

Summary
The transcription factor EB (TFEB) has emerged as a master regulator of lysosomal biogenesis, exocytosis, and autophagy, promoting the clearance of substrates stored in cells. c-Abl is a tyrosine kinase that participates in cellular signaling in physiological and pathophysiological conditions. In this study, we explored the connection between c-Abl and TFEB. Here, we show that under pharmacological and genetic c-Abl inhibition, TFEB translocates into the nucleus promoting the expression of its target genes independently of its well-known regulator, mammalian target of rapamycin complex 1. Active c-Abl induces TFEB phosphorylation on tyrosine and the inhibition of this kinase promotes lysosomal biogenesis, autophagy, and exocytosis. c-Abl inhibition in Niemann-Pick type C (NPC) models, a neurodegenerative disease characterized by cholesterol accumulation in lysosomes, promotes a cholesterol-lowering effect in a TFEB-dependent manner. Thus, c-Abl is a TFEB regulator that mediates its tyrosine phosphorylation, and the inhibition of c-Abl activates TFEB promoting cholesterol clearance in NPC models.
Subject Areas: Biological Sciences, Molecular Biology, Cell Biology
Graphical Abstract
Highlights
-
•
c-Abl is a TFEB regulator that mediates its tyr phosphorylation
-
•
c-Abl inhibition promotes TFEB activity independently of mTORC1
-
•
c-Abl inhibition reduces cholesterol accumulation in NPC1 models
Biological Sciences; Molecular Biology; Cell Biology
Introduction
Lysosomes are essential organelles for the degradation of complex substrates and therefore necessary for maintaining cellular homeostasis. Moreover, in recent years the lysosome has emerged as a signaling organelle able of sensing its external environment and thereby regulating fundamental processes such as cellular clearance and autophagy (Fraldi et al., 2016; Settembre and Ballabio, 2014; Settembre et al., 2013). Therefore, lysosomes are involved in vital cell functions and their dysfunction, due to mutations in genes that encode lysosomal proteins, resulting in more than 50 different lysosomal storage disorders (LSDs) (Fraldi et al., 2016; Parenti et al., 2015).
A key element of lysosome signaling is the basic-helix-loop-helix leucine zipper transcription factor EB (TFEB). TFEB is the master regulator of the coordinated lysosomal expression and regulation (CLEAR) network, covering genes related to autophagy, exocytosis, and lysosomal biogenesis (Palmieri et al., 2011; Sardiello and Ballabio, 2009; Sardiello et al., 2009). TFEB overexpression or activation leads to increased number of lysosomes, autophagic flux, and lysosomal exocytosis, triggering cellular clearance of substrates stored in cells (Medina et al., 2011; Napolitano and Ballabio, 2016; Settembre and Ballabio, 2011; Settembre et al., 2011). This is relevant in diseases in which the degradative capacity of the cells is compromised. In fact, it has been reported that TFEB overexpression promotes cellular clearance ameliorating the phenotypes in a variety of neurodegenerative diseases such as Alzheimer's, Parkinson's, and Huntington's (Ballabio, 2016; Decressac et al., 2013; Napolitano and Ballabio, 2016; Polito et al., 2014; Sardiello et al., 2009; Tsunemi et al., 2012; Xiao et al., 2014; Xiao et al., 2015).
Due to the large number of diseases where lysosomal function is compromised, recent research has focused on elucidating the mechanisms by which TFEB is regulated. TFEB activity is regulated by phosphorylation, mainly by the mammalian target of rapamycin complex 1 (mTORC1), a serine/threonine kinase that controls cell growth and negatively regulates autophagy (Martina et al., 2012; Roczniak-Ferguson et al., 2012; Settembre et al., 2012, 2013). In the presence of nutrients, mTORC1 basally phosphorylates TFEB on serine 142 (S142) (Settembre et al., 2011, 2012) and serine 211 (S211) (Martina et al., 2012; Roczniak-Ferguson et al., 2012), which are in turn dephosphorylated by calcineurin phosphatase in the absence of nutrients (Medina et al., 2015). TFEB phosphorylated at S211 interacts with the cytosolic chaperone 14-3-3 and is retained in the cytoplasm (Martina et al., 2012; Roczniak-Ferguson et al., 2012). In the absence of nutrients, mTORC1 is inactive, and dephosphorylated TFEB translocates into the nucleus promoting the expression of the CLEAR gene network. More recently, other serine/threonine kinases such as protein kinase B (Akt) and protein kinase C have been described as regulators of TFEB activation (Li et al., 2016; Palmieri et al., 2017). Nevertheless, the mechanism of TFEB regulation under different stress conditions, such as those present in different storage diseases, are still not well understood (Martina et al., 2016; Raben and Puertollano, 2016). Interestingly, the possible TFEB regulation by tyrosine phosphorylation has not been explored.
It has been previously described that tyrosine kinase c-Abl inhibition promotes the clearance of alpha-synuclein in models of Parkinson's disease (Hebron et al., 2013). Indeed, our results and others show that inhibiting c-Abl seems to be a good therapeutic option for other neurodegenerative disorders such as Alzheimer's and ALS, where c-Abl is aberrantly activated and the lysosomal function is also compromised (Cancino et al., 2008, 2011; Estrada et al., 2011, 2016; Hebron et al., 2013; Imam et al., 2013; Imamura et al., 2017; Karuppagounder et al., 2014; Katsumata et al., 2012; Pagan et al., 2016; Rojas et al., 2015; Tanabe et al., 2014; Vargas et al., 2018). Interestingly, recent studies suggest a possible connection between c-Abl and TFEB (Gutknecht et al., 2015; Lawana et al., 2017; Ren et al., 2018). Indeed, Gutknecht et al., (2015), have described that c-Abl inhibition induced nuclear translocation of MITF, a transcription factor that is closely related to TFEB, and the up-regulation of its target genes. Similarly, Ren et al., (2018), showed that c-Abl inhibition promoted TFEB nuclear translocation in neurons under MPP + treatment, through a mechanism involving GSK3β. However, it is not known whether TFEB is a direct target of c-Abl and if is phosphorylated in tyrosine by this kinase. Moreover, the effects of inhibiting c-Abl on cholesterol accumulation have not been studied.
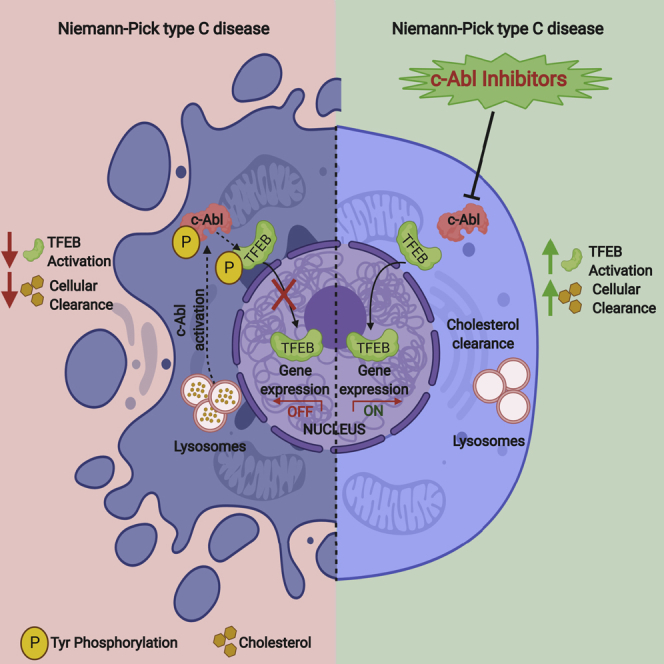
The goal of this study was to evaluate if c-Abl controls cellular clearance through TFEB and the implication of this cell mechanism on alleviating cholesterol storage accumulation in Niemann-Pick type C (NPC) disease. Our data show that c-Abl inhibition promotes increased TFEB nuclear localization — independent of mTORC1 —, and the expression of TFEB target genes. This effect was associated with increased autophagy, lysosome number, and exocytosis. We also found that c-Abl phosphorylates TFEB on tyrosine. We demonstrated that several c-Abl inhibitors promote a reduction in cholesterol accumulation in both in vitro and in vivo NPC models. This increased clearance, TFEB-dependent, reveals the relevance of the axis between c-Abl and TFEB. Our results position the c-Abl/TFEB signaling as a therapeutic target for the treatment of patients with diseases in which the lysosomes are compromised.
Results
c-Abl Inhibition Promotes TFEB Activity
Based on previous data connecting c-Abl with autophagy, lysosomal biogenesis, and cellular clearance, we hypothesized that the tyrosine kinase c-Abl regulates TFEB nuclear translocation (Ertmer et al., 2007; Hebron et al., 2013).
To evaluate this hypothesis, we first examined TFEB-green fluorescent protein (GFP) nuclear localization in HeLa TFEB-GFP cells treated with different c-Abl inhibitors. Imatinib and nilotinib are classic first- and second-generation c-Abl inhibitors that binds to the ATP binding domain. Dasatinib is a tyrosine kinase inhibitor, also used as an c-Abl inhibitor, but it is less specific; the three are FDA-approved drugs (Capdeville et al., 2002; Druker et al., 1996; Hantschel et al., 2008; Maekawa et al., 2007). GNF-2 and its analog GNF-5 are allosteric inhibitors of c-Abl (Iacob et al., 2011). We measured TFEB-GFP nuclear localization using a high-content nuclear translocation assay in a confocal automated microscope. As a positive control for TFEB nuclear translocation, we used 0.3 μM Torin1, an mTORC1 inhibitor, for 3 h. Figures 1A and 1B shows that the different concentrations of c-Abl inhibitors as well as Torin1, promote a significantly increase in TFEB-GFP nuclear signal compared to control conditions (dimethyl sulfoxide [DMSO]), being imatinib and nilotinib the most effective at lower concentrations and noticing significant increases in the nucleus/cytoplasm intensity ratio at 3.33 and 1.11 μM, respectively. We observed the same result at 6 h, 12 hr, and 24 hr (Figure S1A). Treatment with 10 μM imatinib for 3 h promoted TFEB nuclear translocation in HeLa TFEB-GFP cells measured by nucleus cytoplasm fractionation (Figure 1C), confirming our analysis of the high-content nuclear translocation assay. In addition, we tested imatinib in HT22 (a cell line derived from mice hippocampal neurons) and in HEK293 cells (derived from human embryonic kidney) that had been transiently transfected with TFEB-GFP. As expected, we observed that imatinib promoted TFEB nuclear localization (Figures S1B and S1C). These experiments show an increase in TFEB-GFP nuclear translocation when c-Abl is inhibited by using inhibitors that have different inhibition mechanisms.
Figure 1.

c-Abl Inhibition Increases TFEB Nuclear Translocation and Activity
HeLa TFEB-GFP cells were treated with DMSO (control), Torin1 0.3 μM (positive control) and c-Abl inhibitors at different concentrations for 3 h. Then, the cells were fixed and stained with DAPI.
(A) Representative images of the TFEB-GFP translocation assay obtained by confocal automated microscopy and. Scale bars, 10μM.
(B) graph of the ratio value resulting from the average intensity of nuclear TFEB-GFP fluorescence divided by the average cytosolic intensity of TFEB-GFP fluorescence. Black bars represent Torin1 treatment (positive control). Differences are statistically significant compared to control conditions (DMSO). For each condition, 450–800 cells were analyzed (7 images per sample); n = 4 biological independent samples.
