

Abstract
We describe a new catalytic strategy to transcend the energetic limitations of visible light by electrochemically priming a photocatalyst prior to excitation. This new catalytic system is able to productively engage aryl chlorides with reduction potentials hundreds of millivolts beyond the potential of Na0 in productive radical coupling reactions. The aryl radicals produced via this strategy can be leveraged for both carbon–carbon and carbon–heteroatom bond-forming reactions. Through direct comparison, we illustrate the reactivity and selectivity advantages of this approach relative to electrolysis and photoredox catalysis.
Activation of organic molecules through single electron transfer (SET) is a pillar of preparative chemistry. New strategies to induce redox events have the potential to significantly impact organic synthesis.1 In the past decade, visible-light photoredox catalysis has enabled a tremendous array of carbon–carbon and carbon–heteroatom bond-forming reactions.2,3 Unfortunately, blue light (440 nm) possesses sufficient energy for a maximum driving force of only 2.8 eV, and the available energy is further diminished by nonradiative pathways and intersystem crossing.4 Thus, despite catalyst design improvements,5,6 many desirable substrates remain inert to visible-light photoredox catalysis.7 As a result of this limitation, dissolving metal conditions,8 which employ reactive alkali metals in condensed ammonia, remain uniquely potent reductants in the synthetic arsenal9 and are still commonly used despite significant hazards and poor chemoselectivity.10,11 Aiming to provide safer and more scalable conditions for challenging reductions, recent efforts have exploited overcharge protection to unlock deeply reducing cathodic potentials for electroorganic synthesis.12 However, the requisite electrode overpotentials intrinsically limit the functional group tolerance. Furthermore, radical intermediates generated at a cathode are prone to reduction to anions.13 Overall, a new catalytic paradigm to access extremely reducing potentials under mild conditions and without reduction of radical intermediates would address a long-standing challenge in organic synthesis (Figure 1).
Figure 1.

Strategies to induce SET reduction. All potentials shown are relative to SCE. PTH = 10-phenylphenothiazine.
To overcome the energetic limitations of blue photons, König and co-workers recently introduced an appealing approach designed to drive challenging SET events using the energy of two photons rather than one.14,15 This strategy relies on the light-mediated generation and subsequent photochemical excitation of catalytic radical anion intermediates. Although these systems push the limits of photoredox catalysis, they remain many orders of magnitude less reducing than alkali metals. Inspired by photophysical studies suggesting that other organic radical ions can serve as potent photoreductants,16,17 we questioned whether an alternative means of priming a photoredox catalyst with an electron prior to excitation could provide a general catalyst design platform to transcend the energetic limitations of visible light.
We hypothesized that electrochemistry18,19 could offer a more flexible approach than photoreduction to generate electron-primed photoredox catalysts. In addition to providing access to new catalysts, this approach eliminates the complications20 that can arise from the terminal reductants commonly used in photoredox catalysis, such as Et3N. This strategy builds on both long-standing21 and recent22 pioneering efforts combining electrochemistry with photochemistry.23 The majority of these examples take advantage of the desirable features of electrochemistry to generate known photochemically active intermediates or catalysts. However, electrochemical generation of new families of photocatalysts for organic synthesis remains largely unexplored. Recently, Lambert and co-workers reported a new and highly oxidizing photocatalyst (with a calculated potential of +3.3 V vs SCE) that is electrochemically accessible under a mildly oxidizing potential.22d Concurrently, we were exploring the use of electrochemistry to access new, electronically destabilized photocatalysts for challenging reductions.24 Herein we demonstrate that electrochemistry is a viable strategy to generate highly reducing electron-primed photoredox catalysts. We exploit this approach to identify an aryl imide photocatalyst capable of engaging substrates with reduction potentials on par with alkali metals in SET-initiated radical coupling reactions under otherwise mild conditions.
To explore this idea, we targeted the reductive generation of aryl radicals from unactivated precursors. These reactive intermediates are known to participate in a range of synthetically useful carbon–carbon and carbon–heteroatom bond-forming reactions; however, they are typically generated from diazonium salts or aryl iodides using modern photoredox catalysts.25 With the most reducing visible-light photoredox catalysts, aryl bromides are suitable radical precursors.26,27 Unfortunately, aryl chlorides comprise over half of the commercially available aryl halides28 yet are inert under conventional visible-light photoredox catalysis unless they bear electron-withdrawing groups.29–31 This limitation is a result of the combination of thermodynamically challenging SET and the low fragmentation rate due to the relatively strong C(sp2)–Cl bond.32
To assess the viability of the proposed electrophotocatalytic approach, we investigated the dehalogenation of 4-bromobiphenyl (1) because of its reduction potential beyond the standard range of photoredox catalysts (−2.4 V vs SCE) and rapid fragmentation after reduction, as this provides a highfidelity readout for successful SET.32 Using this model reaction, we assessed a series of aryl imides for activity under visible-light irradiation and an appropriate electrochemical potential to reductively activate the imide (Table 1). The radical anion derived from perylene diimide (PDI) can act as an electron-primed photoredox catalyst under two-photon conditions14a and is also well-behaved electrochemically.33 Unfortunately, PDI proved ineffective in the dehalogenation of 1 under these conditions. Photophysical studies have indicated that naphthlene-based analogues (NpDI and NpMI) are more potent photoreductants after they are primed with an electron,17c but they have yet to be leveraged in synthesis. Excitingly, under electrophotocatalytic conditions both NpDI and NpMI promoted the dehalogenation of 1, despite significant electrochemical underpotentials in each case (1.6 and 1.1 V vs SCE respectively). While both NpMI and NpDI are sufficiently potent photoreductants to reduce 1, NpMI promoted dehalogenation significantly more efficiently. However, further stripping down the aromatic core to a phthalimide derivative, PhMI, resulted in a less effective photocatalyst than NpMI. On the basis of these data, we selected NpMI for further study after verifying that no significant conversion was observed in the absence of an applied voltage, light, or the catalyst.
