

Abstract
Dysfunction of the gonadotropin-releasing hormone (GnRH) axis causes a range of reproductive phenotypes resulting from defects in the specification, migration and/or function of GnRH neurons. To identify additional molecular components of this system, we initiated a systematic genetic interrogation of families with isolated GnRH deficiency (IGD). Here, we report 13 families (12 autosomal dominant and one autosomal recessive) with an anosmic form of IGD (Kallmann syndrome) with loss-of-function mutations in TCF12, a locus also known to cause syndromic and non-syndromic craniosynostosis. We show that loss of tcf12 in zebrafish larvae perturbs GnRH neuronal patterning with concomitant attenuation of the orthologous expression of tcf3a/b, encoding a binding partner of TCF12, and stub1, a gene that is both mutated in other syndromic forms of IGD and maps to a TCF12 affinity network. Finally, we report that restored STUB1 mRNA rescues loss of tcf12 in vivo. Our data extend the mutational landscape of IGD, highlight the genetic links between craniofacial patterning and GnRH dysfunction and begin to assemble the functional network that regulates the development of the GnRH axis.
Introduction
Isolated gonadotropin-releasing hormone (GnRH) deficiency (IGD) is a rare genetic disorder, occurring in 1:29 000 males and 1:120 000 females (1) and is characterized by a failure of GnRH secretion or action resulting in failure of puberty, hypogonadotropism and infertility (2,3). Approximately 60% of IGD cases also exhibit anosmia, a feature that defines Kallmann syndrome (KS [MIM: 308700]) and is reflective of shared developmental defect(s) in the joint migration of GnRH and olfactory neurons (4,5). Common precursors of GnRH and olfactory sensory neurons originate in the embryonic olfactory epithelium that is extramural to the central nervous system. Subsequently, both olfactory and GnRH neurons extend their axons into the forebrain; olfactory neurons populate the olfactory system, while their GnRH counterparts continue into the mediobasal hypothalamus. There, they form a neural network that orchestrates the pulsatile release of GnRH secretion that is necessary for the physiologic function of the normal hypothalamic–pituitary–gonadal axis (6).
Over the past three decades, studies of individuals and families with KS have highlighted the extensive genetic heterogeneity of the disorder with ~20 genes identified to date (2,3). These genetic discoveries have, in turn, provided insights into the ontogeny of the genes and proteins governing GnRH neurons and have started to reveal functional modules among the IGD-causing loci based on the developmental and homeostatic roles of their encoded products. Broadly, KS genes can be stratified based on the roles of their protein products in neurogenesis, migration and specification of GnRH neurons and induction of pulsatile hormonal signal release (2). Despite this progress, however, specific genetic drivers of KS have been established in only ~40% of cases, offering an opportunity to identify additional components critical for GnRH neurogenesis and function.
Herein, we report 13 individuals from 12 KS pedigrees with autosomal dominant (inherited or de novo) heterozygous variants and one recessive pedigree with a homozygous variant, all of which are loss-of-function (LoF) mutations in TCF12 (MIM: 600480). This locus, which encodes for transcription factor 12, is a member of the basic helix-loop-helix (bHLH) subfamily of transcription factors (also called E-proteins). TCF12 haploinsufficiency causes human craniosynostosis (MIM: 615314), a rare developmental disorder characterized by premature cranial suture fusion (7). We show that TCF12 dysfunction is physiologically relevant to GnRH axis development in humans; both transient and CRISPR/Cas9 F0 mutant zebrafish models of tcf12 ablation display a decreased axonal length of the developing terminal nerve and disruption of GnRH3 neuronal patterning. Moreover, through the interrogation of a TCF12 socioaffinity network, we show that TCF12 depletion affects the transcriptional regulation of a subset of this network and TCF12-driven pathology can be attenuated upon the re-expression of one of the network members, STUB1 (MIM: 607207), a locus described previously to be mutated in syndromic GnRH deficiency (8). Together, our findings identify a critical role for TCF12 in the ontogeny of GnRH neurons, implicate KS as an allelic disorder of TCF12 haploinsufficiency, expand the observed genetic overlap between disorders of cranial and GnRH axis development and begin to dissect the relationships between genes and their protein products that affect the formation and function of the GnRH axis.
Results
TCF12 LoF mutations identified in KS cases
We performed family-based whole-exome sequencing (WES) analysis in an index family with two affected KS cousins (Fig. 1A). We filtered for shared rare sequence variants (RSVs) conforming to either autosomal recessive or dominant inheritance models. We found no variants under a recessive paradigm nor did we detect any likely pathogenic variants in the known KS or syndromic IGD genes (ANOS1 [MIM: 300836], FGF8 [MIM: 600483], FGFR1 [MIM: 136350], CHD7 [MIM: 608892], NSMF [MIM: 608137], HS6ST1 [MIM: 604846], PROK2 [MIM: 607002], PROKR2 [MIM: 607123], SEMA3A [MIM: 603961], SEMA3E [MIM: 608166], SOX10 [MIM: 602229], FEZF1 [MIM: 613301], CCDC141 [MIM: 616031], FGF17 [MIM: 603725], IL17RD [MIM: 606807], SPRY4 [MIM: 607984], DUSP6 [MIM: 602748], WDR11 [MIM: 606417], AXL [MIM: 109135], FLRT3 [MIM: 604808]). In contrast, autosomal dominant analysis yielded six heterozygous RSVs shared by the cousins (Supplementary Material, Table S1). We assessed the candidacy of these alleles using data from the exome aggregation consortium (ExAC) and genome aggregation database (gnomAD). Given the severe pubertal disruption associated with the KS phenotype and its evolutionary effect on reproductive fitness, we confined our analyses to variants in genes with a high constraint metric for either LoF (pLI >0.9) or missense mutations (Z-score >3) (9). Two RSVs were retained under these stringent criteria: [SRCIN1 (MIM: 610786; NM_025248.2) c.3235G>A, p.Glu1079Lys, and TCF12 (NM_207036.1) c.1528dup, p.Thr510Asnfs*12; Supplementary Material, Table S1]. While the SRCIN1 RSV was present in several public databases [minor allele frequency (MAF) in ExAC: 0.0005985 (72/120310 alleles), MAF in gnomAD: 0.0005217 (144/276036 alleles), the TCF12 frameshifting variant was absent in dbSNP138, the 1000 genomes browser, the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project Exome Variant Server; ExAC and gnomAD (Supplementary Material, Table S2)]. Sanger confirmation of the TCF12 RSV in all available individuals from the index family confirmed the variant in both cousins, in the apparently unaffected father of the proband and in the cousin’s anosmic mother (paternal aunt of index case, Fig. 1A). The allele was absent in the five other family members tested.
Figure 1.
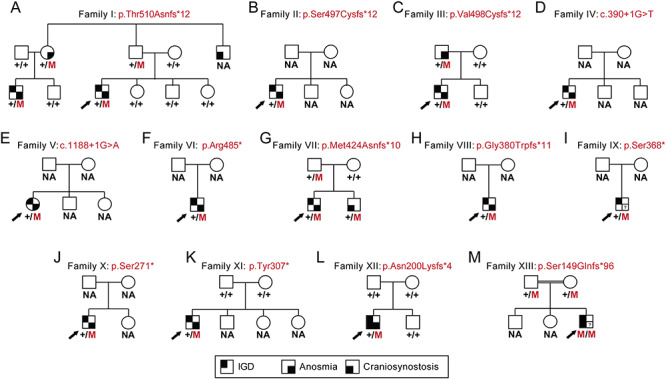
Heterozygous LoF TCF12 mutations in pedigrees with KS. (A–M). Thirteen pedigrees harboring autosomal dominant or autosomal recessive TCF12 mutations. NA, sample not available; M, mutation; ‘+’, WT; IGD, isolated GnRH deficiency; black arrow, index case; square symbol, male; round symbol, female; (?), no data.
We next interrogated WES data from 729 unrelated KS probands (performed on the same Broad Institute sequencing platform as the index family above) for additional high impact TCF12 RSVs (Supplementary Material, Table S2). We identified and Sanger confirmed additional RSVs in 10 unrelated KS individuals [four frameshifts: c.1490_1491del, p.Ser497Cysfs*12 (Family II); c.1491dup, p.Val498Cysfs*12 (Family III); c.1270dup, p.Met424Asnfs*10 (Family VII); c.1136dup, p.Gly380Trpfs*11 (Family VIII); five nonsense changes: c.1453C>T, p.Arg485* (Family VI); c.1103C>G, p.Ser368* (Family IX); c.812C>G, p.Ser271* (Family X); c.920dup, p.Tyr307* (Family XI); and two canonical splice site variants: c.390+1G>T (Family IV) and c.1188+1G>A (Family V); Fig. 1B–K, Fig. 2, Table 1 and Supplementary Material, Table S3]. Similar to Family I, all individuals were negative for mutations in all other known KS genes, harbored novel TCF12 variants that were absent from public databases of healthy controls and segregated with KS, again with incomplete penetrance (Fig. 1; Supplementary Material, Table S2), a common feature of several genetic forms of KS (2).
Figure 2.
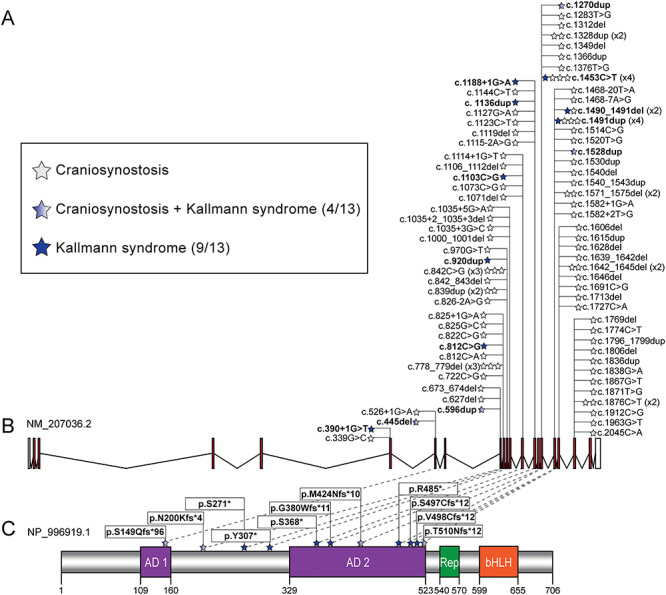
Pathogenic variation in TCF12 results in KS, craniosynostosis and blended craniosynostosis/KS phenotypes. (A) Legend depicting phenotype by case with TCF12 heterozygous LoF variants identified in our cohort. Numbers in parenthesis indicate number of pedigrees from a total of 14. (B) Schematic of the TCF12 locus (GRCh37/hg19, chr15:57210833-57580714; GenBank ID: NM_207036.2) and the allelic series reported from our KS cohort (bold font) and previously reported craniosynostosis studies (82 individuals from refs (7,10,12,14–19). Stars correspond to individuals and are colored according to the legend in panel (A). White boxes, untranslated regions; red boxes, coding exons; black lines, introns. (C) Schematic of the TCF12 protein (GenBank ID: NP_996919.1) and position of variants impacting amino acid sequence. Numbers on the bottom indicate amino acid position; purple boxes, activation domains (AD) 1 and 2; green box, repression domain (Rep); orange box, basic helix-loop-helix domain (bHLH); blue stars, variants identified in cases. Note that the two splice variants found in KS individuals are not shown in the protein schematic.