(C) Western blot and quantification of TFEB-GFP normalized with histone 3 (H3), in a nuclear/cytoplasmic fractionation of HeLa TFEB-GFP cells treated with imatinib 10μM for 3 h. n = 3 independent experiments.
(D) Representative Western blot of endogenous TFEB in a nuclear/cytoplasmic fractionation assay of control human fibroblast treated with imatinib 10μM for 24 hr n = 3 independent experiments.
(E) Representative images of endogenous TFEB in HT22 cells treated with imatinib 10 μM for 24 hr n = 3 independent experiments. Scale bars, 10 μM.
(F) HeLa TFEB-GFP cells were treated with a scramble siRNA and c-Abl siRNA for 72 hr. Representative images of the TFEB-GFP translocation assay and quantification. For each condition 3,000–5,000 cells were analyzed (16 images per sample) n = 4 biological independent samples, Scale bars, 10 μM.
(G) The graph represents q-PCR analysis of c-Abl mRNA levels in HeLa cells treated with the scramble siRNA and siRNA against c-Abl for 24 hr n = 3 independent experiments. (H) The Western blot confirms the reduction of c-Abl protein levels in HeLa cells treated with siRNA against c-Abl for 24 hr n = 3 independent experiments.
(I) The graph shows q-PCR analysis of mRNA levels of different TFEB target genes in HeLa cells treated with the scramble siRNA and a siRNA against c-Abl. n = 3 independent experiments. Statistical analysis with one-way ANOVA followed by Tukey's post-test and Student's t-tests when comparing two experimental groups. ∗p < 0.05, ∗∗p < 0.01,∗∗∗p < 0.001. Data represent mean ± SEM.
Next, we evaluated the effect of c-Abl inhibition on endogenous TFEB. We treated wild type human fibroblasts with imatinib 10 μM for 24 hr and then performed a nucleus cytoplasm fractionation assay. The c-Abl inhibitor clearly increased the levels of endogenous TFEB in the nucleus (Figure 1D). In addition, we performed immunofluorescence experiments to explore the effect of c-Abl inhibition on TFEB nuclear translocation. In the HT22 cell line treated with imatinib for 24 hr, we observed that c-Abl inhibitor increased TFEB nuclear signal (Figure 1E). Consistent with these results, we found an increase on the mRNA levels of TFEB target genes in wild-type (WT) human fibroblasts treated with the c-Abl inhibitors, imatinib and nilotinib, for 24 hr (Figure S1D). Therefore, we suggest that c-Abl inhibition promotes endogenous TFEB nuclear translocation.
To confirm that the pharmacological inhibition of c-Abl mediates the increase in TFEB nuclear localization, we used two strategies. On one hand, we used HeLa c-Abl KO cells that we obtained using the CRISPR/Cas9 tool. We analyzed endogenous TFEB localization by immunofluorescence. HeLa c-Abl KO cells showed increased endogenous TFEB nuclear localization compared with HeLa WT cells (c-Abl WT) (Figure S1E). On the other hand, we used a pool of siRNA against c-Abl and transfected HeLa TFEB-GFP cells for 72 hr and then analyzed TFEB nuclear localization using a high-content nuclear translocation assay. The cells treated with the siRNAs against c-Abl show increased TFEB-GFP nuclear localization compared with cells transfected with a scramble siRNA (Figure 1F), demonstrating that c-Abl depletion promotes TFEB nuclear translocation. The effectiveness of the siRNAs was tested following the levels of c-Abl mRNA and protein (Figures 1G and 1H). Importantly, HeLa cells treated with siRNAs against c-Abl, showed an increased mRNA levels for several TFEB target genes measured by q-PCR (Figure 1I).
Altogether, these experiments demonstrate that under basal conditions c-Abl inhibition promotes TFEB nuclear localization inducing its transcriptional function.
To corroborate that the inhibition of c-Abl activates TFEB, we then analyzed some of the cellular readouts of TFEB activation (Settembre et al., 2013; Settembre and Medina, 2015).
On the one hand, we analyzed the effect of c-Abl inhibition on lysosomes using WT HeLa (control) and HeLa TFEB-knock out (TFEB-KO) cells, in which, as expected, TFEB is not detected by Western blot (Figure 2A). We found increased Lamp1 levels by Western blot in WT HeLa cells treated with imatinib 10 μM for 24 hr (Figure 2B). Nevertheless, we did not observe this increase in Lamp1 levels in TFEB-KO cells treated with imatinib (Figure 2B). In addition, we found an increase in the LysoTracker red marker using FACs in both HeLa cells and in WT human fibroblast treated with imatinib (Figures 2C and 2D, respectively). Concordantly, we observed increased Lamp1 staining by immunofluorescence in human fibroblasts treated with both imatinib 10 μM for 24 hr and with a siRNA against c-Abl (Figure 2E).
Figure 2.

c-Abl Inhibition Increases Lysotracker Positive Organelles and Lysosomal Protein Levels
(A) Representative Western blot of endogenous TFEB in HeLa cells (control), HeLa TFEB-KO cells (TFEB-KO), and HeLa TFEB-GFP cells (TFEB-GFP). GAPDH was used as loading control.
(B) Representative Western blot and quantification of HeLa cells treated with imatinib 10μM for 24 hr using a Lamp1 antibody. n = 3 independent experiments.
(C) Quantitative flow cytometry analysis of lysotracker in HeLa cells treated with imatinib 10μM for 24 hr n = 10,000 cells per conditions.
(D) Quantitative flow cytometry analysis of lysotracker in the human wild type fibroblasts treated with imatinib 10μM for 24 hr n = 10,000 cells per conditions.
(E) Representative immunofluorescence images of lysosomes using Lamp1 antibody in human fibroblast treated with imatinib 10μM for 24 hr, or transfected with a scramble siRNA or a siRNA against c-Abl for 48 hr n = 3 independent experiments. Scale bars, 50 μM.
(F) Representative immunofluorescence images of lysosomes attached to the plasma membrane using the antibody Lamp1-DB4 in HeLa cells treated with imatinib 10μM for 24 hr n = 3 independent experiments. Scale bars, 10 μM.
(G) H4 cells were treated with DMSO (control), Torin1 0.3 μM (positive control) and c-Abl inhibitor imatinib 10 μM for 3 h. The cells were then fixed and stained with DAPI. Representative confocal microscopy images. Scale bars, 10 μM.
(H) Graph of the autophago-lysosome number (spots negative for GFP and positive for RFP). For each condition 450–800 cells were analyzed (19 images per sample). n = 3 biological independent samples. Statistical analysis with one-way ANOVA followed by Tukey's post test and Student's t-tests when comparing two experimental groups. ∗p < 0.05, ∗∗p < 0.01,∗∗∗p < 0.001. Data represent mean ± SEM.
Next, we analyzed the effect of inhibiting c-Abl on lysosome distribution. We treated HeLa cells with imatinib for 24 hr and used an antibody that recognizes the luminal domain of Lamp1. We found that the c-Abl inhibitor promoted an increase in the signal of the antibody in the plasma membrane, suggesting an increase in lysosomes that are attached to this membrane and in the lysosomal exocytic process (Figure 2F).
To measure autophagic flow, we used the stable H4 GFP-red fluorescent protein (mRFP)-LC3 cell line (Figures 2G and 2H). Under conditions that induce the fusion between lysosomes and autophagosomes, the lower pH of lysosomes degrades the GFP signal, being the mRFP-LC3 signal a marker of autophago-lysosome formation. We treated these cells with imatinib for 3 h, and then we quantified the induction of autophagy flux using confocal automated microscopy (Figures 2G and 2H). We observed a trend to increase in the number of autophago-lysosome formation (GFP-negative and mRFP-positive) in the imatinib treated cells as well as in the Torin1 positive control, compared with control conditions (Figures 2G and 2H), suggesting that c-Abl inhibition induces the autophagic flux in these cells.
These results show that c-Abl inhibition increases lysotracker positive organelles and lysosomal protein levels in cells in a TFEB-dependent manner. They also suggest that c-Abl inhibition induces the autophagy flux and increases lysosomal exocytosis. All these effects correlate with TFEB nuclear translocation and activation.
c-Abl Inhibition Activates TFEB Independently of mTORC1 Activity
The regulation of TFEB nuclear localization mediated by mTORC1 and serine phosphorylation is well characterized. Active mTORC1 phosphorylates TFEB on S142 and S211, promoting TFEB cytoplasmic retention by chaperone 14-3-3 (Martina et al., 2012; Roczniak-Ferguson et al., 2012). Conversely, TFEB serine de-phosphorylation promotes its translocation into the nucleus. Therefore, our next question was if TFEB nuclear translocation mediated by inhibition of c-Abl is dependent or independent on mTORC1.
To elucidate if the effect of c-Abl on TFEB depends on mTORC1, we first analyzed the phosphorylation status of endogenous TFEB in cells treated with c-Abl inhibitors by Western blot. As expected, Torin1 promoted an electrophoretic shift on TFEB compared to control conditions. This electrophoretic mobility is due to its de-phosphorylation on S142 and/or S211 as a result of the inhibition of mTORC1 (Martina et al., 2012; Martina and Puertollano, 2013; Roczniak-Ferguson et al., 2012; Settembre et al., 2012) (Figure 3A). Concordantly, in cells under nutrient deprivation induced by starvation media (STV), a condition under which mTORC1 is inhibited, we observed the same electrophoretic shift of the endogenous TFEB compared to control condition (Figure 3A). Interestingly, imatinib, which promoted TFEB nuclear translocation, also induced an electrophoretic shift of the endogenous TFEB compared with the control condition (Figure 3A). Nevertheless, the electrophoretic shift promoted by the inhibition of c-Abl is smaller compared to the conditions in which mTORC1 is inhibited (Figure 3A). Similar results were observed in HeLa TFEB-GFP cells treated with imatinib, in which the electrophoretic shift is induced and there is not a change in TFEB-GFP levels (Figure S2A). These results suggest that c-Abl inhibition promotes TFEB translocation into the nucleus in spite of a TFEB de-phosphorylation status different from that generated by mTORC1 inhibition.
Figure 3.

c-Abl Regulates TFEB Independent of mTORC1 Activity
(A) Western blots of endogenous TFEB in HeLa cells treated with imatinib 10μM for 3 h. Torin1 0.3μM and starvation media (STV) for 3 h were used as a positive control.
(B) Representative Western blot and quantification of TFEB phosphorylated on S142 normalized against GAPDH in HeLa TFEB-GFP cells treated with imatinib 10μM for 3 h and siRNA c-Abl for 48 hr. STV media for 3 h was used as positive control. n = 3 independent experiments.
(C) Representative Western blot and quantification using the 14-3-3 antibody that binds to phosphorylated TFEB on S211. For immunoprecipitated GFP from HeLa TFEB-GFP, cells treated with imatinib 10 μM and Torin1 0.3 μM for 3 h. n = 3 independent experiments.
(D) Representative Western blot and quantification of phospho p70-S6K normalized against GAPDH in HeLa cells treated with imatinib and nilotinib 10μM for 3 h. Torin1 0.3μM and STV media treatment for 3 h were used as positive controls. n = 3 independent experiments. Scale bars, 10 μM.
(E) Representative confocal microscopy images and quantification of TSC2 KO cells transfected with the TFEB-GFP plasmid. Cells were treated with imatinib 10μM for 3 h and with Torin1 0.3μM for 3 h as a positive control. n = 40 cells per conditions. Scale bars, 10 μM.
