

5′ to 3′ cotranslational mRNA decay is dynamically regulated during Arabidopsis seedling development and fine-tunes translation efficiency.
Abstract
RNA turnover is a general process that maintains appropriate mRNA abundance at the posttranscriptional level. Although long thought to be antagonistic to translation, discovery of the 5ʹ to 3ʹ cotranslational mRNA decay pathway demonstrated that both processes are intertwined. Cotranslational mRNA decay globally shapes the transcriptome in different organisms and in response to stress; however, the dynamics of this process during plant development is poorly understood. In this study, we used a multiomics approach to reveal the global landscape of cotranslational mRNA decay during Arabidopsis (Arabidopsis thaliana) seedling development. We demonstrated that cotranslational mRNA decay is regulated by developmental cues. Using the EXORIBONUCLEASE4 (XRN4) loss-of-function mutant, we showed that XRN4 poly(A+) mRNA targets are largely subject to cotranslational decay during plant development. As cotranslational mRNA decay is interconnected with translation, we also assessed its role in translation efficiency. We discovered that clusters of transcripts were specifically subjected to cotranslational decay in a developmental-dependent manner to modulate their translation efficiency. Our approach allowed the determination of a cotranslational decay efficiency that could be an alternative to other methods to assess transcript translation efficiency. Thus, our results demonstrate the prevalence of cotranslational mRNA decay in plant development and its role in translational control.
Over its entire lifetime, any mature cytoplasmic mRNA is in balance between translation, storage, and decay. This equilibrium maintains proper dynamics of gene expression and is crucial to control mRNA homeostasis. Although long thought to be mutually exclusive, there is now a large body of evidence supporting that mRNA translation and decay are interconnected in eukaryotes (Heck and Wilusz, 2018). The impact of codon optimality on mRNA half-life is a clear example of this relationship. Codon optimality is defined as the ribosome-decoding efficiency that depends on tRNA availability. Genome-wide analyses revealed that yeast mRNAs enriched in optimal codons have high ribosome density and are more stable than mRNAs enriched in nonoptimal codons (Presnyak et al., 2015). The finding that codon optimality is a key cis-determinant of transcript stability places the ribosome as a core component linking translation elongation to mRNA degradation.
The most relevant interplay between translation and decay is the 5ʹ to 3ʹ cotranslational decay pathway whereby mRNAs are turned over while they are still engaged in polysomes and being actively translated. This pathway was first described in the yeast Saccharomyces cerevisiae using reporter genes (Hu et al., 2009) and was subsequently found to globally shape the polyadenylated transcriptome in yeast, mammalian cells, and Arabidopsis (Arabidopsis thaliana; Pelechano et al., 2015; Yu et al., 2016; Tuck et al., 2020). For these mRNAs, decapping occurs on polysomes, allowing the 5ʹ to 3ʹ exoribonuclease XRN1/4 to chase the last translating ribosome. The genome-wide effect of cotranslational decay can be revealed by sequencing RNA decay intermediates using high-throughput degradome approaches. Each of these is based on the capture of 5ʹ-monophosphate decay intermediates, including parallel analysis of RNA ends (German et al., 2008), 5ʹP sequencing (Pelechano et al., 2015), degradome sequencing (Addo-Quaye et al., 2008), and genome-wide mapping of uncapped transcripts (GMUCT; Willmann et al., 2014). These approaches reveal that mRNA decay intermediates follow an XRN1/4-dependent, three-nucleotide periodicity. This periodicity can be explained by the fact that XRN1/4 follows the last translating ribosome in a codon-by-codon manner, and since it is a processive enzyme, only degradation intermediates protected by ribosomes can be captured. Consequently, each of these degradome approaches gives a snapshot of cotranslational mRNA degradation and also reveals ribosome dynamics (Pelechano et al., 2015) and how degradation impacts this dynamic. For example, in yeast, 5ʹP sequencing allowed the identification of general translation termination pauses, and novel codon-specific pausing sites were detected, such as that at the rare Pro codon CCG and at the Arg CGA codon (Pelechano et al., 2015). These additional pausing sites are explained by the lowest availability of corresponding tRNAs resulting in the slowing down of the ribosome at these sites. Interestingly, these additional pausing sites were not detected in Arabidopsis flowers, suggesting that there are different ribosome dynamics in plants (Yu et al., 2016).
Metagene degradome analyses also revealed 5ʹP reads accumulation 17 nucleotides upstream of stop codons (UAA, UAG, and UGA). This distance corresponds exactly to a ribosome stalled at the A site. As the termination step is slower than elongation, general 5ʹP reads accumulation can be revealed 17 nucleotides upstream of stop codons and can be used as a proxy for transcriptome-wide cotranslational decay activity. In addition, these approaches also unveil small RNA-guided cleavage sites (Franke et al., 2018), RNA-binding protein footprints (Hou et al., 2014), endonucleolytic cleavage sites (Anderson et al., 2018), exon junction complex footprints (Lee et al., 2020), and ribosome-stalling sites (Hou et al., 2016). For example, degradome analysis in Arabidopsis revealed that AGO7 can bind to a noncleavable miR390 target site on the TAS3 transcript, leading to a ribosome-stalling situation that was suggested to control translation of the TAS3 transcript (Hou et al., 2016). Ribosome stalling at upstream open reading frames is another layer of translational control that can be detected by degradome data. For example, the stalling of three ribosomes can be detected on upstream open reading frames controlling Arabidopsis BZIP3 main open reading frame translation (Hou et al., 2016).
At the physiological level, cotranslational decay was shown to play important roles in responses to various stresses. In Arabidopsis, heat stress triggers 5ʹ-ribosome pausing, the overaccumulation of XRN4 in polysomes, and the 5ʹ to 3ʹ cotranslational decay of around 1,500 transcripts that code for proteins with hydrophobic N termini (Merret et al., 2013, 2015). Recently, following stress, the yeast Lsm1-7/Pat1 complex was shown to trigger the cotranslational decay of stress-induced mRNAs, limiting their translation and preventing a hyperresponse (Garre et al., 2018). Consequently, defects in this pathway trigger the misregulation of translation inhibition under osmotic stress, which correlates with an abnormally high association of stress-induced mRNAs to active polysomes (Garre et al., 2018). Recently, this pathway was also proposed to be important for tubulin autoregulation (Lin et al., 2020). TTC5, a tetratricopeptide protein, recognizes tubulin nascent peptide and triggers the cotranslational decay of its own transcripts to maintain proper tubulin homeostasis. TTC5 mutants have compromised tubulin autoregulation and display chromosome segregation defects during mitosis. Taken together, these different examples support that cotranslational decay plays important roles in translation regulation under normal conditions and in response to different stresses. Nonetheless, if and how the cotranslational decay process reshapes the transcriptomes and proteomes of cells during organism development remains poorly understood.
Transcripts can also be turned over through the cytosolic 5ʹ to 3ʹ mRNA decay pathway, which takes place on ribosome-free mRNAs. This so-called general 5ʹ to 3ʹ cytosolic mRNA turnover process occurs by three stages. Following poly(A) tail shortening (deadenylation), the VARICOSE (VCS)/DCP1/DCP2 decapping complex hydrolyzes the mRNA cap structure. Then, the 5ʹ-phosphate end of the decapped mRNA is attacked by the XRN1/4 exoribonuclease, which digests the body of the transcript. In Arabidopsis, XRN4 can target both deadenylated and polyadenylated fractions, suggesting that part of the degradation could be deadenylation independent (Nagarajan et al., 2019). For cotranslational mRNA decay, the importance of deadenylation is still unclear.
In Arabidopsis, at the whole-organism level, the loss of XRN4 has minimal impact under normal growth conditions. Growth deficiencies were only reported in response to hormones or under stress conditions. Loss-of-function mutants of XRN4 are insensitive to ethylene (Potuschak et al., 2006) and hypersensitive to auxin and abscisic acid treatments (Wawer et al., 2018; Windels and Bucher, 2018). Recently, an xrn4 mutant was found to be defective in the dark-stress response and during nitrogen supply (Nagarajan et al., 2019). However, whether these deficiencies result from a failure of the general cytosolic pathway or of the cotranslational mRNA decay pathway (or a combination of both) remains largely unknown. Only one study distinguished both pathways and reported the exact role of cotranslational decay in the Arabidopsis heat-stress response by analyzing pools of mRNAs associated with ribosomes (Merret et al., 2015).