Table 1.
Summary of clinical features of KS cases harboring heterozygous TCF12 mutations. Variants are named according to GenBank ID: NM_207036.2
| Demographics | Reproductive phenotypes | Non-reproductive phenotypes | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Family | Individual | cDNA | Protein | Gender | Ethnicity | Current age (years) | Micropenis | Cryptorchidism (bilateral) | Hypospadias | Craniosynostosis | Anosmia | Dental | Neurobehavioral phenotypes | Musculoskeletal phenotypes |
| I | 1 | c.1528 dup | p.Thr510 Asnfs*12 | M | White | 18 | + | + | − | − FH | + | − | ASD | Hypermobility |
| I | 2 | c.1528 dup | p.Thr510 Asnfs*12 | M | White | 22 | + | + | − | − FH | + | Missing molars, gap between teeth | ASD | Hypermobility, kyphosis, scoliosis |
| II | 3 | c.1490_1491del | p.Ser497 Cysfs*12 | M | White | 49 | + | + | + | − | + | − | − | Osteoporosis |
| III | 4 | c.1491 dup | p.Val498 Cysfs*12 | M | White | 32 | + | + | − | − | + | − | − | − |
| IV | 5 | c.390+1 G>T | Splice variant | M | White | 61 | + | + | − | − | + | Supernu-merary teeth; gap between teeth | Dyslexia | Hypermobility, osteoporosis |
| V | 6 | c.1188+1 G>A | Splice variant | F | White | 27 | NA | NA | NA | − | + | ND | ND | ND |
| VI | 7 | c.1453 C>T | p.Arg485* | M | White | 36 | + | + | − | − | + | − | − | − |
| VII | 8 | c.1270 dup | p.Met424 Asnfs*10 | M | ND | ND | + | + | + | − FH | + | − | Synkinesis | Clinodactyly, osteopenia, familial Marfan syndrome |
| VIII | 9 | c.1136 dup | p.Gly380 Trpfs*11 | M | Hispanic | 24 | + | + | − | − | + | Gap between teeth | − | Hypermobility, camptodactyly, syndactyly |
| IX | 10 | c.1103 C>G | p.Ser368* | M | White | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| X | 11 | c.812 C>G | p.Ser271* | M | White/Japanese | 20 | + | + | − | − | + | − | Anxiety disorder | − |
| XI | 12 | c.920 dup | p.Tyr307* | M | Algerian | 20 | − | + | − | − | + | − | − | Left brachial plexus palsy |
| XII | 14 | c.596 dup | p.Asn200 Lysfs*4 | M | Hispanic | 19 | + | + | − | + Left coronal | + | − | ASD | − |
| XIII | 13 | c.445 del | p.Ser149 Glnfs*96 | M | Pakistani | 6 | + | + | − | + Bilateral coronal | ND | Small teeth | ID | − |
(+), present; (−), absent; ASD, autism spectrum disorder; F, female; FH, family history of craniosynostosis; ID, intellectual disability; M, male; NA, not applicable; ND, no data.
To test the hypothesis that heterozygous TCF12 LoF mutations impair mRNA stability, we monitored transcript levels in lymphoblastoid cell lines (LCLs) from KS individuals. We performed triplicate quantitative reverse transcription polymerase chain reaction (qRT-PCR) experiments on LCLs from Individuals 1, 4 and 5 who harbor either canonical splice site or frameshifting variants. These changes impact either a N-terminal coding region (exon 6 of 21) or sequence near the stop codon (exon 17 of 21) offering a representative sample of alleles in our cohort. We found a significant reduction of TCF12 mRNA in all KS individuals tested (35–65% decrease versus control; P < 0.05; Supplementary Material, Fig. S1A–C). We validated these results by immunoblotting whole-cell protein lysates from the same LCLs with a TCF12 antibody that detects amino acids 31–160. We found a similar significant reduction of wild-type (WT) TCF12 protein (35–60% decrease in WT protein versus control; P < 0.05; Supplementary Material, Fig. S1D and E) and detected only trace amounts of truncated protein at the expected size in two of three samples upon blot image signal enhancement (Supplementary Material, Fig. S1F). These data indicate that heterozygous LoF variants likely destabilize TCF12 mRNA, reducing WT protein below a critical threshold in a haploinsufficiency pathomechanism.
Overall, our KS cohort was enriched significantly for LoF RSVs in TCF12 compared with gnomAD (rare variant burden test using Fisher’s exact: P = 2.96E-16), supporting further that haploinsufficiency in TCF12 is a likely driver of KS. Additionally, we noted that TCF12 is mutated in autosomal dominant craniosynostosis (typically with incomplete penetrance) (7,10–19), potentially extending the allelic overlap between genes mutated in craniofacial defects and GnRH abnormalities. Prompted by this observation, we re-evaluated five TCF12-positive craniosynostosis cases from a Spanish cohort (15) for GnRH axis deficits. One of these individuals who harbored a de novo frameshift allele [c.596dup, p.Asn200Lysfs*4 (Family XII)], was also confirmed to have pubertal failure and anosmia, consistent with a clinical diagnosis of KS (Fig 1L, Fig. 2, Table 1). Additionally, using the data sharing platform GeneMatcher (20), we identified a male with KS and coronal craniosynostosis who harbors a homozygous frameshift variant [c.445del, p.Ser149Glnfs*96 (Family XIII)] inherited from healthy parents in a consanguineous pedigree (Fig 1M, Fig. 2, Table 1). As an additional step toward determining whether allelism at the TCF12 locus could differentiate GnRH versus craniofacial phenotypes, we plotted the mutational series from this study against point mutations identified in craniosynostosis to date (7,10,12,14–19). We observed that: (a) three LoF changes can cause either phenotype independently (e.g. c.1453C>T, c.1490_1491del, c.1491dup) and (b) four pedigrees in our study (31%) manifest both KS and craniosynostosis phenotypes (Fig. 2). This phenomenon of pleiotropy is not new; we have already documented similar findings for FGFR1 (21–23), SMCHD1 [MIM: 614982] (24) and CHD7 (25,26).
Clinical phenotypes
Index Family I (Fig. 1A and Table 1): two male cousins presented initially at ages 13 (index case) and 17 (cousin) with absent puberty and learning disabilities. Both had micropenis and bilateral cryptorchidism at birth, suggestive of severe in-utero/neonatal GnRH deficiency and absence of GnRH effect during the ‘minipuberty’ of the neonatal period. Both were also anosmic according to the University of Pennsylvania Smell Identification Test (UPSIT) (27). Clinical evaluation confirmed absent puberty, undervirilization, pre-pubertal testicular volume (2 ml bilaterally) and biochemical testing confirmed hypogonadotropic hypogonadism. The index case is now 18 and his cousin is 22 years old. Both have not initiated puberty and currently require hormone replacement. All other family members of pubertal/post-pubertal age reported normal pubertal history. The paternal uncle of the index case was reported to have had craniosynostosis but additional pubertal history or a DNA sample were not available.
Families II-XIII: a summary of clinical findings for the additional families (II–XIII) harboring TCF12 variants are shown in Fig. 1B-M and Table 1. All individuals were sporadic presentations of KS. While craniosynostosis was not reported in any KS probands from Families II–XI from the Massachusetts General Hospital Reproductive Endocrine Unit (MGH REU) cohort, craniosynostosis was noted in three additional pedigrees harboring TCF12 variants. (i) A male sibling of a KS proband in Family VII had craniosynostosis but not KS (Fig. 1G). (ii) A KS individual (Family XII) from a Spanish craniosynostosis cohort (15) had microphallus, bilateral cryptorchidism, anosmia and had not initiated puberty requiring current hormone replacement (Fig. 1L, Table 1). (iii) A child of first-cousin parents (Family XIII) displayed bilateral coronal craniosynostosis in addition to micropenis and bilateral cryptorchidism; his olfaction could not be assessed because of his age and intellectual disability (Fig 1M, Table 1). Notably, 11 of 12 KS males with TCF12 heterozygous LoF variants reported in this study displayed severe neonatal hypogonadism as evidenced by micropenis and bilateral cryptorchidism.
Zebrafish tcf12 LoF models display GnRH axis defects
To investigate the relevance of TCF12 haploinsufficiency to GnRH defects, we turned to Danio rerio (zebrafish), a model that both we and others have used previously to study syndromic genetic disorders affecting GnRH axis development and function, including Bosma arhinia microphthalmia (MIM: 603457) (24), KS (28), Gordon Holmes syndrome (MIM: 212840) (29), CHARGE syndrome (MIM: 214800) (25) and delayed puberty (30). Zebrafish tcf12 message is detectable in anterior structures including the olfactory placode, midbrain and hindbrain at 2 days post-fertilization (dpf) (31). These data are consistent with mouse expression studies: Tcf12 is expressed abundantly in the embryonic ectoderm and neural folds, the precursor regions that gives rise to the olfactory placodes that subsequently generate embryonic olfactory and GnRH neurons (32). Tcf12 mRNA is also highly expressed in the subventricular zone and along the rostral migratory pathway to the olfactory bulb during the first 2 postnatal weeks in mice (32). There is a single zebrafish ortholog, encoding a polypeptide 70% identical and 81% similar to the human protein, which has multiple annotated splice isoforms. Because the previously reported RNA in situ probe maps to the 3′ untranslated sequence shared by at least nine annotated zebrafish transcripts, we performed RT-PCR to determine which splice isoforms are present during early development (Supplementary Material, Fig. S2). We identified three splice isoforms: ENSDART00000161387.2, ENSDART00000131282.2 and ENSDART00000009938.10, encoding 726, 702 and 533 amino acids, respectively. Although all three isoforms share sequence in the C-terminal encoding region that encodes the bHLH domain necessary for dimerization (33,34), the two longer isoforms possess a 5′ region that differs from that of the latter transcript (Supplementary Material, Fig. S2A and B).
