

Abstract
Aim:
The contribution of miRNAs as epigenetic regulators of sexually dimorphic gene expression in the placenta is unknown.
Materials & methods:
382 placentas from the extremely low gestational age newborns (ELGAN) cohort were evaluated for expression levels of 37,268 mRNAs and 2,102 miRNAs using genome-wide RNA-sequencing. Differential expression analysis was used to identify differences in the expression based on the sex of the fetus.
Results:
Sexually dimorphic expression was observed for 128 mRNAs and 59 miRNAs. A set of 25 miRNA master regulators was identified that likely contribute to the sexual dimorphic mRNA expression.
Conclusion:
These data highlight sex-dependent miRNA and mRNA patterning in the placenta and provide insight into a potential mechanism for observed sex differences in outcomes.
Keywords: : epigenetics, epigenome, miRNA, mRNA, placenta, sexual dimorphism
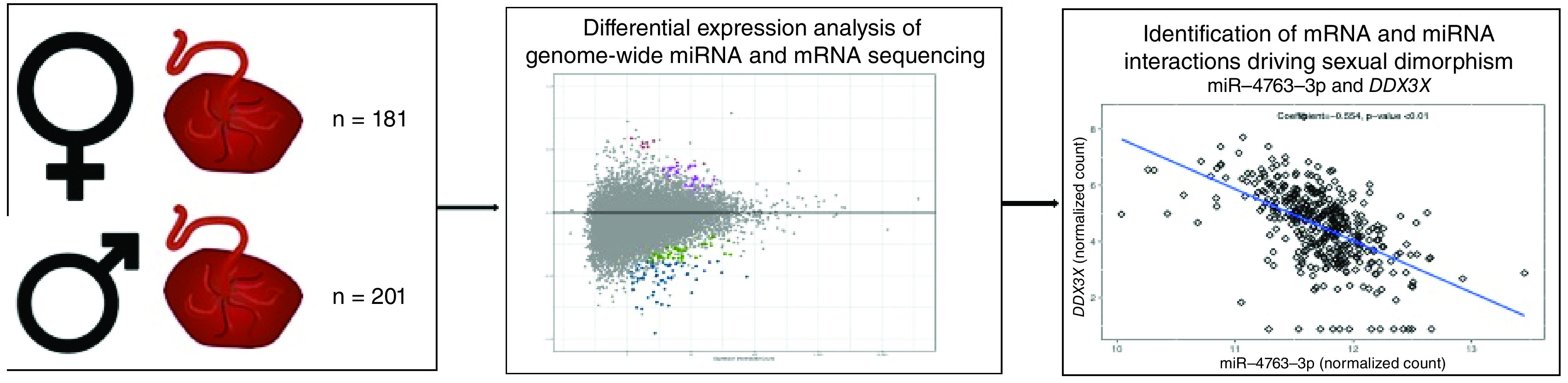
Sexual dimorphism of perinatal, infant and later-in-life outcomes is well documented. For example, fetal sex-specific responses to maternal stress, maternal immune status and exposure to environmental toxicants have been observed [1–5]. Pregnancy complications such as preterm birth and preeclampsia also display sexually dimorphic trends [6–8]. Additionally, later-in-life infant/child health outcomes including neurologic conditions such as cerebral palsy and autism spectrum disorder have sexually dimorphic rates [9–11]. Generally, these differential responses highlight a propensity for male infants to have increased disease risk [5,12,13]. While sexual dimorphism is commonly observed, the underlying biological mechanisms for sex-based differences are understudied.
Sex-specific functioning of the placenta may be a key driver of observed sexual dimorphism [14–16]. The placenta modulates the uterine environment, transports nutrients to the fetus, secretes hormones and removes waste. Thus, the placenta is critical to healthy fetal growth and survival and as such, it sets the foundation for later-life health trajectories [17,18]. Placentas are also known to vary physiologically and morphologically based on sex. For example, human placentas derived from pregnancies of male fetuses (male placenta, herein) or female fetuses (female placenta, herein) display variation in the early placentation process as well as expression of genes involved in immune-disease, the renin-angiotensin system and glucocorticoid receptor genes [19–22]. In fact, an analysis of the transcriptome of 37 late first trimester placentas revealed 58 genes with significantly different expression between sexes where ∼30% of these genes were located on autosomes [23]. Additionally, a meta-analysis of 11 placental gene expression microarray datasets identified over 140 genes that are differentially expressed between male and female term placentas [24]. In this study, the majority (>60%) of these genes were located on autosomes [24]. Given that at the genome-level only sex chromosomes vary between male and female placentas, epigenetic control of the placental transcriptome likely plays a significant role in sex-dependent gene expression [15]. In fact, evidence suggests that indeed epigenetic regulators are key in sex-specific placental gene expression. For example, sexually dimorphic CpG methylation patterns in the placenta have been observed [25]. However, to our knowledge, no studies have assessed the role of miRNAs, another epigenetic regulator, in regulating the sexual dimorphism of placental gene expression. For clarity, herein sexual dimorphism refers to the difference in epigenomic or genomic profiles between placentas derived from a male or female fetus.
miRNAs are a class of single-stranded, noncoding RNA molecules, usually 20–24 nucleotides in length. These molecules play an important role in post-transcription control of gene expression. Specifically, miRNAs induce mRNA degradation or repress translation by binding to the 3′ untranslated region of the target mRNA [26]. Therefore, an often-observed relationship between a miRNA and its target genes is that an increase in miRNA expression corresponds to a subsequent decrease in mRNA levels. However, it is worth noting that in some cases miRNAs may activate gene expression and be positively associated with mRNA expression [26,27]. One proposed mechanism for this positive association is through miRNA-driven chromatin remodeling in enhancer regions [27,28]. A growing body of research has demonstrated the importance of placental miRNAs during pregnancy, including associations between specific miRNAs and preeclampsia, early pregnancy loss and fetal growth restriction as well as response to environmental stressors [29–35]. Therefore, miRNAs are potentially critical regulators of the sex-specific placental functioning that contributes to sex-differences in adverse pregnancy and later-life outcomes.
We hypothesized that sexually dimorphic patterns of mRNAs would be observed in comparing male and female placentas from the extremely low gestational age newborn (ELGAN) cohort. We also anticipated that miRNAs may underlie some of the gene expression changes and that levels of sexually dimorphic miRNAs would correlate with those of mRNA for known gene targets. In order to test these hypotheses, we investigated whether genome-wide mRNA and miRNA expression in the placenta differed based on the sex of the fetus in the ELGAN cohort, which is one of the largest placental multi-OMICS birth cohorts in the USA. This study provides key insights into epigenetic regulation of sexual dimorphism in the placenta.
Methods
The ELGAN study cohort
The ELGAN cohort comprises infants born prior to completing 28 weeks’ gestation and its design, recruitment and sample collection methods have been detailed extensively previously [36]. Briefly, from 2002 to 2004, pregnant women at one of 14 participating institutions were recruited for participation and consent was provided either prior to hospital admission or soon after delivery. A trained research nurse measured demographic and pregnancy variables after delivery using a structured questionnaire. The study was approved by the Institutional Review Board at each of the 14 participating ELGAN sites. A total of 1249 mothers were recruited, resulting in a cohort of 1506 infants. Due to occasional unavailability of research assistants, 1365 placentas were collected. The current study is based on a subsample of 390 placentas in which miRNA and mRNA had been analyzed; though incomplete demographic data and quality assessment/quality control (QA/QC) steps (as detailed below) resulted in a subset of n = 382 placentas included for final statistical analysis of miRNA and mRNA expression. A total of n = 201 placenta samples were derived from a male fetus (male placentas) and n = 181 placentas were derived from a female fetus (female placentas).
Placental tissue collection
Placental tissue collection was carried out using methods previously described [25,37,38]. In brief, placentas were collected upon delivery, placed in a sterilized basin and taken to the sampling room for biopsy. The amnion was pulled back to expose the underlying chorion, representing fetally derived tissue. A sample (<1 g) from the base of the chorion was removed by applying traction to the chorion and underlying trophoblast tissue. Collected samples were immediately placed into a sterile 2 ml cryovial that was quickly submerged in liquid nitrogen, then transferred to a -80°C freezer for long-term storage. The placental samples were processed by first placing the cryotubes containing the placental biopsies on dry ice. Frozen tissue samples were sliced into approximately 0.025 g segments using a sterile dermal curette and washed in phosphate-buffered saline (PBS) (Thermo Fisher Scientific, MA, USA) to reduce any potential blood contamination. To preserve sample integrity, washed samples were then immediately snap frozen in homogenization tubes and placed back on dry ice. Tissue segments were homogenized using a sterile stainless-steel bead (Qiagen, MD, USA) in RLT + lysis buffer (Qiagen) with the TissueLyserII instrument (Qiagen). Then samples were clarified by spinning to collect the bead and cellular debris. Homogenated samples were stored at -80°C until nucleic acid extraction.
mRNA & miRNA extraction & sequencing
RNA molecules 18 nucleotides and greater were extracted using the AllPrep DNA/RNA/miRNA Universal kit (Qiagen). RNA quality was determined using LabChip (Perkin Elmer, MA, USA) instrument to generate RNA integrity numbers (RIN), which ranged from 1 to 3, and DV200 values, which were in acceptable range. Genome-wide mRNA expression was determined using QuantSeq 3′ mRNA-Seq Library Prep Kit (Lexogen, Vienna, Austria), chosen because it can quantify transcripts with lower range RINs [39]. Library preparation was completed according to Lexogen recommendation for lower RIN samples. RNA-sequencing libraries were pooled and sequenced (single-end 50 bp) on one lane of the Illumina Hiseq 2500 (Illumina, CA, USA). Libraries were prepared by automation on Sciclone G3 (Perkin Elmer) to avoid potential batch to batch artifacts. The counts of sequencing reads were aligned to the GENCODE database v30 and organized using Salmon (version 0.11.3) resulting in a total of n = 37,268 unique human RNA transcripts, including protein-coding and non-coding RNAs [40,41]. The aligned count data were used in downstream data processing and statistical analyses as described below.
