

In this study, Tiwari et al. set out to examine the mechanism by which p53 prevents tumor formation. Using both in vivo and genomic analyses, the authors establish that p53 continuously operates at the LINE1 promoter to restrict autonomous copies of these mobile elements in human cells and that this constitutive restriction of these retroelements may help to explain tumor suppression encoded by p53.
Keywords: LINE1, p53, transrepression, tumor suppressors
Abstract
p53 is a potent tumor suppressor and commonly mutated in human cancers. Recently, we demonstrated that p53 genes act to restrict retrotransposons in germline tissues of flies and fish but whether this activity is conserved in somatic human cells is not known. Here we show that p53 constitutively restrains human LINE1s by cooperatively engaging sites in the 5′UTR and stimulating local deposition of repressive histone marks at these transposons. Consistent with this, the elimination of p53 or the removal of corresponding binding sites in LINE1s, prompted these retroelements to become hyperactive. Concurrently, p53 loss instigated chromosomal rearrangements linked to LINE sequences and also provoked inflammatory programs that were dependent on reverse transcriptase produced from LINE1s. Taken together, our observations establish that p53 continuously operates at the LINE1 promoter to restrict autonomous copies of these mobile elements in human cells. Our results further suggest that constitutive restriction of these retroelements may help to explain tumor suppression encoded by p53, since erupting LINE1s produced acute oncogenic threats when p53 was absent.
p53, the most commonly mutated gene in human cancers, has been primarily studied in the context of cellular stress where it directs transcriptional programs specifying apoptosis, cell cycle arrest, and cellular senescence. Accordingly, stimulus-induced transactivation of effector genes is widely thought to account for p53-mediated tumor suppression (Lane and Levine 2010; Bieging et al. 2014; Chen 2016). However, mice collectively lacking essential p53 target genes (p21, noxa, and puma) remain tumor free and, likewise, p53 continues to function as a tumor suppressor even if decoupled from these cellular processes (Brady et al. 2011; Li et al. 2012; Valente et al. 2013). Recently, our laboratory demonstrated that p53 restrains mobile elements in the germline of flies and fish, raising the possibility that, if conserved, modalities related to this tonic activity might contribute to cancer prevention in longer-lived animals (Wylie et al. 2016a,b; Tiwari et al. 2017, 2018). Here, we tested whether retroelement repression by p53 is a conserved property that extends to human somatic cells.
LINE1 (L1) elements are considered the only active retrotransposon in the human genome (Lander et al. 2001). Although the majority of L1 copies are fragmented and/or mutated, ∼100–150 full-length L1 elements are autonomous and retrotransposition competent (Moran et al. 1996; Brouha et al. 2003). These are all members of an evolutionarily young family of L1s designated L1 human specific (L1Hs). Intact L1Hs copies contain an internal promoter located within the 5′UTR (Swergold 1990; Alexandrova et al. 2012) and encode two open reading frames. L1 ORF1p is an RNA chaperone, while ORF2p possesses endonuclease and reverse transcriptase activities (Dombroski et al. 1994; Feng et al. 1996; Martin et al. 2005; Martin 2010; Callahan et al. 2012). While L1s are normally silenced in somatic cells, derepression of these transposons is associated with certain cancers and some neuropathies (Muotri et al. 2010; Hancks and Kazazian 2012; Rodić et al. 2014; Wylie et al. 2016b; Burns 2017). Conceivably, L1 hyperactivity could drive pathologies by destabilizing genomes, promoting mutations, altering gene expression and triggering inflammatory responses (Hancks and Kazazian 2012, 2016; Lee et al. 2012; Tubio et al. 2014; Volkman and Stetson 2014; Chenais 2015; Kemp and Longworth 2015; Scott and Devine 2017; Briggs et al. 2018; Saleh et al. 2019; Rodriguez-Martin et al. 2020).
How p53 loss permits elevated retrotransposon activity is currently unknown, but several lines of evidence suggest that p53 could specify the deposition of epigenetic marks that silence mobile elements. For example, when a synthetic L1 was injected into Zebrafish, the L1-5′UTR was a target for p53-dependent H3K9 trimethylation (Wylie et al. 2016b) and consistent with this, in mouse fibroblasts treated with a DNA demethylating agent, intact p53 was needed to silence repetitive elements (Leonova et al. 2013). Furthermore, since informatic and empirical evidence for p53 binding to retroelements has been reported (Wang et al. 2007; Harris et al. 2009; Zemojtel et al. 2009; Cui et al. 2011), it is plausible that p53 could directly antagonize these elements.
Here, we establish a role for p53 in the silencing of human retroelements and link retroelement derepression caused by p53 loss to oncogenic phenotypes. Using synthetic transposons together with CRISPR methods, we found that L1 activity and de novo transposition were significantly increased when p53 was eliminated. We also observed that p53 binds to the 5′UTR of L1s and established that constitutive occupancy at this internal promoter was required for deposition of repressive histone marks and for transcriptional suppression of these elements. Soon after p53 loss, chromosomal rearrangements associated with L1 sequences were observed. Likewise, without p53, immune response signatures were also detected and, using a retrotransposon antagonist, we demonstrate that retroelement activity underlies this response. Together, these findings establish that tonic repression of L1s is a well-conserved property of p53 that safeguards human somatic cells against potential oncogenic threats presented by these transposons.
Results
p53 suppresses L1 expression in human cell lines
Using CRISPR editing methods a series of p53 knockouts were generated in diverse human cell lines and derepressed L1 activity was observed in both cancer derived (e.g., A375 and U2OS cell lines) as well as normal lung tissue derived HBEC3kt cells (Fig. 1). As shown in Figure 1A (top panel), a common exon found in all p53 isoforms was targeted and genomic sequencing was performed to assign loss-of-function genotypes in derived clones (see Supplemental Table S1). Using Western blot analyses, absence of p53 was verified in these independent knockout clones derived from A375 (Fig. 1C), U2OS (Fig. 1D), HBEC3kt (Fig. 1E, and HCT116 cells (Supplemental Fig. S1E). Furthermore, where available, loss of p53 transcripts was verified in RNA sequencing data sets, consistent with nonsense-mediated decay in independent p53 knockout lines (Fig. 1A, bottom panel). To further verify the specificity of our p53 knockout lines, we computationally identified 114 potential p53 gRNA off target loci, which differed from the p53 target sequence by up to four mismatched (Supplemental Fig. S1M). Less than half of these loci are located in gene bodies (Supplemental Fig. S1N) and, importantly, we confirmed that expression from these was not substantially different in cells exposed to the p53 gRNA and the parental A375 cell line (Supplemental Fig. S1O).
Figure 1.

p53 loss provokes human L1 expression. The p53 gene was mutated using a CRISPR guide RNA targeting the first common exon found in all annotated p53 isoforms, as illustrated in the top panel of A. In A and B, two independently edited p53−/− lines, designated A-3 and A-2, were analyzed. In C and F, independently edited p53−/− lines A-1 and A-2 were analyzed. In the bottom panel of A, normalized RNA sequencing expression levels from wild-type (WT; black) and p53−/− A375 cells (red) are shown. (B) Normalized expression of readthrough transcription from a single, uniquely identifiable L1Hs element in wild-type and p53−/− A375 cells. Western blot in WT, cas9-treated control and p53−/− A375 human melanoma (C), U2OS osteosarcoma (D), and HBEC3kt immortalized normal human lung (E) cells lines for p53 (top panels) and L1ORF1p (middle panels). (Bottom panels) β-Actin is presented as a loading control. Normalized L1-5′UTR mRNA levels in WT, cas9-treated control and two independent p53−/− A375 (F), U2OS (G), and HBEC3kt (H) cell lines assessed by ddPCR. L1-5′UTR transcript levels were normalized to β-actin and relative fold change was calculated with respect to parental wild type. Bar graphs are the average of three independent experiments (n = 3) with error bars representing the standard deviation. p53−/− samples were significantly different from parental wild type (two-tailed t-test P-value < 0.05) and cas9-treated controls (P-value < 0.05).
