained based on this structure. Initial 3D coordinates for HDAC2 were prepared by removing all the crystallographic waters beyond 5 Å from SAHA and adding hydrogen atoms consistent with the physiologic pH, followed by energy minimization with a root mean square deviation (RMSD) cut-off value of 0.3 Å for all heavy atoms. Compounds 7e and 7b were prepared using the builder module of Schrödinger followed by energy minimization with the OPLS3 force field [26]. Then the docking was performed with Glide 7.4 using the default parameters under the standard precision mode. At last, the binding free energies of the protein–ligand complexes were obtained using the MM/GBSA method with OPLS3 force field and a GS/SA continuum solvent model.
Antiproliferative assay
Human breast adenocarcinoma (MDA-MB-231), human cervix carcinoma (HeLa), MCF-10A in DMEM medium supplemented with 115 units/ml of penicillin G, 115 μg/ml of streptomycin and 10% fetal bovine serum (all from Life Technologies, NY, USA). Cells were seeded in 96-well plates (5 × 103 cells/well) containing 50 μl growth medium for 24 h. After medium removal, 100 μl fresh medium containing individual prodrugs and controls at different concentrations was added to each well and incubated at 37°C for 72 h. After 72 h of incubation, 20 μl of resazurin was added for 2 h before recording fluorescence at 560 nm (excitation) and 590 nm (emission) using a Victor microtiter plate fluorometer (Perkin-Elmer, MA, USA). The IC50 was defined as the compound concentration required inhibiting cell proliferation by 50%, in comparison with cells treated with the maximum amount of DMSO (0.25%) and considered as 100% viability.
Results & discussion
SAR exploration
In our efforts to find new HDAC inhibitors, we initially prepared compound 7a and found that it showed inhibition of the target enzyme with IC50 values in the nanomolar range, suggesting that the amide series analogs of belinostat warrant further optimization, even though the inhibitory activity was less potent than the reference drugs SAHA and belinostat. All synthesized target compounds were assessed in a pan-HDAC inhibition assay. Results are presented in Table 1. Because compound 7a still potently inhibited HDAC (266 nM), we then examined the effects of substitution on the phenyl ring of 7a. Introduction of a methyl group at the 2-position of the phenyl ring gave 7b with diminished inhibitory activity (605 nM), whereas potency was improved in the compounds having a 3-methyl (7c, 163 nM) or 4-methyl group (7d, 209 nM). Surprisingly, for the compound bearing a strong electron donating methoxy group at 2-position of the phenyl ring (7e), a low, two-digit nanomolar potency for HDAC inhibition was reached (11.5 nM), which was about threefold more potent than belinostat (31 nM) and 16-fold more potent than SAHA (180 nM). Compound 7f, with a 3-methoxy group was about 14-times less potent (163 nM) than 7e but was more potent than the reference drug SAHA (180 nM). The presence of a 4-methoxy group at the phenyl ring has no positive effect on the inhibition potency (7g, 288 nM). As compound 7e showed low nanomolar binding affinity, we synthesized compound 7h which possesses a strong electron donating amino group at 2-position of the phenyl ring and evaluated its HDAC inhibitory activity. As expected, 7h exhibited a very similar inhibitory potency to 7e (IC50 = 12.5 nM). The naturally occurring trichostatin A, containing a dimethylamino group at 4-position of the phenyl ring, has been shown to be very effective in inhibition of HDAC. Our results have shown the combination of a strong electron donating group in cap group enhances biological activity of the new belinostat analog core. This information led us then to look at the presence of a dimethylamino group at the 4-position. Disappointingly, however, addition of this substituent had little effect on the binding affinity (7i, 223 nM). Substitution with electron withdrawing groups (e.g., fluoro, chloro or bromo 7j-n) at each of the available ring positions, in general, did not result in a significant improvement in activity. These results stressed the importance of the strong electron donating group on the phenyl ring of the cap group. More precisely, the strong electron donating group must be in ‘ortho’ position of the phenyl ring.
Table 1. . Histone deacetylase inhibitory activities of compound 7.