Genome-wide miRNA expression profiles were assessed using the HTG EdgeSeq miRNA Whole Transcriptome Assay (HTG Molecular Diagnostics, AZ, USA). This assay uses next-generation sequencing to measure the expression of n = 2102 human miRNA transcripts. The counts of sequencing reads per miRNA were extracted and used in the analysis. The counts of sequencing reads were aligned to miRBase v20 and organized using Parser (HTG Molecular Diagnostics) [42].
In order to mitigate the potential impacts of RIN scores on the final results, additional quality control steps were included: 1) filtering out universally lowly expressed transcripts (detailed below); 2) including additional surrogate variables in the statistical modeling approach to account for sources of heterogeneity (detailed below); 3) confirming whether published placental miRNA and mRNA transcripts were recapitulated in the data. Specifically, the miRNA data from the present study were compared with the placenta-specific miRNA clusters, C19MC and C14MC, and mRNAs within the list of 50 most abundant mRNAs in an independent placental whole genome RNA-sequencing study by Saban et al. [23,43]. To detail the results of this final QC step, of the 50 mRNAs found to be the most abundant in placenta tissue according to Saban et al., there was a 96% overlap with the transcripts found in this cohort (46 of 50 mRNA transcripts) (Supplementary Table 1). For the miRNAs, there was coverage within our data of 82.3% across both the C19MC and C14MC clusters (79 of 95 miRNAs) (Supplementary Table 1).
Data processing & statistical analysis
To identify miRNAs and mRNAs associated with fetal sex, miRNA and mRNA sequencing data were processed separately using R (v 3.6.2) (cran.r-project.org/). Count data were first filtered to exclude universally lowly expressed transcripts, requiring that >25% of the samples be expressed at signals above the overall median signal intensity, similar to our prior genome-wide mRNA and miRNA analyses [44–48]. This resulted in a total of n = 10,408 mRNA transcripts and n = 1131 miRNA transcripts included in analyses. Of the 390 placentas evaluated, two samples were removed due to having all zero mRNA counts and four samples were removed for missing data on race (n = 384). QA/QC was conducted on the count data using both calculation and visualization of principal components via the prcomp function, and hierarchical clustering, including calculation of distance metrics and visualization, using the hclust function. Two subjects deemed as outliers were removed in quality control on the mRNA data and were therefore removed from both datasets. Following removal of these outliers, the final analysis sample of the same n = 382 subjects for both mRNA and miRNA was constructed.
The DESeq2 package (v 1.24.0) was used to normalize the count data using median signal intensity, resulting in variance stabilized counts [49,50]. The SVA package (v 3.32.1) was used to account for potential batch effects and sources of sample heterogeneity with control probes empirically estimated using default parameters. Three significant surrogate variables were calculated and included as covariates in the statistical model, along with the three other covariates identified in methods described below (Supplementary Figure 1).
To capture potential sources of bias without substantial loss in precision, a parsimonious model was constructed from covariates that were significantly associated (p < 0.05) or had known relationships with placental miRNA and/or mRNA expression levels and fetal sex. This resulted in the inclusion of the following covariates: maternal age (years), maternal race (white/ nonwhite) and newborn gestational age at delivery (days). Statistical methods utilizing negative binomial generalized linear models within DESeq2 were used to identify miRNAs and mRNAs with differential expression according to fetal sex, controlling for covariates listed previously. This method calculated shrunken logarithmic fold changes in expression, which were then divided by their standard error values to produce z-statistics. Resulting z-statistics were compared against standard normal distribution curves to generate Wald test p-values. To account for multiple testing, these p-values were then adjusted using the Benjamini and Hochberg (BH) procedure [51]. All differentially expressed miRNAs and mRNAs were defined as those with false discovery rate < 0.1. miRNAs and mRNAs identified as showing sexually dimorphic expression were evaluated for chromosomal location, according to the GRCh38 genome reference assembly or the 30 gencode hg19 reference genome assembly, respectively. The distribution of miRNAs and mRNAs across chromosomes was evaluated, and the number of sexually dimorphic miRNAs/mRNAs per chromosome was tested for correlation against the total number of miRNAs/mRNAs evaluated per chromosome using the Spearman Rank correlation test. Sex chromosomes were included in this analysis as pseudo-positive controls and to mirror previous studies assessing epigenomic and transcriptomic sexual dimorphism [24,25].
In order to gauge the influence of this cohort being composed of placentas from pregnancies of <28 weeks gestational age on results obtained, significantly sexually dimorphic mRNAs and miRNAs were compared with literature detailing known differences in mRNA expression and miRNA expression over the course of gestation. To briefly detail the studies used for the comparison, Morales-Prieto et al. assessed trophoblast cells isolated from first and third trimester placentae and found n = 46 miRNAs changing over gestation and Gu et al. compared placental villous tissues between first and third trimester placentas and found n = 208 differentially expressed miRNAs [52,53]. For mRNAs, Winn et al. identified 418 genes differentially expressed when comparing basal plate biopsy specimens from mid-gestation (14–24 weeks) to term (37–40 weeks) and Mikheev et al. found n = 764 genes that varied between third and second trimester placenta villous tissue [54,55].
Identification of gene targets of miRNA & correlation between miRNA and mRNA expression
An in silico approach based on experimentally-observed miRNA–mRNA interactions curated from literature coupled with computational predictions was used to identify miRNA-regulators of sexually dimorphic mRNA expression, following methods utilized previously [45,56]. Sexually dimorphic miRNAs were queried within the Ingenuity Knowledge Database (Ingenuity Systems®, CA, USA) for gene targets predicted based on experimental observation or high predicted confidence (cumulative weighted context scores -0.4). Cumulative weighted context scores summarize factors, including binding site type and location, local adenine and uracil content, target site abundance, seed-pairing stability and supplementary pairing, that influence the likelihood of miRNA–mRNA interactions [57]. The gene targets identified were filtered to include only genes for which mRNA was found to be sexually dimorphic as well. The remaining set of miRNAs and mRNAs were deemed to be predicted miRNA–mRNA expression pairs. Thus, an expression pair is defined as a miRNA and a mRNA in which the mRNA is a predicted gene target of the miRNA and both miRNA and mRNA were found to be sexually dimorphic.
In order to test if the predicted expression pairs had correlated expression levels, correlations between the expression level (variance stabilized counts) of the miRNA and mRNA were calculated using the Pearson correlation coefficient and a significance of p ≤ 0.05. The expression pairs that exhibited significant positive or negative correlation between miRNA and mRNA expression were deemed to be those in which the mRNA is likely under miRNA control.
Pathway enrichment analysis of sexually dimorphic mRNAs under miRNA control
To understand the biological implications of the sexually dimorphic mRNAs, canonical pathway and network enrichment analyses were carried out via the Ingenuity Knowledge Database and STRING database (v.11.0). Using Ingenuity Knowledge Database, significantly over-represented canonical pathways were defined as those containing more sexually dimorphic mRNAs than expected by random chance, using a BH-corrected p-value calculated from a right-tailed Fisher's Exact Text (p < 0.05). As an additional methodology to assess for known gene interactions, the STRING database was used to identify functional association networks (“interactomes”) in order to visualize interactions between sexually dimorphic mRNAs. A functional association is defined as a link between two proteins that contribute jointly to a specific biological function [58]. STRING confidence scores range from 0 to 1 and demonstrate the confidence that the predicted interaction is true given all available evidence [59].
Results
Study subject characteristics
General characteristics of the study participants are summarized in Table 1. In this subset of 382 women whose placentas were available for miRNA and mRNA expression analysis, 201 (52.6%) gave birth to male infants and 181 (47.4%) gave birth to female infants. Participating mothers had an overall average maternal age of 29 years. Infants had an average gestational age of 182.5 days (26 weeks), ranging from a minimum of 161 days (23 weeks) to a maximum of 195 days (27.85 weeks). The average gestational age differed significantly between male and females in the cohort (p < 0.05). The majority (89.1%) of study participants were nonsmokers. Other than gestational age, none of the clinical or demographic variables differed significantly (at p < 0.05) between male and female infants in the cohort.
