

Abstract
Pseudouridylation is the most common post-transcriptional modification wherein uridine is isomerized into 5-ribosyluracil (pseudouridine, Ψ). The resulting increase in base stacking and creation of additional hydrogen bonds are thought to enhance RNA stability. Pseudouridine synthases are encoded in humans by 13 genes, two of which are linked to Mendelian diseases: PUS1 and PUS3. Very recently, PUS7 mutations were reported to cause intellectual disability with growth retardation. We describe two families in which two different homozygous PUS7 mutations (missense and frameshift deletion) segregate with a phenotype comprising intellectual disability and progressive microcephaly. Short stature and hearing loss were variable in these patients. Functional characterization of the two mutations confirmed that both result in decreased levels of Ψ13 in tRNAs. Furthermore, the missense variant failed to complement the growth defect of S. cerevisiae pus7Δ trm8Δ mutants. Our results confirm that PUS7 is a bona fide Mendelian disease gene and expand the list of human diseases caused by impaired pseudouridylation.
Keywords: pseudouridylation, microcephaly, PUS7
INTRODUCTION
Ribonucleosides undergo more than 160 modifications (Boccaletto et al. 2017). These modifications influence the stability and function of RNAs, and in some cases are thought to play a key role in the dynamic control of gene expression (Endres et al. 2015; Hoernes et al. 2016; Jackman and Alfonzo 2013; Motorin and Helm 2010; Zhao et al. 2017). The most common of these post-transcriptional modifications (PTMs) is pseudouridylation wherein uridine is isomerized into 5-ribosyluracil (pseudouridine, Ψ) (Charette and Gray 2000). Pseudouridine was first identified in 1951 and was originally referred to as the “fifth” nucleoside before its identity as 5-ribosyluracil was determined later (Cohn 1959; Cohn and Volkin 1951). Pseudouridylation has been documented in all three major RNA species (tRNA, rRNA and mRNA) as well as in snRNA and snoRNA (Andrew et al. 2011; Branlant et al. 1981; Carlile et al. 2014; Grosjean et al. 1995; Li et al. 2015; Lovejoy et al. 2014; SCHATTNER et al. 2006; Schwartz et al. 2014; Sprinzl and Gauss 1982). The reason for the abundance of pseudouridine is incompletely understood but is thought to be driven by its advantageous effect on RNA stability by virtue of improved base stacking and the creation of additional hydrogen bonds (Davis 1995; Spenkuch et al. 2014).
Six families of enzymes are known to synthesize pseudouridine (pseudouridine synthases or PUS enzymes): TruA, TruB, TruD, RsuA, RIuA and Pus10b. With the exception of RsuA, these families are represented in humans by peseudouridine synthases encoded by 13 genes, some of which represent paralogs such as PUS1/PUS1L, TRUB1/TRUB2, and PUS7/PUS7L (Spenkuch et al. 2014). Human PUS enzymes are far less studied than their counterparts in other organisms. However, recent advances in human Mendelian diseases have brought renewed focus on these enzymes and their relevance to clinical medicine. The first human disease linked to PUS genes was myopathy, lactic acidosis, and sideroblastic anemia 1 (MLASA1) (MIM 600462), which was found to be caused by biallelic mutations in PUS1 (Fernandez-Vizarra et al. 2007). In 2016, we described a novel intellectual disability and microcephaly syndrome in three affected siblings who all shared a homozygous truncating variant in PUS3 (Shaheen et al. 2016). In addition, we recently encountered a patient with severe global developmental delay and epilepsy with a homozygous truncating variant in PUS7L (manuscript under review). These findings suggest that PUS genes are attractive candidates for Mendelian diseases in humans and raise interesting questions about the nature and extent of the associated phenotypes.
In this study, we describe two consanguineous families each segregating a different likely deleterious homozygous variant in PUS7 (one missense and one frameshift deletion). The associated phenotype is consistent and comprises severe intellectual disability and progressive microcephaly. Both variants were associated with a specific reduction of Ψ13 in their tRNAs. Our findings are compatible with one study that was published in the course of preparing this manuscript and provide independent confirmation of this novel PUS7-related syndrome (de Brouwer et al. 2018).
MATERIALS AND METHODS
Human Subjects
The two study families were enrolled in an IRB-approved research protocol (RAC#2080006) and informed consent was obtained. All three patients were evaluated by board-certified clinical geneticists, and full phenotypic data were collected from their medical records. In addition to the patients, their parents and available siblings were also collected. Venous blood was collected in EDTA and in sodium heparin tubes for DNA extraction and the establishment of lymphoblastoid cell lines (LCLs), respectively.
Positional Mapping, Exome Sequencing and Variant Interpretation
DNA samples were genotyped using the Axiom SNP Chip platform following the manufacturer’s instructions. Autozygome was mapped based on regions of homozygosity >2Mb as surrogates of autozygosity given the parental consanguinity followed by mapping of the candidate autozygome that is exclusively shared by the affected members using AutoSNPa. Linkage analysis was used to confirm the candidate autozygome and calculate LOD score based on a fully penetrant autosomal recessive model using the EasyLINKAGE package.
