

Abstract
Much of the focus on the transforming growth factor-β (TGFβ) superfamily in cancer has revolved around the TGFβ ligands themselves. However, it is now becoming apparent that deregulated signalling by many of the other superfamily members also has crucial roles in both the development of tumours and metastasis. Furthermore, these signalling pathways are emerging as plausible therapeutic targets. Their roles in tumorigenesis frequently reflect their function in embryonic development or in adult tissue homeostasis, and their influence extends beyond the tumours themselves, to the tumour microenvironment and more widely to complications of cancer such as cachexia and bone loss.
The transforming growth factor-β (TGFβ) superfamily comprises the TGFβs, activins, NODAL, bone morphogenetic proteins (BMPs), growth and differentiation factors (GDFs) and anti-Mullerian hormone (AMH). Over the past three decades much emphasis has been given to the role of the TGFβs themselves in cancer. In this Review, we focus on the other family members, in particular, the BMPs, activins, NODAL and GDFs (collectively referred to as BANGs). These ligands are well known for their functions in early vertebrate development and in adult tissue maintenance1–3, but it is now increasingly apparent that they also have crucial roles in both tumour development and dissemination. A recent surge of papers has revealed that many of their roles in cancer represent a redeployment of their roles in early development, or a perturbation of their roles in tissue homeostasis. In cancer, the role of some of the BANGs is to regulate the balance between the self-renewal and the differentiation of cancer stem cells (CSCs), and, in the case of NODAL, also to increase the plasticity of tumour cells. Moreover, the interplay between the BMPs and their antagonists, which is fundamental for the patterning of early embryos, determines the aggressiveness of primary tumours and the ability of disseminated tumour cells to exit from dormancy and establish metastases. After an introduction to the TGFβ superfamily and an outline of their mechanism of signalling, we briefly discuss the regulation of the BANGs and their functional roles in early vertebrate development. We then focus on recent work that demonstrates how tumour cells hijack the normally well-controlled functions of these ligands to enable the cells to grow at primary sites, and to disseminate and survive at distant sites. In the final section we discuss the therapeutic opportunities that arise from this emerging knowledge.
Signalling by TGFβ superfamily members
There are more than 30 TGFβ superfamily ligands in the human genome, which can be divided into a number of subfamilies on the basis of sequence similarity and function (TABLE 1). They are found in all metazoans and arose with multicellularity, with the activin and BMP families being the most ancient4,5. In all cases, the ligands are synthesized as precursors, with a large prodomain and a carboxy-terminal mature domain, and the mature ligands are cleaved from the precursor by proprotein convertases6,7. The ligands form dimers, which can be homomeric or heteromeric and are held together by disulphide bonds.
Table 1 |.
Ligand-receptor usage in TGFβ superfamily signalling*
| Ligand | Type I receptor | Type II receptor | Co-receptors |
|---|---|---|---|
| Inhibin-α | No type I receptor | ACTRII | ND |
| Activin-βA | ALK4 | ACTRII and ACTRIIB | ND |
| Activin-βB | ALK4 and ALK7 | ACTRII and ACTRIIB | ND |
| Activin-βE | Unknown receptor | Unknown receptor | ND |
| Activin-βC | Unknown receptor | Unknown receptor | ND |
| GDF1 | ALK4 and ALK7 | ACTRII and ACTRIIB | CRIPTO and cryptic |
| GDF3 | ALK4 and ALK7 | ACTRII and ACTRIIB | CRIPTO and cryptic |
| NODAL | ALK4 and ALK7 | ACTRII and ACTRIIB | CRIPTO and cryptic |
| BMP3 | No type I receptor | ACTRIIB | ND |
| BMP3B (also known as GDF10) | ALK4 | ACTRII | ND |
| GDF11 | ALK4 and ALK5 | ACTRII and ACTRIIB | ND |
| Myostatin (also known as GDF8) | ALK4and ALK5 | ACTRIIB | ND |
| GDF9 | ALK4 | BMPR2 | ND |
| TGFβ1 | ALK1‡ and ALK5 | TGFBR2 | β-glycan and endoglin |
| TGFβ2 | ALK1 and ALK5 | TGFBR2 | β-glycan and endoglin |
| TGFβ3 | ALK1 and ALK5 | TGFBR2 | β-glycan and endoglin |
| GDF15 | Unknown | TGFBR2 | ND |
| BMP9 | ALK1 | ACTRII and BMPR2 | ND |
| BMP10 | ALK1 | ACTRII and BMPR2 | ND |
| BMP2 | ALK3 and ALK6 | ACTRII, ACTRIIB and BMPR2 | ND |
| BMP4 | ALK3 and ALK6 | ACTRII, ACTRIIB and BMPR2 | ND |
| GDF5 (also known as BMP14) | ALK3 and ALK6 | ACTRII, ACTRIIB and BMPR2 | ND |
| GDF6 | ALK3 and ALK6 | ACTRII, ACTRIIB and BMPR2 | ND |
| GDF7 | ALK3 and ALK6 | ACTRII, ACTRIIB and BMPR2 | ND |
| BMP5 | ALK2, ALK3 and ALK6 | ACTRII, ACTRIIB and BMPR2 | ND |
| BMP6 | ALK2, ALK3 and ALK6 | ACTRII, ACTRIIB and BMPR2 | ND |
| BMP7 | ALK2, ALK3 and ALK6 | ACTRII, ACTRIIB and BMPR2 | ND |
| BMP8 | ALK2, ALK3 and ALK6 | ACTRII, ACTRIIB and BMPR2 | ND |
| BMP15 | ALK6 | BMPR2 | ND |
| AMH | ALK2 and ALK3 | AMHR2 | ND |
ACTR, activin receptor; ALK, activin receptor-like kinase; AMH, anti-Müllerian hormone; AMHR, AMH receptor; BMP, bone morphogenetic protein; BMPR, BMP receptor; GDF, growth and differentiation factor; ND, not determined; TGFβ, transforming growth factor-β; TGFBR, TGFβ receptor.
The ligands are arranged according to receptor usage. Where multiple type I, type II and co-receptors are listed for a given ligand, multiple permutations are possible.
ALK1 is endothelial specific.
The mechanism of signalling for all the ligands is fundamentally the same (FIG. 1a). Each ligand requires two types of serine/threonine kinase receptors to signal, a type I and a type II (see TABLE 1 for the known ligand–receptor interactions)8. For some ligands, additional co-receptors are required for optimal ligand binding to the type I–type II receptor complex (TABLE 1). In the activated receptor complex the constitutively active type II receptor phosphorylates the type I receptor on several serines and threonines in a highly conserved glycine- and serine-rich domain, close to the membrane-spanning region. This phosphorylation activates the type I receptor kinase and provides a binding site for the downstream substrates, the receptor-regulated SMADs (R-SMADs)9. Although the SMADs are not the only molecules that can transduce TGFβ superfamily signals to the nucleus10, they are by far the best understood, particularly for the BANGs, and thus we focus on this signalling pathway. The traditional view of TGFβ superfamily signalling is that BMPs and GDFs signal through SMAD1, SMAD5 and SMAD8, and that TGFβs, activins and NODAL signal through SMAD2 and SMAD3 (FIG. 1a). However, this view has been revised following the finding that TGFβ induces phosphorylation of SMAD1 and SMAD5 in many cell types in addition to SMAD2 and SMAD3 (REFS 11–14).
Figure 1 |. Signalling downstream of TGFβ superfamily ligands.
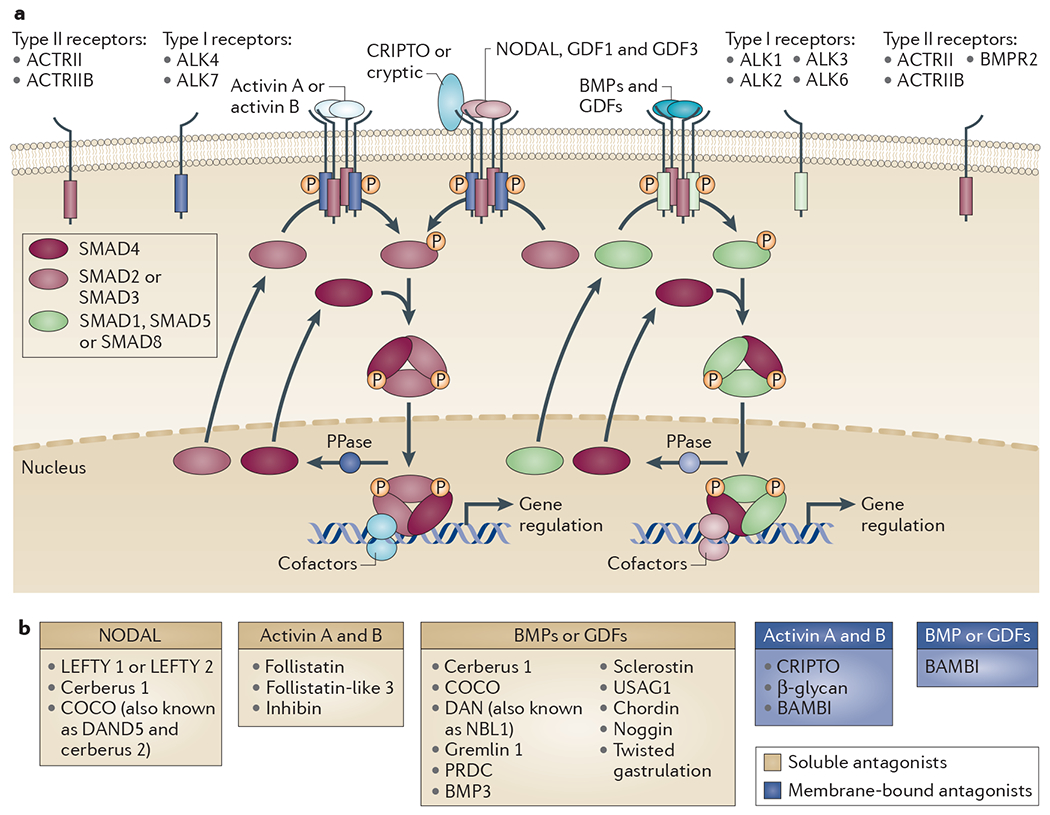
a | The core signalling pathway through the SMADs is shown for the bone morphogenetic proteins (BMPs), activins, NODAL, and growth and differentiation factors (GDFs) (BANGs). The pathways downstream of the transforming growth factor-βs (TGFβs) are not shown. Specific ligands bring together different combinations of type I and type II receptors, as indicated. In humans there are a total of seven type I receptors, known as activin receptor-like kinases (ALK1–7), and five type II receptors, with individual ligands binding different combinations (TABLE 1). Six type I receptors and three type II receptors mediate BANG signalling. The receptors all have a cysteine-rich extracellular domain, a single-pass transmembrane domain and an intracellular kinase domain. NODAL, GDF1 and GDF3 signalling also requires the co-receptors CRIPTO or cryptic, which are members of the EGF–CFC family (named after their epidermal growth factor (EGF)-like motif, and a novel cysteine-rich domain with the founding members CRIPTO, FRL1 and cryptic)21,32. The type I receptors dictate which receptor-regulated SMADs (R-SMADs) are phosphorylated (P) in response to which ligand. ALK1, ALK2, ALK3 and ALK6 phosphorylate SMAD1, SMAD5 and SMAD8, whereas ALK4, ALK5 and ALK7 phosphorylate SMAD2 and SMAD3 (REF. 9).The receptor-mediated phosphorylation of the R-SMADs occurs at their extreme carboxyl termini on two serines in an S-M-S or S-V-S motif. R-SMAD phosphorylation promotes complex formation with SMAD4 and subseguent accumulation in the nucleus. b | TGFβ superfamily ligand antagonists CRIPTO and β-glycan, which are co-receptors for NODAL, GDF1 and GDF3, and TGFβ, respectively (TABLE 1), act as inhibitors of activin signalling109. BAMBI, BMP and activin membrane-bound inhibitor; PPase, phosphatase; PRDC, protein related to DAN and cerberus (also known as gremlin 2); USAG1, uterine sensitization-associated gene 1 protein (also known as SOSTDC1).