Table 1.
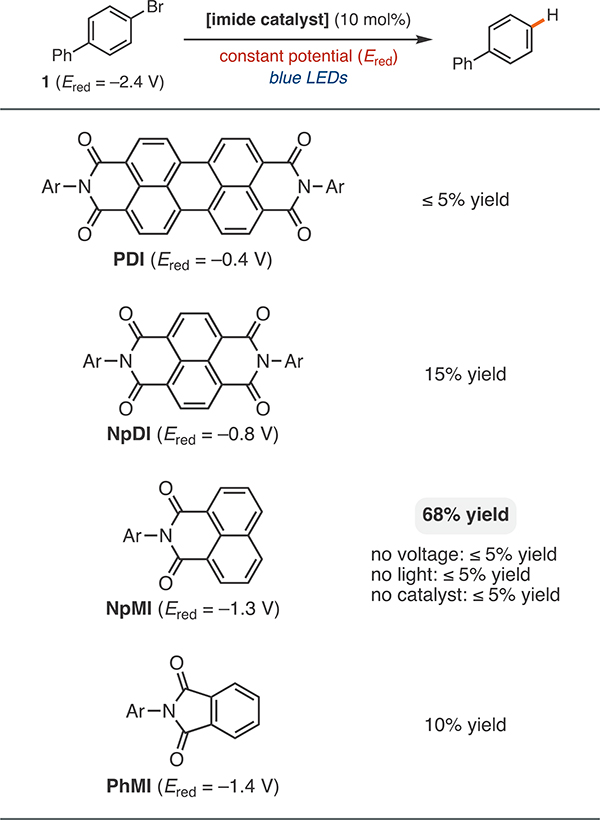 |
All redox potentials given relative to SCE.
Ar = 2,6-diisopropyl-phenyl.
Reactions were conducted on a 0.4 mmol scale in DMF (0.1 M Bu4NPF6) in the presence of 2,4,6-tri-tert-butylphenol (10 mol %) and isopropyl alcohol (1 equiv). See the Supporting Information (SI) for further details.
Having identified a promising electrochemically accessible photocatalyst, we explored whether this system could engage abundant but much more challenging aryl chlorides in radical coupling reactions. We first probed the viability of a photo-Arbuzov process,34 a classic carbon–heteroatom bond-forming reaction that proceeds through an aryl radical intermediate (Table 2). For these studies, we employed more convenient constant-current conditions.35 We found that under simultaneous electrolysis and irradiation, NpMI induced the high-yielding coupling of aryl chlorides with reduction potentials at and beyond the limits of conventional visible-light photoredox catalysis (2–3). To identify the limits of this catalytic system, we next evaluated increasingly electron-rich aryl chloride substrates. Excitingly, aryl chloride substrates bearing electron-donating groups still underwent efficient SET-induced phosphorylation (4–7) even though they possess reduction potentials comparable to that of Na0 (−2.9 V vs SCE). Notably, an exceptionally electron-rich aryl chloride (−3.4 V vs SCE)36 was successfully reduced to produce 7. This result indicates that these conditions provide potency comparable to that of Li0 (−3.3 V vs SCE). To our delight, despite the presence of such a potent reductant, aryl chloride substrates bearing potentially sensitive functional groups7,37 such as esters (8), nitriles (9), carbamates (10), organoboron reagents (11), and heterocycles (12 and 13) all underwent productive SET-induced radical phosphorylation, and the corresponding products were isolated in good to excellent yields.
Table 2.
Scope of Aryl Chloride Phosphorylationa
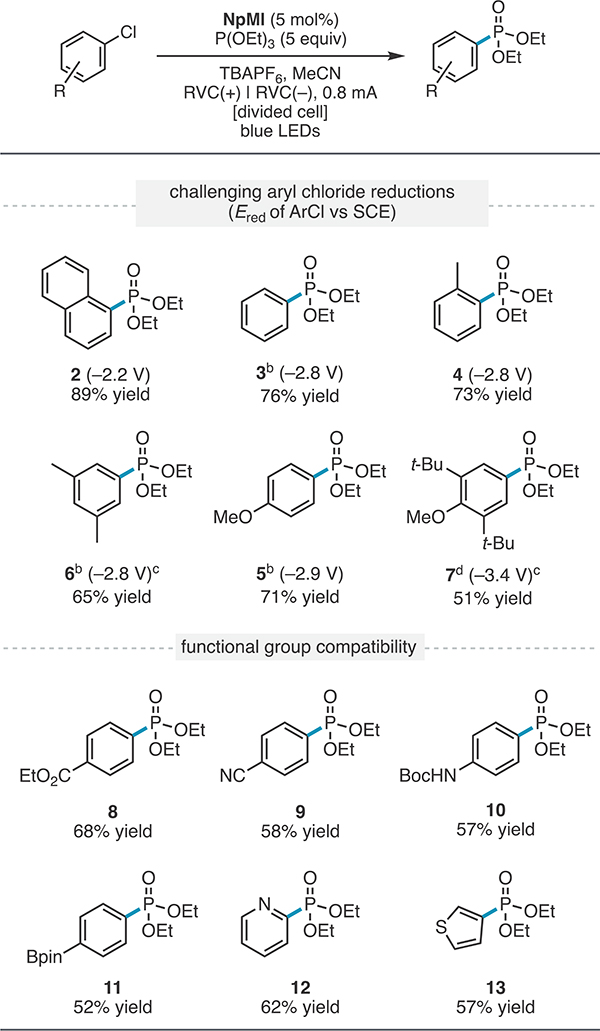 |
Reactions were conducted on a 0.4 mmol scale and run for 8 h. Et3N (2 equiv) was employed in the anode as the counter reaction. See the SI for further experimental details.
