

In wheat and barley anthers, 24-nucleotide reproductive phasiRNAs are abundant in both meiotic and premeiotic stages.
Abstract
Two classes of premeiotic (21-nucleotides [nt]) and meiotic (24-nt) phased small interfering RNAs (phasiRNAs) and their patterns of accumulation have been described in maize (Zea mays) and rice (Oryza sativa) anthers. Their precise function remains unclear, but studies have shown that they support male fertility. The important role of phasiRNAs in anthers underpins our present study to characterize these small RNAs in wheat (Triticum aestivum) and barley (Hordeum vulgare) anthers. We staged anthers at every 0.2 mm of development for one wheat and two barley varieties. We isolated premeiotic (0.2, 0.4, and 0.6 mm), meiotic (0.8, 1.0, and 1.4 mm), and postmeiotic (1.8 mm) anthers, for which we then investigated accumulation patterns of RNAs, including reproductive phasiRNAs. We annotated a total of 12,821 and 2,897 PHAS loci in the wheat and barley genomes, respectively. By comparing the total number of PHAS loci in genomes of maize, rice, barley, and wheat, we identified an expansion of reproductive PHAS loci in the genomes of Poaceae subfamilies from Panicoideae to Oryzoideae and to Poideae. In addition to the two classes of premeiotic (21-nt) and meiotic (24-nt) phasiRNAs, previously described in maize and rice anthers, we characterized a group of 24-nt phasiRNAs that accumulate in premeiotic anthers. The absence of premeiotic 24-nt phasiRNAs in maize and rice suggests a divergence in grass species of the Poideae subfamily. Additionally, we performed a gene coexpression analysis describing the regulation of phasiRNA biogenesis in wheat and barley anthers. We highlight Argonaute 9 (AGO9) and Argonaute 6 (AGO6) as candidate binding partners of premeiotic and meiotic 24-nt phasiRNAs, respectively.
Plants produce small RNAs (sRNAs) of typically 21 to 24 nucleotides (nt). The biogenesis and regulation of sRNAs requires RNA polymerases (Pol), Dicer-like (DCL) proteins, double stranded RNA (dsRNA)-binding (DRB) proteins, RNA-directed RNA polymerases (RDRs), and Argonaute (AGO) proteins (Borges and Martienssen, 2015; Yu et al., 2018). Among the sRNAs expressed in plants, phased small interfering RNAs, or phasiRNAs, are a distinct group of sRNAs characterized as a product of processive cleavage of double-stranded RNAs in regular increments (duplexes of 21 or 24 nt) from a well-defined terminus (Axtell and Meyers, 2018). PhasiRNA biogenesis initiates via microRNA (miRNA)-directed, AGO-catalyzed cleavage of a single-stranded RNA precursor, which is then converted to dsRNA by an RDR protein before being processed into 21- or 24-nt RNA duplexes by a DCL protein. PhasiRNAs originate from both protein-coding and noncoding transcripts (Fei et al., 2013; Komiya, 2017; Yu et al., 2018). PhasiRNAs originating from protein-coding genes are 21-nt long (Komiya, 2017; Yu et al., 2018). A particular group of phasiRNAs, called reproductive phasiRNAs, is specific to male reproductive tissue and its members are either 21- or 24-nt long (Johnson et al., 2009; Song et al., 2012a; Zhai et al., 2015; Fei et al., 2016; Komiya, 2017; Yu et al., 2018). Accumulation of two distinct groups of premeiotic (21-nt) and meiotic (24-nt) reproductive phasiRNAs was found to occur in maize (Zea mays; Zhai et al., 2015) and rice (Oryza sativa; Komiya et al., 2014; Fei et al., 2016) anthers. Subsequent work has demonstrated the widespread prevalence of 24-nt phasiRNAs in angiosperm species (Xia et al., 2019). Whereas pathways governing reproductive phasiRNA production have been described, there remain gaps in our understanding.
Nonomura et al. (2007) identified the MEIOSIS ARRESTED AT LEPTOTENE 1 (MEL1) gene, which encodes an AGO protein, in a male-sterile rice mutant. A mel1 loss-of-function mutant is characterized by an arrest of chromosome condensation at early meiotic stages as well as by irregularly sized and multinucleated and vacuolated pollen mother cells (Nonomura et al., 2007). Komiya et al. (2014) demonstrated that MEL1, also known as OsAGO5c, is a binding partner specific to 21-nt phasiRNAs that are generated from long noncoding RNAs (lncRNAs). Active in premeiotic male reproductive tissues, production of these 21-nt phasiRNAs is dependent on miR2118, RDR6, DCL4, MEL1, and presumably a copy of AGO1 encoding protein to load miR2118 (Song et al., 2012a, 2012b; Komiya et al., 2014; Yu et al., 2018). Although most of the 21-nt phasiRNA loci are associated with male reproductive development, 21-nt phasiRNAs are also involved in other biological processes (Fei et al., 2013). For instance, the cleavage of TAS3 precursor transcripts triggered by miR390 is required for normal development in most, if not all, land plants (Xia et al., 2017). In both cases, RDR6 and DCL4 proteins process the 21-nt phasiRNAs, which complicates the study of this pathway with respect to reproductive development.
A second size class of phasiRNAs, 24-nt phasiRNAs, is apparently specific to male reproductive development in angiosperm species. Although our understanding of this pathway and its function is incomplete, several studies have begun to provide details. First, we know that 24-nt phasiRNA production is dependent on miR2275, RDR6, DCL5, and presumably a copy of AGO1 encoding protein to load miR2275 (Kapoor et al., 2008; Song et al., 2012a, 2012b; Yu et al., 2018). DCL5 is a monocot-specific, Dicer-like gene, that is exclusive to the 24-nt phasiRNA pathway. Recently, a dcl5 loss-of-function mutation was shown to result in developmental defects in maize tapetum (Teng et al., 2020). Although it is not yet confirmed, evidence based on transcriptional regulation suggests that AGO2b (Fei et al., 2016) and AGO18 (Zhai et al., 2015; Fei et al., 2016) might be the binding partners of 24-nt phasiRNA (analogous to MEL1, for 21-nt phasiRNAs.)
The precise function of reproductive phasiRNAs remains unknown, but some studies suggest that they may play a critical role in maintaining fertility under environmental stresses. For example, a mutation in the rice PMS1T transcript (a lncRNA that is a 21-nt phasiRNA precursor, or PHAS transcript) results in photoperiod-sensitive genic male sterility (Fan et al., 2016). Furthermore, the phenotype of temperature-sensitive male sterility was observed in a maize dcl5 loss-of-function mutant (Teng et al., 2020). Thus, both the 21- and 24-nt phasiRNA pathways appear to be vitally involved in reproductive biology and may provide approaches to use self-fertilized grass species in hybrid seed production. Bread wheat (Triticum aestivum) and barley (Hordeum vulgare ssp. vulgare) are two self-fertilized species that respectively rank first and fourth among economically important cereal crops (http://www.fao.org/faostat/). A full understanding of the gene regulatory networks controlling anther development is crucial as it may enable regulation of pollen production and the development of hybrid seed technologies for plant breeders. In this study, we staged anthers by development and performed a time-series analysis of sRNA and RNA transcripts expressed in anther development, in both wheat and barley. We describe a further group of 24-nt phasiRNAs specifically expressed in premeiotic anthers.
RESULTS
A Developmental Time Series of Staged Wheat and Barley Anthers
Anthers develop from undifferentiated meristematic cells into an organized set of tissues with a plethora of functions. To identify premeiotic, meiotic, and early postmeiotic stages of anther development in wheat and barley, anthers were dissected, fixed, and processed for resin embedding, and cross-sectioned. The developmental progression of meiosis was examined at 12 time points corresponding to 0.2- to 3.0-mm-long anthers in bread wheat (‘Fielder’) and two barley varieties: ‘Golden Promise’ (2-row; spring) and ‘Morex’ (6-row; spring). Figure 1 shows a representative anther lobe for each sample and developmental stage. We observed a similar developmental progression between the two barley varieties as well as between wheat and barley. Cell fate specification was observed in 0.2 and 0.4 mm anthers, before observing (by the 0.6-mm stage) the four distinct cell layers, namely the epidermis, endothecium, middle layer, and tapetum (Fig. 1). By 0.6 mm anthers, the microspore mother cells develop from sporogenous tissues. Subsequently, meiosis I and II occur in anthers, at the 0.8- to 1.4-mm stages. The tapetum was at its largest size (in cell width) from 0.8 and 1.0 mm anthers (in length), which corresponds to meiosis I, before degradation, until the tapetum was almost fully gone at the 1.6- to 1.8-mm stage, corresponding to the stage of vacuolated microspores. The vacuolated microspores develop into mature pollen grains by 1.6 to 3.0 mm, when dehiscence occurs and pollen is released. Based on previous observations, we defined anthers to be premeiotic at ≤0.6 mm, meiotic at 0.8 to 1.4 mm, and postmeiotic by 1.6 mm. Thus, we sequenced RNA (sRNA and transcripts) of three premeiotic (0.2, 0.4, and 0.6 mm), three meiotic (0.8, 1.0, and 1.4 mm), and one postmeiotic (1.8 mm) anther stages.
Figure 1.