Receptor-mediated phosphorylation allows the R-SMADs to form heteromeric complexes with another member of the SMAD family, SMAD4 (REF. 9). SMAD4 thus occupies a central position in the signalling pathways downstream of all of the ligands, being required for many, although not all, responses15,16. The R-SMADs also form homomeric complexes, and complexes with other activated R-SMADs12,17. The activated SMAD complexes accumulate in the nucleus, where they directly regulate transcription, both positively and negatively. The amino-terminal domains of SMAD4, and of all the R-SMADs except SMAD2, bind DNA directly, but have a fairly low affinity and low specificity. SMAD3 and SMAD4 recognize the sequence AGAC or its reverse complement, and SMAD1, SMAD5 and SMAD8 seem to preferentially bind GC-rich elements with the sequence GRCGNC. The SMAD complexes bind repeats of these sequences or bind in conjunction with other transcription factors18. The first identified SMAD-interacting transcription factor was FOXH1 (originally called FAST1), which recruits SMAD2–SMAD4 complexes to DNA19. SMAD complexes recruit further chromatin remodelling factors to regulate transcription18. In the nucleus, SMAD phosphatases dephosphorylate the R-SMADs, allowing their export to the cytoplasm. Although several candidates have been proposed, the identity of these phosphatases remains controversial20.
The SMAD pathways were originally thought to be unidirectional and linear, but are actually dynamic networks, as the SMADs shuttle between the cytoplasm and nucleus, in both the absence and the presence of signal (FIG. 1a). In the presence of signal this allows the SMADs to continuously monitor levels of activated receptor, meaning that the level of activated SMADs in the nucleus continuously reflects the levels of activated receptors in the cytoplasm21. TGFβ superfamily ligands frequently function as morphogens, with different doses of ligand eliciting different responses (see below). The nucleocytoplasmic shuttling behaviour of the SMADs, as well as the lack of amplification steps, makes these pathways highly suitable for interpreting morphogen gradients21.
The TGFβ superfamily–SMAD pathways are subject to numerous levels of regulation21,22. One of the most important levels of regulation for the BANGs, which is pertinent to this Review, is the interaction of the ligands with extracellular antagonists that prevent, directly or indirectly, their binding to receptors (FIG. 1b). The output of the signalling pathways thus crucially depends not only on ligand levels, but also on the levels and activities of their antagonists. Ligand levels alone are thus rarely good predictors of signalling activity. In addition, TGFβ superfamily pathways are modulated by other signalling pathways. This modulation occurs at a number of different levels. For example, the R-SMADs themselves are phosphorylated by MAPKs, glycogen synthase kinase 3β (GSK3β) and cyclin-dependent kinases (CDKs) at a number of sites in their middle linker region. Linker phosphorylation is thought to affect the stability of the SMADs, and hence the levels of activated nuclear SMADs. As a result, the activity of the SMADs is regulated by growth factors that signal through the MAPK pathways, by stimuli that regulate GSK3β activity and by the cell cycle23. TGFβ superfamily signalling responses are also modulated by other signalling pathways at the level of promoters or enhancers of target genes; a recent example being the requirement of WNT and activin signalling for the activation of the mesodermal gene mix paired-like homeobox (MIXL1) in human embryonic stem cells (HESCs)24 (see below).
Furthermore, the TGFβ superfamily pathways themselves are known to antagonize each other, and several mechanisms have been uncovered. This can occur at the level of the ligands themselves — for example, GDF3 directly inhibits BMP signalling25 — but also at the level of the SMAD complexes. Antagonism of BMP signalling by TGFβ involves inhibitory complexes formed between phosphorylated SMAD3 and SMAD1 or SMAD5 in response to combined TGFβ and BMP signalling17. In other situations, limiting amounts of SMAD4 may account for the antagonism observed between activin or NODAL and BMP or GDF signalling in early Xenopus laevis embryos26. Finally, the inhibitory SMADs, SMAD6 and SMAD7, which are upregulated in response to most TGFβ superfamily members, inhibit TGFβ superfamily signalling by recruiting E3 ubiquitin ligases to the activated type I receptors to induce their degradation27. This mechanism may explain the antagonism between NODAL and BMP signalling in mouse embryonic stem cells28.
Roles of BANGs in development
Studies predominantly in mice, fish and frogs have revealed that the BANGs have crucial roles in early vertebrate development. We discuss them briefly below to set the scene for a discussion of how these activities are redeployed and perturbed during tumorigenesis in adult animals.
The earliest role of NODAL in the mouse embryo is at the blastocyst stage, where it is responsible for maintaining the determinants of pluripotency, such as Oct4 (also known as Pou5f1) and Nanog29. After implantation, a gradient of NODAL signalling defines the proximal–distal axis, which in turn establishes the anterior–posterior axis of the embryo30. Graded NODAL signalling, in conjunction with WNT and BMP signalling, is subsequently essential for mesoderm and endoderm formation and patterning. At later stages, NODAL is required for left–right axis formation and patterning. In all these contexts, spatial NODAL activity is shaped and regulated by the activity of the antagonists LEFTY and cerberus-related proteins30,31. During these developmental processes, NODAL signalling is important not only for specifying cell fates, but also for governing cell-sorting behaviour, and inducing epithelial-to-mesenchymal transition (EMT)30. Roles for NODAL signalling in pluripotency and differentiation are also evident in HESCs, which express NODAL, GDF1 and GDF3 and the obligate receptors, including the co-receptor CRIPTO (also known as TDGF1)32. Low levels of NODAL are required for self-renewal, and higher levels promote differentiation to mesendoderm in vitro33,34, illustrating an important role for signal strength as a determinant of outcome. The mechanism underlying this has recently been illuminated by the finding that high levels of PI3K activity, which are induced by ligands such as heregulin and insulin-like growth factor 1 (IGF1), are required for activin to induce pluripotency in HESCs, whereas low levels of PI3K are required for activin to induce mesendoderm differentiation24. This probably also applies to NODAL as it shares the same receptors and downstream signalling pathway. Crucial to this mechanism is the ability of PI3K to regulate ERK and WNT signalling. High levels of PI3K suppress these pathways, and, in the absence of PI3K activity, WNT and ERK signalling are activated and activin- or NODAL-induced phosphorylated SMAD2 or SMAD3 cooperates with β-catenin-TCF transcription factor complexes to specifically induce mesendodermal genes, as well as genes required for EMT.
The BMPs also act in gradients in early embryos to establish the embryonic axes and pattern the tissues across them. Similar to NODAL, this spatial activity is moulded by the activity of secreted ligand antagonists22. A good example of this is the ventral-dorsal gradient of BMP activity that is formed before gastrulation in fish and frog embryos, which is dependent on the production of BMP antagonists such as chordin and noggin on the dorsal side22. This gradient is required to specify and pattern the mesoderm and ectoderm. At later stages of development, BMPs and the related GDF ligands are required for the formation of many different organs, such as for the regulation of limb, tooth, kidney, skin, muscle, vascular, haematopoietic and neuronal development, in conjunction with other signalling pathways such as WNT, receptor tyrosine kinases, Hedgehog and Notch35,36. Consistent with these developmental roles, BMPs and GDFs are essential in the adult for tissue homeostasis, regulating somatic stem cells and controlling differentiation, often in conjunction with the same signalling pathways with which they interact in development2 (discussed below). By contrast, NODAL is predominantly expressed in the adult in pathological contexts37.
Roles of BANGs in the tumour parenchyma
Many tumours are phenotypically heterogeneous with only a subpopulation of cells, the CSCs, capable of initiating and sustaining tumorigenesis38. CSCs may originate from the adult somatic stem cell through the perturbation of normal stem cell self-renewal processes, or from more differentiated cells by the reacquisition of stem cell-like characteristics. During tumorigenesis, the normal hierarchical organization of the tissue breaks down owing to the failure of homeostatic processes and varying degrees of differentiation blockade. Furthermore, embryonic programmes specifying stem cell expansion, cellular migration and phenotypic plasticity are often inappropriately reactivated. The central role of BANGs in regulating these processes in embryogenesis and adult homeostasis makes BANG signalling a frequent target for disruption in cancer. The literature suggests particularly important roles for BMPs and NODAL in modulating the tumour parenchyma, whereas the most well-characterized effects of the activins and GDFs target the tumour microenvironment and host organs (discussed below).
Aberrant BMP signalling disrupts stem cell self-renewal and differentiation.
In adult somatic tissues, BMPs have roles at two major levels in the cellular differentiation hierarchy (FIG. 2a). At the apex of this hierarchy, BMPs limit self-renewal of the somatic stem cells, frequently by opposing important regulators of stemness, such as the WNT pathway. Further down the hierarchy, they can specify cell fate and promote differentiation in proliferative progenitor cells. This organizational structure is well-characterized in the intestine (BOX 1), skin and brain1,2,39–42. The BMPs are often produced by specialized mesenchyme, and their activity in the stem cell niche is regulated by transient or highly localized expression of antagonists to permit controlled stem cell self-renewal for tissue maintenance and repair. This delicately balanced process can be disrupted in a number of ways in tumorigenesis (FIG. 2b).
Figure 2 |. Roles for BMPs in normal tissue homeostasis and tumorigenesis.