The reaction was run for 14 h.
Ered was determined by differential pulse voltammetry
0.4 mA current.
Having established the viability of carbon–heteroatom bond-forming reactions from diverse aryl chlorides, we next aimed to intercept the aryl radical intermediate with a heterocycle to form a new carbon–carbon bond (Table 3). We found that the aryl radical intermediates generated under these conditions from neutral to electron-rich aryl chlorides (14–18) could be effectively coupled to N-methylpyrrole, a classic radical trap.38 Again, reductively sensitive functional groups were well-tolerated despite the potency of the photoreductant employed (19–21).
Table 3.
Scope of Aryl Chloride (Hetero)arylationa
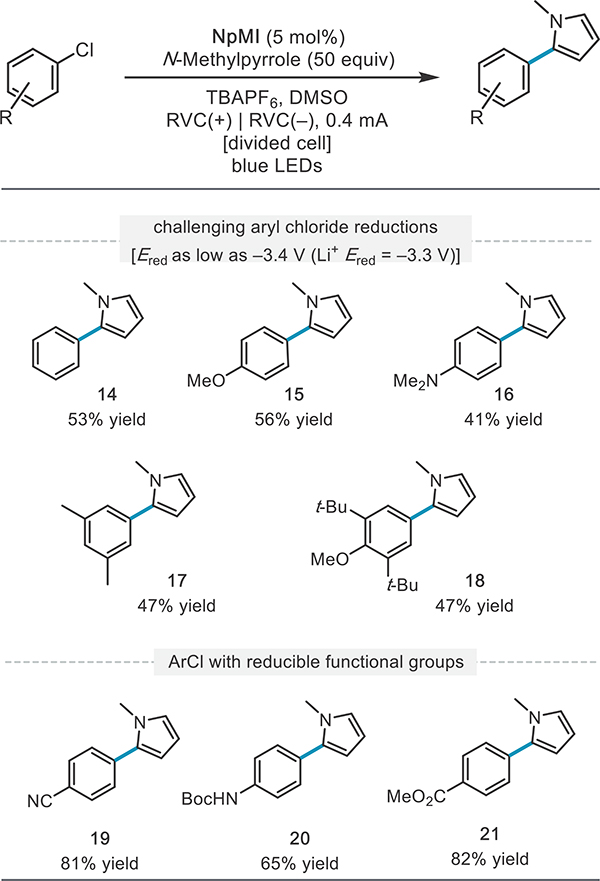 |
Reactions were conducted on a 0.4 mmol scale and run for 27 h. Et3N (2 equiv) was employed in the anode as the counter reaction. See the SI for further experimental details.
With a new catalytic strategy in hand, we next compared its efficacy to those of traditional photochemical and electrochemical approaches for the reductive activation of aryl chlorides (Figure 2). To this end, we investigated the relative yields of N-methylpyrrole coupling and dehalogenation within a subset of aryl chloride substrates ranging from electron-deficient to electron-rich. When the electron-primed photoredox system was used, each substrate delivered the desired product with excellent selectivity for radical coupling over dehalogenation. In contrast, 10-phenylphenothiazine (PTH), an exceptionally reducing photoredox catalyst (−2.1 V vs SCE),26a could only induce the coupling of the electron-deficient aryl chloride. The neutral and electron-rich substrates were unconverted by PTH, consistent with the energetic limitations of visible-light photoredox Direct electrolysis, on the other hand, provided significantly diminished selectivity for coupling of the electron-deficient substrate (2:1) and the other two substrates yielded in exclusively dehalogenation. These results are consistent with over-reduction at the electrode surface that precludes radical coupling reactions at the requisite potentials for aryl chloride reduction.
Figure 2.

Comparison of reactivities and selectivities observed with various methods of generating aryl radical intermediates using N-methylpyrrole as a radical trap. Yields of coupling and dehalogenation were measured relative to an internal standard. The electron-primed photoredox conditions were the same as in Table 3. The photoredox conditions followed pyrrole coupling procedures from the literature employing PTH as a photocatalyst. Several direct electrolysis conditions were attempted. The reported conditions provided the highest yield of A and were standard reaction conditions from Table 3 without catalyst. For further details, see the SI.
We next subjected radical clock 22 to both electron-primed photoredox and direct electrolysis conditions to probe the presence of an aryl radical intermediate and benchmark the rate of its over-reduction (Scheme 1). The aryl radical derived from 22 undergoes radical cyclization with a rate of 8 × 109 s−1.39 As anticipated, NpMI under blue-light irradiation and constant-current electrolysis delivered selective cyclization (54% yield, ≥20:1 selectivity for cyclization over dehalogenation and aryl anion-derived40 isomerization products). This result is fully consistent with the proposed intermediacy of an aryl radical intermediate and high selectivity for radical chemistry instead of over-reduction. Direct electrolysis, however, provided no observable cyclization and generated only dehalogenation and isomerization products consistent with anionic intermediates. This indicates that under the direct electrolysis conditions investigated, any radical reactions with rate constants lower than 109 s−1 will not be viable because of competitive electrochemical reduction of the radical. This is consistent with the facile reduction of aryl radical intermediates at electrode surfaces (phenyl radical Ered = +0.05 V vs SCE).41
Scheme 1.