Exome capture was performed using the TruSeq Exome Enrichment kit (Illumina, San Diego, CA, USA) as per the manufacturer’s instructions. Samples were prepared as an Illumina sequencing library, and in the second step, the sequencing libraries were enriched for the desired target using the Illumina Exome Enrichment protocol. Captured libraries were sequenced using Illumina HiSeq 2000 Sequencer, and the reads mapped against UCSC hg19 (http://genome.ucsc.edu/) by BWA (http://bio-bwa.sourceforge.net/). The SNPs and Indels were detected by SAMTOOLS (http://samtools.sourceforge.net/). The resulting variants were filtered as follows: homozygous=>coding/splicing=>within candidate autozygome=>absent or very rare in Saudi and public exome databases=>predicted to be pathogenic by SIFT/PolyPhen/CADD as previously described (Alkuraya 2013).
Extraction of Total RNA and Purification of tRNA from Human Cells
LCLs were grown at 37°C in 5% CO2 in RPMI 1640 medium containing FBS (15%), penicillin (1 U/mL), streptomycin (1 μg/mL), and amphotericin b (0.5 μg/mL) to a density of ~ 1.0 X 106 cells/mL, and then 2.5 ml was used to generate triplicate cultures. Bulk RNA from ~3.6 X 108 cells was extracted with TRIzol (Life Technologies) according to manufacturer's instructions. Then tRNAVal(AAC) was purified from 1-1.25 mg bulk RNA using a 5' biotinylated oligonucleotide (Integrated DNA Technologies) complementary to nucleotides 53-76, digested with P1 nuclease and phosphatase, and nucleosides were analyzed by HPLC, as previously described (Jackman et al. 2003).
Analysis of Ψ13 of tRNA
Bulk RNA obtained from LCLs was analyzed for Ψ13 by treatment with N-Cyclohexyl-N′-(2-morpholinoethyl)carbodiimide methyl-p-toluenesulfonate (CMCT, also called CMC), followed by primer extension, essentially as previously described (Huang et al. 2016). Incubation of bulk RNA with CMCT was followed by alkaline treatment, and then RNA was annealed with 5’ 32P-labeled primers that hybridized to nucleotides 40-23 of tRNAHis(GTG) or 39-21 of tRNAGlu(CTC), and reverse transcribed with AMV polymerase (Promega). Products were then resolved on a 15% polyacrylamide-7M urea gel and imaged by a phosphorimager.
Yeast Strains and Plasmids
To construct a CEN plasmid expressing Saccharomyces cerevisiae PUS7 from its own promoter, PUS7 DNA was amplified from yeast genomic DNA with primers hybridizing 853 nucleotides upstream and 253 nucleotides downstream of the open reading frame, and then cloned into a [URA3 CEN] vector. Plasmid EMT082-1 expressing the S. cerevisiae Pus7-D478Y variant (homologous to the human PUS7-D503Y variant) was generated by QuickChange PCR according to manufacturer’s instructions (Stratagene), and the resulting plasmid was sequenced before use.
RESULTS
Clinical Report
Index 1 (Family2-IV:3, 16DG0965) is an 8 yr old Saudi girl who presented to clinical genetics at age 6 yrs for evaluation of unexplained intellectual disability, microcephaly and hearing loss. Her pregnancy, delivery and neonatal history was unremarkable. Although her motor development was described as normal, speech delay was evident when she failed to make any words by her second birthday. Hearing loss was suspected due to chronic middle ear effusion but her poor response to ventilation tubes prompted re-evaluation and sensorineural hearing loss was confirmed. However, it was later evident that her speech delay is not isolated, but rather, is also a component of generalized cognitive impairment. Formal IQ assessment failed on multiple occasions due to aggressive behavior but is believed to be in the severe intellectual disability range. She has good general health otherwise and has no history of admissions or epilepsy. Her behavior is described as aggressive and she has a very poor attention span. Her investigations included brain MRI, which was normal but revealed an incidental finding of craniocervical stenosis. Neurosurgical consultation suggested that conservative management is sufficient for the latter finding. Family history is notable for first cousin parents, a sibling with resolved hydrocephalus and two other siblings Figure 1A). Her physical examination revealed severe microcephaly (OFC 45.3 cm, −4.5SD) but normal height (112.5, 33rd centile) and weight (18.3 cm, 20th centile). She was disruptive throughout the clinical examination. Only subtle facial features were noted in the form of mildly upturned nares and everted lower lip.