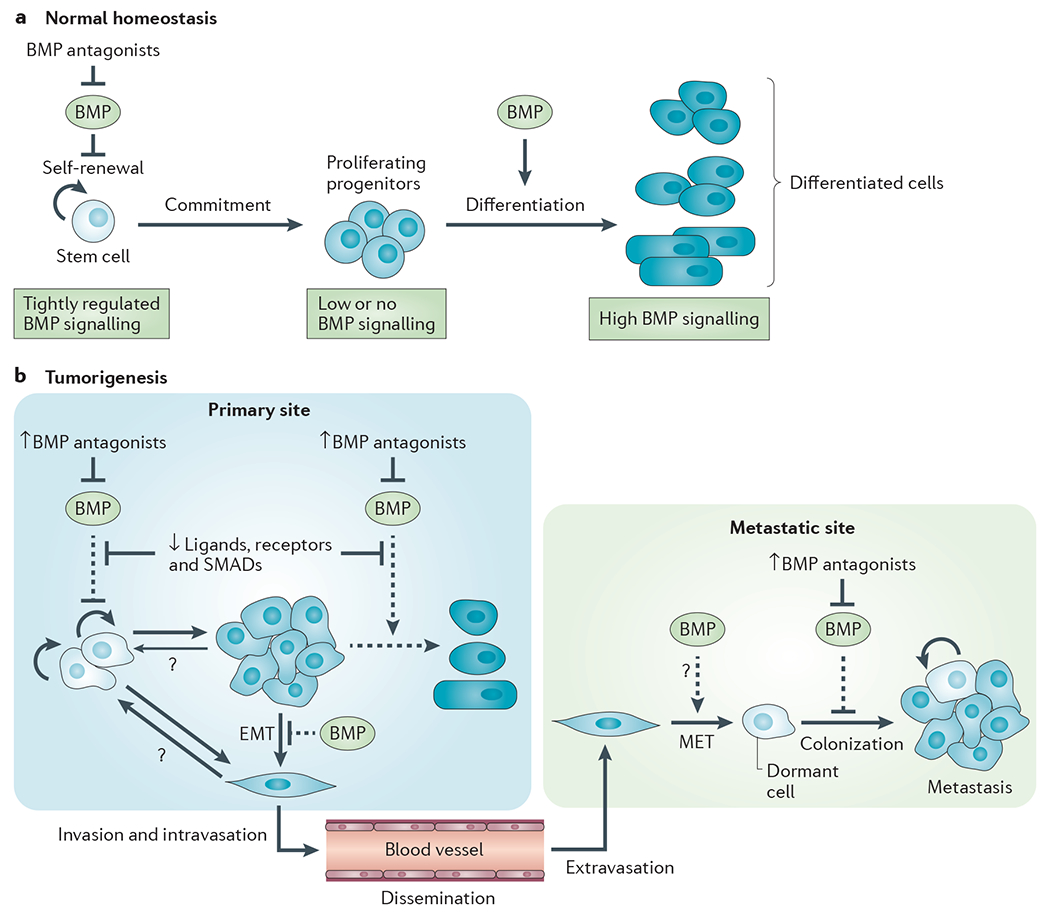
a | Simplified schematic showing roles of bone morphogenetic proteins (BMPs) in normal adult homeostasis in organs such as the intestine, brain and skin. b | Schematic for aberrant BMP signalling in epithelial tumorigenesis. At the primary tumour site, impaired BMP signalling interacts with other oncogenic lesions to promote tumorigenesis. BMP antagonists are frequently overexpressed either by the tumour cells or by the tumour-educated stroma. The BMP ligands, receptors and downstream signalling components can be disabled through genetic or epigenetic targeting, or by aberrant regulation of expression in the dysfunctional tumour environment. As a result of compromised BMP signalling, stem cell self-renewal pathways are hyperactivated, and cellular maturation and differentiation are blocked or incomplete. Furthermore, some tumour cells may respond to oncogenic cues by undergoing an epithelial-to-mesenchymal transition (EMT), leading to increased cell motility, invasiveness and an increased probability of acguiring stem cell-like characteristics. When the activity of the BMP pathway is compromised, one of the natural barriers to EMT is eliminated. Tumour cells can then leave the primary tumour site and disseminate through the circulation to distant organs. Some commonly colonized sites such as the bone and lung express naturally high levels of endogenous BMPs. The local BMPs may promote a mesenchymal-to-epithelial transition (MET) in the newly arrived tumour cell. However, they also maintain the disseminated tumour cells in a dormant state, a barrier to successful metastasis that can be overcome in tumour cells expressing high levels of BMP antagonists. Dashed arrows represent normal functions of BMPs that are compromised in tumorigenesis.
Box 1 |. BMP signalling in the colonic mucosa.
Homeostasis in the colon is regulated by opposing gradients of bone morphogenetic protein (BMP) and WNT pathway activation (see the figure; top part). As in embryogenesis, the BMP signalling gradient and localized BMP signalling domains in the colon are partly established by the balance between BMP ligands and their antagonists. BMP4 and other BMPs are expressed in mesenchymal cells of the intravillus and intercrypt regions, as well as in mesenchymal cells adjacent to the intestinal stem cell. BMP signalling is active in the intestinal stem cells in the crypt base and in the differentiating cells of the villus. There is no BMP signalling in the cells of the proliferative zone, which have very low expression of activin receptor-like kinase 3 (ALK3; also known as BMPR1A). Several BMP antagonists are expressed in subepithelial myofibroblasts at the crypt base, where they contribute to the stem cell niche and override BMP signalling in a regulated manner to permit WNT-driven stem cell self-renewal. BMP signalling in the differentiating zone of the villus is required for proper maturation of the secretory cells and apoptosis of mature colonic cells. There are also important interactions with the Notch and PI3K pathways that are not discussed here. Various lesions in the BMP pathway can contribute to colon cancer (see the figure; lower part). Germline mutations in BMP pathway components serve as initiating lesions that lead to increased stem cell expansion in hereditary polyposis syndromes that progress to carcinoma via a hamartomatous route. In sporadic colorectal cancer, the BMP pathway is typically compromised later in the disease process by somatic mutations or epigenetic silencing events, and the transition from in situ adenoma to invasive carcinoma is promoted.
BMPR, BMP receptor; GREM1, gremlin 1; IE cell, intestinal epithelial cell. Figure is modified, with permission, from REF. 42 © (2007) Proc. Natl Acad. Sci. USA.
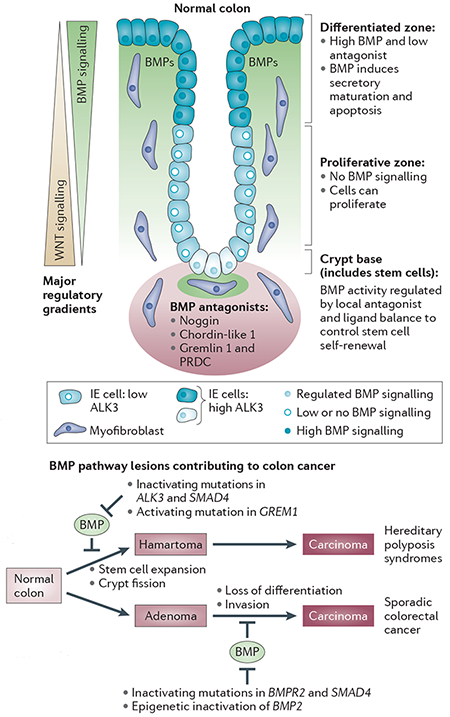
In the colon, two familial polyposis syndromes have been genetically linked to aberrant BMP signalling and altered stem cell dynamics. Juvenile polyposis syndrome (JPS) is an inherited condition in which patients develop hamartomatous polyps in the intestine, which are associated with an increased risk of adenocarcinoma. Germline mutations in the type I receptor activin receptor-like kinase 3 (ALK3; also known as BMPR1A) are seen in 20–25% of JPS cases, with a further 15–20% of cases having mutations in SMAD4 (REF 43). In the clinically-related hereditary mixed polyposis syndrome (HMPS), the causative mutation is a duplication of an upstream region of the GREM1 locus that drives ectopic overexpression of the BMP antagonist gremlin 1 throughout the intestinal epithelium and thus disrupts the tightly controlled ligand–antagonist balance44. Causality was confirmed in mouse models, in which the overexpression of another BMP antagonist, noggin, or the conditional inactivation of ALK3, drove the development of hamartomatous polyps in the intestine41,45. Polyposis was associated with stem cell expansion and crypt fission, reflecting a crucial homeostatic role for BMPs in limiting intestinal stem cell self-renewal. In a recurrent theme of BMP–WNT antagonism46, the underlying molecular mechanism involves BMP blockade of WNT signalling41.
In contrast to these familial syndromes, the evidence for BMP involvement in sporadic colorectal cancer (CRC) points to a role for the loss of BMP signalling in the late adenoma-to-carcinoma transition, rather than as an initiating event1 (BOX 1). Use of nuclear phosphorylated SMAD1, SMAD5 or SMAD8 as a marker showed that BMP signalling is inactivated in ~70% of CRCs at the late adenoma stage or beyond, suggesting that BMPs pose a substantial barrier to tumour progression at this stage1. Somatic mutations in SMAD4 or BMPR2, or epigenetic silencing of BMP2, contribute to breaching this barrier1. As BMPs induce intestinal cell maturation47 and can oppose inducers of EMT48, loss of BMP signalling in adenomas may permit the development of a migratory, invasive cell state through impaired lineage-specific differentiation and failure to maintain the epithelial phenotype.
The ability of BMPs to maintain a hierarchically organized tissue architecture also breaks down in brain cancer. In glioblastoma, this process can occur by reversion from an adult to an early embryonic pattern of BMP signalling. Early in development, BMP signalling through ALK3 promotes proliferative expansion of neural stem cells49. However, in the later maturation phase, upregulation of ALK6 (also known as BMPR1B) qualitatively alters cellular responses so that BMP signalling blocks the proliferation of neural precursors and instead drives their differentiation49. Similar to their role in late embryogenesis, BMPs are also active in the adult brain stem cell niche in the subventricular zone where they limit stem cell self-renewal and promote an astroglial fate50. This control mechanism is retained in some glioblastomas50, but CSCs from nearly 20% of human glioblastomas are rendered unresponsive to the anti-proliferative and differentiation-inducing effects of BMPs by epigenetic silencing of the ALK6 promoter51. In such cases, treatment with BMPs actually expanded the CSC population through the activation of ALK3, suggesting that the glioblastoma cells had reacquired a BMP response pattern that was characteristic of early embryogenesis.
In the skin, endogenous BMP control mechanisms may be overridden by counteracting signals from the tumour-educated stroma. Gremlin 1 is highly expressed in stromal cells of basal cell carcinomas, but not in stromal cells of normal skin, and creates a permissive niche for CSC self-renewal by opposing BMP signalling52. Stromal expression of gremlin 1 was also seen in many breast, lung, colon, pancreatic and oesophageal tumours, suggesting broader relevance52. However, it is clear that normal tissue homeostasis is maintained by a very delicate balance of BANG ligands, antagonists and receptor subtypes, and that perturbation of this balance may have different outcomes depending on the situation. Thus, although the BMPs and related BANGs may inhibit CSC self-renewal in intestinal, brain, skin, liver and breast cancers41,51–55, overproduction of BMP2 and BMP4 in response to a naturally occurring oncogenic fusion gene, CBFA2T3–GLIS2, drives the expansion of haematopoietic progenitors in acute megakaryoblastic leukaemia56, and BMP2, BMP4 and BMP6 made by cancer-associated mesenchymal stem cells may cause the expansion of ovarian CSCs57. Qualitative aspects (for example, the identity of the type I receptor) and quantitative aspects of BMP signalling, as well as the molecular context for BMP signal interpretation, can have major effects on the biological outcome and may contribute to these differences.
Reactivated NODAL signalling promotes phenotypic plasticity and stemness in advanced cancers.