In this study, we monitored the impact of cotranslational mRNA decay during Arabidopsis seedling development. To do so, we assessed the genome-wide impact of XRN4 loss-of-function mutation on poly(A+) mRNAs at the total, polysome, and degradome levels. Through these approaches, we provide evidence that XRN4 mostly catalyzes polyadenylated mRNA degradation in polysomes. We also found that cotranslational decay is dynamically modulated during development and can influence transcript translation efficiency, unveiling the importance of cotranslational mRNA decay during plant development.
RESULTS
XRN4 Differentially Accumulates in Polysomes during Seedling Development
To explore the hypothesis that translation and cotranslational decay are regulated in response to developmental cues, we analyzed two readouts (Fig. 1). First, the global translation activity was assessed by polysome quantification via Suc density gradients at four developmental stages in 3- to 25-d-old seedlings (Fig. 1A). Considering that the cellular activity and hence the ribosome load per cell are most likely to be significantly different between developmental stages, we compared polysome contents from identical quantities of biomass rather than identical quantities of total RNA. Indeed, we observed that polysome content is inversely correlated to seedling age, with the highest levels observed at 3 d, followed by a progressive decrease reaching a minimum at 25 d. Second, considering that XRN4 is proposed to mainly catalyze 5ʹ to 3ʹ cotranslational degradation (Merret et al., 2013; Yu et al., 2016), we used XRN4 accumulation in polysomes as a readout of this mRNA turnover activity. We hence performed immunoblot analysis on input and polysomal fractions using XRN4-specific antibodies (Fig. 1B). Although the enzyme was detected at similar levels in all input fractions, XRN4 accumulation in polysomes differed during development. In 3-d-old seedlings, XRN4 was mostly absent from polysomes and progressively increases to reach a maximum at the 15-d-old stage, a level globally maintained up until 25 d (Fig. 1B). These results suggest that cotranslational decay activity is regulated during seedling development.
Figure 1.

XRN4 differentially accumulates in polysomes during seedling development. A, Polysomal extracts prepared from 3-, 7-, 15-, and 25-d-old seedlings were fractionated on a Suc gradient, and polysome traces were obtained through measurement of OD254nm. Polysome profiling was performed starting from identical quantities of N2-pulverized tissues (e.g. 300 mg of biomass). B, Total proteins extracted from polysomal and input fractions were analyzed by immunoblotting. The four blots were probed with an antibody specific to XRN4. Inputs correspond to an equivalent of 10 mg of tissue powder for all stages. For polysomal fractions, loaded proteins were precipitated from identical volumes of each fraction. Data are representative of at least three replicates.
Cotranslational Decay Efficiency Is Regulated during Seedling Development
To identify XRN4 targets and get a deeper understanding of the dynamics of cotranslational decay, we ran a multiomics approach (Supplemental Fig. S1). For each developmental stage, Columbia-0 (Col-0) wild-type and xrn4-5 loss-of-function (SAIL_681_E01) seedlings were harvested in two biological replicates. As the loss of XRN4 has minimal impact under normal growth conditions, the observed differences between wild-type and mutant plants are a direct consequence of the loss of XRN4 rather than a growth/developmental delay.
Each sample was used to purify (1) total RNAs and (2) RNAs associated with polysomes. Total RNAs were used to run poly(A+) RNA sequencing (RNA-seq) and also in a degradome analysis through a GMUCT/RNA-seq approach. Briefly, GMUCT (also tagged as 5ʹP-seq) consists of the ligation of a 5ʹ-RNA adapter to capture and sequence poly(A+) mRNA molecules that carry a 5ʹ-monophosphate. This also permits the counterselection of mRNAs with a 5ʹ-cap structure. Hence, GMUCT allows specific sequencing of the population of mRNA molecules (full length and decay intermediates) that are in the course of being exonucleolytically degraded from their 5ʹ-end following a decapping step or endonucleolytic cleavage. Considering the very high processivity of 5ʹ-exoribonucleases, only mRNAs with features slowing down the progression of the decay enzyme can be captured. Hence, GMUCT was used mostly to monitor the 5ʹ-cotranslational decay process, where the exoribonuclease digests the transcript chasing the elongating ribosomes. Polysomal RNAs were also purified via a Suc density gradient and sequenced following purification of the polyadenylated fraction (Supplemental Fig. S1).
The total poly(A+) RNA-seq method allows the capture of capped mRNAs free of ribosomes, capped translated mRNAs, and uncapped cotranslational decay intermediates. The polysome poly(A+) RNA-seq method allows the capture of both capped translating mRNAs and uncapped cotranslational decay intermediates. The GMUCT approach captures uncapped cotranslational decay intermediates (Supplemental Fig. S1), hence permitting the cell 5ʹ-degradome to be monitored. For the sequencing approach, the sequence of the biological repeats of each genotype displays a high reproducibility (R2 > 0.94; Supplemental Table S1). Only transcripts with at least 1 read per kilobase by million mapped reads (RPKM)/1 read per million value in at least one wild-type library were kept for further analysis, leaving a total of 23,196 transcripts. Fold changes (FCs) between xrn4-5 and Col-0 were calculated for each transcript at each developmental stage at the total and polysome RNA levels (Fig. 2; Supplemental Fig. S2, A and B). Using the DESeq2 pipeline (Love et al., 2014) and cutoff FCs above 2 or below 0.5, we identified mRNAs that differentially accumulated in the absence of XRN4 in the total and/or polysomal fractions (Fig. 2). Considering that XRN4 is an RNA-decay enzyme, we only focused on up-regulated mRNAs in further analyses. At the total RNA level, one to 13 transcripts were identified as up-regulated in xrn4-5 as compared with Col-0, and at the polysome RNA level, zero to 23 transcripts were identified as up-regulated (Fig. 2). Herein, the pool of misregulated transcripts are referred to as differentially expressed genes. These data are consistent with previous analyses showing that, under normal conditions, only a handful of XRN4 targets can be identified at the total and polysomal poly(A+) RNA levels (Merret et al., 2015).
Figure 2.