Transverse sections of wheat and barley anthers ranging in size from 0.2 mm anther to the stage of pollen maturation. A, Cross-sections of anthers were performed in the middle of anthers (red arrows at left), and one lobe of the anther was captured for images, at right; the anther length was determined as indicated at left. B, Anthers were obtained from the wheat ‘Fielder’ and barley ‘Golden Promise’ and ‘Morex’. Anthers were fixed with a 2% paraformaldehyde:glutaraldehyde solution and embedded using the Monostep HM20 polar resin, sectioned to 1.0 µm and stained using 1.0% toluidine blue O. Scale bars = 20 µm.
A Comprehensive Annotation of Genomic Components Regulating PhasiRNA Biogenesis
To investigate the dynamics of phasiRNA biogenesis, regulation, and accumulation in anthers, we deep-sequenced 126 libraries, evenly distributed across sRNA and poly(A) RNA from seven sequential stages of the same bread wheat (‘Fielder’) and two barley (‘Golden Promise’ and ‘Morex’) varieties. Anthers corresponded to three premeiotic (0.2, 0.4, and 0.6 mm), three meiotic (0.8, 1.0, and 1.4 mm), and one postmeiotic (1.8 mm) developmental stages. PhasiRNA biogenesis requires PHAS precursor transcripts, miRNA triggers, and protein products of the RDR, DRB, DCL, and AGO gene families. To provide a comprehensive overview of phasiRNA regulation in wheat and barley anthers, we performed an annotation of all these components and identified those expressed in anthers.
We first examined the miRNA triggers miR2118 and miR2275, as both families are required to cleave 21-PHAS and 24-PHAS precursor transcripts, respectively. Supplemental Table S1 details the coordinates and abundances of miRNAs annotated from anther tissues. A total of 240 miRNAs were annotated in wheat, of which 71 loci generating miR2118 and 15 loci of miR2275 were identified (Supplemental Table S1). In barley, 100 miRNAs were annotated, of which 12 loci generating miR2118 and four loci of miR2275 were identified. A comparative analysis of miRNA triggers across the wheat subgenomes revealed a bias in their distribution across homeologous chromosomes. First, no miR2118 loci were observed on any homeologs for chromosomes 3, 6, and 7 (Fig. 2, A–C). Second, we observed that 31 of the 71 miR2118 loci (43.7%) came from the B subgenome (Supplemental Table S1). That bias was mainly due to three clusters of miR2118 observed on chromosomes 1B, 4B, and 5B, in which five, nine, and nine miR2118 loci, respectively, were annotated, whereas only one cluster of four loci was observed on chromosome 5 of the A and D homeologs (Fig. 2, A–C). In barley, no miR2118 loci were detected on chromosomes 3H, 5H, 6H, and 7H, although one cluster containing four miR2118 loci was observed on chromosome 4H (Fig. 2D). We did not observe any significant miR2275 clusters in wheat and barley, observing only 15 and four loci, respectively. Even so, the number of miR2275 loci differed between wheat homeologous chromosomes; we annotated six, three, and six miR2275 loci on subgenomes A, B, and D, respectively (Fig. 2, A–C; Supplemental Table S1). In contrast to miR2118, most of miR2275 loci were observed on wheat homeologs of chromosomes 3 and 7 of subgenomes A and D. Thus, our results show that the wheat subgenomes unequally contribute miR2118 and miR2275 loci, as they are mainly located on the B or A and D subgenomes, respectively. In brief, phasiRNA biogenesis can occur since the miRNA families triggering cleavage of PHAS transcripts were detected in anthers of both wheat and barley.
Figure 2.
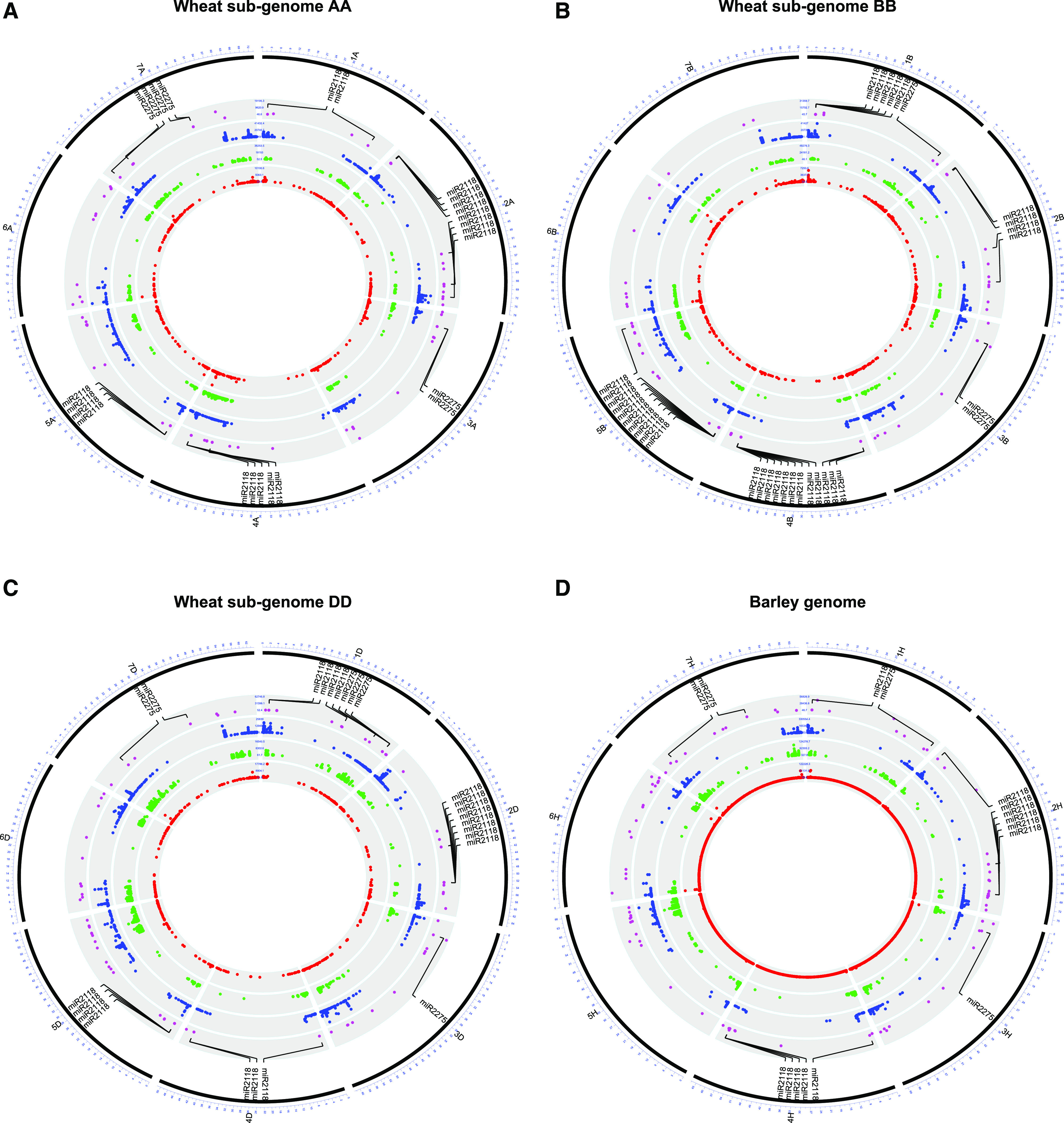
Circular plot showing the distribution and abundance of heterochromatic siRNAs (red), 21-nt (green), and 24-nt (blue) phasiRNAs as well as miRNAs (purple) annotated in wheat subgenomes A (A), B (B), and D (C) and in barley (D) genomes. The genomic distribution of miRNA triggers for 21-PHAS (miR2118) and 24-PHAS (miR2275) transcript cleavage are labeled. Normalized in reads per million mapped, the total abundance includes all sequencing libraries (21 total) covering seven stages of development in three replicates each.
AGO proteins are required to load miRNA triggers and initiate PHAS transcript cleavage in addition to loading 21- or 24-nt phasiRNAs after their biogenesis. Although Komiya et al. (2014) identified the binding partner of 21-nt phasiRNAs, a binding partner for 24-nt phasiRNAs remains elusive. Thus, we carefully annotated all genes encoding AGO proteins in wheat and barley genomes and identified the AGO genes that were expressed in anthers. We found that the wheat and barley genomes, respectively, encode a total of 67 and 22 AGO gene copies, representing nine distinct AGO genes for each species (Fig. 3; Supplemental Table S2). Of the total sets of AGO gene copies that we identified, 47 and 20 were expressed in wheat and barley anthers, respectively, and covered all distinct AGO genes. Our de novo transcript assembly thus led to the characterization of two and eight AGO gene copies in wheat and barley, respectively (Supplemental Table S2). Among the AGO proteins identified in barley, we annotated copies for AGO1 (three copies), AGO2 (two copies), AGO5 (two copies), and AGO10 (one copy).
Figure 3.

Phylogenic tree showing annotated wheat and barley orthologous genes to AGO proteins. Clades of AGO proteins are indicated. Yellow boxes highlight two groups of AGO proteins expanded in wheat and barley. Protein orthologous groups were identified using OrthoFinder and SonicParanoid. Protein alignments and phylogeny analysis were done using MUSCLE and IQ-TREE.