As a first test of the phenotypic relevance of tcf12 function in GnRH neurons, we suppressed tcf12 transiently. Given the potential complexity of the splice isoforms expressed at this locus, we injected a morpholino (MO) targeted against exon 11 (e11i11), thereby affecting all three isoforms. Immunostaining of larval batches at 2 dpf with an anti-GnRH antibody revealed a dose dependent, significant reduction in the length of the terminal nerve axons, a structure that provides the scaffold for migrating GnRH neurons towards the hypothalamus during GnRH development (35) [67% reduction at the highest dose (4 ng), P < 0.0001; Fig. 3A; Supplementary Material, Fig. S2C and F, Table S4]. Additionally, we observed a significant reduction in the size of the olfactory bulb painted by GnRH antibody (22% reduction versus control, P < 0.0001; Supplementary Material, Fig. S4, Table S4). RT-PCR confirmed efficient MO targeting (Supplementary Material, Fig. S2C). Injection of a second MO targeting only the two longest isoforms (e3i3, targeting the exon 3 splice donor), elicited a similar dose-dependent effect on terminal nerve axon length in comparison to that of e11i11 (35% reduction at the highest dose; P < 0.0001; Fig. 3A; Supplementary Material, Fig S2D, E and G, Table S4), and RT-PCR/sequencing showed an excision of exon 3 that introduces a premature termination codon. Importantly, co-injection of human WT TCF12 capped mRNA with either MO resulted in the significant restoration of the terminal nerve axon length and olfactory bulb size in 2 dpf larvae, indicating that these GnRH defects are specific. Injection of WT TCF12 mRNA alone resulted in larvae that were indistinguishable from controls (Fig. 3A and C; Supplementary Material, Fig. S4, Table S4).
Figure 3.
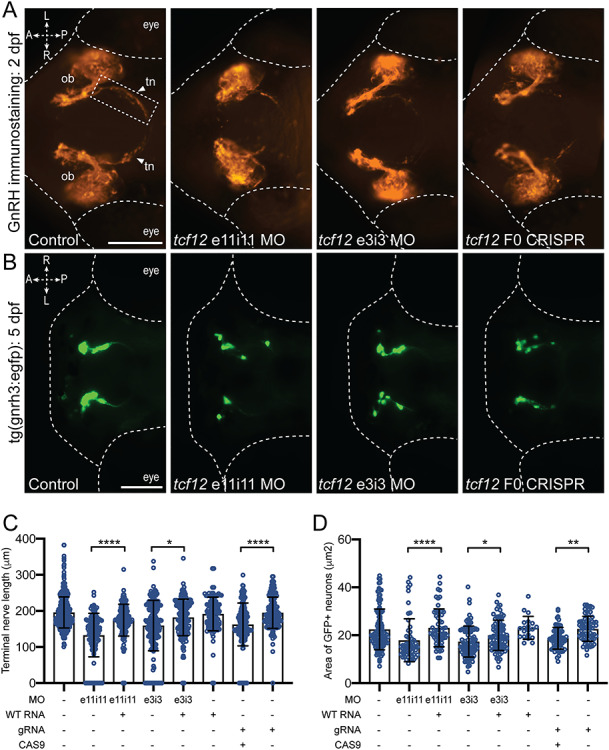
tcf12 suppression or ablation in zebrafish results in GnRH defects. (A) Representative ventral views of fluorescence immunostained (anti-GnRH antibody) tcf12 morphants (MO) or F0 CRISPR mutant embryos at 2 dpf. Arrows point to the terminal nerve axons (dashed rectangle), the length of which was measured to assess the GnRH integrity (see panel ‘C’). Abbreviations: ob, olfactory bulb; tn, terminal nerve; A, anterior; P, posterior; L, left; R, right; scale bar 100 μm, equivalent scale across panels. (B) Representative dorsal views of GFP signal in tg(gnrh3:egfp) tcf12 morphants or F0 CRISPR mutant larvae at 5 dpf; scale bar 100 μm, equivalent scale across panels. (C) Morphometric assessment of terminal nerve length in GnRH antibody stained embryos. (D) Quantitative assessment of GFP signal in gnrh3:gfp neurons was conducted by summing the area of all GFP positive regions in 5 dpf larvae. Statistically significant differences were calculated using a nonparametric Kruskal–Wallis test; (****), P < 0.0001; (**), P < 0.01; (*), P < 0.05; error bars represent standard deviation (s.d.). n = 34–67 embryos per injection batch.
Our immunostaining reagent labels multiple GnRH antigens in zebrafish (GnRH2 and GnRH3), and we wondered whether tcf12 disruption specifically alters GnRH3, the hypophysiotropic form of the zebrafish GnRH axis (36,37). To investigate this, we monitored green fluorescent protein (GFP) in gnrh3:gfp (36) tcf12 morphants using fluorescent imaging of dorsally positioned larvae at 5 dpf, an approach that has been employed previously to model IGD loci (30). Whereas control embryos displayed bilateral groups of organized GFP-positive cells, embryo batches injected with either e3i3 or e11i11 tcf12 MOs displayed GnRH3 reporter cell disorganization that included dispersed localization of individual cells and unilateral asymmetry (Fig. 3B). Quantification of the area marked by GFP, a proxy for the number of GnRH neurons, showed a significant reduction in morphants versus controls (22% reduction in area, P < 0.01 and 25% reduction in area, P < 0.0001 for e11i11 and e3i3 MOs, respectively, Fig. 3B and D; Supplementary Material, Table S4).
To validate our morphant phenotypes, we conducted CRISPR/Cas9-mediated genome editing of the tcf12 locus in the gnrh3:gfp transgenic reporter line. We identified a guide RNA (gRNA) in exon 11 that, similar to the MO placement, was predicted to induce deletions that disrupt all three tcf12 transcripts (Supplementary Material, Fig. S3A). First, we injected embryos at the one-cell stage with tcf12 gRNA and Cas9 protein and established that our reagents produce ~46% mosaicism in F0 mutants (Supplementary Material, Fig. S3B and C). Next, we employed two independent phenotypic assays of GnRH integrity on F0 larvae injected with gRNA and Cas9: (1) GnRH antibody staining on fixed whole-mount larvae at 2 dpf followed by measurement of the terminal nerve axons and olfactory bulb size and (2) quantification of GFP labeling in the anterior structures of 5 dpf larvae. We observed significant reduction in terminal nerve axon length and olfactory bulb size in comparison to controls at the early time point (17% reduction, P < 0.0001 and 18% reduction, P < 0.0001, respectively) but also a reduction in GFP area at 5 dpf (19% reduction, P < 0.0001), both of which were consistent with the pathology of tcf12 morphants (Fig. 3A–D; Supplementary Material, Fig. S4, Table S4). We note that the effect is milder in F0s than morphants for terminal nerve axon length and olfactory bulb size, observations likely explained by differences in reagent efficiency and/or mosaicism. Importantly, larval batches injected with an equivalent dose of gRNA alone resulted in no significant phenotype. Together, these data indicate that tcf12 is involved in the GnRH axis development in zebrafish and likely plays a role in patterning of GnRH3 neurons.
Functional dissection of the TCF12 interaction network
Studies in mice and other vertebrate models have shown that transcriptional regulation of several genes in an orchestrated spatiotemporal manner determines cell fate specification, proliferation, axonal guidance and migration of developing GnRH neurons (38). However, only two bona fide transcription factors (TFs) have been linked to KS in humans to date [SOX10 (39) and FEZF1 (40)], with a third TF (SOX2) mutated in syndromic IGD (41). To determine whether disruption of TCF12 alters the expression of other genes implicated in KS or IGD, we used a web-based platform for genome networks (GeNets) (42) to predict putative TCF12 interactors. This web platform integrates the InWeb protein–protein interaction network (43) and 853 curated pathways from the MSigDB database (44). First, we performed a GeNets analysis using 40 proteins encoded by genes mutated in IGD (Supplementary Material, Table S5). The resulting network output recognized previously known functional interactions between the 40 IGD genes (Supplementary Material, Fig. S5A), thus validating the GeNets approach to IGD. Reassuringly, and consistent with both our genetic and functional data, TCF12 was the only protein identified in Family I predicted to interact with known IGD proteins [SOX2 (41) and SOX10 (39); Fig. 4A; Supplementary Material, Fig. S5A]. To construct an expanded TCF12-affinity network, we then applied an integrative multi-species prediction algorithm (IMP) that integrates data from 3540 data sets spanning >70 000 experimental conditions across seven organisms (45). This step identified TCF3, a closely related bHLH TF and an established binding partner (46), as the top candidate interactor of TCF12. IMP also identified TRIM33 (MIM: 605769), a transcriptional repressor (47), as a TCF12 interactor (Supplementary Material, Fig. S6). By including TCF3 and TRIM33 along with the 40 known IGD/KS genes as the seed/bait in the GeNets platform, we identified a second-order interaction between TCF12 and two IGD genes: CHD7 (25) and STUB1 (8) (Fig. 4A; Supplementary Material, Fig. S5B).
Figure 4.