Figure 1:

PUS7 Mutations Cause Intellectual Disability and Microcephaly. A) upper panel: Families pedigrees of 12DG2083 & 12DG2084 (Family 1) and 16DG0965 (family 2) showing the consanguineous nature of the parents. The index is indicated in each pedigree by grey arrow, and segregation analysis denoted by half black color (carrier). Red star denotes sibling with resolved hydrocephalus. Lower panel the facial photos of the affected individuals in this study. IV:1 (12DG2083) at the age 9 years and 16 years old. Note Triangular face, prominent glabella, arched eyebrows, deep set eyes, infraorbital crease, convergent squint, hypoplastc zygomatic arches, anteverted nostrils and prominent ears with simple helix. IV:2 (12DG2083) at the age of 7 and 14 years old. Note the deep-set eyes and infraorbital crease, downward slanting of eyes low set ears and thick lower lip. IV:3 (16DG0965) at the age of 8 years. Note the microcephaly, of mildly upturned nares and everted lower lip. B) Sequence chromatograms showing the homozygous variants in PUS7 in the index of each family and the heterozygous in the parents. C) Upper panel is the schematic of PUS7 showing the position of domain” TRUD” and the location of the two variants p. (Thr110Argfs*4) and p. (Asp503Tyr). Lower panel showing multisequence alignment of the mutated reside p. (Asp503) showing high conservation down to S. cerevisiae (boxed in gey). D) Genome-wide linkage analysis to the two families revealed a single maximal peak with a LOD score of ~3.4 on chromosome 7.
Index 2 (Family1-IV:1, 12DG2083) was the first child born to a healthy consanguineous Egyptian parents: 25-year-old mother and a 29-year-old father. The couple has no healthy children. The boy presented to clinical genetics initially at the age of 4 7/12 yrs because of developmental and speech delay. He was born at term after an uneventful pregnancy by a normal vaginal delivery. His birth weight was 1500 g (−3 SD) other parameters were not documented. Apart from the physiological jaundice, the neonatal period was uneventful. He sat and walked without support at age 12 and 24 months respectively. He said his first monosyllable words after the age of 36 months. The initial physical examination at age 4 7/12 yrs showed weight 11 kg (−3.5 SD), height 85 cm (−4.9 SD) and OFC 44 cm (−5.7 SD). Triangular face, prominent glabella, arched eyebrows, deep set eyes, infraorbital crease, convergent squint, hypoplastic zygomatic arches, anteverted nostrils and prominent ears with simple helix were noted. He also had behavioral problems including inappropriate stereotypic movements, head nodding, bruxism and sometimes tantrums. He was also noted to be hyperactive with fair eye contact. No epilepsy was reported. EEG recording was normal. Brain MRI showed normal brain architecture. He was admitted in a school for special education. Radiological bone age matched the chronological age. We re-evaluated him at age 16 yrs, he presented with aggressive behavior and Risperidone treatment was introduced. Further, he developed severe sensorineural hearing loss and hearing aid was used. Frequent muscle spasms and fine tremors were observed by the mother. He showed a severe speech delay and his expressive vocabulary was limited to about 30 words. On examination, his weight, height and OFC were 32 kg (− 3 SD) 145 cm (− 4 SD) and 46 cm (− 6 SD), respectively. Normal secondary sexual characters and normal puberty were noted. Neurological examination showed normal tone and reflexes. Oro-dental examination showed high arched palate with narrow vault, short smooth philtrum, everted lower lip wide overjet and deep overbite and pointed chin. No abnormalities were noted for the shape of teeth. His IQ was 44 using Stanford-Bienet test.
Patient 3 ((Family1-IV:2, 12DG2084)) is the younger sib of index 2. Pregnancy and delivery were uneventful. His birth weight was 2500 g (− 2 SD). Other parameters were not documented. He also developed neonatal jaundice. We first examined him at the age of 2 7/12 yrs. He showed developmental delay of his milestones (sat at 10 months and walked at 20 months) and delayed speech. His weight, height and OFC were 8 Kg (−3.5 SD), 75 (−5.5 SD) and 43 (−5.2 SD), respectively. He had deep set eyes and infraorbital crease similar to his brother. In addition, he had downward slanting of eyes and low set ears. He was described as an obedient child. Neurological examination showed normal tone and reflexes. We re-evaluated him at age 14 yrs; on examination, his weight, height and OFC were 21 kg (− 3.7 SD) 125 cm (− 6.6 SD) and 44.5 cm (− 6.7 SD), respectively. He had similar oro-dental findings. In addition, he had mandibular retrognathia, thick lower lip, crowding and retained deciduous teeth. He never developed epilepsy. Psychological evaluation indicated attention deficit hyperactivity but he did not show aggressiveness like his brother. He had speech difficulties with poor language and pronunciation difficulties. He was not able to read or write and was admitted in a special education school with his sib. EEG, Hearing test, Fundus examination abdominal ultrasound showed normal results. His IQ was 48 using Stanford-Bienet test. Brain MRI was performed at age of 4 yrs and repeated at the age 14 yrs, and both studies revealed normal brain architecture.
PUS7 is Mutated in an Autosomal Recessive Syndrome of Intellectual Disability and Microcephaly
Each family was analyzed independently initially. In Family 1, three novel variants that are predicted deleterious in silico were identified within the candidate autozygome of the index: PUS7, GDF9 and TRIM4. In Family 2, however, only one novel variant that is predicted deleterious was identified within the candidate autozygome: PUS7 (NM_019042.3:c.329_332delCTGA). Based on the latter finding, we prioritized PUS7 (NM_019042.3:c.1507G>T, p. (Asp503Tyr) in Family 1 for functional validation. It should be noted that the missense variant p. (Asp503Tyr) is located within the TRUD domain (the pseudouridine catalytic domain of PUS7), predicted to be probably damaging (1) by Polyphen-2, deleterious by SIFT (0), “disease causing” by MutationTaster and has a CADD_phred of 29.4. The truncating variant c.329_332delCTGA is predicted to remove 551 amino acids from the protein, leading to complete absence of the TRUD domain (370-580) (Figure 1C). Both variants fully segregated with the phenotype in each family (Figure 1A). In addition, a LOD score of 3.4 for the locus chr7: 96,488,196-109,035,887 (hgl9) spanning PUS7 was obtained when combining the two families (Figure 1D).
