

Abstract
Huntington’s disease (HD) is caused by an expansion of a poly glutamine (polyQ) stretch in the huntingtin protein (HTT) that is necessary to cause pathology and formation of HTT aggregates. Here we ask whether expanded polyQ is sufficient to cause pathology and aggregate formation. By addressing the sufficiency question, one can identify cellular processes and structural parameters that influence HD pathology and HTT subcellular behavior (i.e. aggregation state and subcellular location). Using Drosophila, we compare the effects of expressing mutant full-length human HTT (fl-mHTT) to the effects of mutant human HTTexon1 and to two commonly used synthetic fragments, HTT171 and shortstop (HTT118). Expanded polyQ alone is not sufficient to cause inclusion formation since full-length HTT and HTTex1 with expanded polyQ are both toxic although full-length HTT remains diffuse while HTTex1 forms inclusions. Further, inclusions are not sufficient to cause pathology since HTT171-120Q forms inclusions but is benign and co-expression of HTT171-120Q with non-aggregating pathogenic fl-mHTT recruits fl-mHTT to aggregates and rescues its pathogenicity. Additionally, the influence of sequences outside the expanded polyQ domain is revealed by finding that small modifications to the HTT118 or HTT171 fragments can dramatically alter their subcellular behavior and pathogenicity. Finally, mutant HTT subcellular behavior is strongly modified by different cell and tissue environments (e.g. fl-mHTT appears as diffuse nuclear in one tissue and diffuse cytoplasmic in another but toxic in both). These observations underscore the importance of cellular and structural context for the interpretation and comparison of experiments using different fragments and tissues to report the effects of expanded polyQ.
Introduction
How does the presence of an expanded polyglutamine repeat (polyQ) in the mutant huntingtin protein (mHTT) lead to Huntington’s disease (OMIM:143100)? The molecular basis of this pathology remains elusive. While the presence of an expanded polyQ domain clearly correlates with pathology, numerous studies have demonstrated that sequences outside the expanded polyQ domain can also influence pathology. Here we ask whether expanded polyQ is sufficient to cause pathology, whether derivatives of full-length HTT (e.g. HTTexon1 (HTTex1) and other fragments) lead to overlapping or distinct pathologies, whether formation of intracellular inclusions is either necessary or sufficient to cause pathology, and whether tissue-specific cellular modifiers can alter HTT behavior.
In man, HTT is expressed globally as a 3144aa protein. Pathogenic versions contain a polyQ-encoding CAG expansion in the N-terminal region beginning at amino acid 17 (1). Numerous studies have shown that small N-terminal fragments of HTT occur in patients as a result of proteolytic processing (2,3) and abnormal RNA splicing (4,5), and staining with antibodies directed at specific epitopes in the HTT protein reveals that small amino-terminal fragments of HTT are the predominant species found in post-mortem patient inclusions, and these fragments are able to cause pathology in experimental systems (e.g. (3,4,6–12)). Indeed, inclusions in post-mortem brain tissue react with N-terminal HTT antibodies (6,13), but not with antibodies that recognize epitopes beyond the first approximately 115 amino acids (2,14). Determining the relative pathogenic contributions of the full-length protein versus specific cleavage or splicing products in mammalian systems is complicated by the natural processing of HTT proteins (15–17). These processing events raise the possibility that observations made using full-length knock-in models or other long-fragment mammalian models may include contributions from short proteolytic or mis-spliced fragments.
In this report, we use a well-validated Drosophila model of HD to ask whether expanded polyQ is intrinsically toxic and to identify possible sources of intrinsic and/or extrinsic modifiers of HTT toxicity. In this model, transgenic human HTT does not undergo proteolytic processing or splicing aberrations (18), the genetic background can be controlled and transgenes can be inserted into a defined chromosomal location to avoid integration-site differences (19). These advantages provide an excellent platform for comparing the pathogenic properties and cellular behavior (i.e. propensity to form aggregates and subcellular location) of different HTT peptides.
Here we report that although challenge with full-length expanded-Q HTT (flHTT-120Q) or with the naturally occurring HTT exon 1 (HTTex1-120Q) fragment causes toxicity, they display distinct intracellular and biochemical behaviors. Additional studies with expanded-Q HTT171 (HTT171-120Q) and shortstop (HTT118-120Q) fragments (14,17,20,21) support the conclusion that even subtle changes in peptide structure can have profound impacts on subcellular behavior and pathology.
Results
Construction of targeted full-length, exon1, 118 & 171
We inserted human fl-HTT as well as HTTex1 (90 amino acids), HTT118 (the ‘shortstop’ fragment) (17) and HTT171 (14) with either 120 or 25 Qs under the control of the yeast UAS promoter into the same chromosomal location (51D) and in the same orientation in a common inbred host Drosophila line, using the phiC31 targeted-insertion system (22). Transgenes under the control of the UAS promoter can be expressed with the Gal4/UAS system (23) by crossing to a driver line that has the yeast Gal4 transcriptional activator under the control of various tissue-specific promoters. Importantly, there is no detectable proteolytic processing of ubiquitously expressed fl-mHTT or intermediate-sized fragments to discrete smaller fragments in Drosophila (18). We also established a randomly-inserted fl-mHTT-expressing line inserted at 55C6 that produces higher levels of lethality than the same construct inserted into the 51D targeting site.
Expanded polyQ is necessary but not sufficient for pathology
Although striatal atrophy is one of the most notable consequences of HD, mammalian HTT is expressed ubiquitously and affects many tissues (1,24,25). To mimic this situation in Drosophila, we expressed different HTT constructs ubiquitously (Fig. 1A). In these experiments, all unexpanded polyQ HTT constructs (25Q) were benign, producing neither detectable phenotypes nor increased lethality. In contrast, only constructs with expanded polyQ exhibited lethality and, all the expanded constructs also increased the incidence of at least some of the following phenotypes in surviving adult flies: failure of abdominal dorsal closure, failure of thoracic dorsal closure and/or rotated male genitalia. In addition, altering levels of HTT expression (by rearing flies with Gal4-driven constructs at different temperatures) caused phenotypes to appear in different tissues supporting the view that multiple cell types are sensitive to expanded polyQ insult. The absence of pathology in the unexpanded-expressing flies demonstrates that expanded polyQ is required to produce pathology. However, the phenotypic severity can be modified by the expanded polyQ’s protein context. For example, at 25°, expression of expanded polyQ (120Q) in the context of both fl-mHTT and HTTex1 is lethal (Fig. 1A) although animals expressing fl-mHTT die in the early pupal phase, while animals expressing HTTex1-120Q die earlier in the larval phase, suggesting that expanded exon1 is more toxic than expanded fl-mHTT. Indeed, at lower levels of expression, HTTex1 120Q remains larval lethal, while some flies expressing fl-mHTT are able to eclose, further illustrating the effect of protein context on polyQ toxicity. Thus, expanded polyQ is necessary to drive HTT-based pathology, and HTTex1-120Q is more toxic than the full-length protein with the identical expanded polyQ domain.