Some interesting observations emerged from our phylogenetic analysis. First, the number and type of AGO proteins encoded by the wheat and barley genomes were consistent if we take into account the hexaploid nature of wheat that results in a triplication of each AGO gene (Fig. 3; Supplemental Table S2). Second, most of the AGO genes annotated in bread wheat were also identified in wheat-related ancestors carrying the A, B, and D genomes. Third, the diversity of wheat, barley, and other grasses is reflected in the evolutionary relationships of the AGO1/2/5 genes (Fig. 3). We observed a division of AGO1 proteins in two subclades grouping AGO1a/b separate from AGO1c/d (Fig. 3). Additionally, we observed an expansion of AGO1 proteins, duplicated from AGO1b, expanding the AGO1 clade to five copies in wheat, Triticum dicoccoides, and barley, compared with four copies in the rice or Brachypodium distachyon genomes. When compared with other grasses, an expansion of some AGO2 and AGO5 genes was also observed (Fig. 3; Supplemental Table S2). In wheat, AGO2b was duplicated in each subgenome expanding the number of copies of that AGO gene to six. In barley, we annotated three copies of the AGO2b gene. When compared with other grass species, the number of copies of AGO5a/b/d is the same. However, AGO5c was triplicated in wheat, bringing the total number of copies to 10, whereas in barley, one AGO5c duplication event was observed. Most AGO genes mentioned above were expressed in anthers of both species.
RDR, DRB, and DCL are three other key protein families involved in phasiRNA biogenesis; thus, we annotated these families as well (Supplemental Fig. S1, A–C; Supplemental Table S2). The RDR6 protein synthesizes the second RNA strand of PHAS transcripts initially sliced by AGO proteins. We found that the RDR6 gene is duplicated in the barley genome. By contrast, we annotated only one RDR6 gene in the wheat genome, which is on chromosome 3 of the B and D homeologs but not on the A homeolog inherited from Triticum urartu. Curiously, we annotated, respectively, two and four RDR6 genes in T. urartu and T. dicoccoides genomes, similar to barley, suggesting an incomplete annotation of RDR genes in the wheat genome. Unfortunately, we did not capture transcripts encoding the RDR6 protein in our de novo transcriptome assembly. Among other RDR genes, RDR1 and RDR3 are duplicated in both wheat and barley. In barley, all RDR1 and RDR3 copies were expressed in anthers. In wheat anthers, five of six RDR1 copies were expressed, in contrast with RDR3, for which only two of seven copies were expressed. Another group of essential proteins involved in phasiRNA biogenesis are the DCL and DRB proteins that, together, process a recursive cleavage of 21- or 24-nt phased sRNA duplexes. A total of 15 and five gene copies, covering five distinct DCL proteins, were annotated in wheat and barley, respectively, which is exactly the same number as DCL genes in rice and maize (each having five copies). In wheat, each copy of these genes is evenly distributed across the A, B, and D subgenomes. Additionally, all annotated DCL genes were expressed in wheat and barley anthers including DCL4 and DCL5 genes that are known to be involved in, respectively, 21- and 24-nt phasiRNA biogenesis. A total of 25 and eight gene copies, covering five distinct DRB proteins, were annotated in wheat and barley, respectively. Both DRB1 and DRB2 were, respectively, duplicated and triplicated in both wheat and barley. DRB4 is a distinct DRB protein clade since most of its members have three or four dsRNA-binding motifs rather than two domains like other DRB proteins. Thus, DRB4 might have a distinct functional activity compared with other DRB proteins.
In summary, we characterized the set of phasiRNA biogenesis components in wheat and barley. We showed that all the machinery needed to process phasiRNAs is expressed in anthers of both species and, thus, phasiRNA biogenesis and functional activity should also be active.
Myriad Stage-Specific phasiRNAs in Wheat and Barley Anthers
To allow accurate and sensitive identification of phasiRNAs that accumulate in anthers, we analyzed the 63 sRNA libraries generated from seven sequential stages of anther development. We identified a total of 12,821 and 2,897 PHAS loci in wheat and barley (detailed in Supplemental Table S3), respectively. Of the PHAS loci identified in barley, 2,699 and 2,824 were observed in cv Golden Promise and cv Morex, respectively, of which 90.7% overlapped between the two varieties (Table 1). PHAS loci annotated in the wheat genome were fairly evenly distributed among homeologous chromosomes; the A, B, and D wheat subgenomes, respectively, encompassed 4,209, 4,079, and 3,930 loci (Table 1). We detected 2.4-fold more 21-PHAS than 24-PHAS loci in these genomes. That ratio was consistent between wheat and barley genomes, barley cultivars, or wheat subgenomes (Table 1). PHAS loci were fairly evenly distributed across all chromosomes in loci corresponding to euchromatic genomic regions in both the wheat and barley genomes (Fig. 2). Overall, fewer than 20 sRNA loci overlapped repetitive regions across nearly 3,000 loci in barley, and not one was detected that overlapped repetitive regions in wheat across almost ∼13,000 loci. This characteristic distinguishes the group of 24-nt phasiRNAs from the plant DCL3-dependent siRNAs, the 24-nt heterochromatic siRNAs, which are mostly derived from repetitive elements, primarily transposable elements.
Table 1. Total number of 21- and 24-PHAS loci annotated in the wheat and barley genomes.
| PHAS Loci | Wheat | Barley | ||||||
|---|---|---|---|---|---|---|---|---|
| Total | AAa | BBa | DDa | Unorderedb | Total | ‘Golden Promise’ | ‘Morex’ | |
| Total number of loci | 12,821 | 4,209 | 4,079 | 3,930 | 603 | 2,897 | 2,699 | 2,824 |
| 21-PHAS | 9,073 | 2,994 | 2,852 | 2,786 | 441 | 2,022 | 1,902 | 1,984 |
| 24-PHAS | 3,748 | 1,215 | 1,227 | 1,144 | 162 | 875 | 797 | 840 |
| Ratio | 2.4 | 2.5 | 2.3 | 2.4 | 2.7 | 2.3 | 2.4 | 2.4 |
The subgenomes in which PHAS loci were identified.
PHAS loci located on unordered DNA scaffolds of the wheat genome. The subgenome of origin could not be designated.
We observed a distinct temporal accumulation of 21- and 24-nt phasiRNAs in anther development. In Figure 4, we show that 21-nt phasiRNAs accumulate in barley anthers by the 0.2 to 0.6 mm stages, during cell fate specification (in 0.2 and 0.4 mm) and the differentiation of the four cell layers (0.6 mm), before decreasing in the early meiotic stage (0.8 or 1.0 mm). At their peak in quantity and diversity (in 0.2 to 0.8 mm), 21-nt phasiRNAs represented more than 90% of all 21-nt sRNAs detected in anthers; this was much higher than the 60% peak proportion of 21-nt reproductive phasiRNAs observed in maize (Zhai et al., 2015). We observed a different phasiRNA accumulation pattern for 24-nt phasiRNAs; in this case, hundreds of 24-nt phasiRNAs remained almost undetectable until the 0.8-mm stage, when anthers enter the early meiotic stage (Fig. 4). At their peak, corresponding to 0.8 to 1.4 mm anthers (with a maximum at 1.0 mm), 24-nt phasiRNAs reached 93% of all 24-nt sRNAs detected in anthers. This was again substantially greater than the 64% peak proportion observed in maize (Zhai et al., 2015). Although their abundance stays detectable, in barley, we observed a significant decrease in 21- and 24-nt phasiRNAs abundance in postmeiotic anthers (1.8-mm stage), as well as a decrease in their proportion of all 21- or 24-nt sRNAs, respectively. After their peak of accumulation, the abundance of wheat 21- and 24-nt phasiRNAs decreased less than that observed in barley.
Figure 4.

The relative accumulation of reproductive phasiRNAs and their miRNA triggers. The triggers of 21- (blue circle) and 24-nt (orange circle) phasiRNAs are, respectively, miR2118 (blue-hatched circle) and miR2275 (orange-hatched circle). These were characterized during the development of anthers in wheat ‘Fielder’ (A) and barley ‘Golden Promise’ (B) and ‘Morex’ (C). n.d., Not detected.
PhasiRNA biogenesis requires miRNA triggers of both the miR2118 and miR2275 families to cleave 21-PHAS and 24-PHAS precursor transcripts, respectively. The abundance of miR2118 family members peaked in premeiotic stages (0.2 to 0.6 mm) and then decreased in early meiotic (0.8 or 1.0 mm) stages (Fig. 4). Thus, good synchrony was observed between abundance peaks of 21-nt phasiRNAs and miR2118. By contrast, expression of miR2275 family members peaked at 0.6 or 0.8 mm, before a drastic decrease that occurred before the burst of 24-nt phasiRNAs that reached their peak at 1.0 mm (Fig. 4). Collectively, miRNA triggers and phasiRNA products showed a coordinated accumulation pattern in wheat and barley varieties, suggesting there is good conservation in the regulation of 21- and 24-nt phasiRNA pathways during anther development in the Triticeae tribe.