Human Mutations in PUS7 Result in Decreased Levels of Ψ13 in tRNAs.
Since S. cerevisiae PUS7 encodes the pseudouridine synthase that catalyzes pseudouridine formation at U13 in yeast cytoplasmic tRNAs, as well as at U35 in pre-tRNATyr(GUA) (BEHM-ANSMANT et al. 2003), we analyzed characterized human tRNAs known to have Ψ13 from the three patient LCLs (IV: 1, IV:2, IV:3) and from control LCLs. We specifically analyzed Ψ13 using the well-established CMCT assay, which detects Ψ as a primer extension pause after CMCT treatment (Bakin and Ofengand 1993; Huang et al. 2016). Our CMCT analysis showed that tRNAHis(GTG) and tRNAGlu(CTC) from LCLs of patients with the Asp503Tyr mutation lacked Ψ13 (Figure 2A), whereas each of two control LCLs had a prominent pause corresponding to the Ψ13 modification. Similarly, tRNAGlu(CTC) from an LCL from a patient with Thr110Argfs*4 mutation lacked Ψ13, compared to a control LCL (Figure 2B).
Figure 2:
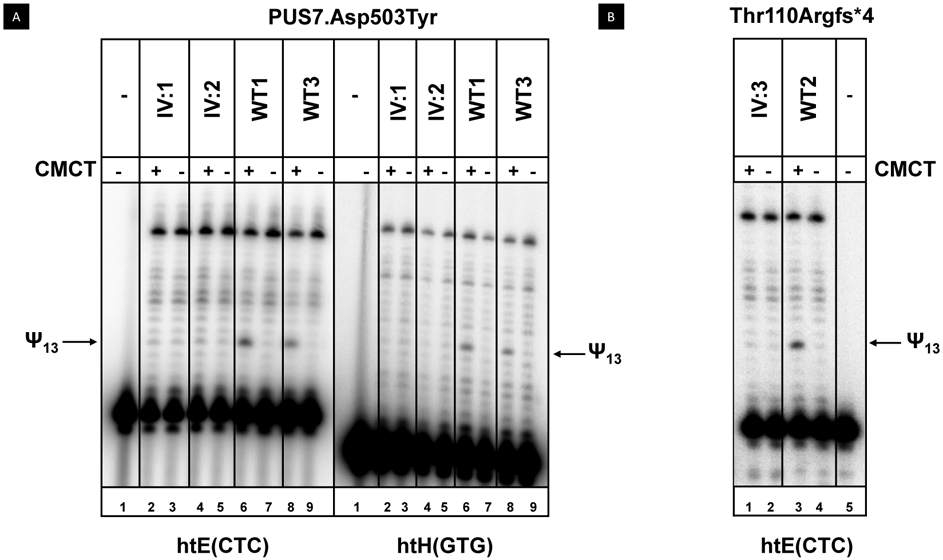
LCLs from patients with PUS7 mutations have reduced levels of Ψ13 in tRNAs. LCLs from patients (IV: 1, IV: 2, IV: 3) and controls (WT 1, WT 2, WT 3) were grown as described and bulk RNA was extracted, modified with CMCT, and analyzed for Ψ13 by primer extension. The reverse transcription pauses corresponding to Ψ13 are marked by arrows. A. Ψ13 is not detected in tRNAGlu(CTC) and tRNAHis(GTG) of LCLs from patients with a PUS7.Asp503Tyr mutation. B. LCLs from patients with a PUS7.Thr110Argfs*4 mutation lack detectable Ψ13 in tRNAGlu(CTC).
To quantify the defect in Ψ13 modification of tRNA due to the Asp503Tyr missense mutation (Figure 1C), we purified tRNAVal(AAC) and analyzed its nucleosides by HPLC. We chose tRNAVal(AAC) because it only had two other pseudouridine residues in addition to Ψ13, and because it was amenable to purification (Shaheen et al. 2015). tRNAVal(AAC) purified from the LCL from patient IV:2 with the Asp503Tyr mutation showed a reduction of 0.88 moles of Ψ relative to that from a WT control (1.75 moles/mole compared to 2.63 moles/mole). By contrast, the levels of the other analyzed tRNAVal(AAC) modifications (m5C, m2G) were very similar between the patient and control LCLs (Figure 3 and Table 1). Thus, our results strongly suggest that PUS7 with either the Asp503Tyr mutation or the Thr110Argfs*4 mutation is severely deficient in the isomerization of U13 to Ψ, linking this defect to the ID, consistent with the known biological effects of PUS7 (see below).