Figure 1.
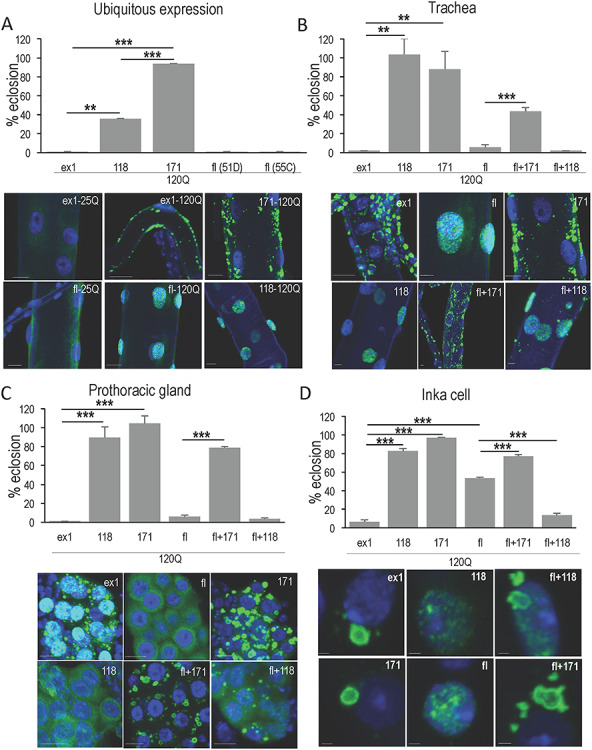
Cellular context influences subcellular behavior and pathology. (A) Ubiquitous expression with da > Gal4 reveals differences in pathogenic potential and subcellular behavior of HTT peptides. Various UAS > HTT/act > GFP males were crossed to females homozygous for the ubiquitously expressed da > Gal-4 driver, and survival to adulthood (eclosion) was monitored. Transgenes included HTT exon1 (ex1-120Q), full-length HTT (fl-120Q), HTT 118, shortstop (118-120Q) and HTT 171 (171-120Q). The percent flies eclosing as adults is calculated in reference to the internal control of non-expressing siblings (i.e. the number of UAS > HTT/+; +/da > Gal4 flies divided by UAS > HTT/+; +/act > GFP flies). Flies expressing unexpanded 25Q versions of these transgenes eclosed at a rate indistinguishable from the non-expressing controls. Peptide composition also influences the subcellular location of HTT transgenes here shown in trachea. HTTex1 with unexpanded polyQ (25 Qs) presents as diffuse cytoplasmic (ex1-25Q) while HTTex1 with 120Qs forms large cytoplasmic aggregates in tracheal cells (ex1-120Q). HTT118 with 120Qs is diffuse nuclear (118-120Q) while HTT171 forms cytoplasmic aggregates (117-120Q). Full-length HTT with 25Q is also diffuse cytoplasmic (fl-25Q) while expansion of the polyQ in full-length HTT converts it to diffuse nuclear in tracheal cells (fl-120Q). Scale bars are 10 μm. Since ubiquitous expression revealed a number of phenotypes that resembled those seen in different tissues, we tested whether multiple tissues exhibit pathology. (B) The toxicity of ex1, fl, 171 and 118 when driven exclusively in the trachea with btl > Gal4 at 25° was evaluated and the subcellular behavior determined by immunofluorescence. Expression of HTTex1-120Q and flHTT-120Q exclusively in trachea produced virtually complete toxicity similar to that seen with ubiquitous expression in (A). Many trachea in both HTTex1 and fl-mHTT expressing animals were visibly defective with gaps and constrictions evident (not shown) even though HTTex1 is clearly cytoplasmic and aggregated while fl-mHTT is diffuse nuclear implying multiple targets for pathology. In contrast, HTT118-120Q is diffuse nuclear and HTT171-120Q is cytoplasmic and aggregated and yet neither exhibits toxicity in trachea. The effect of the HTT171 fragment on fl-mHTT behavior and toxicity was examined by co-expressing both (fl + 171). Notably, the lethality of fl-mHTT is reduced when HTT171-120Q is expressed concurrently and the previously diffuse nuclear flHTT-120Q is now in cytoplasmic aggregates together with HTT171-120Q (fl + 171). (C) To further test the influence of tissue type, the toxicity of ex1, fl, 171 and 118 when driven in the prothoracic gland (PG) with spok > Gal4 at 29° was evaluated, as was the effect of the 171 fragment on fl toxicity in this tissue. In the PG flHTT-120Q is toxic as well and yet it is cytoplasmic and diffuse in these cells (fl). HTTex1-120Q is also toxic and continues to form cytoplasmic aggregates (ex1). HTT118 is relatively benign as it is in trachea although in the PG cells it exhibits a diffuse cytoplasmic distribution (118) and HTT171 is also non-toxic in the PG and occurs in cytoplasmic aggregates (171). When HTT171-120Q is co-expressed with flHTT-120Q, toxicity is significantly suppressed and the two proteins appear together in cytoplasmic aggregates (fl + 171). (D) Since ubiquitous expression produces crytptocephal phenotypes reminiscent of failure to produce edysis-triggering hormone, the toxicity of peptides driven in the 14 ETH producing INKA cells with ETH > Gal4 at 25° was evaluated. As in other tissues, HTTex1 is highly toxic in INKA cells and tends to form large cytoplasmic aggregates, while fl-mHTT exhibits only ~ 47% toxicity and is distributed as diffuse nuclear material. HTT171 exhibits no toxicity and forms large cytoplasmic aggregates similar to HTTex1, while HTT118 produces ~ 20% lethality and is distributed as diffuse nuclear material typically with a single perinucleolar punctum (118). Coexpression of fl-mHTT and HTT171 improves the survival of fl-mHTT expressing animals and recruits fl-mHTT into large cytoplasmic co-aggregates. Thus, both aggregated and diffuse HTT fragments can be toxic, and expanded polyQ (120Q) is not sufficient for either toxicity or aggregation.