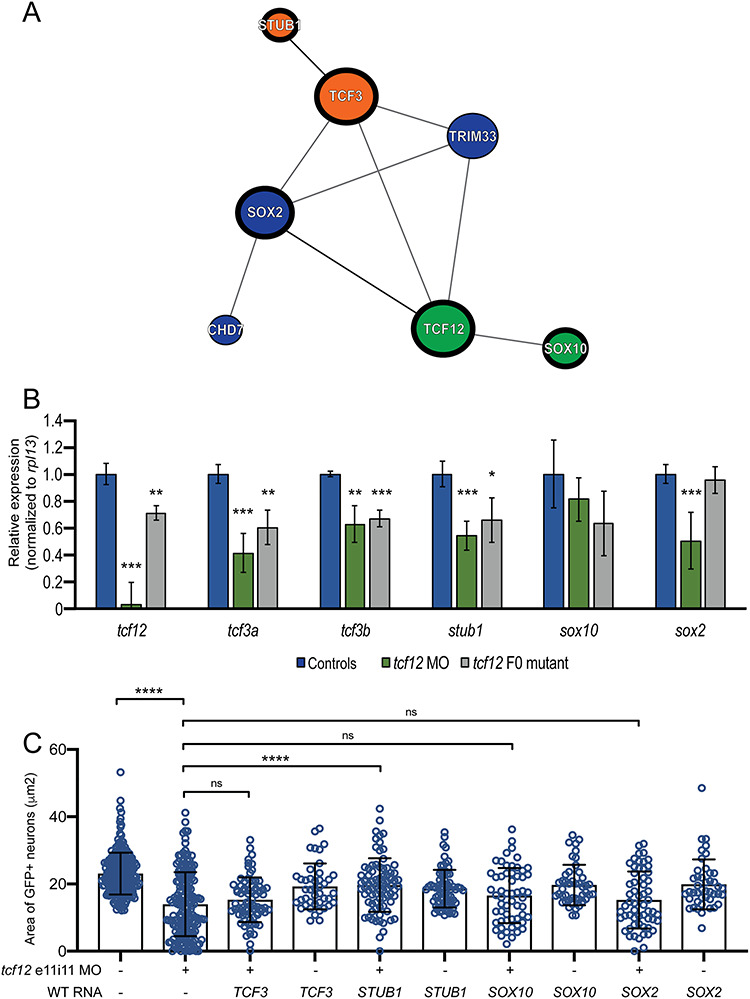
Functional dissection of the TCF12 network. (A) Schematic of the TCF12 protein–protein interaction network according to GeNets (42). Similar colored nodes indicate protein communities more likely to interact with one another than with genes in other modules. (B) qRT-PCR data generated by monitoring gene expression in cDNA derived from heads of uninjected controls, tcf12 morphants (MO) and tcf12 F0 mutants at 2 dpf. P-values are indicated as follows: * < 0.05, ** < 0.01, *** < 0.0001 (Student’s t-test) error bars represent standard deviation (s.d.). (C) In vivo complementation of tcf12 e11i11 MO with WT human mRNA. See Figure 3B for representative tcf12 morphant images; see Supplementary Material, Figure S8 for bright field images and larval length measurements; comparisons are shown versus MO alone; ns, not significant; ****P < 0.0001 (nonparametric Kruskal–Wallis test); error bars represent standard deviation (s.d.). n = 44–71 larvae/batch, repeated).
TCF12 is a member of the widely expressed class I bHLH family of TFs (E-proteins) (48), known to bind to E-box promoter elements with consensus sequence ‘CANNTG’. It has established roles in the regulation of myriad developmental processes including myogenesis, neurogenesis, pancreatic development and lymphopoiesis (32,49,50). To determine whether TCF12 regulates the expression of putative interactors in the GeNets-defined network in a context relevant to the developing GnRH axis, we monitored expression of the orthologous zebrafish loci corresponding to the three first-order interactors (TCF3, SOX10 and SOX2) and a second-order interactor (STUB1) in tcf12 zebrafish morphants and F0 mutants [SOX2 is known to regulate the expression of CHD7 (51)]. In biological replicate experiments using complementary DNA (cDNA) from 2 dpf zebrafish heads, we found that for each of tcf3a/b, stub1 and sox2 mRNA levels were reduced significantly in the tcf12 MO-injected and/or tcf12 F0 mutants compared with controls; sox10 levels were indistinguishable from controls. (Fig. 3B).
Next, we reasoned that if TCF12 regulated expression of any of these four genes directly and this regulation was relevant to the observed GnRH phenotype, then heterologous expression of the human WT transcript of the relevant gene(s) would rescue the GnRH neuronal patterning defect of tcf12 LoF zebrafish models. To test this possibility, we injected gnrh3:egfp embryo batches with 2 ng tcf12 e11i11 MO with or without 12.5 pg mRNA of each of TCF3, SOX2, SOX10 and STUB1. We then allowed larvae to develop to 5 dpf and conducted automated live imaging of GFP signal coupled to a newly developed automated image quantification approach that removes any possibility of investigator bias (see Methods). We first confirmed that our image processing paradigm was highly correlated to manual area measurement using ImageJ (Spearman r = 0.7464; P < 0.0001; Supplementary Material, Fig. S7). We compared gnrh3:gfp fluorescent signal area between tcf12 morphants versus each trans-rescue batch; expression of only one of our four tested genes, STUB1, was sufficient to rescue tcf12-induced GnRH neuronal patterning defects significantly (n = 44–71 embryos/batch, replicated; P < 0.0001 versus MO; Fig. 3C; Supplementary Material, Table S4). Importantly, we observed no major morphological defects in tcf12 e11i11 MO, single mRNA or MO plus mRNA injected batches as indicated by larval length measurements performed on bright field images at 5 dpf (Supplementary Material, Fig. S8). Taken together, our network predictions and functional validation experiments demonstrate that TCF12 is likely critical, either directly or indirectly, for the regulated expression of multiple genes involved in the establishment of the GnRH axis.
Discussion
In this study of 729 pedigrees, we report 14 affected individuals in 13 families with different LoF mutations in TCF12 (12 autosomal dominant and one autosomal recessive), accounting for a minor proportion of the genetic etiology of KS (~1.5%). All individuals presented with a severe form of KS with the majority following an autosomal dominant mode of inheritance likely due to haploinsufficiency of TCF12. The 11 of 12 males in our cohort displayed neonatal hypogonadism, and all adult males have a history of subsequent failure to initiate puberty. These observations emphasize a critical role for TCF12 in both early embryonic and pubertal regulation of GnRH neuronal integrity. We note that our cohort displayed two recurrent comorbidities: hypospadias and autism spectrum disorder (ASD) (2 of 13 males and 3 of 14 KS individuals, respectively, Table 1). Notably, neurodevelopmental phenotypes such as ASD, intellectual disability, developmental delay or learning disability have been reported previously in individuals with pathogenic TCF12 variants, as either a comorbid feature of craniosynostosis (7,11,13,15,17) or as the likely cause of a non-syndromic neurodevelopmental disorder (52,53). In contrast to our knowledge, TCF12 variants have not been described in genetic studies of hypospadias (54,55) and is a rarely documented phenotype in TCF12-related craniosynostosis (19). Although micropenis is a hallmark of KS males, hypospadias is a birth defect of the urethral meatus opening that is not typically associated with GnRH-deficient states such as KS. Our observations suggest additional GnRH-independent roles for TCF12 in androgen biosynthesis or action during early fetal life that determines the development of male external genitalia and thus, evaluation of this locus in hypospadias cohorts would offer greater insight into its possible role in the manifestation of this clinical feature.
In addition to KS, mutations in TCF12 are also linked to craniosynostosis (7,10–19), with no clear phenotype–genotype correlation to differentiate molecularly between the two traits. In both disorders, the proposed mode of action is haploinsufficiency. Here, we show qRT-PCR and immunoblotting studies on LCLs from a subset of cases in our KS cohort and demonstrate that mRNA clearance likely takes place by nonsense-mediated decay. Further, the discovery of individuals with either dominant de novo or inherited recessive frameshifting TCF12 alleles, with both GnRH axis defects and craniosynostosis argues that the differentiator between the two pathologies is not diversity in mutational mechanisms. Additionally, three TCF12 variants identified in our KS cohort [c.1490_1491del, p.Ser497Cysfs*12 (Family II); c.1491dup, p.Val498Cysfs*12 (Family III]; and c.1453C > T, p.Arg485* (Family VI)] have been reported previously in individuals with craniosynostosis and no apparent GnRH phenotypes (7,14). We speculate that cis or trans acting factors and differential mutant versus WT allelic expression during organogenesis at critical developmental windows likely underscore this phenotypic spectrum (56).
It is notable that TCF12 is not the first protein to be associated with both craniofacial abnormalities and GnRH axis defects. In an emerging theme, several syndromic IGD genes (FGFR1, SOX10 and SMCHD1) are known to cause two or more apparently distinct Mendelian disorders. Although the mutational mechanism for FGFR1 is different in craniosynostosis (gain-of-function) (57) and KS (LoF) (21,23), SOX10 and SMCHD1 share mutational mechanisms and directions of variant effect across disorders (24,58). From a mechanistic perspective, it is likely that the observed allelism is imparted by the multiple roles of these proteins in discrete developmental processes, specified in part by their binding partners. For example, a heterodimeric partnership of TCF12 with TWIST1 (MIM: 601622), a class II bHLH TF, and reduced dosage of TCF12-TWIST1 heterodimers has been proposed to underlie the TCF12-related craniosynostosis (7,15). This hypothesis is supported by a recently reported zebrafish model of Saethre–Chotzen syndrome (MIM: 101400) in which neither individual twist1b−/− nor tcf12−/− zebrafish models displayed gross morphological defects as embryos or adults, but nearly half of double mutants had bilateral coronal synostosis (59). We predict that tcf12−/− zebrafish models will not display a GnRH phenotype unless sensitized in a similar paradigm; we propose that either a first-order interactor (Fig. 4A) or another class II bHLH partner might be reasonable candidates to test in future studies. In the context of GnRH specification, TCF12 and related E-proteins can heterodimerize with class II bHLH proneural TFs (e.g. ASCL1, NGN1, NGN2, NEUROD1, NEUROD2) (49,60), initiating their transcriptional activity that is essential for neuronal stem cell differentiation and, in particular, olfactory neurogenesis (61–64). Notably, mice deficient in ASCL1 exhibit olfactory bulb hypoplasia (62), a defect reminiscent of KS. Moreover, ASCL1 also regulates expression of PROK2, mutations in which also cause KS (65).
Consistent with the TCF12 mutational findings in our KS cohort, transient suppression and F0 mutant zebrafish models showed a critical role for TCF12 in the emanation of GnRH neurons. Additionally, initial network analysis showed TCF12 to regulate the expression of STUB1, a gene implicated in GnRH axis establishment. However, it is important to note that our data do not exclude the possibility that TCF12 interacts with TCF3, SOX2 or SOX10 in a GnRH-relevant context through cellular mechanisms other than transcriptional regulation. TCF3 is a known physical interactor of TCF12 (46,66), and we were intrigued to find a reduction of tcf3a/tcf3b in zebrafish models of tcf12 depletion. This is in contrast to a compensatory increase in Tcf3 expression in the cerebellum of P0 Tcf12−/− mice (50), suggesting either lack of compensatory redundancy in teleosts or a discrete spatiotemporal effect that could not be captured in our assay. Further, it is possible that overexpression of multiple interactors (e.g. SOX2 and SOX10) are required to elicit phenotype rescue in the context of tcf12 reduction. Finally, we recognize that our study was limited to the network that emerged from GeNets analysis of IGD genes and did not test for GnRH neuron-specific effects of other known TCF12 interactors such as class II bHLH family members (67).
