

Abstract
PURPOSE
To ascertain the prevalence of recurrent de novo variants among 240 pediatric patients with osteosarcoma (OS; age < 20 years) unselected for family history of cancer.
METHODS
The identification of de novo variants was implemented in 2 phases. In the first, we identified genes with a rare (minor allele frequency < 0.01) de novo variant in > 1 of the 95 case-parent trios examined by whole-exome sequencing (WES) who passed quality control measures. In phase 2, 145 additional patients with OS were evaluated by targeted sequencing to identify rare de novo variants in genes nominated from phase 1. Recurrent rare variants identified from phase 1 and 2 were verified as either de novo or inherited by Sanger sequencing of affected patients and their parents. Categorical and continuous data were analyzed using Fisher exact test and t tests, respectively.
RESULTS
Among 95 case-parent trios who underwent WES, we observed 61 de novo variants in 60 genes among 47 patients, with TP53 identified as the only gene with a pathogenic or likely pathogenic (P/LP) de novo variant in more than one case-parent trio. Among all 240 patients with OS, 13 (5.4%) harbored a P/LP TP53 germline variant, of which 6 (46.2%) were confirmed to be de novo.
CONCLUSION
Apart from TP53, we did not observe any other recurrent de novo P/LP variants in the case-parent trios, suggesting that new mutations in other genes are not a frequent cause of pediatric OS. That nearly half of P/LP TP53 variants in our sample were de novo suggests universal screening for germline TP53 P/LP variants among pediatric patients with OS should be considered.
INTRODUCTION
Osteosarcoma (OS) is the most common primary bone tumor in children and adolescents (age < 20 years) with an age-adjusted incidence of approximately 5.2 cases per million per year.1 Several inherited cancer predisposition syndromes are associated with an increased risk of OS,2 including Li-Fraumeni syndrome (LFS), an autosomal dominant disorder caused by pathogenic germline TP53 variants.3,4 LFS and other inherited syndromes are collectively rare2,5; however, the prevalence of rare pathogenic variants in the OS patient population is reported to be quite substantial. In the largest study to date, 28% of 1,244 patients with OS harbored a pathogenic or likely pathogenic (P/LP) variant in at least one of the 238 cancer-susceptibility genes evaluated.6 Other smaller studies have reported the prevalence of P/LP cancer-susceptibility gene variants in patients with OS to be between 7.1% (three of 42 patients)7 and 17.9% (seven of 39 patients),8 with between 3% and 10% of patients harboring a P/LP variant localized to TP53.9-11
CONTEXT
Key Objective
What is the prevalence of recurrent pathogenic or likely pathogenic (P/LP) rare de novo germline variants in pediatric patients with osteosarcoma (OS)?
Knowledge Generated
TP53 was the only gene with a rare germline de novo variant predicted to be pathogenic in one of 95 patients with OS examined by whole-exome sequencing. Among 240 pediatric patients with OS with either whole-exome or targeted sequencing, 13 (5.4%) harbored a rare P/LP TP53 germline variant, of which six (46%) were confirmed as de novo.
Relevance
A high proportion of rare pathogenic TP53 variants in the pediatric patients with OS may be de novo. Guidelines that recommend clinical TP53 genetic testing among all pediatric patients with OS, regardless of family history or number of primary tumors, should continue to be considered.
The proportion of P/LP germline variants in patients with OS that arise from de novo mutation events rather than familial inheritance remains unclear. However, the apparent discrepancy between the relatively few patients with OS diagnosed with a recognized cancer predisposition syndrome and the high burden of P/LP germline variants in patients with OS can be resolved if the P/LP germline variants are de novo mutations, which would not be associated with a familial pattern of cancer. Indeed, in one study, an estimated 7% to 20% of TP53 variants identified in patients with early-onset cancer were de novo, implying that new mutations may be found in some proportion of those with OS.12 The proportion of TP53 variants that are de novo in patients with OS is reported to be between 33% (one of three variants)13 and 57% (four of seven variants),9 but prior estimates were calculated from patients with OS recruited on the basis of having a family history of cancer or multiple primaries,13 or else from variants presumed to be de novo based solely on an absent family history.9 To date, the prevalence of de novo variants confirmed by genotyping in patients with OS unselected for family history of cancer has yet to be examined and, to our knowledge, no study of OS has examined de novo variants exome-wide.
