

CASE PRESENTATION
Here, we report a 14-year-old man with autism spectrum disorder and growth delay who presented with several days of left shoulder pain and new onset of progressive dyspnea. No palpable masses were detected. Imaging demonstrated a large heterogeneous mass in the upper mediastinum resulting in deviation of the trachea (Fig 1). Enlarged lymphadenopathy was seen in the bilateral supraclavicular regions and bilateral neck level IV, III, and Vb regions. No disease was present below the diaphragm. Bone marrow and CSF studies were negative at the time of diagnosis. A malignant lymphoma was suspected, and a biopsy of an involved lymph node was performed.
FIG 1.

Contrast-enhanced axial computed tomography (CT) imaging of the chest. There is a large, heterogeneous, mildly enhancing mass (circled in green) within the anterior mediastinum. There is mass effect upon the trachea with mild luminal compromise.
Histologic sections of the lymph node revealed effacement of the normal architecture by a diffuse atypical mononuclear cell proliferation with irregular nuclear contours and vesicular chromatin. Scattered larger cells were present showing marked pleomorphism and occasional multinucleation. Mitotic figures were numerous including atypical mitoses. There were a few background inflammatory cells (lymphocytes, eosinophils, and histiocytes) and focal necrosis (Figs 2A and 2B).
FIG 2.

Lymph node and mediastinal involvement by malignant hematopoietic neoplasm with SMARCB1 deletion showing myeloid, B-cell, and T/natural killer (NK)-cell differentiation. (A) Histologic section of left cervical lymph node revealed diffuse proliferation of atypical mononuclear cells with numerous mitotic figures (inset). There were a few background inflammatory cells (hematoxylin and eosin stain, ×400). (B) Histologic sections of the left anterior mediastinal mass showed atypical mononuclear cells with scattered large cells presenting marked pleomorphism and occasional multinucleation (hematoxylin and eosin stain, ×400). (C) Neoplastic cells showed strong expression of CD45 (leukocyte common antigen; CD45 immunohistochemistry, ×400). (D) CD2 immunohistochemistry showed diffuse and strong expression in neoplastic cells, indicating T/NK-cell differentiation (CD2 immunohistochemistry, ×400). (E) CD7 was diffusely and strongly expressed in neoplastic cells (CD7 immunohistochemistry, ×400). (F) A subset of neoplastic cells shows strong expression of CD79a, indicating B-cell differentiation (CD79a immunohistochemistry, ×400). (G) Weak cytoplasmic expression of myeloperoxidase was seen in a subset of cells, and atypical cells also show immunoreactivity to CD13 immunohistochemistry (inset), supporting myeloid differentiation (myeloperoxidase immunohistochemistry, ×1,000; CD13 immunohistochemistry, ×1,000). (H) Immunohistochemistry for myogenin demonstrated that these cells did not show rhabdoid differentiation (myogenin immunohistochemistry, ×400).
Immunohistochemical studies showed that the neoplastic cells expressed CD45 (leukocyte common antigen), supporting hematopoietic cell origin. The neoplastic cells also expressed CD2, CD7, CD79a, myeloperoxidase (dim), and CD13, suggesting T/natural killer (NK)-cell, B-cell and myeloid differentiation (Figs 2C-2G). CD3, CD34, and terminal deoxynucleotidyl transferase were negative (not shown). Neoplastic cells did not express cytokeratin or myogenin (Fig 2H) against an epithelial or rhabdoid origin. On the basis of these findings, the diagnosis of malignant hematopoietic neoplasm with myeloid, B-cell, and T/NK-cell differentiation was rendered.
The patient was treated with the non-Hodgkin lymphoma regimen DA-EPOCH, consisting of etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin. After three cycles of therapy, positron emission tomography (PET) imaging demonstrated a 68% reduction in the size of the mediastinal mass but no notable change in fluorodeoxyglucose avidity (standard uptake value maximum, 11.3 v 10.4 on the initial study). He went on to receive two additional cycles. After the sixth cycle of DA-EPOCH, he exhibited progressive disease, with an interval increase in mass size (approximately 20%) and fluorodeoxyglucose avidity (12.5) on the PET scan.
