

Abstract
Neuroinflammation is a major contributor to secondary neuronal injury that accounts for a significant proportion of final brain cell loss in neonatal hypoxic-ischemic encephalopathy (HIE). However, the immunological mechanisms that underlie HIE remain unclear. MicroRNA-210 (miR-210) is the master “hypoxamir” and plays a key role in hypoxic-ischemic tissue damage. Herein, we report in an animal model of neonatal rats that HIE significantly upregulated miR-210 expression in microglia in the neonatal brain and strongly induced activated microglia. Intracerebroventricular administration of miR-210 antagomir effectively suppressed microglia-mediated neuroinflammation and significantly reduced brain injury caused by HIE. We demonstrated that miR-210 induced microglial M1 activation partly by targeting SIRT1, thereby reducing the deacetylation of the NF-κB subunit p65 and increasing NF-κB signaling activity. Thus, our study identified miR-210 as a novel regulator of microglial activation in neonatal HIE, highlighting a potential therapeutic target in the treatment of infants with hypoxic-ischemic brain injury.
Keywords: neuroinflammation, neonatal hypoxic-ischemic encephalopathy, microRNA-210, microglial activation, SIRT1
Subject terms: Neuroimmunology, Mechanisms of disease
Introduction
Hypoxic-ischemic encephalopathy (HIE) caused by oxygen deprivation to the infant brain is one of the leading causes of neonatal neurologic morbidity and mortality globally.1 The molecular mechanisms and the pathways of brain injury in infants with HIE remain largely elusive. Currently, therapeutic hypothermia as the gold standard of HIE treatment reduces mortality and improves neurological outcomes in the clinical setting.2 However, this treatment has a narrow therapeutic time window of less than 6 h, and nearly half of affected infants treated with hypothermia still die or suffer significant neurologic disability.1–3 Thus, there is an urgent need to elucidate the pathological mechanisms that underlie neonatal hypoxic-ischemic brain injury and to identify potential targets for HIE therapies.
An increasing body of evidence shows that the neuroinflammatory response to acute cerebral ischemia-hypoxia is a major contributor to the pathophysiology of perinatal brain injury.1,4,5 Abundant preclinical evidence supports a role for inflammation in neonatal HIE. Elevated levels of the proinflammatory cytokines IL-6, IL-8, and IL-1β have been found in the cerebrospinal fluid of infants with HIE, which are associated with adverse neurological outcomes and correlate strongly with the likelihood of cerebral palsy.1,4–9 Microglia are resident innate immune cells in the central nervous system (CNS) and play an essential role in the surveillance of and response to invading pathogens and environmental insults. Microglia display a resting phenotype with low expression of the pan-leukocyte marker CD45 in the healthy CNS. However, following pathological insults, pattern recognition receptors, such as toll-like receptors (TLRs), nucleotide-binding oligomerization domain (nod)-like receptors, and retinoic acid-inducible gene-1-like receptors, on the surface of microglia can be activated by injured neuron-released danger-associated molecular pattern molecules (DAMPs), which leads to classical M1 activation with subsequent production of reactive oxygen and nitrogen species as well as proinflammatory cytokines.10 The proinflammatory cytokines IL-1α, TNFα, and C1q secreted by activated neuroinflammatory microglia also induce reactive astrocytes that contribute to the death of neurons and oligodendrocytes.11 Microglia-mediated neuroinflammation has been shown to contribute to various neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, and cerebral ischemia injury.12–14 The inflammatory response is increasingly recognized as a major contributor to secondary neuronal injury in neonatal HIE. Activated microglia are suggested to participate in the inflammatory response by overproduction of proinflammatory cytokines and neurotoxic mediators.15,16 However, the molecular control of microglial M1 activation following neonatal hypoxia-ischemia (HI) remains largely unknown. Understanding the regulatory mechanisms involved in microglial activation thus holds the key to preventing their pathogenic effects in neonatal HIE.
MicroRNAs (miRNAs) are a class of small noncoding RNAs (~22 nucleotides) that bind to the 3′ untranslated region (3′UTR) of their target genes to induce mRNA degradation and/or inhibit translation. Emerging evidence has shown that miRNAs act as novel fine-tuners to modulate development, differentiation, and effector functions of immune cells.17–20 MiR-210 is a “master miRNA” of the hypoxic response and is strongly induced by hypoxia in various cells and tissues.21,22 MiR-210 has been shown to regulate various cellular processes, including immune responses. MiR-210 is induced upon B cell activation and inhibits B cell responses to prevent the production of age-associated autoantibodies.23 MiR-210 has been shown to control pathogenic Th17 cell differentiation in hypoxia by modulating HIF-1α, a key transcription factor of Th17 differentiation, in a negative feedback loop.24 In addition, miR-210 alleviated the proinflammatory responses in liposaccharide (LPS)-induced chondrocytes and the articular cavity of osteoarthritis rats by targeting DR6 and inhibiting the NF-κB signaling pathway.25 Thus, miR-210 seems to be a significant molecular brake on inflammatory responses in adults. However, the functional significance of miR-210 on neuroinflammation in the immature brain following neonatal HI remains unexplored. Recent studies demonstrated that miR-210 expression was significantly upregulated very early within 3 h in neonatal HIE, and inhibition of miR-210 significantly decreased HI-induced brain injury in neonatal rats and improved long-term neurobehavioral function recovery.26,27 These findings identified an exciting and novel regulatory target, miR-210, in neonatal HI brain injury. Mature miR-210 of 22 nt is highly homologous and is identical between humans and rodents. Notably, human studies showed that miR-210 is a novel biomarker in acute cerebral ischemia and congestive heart failure,28,29 suggesting the translational potential of miR-210 in preclinical studies.
In this study, we investigated the role of miR-210 in regulating microglia-mediated neuroinflammation in the Rice-Vannucci neonatal rat model, a well-validated animal model of human neonatal HIE.30 We demonstrated that activated microglia showed a profound induction during HIE and that miR-210 expression was significantly induced in activated microglia following neonatal HI. Inhibition of miR-210 suppressed microglia-mediated inflammatory responses in the immature brain and decreased brain injury in neonatal HIE. In addition, in vitro gain and loss of function studies indicated that miR-210 positively modulated M1 activation of neonatal rat primary microglia and enhanced the innate proinflammatory immune response. We further revealed that sirtuin 1 (SIRT1) was a novel bona fide target of miR-210 in neonatal rat primary microglia, mediating microglial M1 activation partly by modulating deacetylation of the NF-κB subunit p65 and NF-κB signaling activity. Therefore, our study uncovered a novel and important miR-210-mediated mechanism that enhances the activity of the NF-κB signaling pathway in neonatal rat microglia and promotes microglia-mediated neuroinflammation in neonatal HIE.
Results
MiR-210 is upregulated in activated microglia in HIE
To characterize the inflammatory responses induced by acute neonatal HI, we induced HIE or performed a sham operation in postnatal day 7 (P7) Sprague-Dawley rat pups by treating the animals with or without right common carotid artery (CCA) ligation and 8% oxygen and analyzed the various populations of immune cells by flow cytometry (Fig. 1a). Analysis of brain resident and infiltrating immune cells identified a profound induction of activated microglia and peripheral macrophages (CD11b/c+CD45hi) 12 h post-HI in HIE rats, which was reduced at 24 h after HIE treatment (Fig. 1b, c). Low levels of macrophages were detected by using a rat macrophage marker, even though CD11b/c+CD45hi cells may include peripheral macrophages (data not shown). Compared with the sham group, the numbers of granulocytes, NK cells, and B cells were significantly higher in neonatal brains of HIE rats, and CD4+ T and CD8+ T cells began to infiltrate into injured brains at 24 h post-HI (Fig. 1b). However, these populations showed a lower magnitude of induction by HIE than activated microglia. Analysis of peripheral immune cells in the spleen and blood showed no differences during HIE between the two groups (Supplementary Fig. S1a,b). In addition, analysis of the expression of proinflammatory mediators, which are mainly expressed by microglia in the brain, revealed upregulation of these genes in the injured brain, which peaked at 12 h post-HIE induction (Fig. 1d). qPCR analysis of ipsilateral brains harvested from HIE rats revealed an upregulation of global miR-210 expression 12 h post-HI (Fig. 1e). Consistent with the qPCR results, when brain tissues were stained with a double DIG-labeled LNA miR-210 probe, ipsilateral brains from HIE rats showed increased levels of miR-210 as measured by in situ hybridization (Fig. 1f). We next investigated the expression of miR-210 in microglia during HIE. Analysis of brain resident microglia sorted from HIE rat pups showed an upregulation of miR-210 expression in these cells as early as 6 h post-HIE induction (Fig. 1g). Although the levels of miR-210 also increased in other cells, such as astrocytes and neuronal progenitor cells (Fig. 1h), miR-210 levels showed the highest induction in activated microglia after HIE. Collectively, these results indicate that neonatal HIE results in profound activation and proliferation of microglia in the developing brain and strongly induces miR-210 upregulation in activated microglia.
Fig. 1.