Table 1. . Summary of the ELGAN study demographics for the sub cohort used in this analysis (n = 382) consisting of mothers that contributed placentas that were analyzed for sexually dimorphic miRNA and mRNA expression profiles. Subjects with missing data were not included in the percentage calculation.
| Variables | Overall (n = 382) |
Male (n = 201) |
Female (n = 181) |
p-value |
|---|---|---|---|---|
| n (%), or Mean (min–max) |
n (%), or Mean (min–max) |
n (%), or Mean (min–max) |
Chi-square for categorical, Kruskal–Wallis for continuous | |
| Gestational age (days) | 182.5 (161–195) | 181.6 (161–195) | 183.4 (161–195) | 0.04 |
| Maternal age (years) | 29.6 (14.0–45.8) | 30.0 (14.0–45.8) | 29.2 (16.5–45.1) | 0.15 |
| Race | 0.62 | |||
| – White | 235 (61.5) | 126 (62.7) | 109 (60.2) | |
| – Non-white | 147 (34.5) | 75 (37.3) | 72 (39.8) | |
| Ethnicity | 0.50 | |||
| – Non-Hispanic | 354 (92.7) | 188 (93.5) | 166 (91.7) | |
| – Hispanic | 28 (7.3) | 13 (6.5) | 15 (8.3) | |
| Smoking status | 0.61 | |||
| – Yes | 41 (10.9) | 20 (10.1) | 21 (11.8) | |
| – No | 334 (89.1) | 177 (89.9) | 157 (88.2) | |
| – Not reported | 7 | 4 | 3 | |
| Maternal prepregnancy BMI | 0.97 | |||
| – Underweight | 26 (7.1) | 13 (6.7) | 13 (7.4) | |
| – Normal | 198 (53.7) | 104 (53.6) | 94 (53.7) | |
| – Overweight | 66 (17.9) | 34 (17.5) | 32 (18.3) | |
| – Obese | 79 (21.4) | 43 (22.2) | 36 (20.6) | |
| – Not reported | 13 | 7 | 6 |
ELGAN: Extremely low gestational age newborns.
Identification of sexually dimorphic mRNA expression profiles in human placentas
Multivariable modeling approaches used to compare genome-wide mRNA expression profiles in the human placenta resulted in the identification of 128 mRNAs with significant differential expression associated with infant sex (Supplementary Table 2 & Figure 1). These models adjusted for confounding variables such as gestational age. Of these 128 mRNAs identified, 37 displayed increased expression in the male placenta and 91 displayed increased expression in the female placenta. Among the mRNAs increased in the female placenta, the most differentially expressed mRNA was XIST (BH-adjusted p-value = 2.37 × 10-79), known to be expressed exclusively from the inactive X chromosome. Other highly significant mRNAs increased in the female placenta include HDAC1 (BH-adjusted p-value = 8.09 × 10-2) located on chromosome 1 and HDAC8 (BH-adjusted p-value = 3.60 × 10-2) located on the X chromosome. Both of these genes are known to be involved in histone-based epigenetic control. CDKN1B (BH-adjusted p-value = 6.35 × 10-2), located on chromosome 12, was also differentially expressed and increased in female placenta; it is known to be involved in cell cycle progression. Collectively, mRNAs with increased expression in the female placenta were enriched for canonical pathways including EIF2, VEGF and adipogenesis (p-values for pathway enrichment of 2.80 × 10-4 and 7.64 × 10-4, respectively) (Supplementary Table 3).
Figure 1. . MAplot of mean mRNA expression levels versus fold change in expression across the ELGAN placenta samples.
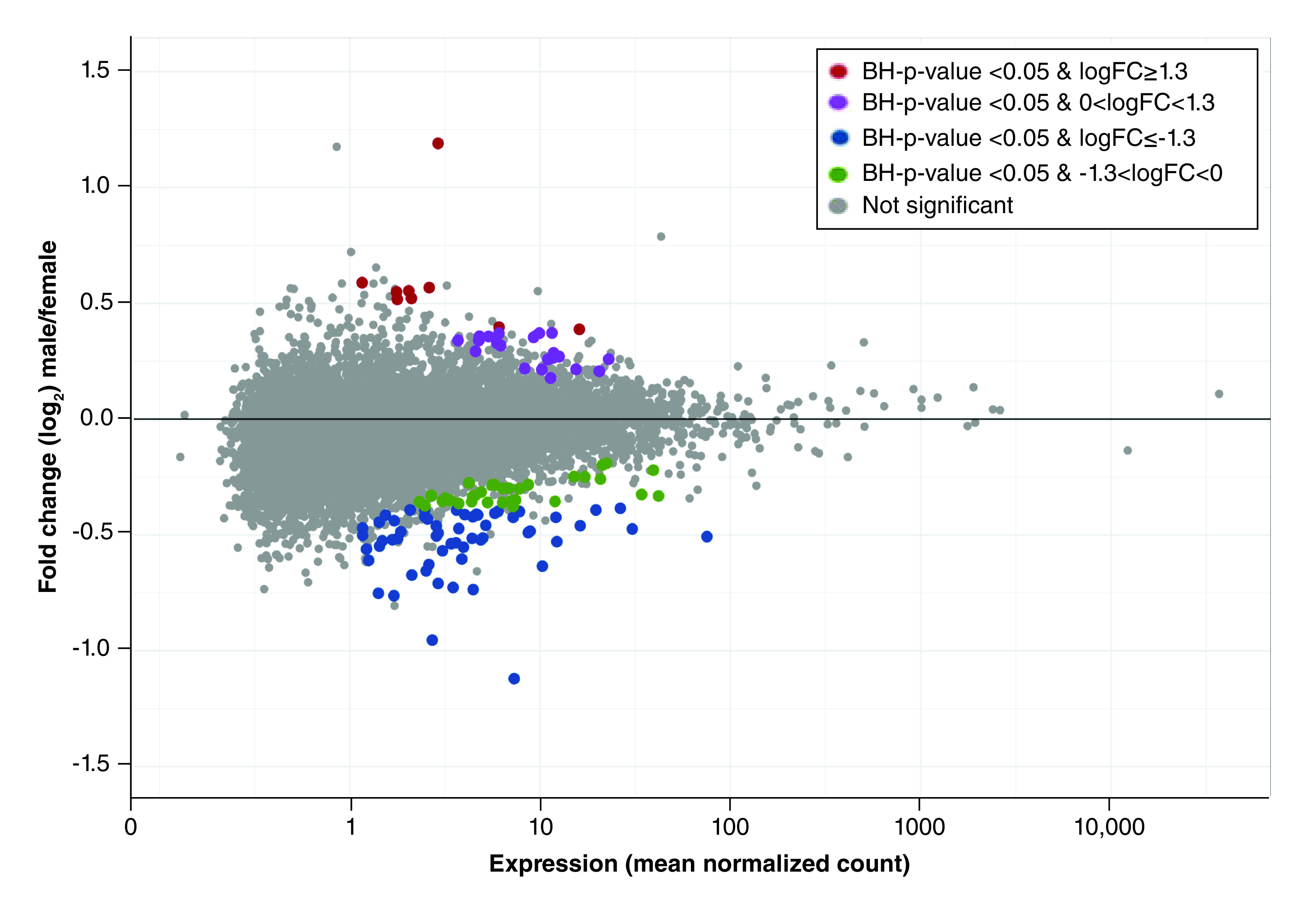
Fold change values represent the ratio of mean normalized mRNA counts in males/females. Fold change values are log2 transformed within the plot (y-axis scale).
BH: Benjamini-Hochberg; ELGAN: Extremely low gestational age newborns.
The differentially expressed mRNAs with highest abundance in the male placenta include TXLNGY (BH-adjusted p-value = 7.63 × 10-149), USP9Y (BH-adjusted p-value = 1.47 × 10-157) and EIF1AY (BH-adjusted p-value = 7.94 × 10-44), all of which are located on the Y chromosome. Other genes of note that were among mRNAs highly expressed in the male placenta include EPRS (BH-adjusted p-value = 9.20 × 10-2) and LARS (BH-adjusted p-value = 9.20 × 10-2), on chromosome 1 and 5, respectively, both of which are involved in the tRNA charging pathway, an enriched canonical pathway (p-value for pathway enrichment = 1.94 × 10-3) (Supplementary Table 3).
Buckberry et al. conducted a meta-transcriptome analysis of 11 publicly available microarray datasets of near-term and term placentas in a total n = 303 placenta samples where gestational age ranged from 263 to 280 days to evaluate sexually dimorphic mRNA expression [24]. Comparing the 128 differentially expressed mRNAs in the present study with Buckberry et al.‘s findings, 19 genes were found to be differentially expressed between sexes in both studies (Supplementary Table 2). Notably, the Buckberry et al. meta-analysis is comprised of placentas at a later gestational age than the ELGAN cohort, thus a comparison to Gonzalez et al, who evaluated sex differences in the first trimester placental transcriptome (n = 37, average gestational age of 83 days), was also conducted [60]. Of the 128 genes identified as sexually dimorphic in the present study, 21 were also identified in Gonzalez et al. (Supplementary Table 2). Overall, 19 genes were identified across all three datasets, although notably each of these datasets represents a different gestational age range.