Pre- and Post-Meiotic Accumulation of 24-nt PhasiRNAs
Although the large majority of miR2275 and 24-nt phasiRNAs reach their peak in 0.8 to 1.0 mm anthers, we examined the abundance of all 24-PHAS loci. We discovered three distinct accumulation patterns of 24-nt phasiRNAs in wheat (Fig. 5A) and barley (Supplemental Fig. S2). For example, in wheat, the group (group a) that accumulates as previously described, 1,611 24-PHAS loci (42.9% of total 24-PHAS loci) were supported by 30.5 million (M) reads, which represent ∼81% of all 24-nt phasiRNA reads (Fig. 5A). The 24-nt phasiRNAs of this cluster are almost completely absent (median ≤5 transcript per millions of mapped reads) in barley ‘Morex’ and wheat anthers smaller than 0.6 mm, and in barley ‘Golden Promise’ anthers smaller than 0.8 mm. An example is shown in Figure 5B for wheat. At their peak of abundance (by 1.0-mm anthers), the median abundance increased greater than 1000-fold, when anthers reached the meiotic stage. Among putative PHAS precursor transcripts of this group, we found one conserved motif that matched perfectly with miR2275 and was present in, as an example in wheat, 84.6% of all PHAS precursors (Fig. 5C).
Figure 5.

Premeiotic 24-nt reproductive phasiRNAs found in wheat ‘Fielder’. We show the accumulation pattern of 24-nt phasiRNAs illustrated by the heatmap in which the scale bar indicates the relative abundance change comparing one column and another as a relative change (A), or as a box plot for an absolute measurement (B). C, two conserved motifs found in putative PHAS transcripts for premeiotic (lower; new motif) and meiotic (upper; miR2275 motif) phasiRNAs. D, Alignment of candidate sRNA triggers for premeiotic 24-PHAS loci. The degree of conservation is denoted by the shading of blue color and consensus sequence is shown with sequence logos. E, Sequence logo denoting conservation of target site (upper). Nucleotide sequence alignment for 100 premeiotic 24-PHAS loci showing the conservation of the candidate miRNA triggers target sites (lower). F, Two tracks showing the abundance (RP10M) of small RNAs in both strands of a representative premeiotic 24-PHAS loci (upper). sRNA sizes are indicated by different color as shown. Two tracks showing the phasing score of each read for both strands in this locus (lower). The red arrow indicates the candidate cleavage site from which the first phasiRNA is generated.
The accumulation pattern for the second group (group b) of 24-PHAS loci is quite distinctive from group a. We observed a peak of abundance at the earliest stage of anther development (0.2 mm) with abundances gradually decreased through development for both barley varieties and in wheat. In wheat, this group consists of 2,058 24-PHAS loci (54.9% of total 24-PHAS loci) and is supported by ∼19% of all 24-nt phasiRNA reads (7.4 m reads). Thus, their abundance peaks in premeiotic anthers and decreases ∼3-fold by meiotic stages. Among putative PHAS precursor transcripts in this group, using wheat as an example, we found one conserved motif with no homology to miR2275, or any other known miRNA, which was present in 87.5% of all PHAS precursors in group b.
To identify the sRNA capable of triggering the 24-nt premeiotic phasiRNAs, we searched for sRNAs that had a strong homology to the conserved motif within our sequenced sRNA libraries. We identified five and four candidate sRNA triggers in wheat (Fig. 5D) and barley, respectively. These candidate triggers are 22-nt in length and start with a 5′ uridine but we could not classify them as miRNAs since no encoding, hairpin structure could be confirmed. However, the candidate triggers aligned exactly to the position of the conserved motif found in 24-nt premeiotic phasiRNAs precursors (Fig. 5E). With the use of wheat as an example, target prediction analysis identified 1,524 (74.1% of all) sRNA-target interaction modules. At a target score cutoff of 4, candidate sRNA triggers were appropriately matched to direct cleavage of 790 (38.4%) 24-nt premeiotic PHAS transcripts. The read distribution and phasing score of a representative 24-nt premeiotic phasiRNA is displayed in Figure 5F. Similar findings were observed in both varieties of barley.
We observed a third group (group c) in the two barley varieties, but this group was less obvious in wheat. Only 19 and 20 PHAS group c loci were detected for barley ‘Golden Promise’ and ‘Morex’, respectively. Although these loci were detectable in premeiotic and meiotic anthers, their peak in abundance was in the postmeiotic stage of anthers, by 1.8 mm (Fig. 5A). Even though few PHAS loci displayed this pattern of read accumulation, representing a low proportion of the total 24-nt phasiRNAs, this cluster accumulating in postmeiotic stages may have a biological function in gametogenesis. In brief, our studies have shown that there are two distinct types of 24-nt phasiRNAs that peak in premeiotic and meiotic wheat and barley anthers, whereas a third category of phasiRNA accumulates in postmeiotic barley anthers.
Stage-Specific PhasiRNAs Are Coexpressed with Specific DRB, DCL, and AGO Genes
The distinct temporal accumulation of 21- and 24-nt phasiRNAs requires precise regulation of PHAS precursor transcription and of the biogenesis components of phasiRNA pathways. Transcripts assembled from 63 poly(A) RNA libraries were used to perform a coexpression analysis to identify the genes that might regulate the phasiRNA groups described above. First, 12, 14, and 15 gene modules were identified in wheat and the barley varieties ‘Golden Promise’ and ‘Morex’, respectively (Fig. 6). Relative to meiotic progression, some modules were associated with distinct phases of anther development. In wheat, for example, a total of two (modules 3 and 5), three (modules 1, 2, and 7), and three (modules 4, 6, and 8) expression modules were strictly associated with premeiotic, meiotic, and postmeiotic anthers, respectively (Fig. 6). Thus, we analyzed genes of these modules corresponding with phasiRNA accumulation, with an emphasis on DCL and DRB genes, and for those encoding candidate AGO binding partners of phasiRNAs.
Figure 6.

Coexpressed genes in anthers of wheat ‘Fielder’ (A) and barley ‘Golden Promise’ (B) and ‘Morex’ (C). For each, we present a heatmap showing relative abundance of gene expression modules and DCL, DRB, and AGO genes annotated in those modules. Coexpression analysis were performed using the R package WGCNA.
These genes are summarized in Table 2 (as well as in Fig. 6), classified into premeiotic, meiotic, or postmeiotic stages. Most of these genes show the same coexpression pattern, relative to meiotic progression, for the wheat and the two barley varieties (Fig. 6; Table 2). Thus, we focused on those showing shared expression patterns. In premeiotic anthers, many AGO genes, namely AGO1b/d, AGO5c, AGO7, AGO9, and AGO10, in addition to four DCL (DCL1, DCL2, DCL3, and DCL4) and three DRB (DRB1, DRB2c, and DRB5) genes, were coexpressed (Fig. 6; Table 2). These genes were coexpressed with two groups of phasiRNAs, namely 21-nt phasiRNAs and premeiotic 24-nt phasiRNAs. Meiotic 24-nt phasiRNA expression was concurrent with numerous AGO genes, namely, AGO2b, AGO5b, AGO6, and AGO18 as well as to DCL5 and DRB4 genes (Fig. 6; Table 2). We found a strong association between the genes mentioned above and anther developmental stages; due to the hexaploid nature of wheat, multiple copies of the same gene showed the same pattern. For example, the wheat genome encodes a total of 10 AGO5c proteins for which seven genes were expressed in anthers and all the corresponding genes were coexpressed with the premeiotic stage of anthers. In wheat, all homeologous copies of the genes reported above showed the same expression pattern in premeiotic (AGO1b/d, AGO5c, AGO9, AGO10, and DCL4) and meiotic (AGO2b, AGO5b, AGO6, AGO18, DCL5, and DRB4) anthers regardless of their total number of copies (three, six, or nine). In barley, the same coherence was observed. For instance, the AGO2b gene in barley was triplicated in its genome and all copies were coexpressed in meiotic anthers. In summary, we found that the accumulation of stage-specific reproductive phasiRNAs corresponds with specific and distinct DCL, DRB, and AGO genes.
Table 2. Coexpressed genes corresponding to anther developmental phases for DCL, DRB, and AGO genes.
Gene Identifiers can be found in Supplemental Table S2.