MiR-210 is upregulated in activated microglia in HIE. The experiments were repeated three times. Representative results are presented. a Schematic representation of the experimental design to induce sham and HIE in P7 Sprague-Dawley rat pups. b Bar graph showing the FACS quantitative analysis of the various populations of immune cells in ipsilateral hemisphere harvested from sham or HIE animals at the indicated time points post-HIE induction. Data are presented as the mean ± SEM (n = 7–8). c Representative FACS plots showing the staining for CD11b/c and CD45 in mononuclear cells isolated from the ipsilateral hemisphere of sham or HIE animals at the indicated time points post-HIE induction. d Dot plots showing the RT-qPCR analysis of proinflammatory molecules in the ipsilateral hemisphere harvested from sham or HIE animals at the indicated time points post-HIE induction. Data are presented as the mean ± SEM (n = 5). e Bar graphs showing the qPCR analysis of global miR-210 expression in the ipsilateral hemisphere of sham or HIE animals. Data are presented as the mean ± SEM (n = 4). f Representative images showing the in situ hybridization staining of brain sections of sham or 12 h HIE animals (n = 3). Scale bar: 400 μm. g Bar graphs showing the qPCR analysis of miR-210 expression in microglia sorted from the ipsilateral hemisphere of sham or HIE animals at the indicated time points post-HIE induction. Data are presented as the mean ± SEM (n = 5). h Bar graphs showing the qPCR analysis of miR-210 expression in other neural cells sorted from the ipsilateral hemisphere of sham or HIE animals at 6 h post-HIE induction. Data are presented as the mean ± SEM (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001
Inhibition of miR-210 prevents microglial activation in vivo and reduces brain injury in HIE
To investigate the role of miR-210 as an in vivo autologous factor regulating activated microglia that mediate neuroinflammation in HIE, we pretreated P6 rat pups with LNA-anti-miR-210 or LNA scramble control by intracerebroventricular injection and then induced HIE in P7 rat pups (Fig. 2a). Administration of LNA-anti-miR-210 one day before HIE substantially reduced the levels of miR-210 in the ipsilateral brains (Fig. 2b). Importantly, inhibition of miR-210 substantially ameliorated cerebral infarction 48 h after HIE (Fig. 2c, d). Consistent with these results, inhibition of miR-210 significantly attenuated the expression of proinflammatory mediators, such as proinflammatory cytokines (TNF-α, IL-1β, and IL-6), inducible nitric oxide synthase (iNOS), and the chemokine MIP-1β in the brain 12 h after HIE (Fig. 2e). To assess the direct effect of miR-210 inhibition on microglial activation in rat pups with HIE, we sacrificed animals at 12, 24, and 48 h post-HIE and isolated mononuclear cells from the brains followed by flow cytometry. Compared with the scramble control treatment, the rat pups treated with LNA-anti-miR-210 had significantly lower numbers of CD11b/c+CD45hi activated microglia and peripheral macrophages in the brains during the course of HIE (Fig. 2f, g). We further sorted microglia from the brains of rat pups treated with LNA-anti-miR-210 or LNA scramble control 6 h after HIE using MACS beads and measured the levels of miR-210 and microglia-related proinflammatory mediators. As shown in Fig. 2h, i, blockage of miR-210 in microglia was associated with deactivation of microglia characterized by significantly downregulated expression of proinflammatory marker genes in microglia. Therefore, in the setting of neonatal HIE, inhibition of miR-210 directly reduces microglial activation and suppresses microglia-mediated neuroinflammation.
Fig. 2.


Inhibition of miR-210 prevents microglial activation in vivo and suppresses HIE. The experiments were repeated three times. Representative results are presented. a Schematic representation of the experimental design to study the role of miR-210 as an autonomous factor in regulating activated microglia induction of HIE in P7 Sprague-Dawley rat pups. ICV intracerebroventricular, MRI magnetic resonance imaging. b Bar graphs showing the qPCR analysis of global miR-210 expression in the ipsilateral hemisphere of 24 h HIE animals. Data are presented as the mean ± SEM (n = 4–6). c Representative MRI images showing the infarct size of the ipsilateral hemisphere of 48 h HIE animals (n = 6). Note that miR-210 inhibition strongly reduced the infarct size in the right ipsilateral brain (areas in white). d Bar graph showing the quantification of MRI images presented in c. Data are presented as the mean ± SEM (n = 6). e Bar graphs showing the RT-qPCR analysis of proinflammatory molecules in the ipsilateral hemisphere harvested from 12 h HIE animals. Data are presented as the mean ± SEM (n = 6–8). f Representative FACS plots showing the staining for CD11b/c and CD45 in mononuclear cells isolated from the ipsilateral hemisphere of HIE animals at the indicated time points post-HIE induction. g Bar graph showing the quantification of the FACS plots presented in f. Data are presented as the mean ± SEM (n = 6). h Bar graphs showing the qPCR analysis of miR-210 expression in microglia sorted from the ipsilateral hemisphere of 12 h HIE animals. Data are presented as the mean ± SEM (n = 6–8). i Bar graphs showing the RT-qPCR analysis of proinflammatory molecule expression in microglia sorted from the ipsilateral hemisphere of 12 h HIE animals. Data are presented as the mean ± SEM (n = 6–8). *P < 0.05, **P < 0.01, ***P < 0.001
Inhibition of miR-210 reduces the loss of neural cells and oligodendrocytes in HIE
Sustained activation of microglia induces the death of neurons and oligodendrocytes by producing excess amounts of inflammatory cytokines (TNF-α, IL-1β, and IL-6) along with nitric oxide (NO) and ROS, which contribute to a major proportion of final brain cell loss in the developing brain after HI insults.31–33 As miR-210 inhibition downregulated microglial activation in HIE, we further investigated whether administration of LNA-anti-miR-210 in vivo affected the death of neurons and oligodendrocytes. Brain cells were isolated from the ipsilateral hemispheres of rat pups treated with either LNA-anti-miR-210 or LNA scramble control 48 h post-HIE and then assessed by intracellular or surface marker staining. We observed that the rats treated with LNA-anti-miR-210 had significantly greater numbers of NeuN+ cells, suggesting that miR-210 inhibition prevents the loss of motor neurons after HIE (Fig. 3a, b). Compared with the scramble control treatment, treatment with the miR-210 inhibitor resulted in increased numbers of A2B5+ neural progenitors (Fig. 3c, d), O4+ prooligodendrocytes (Fig. 3e, f), and MBP+ mature oligodendrocytes (Fig. 3g, h) in the brains of rats pups with HIE. Thus, inhibition of miR-210 protects against the loss of neural cells and oligodendrocytes in neonatal HIE.
Fig. 3.

Inhibition of miR-210 reduces the loss of neural cells and oligodendrocytes in HIE. The experiments were repeated three times. Representative results are presented. a Representative FACS plots showing the intracellular NeuN staining of mononuclear cells isolated from the ipsilateral hemisphere of scramble control or LNA-anti-miR-210-treated animals at 48 h post-HIE induction. b Bar graph showing the quantification of the FACS plots presented in a. Data are presented as the mean ± SEM (n = 7–8). c Representative FACS plots showing A2B5 staining of mononuclear cells isolated from the ipsilateral hemisphere at 48 h post-HIE induction. d Bar graph showing the quantification of the FACS plots presented in c. Data are presented as the mean ± SEM (n = 7–8). e Representative FACS plots showing the staining for oligodendrocyte O4 in mononuclear cells isolated from the ipsilateral hemisphere at 48 h post-HIE induction. f Bar graph showing the quantification of the FACS plots presented in e. Data are presented as the mean ± SEM (n = 7–8). g Representative FACS plots showing the intracellular MBP staining of mononuclear cells isolated from the ipsilateral hemisphere at 48 h post-HIE induction. h Bar graph showing the quantification of the FACS plots presented in g. Data are presented as the mean ± SEM (n = 7–8). *P < 0.05, ***P < 0.001
MiR-210 activates neonatal rat microglia in vitro
To further study the intrinsic requirement for miR-210 in microglial activation, we tested whether gain or loss of miR-210 function increased or reduced, respectively, the expression of activation markers by microglia in the next series of experiments. To this end, we first overexpressed miR-210 in primary microglia from neonatal rat brains using a miR-210 mimic (Fig. 4a). Classically activated microglia are commonly associated with increased expression of surface antigens, including CD11b, CD45, CD80, and CD86, as well as proinflammatory cytokines (TNF-α, IL-1β, and IL-6) and iNOS. Microglia transfected with the negative control showed a relatively “resting” phenotype, with low protein expression of the surface markers CD45, CD80, and CD86. Transfection of microglia with the miR-210 mimic resulted in the upregulation of CD11b, CD45, CD80, and CD86 (Fig. 4b, c). Compared with the negative control, the miR-210 mimic enhanced the gene expression of TNF-α, IL-1β, IL-6, and iNOS at both 24 and 48 h after transfection (Fig. 4d). Interestingly, transfection with the miR-210 mimic significantly downregulated the expression of the antiinflammatory M2 cytokines IL-4 and TGF-β1 in these cells (Fig. 4e). However, miR-210 knockdown in primary microglia from the neonatal rat brain through transfection with LNA-anti-miR-210 (Fig. 4f) significantly reduced the basal levels of key M1 proinflammatory cytokines (TNF-α, IL-1β, IL-6) (Fig. 4g). Additionally, we stimulated neonatal rat primary microglia transfected with either LNA-anti-miR-210 or scramble control with low doses of LPS and IFN-γ, two agents known for microglial M1 activation. Consistent with the results of miR-210 in vitro knockdown, although LPS and IFN-γ stimulation strongly induced the expression of the M1 cytokines TNF-α, IL-1β, and IL-6, inhibition of miR-210 blocked the induction of these proinflammatory cytokines (Fig. 4h). Thus, these data suggest that miR-210 positively modulates M1 activation in neonatal rat primary microglia.
Fig. 4.