It remains unclear whether STUB1 is a direct or indirect target of TCF12. STUB1 was originally characterized as a co-chaperone regulator (68) but can also display E3 and E4 ubiquitin ligase activity implicated in the degradation of misfolded proteins (69,70); it is formally possible that tcf12 phenotype amelioration is due to increased clearance of truncated TCF12 protein induced in our zebrafish models. However, we cannot exclude the possibility that TCF12 potentially lies upstream of STUB1 and regulates its expression directly. Post hoc scanning for regulatory motifs with MotifMap (71) in each of the four genes assessed by qRT-PCR in our tcf12 models identified a predicted TCF12 binding site in a conserved region 782 bp upstream from the human STUB1 transcriptional start site. The other three transcripts had no such features. However, caution is warranted in assigning predictive value to in silico-derived TCF12 binding sites and their potential enrichment as regulatory elements of IGD genes. It will be important to test these predicted regulatory elements biochemically, since the assembly of TF-driven networks will likely inform both our understanding of GnRH axis neurodevelopment, its functional integrity and of the overall genetic architecture of IGD.
In conclusion, through a multiplex family-based WES analysis and additional validation in a large KS cohort, this report validates that rare heterozygous LoF variants in TCF12, a constrained gene for functional LoF mutations, cause two developmental disorders: KS and craniosynostosis. Both the location of mutations and the nature of genetic mutations between these two disorders are not distinct, and pedigrees harboring these mutations display incomplete penetrance. These observations are confirmed by zebrafish functional studies and together define a previously unknown but critical role for the bHLH TFs in GnRH neuronal development. These observations also highlight an emerging class of pleiotropic genes such as FGFR1 (23), SMCHD1 (24), CHD7 (25) and now TCF12 that cause both IGD and distinct craniofacial abnormalities.
Materials and Methods
Human cohorts
KS was defined by: (a) absent or incomplete puberty with evidence of anosmia by UPSIT (27) or by self-report (72), (b) serum testosterone level <100 ng/dL in men or estradiol <20 pg/ml in women in the presence of low or normal levels of serum gonadotropins, (c) otherwise normal anterior pituitary function, (d) normal serum ferritin concentrations and (e) normal magnetic resonance imaging of the hypothalamic–pituitary region. Initially, the index family with two affected cousins with KS referred to the MGH REU from the UK was studied by WES. Putative causal genetic variant(s) were then queried in WES data available from a total of 729 KS subjects participating in the MGH REU genetics study. Phenotypic information on reproductive and non-reproductive features were obtained from medical charts, questionnaires and referring provider’s summaries. In addition, authors reporting prior TCF12 variants in craniosynostosis were contacted (7,15) for potential phenotypic overlap of KS, and the data sharing platform, GeneMatcher (20) was used to identify additional KS cases. All human subjects research was approved by the Massachusetts General Hospital Partners Institutional Review Board. Subjects participated in this study following informed consent.
Whole-exome sequencing
WES was performed in 729 KS individuals including the index northern European family with two male cousins affected with KS (Family I) and Families II–XI at the Broad Institute Genomics Platform as described (24). Briefly, variant calling was performed with genome analysis toolkit (GATK), creating genomic variant call file (gVCF) files for every sample that were then joint called using the GATK genotype gVCF module. Variant quality scores were derived using GATK best practices for indels and single nucleotide variants. The jointly called VCF files were analyzed using the integrative database framework, GEMINI v.0.19.1 (73). Variant annotation was done with the Ensembl variant effect predictor (VEP) against the GRC37/hg19 reference human genome (74). RSVs for both bi-allelic as well as heterozygous variants were required to be either: non-synonymous variants (stop, frameshift or missense) or canonical splice altering variants (± 2 bp from exon–intron boundaries), have a MAF of < 0.01 in the gnomAD browser, a quality depth >5, to be of MEDIUM or HIGH impact as annotated by VEP and predicted as damaging by either PolyPhen-2 or sorting intolerant from tolerant (SIFT). Family-based analysis for Family I was performed for autosomal recessive (bi-allelic mutations) and autosomal dominant (heterozygous) variants. Variants in candidate genes from Family I were then queried in the GEMINI database for ascertaining additional discovery variants in the rest of the KS cohort. Family XII had molecular sequencing analysis for TCF12 as described (15). Trio WES was performed in Family XIII using the SureSelectXT Human All Exon V5 kit and HiSeq sequencing platform (Illumina).
Sanger sequencing
All variants of interest were confirmed using bidirectional Sanger sequencing in all probands and their segregation was established in all family members available. Sanger sequencing was performed by the Center for Computational and Integrative Biology DNA Core Facility at Massachusetts General Hospital (Cambridge, MA).
Quantitative RT-PCR (qRT-PCR) analysis of TCF12 in human cell lines
We extracted total RNA from LCLs derived from KS cases, unaffected family members or unrelated ethnically matched controls using the RNAeasy Mini Kit (Qiagen, Hilden, Germany) and generated cDNA with SuperScript III First-Strand Synthesis System (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) according to manufacturer’s instructions. qRT-PCR with SYBR Green (Takara Bio, Inc., Otsu, Japan) was performed in the StepOne™ Real-Time PCR System (Applied Biosystems Life Technologies) according to the manufacturer’s protocols. We designed primers using Primer 6.0 to test TCF12 levels in the individual with variants in exon 6 (Ex5-F: 5′- GTCGATTAGGAGCCCATGAAG-3′ and Ex7-R: 5′-GGTCTCCTCCTGGAACTTGTA-3′) or exon 17 (Ex16F: 5′-TGCTGGTCACAGTGATATACATAG-3′ and Ex18R: 5′- TTCATCATCTGACTTCATGTCATC-3′). We amplified GAPDH as an endogenous control (F: 5′- GAAGGTGAAGGTCGGAGT-3′ and R: 5′- GAAGATGGTGATGGGATTTC-3′). Each experiment was performed in technical triplicates and repeated at least three times using the following cycling conditions: 10 min at 95°C, 40 cycles at 95°C for 15 s and 60°C for 30 s. Relative mRNA quantity was calculated using the 2-ΔΔCt method and statistical differences were calculated with a Student’s t-test.
TCF12 immunoblotting
We harvested LCLs in radioimmunoprecipitation assay (RIPA) lysis buffer containing 50 mm N-2-hydroxyethylpiperazine-N-ethanesulfonic acid (pH 7.6), 1% Triton X-100, 0.1% sodium dodecyl sulfate (SDS), 50 mm NaCl, 0.5% sodium deoxycholate, 1 mm phenylmethylsulfonyl fluoride (Millipore Sigma), 1× Halt phosphatase inhibitor cocktail (Thermo Scientific) and 1× complete protease inhibitor cocktail (Millipore Sigma). We quantified protein using bicinchoninic acid protein assay kit (Thermo Scientific) according to the manufacturer’s protocol. We first heat denatured 50 μg of protein for each sample in laemmli sample buffer and reducing agent (NuPAGE™, Cat# NP0004; 1X) and migrated the denatured protein on a 4–15% gel using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The bands were then transferred to a polyvinylidene fluoride (PVDF) membrane and incubated overnight in antibody solution (5% milk in PBST) containing mouse anti-TCF12 antibody (Santa Cruz Biotechnology, Cat# sc-28364; 1:600 dilution) and mouse anti-GAPDH antibody (Santa Cruz Biotechnology, Cat# sc-47724; 1:3000 dilution). The PVDF membrane was then incubated in secondary antibody solution (5% milk in PBST; m-IgGκ BP-HRP conjugate, sc-516102; 1:4000 dilution). To detect signal, we used SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Scientific) according to the manufacturer’s protocol and Chemiluminescent bands were visualized using a ChemiDoc XRS imaging system (Bio-Rad). Signal was quantified with Image Studio Lite, and statistical differences were calculated using a Student’s t-test.
Characterization of D. rerio tcf12 splice isoforms
All studies performed with zebrafish were approved by the Duke University Institutional Animal Care and Use Committee. We harvested total RNA from whole zebrafish embryos (n = 20) at 1 dpf obtained from natural matings of WT (AB) adults using Trizol (ThermoFisher) as described (75). Resulting cDNA was generated using oligo-dT priming and SuperScript III-mediated reverse transcription (RT; ThermoFisher). We amplified tcf12 using primer pairs RT-long-F: 5′-GACGCGAGACAAGGAAATCTAC-3′ and RT-long-R: 5′-ACTCTGTCTAACACACGCCATC-3′ or RT-short-F: 5′-TTTATCATGTATTGCGCCTACC-3′ and RT-short-R: 5′-CAAGGTCACCGACTCTGTCTAA-3′, cloned resulting PCR products into the TOPO-TA vector (ThermoFisher), Sanger sequenced resulting clones and aligned them to reference with Sequencher (Gene Codes). Unless indicated otherwise, exons are named according to the isoform with the longest open reading frame (Ensembl ID: ENSDART00000161387.1).