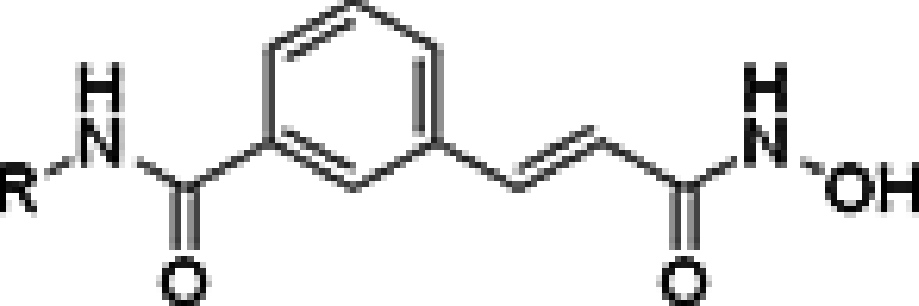 | ||
|---|---|---|
| Compound | R | IC50 (nM)† |
| 7a | Ph | 266 |
| 7b | 2-CH3Ph | 605 |
| 7c | 3-CH3Ph | 163 |
| 7d | 4-CH3Ph | 209 |
| 7e | 2-OCH3Ph | 11.5 |
| 7f | 3-OCH3Ph | 164 |
| 7g | 4-OCH3Ph | 288 |
| 7h | 2-NH2Ph | 12.5 |
| 7i | 4-(CH3)2NH2Ph | 223 |
| 7j | 2-BrPh | 222 |
| 7k | 3-BrPh | 278 |
| 7l | 4-BrPh | 167 |
| 7m | 2-FPh | 259 |
| 7n | 4-ClPh | 250 |
| 7o |  |
204 |
| 7p |  |
417 |
| SAHA | 180 | |
| Belinostat | 31 | |
Assays were performed in replicate (n ≥ 2). Mean values are shown, and the standard deviations are <20% of the mean.
The effect of replacing the phenyl ring by a heterocyclic ring on HDAC inhibition was next investigated. The N-hydroxycinnamamide core and amide CU were unchanged to allow comparison between new compounds and the unsubstituted phenyl analog 7a. The 2-pyridyl analog 7o was of comparable activity as the unsubstituted phenyl analog 7a; however, the 2-thiazole analog 7p was less active, suggesting that a six-membered ring at this position was favorable for the formation of hydrophobic interactions with the rim of the binding pocket.
On the basis of these results, the amide series analogs of belinostat can be summarized as follows:
The sulfonamide moiety is important for belinostat (phenyl-unsubstituted compound) to retain optimal HDAC binding affinity, but it is not an essential requirement for its phenyl-substituted analogs. This is clearly an important observation for ongoing drug design efforts with belinostat because, from the viewpoint of synthesis, the range of modifications of belinostat is notably limited by the necessity of installing a sulfonamide moiety. The inclusion of sulfonamide moiety generally requires the use of extremely corrosive oleum [27] or environmentally unfriendly sulfur dioxide [28] or the use of 3-formayl-N-phenylbenzene sulfonamide as staring material which was not commercially available [29]. By contrast, the introduction of an amide moiety is much easier and simpler, thus bringing more opportunities for modifying the inhibitor to improve the potency and selectivity.
The introduction of a strong electron donating group at the 2-position of the phenyl ring significantly improves the inhibitory activity, as can be seen in the comparison of the IC50 values of 7b and 7e. Despite 7b and 7e having very similar structures (–CH3 vs –OCH3), 7e is significantly more potent than 7b (11.5 vs 605 nM). Most likely, the addition of the oxygen enables the methyl group to pack deeper in the binding pocket. Furthermore, this extension also enables the stabilization of a positive charge due to the electron donating methoxy group, which further increases the compound's affinity for HDAC.
Ring substitution plays a central role in the modulation of the inhibitory activity. Changing the position of an electron donating group on the phenyl ring can dramatically affect the inhibition potency. Indeed, the rank of affinity for the methoxy-substituted series was as follows (the position of the substituent is indicated in parentheses): 7e (2) > 7f (3) > 7g (4). Replacement of the 2-methoxy group by a 2-amino group in 7h retains a high HDAC binding affinity. By contrast, the introduction of a dimethylamino group at the 4-position (7i) did not lead to any positive result.