The purpose of this study was to ascertain the prevalence of recurrent de novo variants among 240 pediatric patients with OS (age < 20 years) unselected for family history of cancer. We focus herein on TP53, the only gene found to have a P/LP de novo variant in more than one of the case-parent trios that formed our discovery set.
METHODS
Study Population and Identification of De Novo Variants
The study sample consisted of 240 pediatric patients with OS (age < 20 years at diagnosis) identified through the Childhood Cancer Research Network of the Children’s Oncology Group and their parents, as described elsewhere.14 A total of 95 patients and their parents underwent whole-exome sequencing (WES) and 145 patients (but not their parents) underwent targeted sequencing. These 240 patients were previously reported as a replication set in a study of rare variant frequency in patients with OS, in which the methods for WES and targeted sequencing of patients were described.6 WES of parental DNA was performed at the same time as that of patients and aligned using the same methods.
The discovery of rare (minor allele frequency < 0.01) de novo variants was implemented in 2 phases. In phase 1, we classified rare variants identified exome-wide in the 95 case-parent trios who underwent WES as either inherited or de novo using SuperNovo (Appendix).15 In brief, SuperNovo uses the genomic variant call format files created by GATK (Broad Institute, Cambridge, MA) to nominate positions that could possibly be de novo and then uses evidence from the original BAM files for every member of the trio to determine the likelihood that each variant is actually a de novo variant and not due to technical artifact (eg, sequencing error, low depth of coverage, or mismapping). Every de novo variant meeting our criteria was examined by hand in Integrative Genomics Viewer (IGV; version 2.4.16; Broad Institute) in the proband and both parents to ensure that the call looked real to an experienced bioinformatician.
Given current estimates,16 we expected to discover approximately one de novo variant per exome per generation and therefore decided a priori to nominate for follow-up only those genes found to have a rare de novo variant in more than one of the 95 case-parent trios sequenced in phase 1. In phase 2, 145 patients with OS who underwent targeted sequencing were examined for rare variants localized to TP53, the only gene nominated from phase 1 for follow-up analyses. Rare TP53 variants identified among patients with OS in phase 1 and 2 were then verified as either de novo or inherited, by Sanger sequencing of the patient and available parent DNA (two case-parent trios from phase 1; eight case-parent trios and three case-parent dyads from phase 2; Appendix and Data Supplement). A variant was confirmed as de novo if it was missing from the DNA of both parents. To calculate the proportion of P/LP variants that were de novo¸ we included all P/LP variants in the denominator regardless of whether there was complete case-parent trio DNA.
Rare Variant Annotation
We classified rare variants (inherited or de novo) as pathogenic or likely pathogenic (P/LP), variant of uncertain significance (VUS), likely benign, or benign, according to the pathogenicity category designated by a badged laboratory in ClinVar (National Center for Biotechnical Information, Bethesda, MD).17 We also reported the im-pact of variants as predicted by SnpEff (Pablo Cingolani, Boston, MA) as high, moderate, or low.18 We further characterized rare TP53 variants using the International Agency for Research on Cancer TP53 database (version R20)19 according to whether they have ever been reported in families with cancer histories consistent with the Li-Fraumeni or Li-Fraumeni–like (LFL) criteria, as well as to describe the affected codon, protein domain function, and the impact of missense mutations (as reported from the Sorting Tolerant From Intolerant20 and Polymorphism Phenotyping, version 2 21 algorithms).
Statistical Analyses
We compared the clinical and demographic characteristics of patients with OS with a P/LP TP53 germline variant to those without the variant, using t tests or Fischer exact test for continuous or categorical variables, respectively, and calculated 95% CIs for P/LP variant proportions, using the Clopper and Pearson procedure.22 All analyses were conducted in R, version 3.6.0.23 We also conducted a gene-enrichment analysis on the genes with a de novo variant identified in phase 1 using STRING software (ELIXIR Infrastructure, Cambridgeshire, UK) with default settings.24 Statistical significance was set at P < .05.