Sexually dimorphic mRNA profiles included genes located on all chromosomes exclusive of chromosome 11, 20, 21 and 22. The largest percentage of mRNAs (28%) were located on sex chromosomes with 28 on the X chromosome and 8 on the Y chromosome. Of the 28 sexually dimorphic genes on the X chromosome, 25 displayed increased expression in the female placenta and 3 displayed increased expression in the male placenta (Supplementary Table 2). Of the 128 genes identified herein, 10 (7.8%) were found to significantly vary in expression over the course of gestation by Winn et al. or Mikheev et al.; and seven of these increased over the course of gestation (Supplementary Table 2) [54,55].
Identification of sexually dimorphic miRNA expression profiles in human placenta
Analysis of genome-wide miRNA expression profiles in the human placenta resulted in the identification of 59 miRNAs with significant differential expression associated with fetal sex (Supplementary Table 4 & Figure 2). Of these, 32 displayed increased expression in male placentas and 27 displayed increased expression in female placentas. Ten of the identified sexually dimorphic miRNAs are known to be placenta-associated (i.e., expressed ubiquitously in the placenta) or placenta-specific (i.e., expressed uniquely in the placenta) (Table 2) [33]. Literature presented in Table 2 was derived and expanded upon from a review by Cai et al. [33]. Specifically, these ten placenta-specific or placenta-associated miRNAs are miR-543, miR-495, miR-323a and miR-323b (all members of the placenta-specific C14MC cluster) as well as miR-23a, miR-15a, miR-30a, miR-155, miR-222 and miR-223. Of these, miR-323b, miR-15a and miR-223 were increased in female placentas, the others displayed increased expression in male placentas. Perhaps surprisingly, no sexually dimorphic miRNAs localized to the C19MC cluster. It is noteworthy that 34 of 46 (73.9%) C19MC cluster miRNAs were identified above the low-expression cut-off and thus were evaluated for sexually dimorphic expression (Supplementary Table 1). In contrast to this, 45 of 49 (91.8%) miRNAs on the C14MC cluster were identified above the low-expression cut-off (Supplementary Table 1).
Figure 2. . MAplot of mean miRNA expression levels versus fold change in expression across human placenta samples.

Fold change values represent the ratio of mean normalized miRNA counts in males/females. Fold change values are log2 transformed within the plot (y-axis scale).
BH: Benjamini-Hochberg.
Table 2. . A list of placenta-specific and placenta-associated miRNAs found to have higher expression in male or female placentas in this study.
| Increased in male (M) or female (F) placenta | Known placental or pregnancy function | Ref. | |
|---|---|---|---|
| Placenta-specific miRNA | |||
| miR-23a | M | Evidence for involvement in pathogenesis of recurrent miscarriage Increased in maternal plasma of preeclampsia patients Increased in placental tissues of preeclampsia patients and thought to contribute to preeclampsia pathogenesis through inducing trophoblast cell apoptosis |
[62] [63] [64] |
| C14MC cluster | Abundant in developing embryo and placenta | [43,65,66] | |
| miR-543 | M | Plays a role in the embryo implantation process | [67] |
| miR-495 | M | Potential biomarker for early detection of preeclampsia | [68] |
| miR-323a | M | Potential biomarker of ectopic pregnancy | [69,70] |
| miR-323b | F | ||
| Placenta-associated miRNA | |||
| miR-15a | F | Associated with phenol and paraben exposure in a sexually dimorphic manner | [71] |
| miR-30a | M | Involved in immune dysregulation of mesenchymal stem cells during preeclampsia | [72] |
| miR-155 | M | Involved in the pathogenesis of preeclampsia through down regulating pro-angiogenic gene targets, regulating endothelial nitric oxide synthase and inhibiting migration of trophoblast cells | [73–76] |
| miR-222 | M | Increased in preeclamptic placentas compared with controls | [35] |
| miR-223 | F | Decreased in preeclamptic placentas compared with controls | [34] |
Placenta-specific miRNAs are expressed uniquely in placental tissue. Placenta-associated miRNAs are expressed ubiquitously in the placenta. Known function is indicated for the miRNAs found to be differentially expressed in this cohort.
The miRNAs showing sexually dimorphic expression were distributed across chromosomes (Supplementary Figure 2). To detail, the highest numbers of sexually dimorphic miRNAs were present on chromosomes X and 1. Of a possible 151 miRNAs evaluated on the X chromosome, a total of 10 sexually dimorphic miRNAs were identified (6.6%). Of the 10 miRNAs identified on the X chromosome, five were increased in male placenta and five were increased in female placenta. Chromosome 1 was also identified to include 10 sexually dimorphic miRNAs of the 190 total miRNAs evaluated (5.3%), again with five increased in female placenta and five increased in male placenta. The number of significant sexually dimorphic miRNAs correlated with the number of miRNAs evaluated per chromosome, demonstrating that there is not a particular enrichment for sexually dimorphic miRNAs on any one chromosome (Spearman Rank rho = 0.81; p < 0.05). However, notably there were no sexually dimorphic miRNAs identified on Chromosomes 4, 5, 6, 10, 13, 20 and 21, all of which had miRNAs evaluated on them. Additionally, three sexually dimorphic miRNAs were identified on Chromosome 7, all with higher expression in female placenta. Conversely, on Chromosome 9, all five miRNAs identified were found with higher expression in male placenta. In total, 13 of the 59 sexually dimorphic miRNAs (22.0%) have been found to significantly vary in expression over the course of gestation by Morales-Prieto et al or Gu et al; nine increased while four decreased over the course of gestation (Supplementary Table 4) [52,53].
Identification of miRNA-controlled mRNAs associated with fetal sex
Computational prediction of mRNA–miRNA expression pairs resulted in the identification of 67 potential miRNA–mRNA pairs including 54 unique miRNAs and 40 unique mRNAs (Supplementary Table 5, Supplementary Figure 3). The correlation between miRNA and mRNA expression in the placenta among these predicted miRNA–mRNA expression pairings was then calculated. The majority (50 of the 67 pairs or 74.6%) indeed displayed significant correlation between miRNA and mRNA expression (Supplementary Table 5). Of these 50 significantly correlated expression pairs, 32 (64.0%) were found to have a negative correlation coefficient, representative of the expected inverse association between miRNA and mRNA expression. Positive correlations were also observed. It should be noted, however, that the associations were stronger among the inversely correlated pairs overall (in terms of p-value and absolute value of correlation coefficient). The six most significantly negatively correlated expression pairs were: (1) miR-6738-5p and DCAF7, (2) miR-4649-5p and MLLT1, (3) miR-6738-5p and HDAC1, (4) miR-939-5p and HIPK2, (5) miR-939-5p and ARID3B, and (6) miR-4763-3p and DDX3X (Figure 3).
Figure 3. . Scatterplots of the most significantly correlated miRNA–mRNA expression pairings.