NODAL is not normally expressed in adult tissues, with the exception of organs that undergo widespread remodelling, such as the placenta, endometrium and lactating mammary gland37,58. However, aggressive tumour cells, which share characteristics with embryonic progenitors in terms of self-renewal and plasticity, have been shown to both secrete and respond to NODAL. This phenomenon was first shown in melanoma cells using blastula stage zebrafish embryos as a biosensor for NODAL59. The amount of NODAL secreted by different melanoma cell lines correlated with tumour aggressiveness. These findings have subsequently been extended to prostate, breast and testicular tumours60–62. In embryonic development, NODAL requires the co-receptor CRIPTO to signal and, indeed, CRIPTO is also widely overexpressed in tumour cells from many different origins. In breast cancer, CRIPTO expression levels correlate with poor prognosis63, and in testicular tumours the amount of NODAL and CRIPTO produced was proportional to invasiveness and number of malignant cells62. The finding that CRIPTO is co-expressed with NODAL in melanoma cells, pancreatic tumour cells and breast carcinoma cells64,65, suggests that in tumour cells NODAL signals via its canonical pathway. Further parallels exist between NODAL secretion in development and in cancer as the same signalling pathway (Notch) that is required for NODAL production during the establishment of left–right asymmetry in vertebrate embryos also facilitates NODAL expression in melanoma66. In contrast to the situation during development, however, in which NODAL signalling is normally tempered by ligand antagonists such as LEFTY and cerberus-related proteins, NODAL signalling in breast carcinoma or melanoma cells seems to be unopposed, as LEFTY is not expressed in these cells65 (FIG. 3). In melanoma this is explained by methylation of the LEFTY gene, which renders it transcriptionally inactive67.
Figure 3 |. Role of NODAL signalling in HESCs and in cancer.
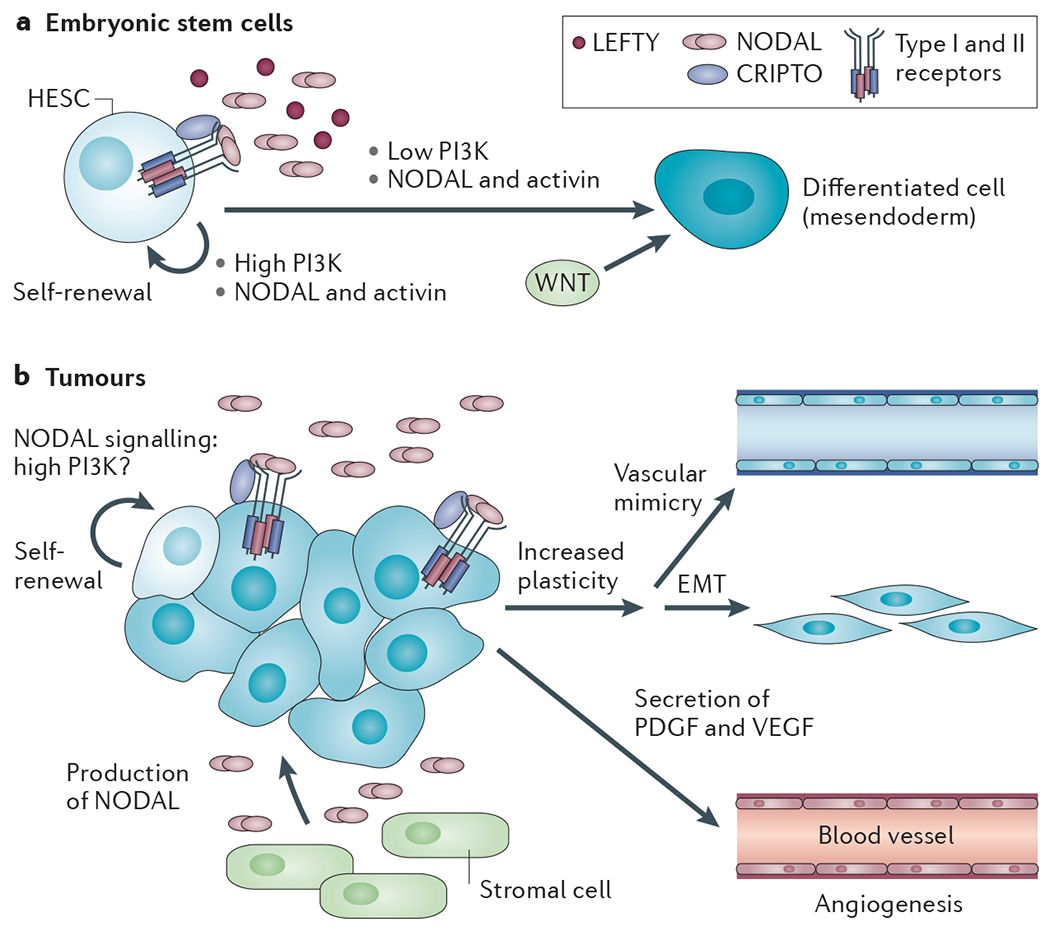
a | Human embryonic stem cells (HESCs) express NODAL (and the related ligands growth and differentiation factor 1 (GDF1) and GDF3), in addition to the relevant type I and type II receptors and CRIPTO, and are thus competent to signal. They also express antagonists such as LEFTY Activin and NODAL signalling induces self-renewal in HESCs in cooperation with high PI3K signalling, but induces differentiation to mesendoderm when PI3K signalling is low or absent, when it cooperates with WNT signalling. b | In many different types of cancer, NODAL is produced both by tumour cells and by stromal cells, such as pancreatic stellate cells. Tumour cells also express CRIPTO, but not LEFTY. NODAL signalling is important for self-renewal of cancer stem cells (CSCs) and this may be influenced by high PI3K signalling in tumours as a result of, for example, high epidermal growth factor (EGF) signalling, mutations in PTEN or mutations in PIK3CA (which encodes the PI3K p110α subunit) itself110. NODAL promotes plasticity of tumour cells — for example, inducing epithelial-to-mesenchymal transit ion (EMT) — or development of a vascular network in the case of aggressive melanoma. NODAL also promotes the secretion of the pro-angiogenic factors platelet-derived growth factor (PDGF) and vascular endothelial growth factor (VEGF), which act on endothelial cells to promote angiogenesis.
The major tumour-promoting role of NODAL signalling in melanoma seems to be to drive cells to a less differentiated, more plastic phenotype59 (FIG. 3). Many aggressive melanomas simultaneously express markers of multiple lineages (mesenchymal, epithelial and endothelial), a feature that is dependent on NODAL signalling and favours functional adaptation of the tumour to hostile growth conditions. In a particularly striking example, NODAL promotes the formation of de novo vascular networks by melanoma cells, a process that is termed vascular mimicry, which in vivo may contribute to the perfusion of rapidly growing tumours66,68. Inhibition of NODAL signalling in melanoma cells reversed this phenotype and inhibited their ability to undergo anchorage-independent growth, to invade extracellular matrix and to form tumours in mice59. In breast cancer, NODAL also potentiates tumorigenesis by promoting tumour vascularization through facilitating endothelial cell migration and tube formation69. This is not a direct effect of NODAL on the endothelial cells, but is rather due to NODAL-induced upregulation of pro-angiogenic factors such as platelet-derived growth factor (PDGF) and vascular endothelial growth factor (VEGF) in the tumour cells69.
A prominent role of NODAL signalling in the developing mouse embryo and in HESCs is to drive pluripotency and self-renewal (FIG. 3), and a similar role for NODAL has recently been demonstrated in pancreatic cancer, where NODAL is required to drive CSC self-renewal64. Pharmacological inhibition of NODAL signalling in the CSCs abolished their self-renewal capacity, and, in combination with a chemotherapeutic agent, gemcitabine, abolished tumorigenicity in a mouse model of pancreatic cancer. The source of NODAL in pancreatic tumours in vivo is not only the tumour cells themselves, but also the pancreatic stellate cells that form a niche for the CSCs, promoting their self-renewal and invasiveness70. These activities of NODAL and CRIPTO in cancer cells, coupled with the absence of NODAL and only low levels of CRIPTO in normal adult tissue, make the NODAL signalling pathway a very attractive target for tumour therapy (discussed below).
BMPs pose natural barriers to tumour progression and metastasis.
The development of metastases requires the expression of new phenotypes in the tumour cell to facilitate escape from the primary site and to permit adaptation to hostile environments. Recent work suggests that many tumour cells of epithelial origin must acquire sufficient phenotypic plasticity so that they can cycle between a proliferative epithelial state and a motile, invasive but non-proliferative mesenchymal state71–73. The plasticity that is necessary for EMT can also enhance stem cell-like properties74. BMPs can oppose inducers of EMT, including TGFβ, in many cell types48,53,75, and may thus present a natural barrier against progression to an invasive state at the primary site (FIG. 2b). Consistent with this idea, BMP signalling in sporadic CRC is frequently inactivated at the late adenoma-to-carcinoma transition when invasion occurs1, and SMAD4 loss promotes invasion and metastasis in prostate cancer76. However, BMPs can stimulate invasion in some in vitro models77.
Common sites for metastatic dissemination, such as lung and bone, are characterized by particularly high levels of BMP expression78,79, which can affect the newly arrived tumour cells in a variety of ways (FIG. 2b). As BMPs can induce mesenchymal-to-epithelial transition (MET)80, it is possible that BMPs may promote reversion to the epithelial state that is a precondition for CSC self-renewal in carcinomas that do not have a constitutive mesenchymal phenotype73. However, the high BMP levels then pose an important protective barrier to further steps in the metastatic colonization process. A screen for genes that promote the post-dissemination phase of breast cancer metastasis to the lung identified the BMP antagonist COCO (also known as DAND5 and cerberus 2) as a key player78. In the absence of COCO, solitary tumour cells in the lung showed active BMP signalling and failed to establish clinically meaningful metastases, suggesting that locally high BMP levels enforce a state of dormancy. Overexpression of COCO allowed a few dormant tumour cells to overcome this BMP barrier, and to establish metastatic outgrowths. Mechanistically, COCO selectively induced a self-renewing stem cell-like phenotype by reversing the BMP-induced repression of core stem cell transcription factors78.
As in the lung, BMPs in the bone can also prevent the colonization of disseminated tumour cells. BMP7 secreted by bone marrow stromal cells maintained the dormancy of prostate cancer cells in bone through the induction of a reversible senescent state in prostate CSCs79. Although the mechanism for escape from BMP7 was not identified, bone-seeking metastatic breast cancer cells have been shown to overexpress the BMP antagonist noggin, which enhances their tumour-initiating and metastatic activity81. Interestingly, BMP7 treatment specifically inhibited the proliferation of prostate cancer cells in the bone, but not in the prostate itself, suggesting that local microenvironmental factors may generate divergent responses at the primary and metastatic sites75. BMP7 also reduced the size of the CSC population and suppressed bone metastasis in a breast cancer model53. However, other studies have shown a stimulatory effect of BMP2 on breast cancer metastasis to the bone82, and BMP4 was implicated in a vicious cycle of prostate cancer and bone stromal cell interaction, leading to enhanced osteoblastic metastases83. Whether these discrepancies are due to differences in BMP ligands, influences of the microenvironment or genetic wiring patterns of the tumours remains to be clarified.