A repeat biopsy confirmed the persistence of a hematopoietic neoplasm. Given the unique histology and aggressive clinical course of the malignancy, material from his biopsy was sent to Memorial Sloan Kettering Cancer Center for genomic profiling through the Make-an-IMPACT program1, an initiative that provides genomic profiling for rare tumors using the MSK-IMPACT targeted next-generation sequencing platform (XXXX).2 Profiling revealed a biallelic loss of SMARCB1, confirmed by loss of INI1 expression by immunohistochemistry (Figs 3A and 3B). A somatic TP53 mutation was also identified. The copy number profile was suggestive of broad copy number gains on chromosomes 6, 9, 19, and X. A targeted RNA-based assay (Archer FusionPlex, ArcherDx, Boulder, CO) did not identify any oncogenic gene fusions. To identify rare oncogenic events in genes not included in the targeted DNA and RNA clinical sequencing assays, whole-exome sequencing was performed but did not identify any additional candidate driver alterations (Data Supplement).
FIG 3.
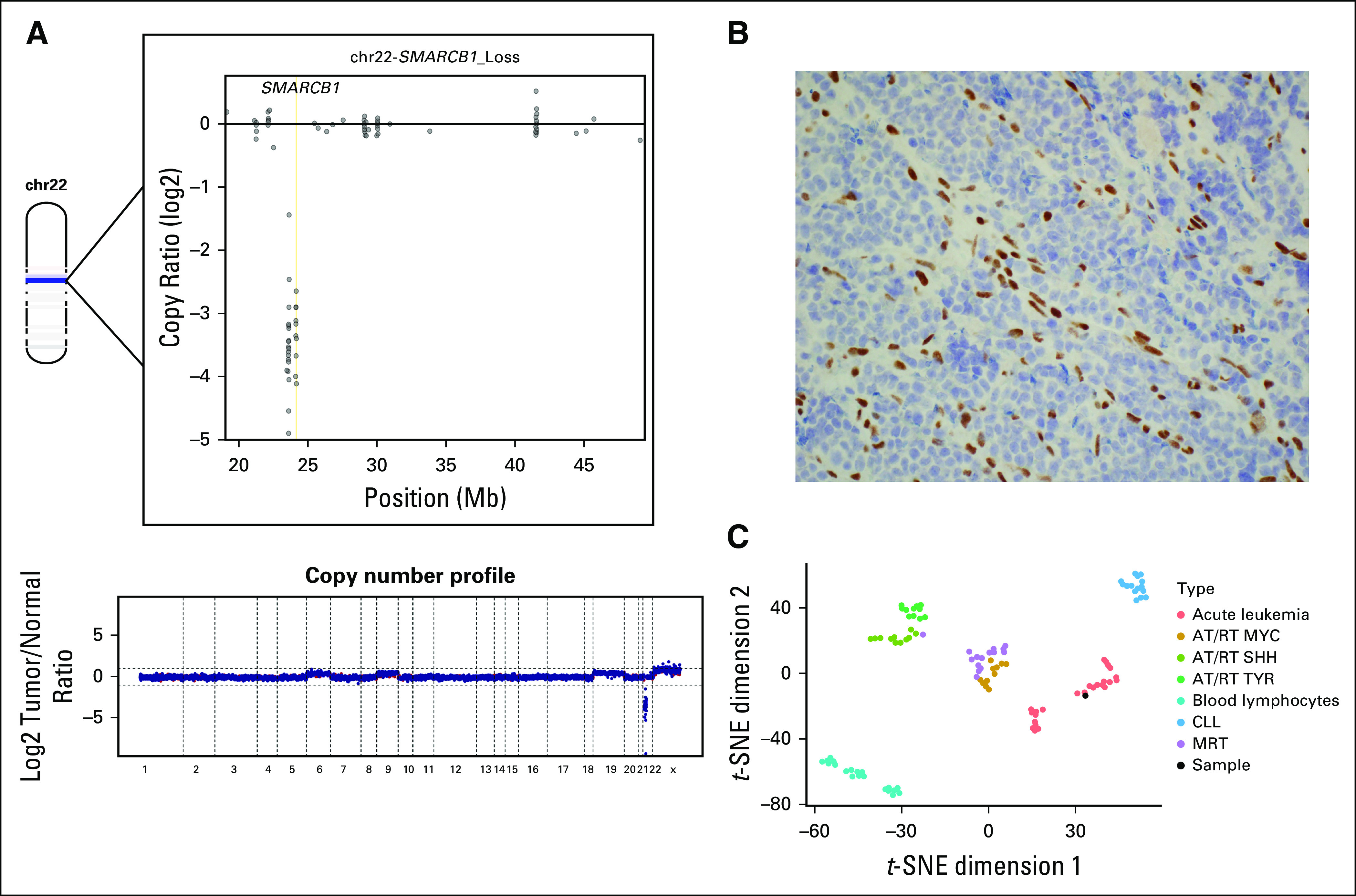
(A) The bottom part of the figure represents the genome wide copy number profile based on log2 copy number coverages at exon locations covered by the MSK IMPACT platform. Chromosome 22 (chr22) is depicted in the upper left part of the figure, with a dark blue band representing the location and loss of SMARCB1. The upper right part of the figure demonstrates log2 copy number coverages at exon locations across chr22, demonstrating complete loss of SMARCB1 at all exon locations. (B) Neoplastic cells showed aberrant loss of INI1 expression, whereas endothelial cells had normal expression of INI1 (INI1/BAF-47 immunohistochemistry, ×400). (C) t-Distributed Stochastic Neighbor Embedding (t-SNE) analysis of methylation array data demonstrated localization of the patient’s sample within the acute leukemia cluster rather than with other SMARCB1-deficient entity clusters. AT/RT, atypical teratoid rhabdoid tumor; CLL, chronic lymphocytic leukemia; MRT, malignant rhabdoid tumor.
DNA-methylation profiling is an effective modality for the classification of SMARCB1-deficient CNS tumors and extracranial malignant rhabdoid tumors.3,4 To assess whether this tumor phenotypically clustered with SMARCB1 CNS or solid tumors, genome-wide methylation profiles were obtained using the Infinium MethylationEPIC/850k platform (Illumina, San Diego, CA) and subjected to a t-Distributed Stochastic Neighbor Embedding dimensionality reduction5 against SMARCB1-deficient entities (atypical teratoid/rhabdoid tumor and malignant rhabdoid tumor) as well as a select cohort of acute and chronic hematopoietic neoplasms (acute leukemia and chronic lymphocytic leukemia) obtained from the University of Heidelberg. Interestingly, the tumor localized to the acute leukemia cluster rather than the SMARCB1-deficient solid/CNS tumors and the control hematopoietic tissue (normal blood lymphocytes and chronic lymphocytic leukemia/small lymphocytic lymphoma samples; Fig 3C).