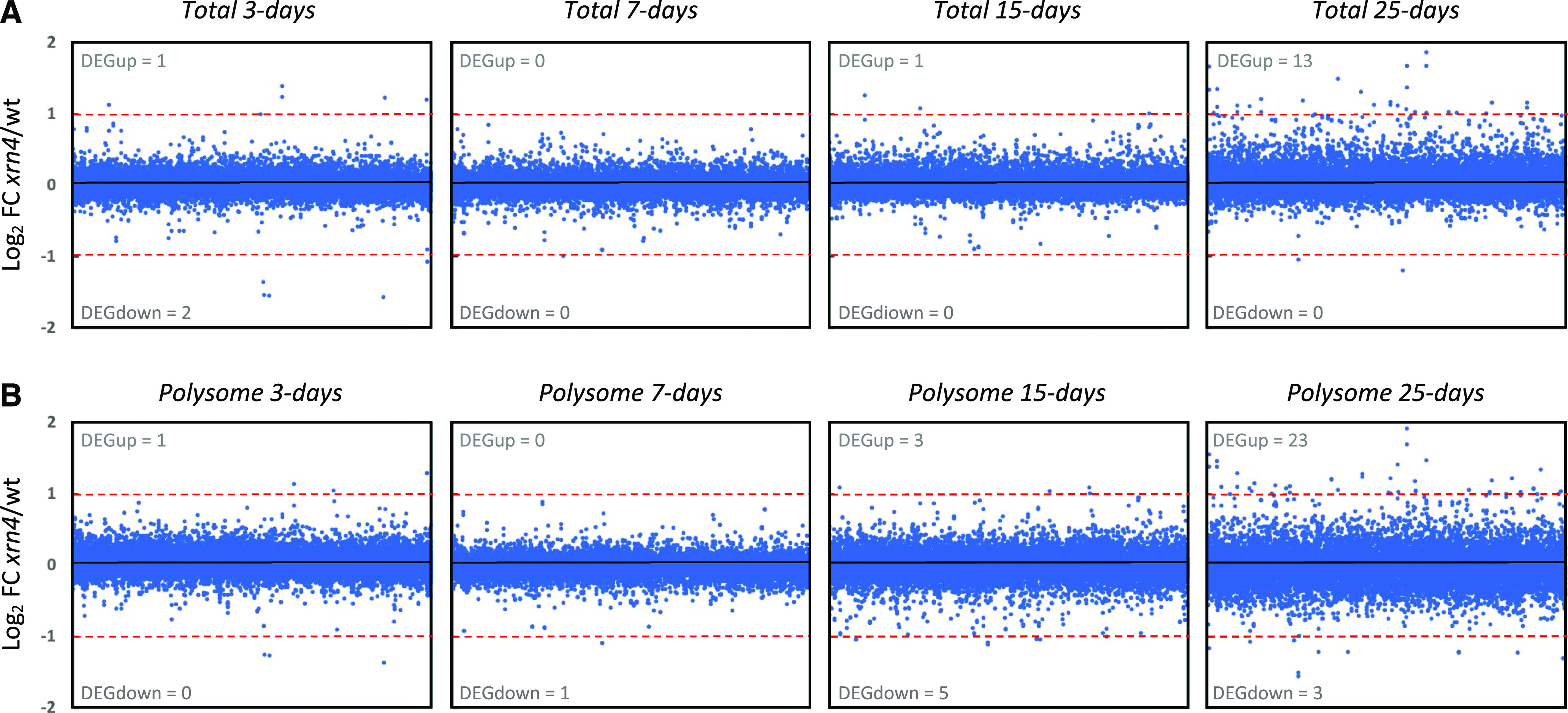
XRN4 loss-of-function mutation has minimal impact during seedling development on total and polysome RNA levels. FCs between xrn4-5 and the Col-0 wild type (wt) were calculated for total (A) and polysome (B) RNA in each condition. The log2 value of the mean is represented in each graph. The number of transcripts significantly regulated in xrn4-5 is reported (as differentially expressed genes [DEG] in gray for upregulated and downregulated transcripts) and was calculated using DESeq2. Dashed red lines mark the |log2(2)| values.
As our main goal in this study was to determine the prevalence of cotranslational 5ʹ to 3ʹ mRNA decay during development, we focused on degradome data. The two main features of cotranslational decay are the accumulation of the 17-nucleotide ribosome footprint at the translation termination site and the three-nucleotide periodicity of fragments resulting from the degradation of mRNA open reading frames (Pelechano et al., 2015; Yu et al., 2016). To assess the reliability of our analysis, a metagene analysis of the 5ʹP reads obtained by GMUCT was performed around stop codons at each developmental stage (Fig. 3, A–E). The relative abundance of reads at each position relative to the stop codon was determined. At all developmental stages, a clear three-nucleotide periodicity pattern was observed as previously described (Yu et al., 2016). Additionally, a clear overaccumulation of reads 17 nucleotides before stop codons was also detected. This accumulation corresponds precisely to the 5′ boundary of the ribosome with its A site stalled at a stop codon (Fig. 3A). Interestingly, differential accumulation at this position was observed during development, reaching a maximum at the 15-d-old stage (Fig. 3D), suggesting that activity of the cotranslational decay pathway is controlled throughout development. Moreover, at the 7- and 15-d-old stages, an additional peak was observed 47 nucleotides before stop codons (Fig. 3, C and D). A similar phenomenon was previously observed in yeast (Pelechano et al., 2015) but was not detected in Arabidopsis flowers (Yu et al., 2016). This peak corresponds to two ribosomes stalled at stop codons, as the distance between the two peaks (30 nucleotides) exactly matches one ribosome footprint (Fig. 3A). To determine if additional peak(s) could also result from ribosome stalling at suboptimal codon(s), we looked for possible enrichment of 5ʹP read ends in coding regions other than the ones surrounding stop codons (Supplemental Fig. S3). We could not identify additional peaks at any codon, other than stop codons, in our four tested developmental conditions. Thus, in our experimental conditions, we found no evidence for ribosome stalling at suboptimal codons. This also confirms that the −47-nucleotide peak, only observed in developmental stages where the cotranslational decay rate was highest, is not associated with a slowing down of ribosomes at a specific codon but rather represents the footprint of two ribosomes stalled at stop codons. Taken together, these observations support that cotranslational decay activity is regulated during development.
Figure 3.

Metagene analyses displaying the abundance of 5ʹP reads relative to stop codons. B, Wild-type (wt) 3-d-old stage. C, Wild-type 7-d-old stage. D, Wild-type 15-d-old stage. E, Wild-type 25-d-old stage. F, xrn4-5 3-d-old stage. G, xrn4-5 7-d-old stage. H, xrn4-5 15-d-old stage. I, xrn4-5 25-d-old stage. Data are means ± sd. The illustrations (A) represent 5ʹP intermediate accumulation at –47 and –17 nucleotides (nt) before stop codons. RPM, Reads per million.
Next, we performed the same analysis using xrn4-5 degradome data (Fig. 3, F–I). For all developmental stages, a decrease in reads accumulation at −17 nucleotides before stop codons was observed, suggesting that the cotranslational decay pathway is severely impaired in this mutant, thus supporting a main role for XRN4 in this pathway. To identify XRN4 cotranslational decay targets, we used the DESeq2 pipeline on Col-0 and xrn4-5 degradome data (Supplemental Fig. S2, A and B) and compared the FCs during development. For each of the differentially accumulated targets, the median FC was systematically higher than 2.5 with a maximum at 3 d (Supplemental Fig. S4A). To extract the most significant targets, a cutoff FC above 2 was applied. Whereas only a handful of XRN4 targets were identified in total and polysome RNA data, the degradome data identified several hundred misregulated mRNAs (Fig. 4, A–D). And consistent with XRN4’s function as a decay enzyme, 98% of differentially expressed genes were up-regulated. The number of cotranslational decay targets increased during development, reaching a maximum at 15 d (479) before dropping again at 25 d (152). The differential accumulation of 5ʹP read ends at 17 nucleotides before stop codons suggests that the repertoire of cotranslational decay targets and/or their decay rates are developmentally regulated. To test this hypothesis, we performed a Venn diagram analysis on lists of transcripts upregulated in the xrn4-5 degradome data (Fig. 4E; Supplemental Table S2). A total of 565 unique targets were identified, with only 47 targets shared by all stages. Each stage was associated with specific targets, with 25, 18, 253, and 37 mRNAs more sensitive to cotranslational decay at the 3-, 7-, 15-, and 25-d-old stages, respectively. Additionally, close developmental stages share more targets than more distant ones. As an example, except for the 47 common targets, 65 common targets were identified between 25- and 15-d-old stages whereas three targets were shared between the 25- and 3-d-old stages (Fig. 4E). In order to determine biological processes targeted by cotranslational decay, a Gene Ontology (GO) analysis was performed on transcripts identified as up-regulated in xrn4-5 in degradome data at all developmental stages. A clustering approach was performed using DAVID software (Supplemental Table S3). Redox signaling processes are mainly affected by XRN4 at the 3-d-old stage. Auxin/growth, Response to stress, and DNA binding processes are preferentially affected at the 7-d-old stage compared with Ribosome/translation, DNA binding, and RNA binding processes that are affected at the 15-d-old stage (Supplemental Table S3). At 25 d, these same GO terms as for the 15-d-old stage are represented but with a lower enrichment score.
Figure 4.
