

Abstract
Nail-patella syndrome (NPS) is a multi-system disorder characterized by hypoplastic nails, hypoplastic patella, skeletal deformities, and iliac horns, which is caused by heterozygous variants of LMX1B. Nephropathy ranging from mild urinary abnormality to end-stage renal disease occurs in some individuals with NPS. Because of the low prevalence of NPS and the lack of longitudinal studies of its kidney involvement, the clinical, pathological, and genetic features characterizing severe nephropathy remain unclear. We conducted a Japanese survey of NPS with nephropathy, and analyzed their clinical course, pathological features, and factors associated with severe renal phenotype. LMX1B gene analysis and luciferase reporter assay were also performed. Among 13 NPS nephropathy cases with genetic validation, 5 patients who had moderate-to-massive proteinuria progressed to advanced chronic kidney disease or end-stage renal disease. Pathological findings in the early phase did not necessarily correlate with renal prognosis. Variants associated with deteriorated renal function including a novel variants were confined to the N-terminal region of the LIM domain and a short sequence in the LMX1B homeodomain, which were distinct from reported variants found in isolated nephropathy without extrarenal manifestation (LMX1B-associated nephropathy). Luciferase reporter analysis demonstrated that variants in patients with severe renal phenotype caused haploinsufficiency, but no dominant-negative effects on promoter activation. A distinct proportion of NPS nephropathy patients progressed to end-stage renal disease in adolescence or young adulthood. Patients with moderate or severe proteinuria, especially those with variants in specific regions of LMX1B, should be monitored for potential deterioration of renal function.
Subject terms: Genetics research, Disease genetics
Introduction
Nail-patella syndrome (NPS) is an autosomal-dominant inherited multi-system disorder characterized by a classic tetrad of absent or hypoplastic finger and toe nails, absent or hypoplastic patella, skeletal deformities involving the elbow joints, and iliac horns [1]. NPS is caused by heterozygous mutations of LMX1B encoding a LIM-homeodomain transcription factor [2].
Although not part of the classic tetrad, kidney disease also occurs in individuals with NPS, and can be a significant threat to quality of life [3]. The renal phenotype of NPS ranges from mild urinary abnormality (hematuria or proteinuria) to end-stage renal disease (ESRD). The reported incidence of kidney involvement varies from 35%−50%, and the risk of ESRD was originally reported to be as high as 15% [4, 5]. However, recent reports suggest that most affected individuals only manifest with an accelerated age-related loss of filtration function, while only a minority of individuals progress to ESRD [5–7]. For example, Sweeney et al. reported that renal replacement therapy was required in only 2 of 120 NPS patients [6], while Bongers et al. reported ESRD in only 1 of 108 NPS patients [7]. Further, Lemley studied 59 NPS patients including 32 adults, none of whom developed ESRD [5].
Histologically, the renal glomeruli of most patients with NPS nephropathy show nonspecific findings by light microscopy, although focal segmental glomerulosclerosis (FSGS) is observed in some cases [5]. Thickening of the glomerular basement membrane (GBM), with an irregular, sharply defined electron-lucent appearance by electron microscopy analysis, is considered a unique feature of NPS nephropathy [8, 9].
With respect to genetic involvement, over 170 heterozygous variants of LMX1B have been reported in NPS patients. Most of these are in the LIM domains (∼80%), with a smaller number in the homeodomain (∼20%) [5, 10]. LIM domain is a zinc-binding, cysteine-rich motif consisting of two tandemly repeated zinc fingers, and the homeodomain is necessary for binding of specific promoter and/or enhancer sequences of DNA [5]. Individuals with an LMX1B variant located in the homeodomain were reported to present significantly more frequent and higher values of proteinuria compared with subjects carrying variants in the LIM domains [7]. However, the evidence for a specific “nephropathy locus” was not confirmed by another cohort [5]. Recent genetic analysis has found that some unique variants in the homeodomain of LMX1B cause isolated nephropathy without extrarenal manifestations (LMX1B-associated nephropathy) [11], while the mechanism behind the organ specificity is still largely unclear.
There are few reports of NPS cases with nephropathy. The low prevalence rate of NPS and the lack of longitudinal studies makes it difficult to determine the precise clinical or genetic features that characterize progression to ESRD [5, 11]. For example, it is unknown whether patients with severe nephropathy always manifest massive proteinuria prior to progression to kidney failure [5]. Further, it is unknown whether a particular variant type is associated with severe nephropathy progressing to ESRD. Herein, we conducted a survey of Japanese patients with nephropathy with NPS to determine the clinical, pathological, and genetic features associated with severe nephropathy.