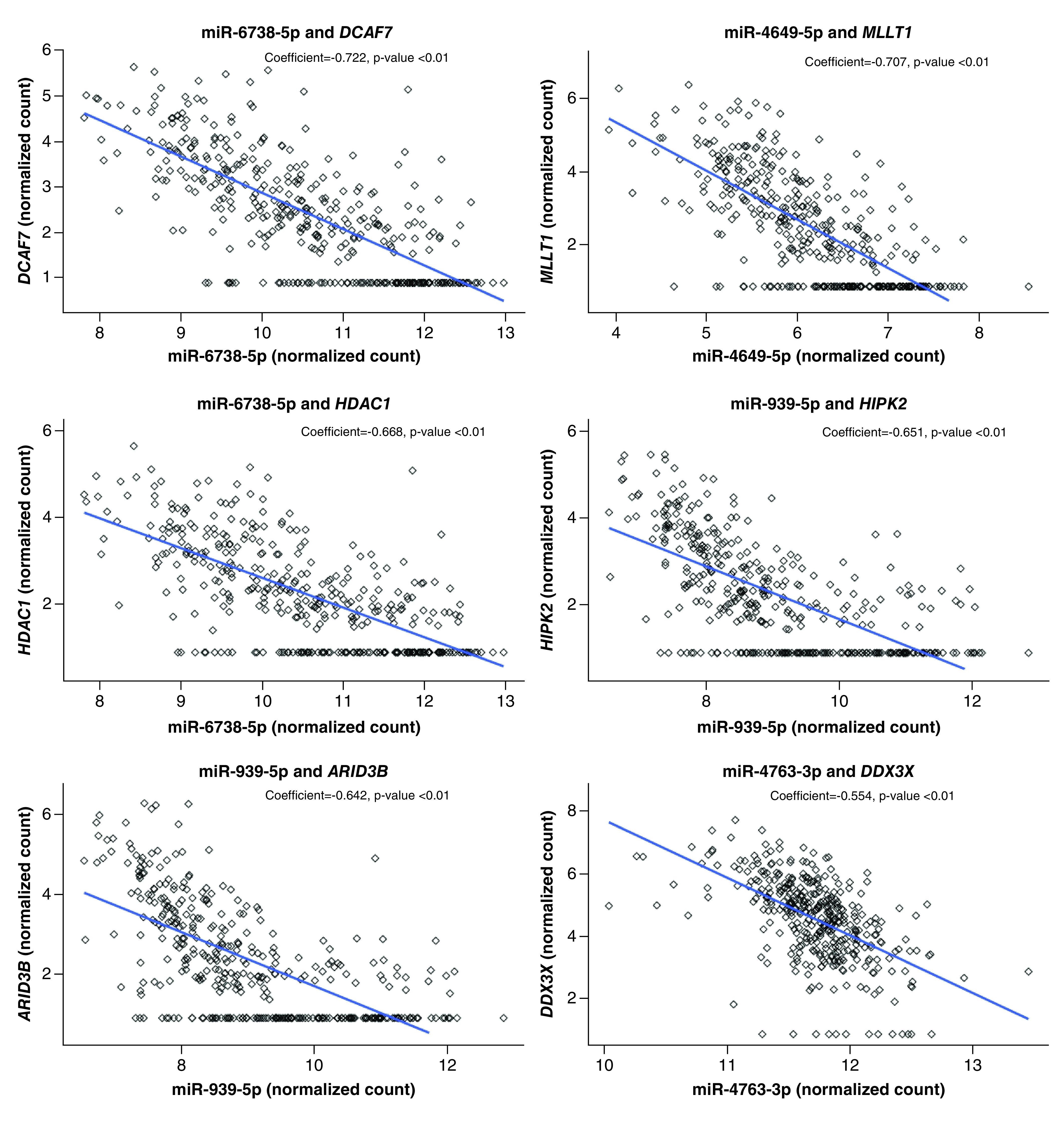
Variance stabilized counts (‘normalized count’) of miRNA are on the x-axis and variance stabilized counts (‘normalized count’) of mRNA are on the y-axis. Correlation coefficient and p value located at the top of the plot.
The 50 significantly correlated mRNA–miRNA expression pairs included 25 unique sexually dimorphic miRNAs and 29 unique sexually dimorphic mRNAs. Thus, these 29 mRNAs were likely under miRNA-control in the placenta as they showed sexually dimorphic expression and had expression levels significantly correlated to those of their targeting miRNAs. Of the 29 miRNA-controlled mRNAs, 23 displayed increased expression in female placenta, and six displayed increased expression in male placenta.
Identification of a female-specific interactome of mRNAs under miRNA control
To understand the biological implications of the mRNAs likely under miRNA control (n = 29), 23 mRNAs that were increased in the female placenta were queried for functional associations. mRNAs that displayed increased expression in the male placenta were not queried for functional association networks due to their low number (n = 6). Of the 23 mRNAs increased in female placenta that are likely under miRNA control, 15 had known functional interactions resulting in a 15-node interactome, including HDAC1, HDAC8, CDKN1B, PDGFRB and LRP6. CDKN1B, PDGFRB and LRP6 are also key genes in the hepatic fibrosis pathway, which was found to be an enriched canonical pathway among transcripts increased in the female placenta (Figure 4, Supplementary Table 3).
Figure 4. . Interactome of sexually dimorphic mRNAs (n = 23) increased in the female placenta that are under miRNA control.
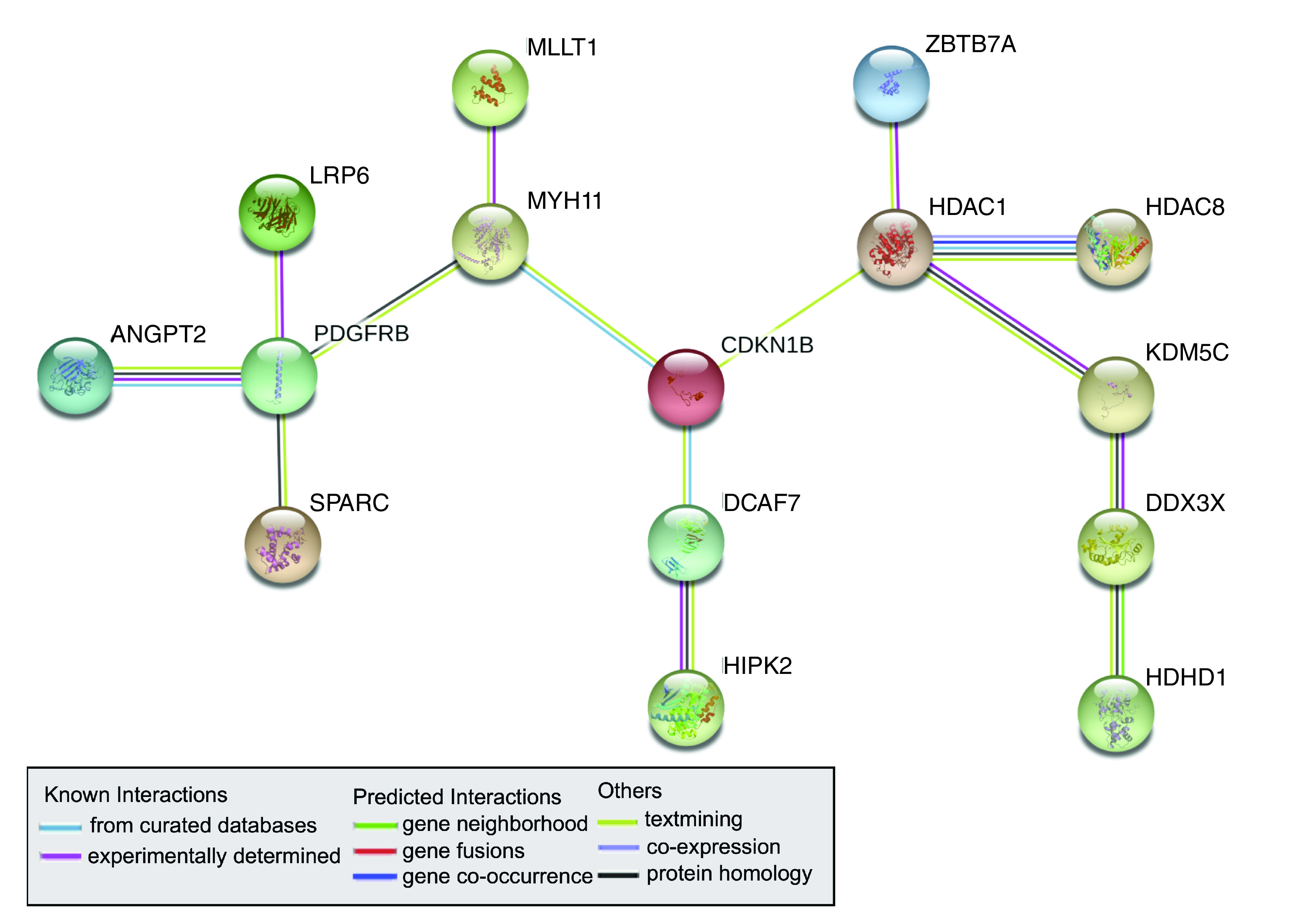
Only mRNAs with interactions are shown (n = 15). Interactome constructed using STRING database (v.11.0) showing gene–gene interactions representing joint contribution to a shared function.
Discussion
Sexual dimorphism, where female or male infants differ in their clinical responses/phenotypes, has been observed in both perinatal and child health outcomes [5–8,12,13]. A growing body of literature is linking these differences in child health outcomes to sex-specific functioning of the placenta [14–16]. Sexual dimorphism in the placenta at the level of gene expression has been observed, yet, the role of miRNAs in modulating these expression differences has not been examined [19–21,24]. In the present study, we set out to investigate the extent to which miRNAs are involved in the epigenetic control of sexually dimorphic gene expression in placentas collected from the ELGAN study. There were three major findings: first, the placenta exhibits sexually dimorphic gene expression with a large number of sexually dimorphic genes located on the X-chromosome. Second, miRNAs also exhibit sexual dimorphism in expression patterns and associations between miRNAs and mRNAs were observed. Specifically, we identified 29 sexually dimorphic mRNAs likely under the control of 25 sexually dimorphic miRNAs. Third, among the sexually dimorphic mRNAs under miRNA control, a gene cluster of 15 genes involved in histone modifications and cell-cycle progression that display increased expression in the female placenta relative to the male was identified. These data highlight the role of the placenta as a sexually dimorphic organ and miRNAs as mediators of this dimorphism.