The introduction of a halogen group at the phenyl ring has no positive effect on binding despite the fact that all compounds with a halogen group showed inhibition of the target enzyme with IC50 values in the nanomolar range.
The replacement of the phenyl ring with a six-membered ring or a five-membered ring results in an opposite effect on the affinity of compound 7a. The relatively low activity of 7p compared with 7a might be due to lack of substitution as well as the smaller, five-membered ring size. This suggests that a substituted aromatic phenyl ring forms the best scaffold for cap group in the amide series.
We then turned our attention to investigating the replacement of the amide group with a more stable ether linkage between the phenyl ring and N-hydroxycinnamamide for further SAR study. Considering that the ether linkage has a spacer one atom shorter than amide, which might decrease the interaction between the surface recognition moiety and the periphery of the HDAC binding pocket, we thought that placing an electron donating amino or methylamino group on the para position of the phenyl ring would presumably enhance the interaction of the surface recognition moiety with the surrounding pocket through hydrophobic interactions. This idea comes from the structural feature of the natural HDAC inhibitor trichostatin A [30].
As shown in Table 2, although compound affinities of the ether series remained in the nanomolar range and the affinities of 12s and 7i were similar (256 vs 223 nM, respectively), none of these compounds with strong electron donating substitutions had a comparable activity to 7h (12.5 nM). These results suggest that the amide moiety would be more promising as the CU compared with the ether, and future efforts to optimize the activity of this type of belinostat analogs should center on the investigation of the reversed amide or ester CUs.
Table 2. . Histone deacetylase inhibitory activities of compound 12.
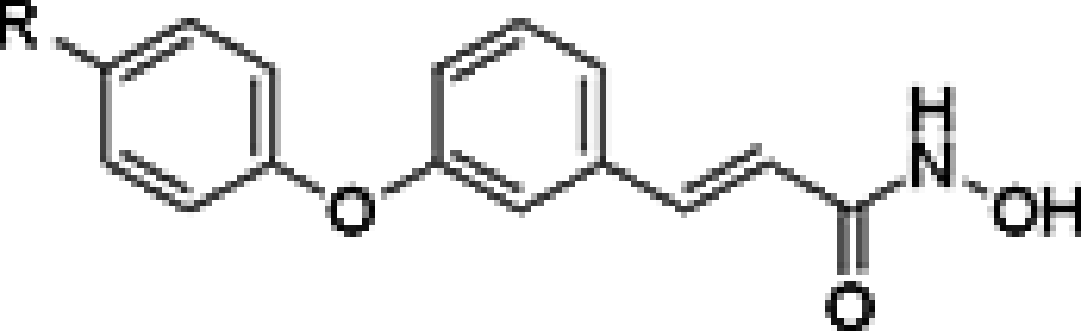 | ||
|---|---|---|
| Compound | R | IC50 (nM)† |
| 12q | NH2 | 321 |
| 12r | CH3NH | 209 |
| 12s | (CH3)2N | 256 |
Assays were performed in replicate (n ≥ 2). Mean values are shown, and the standard deviations are <20% of the mean.