(F) Representative images and quantification of percentage of nuclear TFEB-GFP in HeLa TFEB-GFP cells synchronized with STV media for 1 h. Then, cells were treated with imatinib 10μM for 1 h and Torin1 0.3 μM for 1 h as a positive control and re-fed with normal media plus imatinib and Torin1 for 2 h. n = 3 independent experiments. Scale bars, 10 μM. Statistical analysis with one-way ANOVA followed by Tukey's post-test. ∗p < 0.05, ∗∗p < 0.01,∗∗∗p < 0.001. Data represent mean ± SEM.
Next, we followed the effect of c-Abl inhibition on the levels of TFEB phosphorylated on S142 (TFEB p-S142) by Western blot. As expected, in TFEB-GFP HeLa cells treated with STV, TFEB p-S142 levels were reduced (Figure 3B). We obtained the same results when cells were treated with Torin1 (Figure S2B). However, we did not observe decreased levels of TFEB p-S142 when we treated the cells with imatinib or used a siRNA against c-Abl, indicating that c-Abl inhibition promotes TFEB nuclear translocation independent of S142 phosphorylation levels (Figure 3B). In addition, we measured the levels of S211 phosphorylated TFEB (TFEB p-S211) in HeLa TFEB-GFP cells. We accomplished this by following the binding of the 14-3-3 chaperone to this phosphorylated residue using a specific antibody (Roczniak-Ferguson et al., 2012). Unlike S142 phosphorylation, we observed a decrease in S211 phosphorylation in both Torin1 as well as in imatinib treated TFEB-GFP cells (Figure 3C). Due to TFEB de-phosphorylation on Ser211 is mainly regulated by the phosphatase calcineurin (Medina et al., 2015), we measure TFEB-GFP nuclear translocation in conditions that calcineurin and c-Abl were inhibited. We did not observe a reduction in the TFEB-GFP nuclear translocation induced by the c-Abl inhibitors imatinib or nilotinib, under pharmacological and genetic inhibition of calcineurin, suggesting that this TFEB nuclear translocation induction does not require the calcineurin phosphatase activity (Figures S2C and S2D).
In addition, we measured the levels of TFEB phosphorylated on Serine 138 (S138) since it was recently shown that phosphorylation in this position promotes TFEB nuclear export (Li et al., 2018; Napolitano et al., 2018). As expected, we observed that mTORC1 inhibition promotes full de-phosphorylation in Ser138 (Figure S2E). Interestingly, although c-Abl inhibition promotes only a slight decrease in TFEB Ser138 phosphorylation levels (Figure S2C), TFEB was localized mainly in the nucleus.
These results suggest that c-Abl inhibition with imatinib, promotes a partial and different TFEB de-phosphorylation status compared with the inhibition of mTORC1; S142 phosphorylation is not affected, S138 phosphorylation is partially decreased, while S211 phosphorylation decreases. These results suggest that c-Abl controls TFEB nuclear translocation through a different mechanism than that mediated by mTORC1.
To further analyze if TFEB nuclear translocation induced by inhibition of c-Abl is independent on mTORC1 inhibition, we measured the phosphorylation levels of p70 S6 kinase (phospho-S6K), a well-known mTORC1 target protein. Interestingly, we observed that treatment with the c-Abl inhibitors for 3 h did not change the phosphorylation status of S6K (phospho-S6K) in HeLa cells and in HeLa cells that overexpressed TFEB-GFP (Figures 3D and S2F, respectively). We obtained similar results when HeLa cells were treated with imatinib for 3 h, 6 h and 24 h (Figure S2G), confirming that c-Abl is not modulating mTORC1 activity.
Altogether, these results suggest that c-Abl inhibition contributes to TFEB nuclear translocation by a mechanism that neither involves S142 phosphorylation nor mTORC1 activity.
To corroborate this idea, we evaluated the effect of c-Abl inhibition in cells that have a constitutively active mTORC1, as they are deficient in the intrinsic TSC2 inhibitor of mTORC1. Because active mTORC1 retains TFEB in the cytoplasm, we used this cell line to analyze the TFEB nuclear translocation using a c-Abl inhibitor. We found that imatinib treatment induced TFEB-GFP nuclear translocation in the TCS2 KO cells, as well as with Torin 1 treatment, in spite of mTORC1 over-activation (Figure 3E). To analyze whether c-Abl inhibition also prevents the return of TFEB from the nucleus to the cytosol when nutrients conditions are changed, we synchronized cells using starvation media and then re-fed them in the presence of imatinib and analyzed the intracellular localization of TFEB. We found that imatinib, as well as Torin 1, prevented TFEB-GFP cytoplasmic return in synchronized cells using starvation media and re-fed, whereas in control conditions TFEB returned to the cytosol (Figure 3F).
All together our experiments strongly suggest that c-Abl inhibition promotes TFEB nuclear translocation in spite of mTORC1 over-activation, revealing an mTORC1-independent TFEB regulatory pathway mediated by the c-Abl tyrosine kinase.
c-Abl Phosphorylates TFEB on Tyrosine
Because our results show that inhibition of the tyrosine kinase c-Abl promotes TFEB activation independent of its classic regulator mTORC1, we assessed if this tyrosine kinase could phosphorylate TFEB.
To this end, we used a plasmid encoding a fusion c-Abl-ERT2 protein that contains the c-Abl kinase domain fused to the ligand-binding domain of the estrogen receptor (ER), which binds to tamoxifen with high affinity. When this fusion protein is expressed in cells, it associates with HSP90 chaperones. Upon tamoxifen treatment, the c-Abl-ERT2 protein dissociates from HSP90 and its activity is de-repressed (Figure 4A). As a control, we used a plasmid containing a c-Abl kinase death-ERT2 (c-Abl-ERT2 KD) fusion protein. We overexpressed this c-Abl-ERT2 construction in HeLa TFEB-GFP cells. Remarkably, the TFEB-GFP protein immunoprecipitated from tamoxifen treated cells, in which c-Abl-ERT2 is active, showed a clear band corresponding in size to TFEB-GFP phosphorylated in tyrosine (Figure 4B). This band, corresponding to tyrosine phosphorylated TFEB, was not observed in cells transfected with the control plasmid containing the c-Abl ERT2 KD fusion protein and treated with tamoxifen (Figure 4B). In addition, we treated HeLa TFEB-Flag cells (Figure S3A) and Hela TFEB-GFP cells (Figure S3B) with a c-Abl activator, DPH (Yang et al., 2011), and the immunoprecipitated TFEB showed a tyrosine phosphorylation band, suggesting that the endogenous c-Abl activation promotes the TFEB tyrosine phosphorylation (Figure S3C).
Figure 4.

Active c-Abl Phosphorylates TFEB on Tyrosine
(A) Schematic diagram showing that c-Abl-ERT2 under tamoxifen treatment phosphorylates in tyrosine (P-Tyr) its target proteins.
(B) Representative Western blot of HeLa TFEB-GFP cells transfected with c-Abl-ERT2 and c-Abl-ERT KD plasmids and treated with Tamoxifen for 8 h. GFP was immunoprecipitated using beads-anti-GFP and then used a anti-phospho-tyrosine antibody. n = 3 independent experiments.
(C) Autoradiography of an in vitro phosphorylation assay. TFEB-Flag IP was incubated with human recombinant c-Abl active and ATP-γ-32P for 0 h, 0.5 hr, 1 h, and 2 h. ATP-γ-32P incubation for 2 h without recombinant c-Abl active is showed as control (ct).
(D) Immunoprecipitated (IP) CRKII was incubated with human recombinant c-Abl active and ATP-γ-32P for 2 h.
(E) Tyrosine 75 (Y75) and Y173 are highly conserved across different species and are included in the c-Abl phosphorylation motif YX1-5P, which included the tyrosine (Y) and after one to five different amino acids, it recognize a proline (P).
(F) Representative confocal microscopy images and quantification of subcellular localization of TFEB-GFP mutants and control plasmids. n = 3 independent experiments. Scale bars, 10 μM.
(G) Western blot of HeLa cells transfected with a TFEB-GFP wild type plasmid or with plasmids carrying the Y75 or Y173 mutations. Western blot membranes were incubated with a specific antibody against S211, S138, and S142.
Next, to evaluate if c-Abl can directly phosphorylate TFEB we performed an in vitro phosphorylation assay using TFEB-Flag, recombinant human active c-Abl and ATP-γ-32P. Importantly, we observed the incorporation of radioactivity in TFEB-Flag in a time dependent manner (Figure 4C). As a positive control for c-Abl dependent phosphorylation, we used the well-known c-Abl target CRKII (Figure 4D).
These experiments show that TFEB is phosphorylated on tyrosine by c-Abl kinase.
Then, to analyze which tyrosine on TFEB could be phosphorylated by c-Abl, we performed an in-silico analysis on TFEB. We used the platforms netphos 2.0. to search for tyrosine consensus phosphorylation sites (Blom et al., 1999) and GPS 2.1.2 to find possible c-Abl consensus phosphorylation sites YX1-5P (Cujec et al., 2002; Xue et al., 2011). We identified two tyrosine residues with the highest probability to be phosphorylated by c-Abl: tyrosine 75 and 173 (Y75 and Y173) (Figure 4E). Remarkably, these two tyrosines identified on TFEB are highly conserved in different species (Figure 4E). Then, we carried out site-directed mutagenesis of the TFEB-GFP plasmid in order to change Y75 and Y173 to phenylalanine (F), and analyzed the subcellular localization of the Y75F and Y173F TFEB-GFP mutant proteins. The localization of TFEB-GFP when is transfected into cells is usually cytoplasmic. Interestingly, we observed that the Y173F TFEB-GFP mutant protein has a constitutive nuclear localization compared to the WT and Y75F TFEB-GFP proteins (Figure 4F). Next, we performed in vitro phosphorylation assays using active c-Abl and WT and mutant TFEBs. Even though, we expected decreased incorporation of radioactivity in TFEB mutants, the Y173F TFEB-GFP and Y75F TFEB-GFP proteins incorporated radioactivity at levels similar to that of the wild type TFEB-GFP, probably due to basal phosphorylation of other tyrosines. Slightly less radioactivity was detected in the Y75F TFEB-GFP protein. However, the construct Y75F TFEB-GFP showed lower expression levels in cells transfected for the in vitro phosphorylation assays (Figure S3D).
Then, we analyzed the different serines phosphorylation involved in the TFEB nuclear localization on the mutant TFEBs by Western blot using specific antibodies against phosphorylated TFEB in S211, S138, and S142. Interestingly, and in accordance with our previous results of cells treated with c-Abl inhibitors (Figure 3), we observed a reduction of S211 phosphorylation in the Y173F TFEB-GFP protein (Figure 4G), suggesting a role of Y173 on TFEB cellular localization by influencing S211 phosphorylation.
These results strongly suggest that c-Abl mediates TFEB tyrosine phosphorylation and that TFEB Y173 is relevant for its retention in the cytoplasm.
c-Abl Inhibition Promotes Cellular Clearance through Activation of TFEB
Active TFEB, localized in the nucleus, is crucial for the expression of genes encoding proteins that promote cellular clearance of substrates accumulated in the lysosomes in different LSD (Medina et al., 2011; Spampanato et al., 2013), positioning TFEB as an excellent therapeutic target (Ballabio, 2016; Napolitano and Ballabio, 2016). Our data show that c-Abl promotes TFEB tyrosine phosphorylation and that its inhibition induces TFEB activity. So next, we analyzed if c-Abl inhibition promotes cellular clearance.