Evaluation of genome-wide mRNA expression of the human placenta resulted in the identification of 128 genes with sexually dimorphic expression. In contrast to the current study where genome-wide RNA sequencing was used, prior studies of sexually dimorphic gene expression have used microarray-based approaches [19,24]. In support of the findings, many of the genes identified previously were also identified in the present study. Previous studies also have found that while a large percentage of sexually dimorphic genes are located on sex chromosomes, autosomes also contain significant numbers of sexually dimorphic genes [19,24]. A meta-analysis of 11 microarray datasets conducted by Buckberry et al., overall including n = 303 placenta samples, had similar results as those in the present study. Both studies identified that sexually dimorphic genes were enriched for VEGF and EIF signaling canonical pathways, that XIST is among the most female-biased genes and that EIF1AY is the most male-biased gene [24]. Notably, the VEGF pathway includes many genes implicated in placentation, angiogenesis and adverse pregnancy outcomes such as preeclampsia, which has been shown to vary in severity and incidence by fetal sex [8,77,78]. Additionally, the findings presented here are supported by data from Gonzalez et al.'s study of first trimester-derived placenta samples, which also found that XIST and HDAC8 were the among the most highly expressed genes in the female placenta while EIF1AY was among the most highly expressed genes in the male placenta [60].
In the present study, genes located on the X chromosome constitute a large number of the identified sexually dimorphic genes. Our data are supported by published studies where the same trend was observed [24,60]. The highest percentage of sexually dimorphic genes were found to be located on the X chromosome (n = 28, 21.9%) with 25 of these displaying higher expression in females and three displaying higher expression in males. X chromosome inactivation (XCI), in which one X chromosome is inactivated in females, is the process through which mammals compensate for potential unequal dosage of genes between males and females due to XY and XX sex chromosomes, respectively [15]. Interestingly, XCI can be escaped leading to hyper-expression (biallelic) in females compared with males of X chromosome genes. While the extent to which this process occurs in the human placenta remains controversial, our data, in which we observed that the majority (25 of 28 or 89.3%) of X-chromosome genes were hyper-expressed in the female placenta, are suggestive of the escape of XCI, which also was observed in Gonzales et al. [15,24,60,79].
When assessing miRNAs, sexual dimorphism was observed in a total of 59 miRNAs in the human placenta. It is known that several of these miRNAs play roles in the placenta or are associated with pregnancy outcomes such as preeclampsia (miR-23a, miR-495, miR-222, miR-223) and ectopic pregnancy (miR-323a) [34,35,63,68–70]. Of note, four of the sexually dimorphic miRNAs (miR-543, miR-495, miR-323a, miR-323b) are located in the C14MC miRNA cluster, a maternally-expressed imprinted gene cluster that is preferentially expressed in the placenta [65,80,81]. There is a growing interest in using C14MC and another miRNA cluster (C19MC) as potential biomarkers of pregnancy outcomes such as ectopic pregnancy, preeclampsia and preterm birth, the latter two of which demonstrate sexual dimorphic rates [7,8,65,66,68–70]. In contrast to the C14MC cluster, no sexually dimorphic miRNAs localized to the C19MC cluster. This could result from either a lack of sexual dimorphism among C19MC miRNAs or to lower global expression of C19MC miRNAs. In support of the latter hypothesis, we did find a lower percentage of C19MC miRNAs detected in the ELGAN samples than C14MC (73.9% versus 91.8%). This difference may be explained by the fact that C19MC is thought to increase in global expression over the course of pregnancy, and C14MC is thought to decrease [52]. Thus, the ELGAN placental samples queried in this study may have globally lower expression of C19MC than in term placenta, in which sexual dimorphism of C19MC expression may be more easily detected.
It is also of note that the largest number of sexually dimorphic miRNAs was also found on the X chromosome (n = 10, 16.9%) and five displayed higher expression in female placentas and five displayed increased expression in male placentas. This is suggestive that miRNA genes may also undergo escape from XCI. There is no consensus as to which miRNAs do or do not commonly escape XCI, or if miRNAs escape XCI at all [79]. However, approximately 35 miRNAs do exist in the introns of coding genes that are known to escape XCI, suggestive of their potential to escape [79]. Understanding the mechanisms behind differential expression of these X-linked miRNAs is of particular importance given their known roles in preeclampsia, specifically miR-223 and miR-222, both of which we found to be sexually dimorphic. These two miRNAs are known to target key genes involved in preeclampsia and are differentially expressed in preeclamptic pregnancies [34,35,77,79,82,83]. Interestingly, in the present study, we found that miR-223 was significantly correlated with a computationally-predicted gene target, TUBA1B, which has also been found to be differentially expressed in preeclamptic pregnancies [84]. In sum, sexually dimorphic expression patterns were observed in miRNAs known to play critical processes in the placenta and during pregnancy.
Interestingly, when the miRNA and mRNA data were integrated, evidence of regulation of sexually dimorphic mRNAs by miRNAs was found. Specifically, a set of 29 mRNAs were identified where expression was highly correlated to 25 miRNAs. In total, these represented 50 miRNA–mRNA expression pairs. Both positive and inverse associations between expression levels of miRNA and their computationally predicted targets were found. These findings highlight that the assumption regarding the generalized negative association between mRNA targets and miRNAs is not always observed [26]. Specifically, while an often anticipated relationship between miRNAs and their gene targets is an inverse correlation, positive correlations have been observed elsewhere as well [26]. Of particular interest, an interactome among 15 mRNAs upregulated in the female placenta and predicted to be under miRNA control was identified, including genes such as HDAC1 and HDAC8, involved in histone-based epigenetic control, and CDKN1B, involved in cell-cycle progression. Thus, through the integration of miRNA and mRNA expression data and in silico methods, epigenetic interactions between miRNA and gene expression can be observed.
While this study is novel in its evaluation of sexual dimorphism of placental miRNA expression in relation to gene expression, it is not without limitations. The placentas in the present study are derived from pregnancies ending before 28 weeks of gestational age. Placentas collected from pregnancies ending this early likely are pathologically different than those from later gestational ages. It is also noteworthy that the expression of placental miRNA and mRNA varies throughout gestation, thus both of these factors may contribute in part to some of the findings presented here [53,85,86]. For example, extremely preterm placentas are known to have greater inflammation and more maternal vascular underperfusion compared with placentas derived from other gestational ages, both of which are states that are likely driven in part by changes in gene expression and epigenetics [86]. Additionally, 7.8 and 22.0% of the identified sexually dimorphic mRNAs and miRNAs, respectively, are known to vary in global expression over gestational age [52–55]. Thus, mRNAs and miRNAs with higher levels of expression during the late second trimester to the early third trimester are more likely to have be identified in the present study due to an overall higher signal. Thus, these data may not be generalizable to placentas of all gestational ages. Additionally, it was beyond the scope of this analysis to investigate the relationship between the sex-based expression of miRNAs and mRNAs and sexually dimorphic placental functioning on later in life outcomes. This is a critical area of investigation and future research will aim to relate the findings in the present study with functional and clinical measures in ELGANs.
Future perspective
This research is among the first to identify sexual dimorphism in the expression of miRNAs in the human placenta and to integrate placental miRNA and mRNA data in order to identify the functional effects of miRNA expression. We hypothesize that these differences in baseline genomic and epigenomic patterning by sex may impact susceptibility to exogenous agents such as chemicals in the environment as well as drive later life dimorphism in health outcomes. With further investigation, sexual dimorphic epigenetic markers, such as miRNAs, may serve as biomarkers for susceptibility for diseases or health outcomes known to have different incidence by sex.
Summary points.
Sexual dimorphism of the placenta is thought to be a critical contributor to the sex-based differences observed in perinatal and later-in-life outcomes.
miRNAs are known to play critical roles during pregnancy and are key epigenetic regulators that may modulate sex-specific placental responses to the in utero environment.
To investigate sex-based differences in mRNA and miRNA expression in the placenta we evaluated genome-wide mRNA and miRNA expression in the extremely low gestational age newborns cohort (n = 382).
128 mRNAs and 59 miRNAs showed sexually dimorphic expression in the human placenta with some evidence of escape of X chromosome inactivation.
Through integration of mRNA and miRNA data, a final set of 25 miRNAs was identified that likely contribute to the sexual dimorphic mRNA expression in the placenta.
An interactome of 15 genes likely under miRNA control including HDAC1, HDAC8, and CDKN1B, was identified.
These data provide insight into a potential mechanism for sex differences in infant outcomes and responses to the perinatal environment at the molecular-level in the human placenta.