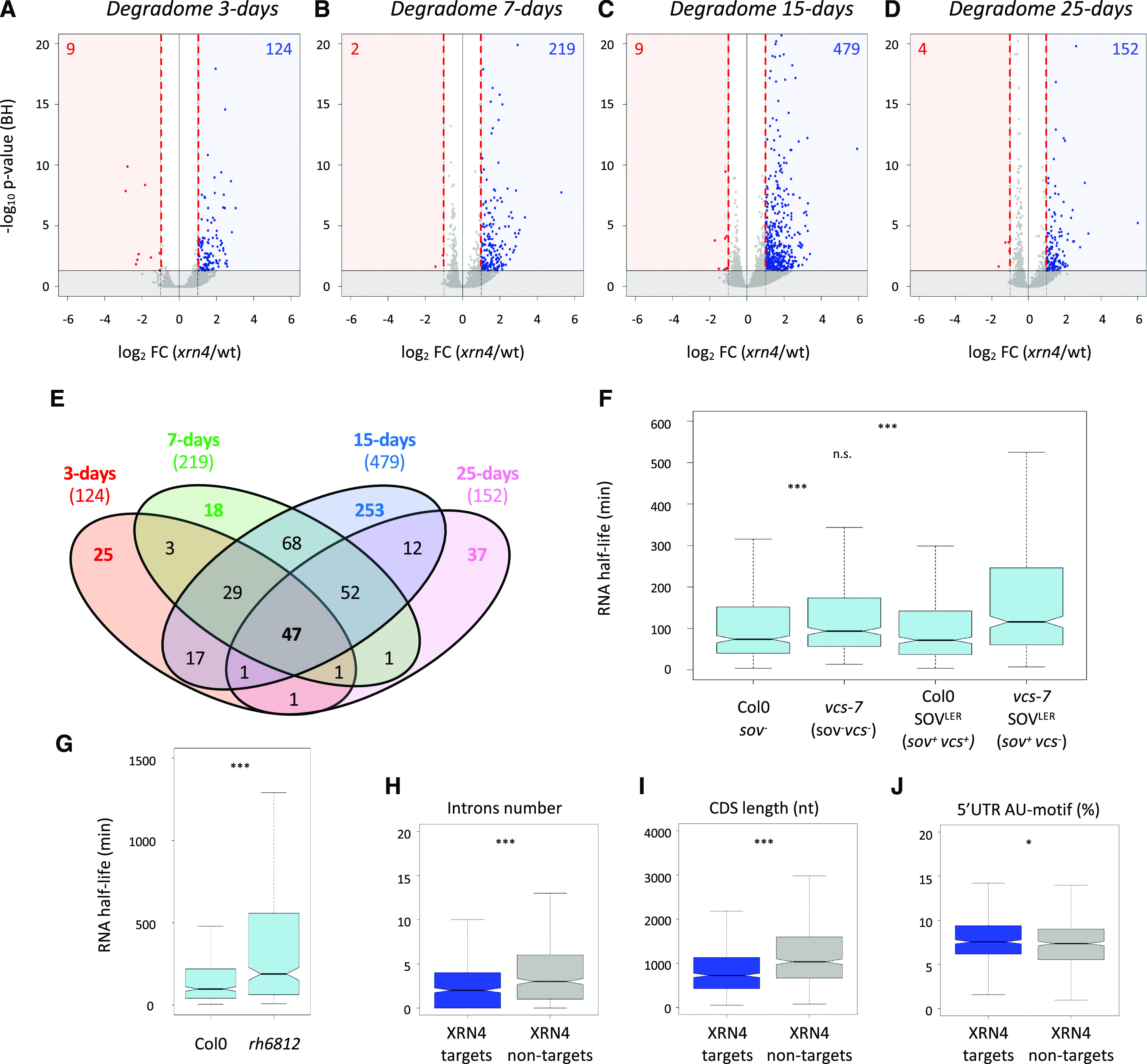
Identification and features of XRN4 cotranslational decay targets during development. A to D, Volcano plots of the change in read abundance in xrn4-5 over wild-type Col-0 (wt). Vertical red dashed lines mark the |log2(2)| values. Log2 FC and Benjamini-Hochberg adjusted P values (BH) were calculated through the DESeq2 pipeline (as DEGs in blue for up-regulated and in red for downregulated transcripts). Horizontal solid black lines demarcate adjusted P values of 0.05. E, Venn diagram of cotranslational decay targets during development. F and G, The majority of XRN4 cotranslational decay targets show longer RNA half-lives in vcs-7 and vcs-7 SOVLER mutants (F) and the rh6812 mutant (G). RNA half-lives were collected from Sorenson et al. (2018; F) or from Chantarachot et al. (2020; G). Only transcripts present in each data set are represented (n = 444/565 for F and n = 390/565 for G). H to J, Intron number (H), CDS length (I), and proportion of AU motifs (J) in the 5ʹ-UTR of transcripts targeted by XRN4 compared with nontargeted random transcripts (n = 565). Asterisks indicate significant difference (***P < 0.001 and *P < 0.05). n.s., Nonsignificant.
Next, we reasoned that genes that we find to be up-regulated in the absence of XRN4 in our degradome data should show an augmented half-life when the 5ʹ to 3ʹ decay system is impaired. VCS is part of the decapping holoenzyme DCP1/DCP2/VCS, and the DEAD box RNA helicases RH6, RH8, and RH12 were recently identified as cofactors of the 5ʹ to 3ʹ cytoplasmic mRNA turnover (Sorenson et al., 2018; Chantarachot et al., 2020). We hence used data from these two recent articles that report genome-wide mRNA half-lives in the wild type and vcs-7 or rh6812 loss-of-function mutants. Of the 565 mRNAs that we found to be up-regulated in xrn4-5, 444 (more than 78%) were detected in the Sorenson et al. (2018) data set, of which 85% have a half-life below 240 min (Fig. 4F; Supplemental Table S2, Col-0 data). Consistently, 77% of the 390 of our cotranslational decay targets that are present in the Chantarachot et al. (2020) data set also show half-lives below 240 min (Fig. 4G; Supplemental Table S2, Col-0 data). This first observation supports that most of the transcripts that are degraded cotranslationally from 5ʹ are intrinsically short-lived. Next, we observed that in the absence of an active decapping enzyme (vcs-7 mutant) or in a background without cofactors of the 5ʹ to 3ʹ decay (rh6812 mutant), their half-lives significantly increase (Fig. 4F compares the Col-0 and vcs-7 box plots, while Fig. 4G compares the Col-0 and rh6812 box plots). Furthermore, the Sorenson et al. (2018) data suggest that these mRNAs are mostly decayed through a 5ʹ to 3ʹ process. Indeed, mRNAs can be decayed either from 5ʹ, following decapping, and/or from 3ʹ, either by the exosome complex or by the 5ʹ to 3ʹ exoribonuclease, SOV. In addition, mRNAs that are not naturally decayed from 5ʹ can be turned down by this pathway in the absence of functional exosome or VCS enzymes. The Col-0 ecotype was found previously to carry a sov-defective allele (Zhang et al., 2010). Hence, to ascertain that mRNAs upregulated in the vcs-7 background are natural targets of the 5ʹ pathway, Sorenson et al. (2018) also used Col-0 Arabidopsis complemented with a functional allele of SOV. mRNAs that we find to be up-regulated in the absence of XRN4 here again show increased half-lives in the absence of VCS, despite the presence of an active SOV (Fig. 4F compares Col-0 and vcs-7 SOVLER). This increase is not seen when VCS is active, such as in Col-0 SOVLER (Fig. 4F compares Col-0 and Col-0 SOVLER). This further supports that XRN4 cotranslational decay targets are actual and specific targets of the 5ʹ to 3ʹ degradation pathway.
A key question is how the 5ʹ to 3ʹ decay pathway and more specifically how the 5ʹ-cotranslational degradation process recognizes its targets among the whole-cell transcriptome. We hence looked for putative cis-elements shared by the mRNAs that we found as XRN4 cotranslational decay targets. To do so, we retrieved the 5ʹ- and 3ʹ-untranslated regions (UTRs) alongside the coding sequences (CDSs) of all targets and looked for common features as compared with a random set of transcripts nontargeted by XRN4. Although no differences were found in 5ʹ- and 3ʹ-UTR lengths, a significant reduction in intron number and CDS length was observed for XRN4 cotranslational decay targets (Fig. 4, H and I; Supplemental Fig. S4, B and C). In addition, AU motifs were found enriched in their 5ʹ-UTRs (Fig. 4J). This observation is consistent with previous studies showing that short-lived mRNAs have fewer introns and AU-rich motifs in their 5ʹ-UTRs (Narsai et al., 2007; Sorenson et al., 2018). Altogether, these data suggest that trans- and cis-elements could regulate cotranslational decay activity.