Materials and methods
Survey of NPS nephropathy in Japan
This study was approved by the ethics committee of the University of Tokyo. To identify patients with NPS nephropathy, we used data prospectively registered in the Japan Renal Biopsy Registry (J-RBR) between July 2007 and June 2015. The J-RBR was established by the Committee for the Standardization of Renal Pathological Diagnosis and the Working Group for the Renal Biopsy Database of the Japanese Society of Nephrology in 2007 [12]. In addition to cases in J-RBR, we collected NPS cases by conducting a questionnaire-based survey. In 2014, questionnaires were sent to representatives of the Japanese Society of Nephrology, the Japanese Society of Pediatric Nephrology, and the Japanese Renal Pathology Society to assess experience in consultation of patients with NPS nephropathy.
Clinical and renal histopathological diagnosis
NPS was defined as having nail hypoplasia or dysplasia with one of the following abnormalities (patellar aplasia/hypoplasia, elbow deformities, iliac horns, or heterozygous variant of LMX1B). Cases were excluded if a patient was diagnosed with other genetic diseases (Meier−Gorlin syndrome, Genitopatellar syndrome, DOOR (deafness, onychodystrophy, osteodystrophy, mentalretardation, and seizures) syndrome, 8 trisomy mosaic syndrome, Coffin-Siris syndrome, Brachymorphism-onychodysplasia-dysphalangism (BOD) syndrome, Rapadilino syndrome). The criteria for NPS were determined by our research group and with support of the Health Labor Sciences Research Grant of Japan. Diagnosis of NPS nephropathy was based on urinary abnormalities (hematuria of any degree or proteinuria; ≥0.15 g/gCre) or decreased renal function (eGFR < 60 mL/min/1.73 m2). In this study, to establish a definitive diagnosis of NPS nephropathy, we only included patients carrying an LMX1B variant. Extrarenal phenotype of the patients were shown in Supplemental Table.
Clinical and pathological data were obtained from the physicians who registered patients diagnosed with NPS nephropathy in J-RBR, or who responded to the questionnaires. CKD stage 3, 4, 5 were defined as GFR < 60, 30, or 15 mL/min/1.73 m2, respectively. Clinical data included extrarenal manifestations, serum data, and urinalysis results at diagnosis of nephropathy or last visits, and the history of renal replacement therapy. Light microcopy and electron microscopy images were reevaluated when the samples were available.
LMX1B gene analysis
LMX1B gene analysis (GenBank accession number: NG_017039.1(NM_002316.3)) was approved by the ethical committee of the University of Tokyo, and performed if patient’s consent was obtained. Sanger sequencing was performed as previously described [13]. The NM_002316.3: c.169G>T, c.668G>A, c.783dupC, c.793G>C, c.793G>T, c.806_811del, and c.819+1G>A variants have been deposited to Global Variome shared LOVD [14] under accession numbers 653378, 653379, 653384, 653381, 653380, 653383, and 653377, and to ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) under accession numbers SCV001190008.1, SCV001190009.1, SCV001190010.1, SCV001190011.1, SCV001190012.1, SCV001190013.1, and SCV001190014.1.
LMX1B reporter assay
The transcriptional activity of LMX1B was analyzed as previously reported [13, 15]. In brief, Cos-1 cells cultured in 24-well plates were transfected with the reporter gene, shPAN1-pBAT14, the indicated amount of each LMX1B expression plasmids, and pRL-CMV (Promega, Madison, WI, USA). After 24 h, the transcriptional activity was assayed using a Dual-Luciferase Reporter Assay System (Promega). The luciferase activities of 5xFar/FLAT-pFOXluc1.prl were normalized to those of pRL-CMV.
Results
Patients with NPS nephropathy
In our Japanese survey of NPS, 13 patients were identified with classic NPS phenotype complicated with nephropathy of varying severity (Table 1). Most patients presented urine abnormalities (hematuria or proteinuria) during infancy or young childhood, mainly in regular urinary screening in 3-year-olds or in yearly urine screening in school.
Table 1.
Clinical, pathological, and genetic characteristics of patients with nail-patella syndrome (NPS) nephropathy.