Next, the effects of p53 loss on L1 activity were tested by inspecting these elements in two ways. First, we examined ORF1p, a commonly used marker for the expression of intact L1 copies. As shown in Figures 1C–E and quantified in Supplemental Figure S1G–I, p53 elimination triggered acute increases in L1 ORF1p expression in A375, U2OS, and HBEC3kt cells. Importantly, neither cas9 exposure alone (Fig. 1C–E; Supplemental Fig. S1G–I) nor introduction of a control gRNA targeting the huAAVS1 locus (Supplemental Fig. S1C,D) was sufficient to trigger ORF1p expression, confirming that p53 loss caused ORF1p derepression in these experiments. As a second approach, we directly inspected L1-derived RNAs using RNA sequencing and/or droplet digital PCR (ddPCR) assays. Using methods adapted from Philippe et al. (2016) and as exemplified in Figure 1B and summarized in Supplemental Figure S1A,B, loss of p53 was associated with elevated expression of individually identifiable L1Hs RNAs in RNA sequencing data sets from A375 cells. Intriguingly, this effect was stochastic, and consistent with (Philippe et al. 2016), different L1Hs elements were clearly derepressed in different clonal p53 knockout cell lines (in Supplemental Fig. S1A,B note red and yellow dots indicating commonly and uniquely derepressed L1Hs elements). Furthermore, the effect triggered by p53 loss was restricted to L1Hs transcripts since ancient L1-, L2-, Alu-, and ERV-derived sequences were not similarly impacted (see Supplemental Fig. S1J–L). Most L1Hs genomic sequences lack a complete 5′ end and are, for this reason, retrotransposition-incompetent. Therefore, to more directly focus on intact L1Hs copies, we also developed a ddPCR assay that selectively targets L1Hs RNAs containing the 5′UTR. Using this assay, 5′UTR L1Hs-derived RNAs were increased approximately fivefold when p53 is lost in A375 cells (Fig. 1F). Similarly, a fourfold to fivefold increase was observed in U2OS and, likewise, approximately fourfold elevation in HBEC3kt cells upon p53 loss (Fig. 1G,H, respectively). Taken together, these results indicate that p53-mediated repression of intact and potentially active retroelements seen in model systems (Wylie et al. 2016b; Tiwari et al. 2017) is conserved in humans and detectable in numerous cultured cell lines. This pattern was not universal since, consistent with previous reports (Coufal et al. 2011), L1s were not activated in HCT116 cells, indicating that additional factors and/or cell-type specific contexts can influence the behavior of L1s when p53 is lost (Supplemental Fig. S1E,F).
p53 inhibits L1-retrotransposition
Results in Figure 1 demonstrate a strong inverse relationship between p53 status and the expression of L1Hs RNA and protein. To determine whether retrotransposition rates are similarly affected, we stably transfected parental and p53 knockout A375 and U2OS cells with a synthetic L1 designated 99-gfp-LRE3 (Ostertag et al. 2000; Kopera et al. 2016). This indicator (see Fig. 2A) encodes a retrotransposition competent copy of L1Hs engineered to permit direct measurements of de novo retrotransposition rates. As illustrated in Figure 2A EGFP expression produced by this synthetic element requires a completed retrotransposition life cycle, including transcription, splicing, reverse transcription, and integration. Using this tool, we observed significantly increased retrotransposition when p53 was removed (Fig. 2B–E) and, importantly, CRISPR/cas9 exposure alone did not trigger increased retrotransposition (Fig. 2B–G). When quantified by flow cytometry, de novo insertions were increased fivefold to sixfold in U2OS p53 knockout cells (Fig. 2E) and increased twofold to threefold in p53 knockout A375 cells (Fig. 2C) relative to parental counterparts or a control transposition indicator lacking the endonuclease, LRE3-H230A-GFP (EN−) (Supplemental Fig. S2C,D). To verify this conclusion, we directly inspected the genomes of wild-type and p53 knockout cells using a ddPCR assay that measures de novo retro-insertions. Importantly, starting copy numbers of the integrated 99-gfp-LRE3 plasmid were similar across genotypes, as verified by genomic PCR using primers that detect unspliced eGFP (Supplemental Fig. S2A,B). As seen in Figure 2, F and G, normalized values of spliced eGFP copies, indicating a complete cycle of retrotransposition, were considerably higher in the genomes of p53 mutant A375 and U2OS cells relative to their wild-type counterparts. Therefore, p53-dependent differences seen in Figure 2, B–E, reflect genuine differences in derived copies of 99-gfp-LRE3 retroinserts, establishing that human p53 restricts retrotransposition in these cells.
Figure 2.
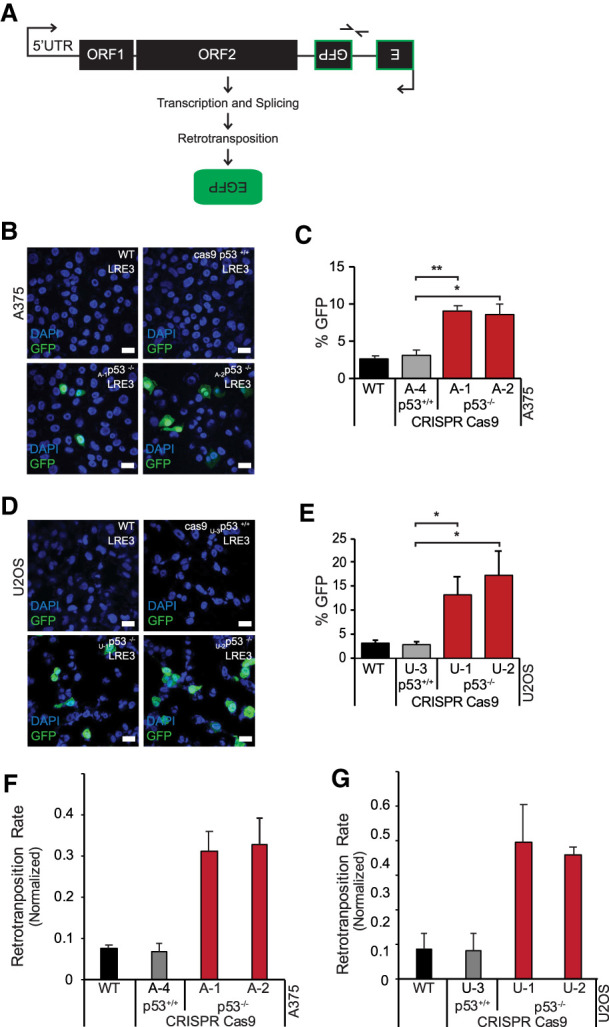
p53 loss is permissive for human L1 de novo retrotransposition. (A) Schematic depiction of the 99-gfp-LRE3 retrotransposition indicator. eGFP fluorescence indicates a complete retrotransposition cycle. Representative confocal images (obtained at 63×; scale bars, 20 μm) of GFP fluorescence in stable wild-type, cas9-treated control, and p53−/− A375 (B) and U2OS (D) 99-gfp-LRE3 integrant cell populations counterstained with DAPI. Quantification (n = 3) of the percentage of GFP-positive cells by flow cytometry for A375 (C) and U2OS (E) cells, error bars represent the standard error of the mean. (*) P-value ≤ 0.05, (**) P-value < 0.005. Droplet digital PCR (ddPCR) quantification (n = 2) of de novo LINE1 integration events in A375 (F) and U2OS (G) cells, normalized to single-copy gene puma abundance. Primer pair 2 (barbed arrows shown in A) specifically detects spliced GFP (indicating a complete retrotransposition life cycle) in genomic DNA. Bar graphs are averages of two biological replicates and error bars are the standard deviation.