To evaluate this point, we first load lysosomes with a substrate. The U18666A drug induces lysosomal cholesterol accumulation by inhibiting the lysosomal protein NPC1 (Lu et al., 2015). Then, we evaluated the effect of imatinib in HeLa cells treated with U18666A for 24 hr. Cholesterol accumulation was followed using the recombinant protein perfringolysin O, a cholesterol-binding bacterial toxin, fused to glutathione S-transferase (GST-PFO). This strategy allowed us to measure cholesterol accumulation in immunofluorescence using anti-GST antibodies. This method for cholesterol visualization has been previously validated (Kwiatkowska et al., 2014).
As expected, we observed intracellular cholesterol accumulation in HeLa cells treated with U18666A (Figure 5A). However, surprisingly pre-treatment with imatinib, decreased the GST-PFO signal, indicating a reduction in cholesterol accumulation induced by U18666A (Figure 5A). To quantify the effect of imatinib on cholesterol accumulation, we used confocal automated microscopy and performed a high-content GST-PFO assay. Interestingly, U18666A treated HeLa cells that had been pre-treated with imatinib showed significantly less cholesterol accumulation (Figure 5B). We also observed that imatinib decreased cholesterol accumulation, followed by GST-PFO staining, in U18666A treated HEK293T cells (Figure S4A).
Figure 5.

c-Abl Inhibitors Reduce Cholesterol Accumulation in a TFEB-Dependent Manner
(A) Representative confocal microscopy images showing cholesterol accumulation. HeLa cells were treated with U18666A 0.5μg/mL and/or imatinib 10μM for 24 hr. Then, GST-PFO (red) immunofluorescence and DAPI (blue) staining were performed. Scale bars, 10 μM.
(B) Quantification of cholesterol accumulation from (A). A high-content GST-PFO assay using confocal automated microscopy was performed. n = 3 independent experiments.
(C) Representative images and quantification of cholesterol accumulation by filipin staining in HT22 cells and Hepa 1–6 cells treated with U18666A 0.5μg/mL and/or imatinib 10μM for 24 hr n = 3 independent experiments. Scale bars, 10 μM.
(D) Representative images and quantification of cholesterol accumulation by filipin staining on hippocampal neurons cultures of 7 DIV from Wild-type (WT) mice and c-Abl KO mice (c-Abl KO) treated with U18666A 0.5μg/mL and/or imatinib 10μM for 24 hr n = 40 neurons per conditions. Scale bars, 10 μM.
(E) HeLa cells; HeLa TFEB-GFP cells and HeLa TFEB-KO were treated with U18666A 0.5 μg/mL for 24 hr and cholesterol accumulation was analyzed by the high-content GST-PFO assay. For each condition 450–800 cells were analyzed (7 images per sample) n = 4 biological independent samples.
(F) Representative images of cholesterol by GST-PFO and quantification of TFEB-GFP translocation assay in HeLa TFEB-GFP cells treated with U18666A 0.5μg/mL and/or imatinib 10μM for 24 hr. For each condition, 450–800 cells were analyzed (7 images per sample) n = 4 biological independent samples. Scale bars, 10 μM.
(G) Representative images of cholesterol accumulation using GST-PFO of HeLa and HeLa TFEB-KO cells treated with U18666A 0.5μg/mL and/or imatinib 10 μM for 24 hr n = 3 independent experiments. Scale bars, 10 μM. (H) Quantification of cholesterol accumulation in (G) n = 3 independent experiments. Statistical analysis with one-way ANOVA followed by Tukey's post-test. ∗p < 0.05, ∗∗p < 0.01,∗∗∗p < 0.001. Data represent mean ± SEM.
To confirm the cholesterol-lowering effect induced by c-Abl inhibition, we treated HT22 (neuronal cell line) and Hepa 1–6 (hepatocyte derived cell line) cells with the U18666A drug and imatinib. Then, we analyzed cholesterol accumulation using the well-known cholesterol-marker filipin (Figure 5C). In agreement with our previous results, we observed that imatinib decreased filipin staining. All these results confirm the imatinib cholesterol-lowering effect in different U18666A treated cell lines using two different techniques to detect cholesterol accumulation (Figure 5C). In addition, GNF-2, an allosteric c-Abl inhibitor, also reduced cholesterol accumulation in HT22 cells, demonstrating that two inhibitors that act on different c-Abl sites have the same effect. These results support the hypothesis that the cholesterol-lowering effects are mediated by c-Abl inhibition (Figure S4B).
Although imatinib and GNF2 are well-known c-Abl inhibitors, to further evaluate if the cholesterol-lowering effect is effectively mediated by c-Abl inhibition, we cultured hippocampal neurons from c-Abl null (c-Abl KO) and WT mice embryos. Seven DIV neurons were treated with U18666A and, as expected, we observed increased filipin staining in WT neurons. Interestingly, in the c-Abl KO hippocampal neurons, we observed a lower filipin staining in spite of U18666A treatment (Figure 5D).
These results show that c-Abl inhibition promotes a decrease in cholesterol accumulation suggesting an increase in cellular clearance in cells loaded with cholesterol using the U18666A drug.
Then, we analyzed the relevance of TFEB on accumulation of cholesterol in U18666A treated cells using WT HeLa (control), HeLa TFEB-GFP and HeLa TFEB-KO cells. High-content GST-PFO assays showed that TFEB-KO cells have higher cholesterol accumulation than cells that express TFEB, indicating that TFEB absence worsens the effect of U18666A (Figure 5E).
To analyze the effect of c-Abl inhibition on TFEB subcellular localization in cells that accumulate cholesterol, we treated HeLa TFEB-GFP cells with U18666A and imatinib and followed TFEB localization using the high-content nuclear translocation assay. We observed that U18666A treatment effectively promotes cholesterol accumulation compared to controls and that it also increases the TFEB nuclear signal (Figure 5F). However, addition of imatinib induces a large increase in TFEB nuclear translocation (Figure 5F). Remarkably, in cells exposed to U18666A, imatinib treatment induces a clear reduction in the accumulation of cholesterol that correlates with TFEB-GFP nuclear translocation (Figures 5F and S4C).
Interestingly, we observed that imatinib did not decrease cholesterol accumulation in HeLa TFEB-KO cells treated with U18666A (Figures 5G and 5H), demonstrating that the decrease in cholesterol accumulation triggered by c-Abl inhibition depends on the presence of TFEB. Moreover, HeLa cells exposed to U18666A showed increased LysoTracker red marker analyzed by FACs (Figure S4D), but co-treatment with imatinib further increased lysotracker levels. Remarkably, we did not observe this increment in HeLa TFEB-KO cells treated with U18666A plus imatinib, suggesting that this increase in lysotracker levels depends on TFEB (Figure S4D).
Altogether, these results demonstrate that c-Abl inhibition induces a decrease in lysosomal cholesterol content and that this effect is mediated by TFEB.
c-Abl Inhibition Promotes Clearance of Cholesterol Accumulation in NPC1 Human Fibroblasts
NPC disease is produced by mutations in the NPC1 or NPC2 genes that encode cholesterol transporter proteins. NPC is characterized by the pathogenic accumulation of cholesterol and other lipids within the late endosomal/lysosomal compartments (Carstea et al., 1997; Kwon et al., 2009; Lloyd-Evans et al., 2008; Naureckiene et al., 2000; Pentchev et al., 1984; Sturley et al., 2004).
To address the relevance of the c-Abl/TFEB axis in NPC pathology, we decided to use fibroblasts from patients with NPC1 that basally accumulate cholesterol, positioning as a good model to analyze clearance. We first analyzed the activation of c-Abl in these cells following phosphorylated-c-Abl (p-c-Abl) and phosphorylated-CRKII (p-CRKII). Interestingly, we observed an increase of both phosphorylated proteins in different fibroblasts of patients with NPC1 compared to WT human fibroblasts (Figure 6A). Next, we treated the different fibroblasts of patients with NPC1 with two c-Abl inhibitors, imatinib and nilotinib, for 24 hr and analyzed cholesterol accumulation with filipin staining. Remarkably, we observed that cholesterol accumulation was reduced in fibroblasts of patients with NPC1 carrying different mutations, when c-Abl inhibitors were used (Figure 6B). The same clearance effect was observed in longer treatments (48 hr) with the c-Abl inhibitors (Figure S5A).
Figure 6.

c-Abl Inhibition Reduces Cholesterol Accumulation in NPC1 Human V1165M Fibroblasts
(A) Representative Western blot of phospho-c-Abl (p-c-Abl) and phosphor-CRKII (p-CRKII) in wild-type and NPC1 human fibroblasts. GAPDH was used as loading control. n = 3 independent experiments. Scale bars, 10 μM.
(B) Representative images and quantification of cholesterol accumulation detected through filipin staining in NPC1 human fibroblast treated with imatinib 10μM and nilotinib 10μM for 24 hr n = 80 cells per conditions.
(C) Representative Western blot of endogenous TFEB in a nuclear/cytoplasmic fractionation assay of V1165M fibroblasts of patients with NPC1 treated with imatinib 10μM for 24 hr n = 3 independent experiments.
(D) Quantitative analysis of lysotracker in V1165M fibroblasts of patients with NPC1 treated with imatinib and nilotinib with 10μM for 24 hr using flow cytometry. n = 10,000 cells per conditions.
(E) q-PCR analysis of different mRNA TFEB target genes in V1165M fibroblasts of patients with NPC1 treated with imatinib and nilotinib 10 μM for 24 hr n = 3 independent experiments.
(F) Representative immunofluorescence images of Lamp1 in V1165M fibroblasts of patients with NPC1 treated with imatinib 10μM for 24 hr n = 3 independent experiments. Scale bars, 50 μM.
(G) Representative images of cholesterol accumulation in NPC1 human fibroblasts treated with imatinib 10 μM or nilotinib 10 μM for 24 hr. Scale bars, 50 μM. (H) Quantification of cholesterol accumulation by high-content GST-PFO assay in NPC1 human fibroblasts treated with imatinib or nilotinib at different concentrations for 24 hr. For each condition 450–800 cells were analyzed (7 images per sample). n = 4 biological independent samples.
(I) Flow cytometry graphs of GST-PFO (cholesterol) in NPC1 human fibroblasts treated with imatinib or nilotinib 10μM for 24 hr (J) Quantitative analysis by flow cytometry of GST-PFO (cholesterol) in (I) n = 10,000 cells per conditions. Statistical analysis with one-way ANOVA followed by Tukey's post-test and Student's t-tests when comparing two experimental groups. ∗p < 0.05, ∗∗p < 0.01,∗∗∗p < 0.001. Data represent mean ± SEM.
Concordantly, we observed a reduction in the GST-PFO signal in Npc1 KO mouse fibroblasts, which accumulate cholesterol at basal levels, under imatinib treatment (Figure S5B). This result supports the idea that c-Abl inhibition promotes cellular cholesterol clearance in NPC disease. Consequently, GNF2 treatment also reduced filipin staining in Npc1 KO fibroblasts (Figure S5C).
In addition, we decided to analyze in more detail the effect on c-Abl inhibition in V1165M fibroblasts of patients with NPC1 because it was one of the NPC1 mutants, where the effect on cholesterol clearance was more clearly seen. We treated the V1165M fibroblasts of patients with NPC1 with imatinib for 24 hr and performed a nucleus cytoplasm fractionation to analyze TFEB subcellular localization. We observed that this mutant presents low basal levels of TFEB in the nucleus and that imatinib treatment clearly induced TFEB translocation to the nucleus (Figure 6C). Concordantly, we observed that in V1165M fibroblasts of patients with NPC1 both c-Abl inhibitors, imatinib and nilotinib, increased the LysoTracker red signal compared with the control condition (Figure 6D). Interestingly, both inhibitors significantly increased the expression of several TFEB target genes such as CTSD, MCOLN1, Lamp1, and TPP1 in this fibroblast of patient with NPC1 measured by q-PCR (Figure 6E). Remarkably, we found that the most important changes were in the genes related to lysosomal biogenesis and exocytosis (Figure 6E), an effect that correlates with increased LAMP1 signal measured by immunofluorescence in V1165M fibroblasts of patients with NPC1 treated with imatinib (Figure 6F).