| Species (Variety) | Anther Developmental Stages | ||
|---|---|---|---|
| Premeiotic | Meiotic | Postmeiotic | |
| Wheat ‘Fielder’ | Tae-AGO1b/d | Tae-AGO2b | Tae-AGO1c |
| Tae-AGO5c | Tae-AGO5b | – | |
| Tae-AGO9 | Tae-AGO6 | – | |
| Tae-AGO10 | Tae-AGO18 | – | |
| Tae-DCL1 | Tae-DCL5 | – | |
| Tae-DCL2 | Tae-DRB4 | – | |
| Tae-DCL4 | – | – | |
| Tae-DRB1 | – | – | |
| Tae-DRB2a/c | – | – | |
| Tae-DRB5 | – | – | |
| Barley ‘Golden Promise’ | Hvu-AGO1b/c/d | Hvu-AGO2b | Hvu-AGO2a |
| Hvu-AGO4 | Hvu-AGO5b | Hvu-AGO5a | |
| Hvu-AGO5c | Hvu-AGO6 | Hvu-DCL4 | |
| Hvu-AGO7 | Hvu-AGO18 | – | |
| Hvu-AGO9 | Hvu-DCL5 | – | |
| Hvu-AGO10 | Hvu-DRB4 | – | |
| Hvu-DCL1 | – | – | |
| Hvu-DCL2 | – | – | |
| Hvu-DCL3 | – | – | |
| Hvu-DCL4 | – | – | |
| Hvu-DRB1 | – | – | |
| Hvu-DRB2a/c | – | – | |
| Hvu-DRB5 | – | – | |
| Barley ‘Morex’ | Hvu-AGO1b/d | Hvu-AGO2b | Hvu-AGO1c |
| Hvu-AGO4 | Hvu-AGO5b | Hvu-AGO2a | |
| Hvu-AGO5c | Hvu-AGO6 | Hvu-DRB2b | |
| Hvu-AGO7 | Hvu-AGO18 | Hvu-DCL4 | |
| Hvu-AGO9 | Hvu-DCL1 | – | |
| Hvu-AGO10 | Hvu-DCL5 | – | |
| Hvu-DCL2 | Hvu-DRB4 | – | |
| Hvu-DCL3 | – | – | |
| Hvu-DCL4 | – | – | |
| Hvu-DRB1 | – | – | |
| Hvu-DRB2c | – | – | |
| Hvu-DRB5 | – | – | |
DISCUSSION
A detailed understanding of morphological development of anthers is necessary to enable studies of the molecular pathways governing their development. Although the morphological development of wheat (Browne et al., 2018; Shunmugam et al., 2018) and barley (Waddington et al., 1983; Gómez and Wilson, 2012) anthers was described previously, in this study, we staged anthers of commonly studied varieties, as they differ from those characterized previously. Additionally, we increased the resolution of developmental stages to every 0.2 mm of development, which provided greater detail and augmented previous results in wheat (Browne et al., 2018) and barley (Gómez and Wilson, 2012). Relative to their length, an approach that aids harvesting of anthers for future molecular studies, we identified premeiotic, meiotic, and postmeiotic anthers. We detected the initiation of meiosis I by 0.8 mm anthers, and meiosis II in 1.4 mm anthers in wheat ‘Fielder’ and both barley varieties. In wheat, our observations were consistent with Shunmugam et al. (2018), who described each phase of meiosis I in 0.7 to 1.2 mm anthers of the wheat Chinese Spring cultivar. Together, our results, and those reported by Shunmugam et al. (2018), suggest a similar developmental kinetic of anthers between, or within, wheat and barley species. However, additional staging of more varieties of each species may allow more precise correspondence of meiotic progression with anther length. To enable this, we performed microscopy analyses to identify premeiotic (0.2, 0.4, and 0.6 mm), meiotic (0.8, 1.0, and 1.4 mm), and postmeiotic (1.8 mm) anthers for which we then investigated accumulation patterns of RNAs, including reproductive phasiRNAs.
In recent years, two classes of premeiotic (21 nt) and meiotic (24 nt) phasiRNAs and their patterns of accumulation in anthers have been described in maize (Zhai et al., 2015) and rice (Johnson et al., 2009; Fei and al., 2016). Their precise function remains unclear but some studies have shown that they play a critical role in supporting male fertility (Fan et al., 2016; Teng et al., 2020). Their important role in anthers underpins our current study to characterize phasiRNAs in wheat and barley anthers, for which there is, to date, only one published sRNA analysis in wheat but nothing in barley. In this study, we annotated a total of 12,821 PHAS loci in the wheat genomes and observed a ratio of 2.4-fold 21-PHAS to 24-PHAS loci. Earlier this year, Zhang et al. (2020) revisited public sRNA data generated from reproductive and nonreproductive wheat tissues. Their goal was to annotate PHAS loci in the wheat genome and compare the PHAS distribution among wheat subgenomes. In nonreproductive wheat tissues, they reported few phasiRNAs (limited 21-nt phasiRNAs and no 24-nt phasiRNAs), illustrating the reproductive enrichment of phasiRNAs. At the same threshold that we used, Zhang et al. (2020) annotated a total of 8,450 nonredundant PHAS loci and a ratio of 1.3-fold 21-PHAS to 24-PHAS loci. Overall, we discovered 4,371 more PHAS loci, perhaps due to our more extensive sampling. Most of the PHAS loci that Zhang et al. (2020) did not annotate were 21-nt phasiRNAs, which mainly accumulate in premeiotic anthers. We attribute our detection of those phasiRNAs to our analysis of isolated anthers dissected from the spike, whereas Zhang et al. (2020) examined sRNAs in spikes rather than anthers, limiting their ability to annotate these several thousands of PHAS loci.
We also annotated 2,897 PHAS loci in the barley genome. Due to the hexaploid nature of the wheat genome, we might expect to find threefold more PHAS loci in wheat than in barley. However, we observed ∼4.4-fold more PHAS loci in wheat than in barley. When the larger depth of sequencing performed for wheat libraries is considered compared with barley, we generated 2.7- (for barley ‘Morex’) or 3.4-fold (for barley ‘Golden Promise’) fewer reads than for wheat, it is possible or even probable that we missed perhaps a few hundred PHAS loci in the barley genome. Regardless, the number of PHAS loci we annotated in the wheat and barley genomes is much higher in comparison with the 639 loci previously annotated in the maize genome (Zhai et al., 2015). We detected 2.4-fold more 21-PHAS than 24-PHAS loci in wheat and barley cultivars, a ratio similar to maize, for which Zhai et al. (2015) reported a ratio of 2.6-fold 21-PHAS to 24-PHAS loci. Fei et al. (2016) annotated 1,843 21-PHAS loci in the rice genome, a similar number to the 2,022 21-PHAS loci that we annotated in the barley genome in this study. Yet, Fei et al. (2016) annotated just 50 24-PHAS loci, which is substantially lower than the number that we annotated in the wheat (3,748) and barley (875) genomes. Fei et al. (2016) analyzed 24-nt phasiRNAs in 0.40- to 0.45-mm rice anthers, corresponding to an early meiotic stage (Zhang et al., 2011). Thus, it is possible that the lower number of 24-PHAS loci reported previously in the rice genome may be due to sampling only at earlier, premeiotic, stages of development (Fei et al., 2016). When comparing the total number of PHAS loci in the genomes of maize, rice, barley, and wheat, we notice an expansion of reproductive PHAS loci in genomes of Poaceae subfamilies from Panicoideae to Oryzoideae and to Poideae.
In addition to the two classes of premeiotic (21-nt) and meiotic (24-nt) phasiRNAs previously described in maize and rice anthers (Zhai et al., 2015; Fei et al., 2016), we also described a group of 24-nt phasiRNAs that accumulate in premeiotic anthers, more like the previously characterized 21-nt phasiRNAs. We observed these uncharacterized premeiotic 24-nt phasiRNAs in both wheat and barley anthers, in which they represented more than 50% of all 24-PHAS loci that we annotated. Premeiotic 24-nt phasiRNAs have not been reported in either maize or rice, despite extensive sampling (i.e. Zhai et al., 2015). We reanalyzed these older sRNA data from maize anthers (Zhai et al., 2015) and we detected no evidence of premeiotic 24-nt phasiRNAs. This apparent absence of premeiotic 24-nt phasiRNAs in maize and rice suggests a divergence in grass species of the Poideae subfamily. Future studies on more grass subfamilies could shed light on the emergence and evolution of this further class of premeiotic 24-nt phasiRNAs.
Initiation of 24-nt meiotic phasiRNA biogenesis canonically occurs at a miR2275 target site (Johnson et al., 2009; Song et al., 2012a). We observed this conserved motif, homologous to miR2275, in PHAS precursors of meiotic 24-nt phasiRNAs. For the premeiotic 24-nt phasiRNAs characterized here, we found that miR2275 is not expressed coincident with their accumulation in anthers, and no motif matching miR2275 was present in their PHAS precursors. We did identify, in these PHAS precursors, a well-conserved motif that lacked confident homology to known miRNAs registered on miRBase and to further miRNAs annotated in wheat or barley. However, we identified a candidate sRNA trigger in wheat (five copies in the genome) and barley (four copies in the genome) with evidence supporting its role in triggering the cleavage of premeiotic 24-nt PHAS transcripts. The poor assembly at the genomic origin of these sRNAs does not allow us to annotate them as miRNAs, since we could not confirm a hairpin precursor sequence at these loci. Thus, it remains unclear how the biogenesis of these previously uncharacterized, premeiotic 24-nt phasiRNA is initiated. Rather than initiation via a miRNA-directed, AGO-catalyzed cleavage of a single-stranded PHAS precursor, phasiRNA biogenesis may be initiated by an as-yet unknown mechanism.
PhasiRNA biogenesis and regulation requires a set of specific proteins. Over the last decade, both the 21- and 24-nt phasiRNA pathways have been described. Functional studies have shown that generation of premeiotic 21-nt phasiRNAs is dependent on miR2118, RDR6, DCL4, and MEL1, and presumably a version of AGO1 (Komiya et al., 2014; Nonomura et al., 2007; Song et al., 2012a, 2012b). Fei et al. (2016) performed a coexpression analysis in premeiotic rice anthers and reported that the expression pattern of OsAGO1d was the same as OsAGO5c, suggesting a functional connection between AGO1d and premeiotic phasiRNAs. Similarly, in this study, we observed the same expression pattern for the AGO1b and AGO5c genes in wheat and barley anthers. Furthermore, the wheat genome contains a total of 10 AGO5c protein-encoding genes of which seven are expressed in anthers. Concordantly, we found that all the gene copies showed the same expression in phase with the peak of 21-nt, premeiotic phasiRNA expression. Moreover, our analysis revealed that AGO7 (only in barley anthers), AGO9, and AGO10 were also coexpressed with AGO1d and AGO5c, although it remains unclear how these AGO proteins may be involved. Perhaps their role is in a developmental process such as the regulation of shoot apical meristems, as by AGO10 (Zhang et al., 2015), or other functions in anther development, independent of a role in the reproductive 21-nt phasiRNA pathway.