The L1-5′UTR is sufficient for p53 repression
Human L1 elements carry an internal RNA polymerase II promoter within their ∼900-bp 5′UTR (Swergold 1990). Previously, we showed p53-dependent deposition of repressive histone marks occurred at this 5′UTR when 99-gfp-LRE3 plasmids were injected into zebrafish embryos (Wylie et al. 2016b), suggesting that p53-dependent suppression of L1s may require the L1-5′UTR. To determine whether the 5′UTR is sufficient for p53 repression, we placed the L1-5′UTR upstream of an eGFP cassette to create an L1 expression reporter (L1-5′UTR-eGFP). Importantly, for these studies, we integrated the L1-5′UTR-eGFP expression reporter at the AAVS1 safe harbor site (Fig. 3A) and, in this way, avoided confounders associated with transient transfections, copy number differences, and position effects. We used genomic PCR to verify single-copy insertions of L1-5′UTR-eGFP at the AAVS1 safe harbor site in our edited A375 and U2OS cells (Supplemental Fig. S3A–C). Using flow cytometry, we monitored L1-5′UTR-eGFP activity and detected significantly higher eGFP expression levels in the p53 knockout A375 and U2OS cells compared with wild-type cells (Fig. 3B,C, Supplemental Fig. S3F,G; Supplemental Tables S2, S3 for the quantification). Likewise, eGFP transcript levels were similarly elevated in p53 knockout A375 and U2OS cells (Supplemental Fig. S3D,E), suggesting that p53 repression occurs at the level of transcription. Together, Figure 3, B and C, and Supplemental Figure S3, D and E indicate that p53 antagonizes the production of L1 RNAs by operating through DNA sequences within the 5′UTR of L1 elements.
Figure 3.

p53-mediated suppression of LINE1s operates at the level of transcription through the 5′UTR. (A) The integrated L1 reporter construct, L1-5′UTR-eGFP, is detectable with primers indicated (barbed arrows). Flow cytometry of GFP in WT, cas9-treated control, and p53−/− A375 (B) or U2OS (C) cells containing the L1-5′UTR-eGFP exposes bright GFP-positive subpopulations in p53−/− cells (see Supplemental Tables S2, S3 for gating parameters). Additional biological replicates are shown in Supplemental Tables S2 and S3; for unstained controls, see Supplemental Figure S3, F and G. ChIP results (D,E) showing H3 normalized H3K27me3 (D) and H3K9me3 (E) ratios at the L1-5′UTR, p21, and puma in wild-type (black) and p53−/− (red) cells. Note that changes in H3K27me3 and H3K9me enrichment are specific to the 5′ UTR of the L1-5′UTR-eGFP and p53 dependent. Error bars represent 95% confidence intervals.
H3K9me3 and H3K27me3 are repressive histone marks characteristic of heterochromatin (Bannister and Kouzarides 2011). These marks are also enriched at repetitive sequences including LINE1 elements (Molaro and Malik 2016; Yang and Wang 2016), but efforts to inspect individual L1 copies are confounded by the absence of uniquely assignable sequences. Therefore, to ask whether p53 might regulate deposition of these marks, we took advantage of the integrated L1-5′UTR-eGFP reporters with primers that uniquely detect this template (Fig. 3A) and, using these, we conducted ChIP experiments to interrogate internal L1 promoter sequences. As seen in Figures 3D and E, when p53 was absent H3K27 trimethylation was reduced by ∼70% and, similarly, H3K9 trimethylation of L1-5′UTR-eGFP was reduced by 50% across the same region. Importantly, these p53-dependent patterns were specific to L1s, since we did not observe differential histone methylation or expression at the canonical p53 target genes p21 and puma (Fig. 3D,E; Supplemental Fig. S3H,I).
p53 directly binds and represses L1-5′UTR
To determine how p53 regulates the deposition of repressive histone marks at the L1-5′UTR, we first assessed the impacts of p53 on known retroelement repressive pathways. Importantly, expression levels of epigenetic modifiers (Supplemental Fig. S4A,C), small RNA effectors (Supplemental Fig. S4B,D), and piRNA precursors (Supplemental Fig. S4E,F) known to repress transposons were not changed in p53 knockout cell lines. Indeed, the piRNA pathway component piwil4 was modestly induced in p53 knockout cells, consistent with a potential adaptive response to hyperactive L1s in these cells. These results indicate that transcriptional regulation of target genes coding for known effectors does not underlie p53 mediated L1 silencing, suggesting that p53 may directly bind and repress these retrotransposons.
The L1 5′UTR contains three predicted p53 binding sites (Smeenk et al. 2008; Harris et al. 2009; Heinz et al. 2010), which we designated as sites I–III (see Fig. 4A). Using in vitro assays, Harris et al. (2009) demonstrated that purified p53 protein bound a synthesized stretch of the L1 5′UTR (bases 427–523) containing both sites I and II. Site III was informatically predicted (see the Materials and Methods). To inspect potential p53 binding to the L1 5′UTR, we conducted ChIP assays in A375 cells containing the uniquely identifiable L1-5′UTR-eGFP reporter. For assay validation, we confirmed robust p53 binding signals at benchmark target genes (e.g., puma and p21) that were present in wild-type samples but absent from p53 knockout cells (Supplemental Fig. S4G,H). As seen in Figure 4B, in these same lysates we detected significant enrichment of p53 signal at the L1 5′UTR (Harris et al. 2009). For these assays, we used primer pairs specific for the L1 expression reporter in the L1 5′UTR and a negative control region at the eGFP 3′ end (see Fig. 4B). Importantly, signals detected in the 5′UTR in wild-type cells were absent from p53−/− cells, while signal detected at the negative control was similar in both cell types. To identify specific sequences needed for p53 binding, we produced a series of L1-5′UTR-eGFP variants that separately eliminated sites I, II, or III (as illustrated in Fig. 4A). Furthermore, like the parental reporters, these were similarly targeted to the identical AAVS1 site and confirmed to a single copy of the L1-5′UTR-eGFP reporter (Supplemental Figs. S3A, S4J). As seen in Figure 4C, deletion of sites I and II led to the complete loss of p53 enrichment at the L1-5′UTR, while deletion of site III had no effect on p53 levels. p53 enrichment at the negative control eGFP 3′ end was similar across all cell lines (Supplemental Fig. S4I).
Figure 4.