Cotranslational Decay Activity during Development Can Influence Translation Efficiency
Hence, our above results suggest that cotranslational decay specificity and efficiency are regulated in response to developmental cues. We next wondered about the molecular role of this regulation. Since cotranslational decay and translation are interrelated, we asked whether the former could control protein production. We rationalized that, for a given mRNA, a cotranslational decay rate higher than the translation rate should result in a decrease in translation efficiency and vice versa. To explore this, we compared variations during development at the polysome and degradome levels in the Col-0 background (Fig. 5A). To limit variations at the polysome and degradome levels, we focused our analysis on 3,366 mRNA transcripts that were detected (RPKM > 1 in all libraries) and remained stable during development at the total RNA level (e.g. FC = 0.66–1.5 among all conditions; Supplemental Fig. S2C). For these transcripts, variation at the polysome RNA level was compared with that at the degradome level. Values were normalized to the 3-d-old stage, and their dynamics were assessed during development (Fig. 5, A and B). Interestingly, based on P values, the median variation of the degradome values appeared more dynamic during development than that of the polysomal values. To test this, we calculated the ratio between degradome and polysome values for each mRNA (Supplemental Fig. S2C). We called this ratio cotranslational decay efficiency, as it reflects the proportion of polysomes associated with uncapped mRNA decay intermediates (degradome data) compared with the total amount (capped and uncapped) of polysome-associated mRNAs (polysome data). A high ratio suggests high cotranslational decay activity resulting in low translation efficiency, whereas a low ratio suggests high translation efficiency. A heat map was generated to observe the variation of this efficiency during development (Fig. 5C).
Figure 5.
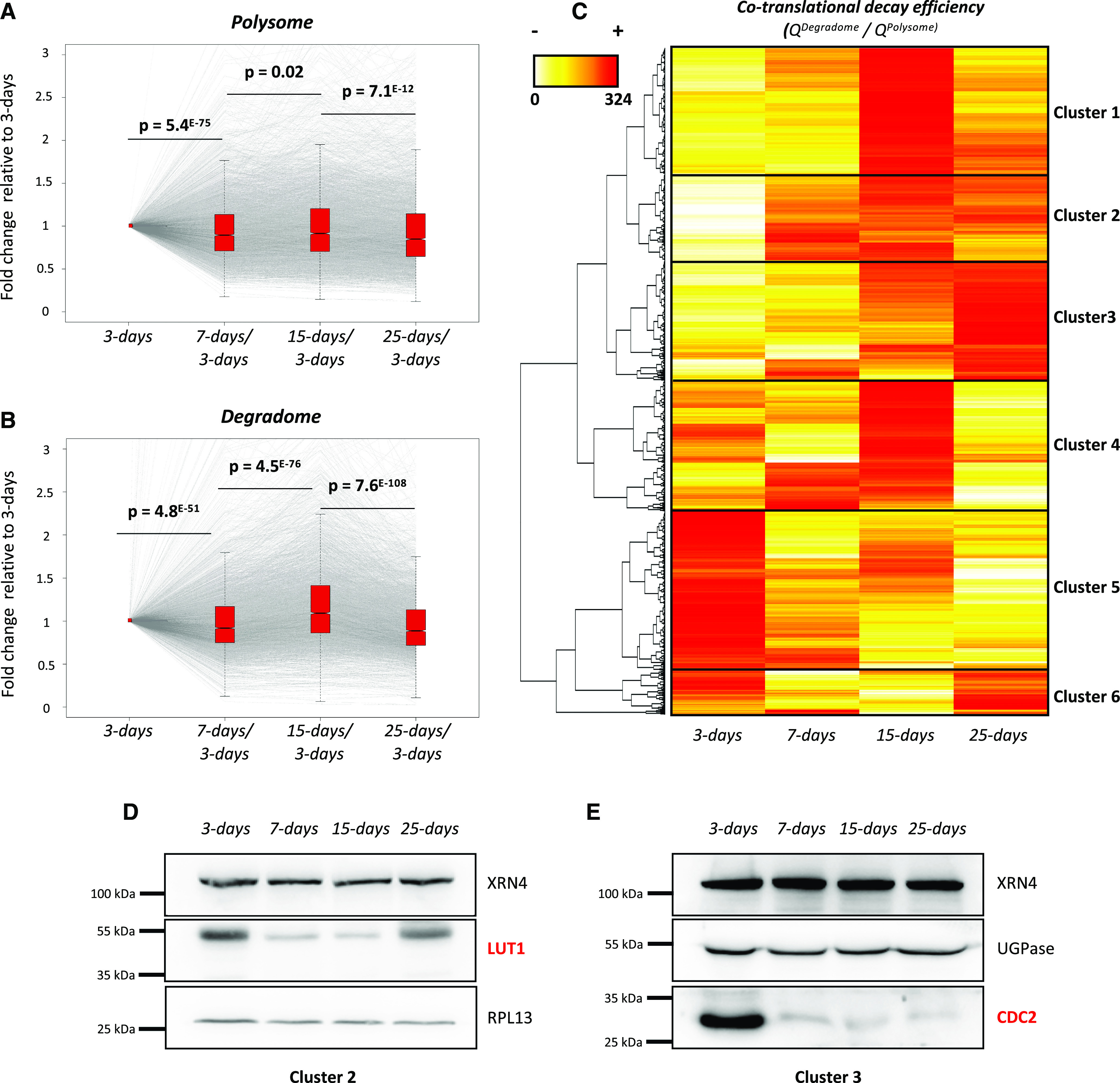
Cotranslational decay is regulated during development and influences protein production. A, Transcript variation at the polysome level during development using 3-d samples as a reference (n = 3,366). B, Transcript variation at the degradome level during development using 3-d samples as a reference (n = 3,366). Gray lines represent individual transcript variation. Transcript distribution is represented by notched box plots, and significance was assessed by P values (nonparametric Wilcoxon test). C, Heat map of cotranslational decay efficiency (ratio in degradome data over polysome RNA-seq data; n = 3,366). Red values correspond to high decay efficiency and yellow values to low decay efficiency. D and E, Immunoblots using LUT1 (D) and CDC2 (E) antibodies. Both candidates were analyzed on distinct SDS-PAGE gels (8% and 10% acrylamide, respectively). RPL13 and UGPase antibodies were used as loading controls. Each immunoblot was performed as two biological replicates.
Interestingly, this efficiency varies between transcripts and is highly modulated during development. Six good resolution clusters were obtained, the behavior of which can be monitored during development. Transcripts from cluster 1 were highly targeted by cotranslational decay at the 15-d-old stage, whereas those from cluster 5 were mostly targeted at the 3-d-old stage. Cluster 3 was also remarkable, as it contained mRNAs that were progressively targeted by cotranslational decay until the 25-d-old stage. As our hypothesis is that cotranslational decay influences translation efficiency, we monitored the mRNA levels during development corresponding to the two proteins, LUT1 and CDC2, that belong to clusters 2 and 3, respectively. These mRNAs were selected as case studies because they show important variation in their cotranslational efficiency, which is mostly due to variations of their degradome values, with stable quantities in total and polysomal fractions during development (Supplemental Fig. S5). Immunoblotting showed that LUT1 levels were highest at the 3-d-old stage and decreased at the 7- and 15-d-old stages, in which the corresponding cotranslational decay efficiency was highest. At the 25-d-old stage, the LUT1 degradome value decreased and, consistently, its protein levels increased compared with those at 7 and 15 d (Fig. 5D; Supplemental Fig. S5). By contrast, CDC2 was mostly detected at the 3-d-old stage, which is consistent with its low cotranslational decay efficiency at this stage (Fig. 5E; Supplemental Fig. S5). Protein level variations are the consequence of both production (translation) and decay-rate variations. Hence, steady-state immunoblotting is not the most accurate readout of translation efficiency. Nonetheless, for LUT1 and CDC2, we observed a perfect correlation between cotranslational efficiency and steady-state protein levels (Fig. 5, D and E), supporting that indeed cotranslational decay may fine-tune translation.