| Case | Sex | Age at onset | Diagnosis at onset | Kidney histology | Age at the last follow-up | Age at ESRD | CKD stage | LMX1B variants | Family (Fig. 1) | Case previously reported | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age of kidney biopsy | LM | EM | Unique GBM changes by EM | ||||||||||
| 1 | M | 20 | Hem | 55 | FGO | ND | ND | 56 | 2 | c.169G>T (p.(Glu57Ter)) | |||
| 2 | M | 0 | Hem | ND | ND | ND | 9 | 1 | c.668G>A (p.(Arg223Gln)) | 1 | |||
| 3 | F | 2 | Prot | 3 | MGA | MEA | + | 10 | 1 | c.668G>A (p.(Arg223Gln)) | 1 | ||
| 4 | F | 17 | Prot | ND | ND | ND | 34 | ND | c.668G>A (p.(Arg223Gln)) | 1 | |||
| 5 | M | 7 | Prot | 9(1st).11(2nd) | CF/GS | MEA | + | 49 | 15 | 5 | c.783dupC (p.(Val262ArgfsTer)) | ||
| 6 | M | 9 | Prot | 14 | MGA | MEA/CF | + | 23 | 4 | c.793G>C (p.(Val265Leu)) | Ref. [13] | ||
| 7 | M | 3 | Prot | ND | ND | ND | 21 | 16 | 5 | c.793G>T (p.(Val265Phe)) | 2 | ||
| 8 | F | 3 | Prot | ND | ND | ND | 20 | 3 | c.793G>T (p.(Val265Phe)) | 2 | |||
| 9 | F | 6 | Hem | ND | ND | ND | 49 | 1 | c.793G>T (p.(Val265Phe)) | 2 | |||
| 10 | M | 8 | Prot | ND | ND | ND | 44 | 2 | c.793G>T (p.(Val265Phe)) | 2 | |||
| 11 | F | ND | Hem | ND | ND | ND | 76 | 2 | c.793G>T (p.(Val265Phe)) | 2 | |||
| 12 | F | 3 | Hem/Prot | 10 | MGA | CF | + | 11 | 1 | c.806_811del (p.(Asn269_Gln270del)) | Ref. [13] | ||
| 13 | F | 7 | NS | 7 | FSGS | FT | − | 25 | 4 | c.819+1G>A | Ref. [16] | ||
Hem Hematuria, Prot Proteinuria, LM light microscopy, EM electron microscopy, NS nephrotic syndrome, MME mesangial matrix expansion, MGA minor glomerular abnormality, FSGS focal segmental glomerulosclerosis, FGO focal glomerular obsolescence, ND not determined, GBM glomerular basement membrane, MEA moth-eaten appearance, CF collagen fibrils in GBM, CKD chronic kidney disease, ESRD end-stage renal disease.
The diagnosis of NPS was difficult in some patients, especially when a family history of NPS was absent. For example, in 1 patient, urinary abnormalities were detected by school urine screening at the age of 9, and the elbow deformity and nail hypoplasia was noted 5 years later by an orthopedist (case #6). Another patient was diagnosed with FSGS, which was treated with steroids and immunosuppressants, while nail abnormality was first recognized at 6 years of age after diagnosis of FSGS, which led to a diagnosis of NPS (case #13). The pedigrees of 2 families with at least 2 NPS patients with nephropathy are shown in Fig. 1.
Fig. 1. Pedigrees of families with nail-patella syndrome (NPS) nephropathy.
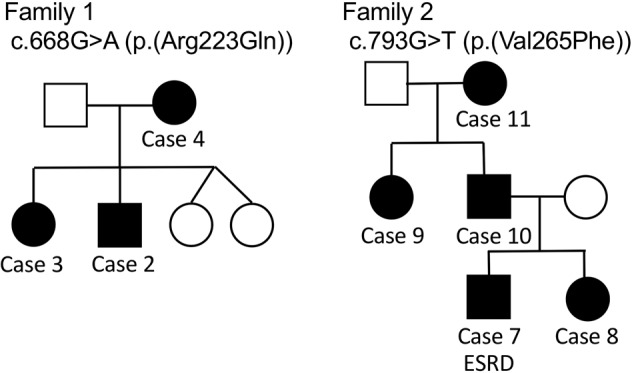
Pedigrees of families with at least 2 NPS patients with nephropathy. Patients who progressed to end-stage renal disease (ESRD) were marked as ESRD.
The majority of patients in the present study were young, and renal function at the last visit was maintained in 8 patients. All the 5 patients with decreased renal function (estimated glomerular filtration rate (eGFR) <60 mL/min/1.73m2) (Table 2) presented with moderate to severe proteinuria (urine protein / creatinine ratio >1.5 g/gCre or urine protein >1.5 g/d) during childhood. Two patients who progressed to ESRD, cases #5, and #7, underwent renal replacement therapy during adolescence or young adulthood (15−16 years old).
Table 2.
Renal phenotype of patients with severe NPS nephropathy.
| Case | Sex | Age at onset of proteinuria | Degree of proteinuria | Age of ESRD | Age at the last follow-up | CKD stage | LMX1B variants |
|---|---|---|---|---|---|---|---|
| 5 | M | 7 | 1.5–2.0 g/day | 15 | 49 | 5 | c.783dupC (p.(Val262ArgfsTer)) |
| 6 | M | 9 | 1.5–2.5 g/day | – | 23 | 4 | c.793G>C (p.(Val265Leu)) |
| 7 | M | 3 | Nephrotic | 16 | 21 | 5 | c.793G>T (p.(Val265Phe)) |
| 8 | F | 3 | Nephrotic | – | 20 | 3 | c.793G>T (p.(Val265Phe)) |
| 13 | F | 7 | Nephrotic | – | 25 | 4 | c.819+1G>A |
Detailed clinical manifestations of patients with severe CKD or ESRD are shown.