Molecular docking
For a better understanding of the interaction between these synthesized small molecule analogs and HDAC, the most active compound 7e and the least active compound 7b were docked to the active site of HDAC2 (PDB ID: 4LXZ.pdb) using Glide 7.4, and compared with the crystal structure of HDAC2 in complex with SAHA (4LXZ.pdb) (Figure 5). The results depicted in Figure 5A & B suggest that both 7e and 7b could bind to the active site of HDAC2, and they exhibited similar binding mode compared with the co-crystallized SAHA. Like the co-crystallized SAHA [31], both compounds chelate zinc in similar trigonal bipyramidal fashion, with virtually identical Zn2+ chelating functional groups, including the hydroxamic acid groups (ZBG) in both compounds. The linker domains’ amide NHs in 7e, 7b and SAHA were found to from hydrogen bonds with Asp104, which could further stabilize the complex. The cap group is usually a hydrophobic and aromatic group, which interacts with the rim of the binding pocket. In 7e the cap group is 2-OCH3Ph, whereas in 7b the cap group is 2-CH3Ph, and the IC50 for these compounds are 11.5 and 650 nM, respectively. Binding of the 7e shows that the –OCH3 is pointed toward the His33, whereas the –CH3 in 7b is pointed away from His33 and exposed to the solvent. The –OCH3 is an electron donating group, which could increase the stability of a positive charge. Isoelectric point pH(I) of histidine is 7.47, and at a pH below its pI histidine carries a net positive charge. This could be the driving force for the orientation of –OCH3 in 7e toward His33, making it a more stable complex than 7b, whereas the orientation of –CH3 in 7b is away from His33 and exposed to solvent, making it a weaker complex. The free energy of binding obtained by MMGB/SA calculations also reflects the stabilities of the complexes observed by docking studies, as the binding energies are −56.66 and −48.23 kcal/mol for 7e and 7b, respectively. Hence the stronger binding of 7e makes it a more active compound (IC50 = 11.5 nM) than 7b (IC50 = 650 nM) as observed from the HDAC binding assay. Docking of SAHA to the binding site of HDAC2 also showed virtually identical binding as the cocrystallized SAHA, and the free energy of binding was −53.46 kcal/mol.
Figure 5. . The molecular binding modes of 7e (yellow), 7b (orange) and SAHA (green) in the active site of HDAC2.

The subsets are (A) comparison of the binding of 7e and SAHA and (B) comparison of the binding of 7e and 7b. Important amino acids in the binding pocket that interact with the compound and/or chelate with the Zn2+ are shown in stick model with carbon in cyan.
In vitro antiproliferative activity & cytotoxicity
To further ascertain the activities of these amide linked analogs of belinostat at the cellular level, compounds 7a, 7c, 7e, 7f, 7h, 7j, 7k, 7m, 7n and 7o were chosen for evaluation of antiproliferative activities in vitro HeLa and MDA-MB-231 cells (Table 3).
Table 3. . Antiproliferative activities of compound 7 against HeLa and MDA-MB-231 cell lines.
| Compound | IC50 (μM)† | ||
|---|---|---|---|
| HeLa | MDA-MB-231 | MCF-10A | |
| 7a | 6.57 | 5.73 | 5.48 |
| 7c | 5.32 | 5.54 | 6.34 |
| 7e | 5.09 | 7.28 | 7.43 |
| 7f | 5.48 | 5.79 | 6.68 |
| 7h | 10.69 | 11.66 | 12.15 |
| 7j | 2.86 | 4.03 | 2.37 |
| 7k | 8.36 | 9.15 | 7.69 |
| 7m | 6.12 | 7.51 | 6.89 |
| 7n | 5.52 | 4.31 | 5.22 |
| 7o | 6.56 | 7.82 | 9.55 |
| belinostat | 3.23 | 2.35 | 0.41 |
Assays were performed in replicate (n ≥ 2). Mean values are shown, and the standard deviations are <20% of the mean.
Among these ten compounds tested, nine showed comparable activity to belinostat with single-digit micromolar IC50 values. It was interesting that although the HDAC inhibition of 7j was inferior, its cellular potency against HeLa (2.86 μM) was comparable and even superior than belinostat (3.23 μM). Furthermore, 7j exhibited much lower toxicity (2.37 μM) than belinostat (0.41 μM) toward MCF-10A, the normal mammary epithelial cells. Unexpectedly, compound 7h, which was very potent in the HDAC inhibition assay at 12.5 nM (Table 1), was found to exert the least inhibitory potency on cell growth, suggesting it may have poor properties, for example, cell permeability, and therefore disqualifying it from further in vivo efficacy testing in the future.
While most of the synthesized compounds did not have better tumor efficacy in our assay, these compounds had a better safety profile than belinostat. As can be seen from Table 3, all the synthetic belinostat analogs showed significantly low cytotoxicity toward the normal breast cell line MCF-10A compared with belinostat. This observation is significant since the development of anticancer drugs target abnormal tumor cell with minimal impact on normal cells is a fundamental objective of cancer research. The result suggests that the sulfonamide CU at belinostat scaffold could be linked to the indiscriminating high toxicity and the replacement of this CU with an amide moiety might improve the safety profile of belinostat.