BANGs sculpt the tumour microenvironment
BANGs not only affect the tumour parenchyma, but they can also fuel an unhealthy dialogue between tumour cells and host cells that fosters the generation of a protumorigenic microenvironment. Tumour cells can interact with mesenchymal cells in the stroma to generate detrimental BMP and antagonist expression patterns52,57,83, and effects of BANGs on the vascular and immune components in the tumour bed are also emerging. The type I receptor ALK1 has a key role in regulating angiogenesis and vasculogenesis in early embryogenesis, mediating a complex interplay of signals from BMP9, BMP10 and the TGFβs84. ALK1 expression is low in adult vasculature, but it increases in neo-angiogenic vessels in wounds and cancer, and may represent an Achilles heel in the angiogenic process84. In preclinical models, ALK1-based ligand traps or antibodies significantly decreased the growth and angiogenesis of tumours85,86, confirming the involvement of ALK1 signalling. NODAL can also increase the tumour blood supply, both through pro-angiogenic effects on endothelial cells69 and through the promotion of vascular mimicry87 (FIG. 3).
Aberrantly activated immune cells are a prominent feature of the tumour ecosystem, resulting from subversive effects of the tumour on the immune response. TGFβs are well known as potent immunosuppressive factors, but activins are also emerging as players88. Like TGFβ, activin A can drive macrophage differentiation towards a tumour-promoting M2 phenotype, as well as skewing T cell differentiation towards T helper 2 (TH2) or regulatory T cell (TReg) fates and thereby suppressing antitumour immune responses88. Activin A is overexpressed in human skin cancer, as well as other tumours, and transgenic expression of activin A in mouse skin enhanced tumorigenesis through effects specifically on the local immune response, by increasing the number of TReg cells and suppressing cytotoxic γδT cells89. Similarly, the divergent BANG GDF15 is overexpressed by many tumours and contributes to immune escape in gliomas by suppressing the cytotoxicity of T cells and natural killer cells90. Thus, aberrant expression of BANGs that signal through SMAD2 and SMAD3 can powerfully modulate the immune response to promote tumour progression.
Roles for BANGs in complications of cancer
Many tumours show increased expression of BANGs91–94, and chemotherapy can also increase BANG production95,96. It is becoming increasingly clear that this aberrant expression of BANGs can have clinical consequences beyond the tumour and its immediate microenvironment. BANGs are important regulators of normal homeostasis in muscle, bone and the haematopoietic system, and tumour- or treatment-induced increases in BANGs of the activin or GDF subfamilies can adversely affect all these tissues. In vitro experiments had generated conflicting views of the possible roles of individual BANGs, particularly in osteogenesis and erythropoiesis95. However, use of ligand traps to probe the integrated effects of endogenous BANGs in vivo has clarified BANG involvement in these tissues in preclinical models, and has generated leads for therapeutic development (see below).
Cachexia is a wasting syndrome that affects the majority of patients with advanced cancers, and may account for up to one-third of cancer-related deaths94. Furthermore, >75% of cancer patients develop anaemia either as a direct result of their cancer, or in response to therapy95. A role for activin in cachexia was first suggested by the phenotype of mice lacking the inhibin-α chain (Inha-knockout mice), which had increased circulating activin levels and which developed gonadal tumours and cachexia97. Subsequently, myostatin (also known as GDF8; originally named for its ability to regulate muscle mass) and GDF15 were also implicated in this process98,99. In an exciting recent advance, an activin receptor IIB (ACTRIIB)-based ligand trap that binds activin and myostatin, not only prevented on-going anorexia and muscular wasting in multiple preclinical models of cancer cachexia, but also fully reversed existing skeletal and heart muscle atrophy, leading to significantly prolonged survival94. Similarly, in the context of cancer anaemia, an ACTRIIA-based ligand trap reduced the anaemia that was induced by treatment with the chemotherapeutic paclitaxel in preclinical models95, confirming an important role for an endogenous ACTRIIA ligand, probably activin A, in suppressing erythropoiesis in vivo.
Bone loss occurs with particularly high incidence in myeloma, lung, breast and prostate cancers, and causes severe pain and also increases the risk of death100. Circulating activin levels are increased in patients with breast and prostate cancer who have bone metastases, as well as in patients with advanced multiple myeloma95. ACTRIIA and ALK3 ligand traps increase bone formation and quality in normal mice, suggesting that endogenous BANG ligands of both the activin family and the BMP2 and BMP4 family negatively regulate bone formation by altering the balance of osteoblast and osteoclast activity101,102. Extending these observations to the cancer setting, an ACTRIIA ligand trap significantly reduced the osteolytic effect of multiple myeloma in a xenograft model93. Antagonism of activins released by tumour-conditioned bone marrow cells also blocks direct tumour-promoting effects93; so, BANG antagonism in this context can be of dual benefit to the patient.
Therapeutic implications and advances
From the discussions above, the reductionist view is that BANGs that signal through SMAD2 and SMAD3 (activins, NODAL and myostatin) are frequently overexpressed and have deleterious effects in cancer development, whereas BANG signalling through SMAD1, SMAD5 and SMAD8 (by BMPs) is frequently inactivated, with consequent loss of tumour suppression. Strategies are under development to reverse both types of aberration (FIG. 4). The promiscuous interrelationship between the BANGs and their receptors creates both opportunities and problems for therapeutic targeting. The highest degree of specificity is achieved by targeting a given ligand with specific antibodies, whereas targeting the receptor kinases with small-molecule antagonists has the broadest effect owing to the high structural relatedness of the ALK kinases103. Building on promising preclinical results, there are currently six BANG-targeted therapeutic agents in early phase clinical trials in cancer patients (TABLE 2).
Figure 4 |. Therapeutic approaches to targeting the BANGs in cancer.
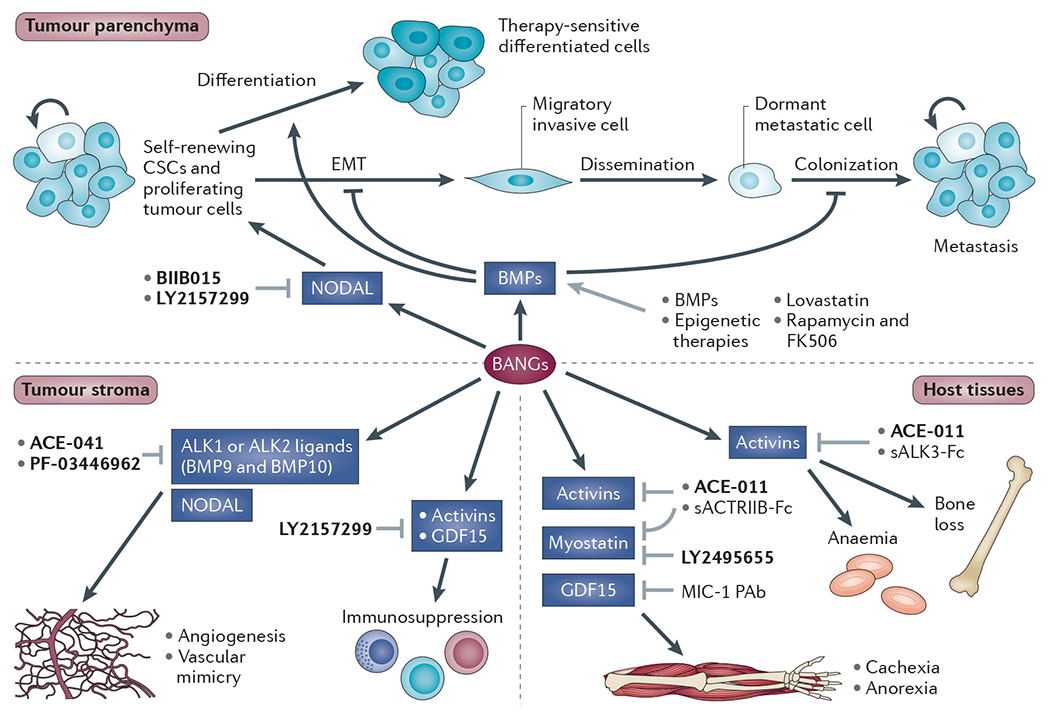
Dysregulation of bone morphogenetic proteins (BMPs), activins, NODAL, and growth and differentiation factors (GDFs) (BANGs) can have deleterious effects on the tumour parenchyma, the tumour stroma and on host tissues that are not directly involved in the tumorigenic process. Broadly, most therapeutic strategies to date have been aimed at either enhancing BMP activity or antagonizing BANGs of the activin and NODAL superfamily. For more details of therapeutic agents under clinical development (shown in bold) see TABLE 2. The other therapeutic agents shown are still at the preclinical development stage. Of these, the BMP ligands (some of which are already US Food and Drug Administration (FDA)-approved for fracture healing and lumbar fusion) have been used to induce cancer stem cell (CSC) differentiation and to restore response to chemotherapeutics50,107. Genetic knockdown of enhancer of zeste homolog 2 (EZH2), a component of the Polycomb repressor complex that is highly expressed in many tumours, restored tumour suppressive BMP responses in glioma cells, suggesting promise for epigenetic therapies that can reverse gene silencing51. Interestingly, some drugs developed in other contexts can activate BMP signalling. Lovastatin, a cholesterol-lowering agent, restored response to 5-fluorouracil by reactivating epigenetically silenced BMP2 in colorectal and gastric cancers111, and the immunosuppressive agents FK506 and rapamycin can also activate BMP signalling112,113. Soluble activin receptor-like kinase 3-Fc (sALK3-Fc) and soluble activin receptor IIB-Fc (sACTRIIB-Fc) are ligand traps that have shown therapeutic promise as BANG antagonists in preclinical studies but that have not been taken into the clinic94,101. The grey arrows indicate interventions that may affect BANG signalling in cancer and cancer-associated processes. MIC-1 PAb, polyclonal antibody to GDF15.
Table 2 |.