Pathological finding of NPS nephropathy
Of the 13 patients with NPS nephropathy, 6 patients underwent kidney biopsy (Table 1). The representative renal pathology of NPS nephropathy is shown in Fig. 2. In light microscopy, there was no significant change in glomeruli detected in the majority of patients, while focal or global sclerosis or focal glomerular obsolescence was observed in 3 patients (Table 1). Segmental or global glomerulosclerosis were found in a patient with severe decline (case #13), and a patient who progressed to ESRD (case #5, Fig. 2a, b). In case #5, crescent formation was observed in a biopsy specimen obtained at 9 years old when he presented nephrotic-range proteinuria. The second biopsy of this patient at 11 years old showed increased sclerotic glomeruli (case #5, Fig. 2a, b). However, glomerular sclerosis was not necessarily observed in patients who later progressed to severe chronic kidney disease (CKD) or ESRD. Renal biopsy of a patient (case #6) who progressed to CKD stage 4 in her twenties showed minor glomerular abnormality at 14 years old (Fig. 2c, d).
Fig. 2. Renal pathology of NPS nephropathy.
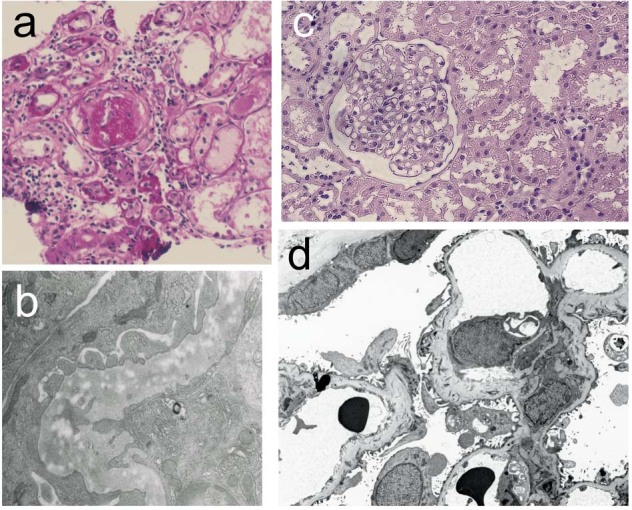
Case #5 (a, b; 11 years old) and case #6 (c, d; 14 years old). In light microscopy, no or only minor glomerular abnormality (c) was detected in some patients, while focal or global sclerosis (a) were detected in others. By electron microscopy, characteristic changes of the glomerular basement membrane (GBM) were observed, including irregular thickening of the GBM with electron-lucent areas (‘moth-eaten appearance’) (b). Fibril clusters in GBM were detected by tannic acid staining (d).
Among the 5 patients with biopsy specimens analyzed by electron microscopy, a characteristic moth-eaten appearance of the GBM or deposition of collagen fibers in the GBM were detected in 4 patients (Table 1, Fig. 2b,d). As these changes were also found in patients with only mild urinary abnormality and normal kidney function (cases # 3 and 12), GBM changes were not associated with deterioration of renal function. In 1 case (case #13), only irregular focal thickening of the GBM, but no collagen fibrils or moth-eaten appearance in the GBM, was observed [16].
LMX1B gene abnormality associated with severe NPS nephropathy
LMX1B gene analysis revealed previously reported variants in 12 patients (Table 1). There was 1 patient with a novel frame shift variant (c.783dupC) (Table1).
In the 5 patients with a moderate-to-severe decline in renal function (CKD stage 3 or 4) or progression to ESRD in young adulthood, 4 variants were identified (Table 2, Fig. 3). Siblings (case # 7 and 8) with a family history of patients with variable renal function (Fig. 1, family 2) carried a c.793G>T (p.(Val265Phe)) variant, and their renal function deteriorated in adolescence. c.819+1G>A is a variant at the first nucleotide of the 5th intron, and was reported to have an increased frequency of kidney involvement [5]. A nonsense variant affecting the N-terminal LIM domain (c.169G>T (p.(Glu57Ter))) was found in 1 patient with hematuria (case #1). The father and uncle of this patient, who had a typical NPS phenotype and were not genetically diagnosed, developed ESRD.
Fig. 3. Variants found in NPS nephropathy and LMX1B-associated nephropathy.