Family History Information
A detailed family history questionnaire was requested of parents. The questionnaire included information on cancer history (type, age at diagnosis), current age (for living relatives), and age (and cause of death, if deceased) for first-, second-, and third-degree relatives. Probands with P/LP rare TP53 germline variants were classified as having LFS if their family histories were consistent with the 2015 modified Chompret criteria (Table 1).25,26 Reported cancer diagnoses in relatives were not confirmed from pathologic reports or other medical records.
TABLE 1.
The Modified Chompret Criteria for TP53 Genetic Testing

RESULTS
Examination of Recurrent De Novo Variants
Among 95 case-parent trios examined by WES (phase 1), we identified 61 de novo variants in 60 genes among 47 patients, of which one variant was pathogenic (localized to HFE), two were LP (both localized to TP53), two were benign (localized to LEPR and FLG), one was a VUS (localized to SMARCA4), and 55 were not reported in ClinVar. SnpEff predicted six and 55 variants to be of high or moderate impact, respectively. Two probands in phase 1 harbored de novo variants in the TP53 gene, and the remainder of de novo variants were present in only one gene (Data Supplement). Apart from TP53, none of the de novo variants localized to one of the 238 cancer-susceptibility genes found to harbor a P/LP rare variant in the Mirabello et al study6 of 1,244 patients with OS.
Phase 2 involved de novo variant discovery in TP53, the only gene nominated from phase 1. Among 145 patients with OS who underwent targeted sequencing, we identified an additional 11 rare TP53 germline variants in 11 patients with OS that were determined to be pathogenic (n = 7; 4.8%; 95% CI, 2.0% to 9.7%) or LP (n = 4; 2.8%; 95% CI, 0.8% to 6.9%). Among all 240 patients with OS (phase 1 and 2), 13 patients (5.4%; 95% CI, 2.9% to 9.1%) harbored either a pathogenic (n = 7; 2.9%; 95% CI, 1.2% to 5.9%) or an LP TP53 germline variant (n = 6; 2.5%; 95% CI, 0.9% to 5.4%; Table 2). Of the 13 rare TP53 variants, six were confirmed de novo by Sanger sequencing (46.2%; 95% CI, 19.2% to 74.9%). The proportion of confirmed de novo TP53 variants among all 240 patients with OS was 2.5% (n = 6 of 240; 95% CI, 0.9% to 5.4%). We note that as many as nine variants may have been de novo, but we could not confirm the inheritance pattern of three variants because of missing parental genotypes.
TABLE 2.
Characterization of 13 P/LP TP53 Germline Variants Discovered in OS Cases (N = 240)

P/LP TP53 variants were distributed between amino acids 107 and 342 (Data Supplement). Of the 13 P/LP TP53 variants discovered, 10 (76.9%; 95% CI, 46.2% to 95.0%) were missense variants that affected the DNA binding domain (six were confirmed de novo variants), one was a missense variant outside of the DNA binding domain (7.7%; 95% CI, 0.1% to 36.0%; tetramerization domain), and two were nonsense variants (15.4%; 95% CI, 1.9% to 4.5%).