MiR-210 activates neonatal rat microglia in vitro. a–e Microglia were isolated from neonatal rat primary microglia culture and treated with negative mimic or miR-210 mimic for 48 h (100 nM). Cells were collected for analysis at the indicated time points. The experiments were repeated three times, and representative results are presented. a Bar graphs showing the qPCR analysis of miR-210 expression in microglia at 24 h post transfection. Data are presented as the mean ± SEM (n = 6). b Representative FACS plots showing the staining for CD11b/c, CD45, CD80, and CD86 in microglia at 24 h post transfection. c Bar graph showing the quantification of the FACS plots presented in b. Data are presented as the mean ± SEM of triplicate cultures. d, e Bar graphs showing the RT-qPCR analysis of proinflammatory M1 markers (d) or antiinflammatory M2 markers (e) of microglia at the indicated time points post transfection. Data are presented as the mean ± SEM (n = 6). f–h Microglia were isolated from neonatal rat primary microglia culture, transfected with LNA scramble control or LNA-anti-miR-210 (100 nM), and recovered overnight (12 h) prior to being treated with or without LPS (0.5 ng/ml) and IFN-γ (0.1 ng/ml) for 12 h. Cells were collected for expression analysis. f Bar graphs showing the qPCR analysis of miR-210 expression in microglia at 24 h post transfection of miR-210 inhibitor. Data are presented as the mean ± SEM (n = 4). g, h Bar graphs showing the RT-qPCR analysis of proinflammatory cytokines (TNF-α, IL-1β, IL-6) in microglia without g or with h M1 stimulation. Data are presented as the mean ± SEM (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001
SIRT1 is a bona fide target of miR-210 in neonatal rat microglia
MiRNAs function through posttranscriptional repression of their target genes. To investigate the mechanism by which miR-210 activated neonatal rat microglia, we performed a predicted target analysis for miR-210 using a bioinformatics method (http://multalin.toulouse.inra.fr/multalin/multalin.html) together with TargetScan analysis. SIRT1, a nicotinamide adenosine dinucleotide-dependent protein deacetylase regulating the transcriptional activity of NF-κB, was predicted as a putative target, with a miR-210-binding sequence within its 3′UTR (Fig. 5a). Next, we performed a luciferase reporter assay to confirm that miR-210 could directly bind the 3′UTR of SIRT1 and downregulate protein expression. The rat pheochromocytoma adherent variant PC12 cells were transfected with a construct of the SIRT1 3′UTR containing the miR-210 binding sequence downstream of firefly luciferase. As shown in Fig. 5b, cotransfection with the miR-210 mimic produced a dose-dependent decrease in luciferase activity up to 50% compared with the negative mimic control. Two additional control reporter constructs were included in the luciferase assay. The mutant construct (pmiR-SIRT1Δseed) and positive control construct (pmiR-TET2) contain a mutation in the seed region and a sequence that is exactly complementary to the seed region, respectively. In contrast to the wild-type SIRT1 construct, there was no inhibition of luciferase activity of the mutant SIRT1 construct by 500 nM miR-210 mimic (Fig. 5b). In addition, 100 nM miR-210 mimic led to ~80% inhibition of the luciferase activity of the positive control construct compared with the control (Fig. 5b). To confirm that SIRT1 is a direct target of miR-210, we performed RISC immunoprecipitation experiments in a rat microglial cell line transfected with miR-210 mimic or a negative mimic. Following IP using the Ago1/2/3 antibody, SIRT1 mRNA was significantly enriched by 1.8-fold in miR-210 mimic-treated microglia (Fig. 5c). To examine whether SIRT1 is also the target of miR-210 in neonatal rat primary microglia, we determined the protein levels of SIRT1 in microglia transfected with either miR-210 mimic or negative mimic and demonstrated that SIRT1 protein levels were significantly reduced in miR-210 mimic-transfected microglia (Fig. 5d, e). However, inhibition of miR-210 in microglia through transfection with LNA-anti-miR-210 increased the protein levels of SIRT1 by 60% (Fig. 5f, g). Thus, SIRT1 is a novel bona fide target of miR-210 in neonatal rat primary microglia.
Fig. 5.

SIRT1 is a bona fide target of miR-210 in neonatal rat microglia. a. Schematic representation of alignment of the predicted miR-210 binding site to the SIRT1 3′UTR is shown for Rattus norvegicus. b The rat PC12 cell line was cotransfected with the indicated constructs (500 ng/well) containing the wild-type or mutant 3′UTR of SIRT1 or the wild-type 3′UTR of TET2 (positive control) in the presence of miR-210 mimic or negative mimic (100 nM, 200 nM, or 500 nM) for 48 h. Bar graph showing the normalized levels of luciferase activity in the transfected PC12 cells. Data are presented as the mean ± SEM of four replicate cultures. c Immortalized rat microglia were transfected with negative mimic or miR-210 mimic (100 nM) and collected for analysis 24 h post transfection. Bar graph showing the RISC-IP analysis of abundance of SIRT1 3′UTR pulled down by Ago1/2/3 antibody. Data are presented as the mean ± SEM (n = 3). d Neonatal rat microglia were transfected with negative mimic or miR-210 mimic (100 nM) and collected for analysis 24 h post transfection. Western blots showing the analysis of SIRT1 protein levels. e Bar graph showing the quantification of the western blots presented in d. Data are presented as the mean ± SEM of triplicate cultures. f Neonatal rat microglia were transfected with LNA scramble control or LNA-anti-miR-210 (100 nM) and collected for analysis 24 h post transfection. Western blots showing the analysis of SIRT1 protein levels. g Bar graph showing the quantification of the western blots presented in f. Data are presented as the mean ± SEM (n = 4). *P < 0.05, **P < 0.01, ***P < 0.001
SIRT1 is a mediator of miR-210-induced M1 activation in neonatal rat microglia
To examine whether SIRT1 is involved in the activation of neonatal rat microglia, we knocked down SIRT1 gene expression in neonatal rat microglia using siRNAs and determined the expression of activation markers of the siRNA-transfected microglia. Our experiments showed that SIRT1 knockdown markedly reduced SIRT1 gene expression (Fig. 6a) and resulted in a significant induction of M1 proinflammatory cytokine expression (Fig. 6b). When neonatal rat microglia were transfected with either SIRT1 siRNA or negative control siRNA and then stimulated with LPS and IFN-γ, SIRT1 knockdown further induced the gene expression of the proinflammatory cytokines IL-1β and IL-6 (Fig. 6c). Consistent with these findings, microglia stimulated with LPS and IFN-γ showed an activated M1 phenotype, with high protein expression of the surface markers CD45, CD11b, CD80, and CD86, while SIRT1 gene knockdown further enhanced the protein expression of the activation markers CD45, CD80, and CD86 (Fig. 6d, e). Given that the NF-κB activity is responsible for the expression of proinflammatory cytokines and microglial M1 activation, we next examined whether SIRT1 mediated miR-210 regulation of NF-κB activity in neonatal rat microglia. A 40% increase in SIRT1 protein levels by inhibition of miR-210 in microglia promoted p65 deacetylation and dramatically downregulated the levels of acetylated NF-κB p65 (Fig. 6f, g). This is an interesting observation because the p65 subunit of NF-κB is highly susceptible to SIRT1 regulation and SIRT1 can rapidly terminate NF-κB p65 transcription by deacetylating NF-κB p65.34,35 In contrast, knockdown of SIRT1 by SIRT1 siRNA reduced SIRT1 by 80% and resulted in stronger NF-κB activation, particularly the significantly elevated protein levels of the acetylated p65 subunit of NF-κB (Fig. 6f, g). Consistent with these findings, we detected increased acetyl-NF-κB p65 binding (~1.5-fold) to the regulatory region of IL-1β in primary microglia when they were treated with SIRT1 siRNA (Fig. 6h). Taken together, our results indicate that SIRT1 acts as a downstream target to mediate miR-210 regulation of M1 activation in neonatal rat microglia, partially through modulating p65 deacetylation and NF-κB activity.
Fig. 6.

SIRT1 is a possible mediator of miR-210 in its regulation of M1 activation in neonatal rat microglia. a, b Neonatal rat primary microglia were treated with nonsilencing control siRNA (siCtrl) or siRNAs specific for rat SIRT1 (siSIRT1) (100 nM) and collected for analysis 24 h post transfection. Bar graph showing the RT-qPCR analysis of the expression of SIRT1 (a) or proinflammatory cytokines (TNF-α, IL-1β, IL-6) (b). Data are presented as the mean ± SEM (n = 6). c–e Neonatal rat microglia were transfected with siCtrl or siSIRT1 (100 nM), recovered overnight (12 h), and then treated with LPS (0.5 ng/ml) and IFN-γ (0.1 ng/ml) for 12 h. Cells were collected for analysis. c Bar graphs showing the RT-qPCR analysis of proinflammatory cytokines (TNF-α, IL-1β, IL-6) in siRNA-transfected microglia. Data are presented as the mean ± SEM (n = 6). d Representative FACS plots showing staining for CD11b/c, CD45, CD80, and CD86 in siRNA-transfected microglia. e Bar graph showing the quantification of the FACS plots presented in d. Data are presented as the mean ± SEM (n = 6). f, g Neonatal rat primary microglia were treated with siCtrl or siSIRT1 or LNA-anti-miR-210 (100 nM) and collected for protein analysis 24 h post transfection. f Western blots showing the analysis of the indicated protein levels in transfected microglia. g Bar graph showing the quantification of the Western blots presented in f. Data are presented as the mean ± SEM of triplicates. h Neonatal rat primary microglia were treated with siCtrl or siSIRT1 (100 nM) and collected for analysis 24 h post transfection. Bar graph showing the ChIP analysis of nuclear binding of acetyl-NF-κB p65 to the IL-1β promoter. Data are presented as the mean ± SEM of triplicates. *P < 0.05, **P < 0.01, ***P < 0.001
Discussion
Based on our findings, we propose a new working model wherein miR-210 positively regulates microglial activation and enhances neuroinflammation in neonatal HIE via activating the NF-κB signaling pathway by targeting SIRT1 and blocking p65 deacetylation (Fig. 7). Following acute neonatal HI insult, DAMPs resulting from primary energy failure activate PPRs, especially TLRs, on the surface of microglia in the neonatal brain. The developing microglia initiate the TLR signaling pathway that leads to NF-κB activation. NF-κB induces the production of proinflammatory molecules, such as TNF-α, IL-1β, IL-6, NO, and ROS, which promote apoptosis of neural cells and cause secondary neurotoxicity. SIRT1 acts as a negative regulator governing the excessive activation of microglia by suppressing the NF-κB signaling pathway through deacetylation of the NF-κB subunit p65 at lysine 310. As a signature of hypoxia, miR-210 is induced by HIF-1α in microglia in response to neonatal HI, which stimulates the NF-κB activity through repression of the NF-κB signaling negative regulator SIRT1. Therefore, through upregulation of NF-κB activity, miR-210 acts as an autologous factor promoting enhanced and sustained activation of microglia and increases microglia-mediated neuroinflammation in neonatal HIE.