Numerous studied have described the 24-nt phasiRNA pathway as dependent on miR2275, RDR6, and DCL5, presumably as well as a paralog of AGO1 (Song et al., 2012a, 2012b; Zhai et al., 2015; Fei et al., 2016; Teng et al., 2020). Until now, no functional studies have identified an AGO protein as the binding partner of 24-nt phasiRNAs. However, some promising candidate AGO genes have been proposed in maize and rice. In rice, Fei et al. (2016) showed that the expression of AGO2b, AGO5b, and AGO18 corresponded with the peak of 24-nt meiotic phasiRNA accumulation. We made the same observation in wheat and barley anthers. We also found a strong correspondence of AGO6 expression with the burst of 24-nt phasiRNA in meiotic anthers. Additionally, the normalized abundance of AGO6 expression is ∼1.5-fold higher than AGO5b or AGO18, and 5- to 10-fold higher than AGO2b. Similar to AGO4 and AGO9, the AGO6 protein was described as a binding partner of 24-nt heterochromatic siRNAs (Borges and Martienssen, 2015) associated with RNA-directed DNA methylation (RdDM). AGO6 thus represents a promising candidate as a binding partner to 24-nt meiotic phasiRNAs.
Among the AGO genes coexpressed with premeiotic 24-nt phasiRNAs, AGO9 is of particular interest, because in Arabidopsis (Arabidopsis thaliana) female reproductive development, AGO9 is a binding partner to heterochromatic 24-nt sRNAs in the silencing of transposable elements (Olmedo-Monfil et al., 2010; Zhang et al., 2015). In both Arabidopsis and maize, AGO9 represses germ cell fate in the somatic cells surrounding the precursors of the gametic cells (Olmedo-Monfil et al., 2010; Singh et al., 2011; Zhang et al., 2015). A loss-of-function ago9 mutant in maize exhibits unreduced (diploid) gametes, abnormal chromatin condensation during meiosis, and hypomethylated repeats in centromeres (Singh et al., 2011; Zhang et al., 2015). Together, the binding affinity of AGO9 with 24-nt heterochromatic sRNAs, its role in the female reproductive development, and the coexpression of AGO9 with premeiotic 24-nt phasiRNAs (in wheat and barley) highlight AGO9 as a strong candidate for the binding partner of premeiotic 24-nt phasiRNAs.
CONCLUSION AND PERSPECTIVES
An efficient and cost-effective hybrid seed program has been difficult to develop in wheat and barley since they are self-fertilized species. The development of genic male-sterile lines may serve as the basis for the development of such a breeding program, via control of the production of pollen. In rice, such breeding programs may use photoperiod- or temperature-sensitive genic male-sterile lines (Ma and Yuan, 2015). Perturbed individual 21-nt reproductive phasiRNAs underlies this trait in rice (Ding et al., 2012; Fan et al., 2016). In maize, loss of the 24-nt reproductive phasiRNAs also confers a conditional male sterile phenotype (Teng et al., 2020). To develop these types of genic male-sterile lines in wheat and/or barley, it will be important to fully characterize the loci and their expression patterns during anther development that yield these reproductive phasiRNAs, and it will be important to understand the genes involved in their biogenesis.
We describe a catalog of PHAS loci in wheat and barley, at high resolution, over the course of anther development. We discovered a large cohort of premeiotic, 24-nt phasiRNAs not previously observed in rice or maize. Questions remain, such as which AGO proteins are the binding partners of premeiotic and meiotic 24-nt phasiRNAs. In the near future, more functional experiments are needed to fully characterize the 24-nt phasiRNA pathway and to precisely describe the biological function of both 21- and 24-nt reproductive phasiRNAs. Ideally, this additional knowledge will enable the modulation of male fertility with varied environmental conditions such as light or temperature.
MATERIAL AND METHODS
Plant Growth Conditions and Tissue Harvesting
Plants of bread wheat (Triticum aestivum ssp. aestivum) ‘Fielder’ (spring) and barley (Hordeum vulgare ssp. vulgare) ‘Golden Promise’ (2-row; spring) and ‘Morex’ (6-row; spring) were grown in a greenhouse under conditions of 20°/18°C day/night, 16-/8-h light/dark, and 50% relative humidity. For both species, we used a stereomicroscope (Mantis Elite-Cam HD, Vision Engineering) to dissect anthers at the following lengths: 0.2, 0.4, 0.6, 0.8, 1.0, 1.2, 1.4, 1.6, 1.8, 2.0, 2.5, and 3.0 mm. Anthers were collected only from middle spikelets of the first three tillers. Only anthers from the first florets, in wheat, or the main spikelets, in barley, were dissected. For histological analysis, 10 anthers were collected per sample. For RNA analysis, per sample, a total of 125 (anther length < 0.8 mm), 75 (anther length between 0.8 to 1.2 mm), and 50 (anther length > 1.2 mm) anthers were collected for RNA isolation. Anthers were harvested in three replicates and, after dissection, samples were immediately frozen in liquid nitrogen and kept at −80°C before RNA isolation.
Tissue Embedding and Microscopy
Fresh young spikelets were fixed (2% [v/v] paraformaldehyde and 2% [v/v] glutaraldehyde plus 0.1% [v/v] Tween20 in 0.1 m PIPES buffer at pH 7.4) overnight and dehydrated through a standard ethanol series (30%, 50%, 70%, 80%, 90%, 100% [v/v] of cold ethanol) before being resin infiltrated and embedded using the Lowicryl Monostep Embedding Media HM20 (no. 14345, Electron Microscopy Sciences) using either heat or UV polymerization. Embedded tissues were sectioned at 1 µm using the Leica Ultracut UCT (Leica Microsystems Inc.) and stained using a 1.0% (v/v) Toluidine Blue O dye (no. 26074-15, Electron Microscopy Sciences). Microscopy images were captured using a ZEISS Axio Zoom.V15 microscope using the PlanNeoFluar Z 2.3×/0.57 FWD 10.6-mm objective lens with a magnification of 260X. Digital images were captured at 2584 × 1936 pixel resolution at 12 bits/channel.
RNA Isolation, Library Construction, and Sequencing
Total RNA was extracted with TRI Reagent (Sigma-Aldrich) following the manufacturer’s instructions. RNA quality was assessed using an Agilent RNA 6000 Nano Kit with the Bioanalyzer 2100 (Agilent Technologies). Only samples with an RNA integrity number ≥7.0 were kept for library construction. sRNA libraries were prepared using the RealSeq-AC miRNA Library Kit for Illumina sequencing (Somagenics), following the manufacturer’s recommendations. Small RNA libraries were size selected for the end product of ∼150 nt using a polyacrylamide/urea gel or the SPRIselect Reagent (Beckman Coulter Life Sciences) magnetic beads for, respectively, wheat and barley. RNA-sequencing (RNA-seq) libraries were constructed using a NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (no. E7765L, New England Biosystems), following the manufacturer’s recommendations to prepare complementary DNA libraries having an insert of ∼200 bp or 300 to 350 bp for wheat and barley, respectively. Single-end sequencing was performed: 76 (3 lanes) and 151 cycles (3 lanes) for, respectively, sRNA-seq and RNA-seq libraries. The sequencing was generated on an Illumina NextSeq 550 instrument (Illumina) at the University of Delaware DNA Sequencing and Genotyping Center.
Bioinformatics Analysis of sRNA-Seq Data
With use of cutadapt v1.9.1 (Martin, 2011), sRNA-seq reads were preprocessed to remove the 3′ adapter; trimmed reads shorter than 15 nt were discarded. These cleaned reads were mapped to the wheat (Triticum_aestivum.IWGSC.dna.toplevel) and barley (Hordeum_vulgare.IBSC_v2.dna.toplevel) reference genomes (ftp://ftp.ensemblgenomes.org/pub/plants/release-44) using ShortStack v3.8.5 (Johnson et al., 2016) with following parameters: -mismatches 0, -bowtie_m 50, -mmap u, -dicermin 19, -dicermax 25, -foldsize 800, and -mincov 0.5 transcripts per million mapped. Results reported by ShortStack were filtered to keep only clusters having a predominant RNA size observed between 20 and 24 nt inclusively. We then annotated classes of miRNAs and phasiRNAs.
First, sRNA reads representative of each cluster were aligned to monocot-derived miRNAs listed in miRBase release 22 (Kozomara and Griffiths-Jones, 2014; Kozomara et al., 2019) using ncbi-blastn v2.9.0+ (Camacho et al., 2009) with following parameters: -strand both, -task blastn-short, -perc_identity 75, -no_greedy, and -ungapped. Homology hits were filtered, and sRNA reads were classed as known miRNAs, based on the following criteria: 1) no more than four mismatches and 2) no more than 2-nt extension or reduction at the 5′ end or 3′ end. Known miRNAs were summarized by family, and when genomes contained multiple miRNA loci per family, miRNAs were ordered per chromosomal position and renamed base on these genomic positions. Small RNA reads with no homology to known miRNA were annotated for new miRNA using the de novo miRNA annotation performed by ShortStack. The secondary structure of new miRNA precursor sequences was drawn using the RNAfold v2.1.9 program (Lorenz et al., 2011). Candidate miRNAs were manually inspected and only those meeting criteria for plant miRNA annotations (Axtell and Meyers, 2018) were kept for downstream analyses. Then, remaining sRNA clusters were analyzed to identify phasiRNA-generating loci based on the ShortStack analysis report. sRNA clusters having a “Phase Score” exceeding or equal to 30 were considered as true positive phasiRNA loci. Genomic regions at these phasiRNAs were identified as PHAS loci and grouped in categories of 21- and 24-PHAS loci based on the length of phasiRNA derived from these loci.