Analyses of Gene Enrichment and Clinical Characteristics
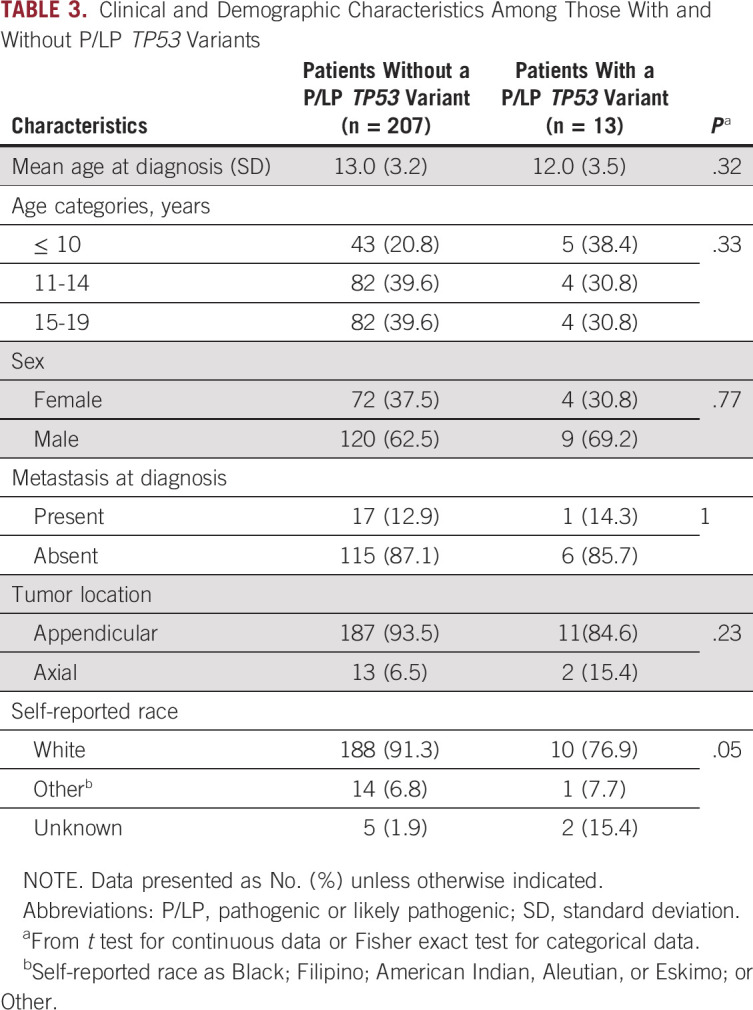
We did not identify a significant association among the set of 60 genes that harbored a rare de novo variant in phase 1 (P = .91; Data Supplement), nor did we find any statistically significant associations between clinical characteristics and the presence of a P/LP TP53 germline variant (Table 3). With regard to family history of cancer, only three of the 13 patients with a P/LP TP53 germline variant returned a questionnaire; all three had reported family histories that were consistent with the modified Chompret criteria,25,26 none of whom had a pathogenic variant that was confirmed as de novo (Table 2).
TABLE 3.
Clinical and Demographic Characteristics Among Those With and Without P/LP TP53 Variants
DISCUSSION
In our whole-exome analysis of 95 pediatric (< 20 years of age) OS case-parent trios, we discovered TP53 was the only gene recurrently affected by P/LP de novo variants. Among all 240 patients (from phase 1 and 2), 13 (5.4%) harbored a P/LP TP53 germline variant, nearly half of which were confirmed to be de novo. The prevalence of patients with a P/LP TP53 germline variant reported herein is similar to the prevalence of 3% to 10% described elsewhere.9-11 The current study also confirms previous findings that a large proportion of pathogenic TP53 germline variants are de novo.9,12,13 However, to our knowledge, this is the first study to confirm by sequencing the prevalence of de novo TP53 germline variants among patients with OS unselected for family history of cancer.
Identifying patients newly diagnosed with cancer who have pathogenic TP53 germline variants has important implications for patient treatment, in part because of the substantially increased risk of secondary cancers. In one report, in nearly 50% of 191 TP53 variant carriers diagnosed with cancer, a subsequent cancer developed after a median follow-up of 10 years.27 Other reports presented similar results,26,28 with the highest risk observed in survivors of childhood cancers.28 Surveillance protocols aimed at early tumor detection show clinical utility among TP53 carriers,29,30 and it is now recommended that such screening be offered to all individuals as soon as an LFS diagnosis is made.31 Moreover, second malignancies in those given radiotherapy are often observed to develop within the radiation field,26,28,32 suggesting that exposure to medical radiation should be limited whenever possible.31,33
The clinical criteria for LFS has been updated over the past several decades to enable identification of TP53 var-iants in patients not meeting the original definition of the syndrome.25,26,34-36 The most current criteria used to recommend TP53 testing encompass four clinical situations suggestive of LFS, (the modified Chompret Criteria26; Table 1) under which pediatric patients with OS would be offered testing if an individual presented with either (1) a familial presentation consistent with the criteria, or (2) with multiple tumors. However, these criteria do not accommodate the possibility of de novo variants in TP53 among first primary patients with OS. In this study, we confirmed previous reports that the prevalence of TP53 P/LP variants in patients with early-onset OS is similar to that observed in patients with early-onset breast cancer,11 all of whom are recommended to receive TP53 genetic testing.26 In addition, the high proportion of de novo variants identified among those that are pathogenic suggests that relying on family history patterns of cancer to identify patients with OS at risk of harboring pathogenic TP53 variants is insufficient. Given these results, it is worthwhile to consider guidelines that recommend clinical TP53 genetic testing among all pediatric patients with OS, regardless of family history or number of primary tumors.