DISCUSSION
The major objective of this study was to monitor the dynamics of cotranslational decay during Arabidopsis seedling development. Using a multifaceted genome-wide approach, we provide evidence that cotranslational decay is highly modulated during development, with maximum activity at the 15-d-old stage. Using the xrn4-5 loss-of-function mutant (Souret et al., 2004; Merret et al., 2013), we found that the majority of cotranslational mRNA decay in seedlings is catalyzed by XRN4, as previously proposed in floral tissue (Yu et al., 2016). However, as the three-nucleotide periodicity is maintained in xrn4-5, as well as a low level of 17-nucleotide 5ʹP reads before stop codons, other 5ʹ to 3ʹ exoribonucleases must also be involved in this process. In yeast, the nuclear Xrn (Xrn2p) was proposed to relocalize to the cytoplasm of the xrn1-null mutant and restore cytosolic mRNA turnover (Johnson, 1997). However, since Arabidopsis has two nuclear 5ʹ-3ʹ exonucleases (XRN2 and XRN3) and since the xrn3 loss-of-function mutant is lethal (Gy et al., 2007), the possibility of nuclear XRNs complementing a deficient cytosolic enzyme is challenging to assess.
The polysome fraction was until recently considered to be essentially composed of actively translating mRNAs and was routinely used to assess mRNA translation efficiency after normalization to total RNA levels (see an example in Bai et al., 2017). The discovery of cotranslational mRNA decay and its conservation in evolutionarily distant eukaryotes (Hu et al., 2009, 2010; Hou et al., 2016; Yu et al., 2016; Simms et al., 2017; Ibrahim et al., 2018) suggests that the proportion of actively translated mRNAs in polysomes might be much lower than initially expected. Translation efficiency is often assessed by ribosome profiling (Ingolia et al., 2009) or translating ribosome affinity purification (Reynoso et al., 2015); however, these approaches do not consider that the mRNAs attached to the ribosomes might be undergoing partial degradation. Thus, these approaches can give rise to misleading conclusions. In yeast, 5′P decay intermediates represent more than 12% of the poly(A+) mRNA fraction (Pelechano et al., 2015). In our study, we found that at least 3,366 transcripts presented variations in degradome data during development and that their cotranslational decay rate could be an efficient way that the cell controls its translation efficiency (Fig. 5). Thus, to take into account the cotranslational decay in translation efficiency measurements, we calculated the ratio between degradome and polysome values for each transcript as proxy (Fig. 5C). This ratio can be determined for each transcript and compared pairwise between conditions or genetic backgrounds. A high ratio would suggest low translation efficiency and a low ratio would suggest high translation efficiency. As a proof of concept, we monitored the accumulation of LUT1 and CDC2 and found that, for both, protein levels varied in an inverse proportional manner to their degradome:polysome mRNA ratios (Fig. 5; Supplemental Fig. S5). We hence propose that this ratio be used as an additional way to determine transcript translation efficiency. We also posit that it would be a more accurate readout than the simple ratio between polysomal and total mRNA levels. The determination of this efficiency will be crucial under conditions where cotranslational decay is highly modulated, such as during development or following stress exposure (Merret et al., 2015; Garre et al., 2018).
In addition to its role in translation efficiency modulation, our analysis allowed the identification of specific XRN4 cotranslational decay targets (Fig. 4). In a landscape analysis of Arabidopsis mRNA half-lives, Sorenson et al. (2018) found that short-lived mRNAs targeted by decapping and 5ʹ to 3ʹ degradation present fewer introns than stable ones (Sorenson et al., 2018). Consistently, this feature is shared by transcripts targeted by cotranscriptional decay in our analysis (Fig. 4H; Supplemental Table S2). Cotranslational decay targets are also enriched in AU motifs in their 5ʹ-UTRs, a motif also known to be shared by unstable transcripts (Narsai et al., 2007). A low intron complexity and the presence of AU-rich motifs could be some of the cis-determinants of cotranslational target recognition. Additionally, we found that the mRNA half-life range of XRN4 cotranslational decay targets increases in mutants involved in 5ʹ to 3ʹ decapping-dependent decay, such as vcs-7 and rh6812, which are key factors of decapping activity (Fig. 4, F and G). The higher stability of these transcripts in these mutants is consistent with decapping being a necessary and crucial step for XRN4 activity. Interestingly, in yeast, DHH1, a homolog of RH6, RH8, and RH12, was proposed to couple mRNA translation to decay (Radhakrishnan et al., 2016). These findings suggest that VCS and RH6, RH8, and RH12 could be trans-determinants of cotranslational target recognition, as recently discussed (Merret and Bousquet-Antonelli, 2020).
Analysis of enriched GO terms associated with cotranslational decay targets revealed six major GO terms (Auxin/growth, Response to stress, DNA binding, Ribosome/translation, RNA binding, and Redox signaling). Published parallel analysis of RNA ends data identified similar GO terms, such as mRNA processing or Ribosome biogenesis, shared by polyadenylated targets of XRN4 (Nagarajan et al., 2019). Recently, it was proposed that XRN4 contributes to root growth under normal conditions and upon salt stress by an unknown mechanism (Kawa et al., 2020). Interestingly, for the Auxin/growth GO term, we identified many genes targeted by the cotranslational decay pathway that are associated with growth regulation (such as Response to auxin [GO:0009733]; Supplemental Table S3), consistent with the described role of XRN4 in root development. As an example, RVE2 (At5g37260), a gene involved in lateral root formation and a VCS substrate (see supplemental table S16 of Kawa et al., 2020), was identified as a cotranslational target of XRN4 at the 7-d stage (Supplemental Table S3). Whereas the respective contributions of both 5ʹ to 3ʹ cytosolic and cotranslational decay were not addressed in these different studies, our results suggest that at least part of these XRN4 targets could be decayed cotranslationally
Cotranslational decay is an evolutionarily conserved mechanism found in many organisms such as yeast (Pelechano et al., 2015), Arabidopsis (Yu et al., 2016), soybean (Glycine max; Hou et al., 2016), barley (Hordeum vulgare; Hou et al., 2016), and mammalian cells (Tuck et al., 2020). This pathway was described as being involved in different stress responses (Merret et al., 2015; Pelechano et al., 2015; Garre et al., 2018); however, analyses were historically only focused on stable lines or specific tissues. Our data demonstrate that 5ʹ to 3ʹ cotranslational decay is dynamically modulated during development and important for the proper regulation of protein expression, thus suggesting that this pathway could be important for plant development and physiology.
MATERIALS AND METHODS
Growth Conditions
Analyses were carried out with Arabidopsis (Arabidopsis thaliana) Col-0 as the wild type and the xrn4-5 mutant (SAIL_681_E01). Seedlings were grown on synthetic Murashige and Skoog medium (Duchefa) containing 1% (w/v) Suc and 0.8% (w/v) plant agar at 22°C under a 16-h-light/8-h-dark regime. Comparable growth conditions were applied for soil culture.
Sampling Procedure for RNA-Seq
To generate two biological replicates, two distinct batches of seeds (generated from different parent plants) were used for each genotype. All samples were generated at the same time as follows. Seeds were sown in vitro on 20 square plates for each replicate and genotype. After 3 d, plantlets from six plates were pooled and harvested to generate 3-d samples. The same procedure was performed at 7 and 15 d to generate 7- and 15-d samples, respectively. Plantlets from the two remaining plates were transferred to soil for an additional 10 d to obtain 25-d samples. In this case, the rosette and the primary root were collected. For each developmental stage, at least 10 plantlets were pooled to avoid individual-specific bias.