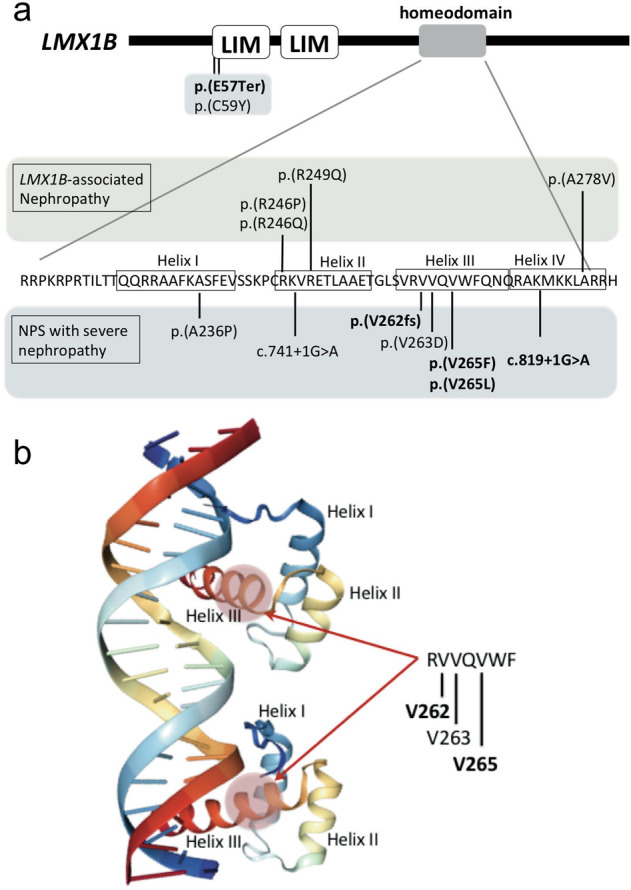
a Variants found in patients with LMX1B-associated nephropathy (isolated nephropathy without extrarenal phenotype) [11] and those in typical NPS with severe nephropathy are shown. Amino acids forming each helix (I to IV) are enclosed in rectangles. Variants in bold represent those found in patients in the present study, and the other variants were described in the discussion section. Missense variants in the homeodomain causing severe NPS nephropathy are located in amino acids in helix I and helix III. By contrast, reported variants causing LMX1B-associated nephropathy are in helix II and IV. b Amino acid alteration in helix III causing severe nephropathy is positioned in the major groove of the DNA, where the homeodomain directly contacts with the DNA bases. 3D structure of homeodomain created by NGL Viewer [25]. The template of this model was from PDB 5HOD (structure of LHX4 complexed with DNA human LHX4 [26]).
To examine the effect of variants in patients with severe nephropathy on LMX1B transactivation, we performed an LMX1B reporter assay. Expression of wild-type or mutant LMX1B was confirmed by Western blot (Fig. 4a). Reporter assay demonstrated that transcriptional activities of c.169G>T (p.(Glu57Ter)), c.783dupC (p.(Val262Argfs)), c.793G>C (p.(Val265Leu)), c.793G>T (p.(Val265Phe) and c.806_811del (p.(Asn269_Gln270del)) mutants were negligible, while that of the wild-type LMX1B significantly increased (Fig. 4b), confirming pathogenicity of these variants. Because these variants were detected heterozygously in patients, the possibility of a dominant-negative effect on the LMX1B-wild type was examined. When the wild type LMX1B was transfected with each mutant in Cos-1 cells, mutants manifested no dominant-negative effect, and there was no additional positive effect on the transcriptional activity of LMX1B-wild type (Fig. 4b). These results suggest that haploinsufficiency, but not a dominant-negative effect, is the main pathogenic mechanism of variants found even in the most severe form of NPS nephropathy.
Fig. 4. Effects of variants on transactivation of LMX1B.
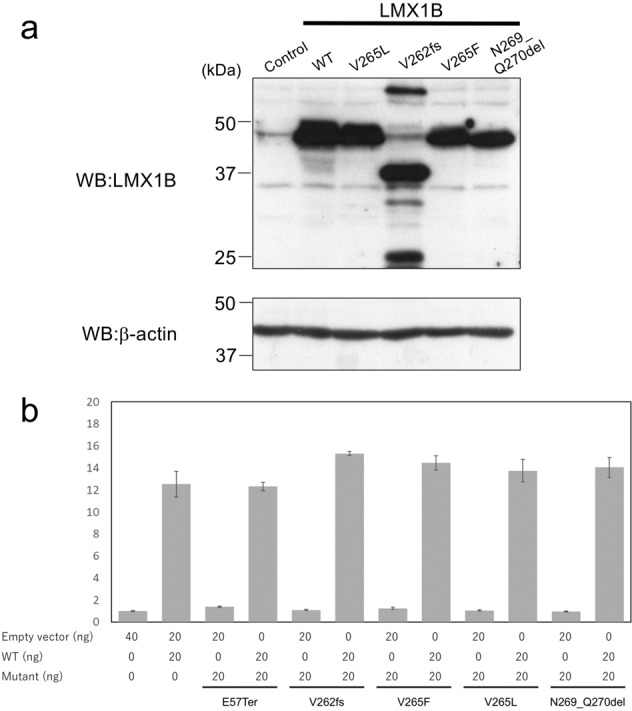
a Western blot analysis of wild-type and mutant LMX1B expressed in Cos-1 cells. b Luciferase reporter assay using wild-type or mutant LMX1B. These mutants lacked transactivation potency, and coexpression of wild-type and mutant LMX1B did not alter reporter activation by wild-type LMX1B.
Discussion
Because of the low prevalence rate of NPS, especially NPS with severe nephropathy, the clinical or genetic features that characterize progression to ESRD are poorly understood. In the present study, we performed a survey focusing on nephropathy complicated with NPS. Our key findings were that (1) a small, but distinct, proportion of patients present with proteinuria and progress to ESRD in young adulthood, (2) pathological findings do not necessarily correlate with renal prognosis, (3) the variants found in cases with deteriorated renal function were mainly in a short region in the LMX1B homeodomain, and (4) the variants found in patients with severe renal phenotype cause haploinsufficiency on its promoter activation.