Transient tcf12 suppression and rescue experiments in zebrafish larvae
We designed splice-blocking MO antisense oligonucleotides (MO; Gene Tools LLC) targeting two tcf12 exons (e3i3: 5′-CCAGAGGAAGTTCCTACCTGATGCT -3′ and e11i11: 5′-TGTGTGGGACAGCATACCCAT-3′, targeting exons 3 and 11, respectively). Embryos were obtained from natural matings of heterozygous transgenic gnrh3:gfp (36) adult fish and were injected with increasing doses of MO (3 ng, 6 ng and 9 ng for e3i3 MO and 1 ng, 2.5 ng and 4 ng for e11i11 MO); we injected a 1 nl cocktail per embryo at the one-to-four cell stage (n = 21–29 embryos/injection). To determine MO efficiency, separate batches of embryos (n = 30/batch) were injected with 6 ng of either MO and then harvested at 1 dpf in Trizol (Invitrogen). We extracted total RNA and conducted RT-PCR as described above to amplify regions flanking tcf12 MO target sites. PCR products were separated on a 2% agarose gel, gel-purified (QiaQuick, Qiagen), sequenced directly using Sanger and aligned to reference with Sequencher (Gene Codes). To rescue MO-induced phenotypes, we cloned the WT human TCF12 (IOH26962; ThermoFisher), TCF3 (MIM: 147141; GC-Y4553; GeneCopoeia), STUB1 (IOH56981; ThermoFisher), SOX10 (MIM: 602229; IOH6892; ThermoFisher) or SOX2 (MIM: 184429; IOH13706; ThermoFisher) open reading frames into a Gateway-compatible pCS2+ destination vector using LR clonase II (ThermoFisher). pCS2+ TCF12 plasmid was linearized with NotI, and the SP6 mMessage mMachine RNA transcription kit (ThermoFisher) was used to generate capped mRNA for embryo injections (75). A 1 nl cocktail of either MO (6 ng e3i3 or 2 ng e11i11) with 12.5 pg mRNA was co-injected into embryo batches at the one-to-four cell stage (n = 35–45 embryos per condition) for rescue experiments.
CRISPR/Cas9 genome editing of tcf12
To induce disruptive deletions in the zebrafish tcf12 locus, we used CRISPR/Cas9 genome editing to target exon 11. First, we identified optimal single gRNA targets with CHOPCHOP (76), and used the GeneArt Precision gRNA Synthesis Kit (ThermoFisher) to synthesize gRNAs as described (24). Next, we injected zebrafish embryos into the cell at the one-cell-stage with a 1 nl cocktail containing 100 pg gRNA and 200 pg Cas9 protein (PNA Bio). To determine gRNA efficiency, we extracted genomic DNA from embryos at 2 dpf and PCR-amplified the region flanking the gRNA target (n = 2 controls and 10 F0 mutants). PCR products were denatured and reannealed slowly, then migrated on a 4–12% pre-cast polyacrylamide gel (ThermoFisher). To estimate mosaicism, we cloned PCR fragments obtained from six F0 mutant embryos into the TOPO-TA vector (ThermoFisher) and conducted Sanger sequencing.
Whole-mount immunostaining of GnRH neurons in zebrafish larvae
To evaluate GnRH secreting neuronal patterning in zebrafish at 2 dpf, we used whole-mount immunostaining as described (24) to visualize relevant structures. Embryos were maintained at 25°C in embryo media (0.3 g/L NaCl, 75 mg/L CaSO4, 37.5 mg/L NaHCO3, 0.003% methylene blue) and at 2 dpf were dechorionated and fixed in 4% paraformaldehyde (PFA) for 2 h. Larvae were then washed twice with PBST to remove residual PFA. Larvae were then washed briefly with 0.1% trypsin in PBS, then washed twice with PBST and then dehydrated with pre-chilled (−20°C) acetone for 15 min. Following dehydration, larvae were washed again with PBST and then blocked for 1 h at room temperature in a solution of 2% bovine serum albumin, 1% dimethyl sulfoxide, 0.5% Triton X-100 and 5% calf serum (PBDT). We used rabbit anti-GnRH antibody (1:500; Sigma-Aldrich, G8294) for primary detection. After overnight incubation of primary antibody, larvae were washed twice with PBDT and then incubated at room temperature for 2 h with Alexa Fluor 555 anti-rabbit secondary antibody (1:500; ThermoFisher). Image acquisition was obtained manually with an AxioZoom V16 microscope and Axiocam 503 monochromatic camera facilitated by Zen Pro software (Zeiss), capturing ventral images of fluorescent immunostaining (2 dpf) and dorsal images of the GFP transgenic reporter (5 dpf). ImageJ [National Institutes of Health (NIH)] software was used to measure the length of the terminal nerve axons.
In vivo assessment of GnRH3 neuronal patterning in zebrafish larvae
To study GnRH3 neuronal patterning zebrafish larvae at 5 dpf, we conducted manual or automated imaging of a transgenic reporter line (gnrh3:gfp) (36). Manual imaging was conducted with an AxioZoom V16 microscope and Axiocam 503 monochromatic camera facilitated by Zen Pro software (Zeiss), capturing dorsal images of GFP signal. For automated imaging, larval batches were positioned and imaged live using the Vertebrate Automated Screening Technology (VAST; software version 1.2.5.4; Union Biometrica) BioImager. Larvae from each experimental condition were anesthetized with 0.2 mg/ml Tricaine prior to being loaded into the sample reservoir. Dorsal and lateral image templates of uninjected controls and morphant larvae were created and acquired images at a >70% minimum similarity for the pattern-recognition algorithms. The larvae were rotated to 0° to acquire a dorsal image via a 10× fluar objective and fluorescent excitation at 470 nm to detect GFP (Axiocam 503 monochromatic camera, Zen Pro software; Zeiss).
Automated image analysis
To perform automated measurement of fluorescent signal in VAST images, each file corresponding to an experimental condition was saved in Carl Zeiss image (CZI) file format in Zen Pro. Images were batch processed by converting each CZI to individual TIFF files that could be opened in ImageJ (NIH). All images were stacked and converted to 8 bit. Threshold was adjusted to 60 (minimum) and 160 (minimum). Images were then measured quantitatively by the highlighted area of the fluorescent signal in each image. We used GraphPad Prism software to calculate r2 values between manual and automated measurements with the ImageJ macro. To perform automated measurement of bright field VAST images, we used FishInspector software (77) to measure the length of zebrafish larvae.
Establishment of the TCF12 protein–protein interaction network
To conduct a network-based analysis of the TCF12 protein–protein interaction network, we used the ‘Broad Institute Web Platform for Genome Networks (GeNets)’ (42) data freeze from 05/25/2019. We created a user-defined ‘seed’ gene set consisting of 40 known IGD genes (including syndromic IGD) genes. GeNets analysis using the InWeb platform (43) generated a series of networks using the machine learning algorithm (Quack) that tests 18 different topological properties for how the seed gene set is interconnected across 853 MSigDB pathways (44) in protein–protein interaction data.
Quantitative RT-PCR of putative TCF12 network genes in zebrafish embryos
We injected zebrafish embryos with a 1 nl injection volume containing either 2 ng tcf12 e11i11 MO (one-to-four cell stage) or 100 pg gRNA and 200 pg Cas9 (one-cell stage). At 2 dpf, we decapitated 30 larvae per condition in two biological replicates using forceps; heads from injected batches and matched uninjected controls were placed on ice prior to total RNA extraction. Total RNA was extracted from heads using Trizol (ThermoFisher) as described above. We generated cDNA according to the manufacturer’s instructions using the QuantiTect Reverse Transcription Kit (Qiagen) using 2 μg of RNA template and by doubling the volumes of all reaction components. The resulting cDNA was used as template for each qRT-PCR reaction (30 ng cDNA template/reaction). qRT-PCR was conducted on an ABI Prism 7900HT System (Applied Biosystems) using the Power SYBR Green PCR Master Mix (Applied Biosystems). The primer sequences are as follows: sox2 F-5′-CATGTCCTATTCGCAGCAAAG-3′ and R-5′-ATACTGATCATGTCCCGCAGGT-3′, sox10 F-5′-AGGAAACTGGCGGATCAATA-3′ and R-5′-TCTTTCTTATGCTGCTTCCTCA-3′, stub1 F-5′-GAGAATTACGAAGAAGCCATCG-3′ and R-5′-CTCTATGCTGTTCCAGCGTTTC-3′, tcf3a F-5′-ATGGGAAACAGAGGAGCAAAC-3′ and R-5′-ACATTAGAAGGAGAGCCCACAG-3′, tcf3b F-5′-AGGAGCACCAGAGGCTTTTAAC-3′ and R-5′-GCTTTCCTTCTTCTCTTCCTCA-3′, tcf12 F-5′-ACAGTCATGCCTCTGATGACC-3′ and R-5′-GCTTTCTGTTCAGGGTTCAGAT-3′, 5′-rpl13 F-5′-CGCTATTGTGGCCAAGCAAG-R; 5′-rpl13 R-5′-TCTTGCGGAGGAAAGCCAAA-3′. Cycling parameters included an initial denaturation step, 95°C for 10 min, 40 cycles of 95°C for 15 s and 60°C for 1 min plus an added dissociation stage. All the samples were analyzed in triplicate. The housekeeping gene rpl13 was used to normalize the expression levels of genes using the 2−ΔΔCT method.
Statistical analysis
Burden testing for TCF12 RSVs seen in KS individuals versus TCF12 RSVs in the gnomAD database was performed using Fisher’s exact test. A Student’s t-test was used to determine statistical differences between human or zebrafish gene expression levels determined by qRT-PCR. A nonparametric Kruskal–Wallis test was used to detect pairwise differences in phenotypes between zebrafish larval batches (terminal nerve axon length at 2 dpf, olfactory bulb area at 2 dpf, GFP area at 5 dpf, larval length at 5 dpf) using GraphPad Prism 8.
Supplementary Material
Supplementary Material is available at HMG online.
Web resources
ChopChop software, https://chopchop.cbu.uib.no/
Ensembl, http://www.ensembl.org/
ExAC Browser, http://exac.broadinstitute.org/
GeNets, https://apps.broadinstitute.org/genets
gnomAD Browser, http://gnomad.broadinstitute.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
OMIM, http://www.omim.org/
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
SIFT, http://sift.jcvi.org/
Supplementary Material
Acknowledgements
We are grateful for the KS families for their support and willingness to participate in our research studies. We also acknowledge Yonathan Zohar (University of Maryland) for providing the gnrh3:egfp transgenic zebrafish. We also thank Kathryn Salnikov (MGH-REU) for critical reading of the manuscript.
Conflict of Interest statement. N.K. is a paid consultant for and holds significant stock of Rescindo Therapeutics, Inc. The other authors have no conflicts of interest to report.
Funding
US National Institutes of Health [grant P50HD028138 to E.E.D., W.F.C. and N.K. and K23HD077043 to R.B.].