Comparison of Radical Clock Outcomesa
aThe reaction was conducted on a 0.4 mmol scale for 20 h.´
Finally, we aimed to gain preliminary insight into the promising chemoselectivity observed with this potent catalytic reductant. Notably, classic photoredox approaches are sensitive to not only the reduction potential of the substrate but also the fragmentation rate of the radical anion formed via SET.25a This is likely due to competition between back electron transfer and the productive fragmentation and coupling. Although back electron transfer can be a hindrance,42 we suspect that this feature also contributes to the excellent chemoselectivity profiles observed in photoredox catalysis. Thus, we wanted to ascertain whether the exceptionally potent photoreductant explored herein exhibited analogous reactivity or whether SET was irreversible.
To this end, we conducted a series of one-pot intermolecular competition experiments between bromo- and chlorobiphenyl (Figure 3). These halogen congeners possess the same reduction potential (−2.4 V vs SCE), but their radical anions exhibit significantly different fragmentation rates.32,43 We subjected a 1:1 mixture of the two aryl halides to NpMI under simultaneous irradiation and a working potential of −1.3 V vs SCE. We found that the initial rate of C(sp2)–Br cleavage was significantly higher than that of C(sp2)–Cl cleavage (krel = 8) despite the fact the two aryl halides possess identical reduction potentials. This observation excludes that conversion is based exclusively on the reduction potential. In stark contrast, the two substrates are converted at similar rates under direct electrolysis conditions (krel = 2).44 Consistent with prior work in photoredox catalysis, PTH promoted the dehalogenation of bromobiphenyl more rapidly than that of the chloride (krel = 15). Taken together, these data indicate that productive conversion is not governed exclusively by the reduction potential under either electron-primed or conventional photoredox catalysis. This observation provides a plausible rationale for the promising chemoselectivity observed under the conditions reported herein relative to deeply reducing direct electrolysis, which predominantly commits to product formation on the basis of the substrate reduction potential.
Figure 3.

Comparison of one-pot competition experiments. krel is given as kBr/kcl and was measured by gas chromatography based on reactions run to low conversion (<20%). The electron-primed photoredox conditions followed from Table 1. The direct electrolysis conditions were those reported in ref 44. Previously reported photoredox conditions were employed 26a. See the SI for details.
Overall, we have demonstrated that electrochemical stimulation is a viable strategy to generate catalytic photoreductants capable of transcending the limits of modern photoredox catalysis. We report effective radical couplings of substrates hundreds of millivolts more challenging to reduce than previous photoredox strategies, including substrates with reduction potentials well beyond that of Na0 and more negative than that of Li0. Crucially, despite accessing such negative potentials, the reactions possess functional group tolerance profiles more consistent with traditional photoredox catalysis than direct electrolysis and result in high selectivity for radical coupling over dehalogenation. Beyond unlocking electronically diverse aryl chlorides as aryl radical precursors, these data lay the foundation for a new catalyst design paradigm. We anticipate that electrochemically priming photocatalysts prior to excitation will allow much more challenging reductions than are feasible with blue light alone and will result in a myriad of transformations inspired by what is feasible under dissolving metal conditions and beyond.
Supplementary Material
ACKNOWLEDGMENTS
We thank Prof. Alison Wendlandt, Prof. Tehshik Yoon, Prof. Daniel Weix, Prof. Bill Morandi, and Sara Alektiar for helpful suggestions and manuscript proofreading. Additionally, we thank the entire Weix group for allowing us to use their GC and the Stahl, Weix, Yoon, and Schomaker groups for sharing their chemical inventory. Dr. Blaise J. Thompson is acknowledged for his assistance with galvanostat design and fabrication. Tracy Drier is acknowledged for electrochemical glassware fabrication. We also acknowledge the invaluable support and helpful suggestions from group members Dylan Holst and Diana Wang throughout this project. This work was financially supported by the Office of the Vice Chancellor for Research and Graduate Education at the University of Wisconsin-Madison with funding from the Wisconsin Alumni Research Foundation. Spectroscopic instrumentation was supported by a generous gift from Paul. J. and Margaret M. Bender, NSF (CHE-1048642), and NIH (1S10 OD020022-1).
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.9b12328.
Experimental procedures, electrochemical characterization, radical clock and competition experiments, comparison of reductive activation methods, constant-current control experiments, product isolation and characterization, and NMR spectra (PDF)
Contributor Information
Nicholas G. W. Cowper, University of Wisconsin-Madison, Madison, Wisconsin.
Colleen P. Chernowsky, University of Wisconsin-Madison, Madison, Wisconsin.
Oliver P. Williams, University of Wisconsin-Madison, Madison, Wisconsin
Zachary K. Wickens, University of Wisconsin-Madison, Madison, Wisconsin.