p53 physically binds to repress the L1-5′UTR The schematic in A shows empirically and computationally defined p53 binding sites in the 5′UTR of L1-5′UTR-eGFP. Note that site I, site II, and site III were independently targeted for deletion. (B) ChIP for p53 and control IgG performed with illustrated primer pairs (1 and 2) that uniquely detect binding at the L1 reporter. Note the absence of signal in p53−/− cells and at the L1-GFP negative control region spanning vector-derived sequences (Ctrl primer pair). Error bars represent 95% confidence intervals. (C) ChIP testing of p53-binding site deletions indicated in A along with the intact wild-type 5′UTR interval. Note, site I and site II contribute to p53 binding, but site III had no effect. Error bars represent 95% confidence intervals. In D, flow cytometry was used to detect for GFP intensities in the intact and mutant L1-5′UTR-eGFP reporters in A375 cells. Note that sites I and site II are required for p53 repression of the L1 expression reporter (see Supplemental Fig. S4L for another biological replicate). (E) Shows that corresponding eGFP mRNA transcript levels mirror GFP intensities seen in these same cells. Bar graphs are averages of three biological replicates. Error bars represent standard error of the mean.
To determine the functional relevance of p53 binding to the 5′UTR we inspected L1-5′UTR-eGFP expression in these same cells, using flow cytometry. As seen in Figure 4D and Supplemental Figure S4L, site I or site II deletions in the L1 5′UTR produced eGFP peaks in wild-type cells that were similar to those observed in p53−/− cells. Deletion of site III, in contrast, showed no such effect, producing eGFP peaks comparable with those observed in wild-type cells expressing the intact reporter (Fig. 4D; Supplemental Fig. S4L). Similarly, deletion of sites I and II led to increased eGFP RNA (Fig. 4E) and protein (Supplemental Fig. S4K), while deletion of site III had no effect on p53 repression. Together, these data expose cis sequences within the 5′UTR of L1 elements that mediate binding to p53 as well as transcriptional repression by p53.
De novo rearrangements in p53−/− cells are associated with retroelements
Genomic instability is a hallmark of cancer, and is commonly associated with loss or mutation of p53 (for review, see Eischen 2016). Using RNA sequencing data sets, we assessed the frequency of gene fusion transcripts, which may be indicative of genomic rearrangements, in A375 wild-type and p53 knockout cell lines. Importantly, early passage p53 knockout cells (<20 doublings) were used for these experiments, enabling us to ask whether genomic instability indicative of p53 mutant human cancers is rapidly induced by the loss of p53. We identified nine fusion transcripts in p53 knockout A375 cell lines that were absent in the parental strain, together with 13 fusion transcripts present in both parental and derived cell lines (Fig. 5A). Both intrachromasomal (n = 5) and interchromosomal (n = 4) fusions were observed (Supplemental Table S4) indicating that genomic stability is rapidly compromised following loss of p53.
Figure 5.

De novo rearrangements detected after p53 loss are associated with retroelements. (A) Circos plot of genomic rearrangements in A375 cells. Genomic rearrangements emerging after p53 loss are indicated (red lines) along with pre-existing rearrangements in the parental A375 cell line (gray lines). Repeat element proximity is indicated by red tiles (outer gray ring) along with annotations for rearrangement junctions (inner gray ring). (B) Genomic rearrangements occur at repeat elements in p53 knockout (n = 9 of 18 breakpoints) but not wild-type (n = 0 of 26 breakpoints) cells. (C) p53 knockout associated genomic rearrangements are commonly associated with LINE and SINE (n = 8 of 18 breakpoints), but not LTR or DNA repeat elements (n = 1 of 18 breakpoints).
To ask whether genomic rearrangements observed in p53 knockout cells were associated with retroelement activity, we inspected rearrangement junctions for the presence of repeat elements. Half (n = 9 of 18) of the de novo rearrangement breakpoints identified in A375 p53 knockout cells spanned an annotated retroelement, yet none of the pre-existing rearrangements were associated with retroelement sequences (Fig. 5B). Intriguingly, virtually all (n = 8 of 9) of these were SINEs or LINEs (Fig. 5C; Supplemental Fig. S5A), suggesting that these elements may have promoted the de novo rearrangements seen after p53 loss. Importantly, these analyses also showed that transposable element-initiated transcripts were not elevated in p53 knockout cell lines (Supplemental Fig. S5B), indicating that p53 does not globally impact transcriptional properties or chromatin features.
Immune response programs are activated in p53−/− cells
We interrogated RNA sequencing data sets from early passage (<20 doublings) A375 p53 knockout cells, allowing us to assess the immediate-early consequences of p53 loss on gene expression programs. For these studies, two independent wild-type (WT-1 and WT-2) and two independent mutant (A-2 and A-3) clones were used. In these RNA sequencing data sets, we identified 35 immunity related gene ontology designated genes that were induced after p53 loss (Fig. 6A). Importantly, we verified the induction of a subset of these genes by RT-ddPCR in p53 knockout clonal cell lines, A-1 and A-2 (Supplemental Fig. S6A). To better understand which immune pathways were induced in the absence of p53, we performed Gene Set Enrichment Analysis on our A375 RNA sequencing data sets (Subramanian et al. 2005). We identified two immune related gene sets that were induced at high significance (P < 0.001) in p53 knockout cells. Specifically, both the Inflammatory Response gene set (Fig. 6B,D) and TNFα signaling via NFKβ gene set (Fig. 6C,E) were profoundly enriched and, as seen in Supplemental Figure S6, B and C, we confirmed a subset of these by RT-ddPCR. These data suggest that induction of inflammatory pathways is an early consequence of p53 loss in vivo, extending previous links between chronic immune activity and advanced p53-driven cancers (Gudkov et al. 2011).
Figure 6.

Immunity associated gene expression programs are induced upon p53 loss. RNA sequencing data sets from two wild-type (WT-1 and WT-2) and two p53 knockout (A-2 and A-3) A375 cell lines were analyzed. The heat map in A displays genes more than fourfold induced in p53 nulls compared with WT controls. Heat maps in B and C similarly represent inflammatory response (B) and TNFα signaling (C) GSEA gene sets, as indicated. Gene set enrichment plots show highly significant enrichments scores for the inflammatory response (D) and TNFα signaling (E) gene sets in p53 knockout A375 cell lines (P < 0.001).
A LINE1 reverse transcriptase antagonist reverses immunity responses triggered by p53 loss
Results in Figures 1 and 6 establish that both retroelement derepression and immune pathway activation are early consequences of p53 loss, raising the possibility that retroelement activity could actually prompt immune activation in this context. Therefore, to test this possibility we treated A375 wild-type and p53 knockout cells with lamivudine (3TC), a potent L1 reverse transcriptase (RT) inhibitor (Jones et al. 2008; Dai et al. 2011; Banuelos-Sanchez et al. 2019), or vehicle (DMSO) for 3 d (Fig. 7A). We found that 3TC treatment of p53 knockout cells returned expression of immunity related genes tlr7, lcn2, itgb6, ifi441, and mmp1 to near wild-type levels (Fig. 7B, cf. open and closed bars). Likewise, the induction of Inflammatory or TNFα signaling genes cxcl8, pde4b, serpine1, tnfsf15, and ereg was fully prevented by treatment of p53 knockout cells with RT inhibitor 3TC (Fig. 7C,D). Altogether, 10 of the 14 tested immune pathway genes induced in p53 knockout cells were fully or substantially rescued by treatment with 3TC (genes unaffected by 3TC are shown in Supplemental Fig. S7A). We further note that induction of piwil4, which is thought to be an effector of the piRNAs was also rescued to wild-type expression levels in 3TC-treated p53 knockout cells (see Supplemental Fig. S7D,E). Importantly, 3TC did not affect growth rates (Supplemental Fig. S7B,C) nor did it affect induced L1-5′UTR RNA transcript levels (Fig. 7E). Taken together, these results indicate that effectors downstream from the L1 reverse transcriptase incite immune responses after p53 loss.