While MSK-IMPACT profiling was being performed, salvage therapy with brentuximab vedotin, nivolumab, and bendamustine was attempted. Unfortunately, the patient experienced a rapid progression of mediastinal disease and died before the availability of the profiling results. No additional tissue sampling or autopsy was performed. The patient’s guardian provided written consent to share his deidentified clinical information in this publication.
DISCUSSION
The SWItch/Sucrose Non-Fermentable (SWI/SNF) chromatin remodeling complex is a large, multiprotein complex, that mobilizes nucleosomes using energy derived from ATP hydrolysis.5 Proteins within the complex serve key roles in transcriptional regulation and tumor suppression.6-8 Mutations in genes encoding subunits of SWI/SNF complex have been found in approximately 20% of all tumor genomes sequenced to date, marking it as one of the most commonly mutated chromatin modulators in human cancer.9 SMARCB1 (also known as INI1 or BAF47) is one of the core component proteins in the SWI/SNF chromatin remodeling complex. Homozygous deletions of SMARCB1 are driver oncogenic events in several solid tumor types, including malignant rhabdoid tumors, renal medullary carcinomas, and a subset of epithelioid sarcomas.10-12
To our knowledge, this is the first report of a hematologic malignancy with confirmed somatic biallelic loss of SMARCB1. Interestingly, mouse models with inactivating SMARCB1 mutations are known to develop T-cell lymphomas.13 However, genomic profiling studies of T-cell lymphomas have not reported recurrent inactivating SMARCB1 mutations.14,15 Deletions of SMARCB1 have also been identified in chronic myeloid leukemia samples but only as heterozygous losses, resulting in the potential reduction of gene dosage but not complete inactivation.16
Our patient had a highly aggressive disease course, a clinical feature shared with SMARCB1-deficient solid tumors. Despite intensive multimodal chemotherapy and radiotherapy, he experienced rapid disease progression and death within 6 months of diagnosis. Genomically, the tumor cells exhibited a low tumor mutational burden, which is congruent with SMARCB1-deficient solid tumors.17 However, the histologic features and DNA-methylation profile identify this tumor as a hematologic malignancy distinct from SMARCB1-deficient solid tumors.