We next asked whether expansion of the polyQ domain in HTT proteins and peptides is sufficient to cause pathology. To test this, we compared the phenotype (lethality) of animals expressing 120Qs when embedded in the HTT118 and the HTT171 peptides. Expression of the HTT118 shortstop fragment with a 120 polyQ expansion allows only 35% survival when expressed ubiquitously (Fig. 1A), while the HTT171 fragment with 120Qs is quite benign, exhibiting less than 6% lethality despite accumulating in tissues (Fig. 1A). Thus, expanded polyQ is necessary but not sufficient to drive pathology.
Since mHTT phenotypes may not be cell autonomous, ubiquitous expression makes it difficult to determine which mHTT-affected tissue(s) gives rise to a particular phenotype. In turn, that uncertainty makes it difficult to correlate a particular phenotype with the behavior (i.e. subcellular localization and/or aggregation) of mHTT in a particular tissue. We used tissue-specific drivers to reveal how the subcellular distribution and aggregation behavior of HTT peptides is correlated with pathology in that specific tissue. In particular, we examined the effects of HTT proteins in trachea (btl > Gal4), prothoracic gland (spok > Gal4), INKA cells (a set of 14 pairs of cells that secrete the ecdysis-triggering hormone, ETH > Gal4, (26)) and neuronal cells (elav > Gal4).
Expanded polyQ is necessary but not sufficient to drive inclusion formation and to dictate subcellular location
The formation of inclusion bodies (IBs) (large microscopically visible intracellular aggregates of mutant HTT) is frequently cited as a hallmark of polyglutamine diseases (6,13,27–29). Despite being known for 20 years, these inclusions continue to generate robust discussions regarding whether they are pathogenic or protective or whether all HTT peptides are toxic or are some preferentially toxic (30) (reviewed in (31))?. To investigate the parameters affecting inclusion formation, subcellular distribution and toxicity, we expressed HTT in specific tissues where both pathology and protein location and aggregation state could be followed in the same cells.
Expression of 55C6 fl-mHTT or HTTex1 in trachea mimics the lethality seen with ubiquitous expression in that virtually no animals eclose (Fig. 1B) except for animals expressing the 51D fl-mHTT insert which exhibits only 20% lethality (not shown). Examination of larvae reveals that approximately 60% of the HTTex1-expressing larvae have defects such as tracheal gaps or constrictions compared to approximately 25 and 30% in the 51D and 55C6 fl-mHTT expressing larvae. Thus, expression of either HTTex1 or full-length exclusively in trachea leads to lethal pathology, and fl-mHTT exhibits a lower level of tracheal defects than HTTex1.
To correlate the tracheal pathology with subcellular behavior of HTT, we examined the subcellular distribution of HTT (Fig. 1B). HTTex1-120Q peptides exhibit a strong propensity to form aggregates in the cytoplasm (Fig. 1A and B), while fl-mHTT proteins remain diffuse but localize to the nucleus (Fig. 1A and B). Despite these differences in subcellular localization and aggregation behavior, both HTTex1-120Q and fl-mHTT cause tracheal disruption although the disruption caused by HTTex1 is more extensive.
To further investigate the influence of flanking peptides, we also examined the behavior of HTT171 and HTT118 in trachea. Like the exon1 fragment, the HTT171 fragment forms cytoplasmic aggregates (Fig. 1B), but unlike exon1, it exhibits little or no toxicity (Fig. 1B). In contrast, the expanded polyQ HTT118 peptide is found as a weakly staining diffuse protein in the nucleus (Fig. 1B) but also does not disrupt tracheal cells. These results demonstrate that peptide context can strongly influence HTT peptide behavior in terms of subcellular location, propensity to form inclusions, and ability to cause cellular pathology; thus, expanded polyQ alone is not sufficient to drive inclusion formation nor to dictate subcellular location.
Inclusion formation is neither necessary nor sufficient for cellular pathology
Despite different subcellular behaviors, both full-length and exon1-expanded Q proteins disrupt trachea (Fig. 1B), although cytoplasmically aggregating HTTex1 is more toxic than diffuse nuclear full-length HTT. Despite the increased toxicity of HTTex1, the tracheal disruptions caused by diffuse nuclear full-length HTT demonstrate that inclusions are not necessary for cellular pathology. These results suggest that multiple avenues to pathology are operative, since the two HTT proteins appear to engage targets in different cellular compartments (i.e. the nucleus and the cytoplasm). Moreover, the fact that the HTT171-120Q fragment forms cytoplasmic inclusions like HTTex1-120Q but remains remarkably non-toxic (Fig. 1B) demonstrates that cytoplasmic inclusions in themselves are not sufficient to cause pathology.
Engineered relocation of full-length HTT is protective
In the studies discussed here, subcellular localization depends on the peptide sequence. It would be helpful to separate these two variables (i.e. location and sequence) by relocating a given peptide without altering its sequence. Studies that manipulate the aggregation status or subcellular localization of a particular pathogenic fragment are challenging, and often require altering the primary amino acid sequence, and thus structure, by synthetic approaches like the inclusion of an NLS or NES (30,32,33).
We find that the HTT171-120Q fragment provides a useful tool to manipulate the subcellular behavior of pathogenic fl-mHTT protein without altering its protein sequence. Specifically, the toxicity of the fl-mHTT is suppressed by co-expression of the HTT171-120Q fragment. When expressed ubiquitously, 90% of HTT171-120Q-expressing flies are viable, while flies expressing fl-mHTT alone are completely lethal, with development arrested in the puparium (Fig. 2A). However, when HTT171-120Q is expressed together with the otherwise lethal flHTT-120Q, approximately 2/3 (68%) of the HTT171 + flHTT-expressing animals are rescued to adult viability despite the fact that the expanded polyQ load is increased by co-expression of the two proteins together (Fig. 2A).
Figure 2.