Fig. 7.
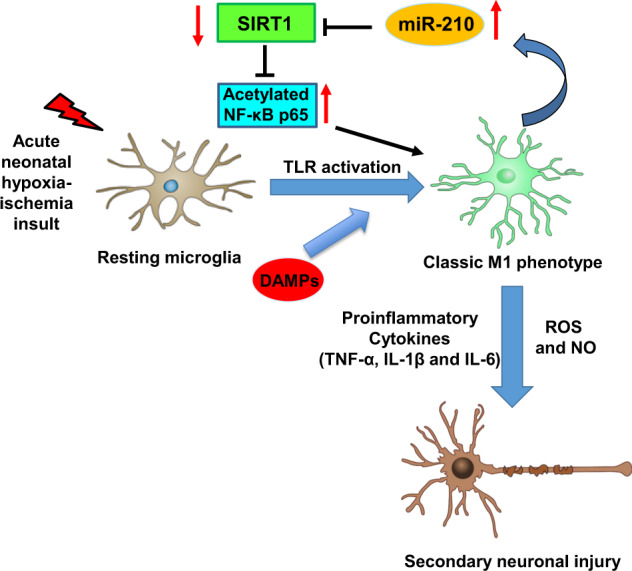
Schematic of a proposed working model wherein miR-210 positively regulates microglia-mediated neuroinflammation via activating the NF-κB signaling pathway by targeting SIRT1 and blocking p65 deacetylation. Upon recognition of DAMPs following acute hypoxic-ischemic insult, developing microglia initiate the TLR signaling pathway that leads to NF-κB activation. NF-κB induces the production of proinflammatory cytokines (such as TNF-α, IL-1β, and IL-6), NO, and ROS, which promotes the apoptotic death of neural cells and causes secondary neurotoxicity. However, SIRT1, as a negative regulator, governs the excessive activation of microglia by deacetylating p65 at lysine 310 and suppressing NF-κB activity. As a HIF-1α-induced master hypoxamir, miR-210 is upregulated in microglia in response to neonatal HI, which rescues NF-κB activity by targeting the NF-κB signaling negative regulator SIRT1. Therefore, through upregulation of NF-κB activity, miR-210, as an autologous factor, promotes sustained activation of microglia and enhances microglia-mediated neuroinflammation in the developing brain
MiR-210 is considered a “master hypoxamir” that is produced during hypoxia in response to HIF-1α transcriptional activity by a variety of cell types, such as cancer cells, neural progenitor cells (NPCs), keratinocytes, endothelial cells, and T lymphocytes.22,24,36–38 As hypoxia is a prominent feature of the physiological and pathological immunological microenvironment that regulates inflammatory pathways in immune cells,39 the role of miR-210 in the regulation of inflammation has gained increased attention. MiR-210 overexpression decreased inflammatory responses and protected against muscle damage upon hindlimb ischemia in mice.40 In murine macrophages, LPS treatment increased the expression of miR-210, which in turn diminished LPS-induced proinflammatory cytokine production by targeting NF-κB1.41 MiR-210 was shown to decrease inflammation in the articular cartilage of osteoarthritis rats by targeting death receptor 6 and inhibiting the NF-κB signaling pathway.25 In addition, miR-210 is abundant in activated T cells, especially in the Th17 lineage of CD4+ T cells. This molecule inhibited Th17 differentiation by a negative feedback regulation of HIF-1α and controlled disease severity of experimental colitis.24 These findings suggest that miR-210 exerts an antiinflammatory role in regulating inflammation. However, HIF-1α-miR-210 signaling has also been shown to play a proinflammatory role in intestinal epithelial cells during the development of acute colitis.42 In psoriasis, HIF-1α was not a downstream target of miR-210 in CD4+ T cells of psoriasis, and upregulated miR-210 in psoriatic skins and CD4+ T cells promoted Th17 differentiation by targeting both STAT6 and LYN.43 Consistently, upregulated miR-210 inhibited FOXP3 expression in CD4+ T cells of psoriasis vulgaris patients, which led to a defect in the immunosuppressive effects of Treg cells.44 Furthermore, TLR3-induced placental miR-210 in cytotrophoblasts suppressed the STAT6/IL-4 antiinflammatory pathway by targeting STAT6, which contributed to the excessive activation of maternal inflammation and led to preeclampsia.45 The discrepancy in the inflammatory regulation of miR-210 may be due to the specific tissue microenvironment and different pathogenesis of inflammatory diseases, resulting in different functional targets that mediate the regulatory effects of miR-210 in different cells. Our previous study provides evidence that inhibition of miR-210 is associated with a reduced proinflammatory response in the adult brain in the acute phase of ischemic stroke.46 Our study is the first to demonstrate increased expression of miR-210 in activated microglia of the neonatal brain in response to hypoxic-ischemic insult (Fig. 1g), where it has a role in promoting the activated state of developing microglia and inflammation, as shown in our in vitro and in vivo gain or loss of function experiments (Figs 2 and 4). Thus, the regulation of miR-210 is tissue- and cell-type specific, and miR-210 may exert distinct functions under different inflammatory conditions.
The capacity of miRNAs to co-ordinately regulate many target genes makes them particularly suitable for regulating complex signaling networks, such as those involved in microglia activation. Indeed, several miRNAs have been shown to play important roles in modulating microglial activation. MiR-124 is directly involved in the maintenance of microglial quiescence in the CNS by targeting the master transcription factor C/EBP-α and its downstream myeloid cell differentiation-associated transcription factor PU.1.47 MiR-424 prevented ischemic brain injury through inhibiting the activation of microglia by targeting cell-cycle activators, including cyclin D1, CDC25A, and CDK6.48 Let-7c-5p inhibited microglial M1 activation post stroke and protected the brain from ischemic damage by suppressing caspase 3 expression.49 However, miR-125b enhanced NF-κB pathway activity by targeting the ubiquitin-editing enzyme A20, which led to persistent microglia activation and motor neuron death in amyotrophic lateral sclerosis.50 In addition, miR-3473b contributed to microglia-mediated neuroinflammation following cerebral ischemia possibly by targeting microglial suppressor of cytokine signaling 3.51 These findings validate the importance of miRNAs in regulating microglial activation in the adult brain. However, expression patterns of microglia phenotypes are age dependent,52 and a recent study suggests the differential impact of miRNAs on microglia of the developing and adult brain.53 Our study is the first to reveal the role and function of miRNAs in microglia in the neonatal brain and demonstrates the importance of miR-210 in regulating microglia-mediated neuroinflammation following neonatal HI. Future studies are needed to identify other miRNA candidates that may contribute to the regulatory network of microglia activation in the developing brain.
Neurogenesis after acute ischemic brain injury is a complex cellular process of many steps, including survival, proliferation/differentiation, migration, and integration of neural cells. Unfavorable microglia-associated proinflammatory responses account for much of the death of brain cells, such as NPCs, neurons, and oligodendrocytes, which greatly impairs neurogenesis post acute ischemic brain injuries.54 In this study, we observed that in addition to reducing microglia-associated neuroinflammation, inhibition of miR-210 also significantly protected against the death of neural cells and oligodendrocytes and reduced neonatal HI brain injury (Fig. 2c, d and 3). However, we cannot exclude an additional contribution from the direct effects of miR-210 inhibition on neural cells in the injured neonatal brain. When we analyzed miR-210 expression in neuron progenitors ex vivo, we found that miR-210 levels were also significantly higher in these cells from HIE animals compared with sham controls (Fig. 1h), suggesting a possible role of miR-210 in neuronal cells during HIE. Indeed, recent studies from our group and others have shown that miR-210 mediates hypoxia-induced neuronal death and inhibits post-HI neurogenesis in vivo.27,55,56 Interestingly, several studies have reported contradictory results showing that miR-210 promotes postischemic neurogenesis in vivo.57–59 A possible mechanism of these controversies is that via enhancing the function of mitochondria, a major target of proinflammatory molecules, miR-210 inhibition not only improves the survival of developing neurons under neuroinflammation but also conversely attenuates the division of neural stem cells during early proliferation.60 In addition to its role in mitochondrial metabolism, several miR-210 targets directly related to neural apoptosis have been identified in vitro. MiR-210 mediated hypoxia-induced apoptotic insults to neuroblastoma cells by targeting Bcl-2,61 an antiapoptotic gene preventing proapoptotic Bax-induced cell apoptosis. However, hypoxia-induced miR-210 played an antiapoptotic role in NPCs via inhibiting BNIP3-induced translocation of apoptosis inducing factor into the nucleus by targeting BNIP3.62 In the PC12 neural cell line under hypoxia, BNIP3 inactivated the PI3K/AKT/mTOR signaling pathway, which participates in cell survival and neuroprotection.63 MiR-210 overexpression also inhibited apoptosis of dorsal root ganglion neurons by targeting EFNA3.64 Thus, these findings suggest that miR-210 may have a dual role in regulating both pro- and antiapoptotic proteins involved in the homeostatic balance between cell survival and apoptosis. However, the roles of miR-210 in HI-induced cell death of neuronal cells still need to be elucidated in vivo. Overall, miR-210 displays distinct functions in the regulation of neurogenesis. MiR-210 likely modulates this process after neonatal HI brain injury through many other mechanisms that have yet to be identified. Further investigation of miR-210-targeting cells and signaling pathways in the neonatal brain is warranted.