Figure 3:

tRNAVal(AAC) purified from LCLs of patients with the PUS7.Asp503Tyr mutation have reduced Ψ relative to that from control LCLs. LCLs were grown and tRNA was purified and analyzed for modifications as described in Materials and Methods. Data derives from Table 1, showing that Ψ levels are reduced in patient LCLs (by 0.88 mole/mole), whereas levels of each of two other modifications are not.
Table 1.
Analysis of modified nucleosides in tRNAVal(AAC) from control (WT3) and patient (IV:2) LCLs.
| Modification a | Moles Expected |
WT 3 | IV:2 |
|---|---|---|---|
| Ψ | 3 | 2.63 ±0.20 | 1.75 ±0.18 |
| m5C | 2 | 1.49 ± 0.15 | 1.27 ±0.20 |
| m2G | 1 | 0.82 ± 0.18 | 0.66 ±0.11 |
Mean and standard deviation based on three individual growths and RNA preparations
Expression of S. cerevisiae pus7-D503Y Does Not Complement the Growth Defect of S. cerevisiae pus7Δtrm8Δ Mutants
We used S. cerevisiae as a model to further analyze the effects of the PUS7-Asp503Tyr missense mutation, since the Thr110Argfs*4 mutation is an apparent loss of function truncating variant. We generated a low copy (CEN) plasmid expressing S. cerevisiae pus7- D478Y (equivalent to human PUS7-D503Y) from its native promoter [CEN URA3 PPUS7-pus7- D478Y] to test its ability to complement the severe slow growth defect of an S. cerevisiae pus7Δ mutant strain in a trm8Δ background which, as reported previously, grows very poorly at 33°C and higher temperatures, due to rapid tRNA decay of tRNAVal(AAC) (Chernyakov et al. 2008). We found that expression of the yeast pus7-D478Y variant failed to detectably rescue the growth defect of the pus7Δ trm8Δ strain (Figure 4). This result indicates that the yeast Pus7- D478Y variant is a complete loss of function mutation, and thus that the importance of the human Thr503 residue is conserved among eukaryotes.
Figure 4:
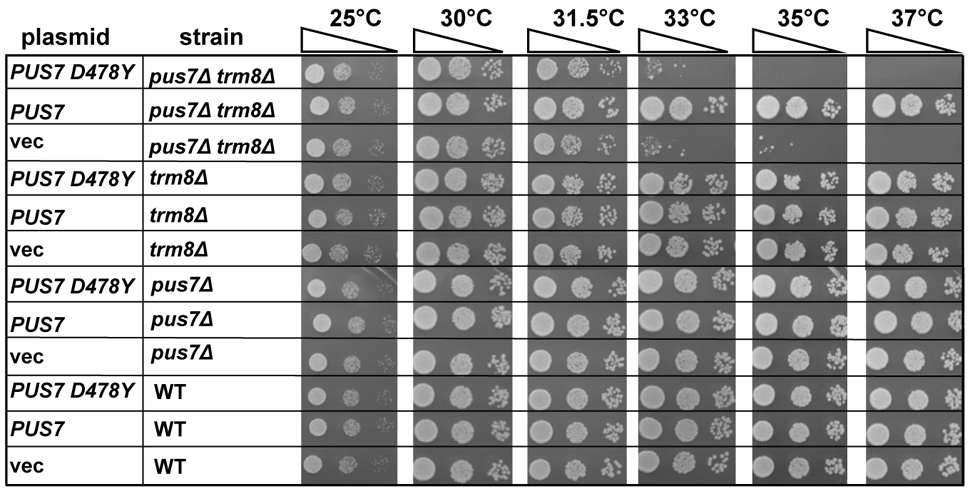
The growth defect of a trm8Δ pus7Δ mutant at high temperature is not complemented by expressing pus7-D478Y. Wild type and trm8Δ pus7Δ strains harboring [CEN URA3 PPUS7-pus7-D503Y] or vector as indicated were grown overnight in S-Ura medium containing dextrose, diluted to OD600 of approximately 0.2 and serially diluted 10-fold, and then 2 μL was spotted onto YPD media, followed by incubation for 3 days at temperatures as indicated.
DISCUSSION
There is a growing appreciation of the role of tRNA modification in human diseases based on revelations from Mendelian disorders (Ramos and Fu 2018; Torres et al. 2014). We previously reported that the most common single gene mutation associated with intellectual disability in Arabia was a point mutation in ADAT3 (Alazami et al. 2013), which encodes an ortholog of yeast TAD3, a subunit of the complex required for I34 modification of substrate tRNAs (Gerber and Keller 1999). Furthermore, we showed that a founder mutation of WDR4 impairs a highly conserved and specific (m7G46) methylation of tRNA with resulting severe encephalopathy and microcephaly (Shaheen et al. 2015). Additional Mendelian disorders include TRM10A- and NSUN2-related syndromes of intellectual disability, microcephaly and short stature (Fahiminiya et al. 2013; Gillis et al. 2014; Igoillo-Esteve et al. 2013; Martinez et al. 2012). TRM10A is the likely human homolog of yeast TRM10 required for m1G9 modification (Jackman et al. 2003), while NSUN2 is required for m5C modification of body residues 48, 49, and 50 in mammals, as well as C34 of the anticodon (Blanco et al. 2014; Martinez et al. 2012).