Figure 7.

A LINE1 antagonist, 3TC, prevents the inflammatory program triggered by p53 loss (A) Schematic of the experimental workflow. Expression of selected immunity (B) and inflammation response (C) genes was quantified using RT-ddPCR. Likewise, targets with overlapping immune pathway designations (D) and 5′ intact L1Hs (E) were similarly analyzed by RT-ddPCR. Wild-type and two p53 knockout lines treated in parallel with vehicle, DMSO, only (filled bars) or 3TC (open bars). Transcript levels were normalized to β-actin and their fold change was calculated relative to their wild-type values (see Supplemental Fig. S6). For B–D, bar graphs plot the average of three biological replicates and error bars represent SEM. For Figure E, bar graphs represent the average of three biological replicates and error bars indicate the standard deviation. Two tailed t-tests were performed for all samples. (*) P-value < 0.05, (**) P-value < 0.005, (***) P-value < 0.0005, (ns) not significant. Note that treatment with the reverse transcriptase inhibitor, 3TC, prevented induction of immune and inflammatory gene sets but did not impact L1Hs RNAs.
Discussion
Here we established p53 as a conserved suppressor of retroelements in unstressed human somatic cells. Accordingly, we further showed that loss of this repressive activity was permissive for L1 eruptions that triggered destabilized genomes and inflammatory responses commonly seen in p53 mutant cancers (Gudkov et al. 2011; Eischen 2016). Using CRISPR methods we generated a series of p53 knockouts in diverse human cell lines and observed generally derepressed L1 activity in both cancer derived (e.g. A375 and U2OS cell lines) as well as normal lung tissue derived HBEC3kt cells (Fig. 1). In p53 deficient cells, elevated L1Hs derived RNAs were also tightly correlated with elevated ORF1 protein and de novo retroinserts (Fig. 2). This effect impacted native and synthetic L1 variants that were integrated and engineered for unique detection. Notably, derepression of individually identifiable L1 retroelements was stochastic and varied between p53 mutant cell lines (Supplemental Fig. S1A,B). Therefore, while p53 loss is a condition that is permissive for L1 eruptions, unknown inciting factors also govern individual elements in this context (Wylie et al. 2016b). Conceivably, p53 restriction could target any step in the L1 retrotransposon lifecycle, but several lines of evidence indicate that suppression occurs at the level of transcription. First, acute induction of these transcripts occurred when p53 was removed (Fig. 1) yet known retroelement silencers were not affected (Supplemental Fig. S4A–F). Second, the internal L1Hs promoter region conferred p53 regulation when fused to reporter templates (Fig. 3B,C). Third, the deposition of repressive histone marks at the internal L1Hs promoter region was clearly p53 dependent (Fig. 3D,E). Together, these results strongly indicate that p53 prevents L1 expression by preventing L1 transcription.
To test the possibility of direct transrepression by p53 at the internal L1 promoter region, we functionally tested three distinct p53 binding sites by independently eliminating each. Two sites, (sites I and II) were clearly essential for binding and repression while a third site (site III) was dispensable for both (Fig. 4C). Surprisingly, loss of either site I or site II completely disrupted binding and completely abolished repression, suggesting that sites I and II are concurrently needed (Fig. 4D,E; Supplemental Fig. S4K,L). This “all-or-none” property suggests that cooperative p53 binding could mediate direct transrepression at the internal L1 promoter. Interestingly, Harris et al. (2009) inspected these same p53 sites and found that site I, but not site II, was needed for stimulus-dependent transcriptional induction. Therefore, a single site can produce opposing outputs and it is likely that additional unknown factors specify the direction of activity in vivo. Several technical considerations could also account for our departures from Harris et al. (2009). For example, Harris et al. (2009) used transient cotransfections to follow episomal reporters as p53 was overexpressed, yet here we tracked integrated reporters after p53 was removed. Second, the reporters in Harris et al. (2009) lack ∼100 bp of the L1 promoter region we used here. Third, Harris et al. (2009) conducted their assays in HCT116 cells, which, for unknown reasons, was the only line of four tested here that did not show L1 derepression after p53 loss. Despite these differences, p53 clearly engages the internal promoter of these transposons and, accordingly, future studies may identify instructive mechanisms that specify inductive versus repressive outcomes.
p53 is commonly studied as a stress-responsive transactivator (Lane and Levine 2010; Chen 2016) and, within this framework, down-regulation of target genes is widely considered to be a secondary effect (for review, see Sullivan et al. 2018). In certain stimulus-dependent contexts, the occlusion of positive transcription factors has been suggested to account for p53 suppression (Bargonetti et al. 1997; Ori et al. 1998; St Clair et al. 2004) but, to our knowledge, the findings presented here constitute the first example of direct and constitutive transrepression mediated by p53 in unstressed cells. Indeed, our data showing that p53 binds and recruits repressive histone marks to LINE1s but not to canonical transactivation targets such as p21 and puma (Fig. 3D,E; Supplemental Figs. S3H,I, S4G,H) are inconsistent with a simple occlusion model of p53 gene repression. Further work is needed to establish precise mechanisms and explore perhaps whether regulatory RNAs and/or physical interactions with epigenetic modifiers are involved. Since direct tonic repression accounts for p53-mediated suppression of LINE1s, and since these transposons appear to constitute drivers that remodel cancer genomes and incite immune responses (Lee et al. 2012; Tubio et al. 2014; Rodriguez-Martin et al. 2020), these observations could have important implications for understanding p53 tumor suppression.
Consistent with this hypothesis, studies in Figures 5–7 illustrate how derepressed retrotransposons present acute oncogenic threats that are typically associated with p53 loss. The prevalence of homologous recombination seen in Figure 5 is one compelling example since, unlike pre-existing alterations, the chromosomal rearrangements occurring after p53 removal were highly associated with retroelements at their junctions (Fig. 5). Furthermore, these de novo rearrangements occurred within 20 cell doublings after CRISPR editing, raising the possibility that p53 continually acts to suppress homologous recombination between distal retrotransposon sequences. Consistent with this possibility, chromatin features are indeed important determinants of recombination frequency (Stapley et al. 2017) and, as seen in Figure 3, D and E, we showed that p53 acts to maintain repressive chromatin marks at LINE1s. Hence, facilitated recombination mediated by derepressed retroelements could represent important early events that destabilize genomes after p53 loss.
Other oncogenic threats posed by L1s relate to inflammatory responses (Leonova et al. 2013; Brégnard et al. 2016; Saleh et al. 2019) and two lines of evidence here show that hyperactive L1s elicit acute inflammatory transcriptional programs in the absence of p53. First, consistent with previous reports (Gudkov et al. 2011) retroelement derepression and immune gene induction both occurred rapidly and simultaneously after p53 loss (Fig. 6). Second, treatment with 3TC definitively prevented the induction of nearly all inflammatory genes (Fig. 7). Since 3TC is a potent inhibitor of the L1 reverse transcriptase (Jones et al. 2008; Dai et al. 2011; Banuelos-Sanchez et al. 2019), these results indicate that L1s can drive inflammatory genes when p53 is abolished. Furthermore, these results also suggest that the trigger eliciting immune signatures lies downstream from the LINE1 enzyme and could possibly involve RNA–DNA hybrids and/or the integration process itself. Taken together, these studies establish direct transrepression of human L1 transposons as a constitutive p53 function that, if compromised, promotes oncogenic features seen in p53 driven cancers.