BANG-targeted therapeutics under clinical development for cancer treatment
| Agent (Company) | Molecular target | Agent type | Biology targeted | Stage of development | Indication or patient population | Clinical trial identifier and status |
|---|---|---|---|---|---|---|
| BIIB015 (Biogen Idec) | CRIPTO | Antibody with ‘toxic payload’: humanized CRIPTO-specific monoclonal (IgG1) conjugated to maytansinoid derivative DM4 | Tumour targeting | Phase I | Relapsed or refractory solid tumours | NCT00674947 (completed; further development discontinued) |
| LY2157299 (Eli Lilly) | ALK4, ALK5 and ALK7 | Small-molecule kinase inhibitor | Tumour targeting | Phase I | Advanced or metastatic cancer | NCT01682187 (complete) |
| Tumour targeting | Phase IB/IIA | Malignant glioma in combination with radiochemotherapy | NCT01220271 (recruiting) | |||
| Tumour targeting | Phase IB/IIA | Metastatic cancer and advanced pancreatic cancer | NCT01373164 (recruiting) | |||
| Tumour targeting | Phase II | Hepatocellular carcinoma | NCT01246986 (recruiting) | |||
| Tumour targeting | Phase II | Recurrent glioblastoma | NCT01582269 (recruiting) | |||
| PF-03446962 (Pfizer) | ALK1 | Fully human ALK1-specific monoclonal antibody (IgG2) | Angiogenesis | Phase I | Advanced solid tumours | NCT00557856 (recruiting) |
| Angiogenesis | Phase II | Malignant pleural mesothelioma and previous cytotoxic chemotherapy | NCT01486368 (recruiting) | |||
| ACE-041 (Dalantercept; Acceleron Pharma) | ALK1 ligands (BMP9 and BMP10) | Ligand trap: human ALK1-Fc fusion (IgG1) | Angiogenesis | Phase I | Advanced solid tumours or refractory multiple myeloma | NCT00996957 (ongoing but not recruiting) |
| Angiogenesis | Phase II | Recurrent or persistent endometrial cancer | NCT01642082 (recruiting) | |||
| Angiogenesis | Phase II | Ovarian, fallopian tube or primary peritoneal cancer | NCT01720173 (planned) | |||
| ACE-011 (Sotatercept; Celgene Corp/Acceleron Pharma) | ACTRIIA ligands (activins, BMP10, myostatin and GDF11) | Ligand trap: human ACTRIIA-Fc (IgG1) | Bone loss | Phase IIA | Patients with multiple myeloma who have osteolytic lesions receiving concomitant MPT | NCT00747123 (completed) |
| Tumour targeting, anaemia and bone loss | Phase I | Relapsed or refractory multiple myeloma treated with dexamethasone and lenalidomide | NCT01562405 (temporarily suspended pending amendment) | |||
| Anaemia | Phase II | Anaemia in solid tumours | NCT01190644 (recruiting) | |||
| Myelofibrosis and anaemia | Phase II | Myelofibrosis and anaemia in myeloproliferative neoplasms | NCT01712308 (planned) | |||
| LY2495655 (Eli Lilly) | Myostatin | Fully humanized myostatin-specific antibody | Cancer-related cachexia | Phase I | Advanced cancer | NCT01524224 (ongoing but not recruiting) |
| Cancer-related cachexia | Phase II | Patients with locally advanced or inoperable metastatic pancreatic cancer receiving chemotherapy (gemcitabine) | NCT01505530 (recruiting) |
ACTRIIA, activin receptor IIA; ALK, activin receptor-like kinase; BANG, BMPs, activins, NODAL, and GDFs; BMP, bone morphogenetic protein; GDF, growth and differentiation factor; igG, immunoglobulin G; MPT, melphalan, prednisolone and thalidomide.
Taking aim at the tumour cell itself, the NODAL-CRIPTO pathway makes a particularly attractive target. NODAL is absent in most normal adult tissues and CRIPTO is only expressed at low levels, but both are frequently reactivated in tumours, creating a viable therapeutic window104. BIIB015 (Biogen-Idec), a CRIPTO-specific monoclonal antibody conjugated to a maytansine toxin, was well-tolerated in a Phase I trial, but further development was discontinued owing to company reprioritization. The kinase inhibitor LY2157299 (Eli Lilly) was originally developed and taken into the clinic as an inhibitor of TGFβ signalling105. However, LY2157299 also inhibits the activity of ALK4 and ALK7 in addition to the TGFβ-specific ALK5. Thus, some of the observed efficacy may be attributable to inhibition of activin or NODAL signalling. Indeed, given the many tumour-promoting effects of activins and NODAL, the combined inhibition of TGFβ, activin and NODAL pathways has appeal as a therapeutic strategy, providing the side effects are tolerable. Early clinical results are encouraging, as the drug is well-tolerated with indications of efficacy in malignant glioma105. Targeting the tumour stroma, an ALK1-specific antibody (PF-03446962; Pfizer) and an ALK1-based ligand trap (ACE-041 (Dalantercept; Acceleron Pharma)) are now in early phase clinical trials as anti-angiogenics in a variety of human solid tumours95. ALK1 was implicated as an escape mechanism in acquired resistance to anti-VEGF therapeutics, so combined antagonism of ALK1 and VEGF may ultimately be desirable86. Finally, on the basis of compelling preclinical data showing that activins and myostatin have deleterious effects on host tissues, an ACTRIIA-based ligand trap (ACE-011 (Sotatercept; Celgene Corp/Acceleron Pharma)) and a myostatin-specific antibody (LY2495655; Eli Lilly) are in clinical trials to treat cancer-induced cachexia, anaemia and bone loss95. Early results from Phase I trials show that both agents are well-tolerated, with evidence for improvement in haemoglobin levels and markers of bone formation–destruction balance (ACE-011), and increases in muscle volume and function (LY2495655).
All the above strategies are based on antagonizing the deleterious effects of BANGs in cancer. However, in contrast to NODAL and activins, many of the actions of BMPs in the carcinogenic process are anti-tumorigenic, and therapeutic strategies are being sought that may restore or enhance these effects. Preclinical data suggest that treatment with BMPs, or strategies to boost the activity of endogenous BMPs, may be effective alone or as adjuncts to chemotherapy. CSCs are intrinsically resistant to therapy106, but treatment of xenografted brain or colon tumours with BMP2 or BMP4 increased tumour cell differentiation, thereby restoring sensitivity to chemotherapeutics50,107. Moreover, reactivation of ALK6 by knockdown of enhancer of zeste homolog 2 (EZH2) restored tumour suppressive effects of endogenous BMPs in glioblastoma cells51, suggesting potential for drugs targeting histone methylation. However, context is crucial, as high BMP4 expression was associated with the induction of EMT and resistance to cisplatin in gastric cancer108. Thus, depending on the tumour type, the specific BANG and the molecular context, it may be desirable to either enhance or block BANG activity to achieve maximum therapeutic benefit. Success in this area will depend on improved understanding of the underlying biology and the identification of good predictive biomarkers.
Conclusions
Having been overshadowed for many years by the focus on the TGFβs in human cancer, the BANGs are now emerging as key players in the tumorigenic process and as viable therapeutic targets. Deregulated BANG signalling can influence all stages of tumorigenesis from initiation, to seeding and growth of metastases. Moreover, because of their essential roles in maintaining normal tissue homeostasis, the consequences of perturbing BANG function are not restricted to the tumour cells themselves and the tumour microenvironment, but have more general effects in the body, as demonstrated by the effects of misregulated BANGs in cachexia, a common complication of cancer. General principles concerning the regulation and in vivo activity of many of the BANGs have emerged from studies in developmental systems. The discovery that the roles of the BANGs in cancer frequently represent the redeployment of their functions in embryonic development suggests that further insights may come via this route. Similarly, it is clear that BANG signals are propagated and interpreted in the context of a complex interactive network of signalling pathways, and network interactions first identified in embryogenesis are also frequently found to be at work in tumorigenesis.
Many questions are still outstanding. Our knowledge of the role of NODAL signalling in cancer is only rudimentary, and much more remains to be learnt regarding how it becomes expressed in tumours and exactly how it contributes to cancer progression. The BMP antagonists are also emerging as key players in tumorigenesis. Their crucial role in ‘reining in’ BMP activity in the context of normal tissue homeostasis is highlighted by the demonstration that increased expression of GREM1 underlies the hereditary syndrome HMPS. Clearly, as in embryogenesis, fairly small changes in flux through BANG signalling pathways can have a major effect on biological outcome. Our understanding of how these antagonists are normally so tightly regulated and what determines their specificity for different BANGs is far from complete. Moreover, some of the genetic lesions in pathway components responsible for certain tumours, such as the epigenetic silencing of ALK6 in glioblastoma, reveal our relative ignorance about how the BANGs signal. From in vitro analysis, the type I receptors ALK3 and ALK6 seem redundant, but signalling through these different receptors evidently has completely different consequences in the context of glioblastoma.
The work on BANGs is now at an exciting stage as we start to understand how these ligands contribute to cancer and await the results of clinical trials to discover whether their activity can be manipulated in patients for therapeutic benefit. Much progress is expected in the next few years.
At a glance.
The bone morphogenetic proteins (BMPs), activins, NODAL, and growth and differentiation factors (GDFs) (collectively referred to as BANGs) have essential roles in early embryonic development and in regulating tissue homeostasis in adults, frequently acting in gradients shaped by the activity of ligand antagonists. Their roles in human cancer frequently constitute a redeployment of their activities in embryonic development or a perturbation of their roles in tissue homeostasis.
Abberant BMP signalling disrupts stem cell self-renewal and differentiation, and can contribute to tumour formation. This may occur, for example, at the level of genetic or epigenetic changes resulting in the overexpression of BMP antagonists, or the loss of BMP receptors, ligands or SMAD4.
NODAL is not expressed in most normal adult tissues, but is expressed, along with the co-receptor CRIPTO, in many different tumours. It promotes phenotypic plasticity, which is important for tumour progression, and can positively regulate cancer stem cell self-renewal.
High BMP signalling provides a natural barrier to tumour progression and metastasis. In the primary tumour, BMP signalling inhibits epithelial-to-mesenchymal transition (EMT), which can prevent tumour invasion. At metastatic sites, high BMP signalling prevents tumour cell colonization by enforcing a dormant state. This can be reversed by tumour-expressed BMP antagonists.
BANGs sculpt the tumour microenvironment by promoting angiogenesis and suppressing immune responses.
Increased expression of BANGs in tumours and as a result of chemotherapy can contribute to severe complications of cancer such as cachexia, anaemia and bone loss.
Strategies are under development to target BANG signalling for therapeutic ends. These include inhibiting NODAL–CRIPTO signalling in the tumour cells, reducing tumour angiogenesis by inhibiting activin receptor-like kinase 1 (ALK1), and inhibiting activin receptors and myostatin to treat cachexia, anaemia and bone loss. Therapies that aim to increase BMP activity are also being developed.
DATABASES
CtinicalTrials.gov: http://www.clinicaltrials.gov NCT00557856 | NCT00674947 | NCT00747123 | NCT00996957 | NCT01190644 | NCT01220271 | NCT01246986 | NCT01373164 | NCT01486368 | NCT01505530 | NCT01524224 | NCT01562405 | NCT01582269 | NCT01642082 | NCT01682187 | NCT01712308 | NCT01720173
FURTHER INFORMATION
Lalage M. Wakefield’s homepage: http://ccr.cancer.gov/staff/staff.asp?profileid=13665
Caroline S. Hill’s homepage: http://www.london-research-institute.org.uk/research/caroline-hill
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
Acknowledgements
The authors thank members of the Hill and Wakefield laboratories, K. Hunter, E. Sahai, M. Sporn, S. Yuspa and Y. Zhang for useful comments on the manuscript. C.S.H. acknowledges Cancer Research UK and the European Commission Network of Excellence EpiGeneSys (HEALTH-F4-2010-257082) for funding. L.M.W. acknowledges the Intramural Research Program of the US National Institutes of Health, National Cancer Institute, Center for Cancer Research for funding.
Glossary
- Mesoderm
The middle germ layer of the developing embryo. Gives rise to the musculoskeletal, vascular and urinogenital systems, and to connective tissue (including that of the dermis).
- Endoderm
The innermost of the three germ layers of the developing embryo. It differentiates to form the linings of two tubes in the body: the digestive tube, which extends the entire length of the body and the respiratory tube. Buds from the digestive tube form the liver, gall bladder and pancreas.
- Cell-sorting behaviour
The process by which a heterogeneous population of cells with different attractive and repellent properties migrate and sort themselves into homogeneous populations.
- Epithelial-to-mesenchymal transition (EMT)
Conversion from an epithelial to a mesenchymal phenotype, which is a normal process in embryonic development. In carcinomas, this transformation results in altered cell morphology, the expression of mesenchymal proteins and increased invasiveness.