Histologically, many patients who underwent renal biopsy presented minor glomerular abnormality by light microscopy. This may be attributed to the relatively young subjects in this study in whom biopsy may be done in the early phase of kidney disease. As renal abnormalities can progress with advancing age (case #5), minor glomerular abnormalities by light microscopy, especially in their childhood or adolescence, do not necessarily predict good prognosis. With respect to electron microscopy findings, characteristic GBM changes or abnormalities in GBM thickness were found in all patients analyzed, including cases with normal kidney function. These findings are comparable with a previous report demonstrating that GBM changes were observed even in NPS patients without any apparent clinical or pathologic renal involvement [17].
Because our data were collected from a renal biopsy registry combined with questionnaires to nephrologists, pediatricians, and pathologists, a strength of our study was the number of younger and more severe referral cases compared with previous reports on NPS patients. Indeed, the present study included 5 patients whose kidney function deteriorated before 25 years of age (eGFR < 60). Among these patients, 2 had already progressed to ESRD requiring hemodialysis or renal transplantation. Clinically, all patients who progressed to CKD stage 3 or 4, or ESRD in young adulthood, manifested with massive proteinuria, if not strictly nephrotic-range proteinuria, since childhood (Table 2). The association with proteinuria and decreased renal function is clearly demonstrated by an intrafamilial phenotypic variability in a family carrying c.793G>T p.(Val265Phe) (Fig. 1, family 2). In this family, the father (case #10) and paternal aunt (case #9) of the affected siblings had only mild or no proteinuria, and maintained a normal kidney function throughout their forties. Thus, patients with moderate or massive proteinuria from childhood require careful observation for possible deterioration of renal function.
To date, only a few studies have reported variants in patients with NPS who progressed to advanced CKD or ESRD (Fig. 3a). A c.176G>A (p.(Cys59Tyr)) variant was found in 2 young patients who progressed to ESRD at the ages of 7 and 16 years [9, 18]. Missense variants in the homeodomain(c.706G>C (p.(Ala236Pro)) and c.788T>A (p.(Val263Asp))) and a splice site variant (c.741+1G>A) were also reported in patients with ESRD [9, 19]. c.793G>T (p.(Val265Phe)) and c.793G>C (p.(Val265Leu)) variants were previously reported, although there was no description of nephropathy [20]. Case #6 (c.793G>C (p.(Val265Leu))) [13], case #12 (c.806_811del (p.(Asn269_Gln270del))) [13], and case #13 (c.819+1G>A) [16], were previously described, and their renal function deteriorated after the original publication. Although an association of the c.819+1G>A variant with nephropathy was reported [5], the patient in the present study is the first case carrying this variant who progressed to advanced CKD. This study also included a patient with a novel variant c.783_784insC that caused p.(Val262ArgfsTer90).
Except for c.169G>T (p.(Glu57Ter)) and c.176G>A (p.(Cys59Tyr)), the variants found in patients with ESRD or a severe reduction in GFR can affect amino acids in the LMX1B homeodomain (Fig. 3a). Bongers et al. reported that individuals with an LMX1B variant located in the homeodomain showed significantly more frequent and higher values of proteinuria compared with subjects carrying variants in the LIM domains [21]. However, the association was not confirmed by another study by Lemley et al.[5]. Considering that the majority (approximately 80%) of variants found in NPS patients, including those without nephropathy, were in the LIM domains, not the homeodomain [5, 18], our findings support the association of variants in the homeodomain with nephropathy complications.
We also found that the majority of variants associated with severe nephropathy fall into a narrow amino acid sequence in its homeodomain (Fig. 3a). X-ray crystallographic and NMR spectroscopic studies [22] have determined the mode of binding of homeodomain proteins with the DNA duplex. Helices I and II are aligned in an antiparallel arrangement above the DNA, and the loop between helix I and II interacts with the DNA backbone. The recognition helix (III/IV) is positioned in the major groove of the DNA, roughly parallel to the groove, where it makes specific contacts with the DNA bases [22, 23]. Dreyer et al. demonstrated that mutant LMX1B proteins affecting each of the helices (p.(Ala236Pro), p.(Arg249Pro), p.(Asn269Lys)) abolish transactivation potency [24]. Among the missense variants found in NPS with severe nephropathy, Ala236 is in helix I, while Val263 and Val265 are in helix III, of the LMX1B homeodomain (Fig. 3a, b). These variants dramatically inhibited its transactivation, but failed to act in a dominant-negative manner on wild-type LMX1B (Fig. 4b), supporting haploinsufficiency as the mechanism underlying its pathogenesis even in severe renal phenotype. Recently, specific variants in the LMX1B homeodomain (c.737G>A (p.(Arg246Gln)), c.737G>C (p.(Arg246Pro)), c.745C>G (p.(Arg249Gln)), and c.833C>T (p.(Ala278Val))) were reported to cause isolated nephropathy without nail, patellar, or skeletal abnormality (LMX1B-associated nephropathy) [11, 15]. Notably, Arg246 and Arg249 are located in helix II, while Ala278 is in helix IV, of the LMX1B homeodomain (Fig. 3a). Because there are no reports of patients carrying c.737G>A (p.(Arg246Gln)), c.737G>C (p.(Arg246Pro)), c.745C>G (p.(Arg249Gln)), or c.833C>T (p.(Ala278Val)) who show typical extrarenal manifestation in addition to nephropathy, these variants are likely distinct from those found in patients with NPS nephropathy. Future analysis of the effects of variants causing LMX1B-associated nephropathy or NPS nephropathy on transcriptome of variable cell types are required to further understand the pathogenic mechanism specific to each variant.