References
- 1. Laitinen E.M., Vaaralahti K., Tommiska J., Eklund E., Tervaniemi M., Valanne L. and Raivio T. (2011) Incidence, phenotypic features and molecular genetics of Kallmann syndrome in Finland. Orphanet J. Rare Dis., 6, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stamou M.I., Cox K.H. and Crowley W.F. Jr. (2016) Discovering genes essential to the hypothalamic regulation of human reproduction using a human disease model: adjusting to life in the "-Omics" era. Endocr. Rev., 2016, 4–22. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 3. Balasubramanian R. and Crowley W.F. Jr. Isolated Gonadotropin-Releasing Hormone (GnRH) Deficiency. 2007 May 23 [Updated 2017 Mar 2]. In: Adam, M.P., Ardinger, H.H., Pagon, R.A., et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1334/.
- 4. Schwanzel-Fukuda M., Bick D. and Pfaff D.W. (1989) Luteinizing hormone-releasing hormone (LHRH)-expressing cells do not migrate normally in an inherited hypogonadal (Kallmann) syndrome. Brain Res. Mol. Brain Res., 6, 311–326. [DOI] [PubMed] [Google Scholar]
- 5. Bick D., Curry C.J., McGill J.R., Schorderet D.F., Bux R.C. and Moore C.M. (1989) Male infant with ichthyosis, Kallmann syndrome, chondrodysplasia punctata, and an Xp chromosome deletion. Am. J. Med. Genet., 33, 100–107. [DOI] [PubMed] [Google Scholar]
- 6. Knobil E., Plant T.M., Wildt L., Belchetz P.E. and Marshall G. (1980) Control of the rhesus monkey menstrual cycle: permissive role of hypothalamic gonadotropin-releasing hormone. Science, 207, 1371–1373. [DOI] [PubMed] [Google Scholar]
- 7. Sharma V.P., Fenwick A.L., Brockop M.S., McGowan S.J., Goos J.A., Hoogeboom A.J., Brady A.F., Jeelani N.O., Lynch S.A., Mulliken J.B. et al. (2013) Mutations in TCF12, encoding a basic helix-loop-helix partner of TWIST1, are a frequent cause of coronal craniosynostosis. Nat. Genet., 45, 304–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shi C.H., Schisler J.C., Rubel C.E., Tan S., Song B., McDonough H., Xu L., Portbury A.L., Mao C.Y., True C. et al. (2014) Ataxia and hypogonadism caused by the loss of ubiquitin ligase activity of the U box protein CHIP. Hum. Mol. Genet., 23, 1013–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O'Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B. et al. (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. di Rocco F., Baujat G., Arnaud E., Renier D., Laplanche J.L., Daire V.C. and Collet C. (2014) Clinical spectrum and outcomes in families with coronal synostosis and TCF12 mutations. Eur. J. Hum. Genet., 22, 1413–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Goos J.A., Fenwick A.L., Swagemakers S.M., McGowan S.J., Knight S.J., Twigg S.R., Hoogeboom A.J., van Dooren M.F., Magielsen F.J., Wall S.A. et al. (2016) Identification of intragenic exon deletions and duplication of TCF12 by whole genome or targeted sequencing as a cause of TCF12-related craniosynostosis. Hum. Mutat., 37, 732–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goumenos A., Tsoutsou E., Traeger-Synodinos J., Petychakis D., Gavra M., Kolialexi A. and Frysira H. (2019) Two novel variants in the TCF12 gene identified in cases with craniosynostosis. Appl. Clin. Genet., 12, 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Le Tanno P., Poreau B., Devillard F., Vieville G., Amblard F., Jouk P.S., Satre V. and Coutton C. (2014) Maternal complex chromosomal rearrangement leads to TCF12 microdeletion in a patient presenting with coronal craniosynostosis and intellectual disability. Am. J. Med. Genet. A, 164A, 1530–1536. [DOI] [PubMed] [Google Scholar]
- 14. Lee E., Le T., Zhu Y., Elakis G., Turner A., Lo W., Venselaar H., Verrenkamp C.A., Snow N., Mowat D. et al. (2018) A craniosynostosis massively parallel sequencing panel study in 309 Australian and New Zealand patients: findings and recommendations. Genet. Med., 20, 1061–1068. [DOI] [PubMed] [Google Scholar]
- 15. Paumard-Hernandez B., Berges-Soria J., Barroso E., Rivera-Pedroza C.I., Perez-Carrizosa V., Benito-Sanz S., Lopez-Messa E., Santos F., Garcia R.,.I.I., Romance A. et al. (2015) Expanding the mutation spectrum in 182 Spanish probands with craniosynostosis: identification and characterization of novel TCF12 variants. Eur. J. Hum. Genet., 23, 907–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Timberlake A.T., Wu R., Nelson-Williams C., Furey C.G., Hildebrand K.I., Elton S.W., Wood J.S., Persing J.A. and Lifton R.P. (2018) Co-occurrence of frameshift mutations in SMAD6 and TCF12 in a child with complex craniosynostosis. Hum. Genome Var., 5, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Topa A., Rohlin A., Andersson M.K., Fehr A., Lovmar L., Stenman G. and Kolby L. (2020) NGS targeted screening of 100 Scandinavian patients with coronal synostosis. Am. J. Med. Genet. A, 182, 348–356. [DOI] [PubMed] [Google Scholar]
- 18. Yilmaz E., Mihci E., Nur B. and Alper O.M. (2019) Coronal craniosynostosis due to TCF12 mutations in patients from Turkey. Am. J. Med. Genet. A, 179, 2241–2245. [DOI] [PubMed] [Google Scholar]
- 19. Yoon J.G., Hahn H.M., Choi S., Kim S.J., Aum S., Yu J.W., Park E.K., Shim K.W., Lee M.G. and Kim Y.O. (2019) Molecular diagnosis of craniosynostosis using targeted next-generation sequencing. Neurosurgery. doi: 10.1093/neuros/nyz470. Online ahead of print. [DOI] [PubMed] [Google Scholar]
- 20. Sobreira N., Schiettecatte F., Valle D. and Hamosh A. (2015) GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat., 36, 928–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dode C., Levilliers J., Dupont J.M., De Paepe A., Le Du N., Soussi-Yanicostas N., Coimbra R.S., Delmaghani S., Compain-Nouaille S., Baverel F. et al. (2003) Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat. Genet., 33, 463–465. [DOI] [PubMed] [Google Scholar]
- 22. Muenke M., Schell U., Hehr A., Robin N.H., Losken H.W., Schinzel A., Pulleyn L.J., Rutland P., Reardon W., Malcolm S. et al. (1994) A common mutation in the fibroblast growth factor receptor 1 gene in Pfeiffer syndrome. Nat. Genet., 8, 269–274. [DOI] [PubMed] [Google Scholar]
- 23. Pitteloud N., Acierno J.S. Jr., Meysing A., Eliseenkova A.V., Ma J., Ibrahimi O.A., Metzger D.L., Hayes F.J., Dwyer A.A., Hughes V.A. et al. (2006) Mutations in fibroblast growth factor receptor 1 cause both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc. Natl. Acad. Sci. U. S. A., 103, 6281–6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shaw N.D., Brand H., Kupchinsky Z.A., Bengani H., Plummer L., Jones T.I., Erdin S., Williamson K.A., Rainger J., Stortchevoi A. et al. (2017) SMCHD1 mutations associated with a rare muscular dystrophy can also cause isolated arhinia and Bosma arhinia microphthalmia syndrome. Nat. Genet., 49, 238–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Balasubramanian R., Choi J.H., Francescatto L., Willer J., Horton E.R., Asimacopoulos E.P., Stankovic K.M., Plummer L., Buck C.L., Quinton R. et al. (2014) Functionally compromised CHD7 alleles in patients with isolated GnRH deficiency. Proc. Natl. Acad. Sci. U. S. A., 111, 17953–17958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vissers L.E., van Ravenswaaij C.M., Admiraal R., Hurst J.A., de Vries B.B., Janssen I.M., van der Vliet W.A., Huys E.H., de Jong P.J., Hamel B.C. et al. (2004) Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat. Genet., 36, 955–957. [DOI] [PubMed] [Google Scholar]
- 27. Doty R.L. (2007) Office procedures for quantitative assessment of olfactory function. Am. J. Rhinol., 21, 460–473. [DOI] [PubMed] [Google Scholar]
- 28. Wang F., Huang G.D., Tian H., Zhong Y.B., Shi H.J., Li Z., Zhang X.S., Wang H. and Sun F. (2015) Point mutations in KAL1 and the mitochondrial gene MT-tRNA(cys) synergize to produce Kallmann syndrome phenotype. Sci. Rep., 5, 13050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Margolin D.H., Kousi M., Chan Y.M., Lim E.T., Schmahmann J.D., Hadjivassiliou M., Hall J.E., Adam I., Dwyer A., Plummer L. et al. (2013) Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N. Engl. J. Med., 368, 1992–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Howard S.R., Guasti L., Ruiz-Babot G., Mancini A., David A., Storr H.L., Metherell L.A., Sternberg M.J., Cabrera C.P., Warren H.R. et al. (2016) IGSF10 mutations dysregulate gonadotropin-releasing hormone neuronal migration resulting in delayed puberty. EMBO Mol. Med., 8, 626–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Thisse C. and Thisse B. (2005) High throughput expression analysis of ZF-models consortium clones. ZFIN Direct Data Submission, (http://zfin.org). [Google Scholar]
- 32. Uittenbogaard M. and Chiaramello A. (2002) Expression of the bHLH transcription factor Tcf12 (ME1) gene is linked to the expansion of precursor cell populations during neurogenesis. Brain Res. Gene Expr. Patterns, 1, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Murre C., McCaw P.S. and Baltimore D. (1989) A new DNA binding and dimerization motif in immunoglobulin enhancer binding, daughterless, MyoD, and myc proteins. Cell, 56, 777–783. [DOI] [PubMed] [Google Scholar]
- 34. Murre C., McCaw P.S., Vaessin H., Caudy M., Jan L.Y., Jan Y.N., Cabrera C.V., Buskin J.N., Hauschka S.D., Lassar A.B. et al. (1989) Interactions between heterologous helix-loop-helix proteins generate complexes that bind specifically to a common DNA sequence. Cell, 58, 537–544. [DOI] [PubMed] [Google Scholar]
- 35. Taroc E.Z.M., Prasad A., Lin J.M. and Forni P.E. (2017) The terminal nerve plays a prominent role in GnRH-1 neuronal migration independent from proper olfactory and vomeronasal connections to the olfactory bulbs. Biol. Open, 6, 1552–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Abraham E., Palevitch O., Gothilf Y. and Zohar Y. (2009) The zebrafish as a model system for forebrain GnRH neuronal development. Gen. Comp. Endocrinol., 164, 151–160. [DOI] [PubMed] [Google Scholar]