In conclusion, we have identified a unique case of an aggressive hematopoietic malignancy characterized by biallelic loss of SMARCB1. Several clinical trials have been initiated that are selectively accruing patients with biallelic loss of SMARCB1, including ongoing studies of the enhancer of zeste homolog 2 (EZH2) inhibitor tazemetostat (ClinicalTrials.gov identifier: NCT02601937). EZH2 expression is upregulated in the setting of biallelic loss of SMARCB1, rendering these tumors potentially vulnerable to target inhibition.18 Results from phase II studies have led to US Food and Drug Administration approval for patients with metastatic or locally advanced epithelioid sarcoma.19 Preliminary pediatric data also demonstrate activity in subsets of other SMARCB1-deficient tumor histologies.20 Our patient experienced disease progression too rapidly to be enrolled into a therapeutic trial. However, this case highlights the potential clinical utility of early genomic and epigenetic profiling of unusual malignant histologies to ensure the correct cancer type diagnosis and to identify potentially actionable alterations that could serve as targets for therapeutic intervention.
ACKNOWLEDGMENT
The authors acknowledge Dalicia Reales, Bernadette Wolf, and Barbara Solit from the Memorial Sloan Kettering Cancer Center Make-an-IMPACT program.
SUPPORT
Supported by Cycle for Survival and by National Cancer Institute Cancer Center Support Grant No. P30 CA008748.
AUTHOR CONTRIBUTIONS
Conception and design: Michael D. Kinnaman, Darcy Hamill, Jonathan Powell, Jamal Benhamida, Neerav Shukla
Collection and assembly of data: Michael D. Kinnaman, Darcy Hamill, Jonathan Powell, Jamal Benhamida, Mariko Yabe, Neerav Shukla
Data analysis and interpretation: Michael D. Kinnaman, Darcy Hamill, Mariko Yabe, Jamal Benhamida, Christian Vokuhl, Christian Koelsche, Andreas von Deimling, Edward Anders Kolb, David B. Solit, Marc Ladanyi, Ahmet Dogan, Neerav Shukla
Provision of study material or patients: Michael D. Kinnaman, Darcy Hamill, Neal Shukla, Jonathan Powell
Administrative support: David B. Solit
Financial support: David B. Solit
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Mariko Yabe
Consulting or Advisory Role: Y-mAb Therapeutics, Janssen Research and Development
Jamal Benhamida
Consulting or Advisory Role: DocDoc
Andreas von Deimling
Consulting or Advisory Role: Bristol Myers Squibb
Research Funding: Bayer
Patents, Royalties, Other Intellectual Property: Patent for IDH1R132H antibody H09administered by the German Cancer Center (DKFZ); patent for BRAFV600E antibody VE1administered by the German Cancer Center (DKFZ)
Travel, Accommodations, Expenses: Roche
Edward Anders Kolb
Travel, Accommodations, Expenses: Roche, Genentech
David B. Solit
Stock and Other Ownership Interests: Loxo
Consulting or Advisory Role: Pfizer, Loxo, Illumina, Vivideon Therapeutics, Lilly, QED Therapeutics, BridgeBio Pharma
Marc Ladanyi
Consulting or Advisory Role: Bristol Myers Squibb, Bayer
Research Funding: Loxo (Inst), Helsinn Therapeutics, Merus NV, Elevation Oncology (Inst)
Ahmet Dogan
Consulting or Advisory Role: Seattle Genetics, Roche, Takeda, EUSA Pharma, Abbvie
Research Funding: Roche, Genentech
Neerav Shukla
Consulting or Advisory Role: Syndax
No other potential conflicts of interest were reported.