REFERENCES
- (1) (a).Ashby EC Single-Electron Transfer, a Major Reaction Pathway in Organic Chemistry. An Answer to Recent Criticisms. Acc. Chem. Res 1988, 21, 414–421. [Google Scholar]; (b) Zhang N; Samanta SR; Rosen BM; Percec V Single Electron Transfer in Radical Ion and Radical-Mediated Organic, Materials and Polymer Synthesis. Chem. Rev 2014, 114, 5848–5958. [DOI] [PubMed] [Google Scholar]; (c) Broggi J; Terme T; Vanelle P Organic Electron Donors as Powerful Single-Electron Reducing Agents in Organic Synthesis. Angew. Chem., Int. Ed 2014, 53, 384–413. [DOI] [PubMed] [Google Scholar]; (d) Girard P; Namy JL; Kagan HB Divalent Lanthanide Derivatives in Organic Synthesis. 1. Mild Preparation of Samarium Iodide and Ytterbium Iodide and Their Use as Reducing or Coupling Agents. J. Am. Chem. Soc 1980, 102, 2693–2698. [Google Scholar]; (e) Eberson L Electron-Transfer Reactions in Organic Chemistry. Adv. Phys. Org. Chem 1982, 18, 79–185. [Google Scholar]
- (2).(a) For selected reviews of visible-light photoredox catalysis, see: Prier CK; Rankic DA; MacMillan DWC Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shaw MH; Twilton J; MacMillan DWC Photoredox Catalysis in Organic Chemistry. J. Org. Chem 2016, 81, 6898–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Romero NA; Nicewicz DA Organic Photoredox Catalysis. Chem. Rev 2016, 116, 10075–10166. [DOI] [PubMed] [Google Scholar]; (d) Skubi KL; Blum TR; Yoon TP Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev 2016, 116, 10035–10074. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) DiRocco DA; Dykstra K; Krska S; Vachal P; Conway DV; Tudge M Late-Stage Functionalization of Biologically Active Heterocycles Through Photoredox Catalysis. Angew. Chem., Int. Ed 2014, 53, 4802–4806. [DOI] [PubMed] [Google Scholar]; (f) Narayanam JMR; Stephenson CRJ Visible Light Photoredox Catalysis: Applications in Organic Synthesis. Chem. Soc. Rev 2011, 40, 102–113. [DOI] [PubMed] [Google Scholar]
- (3).For a special issue on photoredox catalysis in organic chemistry, see: Accounts of Chemical Research 2016, Vol. 49, Issue 10. [Google Scholar]
- (4).Arias-Rotondo DM; McCusker JK The Photophysics of Photoredox Catalysis: A Roadmap for Catalyst Design. Chem. Soc. Rev 2016, 45, 5803–5820. [DOI] [PubMed] [Google Scholar]
- (5).(a) For selected examples of design improvements of organic photoredox catalysts, see: Theriot JC; Lim C-H; Yang H; Ryan MD; Musgrave CB; Miyake GM Organocatalyzed Atom Transfer Radical Polymerization Driven by Visible Light. Science 2016, 352, 1082–1086. [DOI] [PubMed] [Google Scholar]; (b) Speckmeier E; Fischer TG; Zeitler K A Toolbox Approach To Construct Broadly Applicable Metal-Free Catalysts for Photoredox Chemistry: Deliberate Tuning of Redox Potentials and Importance of Halogens in Donor-Acceptor Cyanoarenes. J. Am. Chem. Soc 2018, 140, 15353–15365. [DOI] [PubMed] [Google Scholar]; (c) Joshi-Pangu A; Lévesque F; Roth HG; Oliver SF; Campeau L-C; Nicewicz D; DiRocco DA Acridinium-Based Photocatalysts: A Sustainable Option in Photoredox Catalysis. J. Org. Chem 2016, 81, 7244–7249. [DOI] [PubMed] [Google Scholar]
- (6).(a) For selected progress toward the design of earth-abundant photoredox catalysts, see: Hockin BM; Li C; Robertson N; Zysman-Colman E Photoredox Catalysts Based on Earth-Abundant Metal Complexes. Catal. Sci. Technol 2019, 9, 889–915. [Google Scholar]; (b) Larsen CB; Wenger OS Photoredox Catalysis with Metal Complexes Made from Earth-Abundant Elements. Chem. - Eur. J 2018, 24, 2039–2058. [DOI] [PubMed] [Google Scholar]; (c) Herr P; Glaser F; Büldt LA; Larsen CB; Wenger OS Long-Lived, Strongly Emissive, and Highly Reducing Excited States in Mo(0) Complexes with Chelating Isocyanides. J. Am. Chem. Soc 2019, 141, 14394–14402. [DOI] [PubMed] [Google Scholar]; (d) Stevenson SM; Shores MP; Ferreira EM Photooxidizing Chromium Catalysts for Promoting Radical Cation Cycloadditions. Angew. Chem., Int. Ed 2015, 54 (22), 6506–6510. [DOI] [PubMed] [Google Scholar]; (e) Ahn JM; Peters JC; Fu GC Design of a Photoredox Catalyst That Enables the Direct Synthesis of Carbamate-Protected Primary Amines via Photoinduced, Copper-Catalyzed N-Alkylation Reactions of Unactivated Secondary Halides. J. Am. Chem. Soc 2017, 139, 18101–18106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Roth HG; Romero NA; Nicewicz DA Experimental and Calculated Electrochemical Potentials of Common Organic Molecules for Applications to Single-Electron Redox Chemistry. Synlett 2016, 27, 714–723. [Google Scholar]
- (8) (a).Bouveault L; Blanc G Preparation des Alcools Primaires aú Moyen des Acides Correspondants. C. R. Acad. Sci 1903, 136, 1676–1678. [Google Scholar]; (b) Birch AJ 117 Reduction by Dissolving Metals. Part I. J. Chem. Soc 1944, 430–436. [Google Scholar]