- Mesendoderm
The term given to an embryonic tissue layer that can differentiate into mesoderm and endoderm.
- Ectoderm
The outermost of the three germ layers of the developing embryo. It differentiates to form the nervous system, tooth enamel, the epidermis, hair, nails and the lining of mouth, anus, nostrils and sweat glands.
- Hamartomatous polyps
Intestinal polyps in patients with juvenile polyposis syndrome. They are characterized by increased crypt formation and cell proliferation but otherwise normal epithelial cell maturation, and are associated with an abnormally expanded mesenchymal component with a pronounced inflammatory infiltrate. Unlike intestinal adenomas, they do not show epithelial dysplasia.
- Mesenchymal-to epithelial transition (MET)
The conversion of non-polarized and motile mesenchymal cells into polarized epithelial cells.
- Ligand traps
Chimeric proteins that typically contain the ligand-binding domain of a receptor coupled to the Fc domain of an immunoglobulin. This generates an antibody-like ligand antagonist with the ligand specificity and high affinity of the parent receptor, coupled with the in vivo stability and distribution characteristics of the parent immunoglobulin.
- Osteoblast
A cell responsible for bone formation.
- Osteoclast
A cell that breaks down mineralized bone and that is responsible for bone resorption.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
References
- 1.Hardwick JC, Kodach LL, Offerhaus GJ & van den Brink GR Bone morphogenetic protein signalling in colorectal cancer. Nature Rev. Cancer 8, 806–812 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Oshimori N & Fuchs E The Harmonies played by TGF-β in stem cell biology. Cell Stem Cell 11, 751–764 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu MY & Hill CS TGF-β superfamily signaling in embryonic development and homeostasis. Dev Cell 16, 329–343 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Huminiecki L et al. Emergence, development and diversification of the TGF-β signalling pathway within the animal kingdom. BMC Evol. Biol 9, 28 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pang K, Ryan JF, Baxevanis AD & Martindale MQ Evolution of the TGF-β signaling pathway and its potential role in the ctenophore, Mnemiopsis leidyi. PLoS ONE 6, e24152 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Constam DB & Robertson EJ Regulation of bone morphogenetic protein activity by pro domains and proprotein convertases. J. Cell Biol 144, 139–149 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Constam DB & Robertson EJ SPC4/PACE4 regulates a TGFβ signaling network during axis formation. Genes Dev 14, 1146–1155 (2000). [PMC free article] [PubMed] [Google Scholar]
- 8.Mueller TD & Nickel J Promiscuity and specificity in BMP receptor activation. FEBS Lett. 586, 1846–1859 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Shi Y & Massague J Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 113, 685–700 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Kang JS, Liu C & Derynck R New regulatory mechanisms of TGF-β receptor function. Trends Cell Biol. 19, 385–394 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Goumans MJ et al. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFβ/ALK5 signaling. Mol. Cell 12, 817–828 (2003). [DOI] [PubMed] [Google Scholar]
- 12.Daly AC, Randall RA & Hill CS Transforming growth factor β-induced Smad1/5 phosphorylation in epithelial cells is mediated by novel receptor complexes and is essential for anchorage-independent growth. Mol. Cell. Biol 28, 6889–6902 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu IM et al. TGFβ-stimulated Smad1/5 phosphorylation requires the ALK5 L45 loop and mediates the pro-migratory TGFβ switch. EMBO J. 28, 88–98 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wrighton KH, Lin X, Yu PB & Feng XH Transforming growth factor β can stimulate Smad1 phosphorylation independently of bone morphogenic protein receptors. J. Biol. Chem 284, 9755–9763 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chu GC, Dunn NR, Anderson DC, Oxburgh L & Robertson EJ Differential requirements for Smad4 in TGFβ-dependent patterning of the early mouse embryo. Development 131, 3501–3512 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Levy L & Hill CS Smad4 dependency defines two classes of transforming growth factor β (TGF-β) target genes and distinguishes TGF-β-induced epithelial-mesenchymal transition from its antiproliferative and migratory responses. Mol. Cell. Biol 25, 8108–8125 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gronroos E et al. Transforming growth factor β inhibits bone morphogenetic protein-induced transcription through novel phosphorylated Smad1/5-Smad3 complexes. Mol. Cell. Biol 32, 2904–2916 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ross S & Hill CS How the Smads regulate transcription. Int. J. Biochem. Cell Biol 40, 383–408 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Chen X, Rubock MJ & Whitman M A transcriptional partner for MAD proteins in TGF-β signalling. Nature 383, 691–696 (1996). [DOI] [PubMed] [Google Scholar]
- 20.Bruce DL & Sapkota GP Phosphatases in SMAD regulation. FEBS Lett. 586, 1897–1905 (2012). [DOI] [PubMed] [Google Scholar]
- 21.Schmierer B & Hill CS TGFβ-SMAD signal transduction: molecular specificity and functional flexibility. Nature Rev. Mol. Cell Biol 8, 970–982 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Ramel MC & Hill CS Spatial regulation of BMP activity. FEBS Lett. 586, 1929–1941 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Massague J TGFβ signalling in context. Nature Rev Mol. Cell Biol 13, 616–630 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh AM et al. Signaling network crosstalk in human pluripotent cells: a Smad2/3-regulated switch that controls the balance between self-renewal and differentiation. Cell Stem Cell 10, 312–326 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper explains the underlying mechanism that dictates whether activin signalling in HESCs induces pluripotency or differentiation.
- 25.Levine AJ, Levine ZJ & Brivanlou AH GDF3 is a BMP inhibitor that can activate Nodal signaling only at very high doses. Dev Biol. 325, 43–48 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Candia AF et al. Cellular interpretation of multiple TGF-β signals: intracellular antagonism between activin/BVg1 and BMP-2/4 signaling mediated by Smads. Development 124, 4467–4480 (1997). [DOI] [PubMed] [Google Scholar]
- 27.Itoh S & ten Dijke P Negative regulation of TGF-β receptor/Smad signal transduction. Curr. Opin. Cell Biol 19, 176–184 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Galvin KE, Travis ED, Yee D, Magnuson T & Vivian JL Nodal signaling regulates the bone morphogenic protein pluripotency pathway in mouse embryonic stem cells. J. Biol. Chem 285, 19747–19756 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mesnard D, Guzman-Ayala M & Constam DB Nodal specifies embryonic visceral endoderm and sustains pluripotent cells in the epiblast before overt axial patterning. Development 133, 2497–2505 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Arnold SJ & Robertson EJ Making a commitment: cell lineage allocation and axis patterning in the early mouse embryo. Nature Rev. Mol. Cell Biol 10, 91–103 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Schier AF Nodal signaling in vertebrate development. Annu. Rev Cell Dev Biol 19, 589–621 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Bianco C et al. Role of Cripto-1 in stem cell maintenance and malignant progression. Am. J. Pathol 177, 532–540 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pera MF & Tam PP Extrinsic regulation of pluripotent stem cells. Nature 465, 713–720 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Vallier L et al. Activin/Nodal signalling maintains pluripotency by controlling Nanog expression. Development 136, 1339–1349 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miyazono K, Kamiya Y & Morikawa M Bone morphogenetic protein receptors and signal transduction. J. Biochem 147, 35–51 (2010). [DOI] [PubMed] [Google Scholar]
- 36.Wagner DO et al. BMPs: from bone to body morphogenetic proteins. Sci. Signal 3, mr1 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Strizzi L, Hardy KM, Kirschmann DA, Ahrlund-Richter L & Hendrix MJ Nodal expression and detection in cancer: experience and challenges. Cancer Res. 72, 1915–1920 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Magee JA, Piskounova E & Morrison SJ Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell 21, 283–296 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blanpain C & Fuchs E Epidermal homeostasis: a balancing act of stem cells in the skin. Nature Rev. Mol. Cell Biol 10, 207–217 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bond AM, Bhalala OG & Kessler JA The dynamic role of bone morphogenetic proteins in neural stem cell fate and maturation. Dev Neurobiol. 72, 1068–1084 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.He XC et al. BMP signaling inhibits intestinal stem cell self-renewal through suppression of Wnt-β-catenin signaling. Nature Genet. 36, 1117–1121 (2004). [DOI] [PubMed] [Google Scholar]; This paper demonstrates that conditional inactivation of ALK3 generates a mouse model of JPS and shows important antagonistic actions of BMP and WNT signalling on stem cell self-renewal.
- 42.Kosinski C et al. Gene expression patterns of human colon tops and basal crypts and BMP antagonists as intestinal stem cell niche factors. Proc. Natl Acad. Sci. USA 104, 15418–15423 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Waite KA & Eng C From developmental disorder to heritable cancer: it’s all in the BMP/TGF-β family. Nature Rev Genet. 4, 763–773 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Jaeger E et al. Hereditary mixed polyposis syndrome is caused by a 40-kb upstream duplication that leads to increased and ectopic expression of the BMP antagonist GREM1. Nature Genet. 44, 699–703 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrates that a genetic lesion leading to overexpression of the BMP antagonist gremlin 1 underlies hereditary mixed polyposis syndrome.
- 45.Haramis AP et al. De novo crypt formation and juvenile polyposis on BMP inhibition in mouse intestine. Science 303, 1684–1686 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Itasaki N & Hoppler S Crosstalk between Wnt and bone morphogenic protein signaling: a turbulent relationship. Dev. Dyn 239, 16–33 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Auclair BA, Benoit YD, Rivard N, Mishina Y & Perreault N Bone morphogenetic protein signaling is essential for terminal differentiation of the intestinal secretory cell lineage. Gastroenterology 133, 887–896 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Scheel C et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 145, 926–940 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Panchision DM & McKay RD The control of neural stem cells by morphogenic signals. Curr. Opin. Genet. Dev 12, 478–487 (2002). [DOI] [PubMed] [Google Scholar]
- 50.Piccirillo SG et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 444, 761–765 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Lee J et al. Epigenetic-mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma-initiating cells. Cancer Cell 13, 69–80 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper shows that ALK6 is epigenetically silenced in some human glioblastomas, causing reversion to an early embryonic BMP signalling pattern and the loss of inhibitory effects on the CSC population.
- 52.Sneddon JB et al. Bone morphogenetic protein antagonist gremlin 1 is widely expressed by cancer- associated stromal cells and can promote tumor cell proliferation. Proc. Natl Acad. Sci. USA 103, 14842–14847 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Buijs JT et al. The BMP2/7 heterodimer inhibits the human breast cancer stem cell subpopulation and bone metastases formation. Oncogene 31 , 2164–2174 (2012). [DOI] [PubMed] [Google Scholar]
- 54.Upadhyay G et al. Stem cell antigen-1 enhances tumorigenicity by disruption of growth differentiation factor-10 (GDF10)-dependent TGF-β signaling. Proc. Natl Acad. Sci. USA 108, 7820–7825 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang L et al. BMP4 administration induces differentiation of CD133+ hepatic cancer stem cells, blocking their contributions to hepatocellular carcinoma. Cancer Res. 72, 4276–4285 (2012). [DOI] [PubMed] [Google Scholar]
- 56.Gruber TA et al. An Inv(16)(p 13.3q24.3)-encoded CBFA2T3-GUS2 fusion protein defines an aggressive subtype of pediatric acute megakaryoblastic leukemia. Cancer Cell 22, 683–697 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McLean K et al. Human ovarian carcinoma- associated mesenchymal stem cells regulate cancer stem cells and tumorigenesis via altered BMP production. J. Clin. Invest 121, 3206–3219(2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Quail DF, Siegers GM, Jewer M & Postovit LM Nodal signalling in embryogenesis and tumourigenesis. Int. J. Biochem. Cell Biol 45, 885–898 (2013). [DOI] [PubMed] [Google Scholar]
- 59.Topczewska JM et al. Embryonic and tumorigenic pathways converge via Nodal signaling: role in melanoma aggressiveness. Nature Med. 12, 925–932 (2006). [DOI] [PubMed] [Google Scholar]; This was the first demonstration that NODAL is secreted by tumour cells, with the level of NODAL correlating with tumour aggressiveness.