A distinct group of patients with NPS progressed to ESRD in young adulthood. It is difficult to predict deterioration of renal function of NPS patients using only genetic information because of inter-individual variability, even in patients carrying the same variant in a family. However, patients who have moderate or severe proteinuria during childhood or adolescence, especially those carrying variants in the LMX1B homeodomain, should be monitored for a potential risk of kidney disease progression.
Supplementary information
Acknowledgements
This work was supported by Health and Labour Sciences Research Grants (H26-015 to YH, TI, SK, AA, and MH), and (H29-039 to YH) from the Ministry of Health, Labour and Welfare of Japan. We thank Dr Toshiyuki Fukasawa from Fukasawa Pediatric Clinic, Dr Seiji Tanaka from Kurume University, Dr Toshiharu Ueno from Toranomon Hospital, Dr Kunihiko Aya from Okayama University Medical School, Dr Yu Mihara from Kyoto Prefectural University of Medicine, Dr Hideaki Imamura from Miyazaki University, Dr Ema Takano from Nakaya Hospital, and Dr Noritaka Kawada from Osaka University for cooperating the survey. We also thank Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Funding
This work was supported by Health and Labour Sciences Research Grants (H26-015 to YH, TI, SK, AA, and MH), and (H29-039 to YH) from the Ministry of Health, Labour and Welfare of Japan.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41431-020-0655-3) contains supplementary material, which is available to authorized users.
References
- 1.Mino RA, Mino VH, Livingstone RG. Osseous dysplasia and dystrophy of the nails; review of the literature and report of a case. Am J Roentgenol Radium Ther. 1948;60:633–41. [PubMed] [Google Scholar]
- 2.Dreyer SD, Zhou G, Baldini A, Winterpacht A, Zabel B, Cole W, et al. Mutations in LMX1B cause abnormal skeletal patterning and renal dysplasia in nail patella syndrome. Nat Genet. 1998;19:47–50. doi: 10.1038/ng0598-47. [DOI] [PubMed] [Google Scholar]
- 3.Hawkins CF, Smith OE. Renal dysplasia in a family with multiple hereditary abnormalities including iliac horns. Lancet. 1950;1:803–8. doi: 10.1016/S0140-6736(50)90636-2. [DOI] [PubMed] [Google Scholar]
- 4.Looij BJ, te Slaa RL, Hogewind BL, van de Kamp JJ. Genetic counselling in hereditary osteo-onychodysplasia (HOOD, nail-patella syndrome) with nephropathy. J Med Genet. 1988;25:682–6. doi: 10.1136/jmg.25.10.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lemley KV. Kidney disease in nail-patella syndrome. Pediatr Nephrol. 2009;24:2345–54. doi: 10.1007/s00467-008-0836-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sweeney E, Fryer A, Mountford R, Green A, McIntosh I. Nail patella syndrome: a review of the phenotype aided by developmental biology. J Med Genet. 2003;40:153–62. doi: 10.1136/jmg.40.3.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bongers EM, Huysmans FT, Levtchenko E, de Rooy JW, Blickman JG, Admiraal RJ, et al. Genotype-phenotype studies in nail-patella syndrome show that LMX1B mutation location is involved in the risk of developing nephropathy. Eur J Hum Genet. 2005;13:935–46. doi: 10.1038/sj.ejhg.5201446. [DOI] [PubMed] [Google Scholar]
- 8.Hoyer JR, Michael AF, Vernier RL. Renal disease in nail-patella syndrome: clinical and morphologic studies. Kidney Int. 1972;2:231–8. doi: 10.1038/ki.1972.99. [DOI] [PubMed] [Google Scholar]
- 9.Heidet L, Bongers EM, Sich M, Zhang SY, Loirat C, Meyrier A, et al. In vivo expression of putative LMX1B targets in nail-patella syndrome kidneys. Am J Pathol. 2003;163:145–55. doi: 10.1016/S0002-9440(10)63638-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clough MV, Hamlington JD, McIntosh I. Restricted distribution of loss-of-function mutations within the LMX1B genes of nail-patella syndrome patients. Hum Mutat. 1999;14:459–65. doi: 10.1002/(SICI)1098-1004(199912)14:6<459::AID-HUMU3>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 11.Harita Y, Kitanaka S, Isojima T, Ashida A, Hattori M. Spectrum of LMX1B mutations: from nail-patella syndrome to isolated nephropathy. Pediatr Nephrol. 2017;32:1845–50. doi: 10.1007/s00467-016-3462-x. [DOI] [PubMed] [Google Scholar]