Microglia express a number of receptors involved in the control of innate immune function. Activation of these receptors induces many specific signaling pathways that have been implicated in regulating the activation of microglia. The pattern recognition receptors, especially TLRs, recognize a wide range of danger signals and consequently activate inflammatory cascades in microglia. TLR-4-initiated recruitment of Myd88 leads mainly to the induction of the NF-κB signaling pathway, which plays an essential role in promoting the classic M1 activation of microglia.65 The critical role of the TLR-4-initiated NF-κB signaling pathway in microglia and ischemic brain injury is evidenced by the phenotype of TLR-4-deficient mice, which have a defect in their ability to activate CD11b+ microglial cells and produce TNF-α and IL-6 and are resistant to cerebral ischemic injury.66,67 Notably, SIRT1, as an NAD+-dependent protein deacetylase, can deacetylate p65/RelA, a subunit of the heterodimeric NF-κB protein, and diminish NF-κB signaling by the lysine 310 residue that is essential for full activation of NF-κB transcription potential.34 Our study is the first to demonstrate that SIRT1 is a new bona fide target of miR-210 (Fig. 5) and that microglia-autonomous miR-210 positively regulates TLR-induced NF-κB signaling in neonatal microglia through targeting SIRT1 and inhibiting deacetylation of the lysine 310 residue of p65/RelA (Fig. 6). The pairing level of SIRT1 3′UTR to the miR-210 seed region is not perfect (5 out of 7). However, animal miRNAs can recognize their target mRNAs by using less than perfect pairing in the seed, as well as in the rest of the binding region. The binding site at the SIRT1 3′UTR is conserved in rats and mice (Supplementary Fig. S2). Although the binding site of the miR-210 seed within the SIRT1 3′UTR is less conserved in humans than in rodents (Supplementary Fig. S2), there are still five bases pairing to miR-210 seed (5 out of 7), including two G·U wobble base pairs. In addition, the pairing level in the rest of the binding region in humans is even higher than that in rodents. Therefore, further investigation of SIRT1 as a miR-210-targeting molecule in humans is needed. Notably, we predicted human SIRT6 as a putative target of miR-210-5p using the TargetScan algorithm. Overexpression of SIRT6, a NAD+dependent histone deacetylase, can suppress NF-κB-mediated inflammatory responses in human cells,68 suggesting that human SIRT6 may be an interesting target of miR-210 for future research.
Notably, a single miRNA can target multiple molecules and regulate multiple signaling pathways in different cells, which collectively regulate the same or related pathological effects.69 In addition to SIRT1, miR-210 may also regulate other molecular targets involved in inflammation-associated ischemic brain injury. For instance, miR-210 targeted mitochondrial cytochrome c assembly protein (COX10) and iron–sulfur cluster scaffold homolog (ISCU) and promoted proinflammatory molecule-induced death of young neurons after ischemic brain injury.60 COX10 and ISCU are involved in the biosynthesis of cytochrome c oxidase (complex IV) and the assembly of iron–sulfur clusters, respectively, which enhance mitochondrial function under inflammatory conditions.60,70 Therefore, by targeting multiple molecules, such as SIRT1, COX10, and ISCU, miR-210 can regulate several signaling pathways that collectively promote neuroinflammation-mediated brain damage. Although other miR-210 targets related to inflammation or apoptosis have been reported,44,61,71,72 their functional importance in regulating neuroinflammation and protecting neural cells remains elusive.
In summary, our findings demonstrate for the first time that miR-210 expression was significantly upregulated in activated microglia after neonatal HIE and that the in vivo administration of a miR-210 inhibitor effectively suppressed HIE by controlling microglia-mediated neuroinflammation in the developing brain. The in vitro studies further demonstrated that miR-210 intrinsically promotes M1 activation of neonatal rat primary microglia and enhances the expression of innate proinflammatory cytokines. Importantly, this study has revealed a novel regulatory mechanism of miR-210 in microglial inflammatory activation by targeting the SIRT1 gene to reduce deacetylation of the NF-κB subunit p65 and thereby increase NF-κB signaling activity. Thus, our study provides new insight into a potential therapeutic target of miR-210 in the modulation of microglia-mediated neuroinflammation, which may be beneficial for the treatment of infants with hypoxic-ischemic brain injury. This is of critical importance given the extreme limitation of effective and specific therapeutic interventions currently available other than hypothermia for this important clinical disorder in infants.
Materials and methods
Animals
Pregnant Sprague-Dawley rats were purchased from Charles River Laboratories (Wilmington, MA). Animals were allowed to give birth and were then kept with their pups in a room maintained at 20 ± 2 °C with a 12-h light-dark cycle. Animals were provided ad libitum access to normal rat chow and water. P7 pups of both sexes were subject to the HIE model unless otherwise indicated. P0-P1 pups were used for primary microglial isolation from mixed glial cell cultures. All animal experiments were performed according to protocols that were approved by the Institutional Animal Care and Use Committee of Loma Linda University and followed the guidelines by the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Neonatal hypoxic-ischemic encephalopathy rat model
A modified Rice-Vannucci rat model has been widely used to study neonatal HIE.30 In brief, P7 pups were anesthetized with 2% isoflurane in oxygen. A small incision was made in the neck where the right CCA was exposed, double ligated with a 5.0 silk surgical suture, and then cut between the sutures. The incision was sutured closed, and after recovery for 1 h, the pups were treated with 8% O2 and balanced with 92% nitrogen for 2 h at 37 °C. The pups were returned to their dam for recovery. For the sham control, CCA was exposed but without ligation and the hypoxic treatment. At the various time points indicated during HIE development, experimental animals were terminated for analysis.
Isolation of brain mononuclear cells
Brain mononuclear cells were isolated from neonatal rat brains as previously described.73 Brains were dissected, homogenized and incubated in complete RPMI 1640 medium containing 0.5 mg/mL collagenase D (Roche) and 50 μg/mL DNase I (Sigma) for 45 min at 37 °C. Single-cell suspensions were prepared by passing through a 70-mm strainer and washed by cold complete RPMI 1640 medium. Mononuclear cells were isolated using a 37%/70% Percoll gradient (Sigma). The cells from the interface were collected and washed twice with cold PBS. Cells were immediately used for flow cytometry analysis.
Antibodies and flow cytometry
Fluorochrome-conjugated monoclonal antibodies specific for rat CD4 (Cat#201517; Clone: W3/25), CD8a (Cat#201712; Clone: OX-8), CD11b/c (Cat# 201812; Clone: OX-42), CD45 (Cat#202213; Clone: OX-1), CD45RA (Cat#202315; Clone: OX-33), CD80 (Cat#200205; Clone: 3H5), and CD86 (Cat#200305; Clone: 24 F) were purchased from the BioLegend; antibodies for rat granulocyte marker (Cat#11-0570-82; Clone:HIS48) and macrophage marker (Cat#12-0660-82; Clone:HIS36) were purchased from eBioscience; antibodies for rat CD3 (Cat#563948; Clone:1F4), CD161a (Cat#565413; Clone:10/78), and A2B5 (Cat#565413; Clone:105/A2B5) were purchased from the BD Biosciences; rat oligodendrocyte marker O4 antibody (Cat# FAB1326P; Clone:O4) was purchased from R&D Systems. Rat Fc Block (anti-rat CD32) (Cat#550271; Clone: CD34-485) was purchased from BD Biosciences, and anti-NeuN antibody (Cat#ab177487; Clone: EPR12763) was purchased from Abcam. Fixable Viability Dye eFluor® 506 (Cat#65-0866-14) was purchased from eBioscience. Cells were stained with Fixable Viability Dye first, followed by Fc blocking and surface marker staining, following a standard procedure as described previously. To detect intracellular cytokines, cells were then subjected to intracellular cytokine staining using a Cell Fixation/Permeabilization Kit (BD Biosciences) following the manufacturer’s instructions. An Annexin V Apoptosis Detection Kit (eBioscience) was used to detect apoptotic cell death according to the manufacturer’s instructions. Stained cells were analyzed by using a MACSQuant Analyzer 10 flow cytometer (Miltenyi Biotec). FlowJo software (Tree Star) was used to analyze the data.
Isolation of neural cells
Ipsilateral brain tissues were harvested from neonatal HIE or sham rats and then dissociated to single-cell suspensions using a Neural Tissue Dissociation Kit (Miltenyi Biotec) according to the manufacturer’s instructions. Myelin debris in single-cell suspensions was depleted by using Myelin Removal Beads II (Miltenyi Biotec) according to the manufacturer’s instructions. The unlabeled cell fraction after myelin removal was subjected to magnetic-activated cell sorting (MACS) for subsequent isolation of different neural cell populations, with the following MicroBead kits (Miltenyi Biotec): for microglia, CD11b/c MicroBeads; for astrocytes, Anti-GLAST (ACSA-1) MicroBeads; for immature oligodendrocytes, Anti-O4 MicroBeads; for neuronal progenitor cells, Anti-PSA-NCAM MicroBeads were used for sorting after depletion of A2B5+ glial progenitor cells using Anti-A2B5 MicroBeads. Purified cells were processed immediately for total RNA isolation.