Materials and methods
Generation of p53 knockout cells using CRISPR Cas9
CRISPR cas9 editing (pX458) was used to generate p53 knockout cell lines. p53 knockout clones were identified by Western blot using p53 monoclonal DO-1 (sc-126) and Sanger sequencing was performed to determine the nature of each p53 knockout allele. L1-ORF1p expression levels were examined using anti-L1-ORF1p antibodies. Fiji software (Schindelin et al. 2012) was used to measure the band intensities of L1ORF1p and β-actin from Western blot experiments.
Generation of huAAVS1 targeted L1-5′UTR-EGFP reporter cells
The WT L1 5′UTR and 5′UTR sites I, II, and III deletion mutants were cloned at the FseI and AfeI sites of the huAAVS1-egfp-Hyg donor plasmid (kind gift from Joshua Mendel laboratory, University of Texas Southwestern). Cells were transfected with huAAVS1-TALENL (Addgene 59025), huAAVS1-TALENR (Addgene 59026), and donor plasmids. Three days after transfection, GFP positive single cells were sorted into 96-well plates by FACS. Once confluent, targeted knock-in was verified using PCR primers indicated in Supplemental Table S5. Detailed methods are in the Supplemental Material.
FACS analysis
All FACS analyses were performed on an LSR Fortessa SCC using equal numbers of cells. Live–dead gating was performed based on forward scatter versus side scatter profile. Data analysis and percent GFP-positive cell counts were performed in FloJo. When analyzing GFP signals, untransfected (see Supplemental Fig. S3F,G) cells were used for the baseline gating to identify EGF-positive cells. EGFP fluorescence intensity of WT-L15′UTR-EGFP cells was used to create a second gating to identify bright GFP-positive cells in p53−/−- L15′UTR-EGFP samples (see Supplemental Tables S2, S3).
3TC experiment
WT and p53 knockout A375 cells were treated with DMSO or 10 µM 3TC for 72 h.
RNA expression
Total RNA was extracted with TRIzol (Invitrogen), followed by DNAse treatment using Turbo DNase (Ambion). Droplet digital PCR (ddPCR) was performed using Bio-Rad's EvaGreen system. Gene expression was normalized to β-actin expression, and fold induction was calculated relative to the wild type. See Supplemental Table S5 for the primer sequences used in ddPCR experiments.
Retrotransposition assay
Cells (2 × 105) were transfected with equal amounts of 99-GFP-LRE3 plasmid, as described previously (Ostertag et al. 2000). After 72 h, cells were selected for puromycin resistance. Flow cytometry was used to determine the percentage of EGFP-positive cells 9–10 d after transfection. For GFP integration assays, 2 µg of genomic DNA from each sample was digested for 1 h with EcoRI (NEB) followed by ddPCR using primers detecting unspliced GFP as indicated in Supplemental Table S5. To calculate relative retrotransposition rate, ddPCR was performed on genomic DNA using primers detecting spliced EGFP (see Supplemental Table S5) and normalized against the single-copy gene puma. Detailed methods are in the Supplemental Material.
Statistical analysis
All quantitative data were collected from experiments performed in two to three biological replicates. Two-tailed Student's t-tests were performed to identify statistically significant difference (cutoff P < 0.05).
RNA sequencing and data analysis
RNA was extracted from A375 wild-type or A375 p53−/− cells using Trizol (Invitrogen) followed by DNase treatment using TurboDNase (Ambion). Samples with RNA integrity scores >9.0, was used for sequencing. Sequencing libraries were prepared following the manufacturer's instructions (NEB E7750S with poly(A enrichment module NEB E7490S) and sequenced on an Illumina NextSeq to an average sequencing depth of 40 million 150-bp paired-end reads per sample. Reads were aligned to the hg38 assembly of the human genome with STAR (Dobin et al. 2013). HTseq counts tables were used as input to perform normalization and differential gene expression analysis with Deseq2 (Love et al. 2014; Anders et al. 2015). STAR-Fusion was used to identify potential gene fusion products (Haas et al. 2017). Gene ontology annotations were performed using the DAVID online resource (Huang da et al. 2009a,b). GSEA analyses were performed using the h.all.v7.0.symbols.gmt gene set (Subramanian et al. 2005). Retroelement expression levels were determined using a customized pipeline adapted from (Wylie et al. 2016b). LIONS analytical pipeline was used to measure transposon initiated transcription (Babaian et al. 2019). RNA sequencing data sets were deposited with the Gene Expression Omnibus (GEO) database under accession number GSE159134. Detailed methods are in the Supplemental Material.
p53-binding site prediction
Putative binding sites within the L1 5′UTR were bioinformatically identified using the p53scan program (Smeenk et al. 2008) and Homer's scanMotifGenomeWide module (Heinz et al. 2010). Individual p53 motif files for right and left p53 consensus decamers were used for motif detection.
ChIP ddPCR
Chromatin immunoprecipitation protocol was adapted from Carey et al. (2009). Briefly, A375 cells were fixed with 1% formaldehyde, quenched with glycine, and nuclear lysates were prepared. DNA was sheared to an average of 300–700 bp with a Covaris Sonicator (5 min, 200 cpb, intensity 5). Sonicated lysate was diluted 1:10, aliquoted, and incubated overnight with antibody. Antibody-bound chromatin was precipitated with 25 µL of protein A/G beads (EMD Millipore IP0515ML). Eluted chromatin was treated with RNase and proteinase K, cross-links were reversed, and samples were purified by standard phenol:chloroform extraction and ethanol-precipitated. ChIP enrichment was quantified by ddPCR with EvaGreen (Bio-Rad 1864034) on a QX200 droplet reader (Robin et al. 2014; Wylie et al. 2016b). Antibodies used in these experiments included p53 ChIP (Santa Cruz Biotechnology sc-126), IgG (Diagenode C15400001), H3 (Abcam ab1791), H3K27m3 (Active Motif 39155), and H3K9m3 (Diagenode C15410058). Detailed materials and methods are in the Supplemental Material.
Supplementary Material
Acknowledgments
We thank N.L. and T.S. (University of Texas Southwestern Flow Cytometry) for their assistance in FACS. We thank the McDermott Center Next-Generation Core Facility at University of Texas Southwestern. We thank J.R.P. from the Jerry Shay laboratory at University of Texas Southwestern for intellectual input in p53 KO generation. This work was supported by grants to B.T. and S.D. (Cancer Prevention and Research Institute of Texas [CPRIT] training grant RP160157), A.E.J. (American Cancer Society 128847-PF-15-160-01-DDC), and J.M.A. (National Institutes of Health [NIH] R01GM115682, NIH R01CA222579, and CPRIT RP170086). We acknowledge the assistance of the University of Texas Southwestern Live-Cell Imaging Facility, a shared resource of the Harold C. Simmons Cancer Center, supported in part by National Cancer Institute Cancer Center Support Grant P30 CA142543. We acknowledge the Texas Advanced Computing Center at the University of Texas at Austin (TACC) for providing HPC resources used in the analysis of next-generation sequencing data sets.
Author contributions: B.T. and A.E.J. conceived the studies, performed experiments, analyzed data, and wrote the manuscript. J.M.A. designed the study, analyzed data, and edited the manuscript. C.J.C. performed experiments. S.K.R. assisted in screening reporter cells. S.D. assisted in screening p53 knockout cells.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.343186.120.