Apart from TP53, no gene harbored recurrent de novo P/LP variants in more than one of the 95 case-parent trios who underwent WES. It is possible that a larger sample would reveal recurrent de novo variants in genes affected in single trios in our study or in different genes entirely. However, our study indicates recurrent de novo variants in genes apart from TP53 are not a frequent occurrence in patients with OS. It is also possible that rather than recurrence in a single gene being frequent, de novo variants recurrently affect genes in particular pathways, which is the paradigm in autism.37 We did not identify significant enrichment among the 60 genes identified as harboring a de novo variant in phase 1 of our study. However, additional sequencing of OS case-parent trios followed by pathway analysis will help examine this possibility.
Our study has several strengths, including the large sample size for a rare cancer. Also, by enrolling patients with OS unselected for family history of cancer, we likely avoided biased estimates of de novo variants that may have occurred in studies that enrolled patients on the basis of LFS criteria. Nevertheless, we also note several limitations. First, we could not confirm the inheritance pattern of three variants from case-parent dyads, because of missing parental genotypes. Second, using Sanger sequencing to confirm de novo variants may have failed to detect instances of low-level somatic mosaicism, which is increasingly recognized as a potential mechanism of transmitting mutations.38,39 Third, the true prevalence of P/LP variants may have been underestimated in our study, given that WES could not detect variants that localized to promotor, intronic, or regulatory regions. Finally, we were unable to confirm reported family history through medical records and did not have information on clinical or demographic characteristics of a small proportion of patients, which reduced our ability to compare clinical characteristics and presence of P/LP TP53 variants.
In summary, we report that TP53 was the only gene with a rare de novo variant in more than one OS case-parent trio among 95 evaluated. The high proportion of de novo P/LP TP53 germline variants observed suggests efforts to identify pediatric patients with OS at risk of harboring pathogenic TP53 variants should continue to evolve, including possible universal screening for germline TP53 P/LP variants among pediatric patients with OS. Additional studies with larger sample sizes are needed to refine our observations and determine the extent to which de novo variants in other cancer susceptibility genes contribute to OS etiology.
ACKNOWLEDGMENT
All participants provided informed consent to participate in this research protocol, which was approved by the University of Minnesota Human Research Protection Program (Approval No.: 0701M00961).
Appendix
De Novo Variant Discovery in Phase 1 via SuperNovo
GATK HaplotypeCaller (version 4.1.3.0) was used to call genotypes for all individuals in the study. SuperNovo (https://github.com/PankratzLab/SuperNovo) was then used to make a robust de novo mutation call set. SuperNovo uses the genomic variant call format (gVCF) files created by GATK and the original BAM files for every member of the trio. When considering bases to be viable alleles at a locus, SuperNovo defines viability as an allelic depth of two or more or an allelic fraction > 0.05. In many calculations, SuperNovo uses phred-weighted depths by multiplying the accuracy probability (1 − error probability) of the mapping quality phred score by the accuracy probability of the base quality phred score and then summing these scores for each read at a locus.
Each heterozygous single nucleotide variation with one alternate allele called in the gVCF for each sample was considered a possible de novo mutation if it met all of the following criteria in the BAM alignments:
1. It must be biallelic, defined as having two or more viable alleles, allowing for rare sequencing error but excluding regions with mismapping.
-
2. It must be a heterozygote, defined as:
• A phred-weighted combined depth of the two viable alleles of at least 10.
• A phred-weighted allelic depth of at least four for both viable alleles.