Intracerebroventricular injection
Locked nucleic acid (LNA)-anti-miR-210 and LNA scramble control (Exiqon) were prepared according to the manufacturer’s instructions. Pups were anesthetized with 2% isoflurane and fixed on a stereotaxic apparatus (Stoelting). An incision was made on the skull surface, and the bregma was exposed. LNA-anti-miR-210 (50 pmol) or LNA scramble (50 pmol) in a total volume of 2 μL was injected at a rate of 1 μL/min into the ipsilateral hemisphere (2 mm posterior, 1.5 mm lateral, and 3 mm below the skull surface) 24 h before the HI treatment. The incision was sutured following the procedure, and the pups were returned to their dam.
Magnetic resonance imaging (MRI) analysis
An MRI scan was performed on a Bruker 11.7-T BioSpec (Billerica). A thermostatically controlled water flow system was used to maintain body temperature at 37.0 ± 1.0 °C during the MRI scan. T2-weighted images were acquired 2 days after HIE at the animal imaging facility of Loma Linda University. Infarct volume was quantified using Mango and ImageJ as described previously. The threshold of signal intensity was determined from the contralateral hemisphere in each slice and used for identifying normal brain regions in the HI-affected hemisphere. Irregular regions of interest were drawn to encircle the infarction exhibited as a hyperintensive area within the abnormal brain region in each slice. The whole infarct volume was equal to the summed infarct areas multiplied by the slice thickness. Edema correction was also performed, and the edema-induced space-occupying effect was calculated as previously described. Infarction is expressed as a percentage of infarct volume to the volume of the contralateral hemisphere.
In vitro neonatal rat primary microglia culture
Mixed glial cultures from neonatal brain cortex were prepared as previously described.74 Briefly, brains from neonatal (P0-1) rat pups were collected in cold HBSS, and the meninges were carefully removed from the brain on ice under a dissecting microscope. Brain tissue was dissociated by using a sterile pipette in culture medium for glial cells (DMEM supplemented with 10% FBS, 1 mM l-glutamine, 1 mM sodium pyruvate, 100 U ml−1 penicillin, and 100 mg ml−1 streptomycin). After passage through 70-μm filters and washes, cells were seeded in poly-L-lysine (Sigma)-coated T-75 flasks (one flask per rat pup brain) and then placed in a humidified incubator at 37 °C with 5% CO2. The medium was changed after 5 days in culture and every 3 days thereafter. On days 14–21, microglia were shaken off the mixed brain glial cell cultures using an orbital shaker (150 rpm, 6 h, 37 °C).
In vitro microglia transfection and stimulation assays
Neonatal rat primary microglia isolated from mixed glial cultures were seeded onto poly-l-lysine-pretreated 24-well plates at 2 × 105 cells per well and grown in culture medium for microglia (RPMI 1640 medium supplemented with 10% FCS, 1 mM l-glutamine, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, 50 mM β-mercaptoethanol, 100 U ml−1 penicillin, and 100 mg ml−1 streptomycin). The cells were allowed to attach overnight. A total of 100 nM of miR-210 mimic (Qiagen), negative mimic (Qiagen), LNA-anti-miR-210 (Exiqon), LNA scramble control (Exiqon), On-Target plus rat SIRT1 siRNA (Dharmacon), or control siRNA (Dharmacon) were used for in vitro transfection. Primary microglia transfection was performed with HiPerfect transfection reagent (Qiagen) according to the manufacturer’s instructions. For microglia M1 activation, cells were recovered overnight after 6 h transfection and then stimulated by adding LPS (0.5 ng/ml, Sigma) and IFN-γ (0.1 ng/ml, PeproTech). At the desired time points, microglia were collected for analysis.
Messenger RNA quantitative RT-PCR (mRNA qPCR)
Total RNA from tissues or cells was isolated using TRIzol reagent (Invitrogen) and an RNeasy Mini Kit (Qiagen) following the manufacturer’s instructions. cDNA was prepared using a SuperScript III First-Strand Synthesis Supermix Kit (Invitrogen). Gene expression was measured in a CFX Connect Real-Time PCR Detection System (Bio-Rad) by using iQ SYBR Green Supermix (Bio-Rad) according to the manufacturer’s instructions. GAPDH was used as an internal control. The relative expression of the mRNA of interest was calculated by the 2−ΔΔCT method and is presented as the fold induction relative to the control. Primers are presented in Supplementary Table 1.
MicroRNA quantitative RT-PCR (miRNA qPCR)
Total RNA from cells was isolated using a miRNeasy Mini Kit (Qiagen) and then reverse-transcribed with a miScript II RT Kit (Qiagen) following the manufacturer’s instructions. MiR-210 expression was measured using the miScript SYBR Green PCR Kit with the miScript Primer Assay Kit (Qiagen) according to the manufacturer’s instructions. The results were normalized to the abundance of SNORD61. The relative expression of miR-210 was calculated by the 2−ΔΔCT method and is presented as the fold induction relative to the control.
In situ hybridization (ISH)
The expression of miRNA-210 in brain tissues was determined using miRCURY LNA miRNA ISH optimization Kits (Qiagen), including ISH buffer, controls, and double digoxigenin (DIG)-labeled miR-210 detection probe, following the manufacturer’s instructions. Briefly, after Proteinase K incubation, 10-μm-thick frozen section slides were incubated with 50 µL of a diluted solution (40 nM) of LNA miR-210 probe at 55 °C in a humidified chamber for 1 h. Following hybridization, the slides were washed with 5×, 1×, and 0.2× SSC (5 min each wash) at 55 °C. The slides were then incubated for 15 min in a blocking solution (0.1% Tween-20, 2% sheep serum, and 1% BSA in PBS) followed by incubation for 1 h at room temperature in an anti-DIG-AP antibody solution (1:500, Roche). The slides were washed 3 times in PBS-T and then incubated in freshly prepared AP substrate solution (Roche) for 2 h at 30 °C. After washing with water, nuclei were counterstained with Nuclear Fast Red solution (Vector Laboratories). The slides were then dehydrated and mounted using mounting medium (Sigma) for microscopy.
Prediction of miR-210 targets and luciferase activity assay
Bioinformatics tools were used to predict the miR-210 target site in the SIRT1 RNA sequence with the following settings. Reverse complement (RC) of the sequence (5′ACGCACA-3′) from the seed sequence of miR-210 (3′-UGCGUGU) was generated. We then aligned the RC sequence with SIRT1 3′UTR using sequence alignment software (http://multalin.toulouse.inra.fr/multalin/multalin.html) and identified a single discrete region out of the entire SIRT1 3′UTR where the RC sequence was aligned. In addition, we used this method to predict other targets of miR-210, such as TET2 and ISCU, which were identical to the output results of TargetScan. Thus, we decided to test whether SIRT1 is a true target of miR-210. A 412 bp region (position 173–584) of the 3′UTR of rat SIRT1 mRNA harboring the mature miR-210 target motif was cloned into the pmirGLO vector (Promega) between the NheI (5′) and SalI (3′) sites. The final construct was designated pmirGLO-SIRT1. Using site-directed mutagenesis, the putative interacting site with the miR-210 seed sequence was deleted (Genscript). This deletion construct was named pmirGLO-SIRT1Δseed. A 191 bp region (position 2686–2876) of the 3′UTR of mouse TET2 mRNA harboring a seed sequence that was exactly complementary to mature miR-210 was cloned into the pmirGLO vector (Promega) between the NheI (5′) and SalI (3′) sites, which generated the pmirGLO-TET2 construct to serve as a positive control reporter. Primers for cloning are presented in Supplementary Table 1. For luciferase assays, rat PC12 cells (ATCC) were transfected (500 ng/well) with pmirGLO-SIRT1, pmirGLO-SIRT1Δseed, pmirGLO-TET2, or pmirGLO together with miR-210 mimic (100 nM, 200 nM, or 500 nM, Qiagen) or negative mimic (100 nM, 200 nM, or 500 nM, Qiagen) by using Attractene transfection reagent (Qiagen) following the manufacturer’s instructions. Forty-eight hours post transfection, luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega). The luciferase activity was normalized to Renilla reniformis luciferase activity and expressed relative to control pmirGLO activity (% control).
RNA-induced silencing complex-immunoprecipitation (RISC-IP)
Immortalized rat microglia (Applied Biological Materials) were seeded onto collagen I-pretreated 6-well plates (Thermo Fisher) at 6 × 105 cells per well and transfected with 100 nM of either miR-210 mimic (Qiagen) or negative mimic (Qiagen) by using HiPerfect transfection reagent (Qiagen) according to the manufacturer’s instructions. Cells were harvested 24 h after transfection and washed in ice-cold PBS followed by complete lysis buffer (Active Motif) at 4 °C for 10 min. RISC-IP of the lysate was conducted using the miRNA Target IP Kit (Active Motif) following the manufacturers’ instructions. The RNA was extracted with phenol/chloroform/isoamyl alcohol (25:24:1, Fisher Scientific) once, chloroform once and precipitated and resuspended in RNase-free water. The precipitated RNA was subjected to RT-qPCR using primers specific for the rat SIRT1 3′UTR. β-actin was used as an internal control. The relative abundance of SIRT1 transcript pulled down by Ago1/2/3 antibody was calculated by the 2−ΔΔCT method and is presented as the fold induction relative to the control. Primers are presented in Supplementary Table 1.