Polysome Profile Analysis
Polysome profiles were performed as described previously (Merret et al., 2013). In brief, 400 mg of tissue powder was homogenized with 1.2 mL of lysis buffer (200 mm Tris-HCl, pH 9, 200 mm KCl, 25 mm EGTA, 35 mm MgCl2, 1% [v/v] detergent mix [1% {v/v} Tween 20, 1% {v/v} Triton, 1% {w/v} Brij35, and 1% {v/v} Igepal], 1% [w/v] sodium deoxycholate, 0.5% [w/v] polyoxyethylene tridecyl ether, 5 mm dithiothreitol, 50 μg mL−1 cycloheximide, 50 μg mL−1 chloramphenicol, and 1% [v/v] protease inhibitor cocktail [Sigma-Aldrich]). Crude extract was incubated 10 min on ice. After centrifugation, 900 µL of crude extract was loaded on a 15% to 60% Suc gradient (9 mL). Ultracentrifugation was performed with an SW41 rotor at 180,000g for 3 h. Polysome profile analyses were performed with an ISCO absorbance detector at 254 nm. Twelve fractions of 650 μL were collected. Proteins were extracted from fractions 6 to 12 (corresponding to polysomes). Two volumes of absolute ethanol were added for each fraction. Proteins were precipitated 6 h at 4°C before centrifugation. Pellets were washed and resuspended in 10 μL of Laemmli 4×. For polysomal RNA, extraction was performed as described by Merret et al. (2015) using a Monarch Total RNA Miniprep Kit (New England Biolabs).
Total RNA Extraction
Total RNA was extracted using a Monarch Total RNA Miniprep Kit (New England Biolabs). RNA quality was assessed using an Agilent RNA 6000 Nano kit (Agilent).
RNA Library Preparation
RNA library preparation was performed on total or polysomal RNA using a NEBNext Poly(A) mRNA Magnetic Isolation Module and a NEBNext Ultra II Directional RNA Library Prep Kit (New England Biolabs) according to the manufacturer’s instructions with 1 μg of RNA as a starting point.
GMUCT Assay
The GMUCT library was prepared as described previously with slight modifications (Willmann et al., 2014). Briefly, 50 μg of total RNA was subjected to two poly(A+) purifications. After 5ʹ-adapter ligation, excess of adapter was removed by a new round of poly(A+) purification. Reverse transcription was performed using the SuperScript IV system with the manufacturer’s instructions. cDNAs were amplified with 11 cycles of PCR. Libraries were purified using SPRIselect beads prior to quality control and normalization.
RNA-Seq
Library quality was checked using an Agilent High Sensitivity DNA kit (Agilent). Libraries were normalized, multiplexed, and sequenced on NextSeq 550 (Illumina) in 75-pb single reads.
Bioinformatics Analysis
For total and polysome RNA libraries, after filtering out reads corresponding to chloroplastic, mitochondrial, ribosomal, and small RNA sequences using bowtie2, reads were mapped against the TAIR10 genome using Hisat2 and the gtf TAIR10 annotation file with standard parameters. Reads count by gene was performed by Cufflinks in RPKM. For GMUCT analyses, reads were trimmed to 50 pb using Trimmomatic prior to mapping. Reads count was performed using bedcoverage from the Bedtools suite and normalized by the total of mapped reads (reads per million). For 5ʹP reads abundance, the bam file was converted into a bed file containing only the first nucleotides of each read. Differential expression analyses were performed using the Bioconductor R package DESeq2, with a false discovery rate of 0.05. P values were corrected for multiple tests by the Benjamini-Hochberg rule (adjusted P value). Analysis of codon enrichment was performed as previously described (Yu et al., 2016). For notched plot analysis, a Shapiro test was applied to test normality of the data set. Then, a nonparametric Wilcoxon test was performed between each developmental stage. GO analysis was performed using DAVID software (Huang et al., 2009) with default settings; the six major clusters were retained for analysis. For mRNA features analysis, UTRs, introns, and CDSs were obtained from the Araport11 database and are listed in Supplemental Table S2. RNA half-life data were collected from Data Set_S2 of Sorenson et al. (2018; columns alpha_WT, alpha_sov, alpha_vcs, and alpha_vcs sov) and from supplemental table S4 of Chantarachot et al. (2020) columns mRNA Half-life WT_Col-0 and rh6812) and are presented in Supplemental Table S2. Only transcripts present in each data set were kept for statistical analysis. A Wilcoxon test was systematically performed to test significance (Supplemental Table S5).
Immunoblotting
After electrophoretic separation by SDS-PAGE, proteins were electrotransferred on polyvinylidene fluoride membranes. Immunoblotting was performed in Tris-buffered saline (TBS)-5% (w/v) skim milk/1% (v/v) Tween. Primary antibody was incubated overnight at 4°C under constant agitation. After incubation, membranes were washed six times with TBS-1% Tween. A horseradish peroxidase-coupled secondary antibody was incubated in TBS-5% skim milk/1% Tween for 45 min at room temperature. Membranes were again washed six times with TBS-1% Tween and revealed with the Immobilon-P kit from Millipore. Image acquisition was performed using the Fusion FX imaging system (Vilber). Antibodies against XRN4 (Merret et al., 2013), LUT1 (Agrisera), CDC2 (Agrisera), UGPase (Agrisera), and RPL13 (Merret et al., 2013) were utilized at 1:1,000, 1:1,000, 1:3,000, 1:5,000, and 1:100,000, respectively.
Accession Numbers
The accession numbers for the RNA-seq data reported in this article are NCBI Bioprojects PRJNA604882 for total RNA data, PRJNA604883 for polysome RNA data, and PRJNA604884 for GMUCT data. Further sequence data from this article can be found in the GenBank/EMBL data libraries under the following accession numbers: XRN4 (At1g54490), LUT1 (At3g53130), and CDC2 (At3g48750).
Supplemental Data
The following supplemental materials are available.
Supplemental Figure S1. Representation of the experimental procedure.
Supplemental Figure S2. Representation of the bioinformatics analysis.
Supplemental Figure S3. Enrichment of 5′P read ends at the ribosome boundary of mRNA along mRNA coding regions.
Supplemental Figure S4. Analysis of FC variation and XRN4 cotranslational target UTR features.
Supplemental Figure S5. Values obtained for LUT1 and CDC2 transcripts at the total, polysome, and degradome levels in RNA-seq data.
Supplemental Table S1. Differential expression analysis of 23,196 genes by DESeq2 of total, polysome, and degradome mRNA-seq data, associated with Figures 2 and 4.
Supplemental Table S2. List 565 XRN4 cotranscriptional decay targets identified during development using DESeq2, associated with Figure 4.
Supplemental Table S3. Enriched GO terms associated with XRN4 cotranslational decay targets during development.
Supplemental Table S4. Variation of cotranslational decay efficiency of 3,366 genes exhibiting stability in total RNA level during development, associated with Figure 5.
Supplemental Table S5. Results of statistical analyses used in this article.
Acknowledgments
We thank Drs. Xiang Yu and Brian Gregory (University of Pennsylvania) for advice on GMUCT bioinformatics analysis. We also thank the sequencing facility of the Université de Perpignan Via Domitia BioEnvironnement platform.
Footnotes
This work was supported by the Labex TULIP (grant no. ANR–10–LABX–41 to R.M.), the Institut Universitaire de France (to J.-M.D.), and the Agence Nationale de la Recherche ANR 3'modRN (grant no. ANR–15–CE12–0008 to C.B.-A.).
Articles can be viewed without a subscription.