• A phred-weighted allelic fraction for both viable alleles of at least 0.1.
1. To be considered a possible de novo, there must be a single de novo allele that is not a viable allele at the locus in either parent.
From this pool of all possible de novo variants for a sample, SuperNovo then applies the following set of filters to exclude variants if:
1. The phred-weighted read depth at the site was less than 10 in either parent or the de novo allele is the reference allele, indicating the allele may exist in a parent but was not captured.
2. The region contained other viable de novo alleles that were not at adjacent bases but on the same reads as the variant at least 75% of the time, which is evidence that the reads are simply mismapped from another part of the genome with a similar sequence.
3. The region contained other sites that were triallelic (three or more viable alleles at the same position), indicating that reads in the region are likely mismapped from another part of the genome with a similar sequence.
4. The haplotype for any biallelic variants in the region 150 bp upstream and downstream of the variant were not concordant. For every read that extended far enough to capture the nearby variant, at least 75% of those reads would have to have the other variant, otherwise it would indicate that three or more haplotypes were present, suggesting those haplotypes are likely to be mismapped reads from another locus with a similar sequence.
5. The variant had no obvious biologic significance. We annotated the variants with SnpEff (version 4.3r) and then considered those with impacts of high (loss-of-function variants) or moderate (missense variants).
Every de novo variant meeting all these criteria was then examined by hand in Integrative Genomics Viewer (IGV, version 2.4.16) in the proband and both parents.
Confirmation of De Novo Pathogenic or Likely Pathogenic TP53 Variants by Sanger Sequencing
Rare pathogenic or likely pathogenic TP53 variants discovered in phases 1 and 2 were verified as either de novo or inherited, by Sanger sequencing in the patient and available parent DNA. All polymerase chain reaction (PCR) primers (Data Supplement) were designed using Primer3 software (https://primer3.org/). Amplification reactions contained 5 ng of genomic DNA, 1× Q5 High-Fidelity 2X Master Mix (New England BioLabs, Ipswich, MA), 0.5 µM of each primer pair in a final volume of 25 µL carried out in an Eppendorf Mastercycler Gradient thermocycler (Eppendorf, Hamburg, Germany). After PCR cycling, amplification products were resolved on 2.0% agarose gels with a TrackIt 50bp DNA ladder (Invitrogen, Carlsbad, CA) at 100V for 30 minutes. Amplicons were excised from the agarose gel using a sterile razor blade and purified using the QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany). Purified amplicons at 20 ng with 3.2 pmol of single sequencing primer were sent to University of Minnesota Genomics Center for Sanger sequencing. Sequencing chromatograms were analyzed using Chromas software (Technelysium, South Brisbane, QLD, Australia).
SUPPORT
Collection of samples was supported by the Division of Cancer Epidemiology and Genetics, National Cancer Institute (NCI; Grant No. U01 CA122371 [L.G.S.]). Sequencing was supported by grants to L.G.S. from Hyundai Hope on Wheels and the Zach Sobiech Osteosarcoma Fund. B.J.D. was supported by a pediatric oncology student training award from the Alex’s Lemonade Stand Foundation and the NIH National Institute of Arthritis and Musculoskeletal and Skin Diseases (Grant No. T32 AR050938 “Musculoskeletal Training Grant”). D.M. was supported by grants from the Canadian Institutes for Health Research and a Program Project from the Terry Fox Research Institute. The Children’s Oncology Group is supported by National Clinical Trials Network (NCTN) Operations Center (Grant No. U10CA180886) and the NCTN Statistics & Data Center (Grant No. U10CA180899).
AUTHOR CONTRIBUTIONS
Conception and design: Nathan Pankratz, David Malkin, Logan G. Spector
Financial support: Logan G. Spector
Administrative support: Logan G. Spector
Provision of study material or patients: Logan G. Spector
Collection and assembly of data: Brandon J. Diessner, Nathan Pankratz, Anthony J. Hooten, Lisa Mirabello, Lauren J. Mills, David Malkin, Logan G. Spector
Data analysis and interpretation: Brandon J. Diessner, Nathan Pankratz, Aaron L. Sarver, Lauren J. Mills, Ava C. Kelley, Logan G. Spector
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
David Malkin
Consulting or Advisory Role: Bayer
No other potential conflicts of interest were reported.