Chromatin immunoprecipitation (ChIP)
ChIP assays were performed using the SimpleChIP Plus Sonication Chromatin IP Kit (Cell Signaling) following the manufacturers’ instructions. Briefly, primary microglia transfected with either siSIRT1 or control siRNA were fixed with 1% formaldehyde to crosslink and maintain the DNA/protein interactions. After the reactions were stopped with glycine, the cells were washed with ice-cold PBS. Chromatin extracts were sonicated to produce DNA fragments between 200 and 1000 base pairs. Anti-acetyl-NF-κB p65 ChIP grade antibody (Abcam; Cat#ab19870) or isotype IgG (Abcam; Cat#ab171870) was incubated with the chromatin extracts to precipitate the transcription factor/DNA complexes. Crosslinking was then reversed using a salt solution, and proteins were digested with proteinase K. DNA from chromatin extracts was purified using spin columns and then subjected to real-time PCR analysis. Primers that flank two binding sites of Acetyl-NF-κB p65 at the rat IL-1β promoter are presented in Supplementary Table 1.
Western blot
Total protein was extracted using a lysis buffer containing 20 mM HEPES (pH 7.6), 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, phosphatase inhibitor cocktail (Sigma-Aldrich), and protease inhibitor cocktail (Roche). Protein concentration was measured using the Bradford assay reagent (Bio-Rad). Equal amounts of protein were resolved on a 10% SDS-PAGE gel and then transferred to a nitrocellulose membrane by electrophoresis. The following antibodies were purchased from Cell Signaling and used to blot for the protein of interest: anti-rat NF-κB p65 (Cat#8242; Clone: D14E12), anti-rat Acetyl-NF-κB p65 (Cat#3045; Clone: Lys310), and anti-rat SIRT1 (Cat#8469; Clone: 1F3). Anti-GAPDH (EMD Millipore; Cat#MAB374; Clone: 6C5) was used as an internal control for total protein extracts. Signals were visualized with autoradiography using an ECL system (Thermo Fisher Scientific). The data were analyzed using ImageJ software.
Statistics
Paired comparisons were performed using the Student’s two-tailed t-test. Multiple comparisons were performed using one-way ANOVA followed by Tukey’s test. Data are presented as the mean ± SEM unless otherwise indicated. P < 0.05 was considered significant (*P < 0.05; **P < 0.01; ***P < 0.001).
Supplementary information
Acknowledgements
We thank the animal facility of Loma Linda University (LLU) for providing animal support; the LLU Flow Cytometry Education and Training Core Facility for providing flow cytometry support; and the LLU animal imaging facility for providing MRI support. This work was supported by the National Institutes of Health grants HL118861 (LZ) and NS103017 (LZ).
Author contributions
B.L. designed and conducted the experiments, analyzed the data, and wrote the manuscript. C.D. and L.H. conducted experiments and analyzed data. X.M. conducted experiments. L.Z. conceived and designed the studies, interpreted the data, and wrote the manuscript.
Competing interests
The authors declare no competing interests.
Contributor Information
Bo Li, Email: boli@llu.edu.
Lubo Zhang, Email: lzhang@llu.edu.
Supplementary information
The online version of this article (10.1038/s41423-019-0257-6) contains supplementary material.
References
- 1.Li B, Concepcion K, Meng X, Zhang L. Brain-immune interactions in perinatal hypoxic-ischemic brain injury. Prog. Neurobiol. 2017;159:50–68. doi: 10.1016/j.pneurobio.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davidson JO, Wassink G, van den Heuij LG, Bennet L, Gunn AJ. Therapeutic hypothermia for neonatal hypoxic-ischemic encephalopathy—Where to from here? Front. Neurol. 2015;6:198. doi: 10.3389/fneur.2015.00198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wood T, et al. Treatment temperature and insult severity influence the neuroprotective effects of therapeutic hypothermia. Sci. Rep. 2016;6:23430. doi: 10.1038/srep23430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deng W. Neurobiology of injury to the developing brain. Nat. Rev. Neurol. 2010;6:328–336. doi: 10.1038/nrneurol.2010.53. [DOI] [PubMed] [Google Scholar]
- 5.Hagberg H, et al. The role of inflammation in perinatal brain injury. Nat. Rev. Neurol. 2015;11:192–208. doi: 10.1038/nrneurol.2015.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nelson KB, Dambrosia JM, Grether JK, Phillips TM. Neonatal cytokines and coagulation factors in children with cerebral palsy. Ann. Neurol. 1998;44:665–675. doi: 10.1002/ana.410440413. [DOI] [PubMed] [Google Scholar]
- 7.Bartha AI, et al. Neonatal encephalopathy: association of cytokines with MR spectroscopy and outcome. Pediatric Res. 2004;56:960–966. doi: 10.1203/01.PDR.0000144819.45689.BB. [DOI] [PubMed] [Google Scholar]
- 8.Savman K, Blennow M, Gustafson K, Tarkowski E, Hagberg H. Cytokine response in cerebrospinal fluid after birth asphyxia. Pediatric Res. 1998;43:746–751. doi: 10.1203/00006450-199806000-00006. [DOI] [PubMed] [Google Scholar]
- 9.Grether JK, Nelson KB. Maternal infection and cerebral palsy in infants of normal birth weight. J. Am. Med. Assoc. 1997;278:207–211. [PubMed] [Google Scholar]
- 10.Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int. Rev. Immunol. 2011;30:16–34. doi: 10.3109/08830185.2010.529976. [DOI] [PubMed] [Google Scholar]
- 11.Liddelow SA, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–487. doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.von Bernhardi R, Eugenin-von Bernhardi L, Eugenin J. Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 2015;7:124. doi: 10.3389/fnagi.2015.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weinstein JR, Koerner IP, Moller T. Microglia in ischemic brain injury. Future Neurol. 2010;5:227–246. doi: 10.2217/fnl.10.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rocha-Ferreira E, Hristova M. Antimicrobial peptides and complement in neonatal hypoxia-ischemia induced brain damage. Front. Immunol. 2015;6:56. doi: 10.3389/fimmu.2015.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaur C, Rathnasamy G, Ling EA. Roles of activated microglia in hypoxia induced neuroinflammation in the developing brain and the retina. J. Neuroimmune Pharm. 2013;8:66–78. doi: 10.1007/s11481-012-9347-2. [DOI] [PubMed] [Google Scholar]
- 17.Satoorian T, et al. MicroRNA223 promotes pathogenic T-cell development and autoimmune inflammation in central nervous system in mice. Immunology. 2016;148:326–338. doi: 10.1111/imm.12611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li B, et al. miR-146a modulates autoreactive Th17 cell differentiation and regulates organ-specific autoimmunity. J. Clin. Investig. 2017;127:3702–3716. doi: 10.1172/JCI94012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsitsiou E, Lindsay MA. microRNAs and the immune response. Curr. Opin. Pharm. 2009;9:514–520. doi: 10.1016/j.coph.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baltimore D, Boldin MP, O’Connell RM, Rao DS, Taganov KD. MicroRNAs: new regulators of immune cell development and function. Nat. Immunol. 2008;9:839–845. doi: 10.1038/ni.f.209. [DOI] [PubMed] [Google Scholar]
- 21.Huang X, Le QT, Giaccia AJ. MiR-210–micromanager of the hypoxia pathway. Trends Mol. Med. 2010;16:230–237. doi: 10.1016/j.molmed.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kulshreshtha R, et al. A microRNA signature of hypoxia. Mol. Cell Biol. 2007;27:1859–1867. doi: 10.1128/MCB.01395-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mok Y, et al. MiR-210 is induced by Oct-2, regulates B cells, and inhibits autoantibody production. J. Immunol. 2013;191:3037–3048. doi: 10.4049/jimmunol.1301289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang H, et al. Negative regulation of Hif1a expression and TH17 differentiation by the hypoxia-regulated microRNA miR-210. Nat. Immunol. 2014;15:393–401. doi: 10.1038/ni.2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang D, Cao X, Li J, Zhao G. MiR-210 inhibits NF-kappaB signaling pathway by targeting DR6 in osteoarthritis. Sci. Rep. 2015;5:12775. doi: 10.1038/srep12775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang L, et al. Inhibition of miRNA-210 reverses nicotine-induced brain hypoxic-ischemic injury in neonatal rats. Int. J. Biol. Sci. 2017;13:76–84. doi: 10.7150/ijbs.17278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma Q, et al. Inhibition of microRNA-210 provides neuroprotection in hypoxic-ischemic brain injury in neonatal rats. Neurobiol. Dis. 2016;89:202–212. doi: 10.1016/j.nbd.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Endo K, et al. MicroRNA 210 as a biomarker for congestive heart failure. Biol. Pharm. Bull. 2013;36:48–54. doi: 10.1248/bpb.b12-00578. [DOI] [PubMed] [Google Scholar]
- 29.Zeng L, et al. MicroRNA-210 as a novel blood biomarker in acute cerebral ischemia. Front Biosci. 2011;3:1265–1272. doi: 10.2741/e330. [DOI] [PubMed] [Google Scholar]
- 30.Vannucci RC, Vannucci SJ. Perinatal hypoxic-ischemic brain damage: evolution of an animal model. Dev. Neurosci. 2005;27:81–86. doi: 10.1159/000085978. [DOI] [PubMed] [Google Scholar]