Then, we treated this mutant with different concentrations of imatinib and nilotinib. Remarkably, using the high-content GST-PFO assay, we observed that the inhibitors promote a reduction in cholesterol accumulation in a concentration-depending manner, being imatinib the most effective (Figures 6G and 6H). Concordantly, we observed the reduction of cholesterol accumulation in V1165M fibroblasts of patients with NPC1 treated with these two c-Abl inhibitors measuring the GST-PFO signal by FACs (Figures 6I and 6J). It is important to mention that in Npc1 KO hippocampal neuronal cultures that clearly show cholesterol accumulation by GST-PFO staining, treatment with imatinib reduced this accumulation (Figure S5D).
Remarkably, we observed that the c-Abl inhibitors imatinib and nilotinib, which are FDA-approved drugs, promote a reduction in cholesterol accumulation in different genetic NPC models. Since, in these models cholesterol accumulation is chronic, our results strongly suggest that c-Abl inhibition promotes cellular clearance.
2.6. c-Abl Inhibitors Reduces Cholesterol Accumulation In Vivo
Finally, in order to analyze the in vivo relevance of the c-Abl/TFEB axis, we treated Npc1−/− (NPC) mice with imatinib by administering daily intraperitoneal injections (12.5 mg/Kg) and measured cholesterol accumulation in cerebellum sections by GST-PFO staining. As expected, we observed a clear accumulation of cholesterol in the soma of Purkinje neurons from NPC mice compared with WT mice (Figure 7A). Notably, NPC mice treated with imatinib showed less cholesterol accumulation in Purkinje neurons (Figures 7A and 7B).
Figure 7.

c-Abl Deficiency Reduces Cholesterol Accumulation in NPC In Vivo Models
(A) Representative immunofluorescence showing cholesterol by GST-PFO staining (red) in cerebellum sections from 8-month-old wild-type (WT) and NPC mice treated with vehicle or imatinib 12.5 mg/Kg, every day for 2 weeks by intraperitoneal injection. Arrows show purkinje neurons. Scale bars, 50 μM. (B) Quantification of cholesterol accumulation per Purkinje cell area in (A) (n = 2) (5 images per animal).
(C–E) WT and NPC mice were treated for 4 weeks with vehicle and GNF-2 (5 mg/kg in 40% water, 30% polyethylene glycol 300 and 30% propylene glycol) starting at p28. Motor coordination was assessed weekly by beam test (C), weight was registered during the treatment (D) and (E) Cerebella from vehicle and GNF-2 treated female NPC mice were analyzed at 8 weeks of age for calbindin by immunohistochemistry. Quantification of Purkinje cell area/Purkinje cell layer (PCL) area is shown (n = 9). Scale bars, 100 μM.
(F) WT and NPC mice were treated with GNF-2 (5 mg/Kg) for three weeks starting at p28. Upper panels show filipin staining of cerebellum sections and lower panels show filipin staining of liver sections. n = 3 independent experiments. Scale bars, 50 μM.
(G) Representative immunofluorescence showing cholesterol by GST-PFO (red) and endogenous TFEB (green) in cerebellum sections from 8-month-old wild-type (WT) and c-Abl KO mice treated with vehicle or U18666A 10 mg/Kg, for 2 days by intraperitoneal injections. Arrows show purkinje neurons. Scale bars, 50 μM. Statistical analysis with one-way ANOVA followed by Tukey's post-test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Data represent mean ± SEM.
To strengthen our data, we treated NPC mice with GNF-2, an allosteric c-Abl inhibitor. First, we corroborated that this c-Abl inhibitor improved the behavioral tests of NPC mice as we had previously demonstrated for NPC mice treated with imatinib (Alvarez et al., 2008). Daily intraperitoneal injections of GNF-2 (5 mg/Kg) in NPC mice increased motor coordination, measured by the beam test (Figure 7C); improved weight gain (Figure 7D); and prevented Purkinje cell death in the cerebellum, measured by calbindin staining (Figure 7E). More importantly, filipin staining of cerebellum and liver sections showed that NPC mice treated with GNF-2 accumulate less cholesterol than NPC mice treated with saline solution (Figure 7F).
Finally, to demonstrate that the results obtained previously with c-Abl pharmacological inhibitors are reproduced in a c-Abl genetic inhibition model, we induced cholesterol accumulation in vivo in c-Abl KO mice. With this purpose, we treated WT and c-Abl KO mice with U18666A (10 mg/kg) by intraperitoneal injection an analyzed cholesterol accumulation in Purkinje neurons, one of the first to be affected in NPC mice (Lopez and Scott, 2013; Walkley and Suzuki, 2004). We observed that WT mice treated with U18666A show increased cholesterol accumulation in Purkinje neurons compared with the control mice (Figure 7G). Concordantly, c-Abl KO mice treated with U18666A showed decreased cholesterol accumulation (Figure 7G). Interestingly, and in accordance with our in vitro results, we observed a greater signal for endogenous TFEB in the nucleus of Purkinje cells in c-Abl KO mice treated with U1866A compared with WT mice treated with saline or U18666A (Figure 7G).
Altogether, these results strongly suggest that inhibition of the signaling pathway c-Abl/TFEB promotes a reduction in the accumulation of cholesterol in in vitro and in vivo models of NPC disease.
Discussion
Our results show that c-Abl controls cellular clearance by regulating TFEB activity, opening a potential therapeutic approach for several diseases, where cellular clearance is required. Interestingly, c-Abl inhibition allows us to modulate TFEB skipping the need of modulate the classical TFEB regulator mTORC1, a pleiotropic pathway, which inhibition has harmful consequences for normal cells (Figure 3). By the contrast, c-Abl inhibition has showed to be safe (Pagan et al., 2016, 2019). As a proof of principle, we showed that c-Abl inhibition promotes a reduction in cholesterol accumulation in NPC cells (Figures 5 and 6). Remarkably, we found that inhibition of the tyrosine kinase c-Abl promotes a cholesterol-lowering effect in several NPC1 pharmacological and genetic models demonstrating the involvement of c-Abl in lysosomal cholesterol clearance (Figures 5, 6, and 7).
To demonstrate that c-Abl controls the translocation of TFEB to the nucleus, we used different strategies (Figure 1). First, we used several pharmacological inhibitors, including imatinib, nilotinib and dasatinib, designed to bind to the ATP binding site of c-Abl in the BCR-c-Abl fusion protein produced in patients with chronic myeloid leukemia (CML). In myeloid leukemia c-Abl inhibition for long times has no main secondary effects. In WT cells, c-Abl is the primary target of these drugs. In addition, we included the more recently developed inhibitors GNF2 and GNF5, which bind to the allosteric site of c-Abl (Iacob et al., 2011). We found that all the inhibitors we tested increased TFEB nuclear localization. The more effective inhibitors that promoted TFEB nuclear translocation at lower concentrations were nilotinib and imatinib (Figure 1). Nilotinib was developed based on the chemical structure of the first and classic BCR-c-Abl inhibitor imatinib, optimizing cellular potency and selectivity (Manley et al., 2010). Nevertheless, these inhibitors could also inhibit Arg, a protein with homology to c-Abl and that belongs to the same family of ABL proteins (Hantschel et al., 2008). We cannot discard the possibility that Arg could also have an impact on TFEB activation, a question that should be addressed.
As a second strategy, we used HeLa c-Abl KO cells obtained by CRISPR/Cas9 technoloy, showing increased endogenous TFEB nuclear localization. As a third approach, we used a siRNA against c-Abl and found that two readouts of TFEB activation, nuclear translocation, and the induction of mRNA levels of lysosomal genes were increased. Importantly, our results also show that c-Abl inhibition not only activates overexpressed transfected TFEB but also the endogenous TFEB, indicating that the c-Abl/TFEB axis is a relevant signaling pathway under physiological conditions (Figure 1).
Our results suggest that inhibition of c-Abl promotes several readouts of TFEB activation (Figure 2). We observed an increase in autophago-lysosome formation, lysotracker and Lamp-1 signals and lysosome localization near the plasma membrane. These results suggest an increase in autophagic flux, lysosomal biogenesis and exocytosis in cells treated with imatinib. Concordantly, it has been demonstrated that the inhibition of c-Abl promotes autophagy (Can et al., 2011; Ertmer et al., 2007; Lim et al., 2014), a process that is dysregulated in different LSDs such as NPC, Pompe disease, and MPSIIIA (Lieberman et al., 2012). Because TFEB regulates lysosomal biogenesis, exocytosis, and autophagy (Settembre et al., 2013; Settembre and Medina, 2015), inhibition of the c-Abl/TFEB axis could promote cholesterol clearance through these three biological processes (Medina et al., 2011). It would be interesting to further evaluate whether c-Abl inactivation affects other signaling pathways that start on the lysosome surface, including those related to the release of lysosomal calcium through TRPML1 modulating a series of cellular processes, including the re-formation of lysosomes from hybrid organelles, the fusion between endosomes and lysosomes, autophagosomes and lysosomes, and lysosomes and the plasma membrane in lysosomal exocytosis and biogenesis of the autophagosomes (Ballabio and Bonifacino, 2020). Furthermore, c-Abl could be part of the sensor systems that are associated with the surface of the lysosome. In this sense, as far as we know, there are no reports of association of c-Abl to the surface of lysosomes.
Our results showed that TFEB nuclear translocation, promoted by inhibition of c-Abl, is independent of mTORC1 (Figure 3). Interestingly, the inhibition of c-Abl was able to induce the translocation of TFEB to the nucleus under conditions in which mTORC1 is fully active, including re-fed conditions after nutrient starvation and in cells deficient for TSC2, the negative regulator of mTORC1. This is of great relevance in pathological conditions in which mTORC1 signaling could be compromised such as in certain NPC1 mutations found in patients, in which a negative feedback is lost between NPC1 and mTORC1 because it requires an intact sterol-sensing domain site in NPC1 (Castellano et al., 2017). Furthermore, recent results suggest that mTORC1 regulation by lysosomal cholesterol depends on two proteins, NPC1 and oxysterol-binding protein (OSBP), which transfers cholesterol from the ER to the lysosomal membrane at the ER-lysosome contact sites, activating mTORC1 (Lim et al., 2019). In NPC1-deficient cells, then the transfer of cholesterol to the lysosomal membrane by OSBP would prevail, driving constitutive mTORC1 activation. In this context, our result is encouraging because it is focused on TFEB and not on the inhibition of mTORC1, which is not good target for the treatment of diseases with compromised lysosomes since this kinase fulfills multiple cellular functions (Calderon and Klein, 2018). In addition, a possible regulation of mTORC1 over c-Abl should be also addressed.
Recent studies have shown that inhibition of Akt kinase by compounds such as trehalose induces the nuclear translocation of TFEB independent of mTORC1 (Palmieri et al., 2017). Therefore, regulation of TFEB translocation seems to respond to more than one signaling system, increasing the possibilities for its modulation.