Footnotes
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.futuremedicine.com/doi/suppl/10.2217/epi-2020-0062
Financial & competing interests disclosure
This study was supported by grants from the National Institute of Neurological Disorders and Stroke (U01NS040069; R01NS040069), the Office of the NIH Director (UG3OD023348, UH3OD023348), the National Institute of Child Health and Human Development (R01HD092374), and the National Institute of Nursing Research (K23NR017898). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research
The study was approved by the Institutional Review Board at each of the 14 participating ELGAN sites and informed consent has been obtained from the participants involved. The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
Data sharing statement
Sequencing data have been submitted to National Center for Biotechnology Information (NCBI) Gene Expression Omnibus repository.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Ostlund BD, Conradt E, Crowell SE, Tyrka AR, Marsit CJ, Lester BM. Prenatal stress, fearfulness, and the epigenome: exploratory analysis of sex differences in DNA methylation of the glucocorticoid receptor gene. Front. Behav. Neurosci. 10, 147 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hodyl NA, Stark MJ, Osei-Kumah A, Clifton VL. Prenatal programming of the innate immune response following in utero exposure to inflammation: a sexually dimorphic process? Expert Rev. Clin. Immunol. 7(5), 579–592 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Broberg K, Ahmed S, Engstrom K. et al. Arsenic exposure in early pregnancy alters genome-wide DNA methylation in cord blood, particularly in boys. J. Dev. Orig. Health Dis. 5(4), 288–298 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drake AJ, O'shaughnessy PJ, Bhattacharya S. et al. In utero exposure to cigarette chemicals induces sex-specific disruption of one-carbon metabolism and DNA methylation in the human fetal liver. BMC Med. 13, 18 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenfeld CS, Trainor BC. Environmental health factors and sexually dimorphic differences in behavioral disruptions. Curr. Environ. Health Rep. 1(4), 287–301 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Di Renzo GC, Rosati A, Sarti RD, Cruciani L, Cutuli AM. Does fetal sex affect pregnancy outcome? Gend. Med. 4(1), 19–30 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Ingemarsson I. Gender aspects of preterm birth. BJOG 110(Suppl. 20), S34–S38 (2003). [DOI] [PubMed] [Google Scholar]
- 8.Elsmen E, Kallen K, Marsal K, Hellstrom-Westas L. Fetal gender and gestational-age-related incidence of pre-eclampsia. Acta Obstet. Gynecol. Scand. 85(11), 1285–1291 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Baron-Cohen S, Lombardo MV, Auyeung B, Ashwin E, Chakrabarti B, Knickmeyer R. Why are autism spectrum conditions more prevalent in males? PLoS Biol. 9(6), e1001081 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frondas-Chauty A, Simon L, Branger B. et al. Early growth and neurodevelopmental outcome in very preterm infants: impact of gender. Arch. Dis. Child Fetal Neonatal. Ed. 99(5), F366–F372 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Johnston MV, Hagberg H. Sex and the pathogenesis of cerebral palsy. Dev. Med. Child Neurol. 49(1), 74–78 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Stevenson DK, Verter J, Fanaroff AA. et al. Sex differences in outcomes of very low birthweight infants: the newborn male disadvantage. Arch. Dis. Child Fetal Neonatal Ed. 83(3), F182–F185 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eriksson JG, Kajantie E, Osmond C, Thornburg K, Barker DJ. Boys live dangerously in the womb. Am. J. Hum. Biol. 22(3), 330–335 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clifton VL. Review: Sex and the human placenta: mediating differential strategies of fetal growth and survival. Placenta 31(Suppl.), S33–S39 (2010). [DOI] [PubMed] [Google Scholar]; • Summarizes sexual dimorphism in the placenta and posits hypotheses underlying sexual differences in human development.
- 15.Gabory A, Roseboom TJ, Moore T, Moore LG, Junien C. Placental contribution to the origins of sexual dimorphism in health and diseases: sex chromosomes and epigenetics. Biol. Sex Differ. 4(1), 5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Outlines how epigenetics contribute to the sex-based differences observed in the placenta.
- 16.Rosenfeld CS. Sex-Specific placental responses in fetal development. Endocrinology 156(10), 3422–3434 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burton GJ, Fowden AL, Thornburg KL. Placental origins of chronic disease. Physiol. Rev. 96(4), 1509–1565 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Godfrey KM. The role of the placenta in fetal programming – a review. Placenta 23(Suppl. A), S20–S27 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Sood R, Zehnder JL, Druzin ML, Brown PO. Gene expression patterns in human placenta. Proc. Natl Acad. Sci. USA 103(14), 5478–5483 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Highlights sex differences at the level of the transcriptome.
- 20.Saif Z, Hodyl N, Stark M. et al. Expression of eight glucocorticoid receptor isoforms in the human preterm placenta vary with fetal sex and birthweight. Placenta 36(7), 723–730 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Pringle KG, Sykes SD. et al. Fetal sex affects expression of renin-angiotensin system components in term human decidua. Endocrinology 153(1), 462–468 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Brown Z, Schalekamp-Timmermans S, Tiemeier H, Hofman A, Jaddoe W, Steegers E. Fetal sex specific differences in human placentation: a prospective cohort study. Placenta 35(6), 359–364 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Saben J, Zhong Y, Mckelvey S. et al. A comprehensive analysis of the human placenta transcriptome. Placenta 35(2), 125–131 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buckberry S, Bianco-Miotto T, Bent SJ, Dekker GA, Roberts CT. Integrative transcriptome meta-analysis reveals widespread sex-biased gene expression at the human fetal-maternal interface. Mol. Hum. Reprod. 20(8), 810–819 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Combines 11 datasets from microarray analyses resulting in a total analysis of 303 placenta samples used here as a comparison of the results in the present study.
- 25.Martin E, Smeester L, Bommarito PA. et al. Sexual epigenetic dimorphism in the human placenta: implications for susceptibility during the prenatal period. Epigenomics 9(3), 267–278 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Demonstrates sexually dimorphic CpG methylation patterning in the placenta demonstrating sex-based differences in the placental epigenome.
- 26.O'brien J, Hayder H, Zayed Y, Peng C. Overview of MicroRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. (Lausanne) 9, 402 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vasudevan S. Posttranscriptional upregulation by microRNAs. Wiley Interdiscip. Rev. RNA 3(3), 311–330 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Xiao M, Li J, Li W. et al. MicroRNAs activate gene transcription epigenetically as an enhancer trigger. RNA Biol. 14(10), 1326–1334 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maccani MA, Padbury JF, Marsit CJ. miR-16 and miR-21 expression in the placenta is associated with fetal growth. PloS One 6(6), e21210 2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mayor-Lynn K, Toloubeydokhti T, Cruz AC, Chegini N. Expression profile of microRNAs and mRNAs in human placentas from pregnancies complicated by preeclampsia and preterm labor. Reprod. Sci. 18(1), 46–56 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hosseini MK, Gunel T, Gumusoglu E, Benian A, Aydinli K. MicroRNA expression profiling in placenta and maternal plasma in early pregnancy loss. Mol. Med. Rep. 17(4), 4941–4952 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maccani MA, Avissar-Whiting M, Banister CE, Mcgonnigal B, Padbury JF, Marsit CJ. Maternal cigarette smoking during pregnancy is associated with downregulation of miR-16, miR-21, and miR-146a in the placenta. Epigenetics 5(7), 583–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cai M, Kolluru GK, Ahmed A. Small molecule, big prospects: MicroRNA in pregnancy and its complications. J. Pregnancy 2017, 6972732 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Provides an overview of the importance of miRNAs during pregnancy and in the placenta underlying the importance of understanding sex-based difference in miRNA expression in the placenta.
- 34.Choi SY, Yun J, Lee OJ. et al. MicroRNA expression profiles in placenta with severe preeclampsia using a PNA-based microarray. Placenta 34(9), 799–804 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Hu Y, Li P, Hao S, Liu L, Zhao J, Hou Y. Differential expression of microRNAs in the placentae of Chinese patients with severe pre-eclampsia. Clin. Chem. Lab. Med. 47(8), 923–929 (2009). [DOI] [PubMed] [Google Scholar]
- 36.O'shea TM, Allred EN, Dammann O. et al. The ELGAN study of the brain and related disorders in extremely low gestational age newborns. Early Hum. Dev. 85(11), 719–725 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Addo KA, Bulka C, Dhingra R. et al. Acetaminophen use during pregnancy and DNA methylation in the placenta of the extremely low gestational age newborn (ELGAN) cohort. Environ. Epigenet. 5(2), 2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Onderdonk AB, Delaney ML, Dubois AM, Allred EN, Leviton A. Detection of bacteria in placental tissues obtained from extremely low gestational age neonates. Am. J. Obstet. Gynecol. 198(1), 110.e1-7 (2008). [DOI] [PubMed] [Google Scholar]
- 39.3′ mRNA-Seq Library Prep Ktt User Guide www.lexogen.com/wp-content/uploads/2015/11/015UG009V0211_QuantSeq-Illumina.pdf (2020).