REFERENCES
- 1.Ries LAG, Smith MA, Gurney JG, et al.eds), Malignant bone tumorsin Cancer Incidence and Survival among Children and Adolescents: United States SEER Program 1975-1995 Bethesda, MD: National Cancer Institute, SEER Program; 199999 [Google Scholar]
- 2.Savage SA, Mirabello L. Using epidemiology and genomics to understand osteosarcoma etiology. Sarcoma. 2011;2011:548151. doi: 10.1155/2011/548151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malkin D, Li FP, Strong LC, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233–1238. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- 4.Malkin D. Li-fraumeni syndrome. Genes Cancer. 2011;2:475–484. doi: 10.1177/1947601911413466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Helman LJ, Meltzer P. Mechanisms of sarcoma development. Nat Rev Cancer. 2003;3:685–694. doi: 10.1038/nrc1168. [DOI] [PubMed] [Google Scholar]
- 6.Mirabello L, Zhu B, Koster R, et al. Frequency of pathogenic germline variants in cancer-susceptibility genes in patients with osteosarcoma. JAMA Oncol. 2020;6:724. doi: 10.1001/jamaoncol.2020.0197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gröbner SN, Worst BC, Weischenfeldt J, et al. The landscape of genomic alterations across childhood cancers. Nature. 2018;555:321–327. doi: 10.1038/nature25480. [Erratum: Nature 559:E10, 2018] [DOI] [PubMed] [Google Scholar]
- 8.Zhang J, Walsh MF, Wu G, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med. 2015;373:2336–2346. doi: 10.1056/NEJMoa1508054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McIntyre JF, Smith-Sorensen B, Friend SH, et al. Germline mutations of the p53 tumor suppressor gene in children with osteosarcoma. J Clin Oncol. 1994;12:925–930. doi: 10.1200/JCO.1994.12.5.925. [DOI] [PubMed] [Google Scholar]
- 10.Toguchida J, Yamaguchi T, Dayton SH, et al. Prevalence and spectrum of germline mutations of the p53 gene among patients with sarcoma. N Engl J Med. 1992;326:1301–1308. doi: 10.1056/NEJM199205143262001. [DOI] [PubMed] [Google Scholar]
- 11.Mirabello L, Yeager M, Mai PL, et al. Germline TP53 variants and susceptibility to osteosarcoma. J Natl Cancer Inst. 2015;107:1–4. doi: 10.1093/jnci/djv101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonzalez KD, Buzin CH, Noltner KA, et al. High frequency of de novo mutations in Li-Fraumeni syndrome. J Med Genet. 2009;46:689–693. doi: 10.1136/jmg.2008.058958. [DOI] [PubMed] [Google Scholar]
- 13.Chompret A, Brugières L, Ronsin M, et al. P53 germline mutations in childhood cancers and cancer risk for carrier individuals. Br J Cancer. 2000;82:1932–1937. doi: 10.1054/bjoc.2000.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Musselman JRB, Bergemann TL, Ross JA, et al. Case-parent analysis of variation in pubertal hormone genes and pediatric osteosarcoma: A Children’s Oncology Group (COG) study. Int J Mol Epidemiol Genet. 2012;3:286–293. [PMC free article] [PubMed] [Google Scholar]
- 15. PankratzLab/SuperNovo: https://github.com/PankratzLab/SuperNovo.
- 16.Veltman JA, Brunner HG. De novo mutations in human genetic disease. Nat Rev Genet. 2012;13:565–575. doi: 10.1038/nrg3241. [DOI] [PubMed] [Google Scholar]
- 17.Landrum MJ, Lee JM, Benson M, et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46(D1):D1062–D1067. doi: 10.1093/nar/gkx1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cingolani P, Platts A, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bouaoun L, Sonkin D, Ardin M, et al. TP53 variations in human cancers: New lessons from the IARC TP53 Database and genomics data. Hum Mutat. 2016;37:865–876. doi: 10.1002/humu.23035. [DOI] [PubMed] [Google Scholar]
- 20.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 21.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clopper CJ, Pearson ES. The use of confidence or fiducial limits illustrated in the case of the binomial. Biometrika. 1934;26:404–413. [Google Scholar]
- 23.R Core Team: A language and environment for statistical computing. R Foundation for Statistical Computing. https://www.R-project.org. h ttps://www.r-project.org.