- 37. Abraham E., Palevitch O., Ijiri S., Du S.J., Gothilf Y. and Zohar Y. (2008) Early development of forebrain gonadotrophin-releasing hormone (GnRH) neurones and the role of GnRH as an autocrine migration factor. J. Neuroendocrinol., 20, 394–405. [DOI] [PubMed] [Google Scholar]
- 38. Forni P.E. and Wray S. (2012) Neural crest and olfactory system: new prospective. Mol. Neurobiol., 46, 349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pingault V., Bodereau V., Baral V., Marcos S., Watanabe Y., Chaoui A., Fouveaut C., Leroy C., Verier-Mine O., Francannet C. et al. (2013) Loss-of-function mutations in SOX10 cause Kallmann syndrome with deafness. Am. J. Hum. Genet., 92, 707–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kotan L.D., Hutchins B.I., Ozkan Y., Demirel F., Stoner H., Cheng P.J., Esen I., Gurbuz F., Bicakci Y.K., Mengen E. et al. (2014) Mutations in FEZF1 cause Kallmann syndrome. Am. J. Hum. Genet., 95, 326–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kelberman D., Rizzoti K., Avilion A., Bitner-Glindzicz M., Cianfarani S., Collins J., Chong W.K., Kirk J.M., Achermann J.C., Ross R. et al. (2006) Mutations within Sox2/SOX2 are associated with abnormalities in the hypothalamo–pituitary–gonadal axis in mice and humans. J. Clin. Invest., 116, 2442–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li T., Kim A., Rosenbluh J., Horn H., Greenfeld L., An D., Zimmer A., Liberzon A., Bistline J., Natoli T. et al. (2018) GeNets: a unified web platform for network-based genomic analyses. Nat. Methods, 15, 543–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li T., Wernersson R., Hansen R.B., Horn H., Mercer J., Slodkowicz G., Workman C.T., Rigina O., Rapacki K., Staerfeldt H.H. et al. (2017) A scored human protein–protein interaction network to catalyze genomic interpretation. Nat. Methods, 14, 61–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liberzon A., Birger C., Thorvaldsdottir H., Ghandi M., Mesirov J.P. and Tamayo P. (2015) The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst., 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wong A.K., Krishnan A., Yao V., Tadych A. and Troyanskaya O.G. (2015) IMP 2.0: a multi-species functional genomics portal for integration, visualization and prediction of protein functions and networks. Nucleic Acids Res., 43, W128–W133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sawada S. and Littman D.R. (1993) A heterodimer of HEB and an E12-related protein interacts with the CD4 enhancer and regulates its activity in T-cell lines. Mol. Cell. Biol., 13, 5620–5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ferri F., Parcelier A., Petit V., Gallouet A.S., Lewandowski D., Dalloz M., van den Heuvel A., Kolovos P., Soler E., Squadrito M.L. et al. (2015) TRIM33 switches off Ifnb1 gene transcription during the late phase of macrophage activation. Nat. Commun., 6, 8900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Belle I. and Zhuang Y. (2014) E proteins in lymphocyte development and lymphoid diseases. Curr. Top. Dev. Biol., 110, 153–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fischer B., Azim K., Hurtado-Chong A., Ramelli S., Fernandez M. and Raineteau O. (2014) E-proteins orchestrate the progression of neural stem cell differentiation in the postnatal forebrain. Neural Dev., 9, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ravanpay A.C. and Olson J.M. (2008) E protein dosage influences brain development more than family member identity. J. Neurosci. Res., 86, 1472–1481. [DOI] [PubMed] [Google Scholar]
- 51. Engelen E., Akinci U., Bryne J.C., Hou J., Gontan C., Moen M., Szumska D., Kockx C., van Ijcken W., Dekkers D.H. et al. (2011) Sox2 cooperates with Chd7 to regulate genes that are mutated in human syndromes. Nat. Genet., 43, 607–611. [DOI] [PubMed] [Google Scholar]
- 52. Piard J., Roze V., Czorny A., Lenoir M., Valduga M., Fenwick A.L., Wilkie A.O. and Maldergem L.V. (2015) TCF12 microdeletion in a 72-year-old woman with intellectual disability. Am. J. Med. Genet. A, 167A, 1897–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tammimies K., Marshall C.R., Walker S., Kaur G., Thiruvahindrapuram B., Lionel A.C., Yuen R.K., Uddin M., Roberts W., Weksberg R. et al. (2015) Molecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with autism spectrum disorder. JAMA, 314, 895–903. [DOI] [PubMed] [Google Scholar]
- 54. Geller F., Feenstra B., Carstensen L., Pers T.H., van Rooij I.A., Korberg I.B., Choudhry S., Karjalainen J.M., Schnack T.H., Hollegaard M.V. et al. (2014) Genome-wide association analyses identify variants in developmental genes associated with hypospadias. Nat. Genet., 46, 957–963. [DOI] [PubMed] [Google Scholar]
- 55. Kalfa N., Gaspari L., Ollivier M., Philibert P., Bergougnoux A., Paris F. and Sultan C. (2019) Molecular genetics of hypospadias and cryptorchidism recent developments. Clin. Genet., 95, 122–131. [DOI] [PubMed] [Google Scholar]
- 56. Kousi M. and Katsanis N. (2015) Genetic modifiers and oligogenic inheritance. Cold Spring Harb. Perspect. Med., 5, a017145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wilkie A.O., Patey S.J., Kan S.H., van den Ouweland A.M. and Hamel B.C. (2002) FGFs, their receptors, and human limb malformations: clinical and molecular correlations. Am. J. Med. Genet. A, 112, 266–278. [DOI] [PubMed] [Google Scholar]
- 58. Bondurand N., Dastot-Le Moal F., Stanchina L., Collot N., Baral V., Marlin S., Attie-Bitach T., Giurgea I., Skopinski L., Reardon W. et al. (2007) Deletions at the SOX10 gene locus cause Waardenburg syndrome types 2 and 4. Am. J. Hum. Genet., 81, 1169–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Teng C.S., Ting M.C., Farmer D.T., Brockop M., Maxson R.E. and Crump J.G. (2018) Altered bone growth dynamics prefigure craniosynostosis in a zebrafish model of Saethre–Chotzen syndrome. Elife, 7, e37024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Holmberg J., Hansson E., Malewicz M., Sandberg M., Perlmann T., Lendahl U. and Muhr J. (2008) SoxB1 transcription factors and notch signaling use distinct mechanisms to regulate proneural gene function and neural progenitor differentiation. Development, 135, 1843–1851. [DOI] [PubMed] [Google Scholar]
- 61. Kim E.J., Battiste J., Nakagawa Y. and Johnson J.E. (2008) Ascl1 (Mash1) lineage cells contribute to discrete cell populations in CNS architecture. Mol. Cell. Neurosci., 38, 595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Murray R.C., Navi D., Fesenko J., Lander A.D. and Calof A.L. (2003) Widespread defects in the primary olfactory pathway caused by loss of Mash1 function. J. Neurosci., 23, 1769–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cau E., Casarosa S. and Guillemot F. (2002) Mash1 and Ngn1 control distinct steps of determination and differentiation in the olfactory sensory neuron lineage. Development, 129, 1871–1880. [DOI] [PubMed] [Google Scholar]
- 64. Cau E., Gradwohl G., Fode C. and Guillemot F. (1997) Mash1 activates a cascade of bHLH regulators in olfactory neuron progenitors. Development, 124, 1611–1621. [DOI] [PubMed] [Google Scholar]
- 65. Zhang C., Ng K.L., Li J.D., He F., Anderson D.J., Sun Y.E. and Zhou Q.Y. (2007) Prokineticin 2 is a target gene of proneural basic helix-loop-helix factors for olfactory bulb neurogenesis. J. Biol. Chem., 282, 6917–6921. [DOI] [PubMed] [Google Scholar]
- 66. Barndt R.J., Dai M. and Zhuang Y. (2000) Functions of E2A-HEB heterodimers in T-cell development revealed by a dominant negative mutation of HEB. Mol. Cell. Biol., 20, 6677–6685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Massari M.E. and Murre C. (2000) Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol. Cell. Biol., 20, 429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ballinger C.A., Connell P., Wu Y., Hu Z., Thompson L.J., Yin L.Y. and Patterson C. (1999) Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol. Cell. Biol., 19, 4535–4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jiang J., Ballinger C.A., Wu Y., Dai Q., Cyr D.M., Hohfeld J. and Patterson C. (2001) CHIP is a U-box-dependent E3 ubiquitin ligase: identification of Hsc70 as a target for ubiquitylation. J. Biol. Chem., 276, 42938–42944. [DOI] [PubMed] [Google Scholar]
- 70. Murata S., Minami Y., Minami M., Chiba T. and Tanaka K. (2001) CHIP is a chaperone-dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep., 2, 1133–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Xie X., Rigor P. and Baldi P. (2009) MotifMap: a human genome-wide map of candidate regulatory motif sites. Bioinformatics, 25, 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lewkowitz-Shpuntoff H.M., Hughes V.A., Plummer L., Au M.G., Doty R.L., Seminara S.B., Chan Y.M., Pitteloud N., Crowley W.F. Jr. and Balasubramanian R. (2012) Olfactory phenotypic spectrum in idiopathic hypogonadotropic hypogonadism: pathophysiological and genetic implications. J. Clin. Endocrinol. Metab., 97, E136–E144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Paila U., Chapman B.A., Kirchner R. and Quinlan A.R. (2013) GEMINI: integrative exploration of genetic variation and genome annotations. PLoS Comput. Biol., 9, e1003153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R., Thormann A., Flicek P. and Cunningham F. (2016) The Ensembl variant effect predictor. Genome Biol., 17, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Niederriter A.R., Davis E.E., Golzio C., Oh E.C., Tsai I.C. and Katsanis N. (2013) In vivo modeling of the morbid human genome using Danio rerio. J. Vis. Exp., e50338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Labun K., Montague T.G., Gagnon J.A., Thyme S.B. and Valen E. (2016) CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res., 44, W272–W276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Teixido E., Kiessling T.R., Krupp E., Quevedo C., Muriana A. and Scholz S. (2019) Automated morphological feature assessment for Zebrafish embryo developmental toxicity screens. Toxicol. Sci., 167, 438–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.