- (9).(a) Although not as reducing as dissolving metal conditions, SmI2/HMPA can promote challenging reductions and has been broadly employed in synthesis. For reviews, see: Edmonds DJ; Johnston D; Procter DJ Samarium(II)-Iodide-Mediated Cyclizations in Natural Product Synthesis. Chem. Rev 2004, 104, 3371–3404. [DOI] [PubMed] [Google Scholar]; (b) Nicolaou KC; Ellery SP; Chen JS Samarium Diiodide Mediated Reactions in Total Synthesis. Angew. Chem., Int. Ed 2009, 48, 7140–7165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) For recent examples where dissolving metal conditions were crucial to a successful synthesis, see: Chuang KV; Xu C; Reisman SE A 15-Step Synthesis of (+)-Ryanodol. Science 2016, 353, 912–915. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liu Y-T; Li L-P; Xie J-H; Zhou Q-L Divergent Asymmetric Total Synthesis of Mulinane Diterpenoids. Angew. Chem., Int. Ed 2017, 56, 12708–12711. [DOI] [PubMed] [Google Scholar]; (c) He C; Stratton TP; Baran PS Concise Total Synthesis of Herqulines B and C. J. Am. Chem. Soc 2019, 141, 29–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).For an example of large-scale use of dissolving metal conditions and an illustration of the necessary engineering controls, see: Joshi DK; Sutton JW; Carver S; Blanchard JP Experiences with Commercial Production Scale Operation of Dissolving Metal Reduction Using Lithium Metal and Liquid Ammonia. Org. Process Res. Dev 2005, 9, 997–1002. [Google Scholar]
- (12).Peters BK; Rodriguez KX; Reisberg SH; Beil SB; Hickey DP; Kawamata Y; Collins M; Starr J; Chen L; Udyavara S; Klunder K; Gorey TJ; Anderson SL; Neurock M; Minteer SD; Baran PS Scalable and Safe Synthetic Organic Electroreduction Inspired by Li-Ion Battery Chemistry. Science 2019, 363, 838–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13) (a).Nelleborg P; Lund H; Eriksen J Photochemical vs. Electrochemical Electron-Transfer Reactions. One-Electron Reduction of Aryl Halides by Photoexcited Anion Radicals. Tetrahedron Lett 1985, 26, 1773–1776. [Google Scholar]; (b) Costentin C; Robert M; Savéant J-M Fragmentation of Aryl Halide π Anion Radicals. Bending of the Cleaving Bond and Activation vs Driving Force Relationships. J. Am. Chem. Soc 2004, 126, 16051–16057. [DOI] [PubMed] [Google Scholar]
- (14) (a).Ghosh I; Ghosh T; Bardagi JI; König B Reduction of Aryl Halides by Consecutive Visible Light-Induced Electron Transfer Processes. Science 2014, 346 (6210), 725–728. [DOI] [PubMed] [Google Scholar]; (b) Ghosh I; König B Chromoselective Photocatalysis: Controlled Bond Activation through Light-Color Regulation of Redox Potentials. Angew. Chem., Int. Ed 2016, 55 (27), 7676–7679. [DOI] [PubMed] [Google Scholar]; (c) Neumeier M; Sampedro D; Májek M; de la Peña O’Shea VA; Jacobi von Wangelin A; Pérez-Ruiz R Dichromatic Photocatalytic Substitutions of Aryl Halides with a Small Organic Dye. Chem. - Eur. J 2018, 24, 105–108. [DOI] [PubMed] [Google Scholar]
- (15).For a recent example of high-power laser irradiation allowing excitation of the excited state of a photoredox catalyst to produce solvated electrons, see: Kerzig C; Guo X; Wenger OS Unexpected Hydrated Electron Source for Preparative Visible-Light Driven Photoredox Catalysis. J. Am. Chem. Soc 2019, 141, 2122–2127. [DOI] [PubMed] [Google Scholar]
- (16).(a) For reviews covering photochemically active radical anions, see: La Porte NT; Martinez JF; Chaudhuri S; Hedström S; Batista VS; Wasielewski MR Photoexcited Radical Anion Super-Reductants for Solar Fuels Catalysis. Coord. Chem. Rev 2018, 361, 98–119. [Google Scholar]; (b) Fox MA The Photoexcited States of Organic Anions. Chem. Rev 1979, 79, 253–273. [Google Scholar]
- (17).(a) For relevant examples of photophysical studies of radical anions, see: Fukuzumi S; Ohkubo K; Chen Y; Pandey RK; Zhan R; Shao J; Kadish KM Photophysical and Electrochemical Properties of New Bacteriochlorins and Characterization of Radical Cation and Radical Anion Species. J. Phys. Chem. A 2002, 106, 5105–5113. [Google Scholar]; (b) Fujitsuka M; Kim SS; Lu C; Tojo S; Majima T Intermolecular and Intramolecular Electron Transfer Processes from Excited Naphthalene Diimide Radical Anions. J. Phys. Chem. B 2015, 119, 7275–7282. [DOI] [PubMed] [Google Scholar]; (c) Gosztola D; Niemczyk MP; Svec W; Lukas AS; Wasielewski MR Excited Doublet States of Electrochemically Generated Aromatic Imide and Diimide Radical Anions. J. Phys. Chem. A 2000, 104, 6545–6551. [Google Scholar]