Our study demonstrates that TFEB activity is negatively regulated by tyrosine phosphorylation. We found that the activity of c-Abl is sufficient to promote TFEB tyrosine phosphorylation. Indeed, our results showed that the Y173F TFEB-GFP mutant protein has a constitutive nuclear localization, suggesting that Y173 is relevant to retain TFEB in the cytoplasm (Figure 4). Interestingly, this tyrosine is highly conserved in TFEB from different species.
Notably, we found that under conditions in which c-Abl is inhibited, TFEB translocates into the nucleus when it is phosphorylated in S142 and S138 but not on S211 (Figures 3 and 4). This difference would probably reflect a decrease in the interaction between TFEB and the 14-3-3 chaperone, which retains TFEB in the cytoplasm when TFEB is phosphorylated on S211 (Roczniak-Ferguson et al., 2012). It is possible to propose that the reduction of TFEB tyrosine phosphorylation, as a consequence of c-Abl inhibition, would favor de-phosphorylation on TFEB-S211. Indeed, we found that the Y173F TFEB-GFP protein exhibit less phosphorylation on S211, whereas S142 and S138 did not change when wild type TFEB-GFP protein or Y75F TFEB-GFP protein were expressed (Figure 4). In addition, we observed that the TFEB nuclear localization induced by the c-Abl inhibitors is independent of the calcineurin phosphatase activity. However, additional studies are required to solve this point and to determine how c-Abl inhibition promotes TFEB de-phosphorylation on S211. Also, it will be necessary to analyze if other proteins that conform the MiTF-TFe family such as MITF, TFE3, and TFEC, are possible targets of c-Abl and if their cellular localization could be regulated by tyrosine phosphorylation. Nevertheless, these findings reinforce the idea that tyrosine phosphorylation levels could have an impact on the serine phosphorylation state of signaling proteins. This priming phenomenon has been previously described in other contexts (Kosten et al., 2014).
The relevance of TFEB in NPC disease has not been totally explored yet. Previously, it was shown that the mTORC1 inhibitor Torin1 promotes clearance of cholesterol accumulated in lysosomes (Wang et al., 2015). However, Torin1 has been shown to be toxic. In addition, mTORC1 has been shown to be over-activated in NPC cells (Castellano et al., 2017), and TFEB preferentially localized in the cytoplasm of NPC null CHO cells (Castellano et al., 2017). Our results show that in HeLa cells overexpressing TFEB, U18666A treatment slightly increases TFEB nuclear translocation (Figure 5). This result is consistent with data from the literature that shows the promotion of TFEB transfer to the nucleus by mTORC1-independent signals including, among others, oxidative stress (Ballabio and Bonifacino, 2020; Raben and Puertollano, 2016). Previous results from our laboratory show that already from 1 hr of treatment with U18666A it is possible to visualize an increase in oxidative stress markers (Klein et al., 2011). However, after imatinib treatment, TFEB nuclear localization is increased further. Most importantly, in NPC1 human fibroblasts, the c-Abl inhibitors promoted a reduction in cholesterol accumulation, independent of the specific NPC1 mutation. Further studies are required to evaluate the effect of c-Abl inhibition on NPC2 mutants, which are responsible for 5% of NPC cases (Patterson et al., 2012), as well as in models of other lysosomal disorders, to find the relevance of this axis in other diseases.
Remarkably, under basal conditions, NPC1 human fibroblasts show very low levels of TFEB in the nucleus, which are greatly increased after c-Abl inhibition (Figure 6). These experiments demonstrate that in the context of NPC disease, c-Abl inhibition promotes TFEB nuclear translocation promoting an increase in Lamp1 protein levels and a TFEB-dependent increase in LysoTracker red staining in Hela-U18666A treated cells. Concordantly, mRNA levels of several TFEB target genes, most of them related with lysosomal biogenesis and exocytosis, were recovered in NPC1 human fibroblasts treated with imatinib or nilotinib (Figure 6). Further studies are needed to analyze if in specific contexts, TFEB promotes the fine-tune regulation of particular genes. Remarkably, we demonstrated that the cholesterol-lowering effect of c-Abl inhibition is abolished in HeLa TFEB-KO cells (Figure 5). These results confirm the relevance of TFEB in cellular clearance induced by c-Abl inhibition in NPC cells.
Importantly, we observed similar results in NPC in vivo models and also that imatinib treatment prevented cholesterol accumulation in NPC mice (Figure 7). We described previously that this c-Abl inhibitor prevents neuronal death and improves performance in different behavioral test and increases lifespan of this NPC mice model (Alvarez et al., 2008). Therefore, the cholesterol-lowering effect of imatinib could be crucial to this improvement in the NPC mice. Then, it was important to corroborate these results with other c-Abl inhibitor. We chose GNF-2 because even though is not an approved FDA drug, and our analysis showed that it has a better penetrance of the blood brain barrier (data not shown) than imatinib. In agreement with our previous data with imatinib, we observed that NPC mice treated with GNF-2 have a better performance in the behavioral tests. Importantly, this improved performance correlates with decreased cholesterol accumulation in Purkinje neurons. Similar results on cholesterol accumulation were observed in c-Abl KO mice treated with U18666A, in which endogenous TFEB was found mainly in the nucleus of Purkinje cells (Figure 7).
c-Abl can be activated through many different extrinsic ligands that activate the epidermal growth factor receptor (EGFR), the platelet-derived growth factor receptor and the vascular EGFR, among others (Wang, 2014). c-Abl also can be activated by intrinsic signals such as oxidative stress and DNA damage. Interestingly, because c-Abl is a tyrosine kinase that participates in several signaling pathways that induce neuronal dysfunction and death, its inhibition also generates beneficial effects in neurodegenerative diseases including decreased activation of the proapoptotic p73 transcription factor, HDAC2 activity and APP processing, as it has already been shown in AD and NPC models (Cancino et al., 2008; Contreras et al., 2016; Estrada et al., 2016; Gonzalez-Zuniga et al., 2014; Klein et al., 2011; Yanez et al., 2016). In addition, the inhibition of c-Abl decreases protein accumulation of misfolded α-synuclein and mutant SOD1 in Parkinson and ALS (Imamura et al., 2017), respectively. Imatinib and nilotinib are FDA-approved c-Abl inhibitors and their chronic use in patients with CML is safe and has few side effects. Moreover, the results from a small group of patients with Parkinson's disease and Dementia with Lewy bodies that had been treated with nilotinib for 24 weeks, suggested a possible beneficial effect of this drug in neurodegenerative diseases (Pagan et al., 2016). Altogether, these results position c-Abl inhibitors as an interesting strategy for the treatment of diseases in which lysosomes and TFEB have been proposed as therapeutic targets, such as Pompe, Gaucher, MPSIV, and Batten disease, among others (Ballabio, 2016; Napolitano and Ballabio, 2016), and depending on the signal activated, only a fraction of c-Abl may be active in the cells, and as a consequence, different signaling pathways will be activated.
Finally, our data revealed an mTORC1-independent mechanism for lysosomal clearance through the activation of TFEB by inhibiting the tyrosine kinase c-Abl. Our results show that c-Abl is a very good target for treatment of lysosomal storage diseases. This mechanism is relevant in the context of the NPC LSD in which this kinase is active. The inhibition of c-Abl using FDA-approved drugs promotes the activation of TFEB and cellular clearance, opening the possibility of treating patients with NPC, as well as other patients suffering from other neurodegenerative LSDs.
Limitations of the Study
In our current study, we performed in vitro phosphorylation assays using tyrosine mutants on TFEB plasmids. We could not observe differences in the radioactivity incorporation on TFEB in spite of our results suggested that Y173 could be a main target of c-Abl phosphorylation. This is probably due to basal phosphorylation of other tyrosines. This experiment by itself is not enough to discard that Y173 is phosphorylated by c-Abl. Further studies are necessary to dissect more deeply the role of Y173 on TFEB signaling.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to the lead contact Silvana Zanlungo (szanlungo@uc.cl).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability
Further information about data supporting the conclusions of this manuscript will be made available by the Lead Contact to any qualified researcher.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Douglas Green (St. Jude Children's Research Hospital, Memphis, TN, USA) and Patricia Burgos (Universidad Austral de Chile, Valdivia and Universidad San Sebastián, Santiago, Chile) for the donation of the c-Abl ERT2 and c-Abl ERT2 KD plasmids and the H4 GFP-mRFP-LC3 cells, respectively.
This study was supported by grants from the Fondo Nacional de Desarrollo Científico y Tecnológico (FONDECYT) (grant numbers 1161065 and 1201668 to A.R.A, and 1150186 and 1190334 to S.Z); from CONICYT-Chile Grant Redes 150082 (SZ); from the University of Pennsylvania Orphan Disease Center and a donation from the University of Notre Dame’s Warren Family Research Center for Drug Discovery. From European Union’s Horizon 2020 research and innovation program (RISE) under the Marie Skłodowska-Curie grant agreement No 734825 (P.S.C, A.D.K, F.M.P, and S.Z). From the Fomento al Desarrollo Científico y Tecnológico FONDEF D10E1077 to A.R.A and S.Z, and Proyecto Basal AFB170005 (A.R.A.). P.S.C acknowledges support from CONICYT, Beca doctorado nacional 21140469. This work was support by the Advanced Microscopy Facility UC. The authors acknowledge the services provided by UC CIBEM Animal Facility funded by PIA CONICYT ECM-07. Graphical abstract created with BioRender.com.
Author Contributions
P.S.C., A.R.A., and S.Z. conceived of the study. P.S.C., D.L.M., A.B., A.R.A., and S.Z., designed experiments. P.S.C., P.J.T., L.G-H., I.P., C.S., G.N., M.M., M.LH., C.V., D.W., and A.D.K. performed experiments. P.S.C., P.J.T., L.G-H., I.P., M.LH., and C.S. performed quantitative analysis of the results. P.S.C., A.M., E.B., F.M.P., J.C., N.L., and A.S. generated reagents. P.S.C., A.B., A.R.A., and S.Z. wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: November 20, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101691.
Contributor Information
Alejandra R. Alvarez, Email: aalvarez@bio.puc.cl.
Silvana Zanlungo, Email: szanlungo@uc.cl.