Pseudouridine is the most abundant PTM of non-coding RNA observed to date across all domains of life (Cantara et al. 2010; Charette and Gray 2000). The additional NH group in pseudouridine at position 3 permits additional hydrogen bonding with other molecules, and this is thought to play a role in increasing the stability of RNA (Auffinger and Westhof 1998). In human tRNAs, pseudouridines are found at 13 different positions (residues 13, 20a, 20b, 31, 32, 27, 28, 35, 38, 39, 54, 55, and e2), and different pseudouridylases are required for modification at each of the different sites or sets of sites. Despite the abundance of this PTM of tRNA, its physiological relevance in humans has been limited to the reported phenotypes associated with mutations in PUS1 and PUS3 (Fernandez-Vizarra et al. 2007; Shaheen et al. 2016).
During the preparation of this manuscript, de Brouwer and colleagues described three families in which patients with intellectual disability, microcephaly and short stature harbored biallelic mutations in PUS7. They also demonstrated that these mutations are associated with impaired pseudouridylation (de Brouwer et al. 2018). The two families described here have different PUS7 mutations, one of which is a point mutation in a highly conserved residue involved in a salt bridge in the E. coll TruD homolog (Kaya et al. 2004). While the phenotype we present in the three study patients supports the consistent involvement of cognition and postnatal brain growth and lack of epilepsy in PUS7 deficiency, it also suggests that other aspects are more variable. For example, index 1, whose mutation predicts a large PUS7 truncation, lacked the growth deficiency that was reported as a consistent feature by de Brouwer. In addition, we note the presence of sensorineural hearing loss in two of our patients. Unfortunately, the latter feature was not specifically commented on by de Brouwer so it remains to be seen how common this, and indeed other, clinical features are in PUS7-related syndrome.
We and others have previously emphasized the predilection of Mendelian diseases caused by tRNA modification genes to CNS involvement and how this suggests the vulnerability of the brain to any perturbation of tRNA modification, presumably through its deleterious effect on protein synthesis (Kirchner and Ignatova 2015; Ramos and Fu 2018; Shaheen et al. 2015; Torres et al. 2014). Indeed, a number of recent reviews have emphasized the emerging role of protein translation in neurological disorders in view of the expanding list of neurodevelopmental disorders caused by mutations in various components of protein translation and its regulation (Kapur and Ackerman 2018; Tahmasebi et al. 2018). Although the benefit of discovering these disease-gene links, including the one described in this study, is currently limited to establishing an accurate molecular diagnosis and prevention through informed reproductive choices, it is likely that these revelations will inform the development of therapeutics in the future.
In conclusion, we confirm that biallelic PUS7 mutations in humans produce a syndrome of intellectual disability, progressive microcephaly and other variable features. The accompanying consistent defect in pseudouridylation provides hints at the potential pathogenesis of this syndrome. Additionally, this may also serve as a very helpful assay for the proper classification of variants of unknown significance that will inevitably be encountered in this gene as exome/genome sequencing of children with intellectual disability will incorporate it in their annotation.
ACKNOWLEDGEMENTS
We thank the study family for their enthusiastic participation. We also thank Mais Hashem, Niema Ibrahim and Firdous Abdulwahab for their help in coordinating the recruitment of the families and the Sequencing and Genotyping Core Facilities at KFSHRC for their technical help. This work was supported by the King Salman Center for Disability Research (F.S.A.), the Saudi Human Genome Program (F.S.A.), and by National Institutes of Health grant GM052347 (E.M.P.). M.T. was partially supported by NIH Training Grant in Cellular, Biochemical, and Molecular Sciences GM068411.