- (18).(a) For general reviews of electroorganic synthesis, see: Yan M; Kawamata Y; Baran PS Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance. Chem. Rev 2017, 117, 13230–13319. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yoshida J; Kataoka K; Horcajada R; Nagaki A Modern Strategies in Electroorganic Synthesis. Chem. Rev 2008, 108, 2265–2299. [DOI] [PubMed] [Google Scholar]; (c) Wiebe A; Gieshoff T; Möhle S; Rodrigo E; Zirbes M; Waldvogel SR Electrifying Organic Synthesis. Angew. Chem., Int. Ed 2018, 57, 5594–5619. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Moeller KD Using Physical Organic Chemistry To Shape the Course of Electrochemical Reactions. Chem. Rev 2018, 118, 4817–4833. [DOI] [PubMed] [Google Scholar]
- (19).(a) For reviews specifically focused on electrocatalysis, see: Francke R; Little RD Redox Catalysis in Organic Electrosynthesis: Basic Principles and Recent Developments. Chem. Soc. Rev 2014, 43, 2492–2521. [DOI] [PubMed] [Google Scholar]; (b) Meyer TH; Finger LH; Gandeepan P; Ackermann L Resource Economy by Metallaelectrocatalysis: Merging Electrochemistry and C-H Activation. Trends Chem 2019, 1, 63–76. [Google Scholar]
- (20).Et3N can promote premature radical quenching through hydrogen atom transfer and back electron transfer to Et3N•+. For details, see ref 2c. [Google Scholar]
- (21) (a).Lund H; Simonet J Anion Radicals and Dianions as Electron Transfer Reagents. J. Electroanal. Chem 1975, 65, 205–218. [Google Scholar]; (b) Shukla SS; Rusling JF Photoelectrocatalytic Reduction of 4-Chlorobiphenyl Using Anion Radicals and Visible Light. J. Phys. Chem 1985, 89, 3353–3358. [Google Scholar]; (c) Lund H; Carlsson HS; Nishida T; Enzell CR; Matsuno T Photochemistry of Radical Ions. Acta Chem. Scand 1978, 32b, 505–509. [Google Scholar]; (d) Eriksen J; Lund H; Nyvad AI; Yamato T; Mitchell RH; Dingle TW; Williams RV; Mahedevan R Electron-Transfer Fluorescence Quenching of Radical Ions. Acta Chem. Scand 1983, 37b, 459–466. [Google Scholar]; (e) Scheffold R; Orlinski R Synthesis and Reactions of Porphine-Type Metal Complexes. 15. Carbon-Carbon Bond Formation by Light Assisted B12-Catalysis. Nucleophilic Acylation of Michael Olefins. J. Am. Chem. Soc 1983, 105, 7200–7202. [Google Scholar]
- (22) (a).Li T; Kasahara T; He J; Dettelbach KE; Sammis GM; Berlinguette CP Photoelectrochemical Oxidation of Organic Substrates in Organic Media. Nat. Commun 2017, 8, 390. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yan H; Hou Z-W; Xu H-C Photoelectrochemical C-H Alkylation of Heteroarenes with Organotrifluoroborates. Angew. Chem., Int. Ed 2019, 58, 4592–4595. [DOI] [PubMed] [Google Scholar]; (c) Wang F; Stahl SS Merging Photochemistry with Electrochemistry: Functional-Group Tolerant Electrochemical Amination of C(Sp3)-H Bonds. Angew. Chem., Int. Ed 2019, 58, 6385–6390. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Huang H; Strater ZM; Rauch M; Shee J; Sisto TJ; Nuckolls C; Lambert TH Electrophotocatalysis with a Trisaminocyclopropenium Radical Dication. Angew. Chem., Int. Ed 2019, 58, 13318–13322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).For a highlight of recent efforts combining electrochemical and photochemical stimulation for organic synthesis, see: Capaldo L; Quadri LL; Ravelli D Merging Photocatalysis with Electrochemistry: The Dawn of a New Alliance in Organic Synthesis. Angew. Chem., Int. Ed 2019, 58, 17508–17510. [DOI] [PubMed] [Google Scholar]
- (24).As we were preparing this Communication, Lin, Lambert and co-workers uploaded to ChemRxiv a manuscript disclosing a similar concept. See: Kim H; Kim H; Lambert T; Lin S Reductive Electrophotocatalysis: Merging Electricity and Light to Achieve Extreme Reduction Potentials. ChemRxiv, October 8, 2019, ver. 2. DOI: 10.26434/chemrxiv.9936623.v2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25) (a).Ghosh I; Marzo L; Das A; Shaikh R; König B Visible Light Mediated Photoredox Catalytic Arylation Reactions. Acc. Chem. Res 2016, 49, 1566–1577. [DOI] [PubMed] [Google Scholar]; (b) Wang C-S; Dixneuf PH; Soulé JF Photoredox Catalysis for Building C-C Bonds from C(Sp2)-H Bonds. Chem. Rev 2018, 118, 7532–7585. [DOI] [PubMed] [Google Scholar]
- (26) (a).Discekici EH; Treat NJ; Poelma SO; Mattson KM; Hudson ZM; Luo Y; Hawker CJ; Read de Alaniz J A Highly Reducing Metal-Free Photoredox Catalyst: Design and Application in Radical Dehalogenations. Chem. Commun 2015, 51, 11705–11708. [DOI] [PubMed] [Google Scholar]; (b) Devery JJ; Nguyen JD; Dai C; Stephenson CR J. Light-Mediated Reductive Debromination of Unactivated Alkyl and Aryl Bromides. ACS Catal 2016, 6, 5962–5967. [Google Scholar]; (c) Boyington AJ; Riu M-LY; Jui NT Anti-Markovnikov Hydroarylation of Unactivated Olefins via Pyridyl Radical Intermediates. J. Am. Chem. Soc 2017, 139, 6582–6585. [DOI] [PubMed] [Google Scholar]
- (27).(a) tert-BuOK in combination with a nitrogen base can reductively generate aryl radical intermediates. See: Cheng Y; Gu X; Li P Visible-Light Photoredox in Homolytic Aromatic Substitution: Direct Arylation of Arenes with Aryl Halides. Org. Lett 2013, 15, 2664–2667. [DOI] [PubMed] [Google Scholar]; (b) Cheng Y; Gu X; Li P Visible-Light Photoredox in Homolytic Aromatic Substitution: Direct Arylation of Arenes with Aryl Halides. Org. Lett 2013, 15, 2664–2667. [DOI] [PubMed] [Google Scholar]