Supplemental Information
References
- Alvarez A.R., Klein A., Castro J., Cancino G.I., Amigo J., Mosqueira M., Vargas L.M., Yevenes L.F., Bronfman F.C., Zanlungo S. Imatinib therapy blocks cerebellar apoptosis and improves neurological symptoms in a mouse model of Niemann-Pick type C disease. FASEB J. 2008;22:3617–3627. doi: 10.1096/fj.07-102715. [DOI] [PubMed] [Google Scholar]
- Ballabio A. The awesome lysosome. EMBO Mol. Med. 2016;8:73–76. doi: 10.15252/emmm.201505966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballabio A., Bonifacino J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2020;21:101–118. doi: 10.1038/s41580-019-0185-4. [DOI] [PubMed] [Google Scholar]
- Blom N., Gammeltoft S., Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 1999;294:1351–1362. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- Calderon J.F., Klein A.D. Controversies on the potential therapeutic use of rapamycin for treating a lysosomal cholesterol storage disease. Mol. Genet. Metab. Rep. 2018;15:135–136. doi: 10.1016/j.ymgmr.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Can G., Ekiz H.A., Baran Y. Imatinib induces autophagy through BECLIN-1 and ATG5 genes in chronic myeloid leukemia cells. Hematology. 2011;16:95–99. doi: 10.1179/102453311X12902908412039. [DOI] [PubMed] [Google Scholar]
- Cancino G.I., Perez de Arce K., Castro P.U., Toledo E.M., von Bernhardi R., Alvarez A.R. c-Abl tyrosine kinase modulates tau pathology and Cdk5 phosphorylation in AD transgenic mice. Neurobiol. Aging. 2011;32:1249–1261. doi: 10.1016/j.neurobiolaging.2009.07.007. [DOI] [PubMed] [Google Scholar]
- Cancino G.I., Toledo E.M., Leal N.R., Hernandez D.E., Yevenes L.F., Inestrosa N.C., Alvarez A.R. STI571 prevents apoptosis, tau phosphorylation and behavioural impairments induced by Alzheimer's beta-amyloid deposits. Brain. 2008;131:2425–2442. doi: 10.1093/brain/awn125. [DOI] [PubMed] [Google Scholar]
- Capdeville R., Buchdunger E., Zimmermann J., Matter A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat. Rev. Drug Discov. 2002;1:493–502. doi: 10.1038/nrd839. [DOI] [PubMed] [Google Scholar]
- Carstea E.D., Morris J.A., Coleman K.G., Loftus S.K., Zhang D., Cummings C., Gu J., Rosenfeld M.A., Pavan W.J., Krizman D.B. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 1997;277:228–231. doi: 10.1126/science.277.5323.228. [DOI] [PubMed] [Google Scholar]
- Castellano B.M., Thelen A.M., Moldavski O., Feltes M., van der Welle R.E., Mydock-McGrane L., Jiang X., van Eijkeren R.J., Davis O.B., Louie S.M. Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science. 2017;355:1306–1311. doi: 10.1126/science.aag1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras P.S., Gonzalez-Zuniga M., Gonzalez-Hodar L., Yanez M.J., Dulcey A., Marugan J., Seto E., Alvarez A.R., Zanlungo S. Neuronal gene repression in Niemann-Pick type C models is mediated by the c-Abl/HDAC2 signaling pathway. Biochim. Biophys. Acta. 2016;1859:269–279. doi: 10.1016/j.bbagrm.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cujec T.P., Medeiros P.F., Hammond P., Rise C., Kreider B.L. Selection of v-abl tyrosine kinase substrate sequences from randomized peptide and cellular proteomic libraries using mRNA display. Chem. Biol. 2002;9:253–264. doi: 10.1016/s1074-5521(02)00098-4. [DOI] [PubMed] [Google Scholar]
- Decressac M., Mattsson B., Weikop P., Lundblad M., Jakobsson J., Bjorklund A. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc. Natl. Acad. Sci. U S A. 2013;110:E1817–E1826. doi: 10.1073/pnas.1305623110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druker B.J., Tamura S., Buchdunger E., Ohno S., Segal G.M., Fanning S., Zimmermann J., Lydon N.B. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat. Med. 1996;2:561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- Ertmer A., Huber V., Gilch S., Yoshimori T., Erfle V., Duyster J., Elsasser H.P., Schatzl H.M. The anticancer drug imatinib induces cellular autophagy. Leukemia. 2007;21:936–942. doi: 10.1038/sj.leu.2404606. [DOI] [PubMed] [Google Scholar]
- Estrada L.D., Chamorro D., Yanez M.J., Gonzalez M., Leal N., von Bernhardi R., Dulcey A.E., Marugan J., Ferrer M., Soto C. Reduction of blood amyloid-beta oligomers in Alzheimer's disease transgenic mice by c-abl kinase inhibition. J. Alzheimer's Dis. 2016;54:1193–1205. doi: 10.3233/JAD-151087. [DOI] [PubMed] [Google Scholar]
- Estrada L.D., Zanlungo S.M., Alvarez A.R. C-Abl tyrosine kinase signaling: a new player in AD tau pathology. Curr. Alzheimer Res. 2011;8:643–651. doi: 10.2174/156720511796717249. [DOI] [PubMed] [Google Scholar]
- Fraldi A., Klein A.D., Medina D.L., Settembre C. Brain disorders due to lysosomal dysfunction. Annu. Rev. Neurosci. 2016;39:277–295. doi: 10.1146/annurev-neuro-070815-014031. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Zuniga M., Contreras P.S., Estrada L.D., Chamorro D., Villagra A., Zanlungo S., Seto E., Alvarez A.R. c-Abl stabilizes HDAC2 levels by tyrosine phosphorylation repressing neuronal gene expression in Alzheimer's disease. Mol. Cell. 2014;56:163–173. doi: 10.1016/j.molcel.2014.08.013. [DOI] [PubMed] [Google Scholar]
- Gutknecht M., Geiger J., Joas S., Dorfel D., Salih H.R., Muller M.R., Grunebach F., Rittig S.M. The transcription factor MITF is a critical regulator of GPNMB expression in dendritic cells. Cell Commun. signaling. 2015;13:19. doi: 10.1186/s12964-015-0099-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hantschel O., Rix U., Superti-Furga G. Target spectrum of the BCR-ABL inhibitors imatinib, nilotinib and dasatinib. Leuk. Lymphoma. 2008;49:615–619. doi: 10.1080/10428190801896103. [DOI] [PubMed] [Google Scholar]
- Hebron M.L., Lonskaya I., Moussa C.E. Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of alpha-synuclein in Parkinson's disease models. Hum. Mol. Genet. 2013;22:3315–3328. doi: 10.1093/hmg/ddt192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacob R.E., Zhang J., Gray N.S., Engen J.R. Allosteric interactions between the myristate- and ATP-site of the Abl kinase. PLoS One. 2011;6:e15929. doi: 10.1371/journal.pone.0015929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imam S.Z., Trickler W., Kimura S., Binienda Z.K., Paule M.G., Slikker W., Jr., Li S., Clark R.A., Ali S.F. Neuroprotective efficacy of a new brain-penetrating C-Abl inhibitor in a murine Parkinson's disease model. PLoS One. 2013;8:e65129. doi: 10.1371/journal.pone.0065129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura K., Izumi Y., Watanabe A., Tsukita K., Woltjen K., Yamamoto T., Hotta A., Kondo T., Kitaoka S., Ohta A. The Src/c-Abl pathway is a potential therapeutic target in amyotrophic lateral sclerosis. Sci. Transl. Med. 2017;9:eaaf3962. doi: 10.1126/scitranslmed.aaf3962. [DOI] [PubMed] [Google Scholar]
- Karuppagounder S.S., Brahmachari S., Lee Y., Dawson V.L., Dawson T.M., Ko H.S. The c-Abl inhibitor, nilotinib, protects dopaminergic neurons in a preclinical animal model of Parkinson's disease. Sci. Rep. 2014;4:4874. doi: 10.1038/srep04874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsumata R., Ishigaki S., Katsuno M., Kawai K., Sone J., Huang Z., Adachi H., Tanaka F., Urano F., Sobue G. c-Abl inhibition delays motor neuron degeneration in the G93A mouse, an animal model of amyotrophic lateral sclerosis. PLoS One. 2012;7:e46185. doi: 10.1371/journal.pone.0046185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein A., Maldonado C., Vargas L.M., Gonzalez M., Robledo F., Perez de Arce K., Munoz F.J., Hetz C., Alvarez A.R., Zanlungo S. Oxidative stress activates the c-Abl/p73 proapoptotic pathway in Niemann-Pick type C neurons. Neurobiol. Dis. 2011;41:209–218. doi: 10.1016/j.nbd.2010.09.008. [DOI] [PubMed] [Google Scholar]
- Kosten J., Binolfi A., Stuiver M., Verzini S., Theillet F.X., Bekei B., van Rossum M., Selenko P. Efficient modification of alpha-synuclein serine 129 by protein kinase CK1 requires phosphorylation of tyrosine 125 as a priming event. ACS Chem. Neurosci. 2014;5:1203–1208. doi: 10.1021/cn5002254. [DOI] [PubMed] [Google Scholar]
- Kwiatkowska K., Marszalek-Sadowska E., Traczyk G., Koprowski P., Musielak M., Lugowska A., Kulma M., Grzelczyk A., Sobota A. Visualization of cholesterol deposits in lysosomes of Niemann-Pick type C fibroblasts using recombinant perfringolysin O. Orphanet J. Rare Dis. 2014;9:64. doi: 10.1186/1750-1172-9-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon H.J., Abi-Mosleh L., Wang M.L., Deisenhofer J., Goldstein J.L., Brown M.S., Infante R.E. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell. 2009;137:1213–1224. doi: 10.1016/j.cell.2009.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawana V., Singh N., Sarkar S., Charli A., Jin H., Anantharam V., Kanthasamy A.G., Kanthasamy A. Involvement of c-abl kinase in microglial activation of NLRP3 inflammasome and impairment in autolysosomal system. J. Neuroimmune Pharmacol. 2017;12:624–660. doi: 10.1007/s11481-017-9746-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Friedrichsen H.J., Andrews S., Picaud S., Volpon L., Ngeow K., Berridge G., Fischer R., Borden K.L.B., Filippakopoulos P. A TFEB nuclear export signal integrates amino acid supply and glucose availability. Nat. Commun. 2018;9:2685. doi: 10.1038/s41467-018-04849-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Xu M., Ding X., Yan C., Song Z., Chen L., Huang X., Wang X., Jian Y., Tang G. Protein kinase C controls lysosome biogenesis independently of mTORC1. Nat. Cell Biol. 2016;18:1065–1077. doi: 10.1038/ncb3407. [DOI] [PubMed] [Google Scholar]
- Lieberman A.P., Puertollano R., Raben N., Slaugenhaupt S., Walkley S.U., Ballabio A. Autophagy in lysosomal storage disorders. Autophagy. 2012;8:719–730. doi: 10.4161/auto.19469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim C.Y., Davis O.B., Shin H.R., Zhang J., Berdan C.A., Jiang X., Counihan J.L., Ory D.S., Nomura D.K., Zoncu R. ER-lysosome contacts enable cholesterol sensing by mTORC1 and drive aberrant growth signalling in Niemann-Pick type C. Nat. Cell Biol. 2019;21:1206–1218. doi: 10.1038/s41556-019-0391-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim Y.M., Lim H., Hur K.Y., Quan W., Lee H.Y., Cheon H., Ryu D., Koo S.H., Kim H.L., Kim J. Systemic autophagy insufficiency compromises adaptation to metabolic stress and facilitates progression from obesity to diabetes. Nat. Commun. 2014;5:4934. doi: 10.1038/ncomms5934. [DOI] [PubMed] [Google Scholar]
- Lloyd-Evans E., Morgan A.J., He X., Smith D.A., Elliot-Smith E., Sillence D.J., Churchill G.C., Schuchman E.H., Galione A., Platt F.M. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 2008;14:1247–1255. doi: 10.1038/nm.1876. [DOI] [PubMed] [Google Scholar]
- Lopez M.E., Scott M.P. Genetic dissection of a cell-autonomous neurodegenerative disorder: lessons learned from mouse models of Niemann-Pick disease type C. Dis. Models Mech. 2013;6:1089–1100. doi: 10.1242/dmm.012385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F., Liang Q., Abi-Mosleh L., Das A., De Brabander J.K., Goldstein J.L., Brown M.S. Identification of NPC1 as the target of U18666A, an inhibitor of lysosomal cholesterol export and Ebola infection. eLife. 2015;4:e12177. doi: 10.7554/eLife.12177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa T., Ashihara E., Kimura S. The Bcr-Abl tyrosine kinase inhibitor imatinib and promising new agents against Philadelphia chromosome-positive leukemias. Int. J. Clin. Oncol. 2007;12:327–340. doi: 10.1007/s10147-007-0699-1. [DOI] [PubMed] [Google Scholar]