Footnotes
Authors declare no conflict of interest
REFERENCES
- Alazami AM, Hijazi H, Al-Dosari MS, Shaheen R, Hashem A, Aldahmesh MA, Mohamed JY, Kentab A, Salih MA, Awaji A, Masoodi TA, Alkuraya FS (2013) Mutation in ADAT3, encoding adenosine deaminase acting on transfer RNA, causes intellectual disability and strabismus. J Med Genet 50: 425–30. doi: 10.1136/jmedgenet-2012-101378 [DOI] [PubMed] [Google Scholar]
- Alkuraya FS (2013) The application of next-generation sequencing in the autozygosity mapping of human recessive diseases. Human genetics 132: 1197–1211. [DOI] [PubMed] [Google Scholar]
- Andrew TY, Ge J, Yu Y-T (2011) Pseudouridines in spliceosomal snRNAs. Protein & cell 2: 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auffinger P, Westhof E (1998) Effects of pseudouridylation on tRNA hydration and dynamics: a theoretical approach. Modification and Editing of RNA: 103–112. [Google Scholar]
- Bakin A, Ofengand J (1993) Four newly located pseudouridylate residues in Escherichia coli 23S ribosomal RNA are all at the peptidyltransferase center: analysis by the application of a new sequencing technique. Biochemistry 32: 9754–9762. [DOI] [PubMed] [Google Scholar]
- BEHM-ANSMANT I, URBAN A, MA X, YU Y-T, MOTORIN Y, BRANLANT C (2003) The Saccharomyces cerevisiae U2 snRNA: pseudouridine-synthase Pus7p is a novel multisite-multisubstrate RNA: Ψ-synthase also acting on tRNAs. Rna 9: 1371–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco S, Dietmann S, Flores JV, Flussain S, Kutter C, Flumphreys P, Lukk M, Lombard P, Treps L, Popis M, Kellner S, Holter SM, Garrett L, Wurst W, Becker L, Klopstock T, Fuchs H, Gailus-Durner V, Hrabe de Angelis M, Karadottir RT, Helm M, Ule J, Gleeson JG, Odom DT, Frye M (2014) Aberrant methylation of tRNAs links cellular stress to neuro-developmental disorders. EMBO J. doi: 10.15252/embj.201489282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccaletto P, Machnicka MA, Purta E, Piątkowski P, Bagiński B, Wirecki TK, de Crécy-Lagard V, Ross R, Limbach PA, Kotter A (2017) MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic acids research 46: D303–D307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branlant C, Krol A, Machatt MA, Pouyet J, Ebel J-P, Edwards K, Kössel H (1981) Primary and secondary structures of Escherichia coli MRE 600 23S ribosomal RNA. Comparison with models of secondary structure for maize chloroplast 23S rRNA and for large portions of mouse and human 16S mitochondrial rRNAs. Nucleic Acids Research 9: 4303–4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantara WA, Crain PF, Rozenski J, McCloskey JA, Harris KA, Zhang X, Vendeix FA, Fabris D, Agris PF (2010) The RNA modification database, RNAMDB: 2011 update. Nucleic acids research: gkq1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlile TM, Rojas-Duran MF, Zinshteyn B, Shin H, Bartoli KM, Gilbert WV (2014) Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 515: 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charette M, Gray MW (2000) Pseudouridine in RNA: what, where, how, and why. IUBMB life 49: 341–351. [DOI] [PubMed] [Google Scholar]
- Chernyakov I, Whipple JM, Kotelawala L, Grayhack EJ, Phizicky EM (2008) Degradation of several hypomodified mature tRNA species in Saccharomyces cerevisiae is mediated by Met22 and the 5′−3′ exonucleases Rat1 and Xrn1. Genes & development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn WE (1959) 5-Ribosyl uracil, a carbon-carbon ribofuranosyl nucleoside in ribonucleic acids. Biochimica et biophysica acta 32: 569–571. [DOI] [PubMed] [Google Scholar]
- Cohn WE, Volkin E (1951) Nucleoside-5′-phosphates from ribonucleic acid. Nature 167: 483. [Google Scholar]
- Davis DR (1995) Stabilization of RNA stacking by pseudouridine. Nucleic acids research 23: 5020–5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Brouwer APM, Abou Jamra R, Kortel N, Soyris C, Polla DL, Safra M, Zisso A, Powell CA, Rebelo-Guiomar P, Dinges N, Morin V, Stock M, Hussain M, Shahzad M, Riazuddin S, Ahmed ZM, Pfundt R, Schwarz F, de Boer L, Reis A, Grozeva D, Raymond FL, Riazuddin S, Koolen DA, Minczuk M, Roignant JY, van Bokhoven H, Schwartz S (2018) Variants in PUS7 Cause Intellectual Disability with Speech Delay, Microcephaly, Short Stature, and Aggressive Behavior. Am J Hum Genet 103: 1045–1052. doi: 10.1016/j.ajhg.2018.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endres L, Dedon PC, Begley TJ (2015) Codon-biased translation can be regulated by wobble-base tRNA modification systems during cellular stress responses. RNA biology 12: 603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahiminiya S, Almuriekhi M, Nawaz Z, Staffa A, Lepage P, Ali R, Hashim L, Schwartzentruber J, Abu Khadija K, Zaineddin S, Gamal H, Majewski J, Ben-Omran T (2013) Whole exome sequencing unravels disease-causing genes in consanguineous families in Qatar. Clin Genet, doi: 10.1111/cge.12280 [DOI] [PubMed] [Google Scholar]
- Fernandez-Vizarra E, Berardinelli A, Valente L, Tiranti V, Zeviani M (2007) Nonsense mutation in pseudouridylate synthase 1 (PUS1) in two brothers affected by myopathy, lactic acidosis and sideroblastic anaemia (MLASA). Journal of medical genetics 44: 173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber AP, Keller W (1999) An adenosine deaminase that generates inosine at the wobble position of tRNAs. Science 286: 1146–9. [DOI] [PubMed] [Google Scholar]