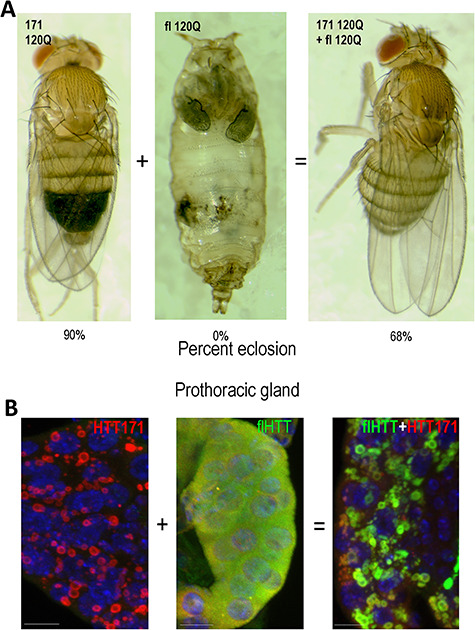
Co-expression of HTT171 with pathogenic fl-mHTT suppresses multiple pathogenic events. (A) When expressed ubiquitously with da > Gal4 at 25°, 90% of HTT171-120Q survive to adulthood (left panel) while 0% of fl-mHTT (55C) expressing flies survive (center panel). However, co-expression of fl plus 171 suppresses the lethality, raising the survival from 0 to 68% when ubiquitously expressed. The pupa in the center panel (fl 120Q) shows a typical cryptocephal phenotype. (B) When co-expressed, toxic fl-mHTT is sequestered into non-toxic HTT171 aggregates. Prothoracic glands expressing either HTT171-120Q or fl-mHTT or both (flHTT+HTT171) were stained with an N-terminal HTT antibody (red) and a C-terminal HTT antibody (green) and DAPI to reveal nuclei (blue). The left panel expressing HTT171 alone reveals a strongly aggregated N-terminal signal and noC-terminal signal. The center panel expressing fl-mHTT alone reveals a strong diffuse cytoplasmic C-terminal HTT signal (green) as well as diffuse N-terminal signal (red). In the right panel, a PG co-expressing HTT171 and fl-mHTT reveals that the previously diffuse fl signal is now co-localized with aggregated HTT171. Scale bars are 2 μm.
Ubiquitous expression of flHTT-120Q causes a range of pathologies, each of which could potentially lead to lethality. For example, when fl-mHTT is expressed at high levels (29°), a ‘failure to metamorphose’ phenotype reminiscent of failure to secrete ecdysone from the prothoracic gland predominates, while as the level of fl-mHTT expression is decreased (25°), developmentally arrested pupae exhibit a cryptocephal phenotype (e.g. Fig. 2A) reminiscent of dysfunctional secretion of ecdysis-triggering hormone (ETH) (26,34). At even lower levels of expression (20°), late-stage pupae with dorsal-closure defects (clefting) predominate (Fig. S1). The clefting phenotype is typical of disruptions of the developmentally programmed apoptosis of larval epidermal cells (LEC) (35,36). Ubiquitous co-expression of HTT171-120Q with pathogenic fl-mHTT at 25° appears to rescue all of the phenotypes occurring at 25° or lower (Fig. 2A). Thus, the less-pathogenic HTT171-120Q fragment exhibits epistatic influence over the pathogenic fl-mHTT.
Intrinsic and extrinsic factors affect pathology and aggregation
To better understand the mechanisms that drive fl-mHTT pathogenicity, we examined the subcellular behavior and pathogenic effects of HTT in trachea (btl > Gal4), INKA cells (ETH > Gal4), prothoracic gland (spok > Gal4) and neuronal cells (elav > Gal4), where specific pathologies can be ascribed to dysfunction in those particular cell types.
Tracheal expression and rescue
When expressed in trachea using the tracheal-specific driver btl > Gal4, both HTTex1 and fl-mHTT cause tracheal disruptions (60 and 30%, respectively) while HTT171-120Q causes very little disruption (<10%) (Fig. 1B). At the subcellular level, both HTTex1 and the HTT171 peptides are found in cytoplasmic inclusions, while the full-length protein is nuclear and diffuse as described earlier. However, when fl-mHTT is expressed together with HTT171-120Q, the nucleus is devoid of fl-mHTT, which localizes instead in the HTT171 cytoplasmic aggregates (Fig. 1B). Co-expression also leads to reduced lethality compared to expression of fl-mHTT alone (i.e. 5% survival with fl-mHTT vs. 43% with fl + 171, respectively) (Fig. 1B). In contrast, co-expression of the 118 fragment did not significantly alter survival compared to fl-mHTT alone (i.e. 5.7% with fl-mHTT alone vs. 2% with fl + 118) (Fig. 1B). The increased expanded polyQ load when fl-mHTT was co-expressed with HTT118 was associated with occasional aggregates appearing in the cytoplasm but most of the material remained in the nucleus (Fig. 1B). Thus, full-length protein that engages a target in the nucleus is significantly less toxic when relocated to cytoplasmic aggregates in trachea.
Prothoracic gland expression and rescue
The previous experiments suggest two alternative interpretations. One is that diffuse fl-mHTT is toxic when in the nucleus and that removing it from the nucleus suppresses that toxicity. The other is that diffuse fl-mHTT is toxic regardless of its location and that sequestering into aggregates renders it non-toxic. The prothoracic gland (PG) of Drosophila provides a means to distinguish these two options. The prothoracic gland is a neuroendocrine gland that secretes ecdysone. Expression of fl-mHTT in the PG of the 3rd instar larvae causes toxicity (~95% vs. 100% for ex1) and yet is found diffuse in the cytoplasm rather than the nucleus as was seen in trachea (Fig. 1C). Thus, either diffuse fl-mHTT is toxic in the cytoplasm of some cells (e.g. PG cells) and in the nucleus of other cells (e.g. trachea) or there is an as-yet-to-be-described developmental relocation to the nucleus shortly before toxicity is evident in the PG. Co-expression with HTT171 suppresses the lethality caused by fl-mHTT expression and yields ~ 80% survival. Immunofluorescent staining using an N-terminal HTT antibody (red) as well as an antibody against a portion of HTT that is not found in the HTT171 fragment (green) demonstrates that the diffuse full-length protein has been captured into cytoplasmic aggregates formed by HTT171 (Fig. 2B) with accompanying rescue of lethality (Fig. 2A). Similar immunofluorescent studies show that pathogenic fl-mHTT is relocated from whatever compartment it is in (i.e. nucleus or cytoplasm) to join HTT171 aggregates in the cytoplasm, with accompanying loss of toxicity in trachea (Fig. 1B), PG (Fig. 1C), INKA cells (Fig. 1D) and neurons (Fig. 3). Thus, capture into HTT171 co-aggregates renders fl-mHTT non-toxic. Further, subcellular distribution of mHTT is dictated by cellular context since fl-mHTT is nuclear in some cell types but cytoplasmic in others.
Figure 3.