Conclusion
The optimization of an approved HDAC inhibitor belinostat and a focused SAR study designed to understand the impact of changing the CU from sulfonamide to amide has been described. We utilized scaffold hybridization techniques to combine the attractive features from distinct chemical scaffolds of some relatively successful HDAC inhibitors. The replacement of the sulfonamide of belinostat with an amide moiety led to a slight decrease in the HDAC inhibition potency, which was, however, substantially improved by introducing an additional methoxy or amino substituent at the 2-position of the phenyl ring. Compound 7e possessed superior HDAC inhibitory activity with an IC50 value of 11.5 nM in the pan-HDAC assay compared with belinostat, which could be a good candidate for further pharmacological studies.
Future perspective
While belinostat was synthetically challenging to access due to the presence of a sulfonamide moiety, the resulting analogs featured higher synthetic tractability and therefore represented a better starting point for chemistry optimization. In this manuscript, we have modified only the sulfonamide of the scaffold with the replacement of amide group as a first-stage optimization study. It is our purpose now to perform further modifications of the structure of the best compound 7e, with the aim of obtaining derivatives that are characterized by an improved HDAC inhibition activity.
Summary points.
A series of belinostat analogs were designed and synthesized as histone deacetylase inhibitors.
The most potent compound 7e showed an IC50 value of 11.5 nM against histone deacetylase.
Molecular docking studies supported the experimental observations.
Modification on sulfonamide of belinostat is a strategy to search for improved derivatives.
Footnotes
Financial & competing interests disclosure
This work was supported by the Science and Technology Support Program of Jiangsu Province (BE2017643) (LM Zhao), the Open Project Program of the State Key Laboratory for Chemistry and Molecular Engineering of Medicinal Resources, Guangxi Normal University (CMEMR2017-B11) (LM Zhao) and NIMHD grant number 2G12MD007595 (G Wang). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Bailly C. Cell-targeted cytotoxics: a new generation of cytotoxic agents for cancer treatment. Phytochem. Rev. 13(1), 171–181 (2014). [Google Scholar]
- 2.Paris M, Porcelloni M, Binaschi M, Fattori D. Histone deacetylase inhibitors: from bench to clinic. J. Med. Chem. 51(6), 1505–1529 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Mottamal M, Zheng S, Huang TL, Wang G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 20(3), 3898–3941 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin RJ, Nagy L, Inoue S, Shao W, Miller WH, Jr, Evans RM. Role of the histone deacetylase complex in acute promyelocytic leukemia. Nature 391(6669), 811–814 (1998). [DOI] [PubMed] [Google Scholar]
- 5.Kouzarides T. Histone acetylases and deacetylases in cell proliferation. Curr. Opin. Genet. Dev. 9(1), 40–48 (1999). [DOI] [PubMed] [Google Scholar]
- 6.Marks PA, Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 25(1), 84–90 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Prince HM, Dickinson M, Khot A. Romidepsin for cutaneous T-cell lymphoma. Future Oncol. 9(12), 1819–1827 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Lee HZ, Kwitkowski VE, Del Valle PL. et al. FDA approval: belinostat for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma. Clin. Cancer Res. 21(12), 2666–2670 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Garnock-Jones KP. Panobinostat: first global approval. Drugs 75(6), 695–704 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Chan TS, Tse E, Kwong YL. Chidamide in the treatment of peripheral T-cell lymphoma. Onco Targets Ther. 10, 347–352 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raju TN. The Nobel chronicles. 1988: James Whyte Black, (b 1924), Gertrude Elion (1918-99), and George H Hitchings (1905-98). Lancet 355(9208), 1022 (2000). [DOI] [PubMed] [Google Scholar]
- 12.Pontiki E, Hadjipavlou-Litina D. Histone deacetylase inhibitors (HDACIs). Structure–activity relationships: history and new QSAR perspectives. Med. Res. Rev. 32(1), 1–165 (2012). [DOI] [PubMed] [Google Scholar]; •• Important review on histone deacetylase (HDAC) inhibitors and their structure–activity relationships.