- 24.Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tinat J, Bougeard G, Baert-Desurmont S, et al. 2009 version of the Chompret criteria for Li Fraumeni syndrome. J Clin Oncol. 2009;27:e108–e109, author reply e110. doi: 10.1200/JCO.2009.22.7967. [DOI] [PubMed] [Google Scholar]
- 26.Bougeard G, Renaux-Petel M, Flaman JM, et al. Revisiting Li-Fraumeni syndrome from TP53 mutation carriers. J Clin Oncol. 2015;33:2345–2352. doi: 10.1200/JCO.2014.59.5728. [DOI] [PubMed] [Google Scholar]
- 27.Mai PL, Best AF, Peters JA, et al. Risks of first and subsequent cancers among TP53 mutation carriers in the National Cancer Institute Li-Fraumeni syndrome cohort. Cancer. 2016;122:3673–3681. doi: 10.1002/cncr.30248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hisada M, Garber JE, Fung CY, et al. Multiple primary cancers in families with Li-Fraumeni syndrome. J Natl Cancer Inst. 1998;90:606–611. doi: 10.1093/jnci/90.8.606. [DOI] [PubMed] [Google Scholar]
- 29.Villani A, Tabori U, Schiffman J, et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: A prospective observational study. Lancet Oncol. 2011;12:559–567. doi: 10.1016/S1470-2045(11)70119-X. [DOI] [PubMed] [Google Scholar]
- 30.Villani A, Shore A, Wasserman JD, et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: 11 Year follow-up of a prospective observational study. Lancet Oncol. 2016;17:1295–1305. doi: 10.1016/S1470-2045(16)30249-2. [DOI] [PubMed] [Google Scholar]
- 31.Kratz CP, Achatz MI, Brugières L, et al. Cancer screening recommendations for individuals with Li-Fraumeni syndrome. Clin Cancer Res. 2017;23:e38–e45. doi: 10.1158/1078-0432.CCR-17-0408. [DOI] [PubMed] [Google Scholar]
- 32.Limacher JM, Frebourg T, Natarajan-Ame S, et al. Two metachronous tumors in the radiotherapy fields of a patient with Li-Fraumeni syndrome. Int J Cancer. 2001;96:238–242. doi: 10.1002/ijc.1021. [DOI] [PubMed] [Google Scholar]
- 33.Somasundaram N, Chan SH, Quek R, et al. From Basic Research to Precision Medicine. Cambridge, MA: Academic Press/Elsevier; 2019. Advances in sarcoma genomics and therapeutic management, in Dammacco F, Silvestris F, (eds), Oncogenomics; pp. 609–621. [Google Scholar]
- 34.Li FP, Fraumeni JF, Jr, Mulvihill JJ, et al. A cancer family syndrome in twenty-four kindreds. Cancer Res. 1988;48:5358–5362. [PubMed] [Google Scholar]
- 35.Birch JM, Hartley AL, Tricker KJ, et al. Prevalence and diversity of constitutional mutations in the p53 gene among 21 Li-Fraumeni families. Cancer Res. 1994;54:1298–1304. [PubMed] [Google Scholar]
- 36.Chompret A, Abel A, Stoppa-Lyonnet D, et al. Sensitivity and predictive value of criteria for p53 germline mutation screening. J Med Genet. 2001;38:43–47. doi: 10.1136/jmg.38.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Roak BJ, Vives L, Girirajan S, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gajecka M. Unrevealed mosaicism in the next-generation sequencing era. Mol Genet Genomics. 2016;291:513–530. doi: 10.1007/s00438-015-1130-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Campbell IM, Yuan B, Robberecht C, et al. Parental somatic mosaicism is underrecognized and influences recurrence risk of genomic disorders. Am J Hum Genet. 2014;95:173–182. doi: 10.1016/j.ajhg.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]