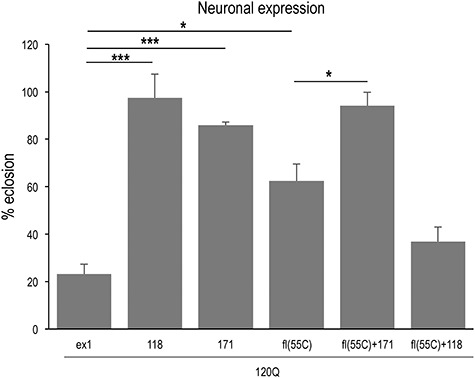
Toxicity caused by neuronal expression of fl-mHTT is suppressed by co-expression of 171. When expressed at 29° in all neurons with elav > Gal4, the toxicity of HTTex1 and fl-mHTT (55C) is evident (77 and 38% lethality, respectively). As in other tissues, co-expression of HTT171 suppresses fl-mHTT toxicity, in this case to 6% (fl(55C) + 171(51D)). As a control, co-expression of HTT118 with fl-mHTT fails to rescue and indeed exacerbates lethality (64%) in this experiment.
INKA cell expression and rescue
When animals are challenged by ubiquitous expression of fl-mHTT, they are able to initiate metamorphosis and then die as pupae with a cryptocephal phenotype (Fig. 2A). These phenotypes are reminiscent of those in mutants that affect the cells responsible for secretion of ecdysis-triggering hormone (ETH), which is required for pupal molting and metamorphosis (26). As another test to correlate subcellular behavior with pathology, we used the ETH > Gal4 driver to express both fl and HTTex1 in the 14 secretory INKA cells that produce ETH (26). These crosses resulted in phenotypes consistent with the cryptocephal phenotype observed when the same transgenes were expressed ubiquitously with da > Gal4 at intermediate levels, suggesting that some of the lethality and pathology observed can be attributed to INKA-cell dysfunction.
When expressed in INKA cells, fl-mHTT consistently exhibits diffuse staining in the nucleus (27/27) with occasional appearance of small puncta and approximately 47% lethality while HTTex1 forms aggregates in the cytoplasm with variable levels of diffuse staining in the nucleus (27/27) and 94% lethality (Fig. 1D). Expression of HTT171 alone exhibits no lethality and exclusively cytoplasmic aggregates (20/20), while expression of HTT118 exhibits 18% lethality and low levels of diffuse staining in the nucleus with one small perinucleolar punctum in each cell (14/14) (Fig. 1D). When the HTT171 fragment is expressed together with fl-mHTT, pupal lethality is suppressed (23% vs. 47% lethality), and the diffuse nuclear fl-mHTT is incorporated into cytoplasmic inclusions (7/7) (Fig. 1D). In contrast, co-expression of the 118 fragment with fl-mHTT exhibits diffuse nuclear and cytoplasmic aggregated material (11/11) and exacerbates lethality (86% vs. 47% lethality). Again, cytoplasmically aggregating HTT171-120Q is able to relocate flHTT-120Q to cytoplasmic aggregates and suppress its toxicity.
Neuronal rescue
To determine whether the rescue by sequestration also occurs in neurons, we tested the ability of HTT171 co-expressed with fl-mHTT to rescue neuronal toxicity. When expressed in neurons using the elav > Gal4 driver, both HTTex1-120Q and fl-mHTT produce some lethality (e.g. 77 and 38%, respectively, at 29°C), while HTT171-120Q produces less than 14% lethality (Fig. 3). However, co-expression of HTT171-120Q suppresses the lethality of fl-mHTT transgenes (i.e. 38% lethality with fl-mHTT is reduced to 6% by coexpression of HTT171-120Q). In contrast, co-expression of the HTT118 fragment with fl-mHTT does not suppress lethality (and may exacerbate it) (Fig. 3). Thus, although the specific neurons that lead to lethality are unknown, co-expression of HTT171-120Q with fl-mHTT suppresses neuronal lethality in a manner similar to the suppression seen with the other tissues above.
Intrinsic factors affecting HTT properties
The studies described above demonstrate that while expanded polyQ is necessary for pathology, it alone is not sufficient. Studies from our laboratory and others demonstrate that sequences adjacent to the expanded polyQ domain can influence the pathology caused by expanded polyQ and that extensive intramolecular interactions can occur within the HTT protein, especially in the amino-terminal region (18,37–42). These intramolecular interactions could potentially affect how expanded polyQ HTT engages cellular targets including clearance machinery such as the proteasome and autophagy pathways (40,42). These putative intramolecular interactions are significant for two reasons. Different flanking sequences on particular N-terminal proteolytic fragments of HTT can influence the pathogenicity (18,43,44), and the inclusion of various ‘tags’ when developing different HTT transgenes for model studies may influence HTT behavior (45,46).
To document the effect of local peptide changes on HTT fragment behavior, we explored the behavior of two HTT fragments. The first is the ‘N171’ fragment (14), which, compared to HTT exon 1, contains 81 additional amino acids from the HTT protein. The second is the 118 aa short-stop fragment that is only 28 amino acids longer than HTT exon1 and a mere 10 amino acids longer than the aspartate endopeptidase fragment (108, (2)), both of which are highly toxic in the Drosophila model used here (17,18). Yet, despite these seemingly minor differences, short-stop expression is surprisingly benign in both mice and flies (17,18).
Analysis of HTT171
The N171 fragment has been used extensively in animal models and is reportedly highly toxic (16,20,21,43,47). When inserted into mice (Jackson Labs (B6C3-°©- Tg(HD82Gln)81Dbo/J), rats (48) and monkeys (49), N171 is highly toxic, although some reports in mice suggest that it may be cleaved to generate a ~ 90 aa fragment (14,50). In our hands, however, a transgene expressing just the first 171 amino acids of human HTT with 120Qs in Drosophila is benign. A close examination of the literature reveals a potential explanation for this. The original synthetic N171 fragment was generated using a convenient XhoI restriction site that adds an additional 11 amino acids to HTT171 (47). Most of the N171 fragments used in other studies appear to encode these extra 11 vector amino acids at the C-terminus (…PRLQLEPSCLFLRLLVV*—native HTT amino acids in italics) (14,47). In contrast, our fragment terminates precisely at amino acid 171 of the HTT protein (i.e. …PRLQLE*). This difference raises the possibility that relatively minor changes in peptide context may drastically affect HTT behavior. To test this hypothesis, we added short sequences to the C-terminus of HTT171 fragment to create two novel fragments. The added sequences included a 9-amino-acid hemagglutinin tag (HA) (…YPYDVPDYA*) (51) and a 9-amino-acid tetracysteine ‘Lumio’ tag (LUM) (…GCCPGCCGG*) (52). We chose HA and LUM because it is often advantageous to place an epitope tag on the HTT peptide and these are two widely used tags with very different chemical characteristics that are similar in size to the random 11 amino acids of N171.