- 40.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14(4), 417–419 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harrow J, Frankish A, Gonzalez JM. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22(9), 1760–1774 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. NAR 39, D152–D157 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Malnou EC, Umlauf D, Mouysset M, Cavaille J. Imprinted MicroRNA gene clusters in the evolution, development, and functions of mammalian placenta. Front. Genet. 9, 706 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rager JE, Auerbach SS, Chappell GA, Martin E, Thompson CM, Fry RC. Benchmark dose modeling estimates of the concentrations of inorganic arsenic that induce changes to the neonatal transcriptome, proteome, and epigenome in a pregnancy cohort. Chem. Res. Toxicol. 30(10), 1911–1920 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rager JE, Bailey KA, Smeester L. et al. Prenatal arsenic exposure and the epigenome: altered microRNAs associated with innate and adaptive immune signaling in newborn cord blood. Environ. Mol. Mutagen. 55(3), 196–2008 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rager JE, Moeller BC, Miller SK. et al. Formaldehyde-associated changes in microRNAs: tissue and temporal specificity in the rat nose, white blood cells, and bone marrow. Toxicol. Sci. 138(1), 36–46 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klaren WD, Ring C, Harris MA. et al. Identifying attributes that influence in vitro-to-in vivo concordance by comparing in vitro Tox21 bioactivity versus in vivo drugmatrix transcriptomic responses across 130 chemicals. Toxicol Sci. 167(1), 157–171 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rager JE, Ring CL, Fry RC. et al. High-throughput screening data interpretation in the context of in vivo transcriptomic responses to oral Cr(VI) exposure. Toxicol. Sci. 158(1), 199–212 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 11(10), R106 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15(12), 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289–300 (1995). [Google Scholar]
- 52.Morales-Prieto DM, Chaiwangyen W, Ospina-Prieto S. et al. MicroRNA expression profiles of trophoblastic cells. Placenta 33(9), 725–734 (2012). [DOI] [PubMed] [Google Scholar]
- 53.Gu Y, Sun J, Groome LJ, Wang Y. Differential miRNA expression profiles between the first and third trimester human placentas. Am. J. Physiol. Endocrinol. Metab. 304(8), E836–E843 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Winn VD, Haimov-Kochman R, Paquet AC. et al. Gene expression profiling of the human maternal-fetal interface reveals dramatic changes between midgestation and term. Endocrinology 148(3), 1059–1079 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Mikheev AM, Nabekura T, Kaddoumi A. et al. Profiling gene expression in human placentae of different gestational ages: an OPRU* network and UW SCOR† study. Reprod. Sci. 15(9), 866–877 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rager JE, Moeller BC, Doyle-Eisele M, Kracko D, Swenberg JA, Fry RC. Formaldehyde and epigenetic alterations: microRNA changes in the nasal epithelium of nonhuman primates. Environ. Health Perspect. 121(3), 339–344 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife 4, e05005 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Szklarczyk D, Gable AL, Lyon D. et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. NAR 47, D607–D613 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Von Mering C, Jensen LJ, Snel B. et al. STRING: known and predicted protein–protein associations, integrated and transferred across organisms. Nucleic Acids Res. 33(Database issue), D433–D437 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gonzalez TL, Sun T, Koeppel AF. et al. Sex differences in the late first trimester human placenta transcriptome. Biol. Sex. Differ. 9, 4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Demonstrates sex differences in the placenta transcriptome utilizing genome-wide RNA-sequencing on 37 late first trimester placentas.
- 61.Cai M, Kolluru GK, Ahmed A. et al. Small molecule, big prospects: MicroRNA in pregnancy and its complications. J. Pregnancy (2017). 10.1155/2017/6972732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang Q, Gu W-W, Gu Y. et al. Association of the peripheral blood levels of circulating microRNAs with both recurrent miscarriage and the outcomes of embryo transfer in an in vitro fertilization process. J. Transl. Med. 16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li H, Ge Q, Guo L, Lu Z. Maternal plasma miRNAs expression in preeclamptic pregnancies. Biomed. Res. Int. 2013, 970265 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li L, Hou A, Gao X. et al. Lentivirus-mediated miR-23a overexpression induces trophoblast cell apoptosis through inhibiting X-linked inhibitor of apoptosis. Biomed. Pharmacother. 94, 412–417 (2017). [DOI] [PubMed] [Google Scholar]
- 65.Morales-Prieto DM, Ospina-Prieto S, Chaiwangyen W, Schoenleben M, Markert UR. Pregnancy-associated miRNA-clusters. J. Reprod. Immunol. 97(1), 51–61 (2013). [DOI] [PubMed] [Google Scholar]
- 66.Wommack JC, Trzeciakowski JP, Miranda RC, Stowe RP, Ruiz RJ. Micro RNA clusters in maternal plasma are associated with preterm birth and infant outcomes. PloS One 13(6), (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang P, Wu Z, Ma C, Pan N, Wang Y, Yan L. Endometrial miR-543 is downregulated during the implantation window in women with endometriosis-related infertility. Reprod. Sci. 26(7), 900–908 (2019). [DOI] [PubMed] [Google Scholar]
- 68.Motawi TMK, Sabry D, Maurice NW, Rizka SM. Role of mesenchymal stem cells exosomes derived microRNAs; miR-136, miR-494 and miR-495 in pre-eclampsia diagnosis and evaluation. Arch. Biochem. Biophys. 659, 13–21 (2018). [DOI] [PubMed] [Google Scholar]
- 69.Kontomanolis EN, Kalagasidou S, Fasoulakis Z. MicroRNAs as potential serum biomarkers for early detection of ectopic pregnancy. Cureus 10(3), e2344 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhao Z, Zhao Q, Warrick J. et al. Circulating microRNA miR-323-3p as a biomarker of ectopic pregnancy. Clin.Chem. 58(5), 896–905 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Larocca J, Binder AM, Mcelrath TF, Michels KB. First-trimester urine concentrations of phthalate metabolites and phenols and placenta miRNA expression in a cohort of U.S. women. Environ. Health Perspect. 124(3), 380- 387 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hu E, Ding L, Miao H. et al. MiR-30a attenuates immunosuppressive functions of IL-1β-elicited mesenchymal stem cells via targeting TAB3. FEBS Lett. 589(24), 3899–3907 (2015). [DOI] [PubMed] [Google Scholar]
- 73.Dai Y, Qiu Z, Diao Z. et al. MicroRNA-155 inhibits proliferation and migration of human extravillous trophoblast derived HTR-8/SVneo cells via down-regulating cyclin D1. Placenta 33(10), 824–829 (2012). [DOI] [PubMed] [Google Scholar]
- 74.Cheng W, Liu T, Jiang F. et al. microRNA-155 regulates angiotensin II type 1 receptor expression in umbilical vein endothelial cells from severely pre-eclamptic pregnant women. Int. J. Mol. Med. 27(3), 393–399 (2011). [DOI] [PubMed] [Google Scholar]
- 75.Li X, Li C, Dong X, Gou W. MicroRNA-155 inhibits migration of trophoblast cells and contributes to the pathogenesis of severe preeclampsia by regulating endothelial nitric oxide synthase. Mol. Med. Rep. 10(1), 550–554 (2014). [DOI] [PubMed] [Google Scholar]
- 76.Zhang Y, Diao Z, Su L. et al. MicroRNA-155 contributes to preeclampsia by down-regulating CYR61. Am. J. Obstet. Gynecol. 202(5), 466.e461–e467 (2010). [DOI] [PubMed] [Google Scholar]
- 77.Andraweera PH, Dekker GA, Roberts CT. The vascular endothelial growth factor family in adverse pregnancy outcomes. Hum. Reprod. Update 18(4), 436–457 (2012). [DOI] [PubMed] [Google Scholar]
- 78.Muralimanoharan S, Maloyan A, Myatt L. Evidence of sexual dimorphism in the placental function with severe preeclampsia. Placenta 34(12), 1183–1189 2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Buckberry S, Bianco-Miotto T, Roberts CT. Imprinted and X-linked non-coding RNAs as potential regulators of human placental function. Epigenetics 9(1), 81–89 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Malnou EC, Umlauf D, Mouysset M, Cavaillé J. Imprinted MicroRNA gene clusters in the evolution, development, and functions of mammalian placenta. Front. Genet. 9(706), 2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fu G, Brkić J, Hayder H, Peng C. MicroRNAs in human placental development and pregnancy complications. Int. J. Mol. Sci. 14, 5519–5544 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Challis JR, Lockwood CJ, Myatt L, Norman JE, Strauss JF, 3rd, Petraglia F. Inflammation and pregnancy. Reprod. Sci. 16(2), 206–215 (2009). [DOI] [PubMed] [Google Scholar]
- 83.Levy R. The role of apoptosis in preeclampsia. Isr. Med. Assoc. J. 7(3), 178–181 (2005). [PubMed] [Google Scholar]
- 84.Ruan Y, Li Y, Liu Y, Zhou J, Wang X, Zhang W. Investigation of optimal pathways for preeclampsia using network-based guilt by association algorithm. Exp. Ther. Med. 4139–4143 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Uuskula L, Mannik J, Rull K. et al. Mid-gestational gene expression profile in placenta and link to pregnancy complications. PloS One 7(11), e49248 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dogan K, Salihoglu O, Sever N, Tombul T, Sari E, Ya L. Do placental histopathologic characteristics differ with gestational ages in preterm and term deliveries? Fetal Pediatr. Pathol. 34(6), 365–374 (2015). [DOI] [PubMed] [Google Scholar]