REFERENCES
- 1. Memorial Sloan Kettering Cancer Center: Make an IMPACT research program. https://www.mskcc.org/research-programs/molecular-oncology/make-impact.
- 2.Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–713. doi: 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Capper D, Jones DTW, Sill M, et al. DNA methylation-based classification of central nervous system tumours. Nature. 2018;555:469–474. doi: 10.1038/nature26000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koelsche C, Hartmann W, Schrimpf D, et al. Array-based DNA-methylation profiling in sarcomas with small blue round cell histology provides valuable diagnostic information. Mod Pathol. 2018;31:1246–1256. doi: 10.1038/s41379-018-0045-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu JI, Lessard J, Crabtree GR. Understanding the words of chromatin regulation. Cell. 2009;136:200–206. doi: 10.1016/j.cell.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kowenz-Leutz E, Leutz A. A C/EBP β isoform recruits the SWI/SNF complex to activate myeloid genes. Mol Cell. 1999;4:735–743. doi: 10.1016/s1097-2765(00)80384-6. [DOI] [PubMed] [Google Scholar]
- 7.Reisman D, Glaros S, Thompson EA. The SWI/SNF complex and cancer. Oncogene. 2009;28:1653–1668. doi: 10.1038/onc.2009.4. [DOI] [PubMed] [Google Scholar]
- 8.Kim KH, Roberts CWM. Mechanisms by which SMARCB1 loss drives rhabdoid tumor growth. Cancer Genet. 2014;207:365–372. doi: 10.1016/j.cancergen.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kadoch C, Hargreaves DC, Hodges C, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet. 2013;45:592–601. doi: 10.1038/ng.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kohashi K, Tanaka Y, Kishimoto H, et al. Reclassification of rhabdoid tumor and pediatric undifferentiated/unclassified sarcoma with complete loss of SMARCB1/INI1 protein expression: Three subtypes of rhabdoid tumor according to their histological features. Mod Pathol. 2016;29:1232–1242. doi: 10.1038/modpathol.2016.106. [DOI] [PubMed] [Google Scholar]
- 11.Hornick JL, Dal Cin P, Fletcher CDM. Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol. 2009;33:542–550. doi: 10.1097/PAS.0b013e3181882c54. [DOI] [PubMed] [Google Scholar]
- 12.Cheng JX, Tretiakova M, Gong C, et al. Renal medullary carcinoma: Rhabdoid features and the absence of INI1 expression as markers of aggressive behavior. Mod Pathol. 2008;21:647–652. doi: 10.1038/modpathol.2008.44. [DOI] [PubMed] [Google Scholar]
- 13.Roberts CWM, Leroux MM, Fleming MD, et al. Highly penetrant, rapid tumorigenesis through conditional inversion of the tumor suppressor gene Snf5. Cancer Cell. 2002;2:415–425. doi: 10.1016/s1535-6108(02)00185-x. [DOI] [PubMed] [Google Scholar]
- 14.Heavican TB, Bouska A, Yu J, et al. Genetic drivers of oncogenic pathways in molecular subgroups of peripheral T-cell lymphoma. Blood. 2019;133:1664–1676. doi: 10.1182/blood-2018-09-872549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sandell RF, Boddicker RL, Feldman AL. Genetic landscape and classification of peripheral T cell lymphomas. Curr Oncol Rep. 2017;19:28. doi: 10.1007/s11912-017-0582-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grand F, Kulkarni S, Chase A, et al. Frequent deletion of hSNF5/INI1, a component of the SWI/SNF complex, in chronic myeloid leukemia. Cancer Res. 1999;59:3870–3874. [PubMed] [Google Scholar]
- 17.Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knutson SK, Warholic NM, Wigle TJ, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci USA. 2013;110:7922–7927. doi: 10.1073/pnas.1303800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoy SM. Tazemetostat: First approval. Drugs. 2020;80:513–521. doi: 10.1007/s40265-020-01288-x. [DOI] [PubMed] [Google Scholar]
- 20.Chi SN, Bourdeaut F, Laetsch TW, et al. Phase I study of tazemetostat, an enhancer of zeste homolog-2 inhibitor, in pediatric pts with relapsed/refractory integrase interactor 1-negative tumors. J Clin Oncol. 2020;38(suppl; abstr 10525) [Google Scholar]