- 31.Rathnasamy G, Ling EA, Kaur C. Iron and iron regulatory proteins in amoeboid microglial cells are linked to oligodendrocyte death in hypoxic neonatal rat periventricular white matter through production of proinflammatory cytokines and reactive oxygen/nitrogen species. J. Neurosci. 2011;31:17982–17995. doi: 10.1523/JNEUROSCI.2250-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kinney HC. Human myelination and perinatal white matter disorders. J. Neurol. Sci. 2005;228:190–192. doi: 10.1016/j.jns.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 33.Liu F, McCullough LD. Inflammatory responses in hypoxic ischemic encephalopathy. Acta Pharm. Sin. 2013;34:1121–1130. doi: 10.1038/aps.2013.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yeung F, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu TF, McCall CE. Deacetylation by SIRT1 Reprograms Inflammation and Cancer. Genes Cancer. 2013;4:135–147. doi: 10.1177/1947601913476948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiong L, et al. DNA demethylation regulates the expression of miR-210 in neural progenitor cells subjected to hypoxia. FEBS J. 2012;279:4318–4326. doi: 10.1111/febs.12021. [DOI] [PubMed] [Google Scholar]
- 37.Fasanaro P, et al. MicroRNA-210 modulates endothelial cell response to hypoxia and inhibits the receptor tyrosine kinase ligand Ephrin-A3. J. Biol. Chem. 2008;283:15878–15883. doi: 10.1074/jbc.M800731200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Biswas S, et al. Hypoxia inducible microRNA 210 attenuates keratinocyte proliferation and impairs closure in a murine model of ischemic wounds. Proc. Natl Acad. Sci. USA. 2010;107:6976–6981. doi: 10.1073/pnas.1001653107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taylor CT, Colgan SP. Regulation of immunity and inflammation by hypoxia in immunological niches. Nat. Rev. Immunol. 2017;17:774–785. doi: 10.1038/nri.2017.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zaccagnini G, et al. Overexpression of miR-210 and its significance in ischemic tissue damage. Sci. Rep. 2017;7:9563. doi: 10.1038/s41598-017-09763-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qi J, et al. microRNA-210 negatively regulates LPS-induced production of proinflammatory cytokines by targeting NF-kappaB1 in murine macrophages. FEBS Lett. 2012;586:1201–1207. doi: 10.1016/j.febslet.2012.03.011. [DOI] [PubMed] [Google Scholar]
- 42.Bakirtzi K, et al. Neurotensin promotes the development of colitis and intestinal angiogenesis via Hif-1alpha-miR-210 Signaling. J. Immunol. 2016;196:4311–4321. doi: 10.4049/jimmunol.1501443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu R, et al. MicroRNA-210 overexpression promotes psoriasis-like inflammation by inducing Th1 and Th17 cell differentiation. J. Clin. Investig. 2018;128:2551–2568. doi: 10.1172/JCI97426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao M, et al. Up-regulation of microRNA-210 induces immune dysfunction via targeting FOXP3 in CD4(+) T cells of psoriasis vulgaris. Clin. Immunol. 2014;150:22–30. doi: 10.1016/j.clim.2013.10.009. [DOI] [PubMed] [Google Scholar]
- 45.Kopriva SE, Chiasson VL, Mitchell BM, Chatterjee P. TLR3-induced placental miR-210 down-regulates the STAT6/interleukin-4 pathway. PLoS One. 2013;8:e67760. doi: 10.1371/journal.pone.0067760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang L, Ma Q, Li Y, Li B, Zhang L. Inhibition of microRNA-210 suppresses pro-inflammatory response and reduces acute brain injury of ischemic stroke in mice. Exp. Neurol. 2018;300:41–50. doi: 10.1016/j.expneurol.2017.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ponomarev ED, Veremeyko T, Barteneva N, Krichevsky AM, Weiner HL. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-alpha-PU.1 pathway. Nat. Med. 2011;17:64–70. doi: 10.1038/nm.2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao H, et al. MiRNA-424 protects against permanent focal cerebral ischemia injury in mice involving suppressing microglia activation. Stroke. 2013;44:1706–1713. doi: 10.1161/STROKEAHA.111.000504. [DOI] [PubMed] [Google Scholar]
- 49.Ni J, et al. MicroRNA let-7c-5p protects against cerebral ischemia injury via mechanisms involving the inhibition of microglia activation. Brain Behav. Immun. 2015;49:75–85. doi: 10.1016/j.bbi.2015.04.014. [DOI] [PubMed] [Google Scholar]
- 50.Parisi C, et al. MicroRNA-125b regulates microglia activation and motor neuron death in ALS. Cell Death Differ. 2016;23:531–541. doi: 10.1038/cdd.2015.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang X, et al. miRNA-3473b contributes to neuroinflammation following cerebral ischemia. Cell Death Dis. 2018;9:11. doi: 10.1038/s41419-017-0014-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Butovsky O, et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014;17:131–143. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Varol D, et al. Dicer deficiency differentially impacts microglia of the developing and adult brain. Immunity. 2017;46:1030–1044 e1038. doi: 10.1016/j.immuni.2017.05.003. [DOI] [PubMed] [Google Scholar]
- 54.Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302:1760–1765. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]
- 55.Sun JJ, et al. MiRNA-210 induces the apoptosis of neuronal cells of rats with cerebral ischemia through activating HIF-1alpha-VEGF pathway. Eur. Rev. Med. Pharm. Sci. 2019;23:2548–2554. doi: 10.26355/eurrev_201903_17403. [DOI] [PubMed] [Google Scholar]
- 56.Ma Q., Dasgupta C., Li Y., & Huang L., Zhang L. MicroRNA-210 downregulates ISCU and induces mitochondrial dysfunction and neuronal death in neonatal hypoxic-ischemic brain injury. Mol. Neurobiol. 2019. 10.1007/s12035-019-1491-8 [DOI] [PMC free article] [PubMed]
- 57.Qiu J, et al. Neuroprotective effects of microRNA-210 on hypoxic-ischemic encephalopathy. Biomed. Res Int. 2013;2013:350419. doi: 10.1155/2013/350419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zeng L, et al. MicroRNA-210 overexpression induces angiogenesis and neurogenesis in the normal adult mouse brain. Gene Ther. 2014;21:37–43. doi: 10.1038/gt.2013.55. [DOI] [PubMed] [Google Scholar]
- 59.Meng Z. Y., et al. MicroRNA-210 promotes accumulation of neural precursor cells around ischemic foci after cerebral ischemia by regulating the SOCS1-STAT3-VEGF-C pathway. J. Am. Heart Assoc.7, e005052 (2018) [DOI] [PMC free article] [PubMed]
- 60.Voloboueva LA, Sun X, Xu L, Ouyang YB, Giffard RG. Distinct effects of miR-210 reduction on neurogenesis: increased neuronal survival of inflammation but reduced proliferation associated with mitochondrial enhancement. J. Neurosci. 2017;37:3072–3084. doi: 10.1523/JNEUROSCI.1777-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chio CC, et al. MicroRNA-210 targets antiapoptotic Bcl-2 expression and mediates hypoxia-induced apoptosis of neuroblastoma cells. Arch. Toxicol. 2013;87:459–468. doi: 10.1007/s00204-012-0965-5. [DOI] [PubMed] [Google Scholar]
- 62.Wang F, et al. miR-210 suppresses BNIP3 to protect against the apoptosis of neural progenitor cells. Stem Cell Res. 2013;11:657–667. doi: 10.1016/j.scr.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 63.Luan Y, Zhang X, Zhang Y, Dong Y. MicroRNA-210 Protects PC-12 Cells Against Hypoxia-Induced Injury by Targeting BNIP3. Front. Cell. Neurosci. 2017;11:285. doi: 10.3389/fncel.2017.00285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hu YW, Jiang JJ, Yan G, Wang RY, Tu GJ. MicroRNA-210 promotes sensory axon regeneration of adult mice in vivo and in vitro. Neurosci. Lett. 2016;622:61–66. doi: 10.1016/j.neulet.2016.04.034. [DOI] [PubMed] [Google Scholar]
- 65.ElAli A, Rivest S. Microglia ontology and signaling. Front. Cell Dev. Biol. 2016;4:72. doi: 10.3389/fcell.2016.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hyakkoku K, et al. Toll-like receptor 4 (TLR4), but not TLR3 or TLR9, knock-out mice have neuroprotective effects against focal cerebral ischemia. Neuroscience. 2010;171:258–267. doi: 10.1016/j.neuroscience.2010.08.054. [DOI] [PubMed] [Google Scholar]
- 67.Ginhoux F, Lim S, Hoeffel G, Low D, Huber T. Origin and differentiation of microglia. Front. Cell. Neurosci. 2013;7:45. doi: 10.3389/fncel.2013.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu Y, et al. Overexpression of Sirtuin 6 suppresses cellular senescence and NF-kappaB mediated inflammatory responses in osteoarthritis development. Sci. Rep. 2015;5:17602. doi: 10.1038/srep17602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 70.Chen Z, Li Y, Zhang H, Huang P, Luthra R. Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene. 2010;29:4362–4368. doi: 10.1038/onc.2010.193. [DOI] [PubMed] [Google Scholar]
- 71.Wang Y, Ni H, Zhang W, Wang X, Zhang H. Downregulation of miR-210 protected bupivacaine-induced neurotoxicity in dorsal root ganglion. Exp. Brain Res. 2016;234:1057–1065. doi: 10.1007/s00221-015-4513-4. [DOI] [PubMed] [Google Scholar]
- 72.Kelly TJ, Souza AL, Clish CB, Puigserver P. A hypoxia-induced positive feedback loop promotes hypoxia-inducible factor 1alpha stability through miR-210 suppression of glycerol-3-phosphate dehydrogenase 1-like. Mol. Cell. Biol. 2011;31:2696–2706. doi: 10.1128/MCB.01242-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mohammad MG, et al. Immune cell trafficking from the brain maintains CNS immune tolerance. J. Clin. Investig. 2014;124:1228–1241. doi: 10.1172/JCI71544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tamashiro T. T., Dalgard C. L., & Byrnes K. R. Primary microglia isolation from mixed glial cell cultures of neonatal rat brain tissue. J. Vis. Exp.66, e3814 (2012). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.