Although ubiquitous expression of the true HTT171-120Q in Drosophila is benign in our hands at multiple temperatures (Figs 1A; 4A and B), addition of the HA tag and testing at 22.5°C increased the lethality to 70%, while appending the LUM tag resulted in complete lethality even when expressed at lower levels (Fig. 4A). The addition of these same tags to the C-termini of exon1 and other fragments (HTT90HA/LUM), 108HA, 469HA/LUM and 586HA/LUM) did not demonstrably alter the behavior or toxicity of those fragments (18) indicating that the tags themselves are not inherently toxic. Consistent with the increased lethality, inclusion of C-terminal tags on the HTT171 fragment also reduces longevity (Fig. 5B). Namely, when expressed ubiquitously at 22.5°C, female flies expressing the precise HTT171 fragment die with a t1/2 of about 20 days while HTT171HA flies die with a t1/2 of 9 days and HTT171LUM flies do not survive to adulthood at all (Fig. 4B).
Figure 4.
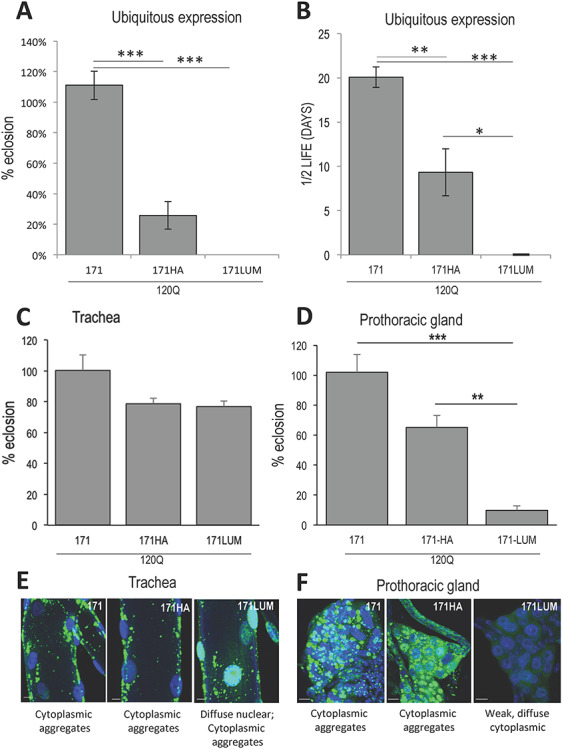
The influence of peptide modifications on HTT171 fragment behavior. (A) When expressed ubiquitously with da > Gal4, the HTT171-120Q fragment exhibits little or no lethality, but appending either an HA (…YPYDVPDYA*) or a LUM tag (…GCCPGCCGG*) to the C-terminus greatly increases the toxicity of the fragment. (B) Surviving adult flies expressing an HA-tagged HTT171 transgene exhibit about half the life-span of those expressing untagged 171. Flies expressing LUM-tagged HTT171 do not survive to adulthood. (C) Flies expressing HA- or LUM-tagged 171 in tracheal cells show only modest increases in lethality that do not rise to the level of significance (P = 0.12; 0.09), but the inclusion of tags changes the subcellular behavior significantly. (D) In contrast, in the prothoracic gland, expression of LUM-tagged 171 shows significant increase in toxicity compared to 171 alone, while the HA-tagged 171 borders on significance (P = 0.051). (E) In trachea, pure 171 forms cytoplasmic aggregates, while inclusion of HA causes some HTT to become nuclear diffuse, and inclusion of the LUM tag causes the bulk of the HTT to appear as diffuse nuclear material with some cytoplasmic aggregates remaining when expressed with btl > Gal4 at 29°C. (F) In the prothoracic gland, addition of the LUM tag causes aggregated-cytoplasmic HTT to become weakly staining diffuse-cytoplasmic material while HTT171HA remains as extensive aggregates in the cytoplasm with a haze of diffuse staining as well. Scale bars are 10 μm.
Figure 5.
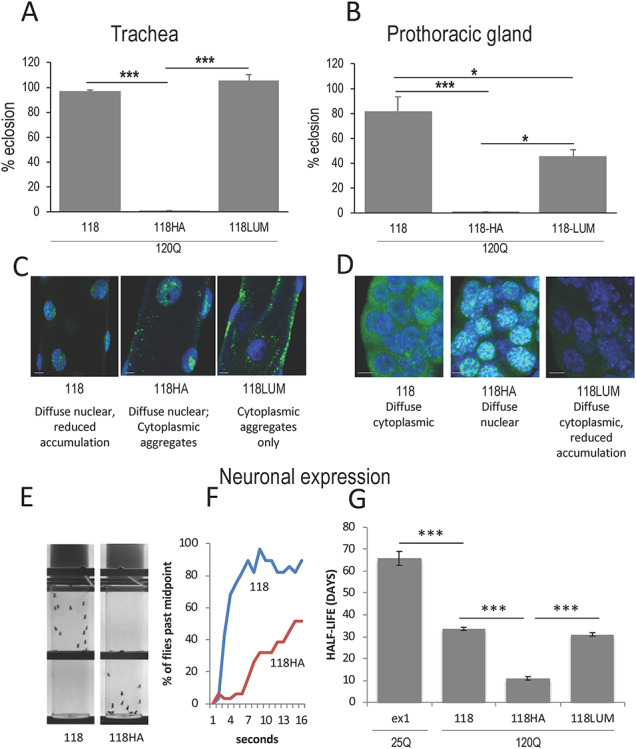
The influence of peptide modifications on HTT118 fragment behavior. (A) When expressed in trachea (btl > Gal4) (29°), the HTT118-120Q fragment does not cause lethality but addition of an HA tag renders it lethal, while addition of a LUM tag has only a modest effect on toxicity of the fragment. (B) Expression of 118 in the PG with spok > Gal4 drives modest lethality, while expression of an HA-tagged HTT118 with spok > Gal4 allows no survivors, and the LUM tag reduces viability by about 50%. (C, D) Subcellular location of 118, 118HA and 118LUM in trachea (C) and prothoracic gland (D). Note: HTT118 is diffuse nuclear in tracheal cells and diffuse cytoplasmic in PG cells and converts to diffuse nuclear with the addition of an HA tag in PG cells. Addition of a LUM tag does not affect location much in PG, but does seem to increase clearance of the peptide (note the weak staining). (E, F) When expressed at lower levels in neurons (i.e. elav > Gal4 @ 25°), a strong difference in the propensity for flies to exhibit negative geotaxis (climbing behavior) is seen when comparing HTT118 to HTT118HA in vials 4 s after the flies have been shaken down (10 days post-eclosion). (E) A frame from a video tracking shows a point at which all the 118 flies are in the top half of the vial while all the 118HA flies are still in the bottom half. (F) The progress of the tagged vs. non-tagged HTT-118-expressing flies tracked by video monitoring demonstrates the difference in climbing speed. (G) Flies expressing the HTT 118120Q fragment under the influence of elav > Gal4 show reduced longevity which is exacerbated by the addition of an HA tag to the transgene but not by the addition of LUM.