- 60.Lawrence MG et al. Reactivation of embryonic nodal signaling is associated with tumor progression and promotes the growth of prostate cancer cells. Prostate 71, 1198–1209 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Strizzi L et al. Potential for the embryonic morphogen Nodal as a prognostic and predictive biomarker in breast cancer. Breast Cancer Res. 14, R75 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Spiller CM et al. Endogenous Nodal signaling regulates germ cell potency during mammalian testis development. Development 139, 4123–4132 (2012). [DOI] [PubMed] [Google Scholar]
- 63.Bianco C & Salomon DS Targeting the embryonic gene Cripto-1 in cancer and beyond. Expert Opin. Then Pat 20, 1739–1749 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lonardo E et al. Nodal/Activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell 9, 433–446 (2011). [DOI] [PubMed] [Google Scholar]; This was the first demonstration that NODAL and activin signalling promotes self-renewal of CSCs.
- 65.Postovit LM et al. Human embryonic stem cell microenvironment suppresses the tumorigenic phenotype of aggressive cancer cells. Proc. Natl Acad. Sci. USA 105, 4329–4334 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hardy KM et al. Regulation of the embryonic morphogen Nodal by Notch4 facilitates manifestation of the aggressive melanoma phenotype. Cancer Res. 70, 10340–10350 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Costa FF et al. Epigenetically reprogramming metastatic tumor cells with an embryonic microenvironment. Epigenomics 1, 387–398 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Seftoi RE et al. Tumor cell vasculogenic mimicry: from controversy to therapeutic promise. Am. J. Pathol 181, 1115–1125 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Quail DF et al. Embryonic protein nodal promotes breast cancer vascularization. Cancer Res. 72, 3851–3863 (2012). [DOI] [PubMed] [Google Scholar]
- 70.Lonardo E, Frias-Aldeguer J, Hermann PC & Heeschen C Pancreatic stellate cells form a niche for cancer stem cells and promote their self-renewal and invasiveness. Cell Cycle 11, 1282–1290 (2012). [DOI] [PubMed] [Google Scholar]
- 71.Ocana OH et al. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer prrxl. Cancer Cell 22, 709–724 (2012). [DOI] [PubMed] [Google Scholar]
- 72.Tsai JH, Donaher JL, Murphy DA, Chau S & Yang J Spatiotemporal regulation of epithelial- mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 22, 725–736 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brabletz T To differentiate or not-routes towards metastasis. Nature Rev. Cancer 12, 425–436 (2012). [DOI] [PubMed] [Google Scholar]
- 74.Polyak K & Weinberg RA Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nature Rev. Cancer 9, 265–273 (2009). [DOI] [PubMed] [Google Scholar]
- 75.Buijs JT et al. BMP7, a putative regulator of epithelial homeostasis in the human prostate, is a potent inhibitor of prostate cancer bone metastasis in vivo. Am. J. Pathol 171, 1047–1057 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ding Z et al. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature 470, 269–273 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bailey JM, Singh PK & Hollingsworth MA Cancer metastasis facilitated by developmental pathways: Sonic hedgehog, Notch, and bone morphogenic proteins. J. Cell Biochem 102, 829–839 (2007). [DOI] [PubMed] [Google Scholar]
- 78.Gao H et al. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell 150, 764–779 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper shows that tumour cells can upregulate BANG antagonists to overcome organ-specific BMP barriers to metastatic colonization.
- 79.Kobayashi A et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem- like cells in bone. J. Exp. Med 208, 2641–2655 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Samavarchi-Tehrani P et al. Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell 7, 64–77 (2010). [DOI] [PubMed] [Google Scholar]
- 81.Tarragona M et al. Identification of NOG as a specific breast cancer bone metastasis- supporting gene. J. Biol. Chem 287, 21346–21355 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Katsuno Y et al. Bone morphogenetic protein signaling enhances invasion and bone metastasis of breast cancer cells through Smad pathway. Oncogene 27, 6322–6333 (2008). [DOI] [PubMed] [Google Scholar]
- 83.Nishimori H, Ehata S, Suzuki HI, Katsuno Y & Miyazono K Prostate cancer cells and bone stromal cells mutually interact with each other through bone morphogenetic protein- mediated signals. J. Biol Chem 287, 20037–20046 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cunha SI & Pietras K ALK1 as an emerging target for antiangiogenic therapy of cancer. Blood 117, 6999–7006(2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cunha SI et al. Genetic and pharmacological targeting of activin receptor-like kinase 1 impairs tumor growth and angiogenesis. J. Exp. Med 207, 85–100 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study implicated ALK1 ligands as key players in tumour angiogenesis.
- 86.Hu-Lowe DD et al. Targeting activin receptor-like kinase 1 inhibits angiogenesis and tumorigenesis through a mechanism of action complementary to anti-VEGF therapies. Cancer Res. 71, 1362–1373 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Strizzi L et al. Nodal as a biomarker for melanoma progression and a new therapeutic target for clinical intervention. Expert Rev. Dermatol 4, 67–78 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hedger MP, Winnall WR, Phillips DJ & de Kretser DM The regulation and functions of activin and follistatin in inflammation and immunity. Vitam. Norm 85, 255–297 (2011). [DOI] [PubMed] [Google Scholar]
- 89.Antsiferova M et al. Activin enhances skin tumourigenesis and malignant progression by inducing a pro-tumourigenic immune cell response. Nature Corrirriun. 2, 576 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Roth P et al. GDF-15 contributes to proliferation and immune escape of malignant gliomas. Clin. Cancer Res 16, 3851–3859 (2010). [DOI] [PubMed] [Google Scholar]
- 91.Corre J et al. Bioactivity and prognostic significance of growth differentiation factor GDF 15 secreted by bone marrow mesenchymal stem cells in multiple myeloma. Cancer Res. 72, 1395–1406 (2012). [DOI] [PubMed] [Google Scholar]
- 92.Harada K et al. Serum immunoreactive activin A levels in normal subjects and patients with various diseases. J. Clin. Endocrinol. Metab 81, 2125–2130 (1996). [DOI] [PubMed] [Google Scholar]
- 93.Vallet S et al. Activin A promotes multiple myeloma- induced osteolysis and is a promising target for myeloma bone disease. Proc. Natl Acad. Sci. USA 107, 5124–5129 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhou X et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 142, 531–543 (2010). [DOI] [PubMed] [Google Scholar]; This important preclinical study showed that neutralization of ACTRIIB ligands leads to dramatically prolonged survival in cancer-bearing mice by reversing adverse effects of BANGs on host tissues.
- 95.Fields SZ et al. Activin receptor antagonists for cancer-related anemia and bone disease. Expert Opin. Investig. Drugs 22, 87–101 (2013). [DOI] [PubMed] [Google Scholar]
- 96.Pellagatti A et al. Lenalidomide inhibits the malignant clone and up-regulates the SPARC gene mapping to the commonly deleted region in 5q- syndrome patients. Proc. Natl Acad. Sci. USA 104, 11406–11411 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Matzuk MM et al. Development of cancer cachexia- like syndrome and adrenal tumors in inhibin-deficient mice. Proc. Natl Acad. Sci. USA 91, 8817–8821 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Johnen H et al. Tumor-induced anorexia and weight loss are mediated by the TGF-β superfamily cytokine MIC-1. Nature Med. 13, 1333–1340 (2007). [DOI] [PubMed] [Google Scholar]
- 99.Zimmers TA et al. Induction of cachexia in mice by systemically administered myostatin. Science 296, 1486–1488 (2002). [DOI] [PubMed] [Google Scholar]
- 100.Saad F et al. Pathologic fractures correlate with reduced survival in patients with malignant bone disease. Cancer 110, 1860–1867 (2007). [DOI] [PubMed] [Google Scholar]
- 101.Baud’huin M et al. A soluble bone morphogenetic protein type IA receptor increases bone mass and bone strength. Proc. Natl Acad. Sci. USA 109, 12207–12212 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pearsall RS et al. A soluble activin type IIA receptor induces bone formation and improves skeletal integrity. Proc. Natl Acad. Sci. USA 105, 7082–7087 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Vogt J, Traynor R & Sapkota GP The specificities of small molecule inhibitors of the TGFβ and BMP pathways. Cell. Signal 23, 1831–1842 (2011). [DOI] [PubMed] [Google Scholar]
- 104.Rangel MC et al. Role of Cripto-1 during epithelial-to-mesenchymal transition in development and cancer. Am. J. Pathol 180, 2188–2200 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Akhurst RJ & Hata A Targeting the TGFβ signalling pathway in disease. Nature Rev. Drug Discov 11, 790–811 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Alison MR, Lin WR, Urn SM & Nicholson LJ Cancer stem cells: in the line of fire. Cancer Treat. Rev 38, 589–598(2012). [DOI] [PubMed] [Google Scholar]
- 107.Lombardo Y et al. Bone morphogenetic protein 4 induces differentiation of colorectal cancer stem cells and increases their response to chemotherapy in mice. Gastroenterology 140, 297–309 (2011). [DOI] [PubMed] [Google Scholar]
- 108.Ivanova T et al. Integrated epigenomics identifies BMP4 as a modulator of cisplatin sensitivity in gastric cancer. Gut 62, 22–33 (2013). [DOI] [PubMed] [Google Scholar]
- 109.Antsiferova M & Werner S The bright and the dark sides of activin in wound healing and cancer. J. Cell Sci 125, 3929–3937 (2012). [DOI] [PubMed] [Google Scholar]
- 110.Weigelt B & Downward J Genomic determinants of PI3K pathway inhibitor response in Cancer. Front. Oncol 2, 109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kodach LL et al. Statins augment the chemosensitivity of colorectal cancer cells inducing epigenetic reprogramming and reducing colorectal cancer cell ‘stemness’ via the bone morphogenetic protein pathway. Gut 60, 1544–1553 (2011). [DOI] [PubMed] [Google Scholar]
- 112.Kugimiya F et al. Mechanism of osteogenic induction by FK506 via BMP/Smad pathways. Biochem. Biophys. Res. Commun 338, 872–879 (2005). [DOI] [PubMed] [Google Scholar]
- 113.van der Poel HG, Hanrahan C, Zhong H & Simons JW Rapamycin induces Smad activity in prostate cancer cell lines. Urol. Res 30, 380–386 (2003). [DOI] [PubMed] [Google Scholar]