- 12.Sugiyama H, Yokoyama H, Sato H, Saito T, Kohda Y, Nishi S, et al. Japan Renal Biopsy Registry: the first nationwide, web-based, and prospective registry system of renal biopsies in Japan. Clin Exp Nephrol. 2011;15:493–503. doi: 10.1007/s10157-011-0430-4. [DOI] [PubMed] [Google Scholar]
- 13.Sato U, Kitanaka S, Sekine T, Takahashi S, Ashida A, Igarashi T. Functional characterization of LMX1B mutations associated with nail-patella syndrome. Pediatr Res. 2005;57:783–8. doi: 10.1203/01.PDR.0000157674.63621.2C. [DOI] [PubMed] [Google Scholar]
- 14.Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat. 2011;32:557–63. doi: 10.1002/humu.21438. [DOI] [PubMed] [Google Scholar]
- 15.Isojima T, Harita Y, Furuyama M, Sugawara N, Ishizuka K, Horita S, et al. LMX1B mutation with residual transcriptional activity as a cause of isolated glomerulopathy. Nephrol Dial Transpl. 2014;29:81–88. doi: 10.1093/ndt/gft359. [DOI] [PubMed] [Google Scholar]
- 16.Nakata T, Ishida R, Mihara Y, Fujii A, Inoue Y, Kusaba T, et al. Steroid-resistant nephrotic syndrome as the initial presentation of nail-patella syndrome: a case of a de novo LMX1B mutation. BMC Nephrol. 2017;18:100. doi: 10.1186/s12882-017-0516-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bennett WM, Musgrave JE, Campbell RA, Elliot D, Cox R, Brooks RE, et al. The nephropathy of the nail-patella syndrome. Clinicopathologic analysis of 11 kindred. Am J Med. 1973;54:304–19. doi: 10.1016/0002-9343(73)90025-9. [DOI] [PubMed] [Google Scholar]
- 18.Bongers EM, Gubler MC, Knoers NV. Nail-patella syndrome. Overview on clinical and molecular findings. Pediatr Nephrol. 2002;17:703–12. doi: 10.1007/s00467-002-0911-5. [DOI] [PubMed] [Google Scholar]
- 19.Ghoumid J, Petit F, Holder-Espinasse M, Jourdain AS, Guerra J, Dieux-Coeslier A, et al. Nail-Patella Syndrome: clinical and molecular data in 55 families raising the hypothesis of a genetic heterogeneity. Eur J Hum Genet. 2016;24:44–50. doi: 10.1038/ejhg.2015.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dunston JA, Hamlington JD, Zaveri J, Sweeney E, Sibbring J, Tran C, et al. The human LMX1B gene: transcription unit, promoter, and pathogenic mutations. Genomics. 2004;84:565–76. doi: 10.1016/j.ygeno.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 21.Bongers EM, de Wijs IJ, Marcelis C, Hoefsloot LH, Knoers NV. Identification of entire LMX1B gene deletions in nail patella syndrome: evidence for haploinsufficiency as the main pathogenic mechanism underlying dominant inheritance in man. Eur J Hum Genet. 2008;16:1240–4. doi: 10.1038/ejhg.2008.83. [DOI] [PubMed] [Google Scholar]
- 22.Gehring WJ, Qian YQ, Billeter M, Furukubo-Tokunaga K, Schier AF, Resendez-Perez D, et al. Homeodomain-DNA recognition. Cell. 1994;78:211–23. doi: 10.1016/0092-8674(94)90292-5. [DOI] [PubMed] [Google Scholar]
- 23.Gehring WJ. The homeobox in perspective. Trends Biochem Sci. 1992;17:277–80. doi: 10.1016/0968-0004(92)90434-B. [DOI] [PubMed] [Google Scholar]
- 24.Dreyer SD, Morello R, German MS, Zabel B, Winterpacht A, Lunstrum GP, et al. LMX1B transactivation and expression in nail-patella syndrome. Hum Mol Genet. 2000;9:1067–74. doi: 10.1093/hmg/9.7.1067. [DOI] [PubMed] [Google Scholar]
- 25.Rose AS, Bradley AR, Valasatava Y, Duarte JM, Prlic A, Rose PW. NGL viewer: web-based molecular graphics for large complexes. Bioinformatics. 2018;34:3755–8. doi: 10.1093/bioinformatics/bty419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yin Y, Morgunova E, Jolma A, Kaasinen E, Sahu B, Kund-Sayeed S. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science. 2017;356:eaaj2239. doi: 10.1126/science.aaj2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.