To better understand the functional effects of these small changes in peptide structure, we expressed these tagged transgenes in specific tissues and evaluated their effects on subcellular localization, aggregation and pathology. Compared to HTT171, the addition of tags to HTT171 constructs causes changes in subcellular behavior in tracheal cells (Fig. 4E). The addition of a LUM tag converts the normally cytoplasmic HTT171 to a primarily diffuse nuclear peptide with some residual cytoplasmic aggregates remaining (Figs 4E; S2). On the other hand, HTT171 with an HA tag remains primarily as cytoplasmic aggregates with minor diffuse nuclear staining (Figs 4E; S2). Unlike the ubiquitous expression of either tagged HTT171 construct, expression in trachea only (Fig. 4C) is relatively benign (i.e. it produces only a slight increase in lethality that does not rise to the level of statistical significance).
When HTT171 constructs are expressed in the prothoracic gland at 29°C, (Fig. 4D), the addition of the HA tag increases lethality, and the LUM tag increases it even more (i.e. 100, 65 and 10% eclosion, respectively, for HTT171, HTT171-HA and HTT171-LUM). Accompanying this increased lethality is a change in subcellular behavior. The HTT171-HA fragment continues to accumulate in cytoplasmic aggregates albeit with a haze of diffuse staining, while the HTT171-LUM fragment accumulates as weak-diffuse material in the cytoplasm (Fig. 4F). Interestingly, both HA- and LUM- tagged HTT171 fragments exhibit increased PG pathology with the HA tagged HTT171 causing ~ 35% lethality while the diffuse, cytoplasmic LUM tagged HTT171 causes ~ 90% lethality when expressed in prothoracic gland cells. Thus, small changes to the HTT171 fragment can alter both its subcellular behavior and its level of pathology. Furthermore, both aggregated as well as diffuse cytoplasmic distributions can correlate with elevated pathogenesis.
Analysis of the HTT118 ‘shortstop’ fragment
The shortstop fragment arose from spontaneous truncation or recombination of a full-length Htt gene during multiplication of a yeast artificial chromosome (YAC) before injection into mice (17). The fragment terminates at the end of exon 2 at aa 118 (20). The majority of subsequent studies using this fragment have employed clones obtained from the founder laboratory. The ‘shortstop’ fragment exhibited unexpectedly low toxicity in mice (17) and also exhibits relatively low toxicity in Drosophila. For example, flies ubiquitously expressing the HTT118-120Q fragment survive better than ones expressing HTTex1-120Q (i.e. 100% vs. 0% at 22.5° and 36% survival vs. 0% at 25°C, Fig. 1A).
When HTT118-120Q is expressed exclusively in either trachea or the PG, little or no lethality is observed (Fig. 5A and B). In contrast, addition of a 9 amino acid HA tag renders the HTT118 fragment completely lethal in either trachea or the PG (Fig. 5A and B). Accompanying this increase in lethality are major but opposite changes in subcellular location. In trachea, the addition of the HA tag converts the weakly staining HTT118 fragment from diffuse nuclear to a mix of diffuse nuclear and cytoplasmic aggregated (Figs 5C; S2) while in the PG, HTT118 exhibits robust diffuse cytoplasmic staining but converts to diffuse nuclear with the addition of the HA tag (Figs 5D; S2). These observations underscore the role of tissue context and its interplay with peptide structure in dictating both subcellular behavior and toxicity.
In contrast, addition of a 9 amino acid LUM tag had no effect on tracheal lethality and the tag causes the fragment to be absent from the nucleus forming aggregates only in the cytoplasm (Figs 5A and C; S2). In the PG, there is an approximately 40% increase in lethality accompanying the addition of the LUM tag despite the fact that the peptide exhibits reduced levels and is primarily diffuse in the cytoplasm (Fig. 5B and D). Thus, expansion of polyQ in the context of the HTT118 peptide exhibits much lower pathology than other fragments, but small changes in the physical structure of the peptide surrounding the expanded polyQ can significantly affect the pathology and the subcellular behavior of the peptide.
As an example of how these structural changes can also affect the HTT118-120Q behavior in neurons, we expressed these fragments using the elav > Gal4 neuronal driver. When expressed in neurons at 25°C, no overt lethality was found with any of the three HTT118 constructs (not shown). However, we noted obvious defects in motor function and longevity with the HA-tagged transgenes. For example, addition of the HA tag, (HTT118-HA), profoundly affects motor function as measured by climbing behavior (Fig. 5E and F). Similarly, the longevity of HTT118-HA expressing animals is about 1/4th that of HTT118 alone [i.e. from a t1/2 of 30 days for the unmodified HTT118 fragment to less than 10 days when HA is appended (Fig. 5G)]. Again, these results demonstrate that small changes to peptide structure can have profound effects on various aspects of HTT peptide pathology and subcellular behavior and underscores how HTT-peptide behavior is strongly affected by extrinsic factors, such as the specific cellular environment (Figs. 4C–F; S2).
Discussion
HD is a dominant progressive neurodegenerative disorder with hallmarks that include the formation of intracellular inclusions/aggregates and preferential degeneration of specific portions of the brain. HTT is expressed ubiquitously, and while neurodegeneration is the most debilitating aspect of the disease, dysfunction in other organs is clearly present (1,24,25).
Much effort has been expended investigating whether a particular toxic species is responsible for HD pathology and determining whether its aggregation state is critical for toxicity. Ubiquitous HTT expression complicates the analysis, since the answer may not be the same in all cell types. Moreover, direct data comparisons are difficult since many of these studies are performed in different systems (different cultured cells or organisms) with different transgenic, expanded polyQ, HTT transgenes inserted in different chromosomal locations using different genetic backgrounds. In addition, one cannot avoid proteolytic processing of transgenic HTT fragments in mammalian systems (2,3). Thus, determining whether pathology is caused by the engineered transgene or by its proteolytic products is challenging (16). Here we address those challenges by using a Drosophila model to systematically compare the pathology and biophysical behavior of full-length HTT and HTTex1 transgenes as well as two other often-used amino-terminal fragments, HTT171 and HTT118. To facilitate comparisons, transgenes were inserted into a single chromosomal location and expressed in a common genetic background. The consequence of expressing these transgenes in different tissues is summarized in Fig. S2.