- Manley P.W., Drueckes P., Fendrich G., Furet P., Liebetanz J., Martiny-Baron G., Mestan J., Trappe J., Wartmann M., Fabbro D. Extended kinase profile and properties of the protein kinase inhibitor nilotinib. Biochim. Biophys. Acta. 2010;1804:445–453. doi: 10.1016/j.bbapap.2009.11.008. [DOI] [PubMed] [Google Scholar]
- Martina J.A., Chen Y., Gucek M., Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8:903–914. doi: 10.4161/auto.19653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina J.A., Diab H.I., Brady O.A., Puertollano R. TFEB and TFE3 are novel components of the integrated stress response. EMBO J. 2016;35:479–495. doi: 10.15252/embj.201593428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina J.A., Puertollano R. Rag GTPases mediate amino acid-dependent recruitment of TFEB and MITF to lysosomes. J. Cell Biol. 2013;200:475–491. doi: 10.1083/jcb.201209135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina D.L., Di Paola S., Peluso I., Armani A., De Stefani D., Venditti R., Montefusco S., Scotto-Rosato A., Prezioso C., Forrester A. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015;17:288–299. doi: 10.1038/ncb3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina D.L., Fraldi A., Bouche V., Annunziata F., Mansueto G., Spampanato C., Puri C., Pignata A., Martina J.A., Sardiello M. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev. Cell. 2011;21:421–430. doi: 10.1016/j.devcel.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano G., Ballabio A. TFEB at a glance. J. Cell Sci. 2016;129:2475–2481. doi: 10.1242/jcs.146365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano G., Esposito A., Choi H., Matarese M., Benedetti V., Di Malta C., Monfregola J., Medina D.L., Lippincott-Schwartz J., Ballabio A. mTOR-dependent phosphorylation controls TFEB nuclear export. Nat. Commun. 2018;9:3312. doi: 10.1038/s41467-018-05862-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naureckiene S., Sleat D.E., Lackland H., Fensom A., Vanier M.T., Wattiaux R., Jadot M., Lobel P. Identification of HE1 as the second gene of Niemann-Pick C disease. Science. 2000;290:2298–2301. doi: 10.1126/science.290.5500.2298. [DOI] [PubMed] [Google Scholar]
- Pagan F., Hebron M., Valadez E.H., Torres-Yaghi Y., Huang X., Mills R.R., Wilmarth B.M., Howard H., Dunn C., Carlson A. Nilotinib effects in Parkinson's disease and Dementia with Lewy bodies. J. Parkinson's Dis. 2016;6:503–517. doi: 10.3233/JPD-160867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagan F.L., Hebron M.L., Wilmarth B., Torres-Yaghi Y., Lawler A., Mundel E.E., Yusuf N., Starr N.J., Arellano J., Howard H.H. Pharmacokinetics and pharmacodynamics of a single dose Nilotinib in individuals with Parkinson's disease. Pharmacol. Res. Perspect. 2019;7:e00470. doi: 10.1002/prp2.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri M., Impey S., Kang H., di Ronza A., Pelz C., Sardiello M., Ballabio A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011;20:3852–3866. doi: 10.1093/hmg/ddr306. [DOI] [PubMed] [Google Scholar]
- Palmieri M., Pal R., Nelvagal H.R., Lotfi P., Stinnett G.R., Seymour M.L., Chaudhury A., Bajaj L., Bondar V.V., Bremner L. mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat. Commun. 2017;8:14338. doi: 10.1038/ncomms14338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parenti G., Andria G., Ballabio A. Lysosomal storage diseases: from pathophysiology to therapy. Annu. Rev. Med. 2015;66:471–486. doi: 10.1146/annurev-med-122313-085916. [DOI] [PubMed] [Google Scholar]
- Patterson M.C., Hendriksz C.J., Walterfang M., Sedel F., Vanier M.T., Wijburg F., Group N.-C.G.W. Recommendations for the diagnosis and management of Niemann-Pick disease type C: an update. Mol. Genet. Metab. 2012;106:330–344. doi: 10.1016/j.ymgme.2012.03.012. [DOI] [PubMed] [Google Scholar]
- Pentchev P.G., Boothe A.D., Kruth H.S., Weintroub H., Stivers J., Brady R.O. A genetic storage disorder in BALB/C mice with a metabolic block in esterification of exogenous cholesterol. J. Biol. Chem. 1984;259:5784–5791. [PubMed] [Google Scholar]
- Polito V.A., Li H., Martini-Stoica H., Wang B., Yang L., Xu Y., Swartzlander D.B., Palmieri M., di Ronza A., Lee V.M. Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Mol. Med. 2014;6:1142–1160. doi: 10.15252/emmm.201303671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raben N., Puertollano R. TFEB and TFE3: linking lysosomes to cellular adaptation to stress. Annu. Rev. Cell. Dev. Biol. 2016;32:255–278. doi: 10.1146/annurev-cellbio-111315-125407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y., Chen J., Wu X., Gui C., Mao K., Zou F., Li W. Role of c-abl-GSK3beta signaling in MPP+-Induced autophagy-lysosomal dysfunction. Toxicol. Sci. 2018;165:232–243. doi: 10.1093/toxsci/kfy155. [DOI] [PubMed] [Google Scholar]
- Roczniak-Ferguson A., Petit C.S., Froehlich F., Qian S., Ky J., Angarola B., Walther T.C., Ferguson S.M. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signaling. 2012;5:ra42. doi: 10.1126/scisignal.2002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas F., Gonzalez D., Cortes N., Ampuero E., Hernandez D.E., Fritz E., Abarzua S., Martinez A., Elorza A.A., Alvarez A. Reactive oxygen species trigger motoneuron death in non-cell-autonomous models of ALS through activation of c-Abl signaling. Front. Cell. Neurosci. 2015;9:203. doi: 10.3389/fncel.2015.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardiello M., Ballabio A. Lysosomal enhancement: a CLEAR answer to cellular degradative needs. Cell cycle. 2009;8:4021–4022. doi: 10.4161/cc.8.24.10263. [DOI] [PubMed] [Google Scholar]
- Sardiello M., Palmieri M., di Ronza A., Medina D.L., Valenza M., Gennarino V.A., Di Malta C., Donaudy F., Embrione V., Polishchuk R.S. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–477. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- Settembre C., Ballabio A. TFEB regulates autophagy: an integrated coordination of cellular degradation and recycling processes. Autophagy. 2011;7:1379–1381. doi: 10.4161/auto.7.11.17166. [DOI] [PubMed] [Google Scholar]
- Settembre C., Ballabio A. Lysosomal adaptation: how the lysosome responds to external cues. Cold Spring Harb. Perspect. Biol. 2014;6:a016907. doi: 10.1101/cshperspect.a016907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C., Di Malta C., Polito V.A., Garcia Arencibia M., Vetrini F., Erdin S., Erdin S.U., Huynh T., Medina D., Colella P. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C., Fraldi A., Medina D.L., Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013;14:283–296. doi: 10.1038/nrm3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C., Medina D.L. TFEB and the CLEAR network. Methods Cell Biol. 2015;126:45–62. doi: 10.1016/bs.mcb.2014.11.011. [DOI] [PubMed] [Google Scholar]
- Settembre C., Zoncu R., Medina D.L., Vetrini F., Erdin S., Erdin S., Huynh T., Ferron M., Karsenty G., Vellard M.C. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31:1095–1108. doi: 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spampanato C., Feeney E., Li L., Cardone M., Lim J.A., Annunziata F., Zare H., Polishchuk R., Puertollano R., Parenti G. Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol. Med. 2013;5:691–706. doi: 10.1002/emmm.201202176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturley S.L., Patterson M.C., Balch W., Liscum L. The pathophysiology and mechanisms of NP-C disease. Biochim. Biophys. Acta. 2004;1685:83–87. doi: 10.1016/j.bbalip.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Tanabe A., Yamamura Y., Kasahara J., Morigaki R., Kaji R., Goto S. A novel tyrosine kinase inhibitor AMN107 (nilotinib) normalizes striatal motor behaviors in a mouse model of Parkinson's disease. Front. Cell. Neurosci. 2014;8:50. doi: 10.3389/fncel.2014.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunemi T., Ashe T.D., Morrison B.E., Soriano K.R., Au J., Roque R.A., Lazarowski E.R., Damian V.A., Masliah E., La Spada A.R. PGC-1alpha rescues Huntington's disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci. transl. Med. 2012;4:142ra197. doi: 10.1126/scitranslmed.3003799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas L.M., Cerpa W., Munoz F.J., Zanlungo S., Alvarez A.R. Amyloid-beta oligomers synaptotoxicity: the emerging role of EphA4/c-Abl signaling in Alzheimer's disease. Biochim. Biophys. Acta. 2018;1864:1148–1159. doi: 10.1016/j.bbadis.2018.01.023. [DOI] [PubMed] [Google Scholar]
- Walkley S.U., Suzuki K. Consequences of NPC1 and NPC2 loss of function in mammalian neurons. Biochim. Biophys. Acta. 2004;1685:48–62. doi: 10.1016/j.bbalip.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Wang J.Y. The capable ABL: what is its biological function? Mol. Cell. Biol. 2014;34:1188–1197. doi: 10.1128/MCB.01454-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Gao Q., Yang M., Zhang X., Yu L., Lawas M., Li X., Bryant-Genevier M., Southall N.T., Marugan J. Up-regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proc. Natl. Acad. Sci. U S A. 2015;112:E1373–E1381. doi: 10.1073/pnas.1419669112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Q., Yan P., Ma X., Liu H., Perez R., Zhu A., Gonzales E., Burchett J.M., Schuler D.R., Cirrito J.R. Enhancing astrocytic lysosome biogenesis facilitates Abeta clearance and attenuates amyloid plaque pathogenesis. J. Neurosci. 2014;34:9607–9620. doi: 10.1523/JNEUROSCI.3788-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Q., Yan P., Ma X., Liu H., Perez R., Zhu A., Gonzales E., Tripoli D.L., Czerniewski L., Ballabio A. Neuronal-Targeted TFEB accelerates lysosomal degradation of APP, reducing abeta generation and amyloid plaque pathogenesis. J. Neurosci. 2015;35:12137–12151. doi: 10.1523/JNEUROSCI.0705-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y., Liu Z., Cao J., Ma Q., Gao X., Wang Q., Jin C., Zhou Y., Wen L., Ren J. Gps 2.1: enhanced prediction of kinase-specific phosphorylation sites with an algorithm of motif length selection. Protein Eng. Des. Selection. 2011;24:255–260. doi: 10.1093/protein/gzq094. [DOI] [PubMed] [Google Scholar]
- Yanez M.J., Belbin O., Estrada L.D., Leal N., Contreras P.S., Lleo A., Burgos P.V., Zanlungo S., Alvarez A.R. c-Abl links APP-BACE1 interaction promoting APP amyloidogenic processing in Niemann-Pick type C disease. Biochim. Biophys. Acta. 2016;1862:2158–2167. doi: 10.1016/j.bbadis.2016.08.016. [DOI] [PubMed] [Google Scholar]
- Yang J., Campobasso N., Biju M.P., Fisher K., Pan X.Q., Cottom J., Galbraith S., Ho T., Zhang H., Hong X. Discovery and characterization of a cell-permeable, small-molecule c-Abl kinase activator that binds to the myristoyl binding site. Chem. Biol. 2011;18:177–186. doi: 10.1016/j.chembiol.2010.12.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Further information about data supporting the conclusions of this manuscript will be made available by the Lead Contact to any qualified researcher.