- Gillis D, Krishnamohan A, Yaacov B, Shaag A, Jackman JE, Elpeleg O (2014) TRMT10A dysfunction is associated with abnormalities in glucose homeostasis, short stature and microcephaly. J Med Genet 51: 581–6. doi: 10.1136/jmedgenet-2014-102282 [DOI] [PubMed] [Google Scholar]
- Grosjean H, Sprinzl M, Steinberg S (1995) Posttranscriptionally modified nucleosides in transfer RNA: their locations and frequencies. Biochimie 77: 139–141. [DOI] [PubMed] [Google Scholar]
- Hoernes TP, Hüttenhofer A, Erlacher MD (2016) mRNA modifications: Dynamic regulators of gene expression? RNA biology 13: 760–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Karijolich J, Yu Y-T (2016) Detection and quantification of RNA 2′-O-methylation and pseudouridylation. Methods 103: 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igoillo-Esteve M, Genin A, Lambert N, Desir J, Pirson I, Abdulkarim B, Simonis N, Drielsma A, Marselli L, Marchetti P, Vanderhaeghen P, Eizirik DL, Wuyts W, Julier C, Chakera AJ, Ellard S, Hattersley AT, Abramowicz M, Cnop M (2013) tRNA methyltransferase homolog gene TRMT10A mutation in young onset diabetes and primary microcephaly in humans. PLoS Genet 9: e1003888. doi: 10.1371/journal.pgen.1003888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman JE, Alfonzo JD (2013) Transfer RNA modifications: nature's combinatorial chemistry playground. Wiley Interdisciplinary Reviews: RNA 4: 35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman JE, Montange RK, Malik HS, Phizicky EM (2003) Identification of the yeast gene encoding the tRNA mlG methyltransferase responsible for modification at position 9. RNA 9: 574–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur M, Ackerman SL (2018) mRNA translation gone awry: translation fidelity and neurological disease. Trends in Genetics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaya Y, Del Campo M, Ofengand J, Malhotra A (2004) Crystal structure of TruD, a novel pseudouridine synthase with a new protein fold. Journal of Biological Chemistry 279: 18107–18110. [DOI] [PubMed] [Google Scholar]
- Kirchner S, Ignatova Z (2015) Emerging roles of tRNA in adaptive translation, signalling dynamics and disease. Nature Reviews Genetics 16: 98. [DOI] [PubMed] [Google Scholar]
- Li X, Zhu P, Ma S, Song J, Bai J, Sun F, Yi C (2015) Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome. Nature chemical biology 11: 592. [DOI] [PubMed] [Google Scholar]
- Lovejoy AF, Riordan DP, Brown PO (2014) Transcriptome-wide mapping of pseudouridines: pseudouridine synthases modify specific mRNAs in S. cerevisiae. PloS one 9: e110799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FJ, Lee JH, Lee JE, Blanco S, Nickerson E, Gabriel S, Frye M, Al-Gazali L, Gleeson JG (2012) Whole exome sequencing identifies a splicing mutation in NSUN2 as a cause of a Dubowitz-like syndrome. J Med Genet 49: 380–5. doi: 10.1136/jmedgenet-2011-100686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motorin Y, Helm M (2010) tRNA stabilization by modified nucleotides. Biochemistry 49: 4934–4944. [DOI] [PubMed] [Google Scholar]
- Ramos J, Fu D (2018) The emerging impact of tRNA modifications in the brain and nervous system. Biochimica et Biophysica Acta (BBA)-Gene Regulatory Mechanisms. [DOI] [PubMed] [Google Scholar]
- SCHATTNER P, BARBERAN-SOLER S, LOWE TM (2006) A computational screen for mammalian pseudouridylation guide H/ACA RNAs. Rna 12: 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Bernstein DA, Mumbach MR, Jovanovic M, Herbst RH, León-Ricardo BX, Engreitz JM, Guttman M, Satija R, Lander ES (2014) Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell 159: 148–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen R, Abdel-Salam GM, Guy MP, Alomar R, Abdel-Hamid MS, Afifi HH, Ismail SI, Emam BA, Phizicky EM, Alkuraya FS (2015) Mutation in WDR4 impairs tRNA m 7 G 46 methylation and causes a distinct form of microcephalic primordial dwarfism. Genome biology 16: 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen R, Han L, Faqeih E, Ewida N, Alobeid E, Phizicky EM, Alkuraya FS (2016) A homozygous truncating mutation in PUS3 expands the role of tRNA modification in normal cognition. Human genetics 135: 707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spenkuch F, Motorin Y, Helm M (2014) Pseudouridine: Still mysterious, but never a fake (uridine)! RNA biology 11: 1540–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprinzl M, Gauss DH (1982) Compilation of sequences of tRNA genes. Nucleic acids research 10: r57. [PMC free article] [PubMed] [Google Scholar]
- Tahmasebi S, Khoutorsky A, Mathews MB, Sonenberg N (2018) Translation deregulation in human disease. Nature Reviews Molecular Cell Biology: 1. [DOI] [PubMed] [Google Scholar]
- Torres AG, Batlle E, de Pouplana LR (2014) Role of tRNA modifications in human diseases. Trends in molecular medicine 20: 306–314. [DOI] [PubMed] [Google Scholar]
- Zhao BS, Roundtree IA, He C (2017) Post-transcriptional gene regulation by mRNA modifications. Nature reviews Molecular cell biology 18: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]