Identification of Candidate 24-nt Premeiotic PhasiRNA Triggers
We used MEME v5.1.0 (Bailey et al., 2009; Bailey et al., 2015) to identify and visualize conserved nucleotide motifs within 24-PHAS transcripts. The consensus motif sequence was used as query to search candidate small RNA triggers of 24-nt, premeiotic phasiRNAs using CD‐HIT v4.8.1 (Li and Godzik, 2006) with following options: -n 8, -d 0, -g 1, -T 10, and -c 0.8. sRNAs were considered as candidate triggers when the meet following criteria: 1) 22-nt in length, 2) ≥10 raw reads, and 3) an uracil as 5′-end nucleotides.
We performed target prediction for candidate sRNA triggers on premeiotic 24-PHAS precursor transcripts using sPARTA v1.26 (Kakrana et al., 2014). The sRNA-target interactions having score ≤4 were considered valid and used for subsequent analysis. To visualize the conservation of the motif among PHAS transcripts, we performed a multiple sequence alignment using MUSCLE v3.8.1551 (Edgar, 2004) with default parameters. We visualize the alignment using Jalview v2.11 (Waterhouse et al., 2009).
Bioinformatics Analysis of RNA-seq Data
Using cutadapt v1.9.1, RNA-seq reads were preprocessed at the 3′ end using a Phred quality score ≥20 and trimmed reads shorter than 25 nt were discarded. Clean reads were mapped to wheat and barley reference genomes using HISAT2 v2.1.0 (Kim et al., 2015). A de novo reference-guide transcript assembly was performed using StringTie v2.0.1 (Pertea et al., 2015; Pertea et al., 2016) with following parameters: -c 10, -f 0.2, and -m 200. Anther-specific transcriptomes expressed in wheat and barley were then reconstructed with StringTie–merge (run with -f 0.2 and -m 200 parameters).
Transcript assemblies were compared with wheat and barley reference annotations using gffcompare utility (http://github.com/gpertea/gffcompare) and parsed in two groups of known and unknown gene/transcript loci. Unknown transcripts were analyzed in three steps. First, we annotated transcripts having homology to Rfam v14.1 sequences (Kalvari et al., 2018a, 2018b) using the Infernal v1.1.2 program (Nawrocki and Eddy, 2013) with default parameters. Second, transcripts without a confident match to Rfam were analyzed to annotate lncRNA by combining CREMA v1.0 (Simopoulos et al., 2018) and FEELnc v0.1.1 (Wucher et al., 2017) programs with default parameters. Third, remaining transcripts were analyzed to identify open reading frames (ORFs) using Transdecoder v5.5.0 (https://github.com/TransDecoder/TransDecoder.git). ORFs containing a CDS ≥ 150 amino acids were aligned to the SwissProt (https://www.uniprot.org/downloads; download: 2019-01-10) protein database using the blastp option of diamond v0.9.29.130 (Buchfink et al., 2015) with following options: -e 1e-100,–matrix BLOSUM62, and–more-sensitive. The final annotated assembly was used to support interpretations of sRNA regulation during anther development.
Abundance Analyses of RNA
Before performing abundance analyses, we generated a read-count matrix for sRNA- and RNA-seq experiments in wheat and barley. The read-count matrix produced by ShortStack was used to perform sRNA abundance analyses. For RNA-seq assembly, reads mapping to genes of wheat and barley assemblies were counted using StringTie (run with -e and -B parameters) and a read-count matrix was extracted using a python helper script (prepDE.py) provided with StringTie. Before preforming the expression analysis, we filtered the read-count matrices and kept only sRNA clusters or genes with, respectively, ≥5 and ≥10 reads per million over a minimum of three samples and normalized read count abundance for library size with the trimmed mean of M values method by using edgeR (Robinson et al., 2010; McCarthy et al., 2012). To assess the degree of uniformity among replicates of anther at distinct developmental stages, a multidimensional scaling analysis was performed with edgeR. The trimmed mean of M values normalized transcript count per million (TPM) was generated and used to identify coexpressed genes using the weighted correlation network analysis R package (Langfelder and Horvath, 2008) to perform a weighted correlation network analysis.
Orthology and Phylogeny Analysis of Protein Genes Involved in sRNA Pathways
To study protein-coding genes involved in sRNA pathways, we used OrthoFinder v2.3.11 (Emms and Kelly, 2015) and SonicParanoid v1.2.6 (Cosentino and Iwasaki, 2019) to perform a gene orthology inference in wheat and barley. Proteomes of three dicots (Arabidopsis [Arabidopsis thaliana), Glycine max and Solanum lycopersicum) and ten monocots (Aegilops tauschii, Brachypodium distachyon, barley, Oryza sativa ssp. indica, O. sativa ssp. japonica, wheat, Triticum turgidum ssp. dicoccoides, T. turgidum ssp. durum, Triticum urartu, and Zea mays) were included to the analysis. Additionally, unknown ORFs derived from de novo transcriptome assemblies in wheat and barley anther were added to these analyses. RDR, DCL, DRB, and AGO reference protein sequences of Arabidopsis, Z. mays and O. sativa (reported by Zhang et al., 2015 or available on UniProt database) were used to identify orthologous groups of these protein families. Retrieved protein sequences were aligned using MUSCLE v3.8.1551 (Edgar, 2004) with default parameters. Protein alignment was trimmed with trimAL v1.4.rev15 (Capella-Gutiérrez et al., 2009) with the following parameter: -gappyout. Protein alignments were used to infer phylogenetic trees by maximum likelihood using IQ-TREE v1.6.12 (Minh et al., 2013; Nguyen et al., 2015; Kalyaanamoorthy et al., 2017) with the following parameters: -alrt 1000 and -bb 1000. Finally, we used iTOL v4 (Letunic and Bork, 2019) to draw consensus trees.
Data Visualization
Various plots were produced to visualize data using R programs. Circular plots were drawn using OmicCircos v.1.24.0 (Ying and Chunhua, 2015) to draw the chromosomal distribution and total abundance of phasiRNA and miRNA loci. Programs pheatmap v.1.0.10 (https://rdrr.io/cran/pheatmap/) and ggpubr v.0.2.4.999 (Wickham, 2016; Kassambara, 2018) were used to visualize abundance changes in phasiRNA through the development of anther.
Data Availability
The complete set of raw sRNA-seq and RNA-seq reads were deposited in the Sequence Read Archive under SRA accession number PRJNA636099.
Accession Numbers
Sequence data from this article can be found in the GenBank/EMBL data libraries under accession numbers PRJNA636099.
Supplemental Data
The following supplemental materials are available.
Supplemental Figure S1. Phylogenic trees showing annotated wheat and barley orthologous genes to RDR, DCL, and DRB proteins.
Supplemental Figure S2. Subgroups of 24-nt phasiRNAs having distinct accumulation patterns through anther development for barley.
Supplemental Table S1. Coordinates and abundance of all miRNA detected in wheat ‘Fielder’ and in barley ‘Golden Promise’ and ‘Morex’.
Supplemental Table S2. Summary of all RDR, DRB, DCL, and AGO protein genes encoded by the wheat and barley genome in addition to names proposed according to their evolutionary relation to other species determinate by a phylogenic analysis.
Supplemental Table S3. Coordinates and abundance of all PHAS loci detected in wheat ‘Fielder’ and in barley ‘Golden Promise’ and ‘Morex’.
Acknowledgments
We thank members of the Meyers lab, as well as Graham Moore and Azahara Martin (John Innes Centre), for helpful discussions and Joanna Friesner (Donald Danforth Plant Science Center) for assistance with editing. We thank Mayumi Nakano (Donald Danforth Plant Science Center) for assistance with data handling. We wish to thank the Advanced Bioimaging Laboratory at the Donald Danforth Plant Science Center for support with sample preparation and imaging.
Footnotes
This work was supported by USDA | National Institute of Food and Agriculture (“BTT EAGER” award no. 2018–09058 to B.C.M.), as well as resources from the Donald Danforth Plant Science Center, the University of Missouri–Columbia, Calcul Québec, and Compute Canada.
Articles can be viewed without a subscription.