- (28).Weix and co-workers recently conducted an analysis concluding that there are more commercially available ArCl than ArI, ArBr, and ArOH combined. See: Huang L; Ackerman LKG; Kang K; Parsons AM; Weix DJ LiCl-Accelerated Multimetallic Cross-Coupling of Aryl Chlorides with Aryl Triflates. J. Am. Chem. Soc 2019, 141, 10978–10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).For examples of photoredox catalysts reductively activating aryl chlorides bearing electron-withdrawing groups, see ref 14a, ref 25a, and the following reference: Ghosh I; Shaikh RS; König B Sensitization-Initiated Electron Transfer for Photoredox Catalysis. Angew. Chem., Int. Ed 2017, 56, 8544–8549. [DOI] [PubMed] [Google Scholar]
- (30).For an example of a cocatalytic system composed of highly reducing DyI2 and an organic dye photochemically reducing aryl chlorides, see: Meyer AU; Slanina T; Heckel A; König B Lanthanide Ions Coupled with Photoinduced Electron Transfer Generate Strong Reduction Potentials from Visible Light. Chem. - Eur. J 2017, 23, 7900–7904. [DOI] [PubMed] [Google Scholar]
- (31).(a) For examples of UV-light-promoted dichlorination of aryl chlorides, see: Yin H; Jin Y; Hertzog JE; Mullane KC; Carroll PJ; Manor BC; Anna JM; Schelter EJ The Hexachlorocerate(III) Anion: A Potent, Benchtop Stable, and Readily Available Ultraviolet A Photosensitizer for Aryl Chlorides. J. Am. Chem. Soc 2016, 138, 16266–16273. [DOI] [PubMed] [Google Scholar]; (b) Matsubara R; Yabuta T; Md Idros U; Hayashi M; Ema F; Kobori Y; Sakata K UVA- and Visible-Light-Mediated Generation of Carbon Radicals from Organochlorides Using Nonmetal Photocatalyst. J. Org. Chem 2018, 83, 9381–9390. [DOI] [PubMed] [Google Scholar]
- (32).Costentin C; Robert M; Saveant J-M. Fragmentation of Aryl Halide π Anion Radicals. Bending of the Cleaving Bond and Activation vs Driving Force Relationships. J. Am. Chem. Soc 2004, 126, 16051–16057. [DOI] [PubMed] [Google Scholar]
- (33).Sun G; Ren S; Zhu X; Huang M; Wan Y Direct Arylation of Pyrroles via Indirect Electroreductive C-H Functionalization Using Perylene Bisimide as an Electron-Transfer Mediator. Org. Lett 2016, 18, 544–547. [DOI] [PubMed] [Google Scholar]
- (34).Plumb JB; Obrycki R; Griffin CE Phosphonic Acids and Esters. XVI. Formation of Dialkyl Phenylphosphonates by the Photoinitiated Phenylation of Trialkyl Phosphites1,2. J. Org. Chem 1966, 31 (8), 2455–2458. [Google Scholar]
- (35).Both constant current and constant cell potential conditions allow the working electrode potential to vary over the reaction. We found that the working potential dropped over the course of a standard reaction. Control experiments verified that in each stage of the reaction both light and electrolysis were necessary. See the SI for details. [Google Scholar]
- (36).The reduction wave for LiClO4 in DMSO begins before reduction of the aryl chloride leading to 7. Differential pulse voltammetry deconvoluted these features and provided an Ered of −3.4 V vs SCE. See the SI for details. [Google Scholar]
- (37).Each of these functional groups possess an Ered less negative than the substrates productively engaged in radical coupling. [Google Scholar]
- (38).Beveridge S; Huppatz JL The Pschorr Cyclization. A Novel Pschorr Reaction with a Pyrrole Derivative: Synthesis of a Pyrrolo[1,2-c]Quinazoline and a New Route to Arylpyrroles. Aust. J. Chem 1970, 23, 781–789. [Google Scholar]
- (39).Newcomb M Radical Kinetics and Clocks In Encyclopedia of Radicals in Chemistry, Biology and Materials; Wiley, 2012. [Google Scholar]
- (40).Kimura M; Miyahara H; Moritani N; Sawaki Y Electroreductive Dehalogenation of Chlorinated Aromatic Ethers. Unexpected Electrogenerated Base-Catalyzed Reactions. J. Org. Chem 1990, 55 (12), 3897–3902. [Google Scholar]
- (41).Andrieux CP; Pinson J The Standard Redox Potential of the Phenyl Radical/Anion Couple. J. Am. Chem. Soc 2003, 125, 14801–14806. [DOI] [PubMed] [Google Scholar]
- (42).Ruccolo S; Qin Y; Schnedermann C; Nocera DG General Strategy for Improving the Quantum Efficiency of Photoredox Hydroamidation Catalysis. J. Am. Chem. Soc 2018, 140, 14926–14937. [DOI] [PubMed] [Google Scholar]
- (43).Takeda N; Poliakov PV; Cook AR; Miller JR Faster Dissociation: Measured Rates and Computed Effects on Barriers in Aryl Halide Radical Anions. J. Am. Chem. Soc 2004, 126, 4301–4309. [DOI] [PubMed] [Google Scholar]
- (44).Ke J; Wang H; Zhou L; Mou C; Zhang J; Pan L; Chi YR Hydrodehalogenation of Aryl Halides through Direct Electrolysis. Chem. - Eur. J 2019, 25, 6911–6914. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.