References
- Alexandrova EA, Olovnikov IA, Malakhova GV, Zabolotneva AA, Suntsova MV, Dmitriev SE, Buzdin AA. 2012. Sense transcripts originated from an internal part of the human retrotransposon LINE-1 5′ UTR. Gene 511: 46–53. 10.1016/j.gene.2012.09.026 [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. 2015. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31: 166–169. 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babaian A, Thompson IR, Lever J, Gagnier L, Karimi MM, Mager DL. 2019. LIONS: analysis suite for detecting and quantifying transposable element initiated transcription from RNA-seq. Bioinformatics 35: 3839–3841. 10.1093/bioinformatics/btz130 [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T. 2011. Regulation of chromatin by histone modifications. Cell Res 21: 381–395. 10.1038/cr.2011.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banuelos-Sanchez G, Sanchez L, Benitez-Guijarro M, Sanchez-Carnerero V, Salvador-Palomeque C, Tristan-Ramos P, Benkaddour-Boumzaouad M, Morell S, Garcia-Puche JL, Heras SR, et al. 2019. Synthesis and characterization of specific reverse transcriptase inhibitors for mammalian LINE-1 retrotransposons. Cell Chem Biol 26: 1095–1109.e14. 10.1016/j.chembiol.2019.04.010 [DOI] [PubMed] [Google Scholar]
- Bargonetti J, Chicas A, White D, Prives C. 1997. p53 represses Sp1 DNA binding and HIV-LTR directed transcription. Cell Mol Biol (Noisy-le-grand) 43: 935–949. [PubMed] [Google Scholar]
- Bieging KT, Mello SS, Attardi LD. 2014. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer 14: 359–370. 10.1038/nrc3711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, Kenzelmann Broz D, Basak S, Park EJ, McLaughlin ME, et al. 2011. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 145: 571–583. 10.1016/j.cell.2011.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brégnard C, Guerra J, Déjardin S, Passalacqua F, Benkirane M, Laguette N. 2016. Upregulated LINE-1 activity in the Fanconi anemia cancer susceptibility syndrome leads to spontaneous Pro-inflammatory cytokine production. EBioMedicine 8: 184–194. 10.1016/j.ebiom.2016.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs EM, Ha S, Mita P, Brittingham G, Sciamanna I, Spadafora C, Logan SK. 2018. Long interspersed nuclear element-1 expression and retrotransposition in prostate cancer cells. Mob DNA 9: 1 10.1186/s13100-017-0106-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouha B, Schustak J, Badge RM, Lutz-Prigge S, Farley AH, Moran JV, Kazazian HH Jr. 2003. Hot L1s account for the bulk of retrotransposition in the human population. Proc Natl Acad Sci 100: 5280–5285. 10.1073/pnas.0831042100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns KH. 2017. Transposable elements in cancer. Nat Rev Cancer 17: 415–424. 10.1038/nrc.2017.35 [DOI] [PubMed] [Google Scholar]
- Callahan KE, Hickman AB, Jones CE, Ghirlando R, Furano AV. 2012. Polymerization and nucleic acid-binding properties of human L1 ORF1 protein. Nucleic Acids Res 40: 813–827. 10.1093/nar/gkr728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey MF, Peterson CL, Smale ST. 2009. Chromatin immunoprecipitation (ChIP). Cold Spring Harb Protoc 2009: pdb.prot5279. [DOI] [PubMed] [Google Scholar]
- Chen J. 2016. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb Perspect Med 6: a026104 10.1101/cshperspect.a026104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenais B. 2015. Transposable elements in cancer and other human diseases. Curr Cancer Drug Targets 15: 227–242. 10.2174/1568009615666150317122506 [DOI] [PubMed] [Google Scholar]
- Coufal NG, Garcia-Perez JL, Peng GE, Marchetto MC, Muotri AR, Mu Y, Carson CT, Macia A, Moran JV, Gage FH. 2011. Ataxia telangiectasia mutated (ATM) modulates long interspersed element-1 (L1) retrotransposition in human neural stem cells. Proc Natl Acad Sci 108: 20382–20387. 10.1073/pnas.1100273108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui F, Sirotin MV, Zhurkin VB. 2011. Impact of Alu repeats on the evolution of human p53 binding sites. Biol Direct 6: 2 10.1186/1745-6150-6-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai L, Huang Q, Boeke JD. 2011. Effect of reverse transcriptase inhibitors on LINE-1 and Ty1 reverse transcriptase activities and on LINE-1 retrotransposition. BMC Biochem 12: 18 10.1186/1471-2091-12-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombroski BA, Feng Q, Mathias SL, Sassaman DM, Scott AF, Kazazian HH Jr., Boeke JD. 1994. An in vivo assay for the reverse transcriptase of human retrotransposon L1 in Saccharomyces cerevisiae. Mol Cell Biol 14: 4485–4492. 10.1128/MCB.14.7.4485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eischen CM. 2016. Genome stability requires p53. Cold Spring Harb Perspect Med 6: a026096 10.1101/cshperspect.a026096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Moran JV, Kazazian HH Jr., Boeke JD. 1996. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell 87: 905–916. 10.1016/S0092-8674(00)81997-2 [DOI] [PubMed] [Google Scholar]
- Gudkov AV, Gurova KV, Komarova EA. 2011. Inflammation and p53: a tale of Two stresses. Genes Cancer 2: 503–516. 10.1177/1947601911409747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, Dobin A, Stransky N, Li B, Yang X, Tickle T, Bankapur A, Ganote C, Doak TG, Pochet N, et al. 2017. STAR-Fusion: fast and accurate fusion transcript detection from RNA-seq. bioRxiv 120295. [Google Scholar]
- Hancks DC, Kazazian HH Jr. 2012. Active human retrotransposons: variation and disease. Curr Opin Genet Dev 22: 191–203. 10.1016/j.gde.2012.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancks DC, Kazazian HH Jr. 2016. Roles for retrotransposon insertions in human disease. Mob DNA 7: 9 10.1186/s13100-016-0065-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris CR, Dewan A, Zupnick A, Normart R, Gabriel A, Prives C, Levine AJ, Hoh J. 2009. p53 responsive elements in human retrotransposons. Oncogene 28: 3857–3865. 10.1038/onc.2009.246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589. 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. 2009a. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13. 10.1093/nar/gkn923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. 2009b. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57. 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- Jones RB, Garrison KE, Wong JC, Duan EH, Nixon DF, Ostrowski MA. 2008. Nucleoside analogue reverse transcriptase inhibitors differentially inhibit human LINE-1 retrotransposition. PLoS One 3: e1547 10.1371/journal.pone.0001547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp JR, Longworth MS. 2015. Crossing the LINE toward genomic instability: LINE-1 retrotransposition in cancer. Front Chem 3: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopera HC, Larson PA, Moldovan JB, Richardson SR, Liu Y, Moran JV. 2016. LINE-1 cultured cell retrotransposition assay. Methods Mol Biol 1400: 139–156. 10.1007/978-1-4939-3372-3_10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860–921. 10.1038/35057062 [DOI] [PubMed] [Google Scholar]