References
- Addo-Quaye C, Eshoo TW, Bartel DP, Axtell MJ(2008) Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr Biol 18: 758–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SJ, Kramer MC, Gosai SJ, Yu X, Vandivier LE, Nelson ADL, Anderson ZD, Beilstein MA, Fray RG, Lyons E, et al. (2018) N6-Methyladenosine inhibits local ribonucleolytic cleavage to stabilize mRNAs in Arabidopsis. Cell Rep 25: 1146–1157.e3 [DOI] [PubMed] [Google Scholar]
- Bai B, Peviani A, van der Horst S, Gamm M, Snel B, Bentsink L, Hanson J(2017) Extensive translational regulation during seed germination revealed by polysomal profiling. New Phytol 214: 233–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chantarachot T, Sorenson RS, Hummel M, Ke H, Kettenburg AT, Chen D, Aiyetiwa K, Dehesh K, Eulgem T, Sieburth LE, et al. (2020) DHH1/DDX6-like RNA helicases maintain ephemeral half-lives of stress-response mRNAs. Nat Plants 6: 675–685 [DOI] [PubMed] [Google Scholar]
- Franke KR, Schmidt SA, Park S, Jeong DH, Accerbi M, Green PJ(2018) Analysis of Brachypodium miRNA targets: Evidence for diverse control during stress and conservation in bioenergy crops. BMC Genomics 19: 547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garre E, Pelechano V, Sánchez Del Pino M, Alepuz P, Sunnerhagen P(2018) The Lsm1-7/Pat1 complex binds to stress-activated mRNAs and modulates the response to hyperosmotic shock. PLoS Genet 14: e1007563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German MA, Pillay M, Jeong DH, Hetawal A, Luo S, Janardhanan P, Kannan V, Rymarquis LA, Nobuta K, German R, et al. (2008) Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends. Nat Biotechnol 26: 941–946 [DOI] [PubMed] [Google Scholar]
- Gy I, Gasciolli V, Lauressergues D, Morel J-B, Gombert J, Proux F, Proux C, Vaucheret H, Mallory AC(2007) Arabidopsis FIERY1, XRN2, and XRN3 are endogenous RNA silencing suppressors. Plant Cell 19: 3451–3461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck AM, Wilusz J(2018) The interplay between the RNA decay and translation machinery in eukaryotes. Cold Spring Harb Perspect Biol 10: a032839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou CY, Lee WC, Chou HC, Chen AP, Chou SJ, Chen HM(2016) Global analysis of truncated RNA ends reveals new insights into ribosome stalling in plants. Plant Cell 28: 2398–2416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou CY, Wu MT, Lu SH, Hsing YI, Chen HM(2014) Beyond cleaved small RNA targets: Unraveling the complexity of plant RNA degradome data. BMC Genomics 15: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Petzold C, Coller J, Baker KE(2010) Nonsense-mediated mRNA decapping occurs on polyribosomes in Saccharomyces cerevisiae. Nat Struct Mol Biol 17: 244–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Sweet TJ, Chamnongpol S, Baker KE, Coller J(2009) Co-translational mRNA decay in Saccharomyces cerevisiae. Nature 461: 225–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Sherman BT, Lempicki RA(2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
- Ibrahim F, Maragkakis M, Alexiou P, Mourelatos Z(2018) Ribothrypsis, a novel process of canonical mRNA decay, mediates ribosome-phased mRNA endonucleolysis. Nat Struct Mol Biol 25: 302–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS(2009) Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324: 218–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AW.(1997) Rat1p and Xrn1p are functionally interchangeable exoribonucleases that are restricted to and required in the nucleus and cytoplasm, respectively. Mol Cell Biol 17: 6122–6130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawa D, Meyer AJ, Dekker HL, Abd-El-Haliem AM, Gevaert K, Van De Slijke E, Maszkowska J, Bucholc M, Dobrowolska G, De Jaeger G, et al. (2020) SnRK2 protein kinases and mRNA decapping machinery control root development and response to salt. Plant Physiol 182: 361–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Hou B, Hou C, Tsao S, Kao P, Chen H, Biotechnology A(2020) Widespread exon junction complex footprints in the RNA degradome mark mRNA degradation before steady-state translation. Plant Cell 32: 904–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Gasic I, Chandrasekaran V, Peters N, Shao S, Mitchison TJ, Hegde RS(2020) TTC5 mediates autoregulation of tubulin via mRNA degradation. Science 367: 100–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S(2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merret R, Bousquet-Antonelli C(2020) Immunity gate-keepers. Nat Plants 6: 608–609 [DOI] [PubMed] [Google Scholar]
- Merret R, Descombin J, Juan YT, Favory JJ, Carpentier MC, Chaparro C, Charng YY, Deragon JM, Bousquet-Antonelli C(2013) XRN4 and LARP1 are required for a heat-triggered mRNA decay pathway involved in plant acclimation and survival during thermal stress. Cell Rep 5: 1279–1293 [DOI] [PubMed] [Google Scholar]
- Merret R, Nagarajan VK, Carpentier MC, Park S, Favory JJ, Descombin J, Picart C, Charng YY, Green PJ, Deragon JM, et al. (2015) Heat-induced ribosome pausing triggers mRNA co-translational decay in Arabidopsis thaliana. Nucleic Acids Res 43: 4121–4132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan VK, Kukulich PM, von Hagel B, Green PJ(2019) RNA degradomes reveal substrates and importance for dark and nitrogen stress responses of Arabidopsis XRN4. Nucleic Acids Res 47: 9216–9230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narsai R, Howell KA, Millar AH, O’Toole N, Small I, Whelan J(2007) Genome-wide analysis of mRNA decay rates and their determinants in Arabidopsis thaliana. Plant Cell 19: 3418–3436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelechano V, Wei W, Steinmetz LM(2015) Widespread co-translational RNA decay reveals ribosome dynamics. Cell 161: 1400–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potuschak T, Vansiri A, Binder BM, Lechner E, Vierstra RD, Genschik P(2006) The exoribonuclease XRN4 is a component of the ethylene response pathway in Arabidopsis. Plant Cell 18: 3047–3057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presnyak V, Alhusaini N, Chen YH, Martin S, Morris N, Kline N, Olson S, Weinberg D, Baker KE, Graveley BR, et al. (2015) Codon optimality is a major determinant of mRNA stability. Cell 160: 1111–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan A, Chen YH, Martin S, Alhusaini N, Green R, Coller J(2016) The DEAD-box protein Dhh1p couples mRNA decay and translation by monitoring codon optimality. Cell 167: 122–132.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynoso MA, Juntawong P, Lancia M, Blanco FA, Bailey-Serres J, Zanetti ME(2015) Translating ribosome affinity purification (TRAP) followed by RNA sequencing technology (TRAP-SEQ) for quantitative assessment of plant translatomes. Methods Mol Biol 1284: 185–207 [DOI] [PubMed] [Google Scholar]
- Simms CL, Yan LL, Zaher HS(2017) Ribosome collision is critical for quality control during no-go decay. Mol Cell 68: 361–373.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorenson RS, Deshotel MJ, Johnson K, Adler FR, Sieburth LE(2018) Arabidopsis mRNA decay landscape arises from specialized RNA decay substrates, decapping-mediated feedback, and redundancy. Proc Natl Acad Sci USA 115: E1485–E1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souret FF, Kastenmayer JP, Green PJ(2004) AtXRN4 degrades mRNA in Arabidopsis and its substrates include selected miRNA targets. Mol Cell 15: 173–183 [DOI] [PubMed] [Google Scholar]
- Tuck AC, Rankova A, Arpat AB, Liechti LA, Hess D, Iesmantavicius V, Castelo-Szekely V, Gatfield D, Bühler M(2020) Mammalian RNA decay pathways are highly specialized and widely linked to translation. Mol Cell 77: 1222–1236.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wawer I, Golisz A, Sulkowska A, Kawa D, Kulik A, Kufel J(2018) mRNA decapping and 5′-3′ decay contribute to the regulation of ABA signaling in Arabidopsis thaliana. Front Plant Sci 9: 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willmann MR, Berkowitz ND, Gregory BD(2014) Improved genome-wide mapping of uncapped and cleaved transcripts in eukaryotes: GMUCT 2.0. Methods 67: 64–73 [DOI] [PubMed] [Google Scholar]
- Windels D, Bucher E(2018) The 5′-3′ exoribonuclease XRN4 regulates auxin response via the degradation of auxin receptor transcripts. Genes (Basel) 9: E638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Willmann MR, Anderson SJ, Gregory BD(2016) Genome-wide mapping of uncapped and cleaved transcripts reveals a role for the nuclear mRNA cap-binding complex in cotranslational RNA decay in Arabidopsis. Plant Cell 28: 2385–2397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Murphy C, Sieburth LE(2010) Conserved RNaseII domain protein functions in cytoplasmic mRNA decay and suppresses Arabidopsis decapping mutant phenotypes. Proc Natl Acad Sci USA 107: 15981–15985 [DOI] [PMC free article] [PubMed] [Google Scholar]