- 13.Rajak H, Singh A, Raghuwanshi K. et al. A structural insight into hydroxamic acid based histone deacetylase inhibitors for the presence of anticancer activity. Curr. Med. Chem. 21(23), 2642–2664 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y, Yang P, Chou CJ, Liu C, Wang X, Xu W. Development of N-hydroxycinnamamide-based histone deacetylase inhibitors with an indole-containing cap group. ACS Med. Chem. Lett. 4(2), 235–238 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tu S, Yuan H, Hu J, Zhao C, Chai R, Cao H. Design, synthesis and biological evaluation of nitro oxide donating N-hydroxycinnamamide derivatives as histone deacetylase inhibitors. Chem. Pharm. Bull. 62(12), 1185–1191 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Li X, Inks ES, Li X. et al. Discovery of the first N-hydroxycinnamamide-based histone deacetylase 1/3 dual inhibitors with potent oral antitumor activity. J. Med. Chem. 57(8), 3324–3341 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Important paper on N-hydroxycinnamamide-based HDAC inhibitors.
- 17.Zang J, Shi B, Liang X, Gao Q, Xu W, Zhang Y. Development of N-hydroxycinnamamide-based HDAC inhibitors with improved HDAC inhibitory activity and in vitro antitumor activity. Bioorg. Med. Chem. 25(9), 2666–2675 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Chen C, Hou X, Wang G. et al. Design, synthesis and biological evaluation of quinoline derivatives as HDAC class I inhibitors. Eur. J. Med. Chem. 133, 11–23 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Yuan Z, Sun Q, Li D. et al. Design, synthesis and anticancer potential of NSC-319745 hydroxamic acid derivatives as DNMT and HDAC inhibitors. Eur. J. Med. Chem. 134, 281–292 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Wang DF, Wiest O, Helquist P, Lan-Hargest HY, Wiech NL. On the function of the 14 Å long internal cavity of histone deacetylase-like protein: implications for the design of histone deacetylase inhibitors. J. Med. Chem. 47(13), 3409–3417 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Allen FH. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Cryst. B 58(3), 380–388 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Brameld KA, Kuhn B, Reuter DC, Stahl M. Small molecule conformational preferences derived from crystal structure data. A medicinal chemistry focused analysis. J. Chem. Inf. Model. 48(1), 1–24 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Monneret C. Histone deacetylase inhibitors. Eur. J. Med. Chem. 40(1), 1–13 (2005). [DOI] [PubMed] [Google Scholar]; • Important review on HDAC inhibitors and future challenges.
- 24.Murray PM, Bower JF, Cox DK, Galbraith EK, Parker JS, Sweeney JB. A robust first-pass protocol for the Heck-Mizoroki reaction. Org. Process Res. Dev. 17(3), 397–405 (2013). [Google Scholar]
- 25.Schrödinger Release 2017–1: Glide, Schrödinger, LLC, New York, NY, USA: (2017). https://www.schrodinger.com/glide [Google Scholar]
- 26.Harder E, Damm W, Maple J. et al. OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 12(1), 281–296 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Finn PW, Bandara M, Butcher C. et al. Novel sulfonamide derivatives as inhibitors of histone deacetylase. Helv. Chim. Acta 88(7), 1630–1657 (2005). [Google Scholar]
- 28.Yang L, Xue X, Zhang Y. Simple and efficient synthesis of belinostat. Synth. Commun. 40(17), 2520–2524 (2010). [Google Scholar]
- 29.Wang Q, Luo J, Cao Y, Zhang L, Li C, Yuan Q. A process for preparing belinostat cis-isomer. CN 105367455 A (2016). https://worldwide.espacenet.com/searchResults?DB=EPODOC&submitted=true&locale=en_EP&ST=singleline&compact=false&query=CN105367455
- 30.Codd R, Braich N, Liu J, Soe CZ, Pakchung AAH. Zn(II)-dependent histone deacetylase inhibitors: suberoylanilide hydroxamic acid and trichostatin A. Int. J. Biochem. Cell B. 41(4), 736–739 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Lauffer BE, Mintzer R, Fong R. et al. Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J. Biol. Chem. 288(37), 26926–26943 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]