- Lane D, Levine A. 2010. p53 research: the past thirty years and the next thirty years. Cold Spring Harb Perspect Biol 2: a000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Iskow R, Yang L, Gokcumen O, Haseley P, Luquette LJ, Lohr JG, Harris CC, Ding L, Wilson RK, et al. 2012. Landscape of somatic retrotransposition in human cancers. Science 337: 967–971. 10.1126/science.1222077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonova KI, Brodsky L, Lipchick B, Pal M, Novototskaya L, Chenchik AA, Sen GC, Komarova EA, Gudkov AV. 2013. p53 cooperates with DNA methylation and a suicidal interferon response to maintain epigenetic silencing of repeats and noncoding RNAs. Proc Natl Acad Sci USA 110: E89–E98. 10.1073/pnas.1216922110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R, Gu W. 2012. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 149: 1269–1283. 10.1016/j.cell.2012.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SL. 2010. Nucleic acid chaperone properties of ORF1p from the non-LTR retrotransposon, LINE-1. RNA Biol 7: 706–711. 10.4161/rna.7.6.13766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SL, Cruceanu M, Branciforte D, Wai-Lun Li P, Kwok SC, Hodges RS, Williams MC. 2005. LINE-1 retrotransposition requires the nucleic acid chaperone activity of the ORF1 protein. J Mol Biol 348: 549–561. 10.1016/j.jmb.2005.03.003 [DOI] [PubMed] [Google Scholar]
- Molaro A, Malik HS. 2016. Hide and seek: how chromatin-based pathways silence retroelements in the mammalian germline. Curr Opin Genet Dev 37: 51–58. 10.1016/j.gde.2015.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran JV, Holmes SE, Naas TP, DeBerardinis RJ, Boeke JD, Kazazian HH Jr. 1996. High frequency retrotransposition in cultured mammalian cells. Cell 87: 917–927. 10.1016/S0092-8674(00)81998-4 [DOI] [PubMed] [Google Scholar]
- Muotri AR, Marchetto MCN, Coufal NG, Oefner R, Yeo G, Nakashima K, Gage FH. 2010. L1 retrotransposition in neurons is modulated by MeCP2. Nature 468: 443–446. 10.1038/nature09544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ori A, Zauberman A, Doitsh G, Paran N, Oren M, Shaul Y. 1998. p53 binds and represses the HBV enhancer: an adjacent enhancer element can reverse the transcription effect of p53. Embo j 17: 544–553. 10.1093/emboj/17.2.544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostertag EM, Prak ET, DeBerardinis RJ, Moran JV, Kazazian HH Jr. 2000. Determination of L1 retrotransposition kinetics in cultured cells. Nucleic Acids Res 28: 1418–1423. 10.1093/nar/28.6.1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe C, Vargas-Landin DB, Doucet AJ, van Essen D, Vera-Otarola J, Kuciak M, Corbin A, Nigumann P, Cristofari G. 2016. Activation of individual L1 retrotransposon instances is restricted to cell-type dependent permissive loci. Elife 5: e13926 10.7554/eLife.13926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin JD, Ludlow AT, Batten K, Magdinier F, Stadler G, Wagner KR, Shay JW, Wright WE. 2014. Telomere position effect: regulation of gene expression with progressive telomere shortening over long distances. Genes Dev 28: 2464–2476. 10.1101/gad.251041.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodić N, Sharma R, Sharma R, Zampella J, Dai L, Taylor MS, Hruban RH, Iacobuzio-Donahue CA, Maitra A, Torbenson MS, et al. 2014. Long interspersed element-1 protein expression is a hallmark of many human cancers. Am J Pathol 184: 1280–1286. 10.1016/j.ajpath.2014.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Martin B, Alvarez EG, Baez-Ortega A, Zamora J, Supek F, Demeulemeester J, Santamarina M, Ju YS, Temes J, Garcia-Souto D, et al. 2020. Pan-cancer analysis of whole genomes identifies driver rearrangements promoted by LINE-1 retrotransposition. Nat Genet 52: 306–319. 10.1038/s41588-019-0562-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh A, Macia A, Muotri AR. 2019. Transposable elements, inflammation, and neurological disease. Front Neurol 10: 894 10.3389/fneur.2019.00894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9: 676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott EC, Devine SE. 2017. The role of somatic L1 retrotransposition in human cancers. Viruses 9: 131 10.3390/v9060131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeenk L, van Heeringen SJ, Koeppel M, van Driel MA, Bartels SJ, Akkers RC, Denissov S, Stunnenberg HG, Lohrum M. 2008. Characterization of genome-wide p53-binding sites upon stress response. Nucleic Acids Res 36: 3639–3654. 10.1093/nar/gkn232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapley J, Feulner PGD, Johnston SE, Santure AW, Smadja CM. 2017. Variation in recombination frequency and distribution across eukaryotes: patterns and processes. Philos Trans R Soc Lond B Biol Sci 372: 20160455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Clair S, Giono L, Varmeh-Ziaie S, Resnick-Silverman L, Liu WJ, Padi A, Dastidar J, DaCosta A, Mattia M, Manfredi JJ. 2004. DNA damage-induced downregulation of Cdc25C is mediated by p53 via two independent mechanisms: one involves direct binding to the cdc25C promoter. Mol Cell 16: 725–736. 10.1016/j.molcel.2004.11.002 [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan KD, Galbraith MD, Andrysik Z, Espinosa JM. 2018. Mechanisms of transcriptional regulation by p53. Cell Death Differ 25: 133–143. 10.1038/cdd.2017.174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swergold GD. 1990. Identification, characterization, and cell specificity of a human LINE-1 promoter. Mol Cell Biol 10: 6718–6729. 10.1128/MCB.10.12.6718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari B, Kurtz P, Jones AE, Wylie A, Amatruda JF, Boggupalli DP, Gonsalvez GB, Abrams JM. 2017. Retrotransposons mimic germ plasm determinants to promote transgenerational inheritance. Curr Biol 27: 3010–3016.e3. 10.1016/j.cub.2017.08.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari B, Jones AE, Abrams JM. 2018. Transposons, p53 and genome security. Trends Genet 34: 846–855. 10.1016/j.tig.2018.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubio JMC, Li Y, Ju YS, Martincorena I, Cooke SL, Tojo M, Gundem G, Pipinikas CP, Zamora J, Raine K, et al. 2014. Mobile DNA in cancer. extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science 345: 1251343 10.1126/science.1251343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente LJ, Gray DH, Michalak EM, Pinon-Hofbauer J, Egle A, Scott CL, Janic A, Strasser A. 2013. p53 efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, puma, and noxa. Cell Rep 3: 1339–1345. 10.1016/j.celrep.2013.04.012 [DOI] [PubMed] [Google Scholar]
- Volkman HE, Stetson DB. 2014. The enemy within: endogenous retroelements and autoimmune disease. Nat Immunol 15: 415–422. 10.1038/ni.2872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Zeng J, Lowe CB, Sellers RG, Salama SR, Yang M, Burgess SM, Brachmann RK, Haussler D. 2007. Species-specific endogenous retroviruses shape the transcriptional network of the human tumor suppressor protein p53. Proc Natl Acad Sci 104: 18613–18618. 10.1073/pnas.0703637104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylie A, Jones AE, Abrams JM. 2016a. p53 in the game of transposons. Bioessays 38: 1111–1116. 10.1002/bies.201600115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylie A, Jones AE, D'Brot A, Lu WJ, Kurtz P, Moran JV, Rakheja D, Chen KS, Hammer RE, Comerford SA, et al. 2016b. p53 genes function to restrain mobile elements. Genes Dev 30: 64–77. 10.1101/gad.266098.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Wang PJ. 2016. Multiple LINEs of retrotransposon silencing mechanisms in the mammalian germline. Semin Cell Dev Biol 59: 118–125. 10.1016/j.semcdb.2016.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemojtel T, Kielbasa SM, Arndt PF, Chung HR, Vingron M. 2009. Methylation and deamination of cpGs generate p53-binding sites on a genomic scale. Trends Genet 25: 63–66. 10.1016/j.tig.2008.11.005 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.