References
- Axtell MJ, Meyers BC(2018) Revisiting criteria for plant miRNA annotation in the era of big data. Plant Cell 30: 272–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS(2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37: W202-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Johnson J, Grant CE, Noble WS(2015) The MEME suite. Nucleic Acids Res 43(W1): W39-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges F, Martienssen RA(2015) The expanding world of small RNAs in plants. Nat Rev Mol Cell Biol 16: 727–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne RG, Iacuone S, Li SF, Dolferus R, Parish RW(2018) Anther morphological development and stage determination in Triticum aestivum. Front Plant Sci 9: 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B, Xie C, Huson DH(2015) Fast and sensitive protein alignment using DIAMOND. Nat Methods 12: 59–60 [DOI] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL(2009) BLAST+: architecture and applications. BMC Bioinformatics 10: 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T(2009) trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25: 1972–1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosentino S, Iwasaki W(2019) SonicParanoid: Fast, accurate and easy orthology inference. Bioinformatics 35: 149–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Lu Q, Ouyang Y, Mao H, Zhang P, Yao J, Xu C, Li X, Xiao J, Zhang Q(2012) A long noncoding RNA regulates photoperiod-sensitive male sterility, an essential component of hybrid rice. Proc Natl Acad Sci USA 109: 2654–2659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC.(2004) MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emms DM, Kelly S(2015) OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol 16: 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez JF, Wilson ZA(2012) Non-destructive staging of barley reproductive development for molecular analysis based upon external morphology. J Exp Bot 63: 4085–4094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Yang J, Mathioni SM, Yu J, Shen J, Yang X, Wang L, Zhang Q, Cai Z, Xu C, et al. (2016) PMS1T, producing phased small-interfering RNAs, regulates photoperiod-sensitive male sterility in rice. Proc Natl Acad Sci USA 113: 15144–15149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei Q, Xia R, Meyers BC(2013) Phased, secondary, small interfering RNAs in posttranscriptional regulatory networks. Plant Cell 25: 2400–2415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei Q, Yang L, Liang W, Zhang D, Meyers BC(2016) Dynamic changes of small RNAs in rice spikelet development reveal specialized reproductive phasiRNA pathways. J Exp Bot 67: 6037–6049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson C, Kasprzewska A, Tennessen K, Fernandes J, Nan GL, Walbot V, Sundaresan V, Vance V, Bowman LH(2009) Clusters and superclusters of phased small RNAs in the developing inflorescence of rice. Genome Res 19: 1429–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson NR, Yeoh JM, Coruh C, Axtell MJ(2016) Improved placement of multi-mapping small RNAs. G3 (Bethesda) 6: 2103–2111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakrana A, Hammond R, Patel P, Nakano M, Meyers BC(2014) sPARTA: A parallelized pipeline for integrated analysis of plant miRNA and cleaved mRNA data sets, including new miRNA target-identification software. Nucleic Acids Res 42: e139–e139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalvari I, Argasinska J, Quinones-Olvera N, Nawrocki EP, Rivas E, Eddy SR, Bateman A, Finn RD, Petrov AI(2018a) Rfam 13.0: Shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res 46: D335–D342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalvari I, Nawrocki EP, Argasinska J, Quinones-Olvera N, Finn RD, Bateman A, Petrov AI(2018b) Non-coding RNA analysis using the Rfam database. Curr Protoc Bioinformatics 62: e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS(2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14: 587–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor M, Arora R, Lama T, Nijhawan A, Khurana JP, Tyagi AK, Kapoor S(2008) Genome-wide identification, organization and phylogenetic analysis of Dicer-like, Argonaute and RNA-dependent RNA Polymerase gene families and their expression analysis during reproductive development and stress in rice. BMC Genomics 9: 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassambara A. (2018) ggpubr: “ggplot2” Based publication ready plots. R package, version 0.2. https://rpkgs.datanovia.com/ggpubr/
- Kim D, Langmead B, Salzberg SL(2015) HISAT: A fast spliced aligner with low memory requirements. Nat Methods 12: 357–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiya R.(2017) Biogenesis of diverse plant phasiRNAs involves an miRNA-trigger and Dicer-processing. J Plant Res 130: 17–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiya R, Ohyanagi H, Niihama M, Watanabe T, Nakano M, Kurata N, Nonomura K(2014) Rice germline-specific Argonaute MEL1 protein binds to phasiRNAs generated from more than 700 lincRNAs. Plant J 78: 385–397 [DOI] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S(2014) miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42: D68–D73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara A, Birgaoanu M, Griffiths-Jones S(2019) miRBase: From microRNA sequences to function. Nucleic Acids Res 47: D155–D162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S(2008) WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics 9: 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Bork P(2019) Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res 47: W256–W259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Godzik A(2006) Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22: 1658–1659 [DOI] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Höner Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL(2011) ViennaRNA Package 2.0. Algorithms Mol Biol 6: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma GH, Yuan LP(2015) Hybrid rice achievements, development and prospect in China. J Integr Agric 14: 197–205 [Google Scholar]
- Martin M.(2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17: 10–12 [Google Scholar]
- McCarthy DJ, Chen Y, Smyth GK(2012) Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res 40: 4288–4297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh BQ, Nguyen MA, von Haeseler A(2013) Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol 30: 1188–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki EP, Eddy SR(2013) Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29: 2933–2935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ(2015) IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32: 268–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonomura K, Morohoshi A, Nakano M, Eiguchi M, Miyao A, Hirochika H, Kurata N(2007) A germ cell specific gene of the ARGONAUTE family is essential for the progression of premeiotic mitosis and meiosis during sporogenesis in rice. Plant Cell 19: 2583–2594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmedo-Monfil V, Durán-Figueroa N, Arteaga-Vázquez M, Demesa-Arévalo E, Autran D, Grimanelli D, Slotkin RK, Martienssen RA, Vielle-Calzada JP(2010) Control of female gamete formation by a small RNA pathway in Arabidopsis. Nature 464: 628–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL(2016) Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc 11: 1650–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL(2015) StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33: 290–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK(2010) edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simopoulos CMA, Weretilnyk EA, Golding GB(2018) Prediction of plant lncRNA by ensemble machine learning classifiers. BMC Genomics 19: 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Goel S, Meeley RB, Dantec C, Parrinello H, Michaud C, Leblanc O, Grimanelli D(2011) Production of viable gametes without meiosis in maize deficient for an ARGONAUTE protein. Plant Cell 23: 443–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Li P, Zhai J, Zhou M, Ma L, Liu B, Jeong DH, Nakano M, Cao S, Liu C, et al. (2012a) Roles of DCL4 and DCL3b in rice phased small RNA biogenesis. Plant J 69: 462–474 [DOI] [PubMed] [Google Scholar]
- Song X, Wang D, Ma L, Chen Z, Li P, Cui X, Liu C, Cao S, Chu C, Tao Y, et al. (2012b) Rice RNA-dependent RNA polymerase 6 acts in small RNA biogenesis and spikelet development. Plant J 71: 378–389 [DOI] [PubMed] [Google Scholar]
- Shunmugam ASK, Bollina V, Dukowic-Schulze S, Bhowmik PK, Ambrose C, Higgins JD, Pozniak C, Sharpe AG, Rozwadowski K, Kagale S(2018) MeioCapture: An efficient method for staging and isolation of meiocytes in the prophase I sub-stages of meiosis in wheat. BMC Plant Biol 18: 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng C, Zhang H, Hammond R, Huang K, Meyers BC, Walbot V(2020) Dicer-like 5 deficiency confers temperature-sensitive male sterility in maize. Nat Commun 11: 2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddington SR, Cartwright PM, Wall PC(1983) A quantitative scale of spike initial and pistil development in barley and wheat. Ann Bot (Lond) 51: 119–130 [Google Scholar]
- Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ(2009) Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 25: 1189–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H.(2016) ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag, New York [Google Scholar]
- Wucher V, Legeai F, Hédan B, Rizk G, Lagoutte L, Leeb T, Jagannathan V, Cadieu E, David A, Lohi H, et al. (2017) FEELnc: A tool for long non-coding RNA annotation and its application to the dog transcriptome. Nucleic Acids Res 45: e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia R, Chen C, Pokhrel S, Ma W, Huang K, Patel P, Wang F, Xu J, Liu Z, Li J, Meyers BC(2019) 24-Nt reproductive phasiRNAs are broadly present in angiosperms. Nat Commun 10: 627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia R, Xu J, Meyers BC(2017) The emergence, evolution, and diversification of the miR390-TAS3-ARF pathway in land plants. Plant Cell 29: 1232–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying H, Chunhua Y (2015) OmicCircos: High-quality circular visualization of omics data. R package version. 1(0). https://bioconductor.org/packages/release/bioc/html/OmicCircos.html
- Yu Y, Zhou Y, Zhang Y, Chen Y(2018) Grass phasiRNAs and male fertility. Sci China Life Sci 61: 148–154 [DOI] [PubMed] [Google Scholar]
- Zhai J, Zhang H, Arikit S, Huang K, Nan GL, Walbot V, Meyers BC(2015) Spatiotemporally dynamic, cell-type-dependent premeiotic and meiotic phasiRNAs in maize anthers. Proc Natl Acad Sci USA 112: 3146–3151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Luo X, Zhu L(2011) Cytological analysis and genetic control of rice anther development. J Genet Genomics 38: 379–390 [DOI] [PubMed] [Google Scholar]
- Zhang H, Xia R, Meyers BC, Walbot V(2015) Evolution, functions, and mysteries of plant ARGONAUTE proteins. Curr Opin Plant Biol 27: 84–90 [DOI] [PubMed] [Google Scholar]
- Zhang R, Huang S, Li S, Song G, Li Y, Li W, Li J, Gao J, Gu T, Li D, et al. (2020) Evolution of PHAS loci in the young spike of Allohexaploid wheat. BMC Genomics 21: 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The complete set of raw sRNA-seq and RNA-seq reads were deposited in the Sequence Read Archive under SRA accession number PRJNA636099.