Our experiments suggest several conclusions relevant to the relationship between HTT-peptide pathogenesis and the HTT-peptide intracellular behavior. For example, a particular HTT peptide’s subcellular location is strongly dependent on the cellular environment [i.e. a given peptide will locate to different places in different cell types—compare the location of fl-HTT in trachea and PG (Figs. 1B and C; S2)]. Despite these differences in location, the full length protein is toxic in both tissues. Thus, a given protein can cause cellular disruption whether it is located in the cytoplasm or the nucleus. An alternative is that HTT peptides change subcellular location shortly before pathology is manifest when such changes are difficult to detect. However, in the absence of data suggesting major temporal changes in subcellular location, it would appear that multiple cellular targets leading to pathology exist (e.g. fl-HTT causes disruption in tracheal and INKA cells where it is nuclear and also causes disruption in PG cells where it is cytoplasmic).
The data also speak to the role of aggregation in the pathogenic process and whether preventing aggregation is a viable therapeutic objective. Clearly, both diffuse and aggregation-prone HTT can be pathogenic (e.g. compare fl-HTT and HTTex1). This is the first study we are aware of where the relative toxicity of fl-HTT can be compared to that of HTTex1 and other N-terminal fragments in a setting where the secondary production of N-terminal fragments by proteolytic processing from the fl-HTT does not occur and where all transgenes are expressed from the same chromosomal location. Other studies find that mice expressing fl-mHTT eventually develop inclusions and pathology, but since those inclusions are composed of small N-terminal HTT fragments, it is difficult to determine if the fl-mHTT protein drives the pathology or if the secondary small fragments produced from it are responsible (15,17). We have found that processing of HTT to small discreet fragments does not occur in Drosophila (18) and therefore, our data demonstrate that the fl-HTT protein is itself pathogenic, although less so than mHTTex1 and that formation of inclusions is not essential for its pathogenicity.
In addition, we find that aggregates per se need not be pathogenic. For example, compare HTTex1-120Q and HTT171-120Q (Fig. S2). Both form cytoplasmic aggregates and yet mHTTex1 is highly toxic, while mHTT171 is remarkably benign (Fig. 1). Indeed, studies with the HTT171 fragment demonstrate that the toxicity of diffuse fl-mHTT can be rescued by incorporation into benign co-aggregates and that it is the aggregated status per se and not the new location of the pathogenic fragment from a different subcellular compartment that is key to the rescue of toxicity [e.g. diffuse nuclear fl-mHTT (trachea) or diffuse cytoplasmic fl-mHTT (prothoracic gland) are both pathogenic but both are suppressed by incorporation into cytoplasmic HTT171 co-aggregates (Fig. 1B and C)].
In addition to cell type, a second feature that can strongly influence HTT behavior is the peptide’s primary sequence. A number of studies have described intramolecular interactions in HTT (18,37–42,53). Our data suggest that intramolecular interactions can have profound effects on pathogenicity, subcellular location, turn-over and aggregation. For example, both fl-HTT and HTTex1 with 25Qs are diffuse and cytoplasmic. Expansion of the polyQ in HTTex1-120Q causes it to form cytoplasmic aggregates. However, addition of just 28 additional aas in the context of those 120Qs (i.e. HTT118) virtually eliminates aggregation and leads to diffuse and reduced staining in nucleus or cytoplasm depending on cell type. Addition of 53 more amino acids (i.e. HTT171) leads to increased accumulation and aggregates in the cytoplasm but little or no pathology. The fact that expanded polyQ is necessary but not sufficient to produce either pathology or aggregates demonstrates that peptide context is important for these properties. These observations suggest that intramolecular interactions influence an HTT peptide’s pathogenicity and its propensity to form aggregates, as well as its ability to engage cellular targets including perhaps components involved in clearance.
Studies in mammalian systems suggest that fl-mHTT (~350 kDa) is processed into several N-terminal fragments, including an exon1-like fragment with an expanded polyQ tract (3,38,54,55). These observations have generated discussions about whether particular fragments are especially critical to HD pathology and whether appropriate models for studying HD should focus on the full-length protein or on various fragments (e.g. (56). In addition, other studies have suggested that N-terminal fragments of HTT are more prone to aggregation and are more toxic than full-length HTT (43,44,47,57).
This study addresses these issues by comparing fragment toxicity and behavior in a highly controlled setting. With respect to relative toxicity of HTT and its proteolytic and splicing fragments, we find that the naturally occurring HTT exon1 peptide is uniquely toxic among the HTT fragments. The 108 aa endopeptidase fragment runs a close second in toxicity and aggregation (18). The fact that exon1 can occur directly as a result of mis-splicing without being derived from a fl-HTT protein demonstrates that early conformational events in the context of fl-mHTT are not necessary for the development of pathogenic structures in HTTex1 fragments (56). In agreement with other studies, (17,20), we find that the HTT118 shortstop fragment is not prone to aggregation and is much less toxic than either HTTex1 or fl-mHTT. In contrast, the HTT171 fragment is highly prone to aggregation and yet is particularly non-toxic when compared to other fragments and full-length. These data point to HTTex1 as a uniquely toxic species.
In no case does unexpanded polyQ HTT exhibit pathology in any assay we have conducted. This demonstrates that expanded polyQ is necessary for HD pathogenesis.
Clearly, both diffuse and aggregated peptides can cause pathology, and pathogenic targets exist in both the nucleus and the cytoplasm that can be engaged by non-aggregating HTT species. The failure of fl-HTT-120Q to form inclusions in our studies is consistent with the early observations that inclusions in post-mortem samples reacted with N-terminally targeted antibodies but not with antibodies that recognize longer HTT proteins (i.e. epitopes beyond aa 115) (16,58). These experiments do not rule out the possibility that full length mHTT might be recruited to aggregates by shorter species but be proteolytically cleaved in the process, thus leaving aggregates free of epitopes C-terminal to the expanded polyQ. In either case, initiation of inclusions appears to be a property of selected N-terminal peptides such as HTTex1, HTT171 and selected others (18).
The influence of HTT peptide composition
It has been demonstrated repeatedly that expanded polyQ is associated with both inclusion formation and pathogenesis (59,60). Experiments involving expanded polyQ with minimal other surrounding amino acids (61–63) suggest that polyQ alone may be sufficient for pathology. We find that HTT fragments containing 120Qs exhibit varying degrees of toxicity (18) ranging from HTTex1-120Q, which is extremely toxic, to the HTT171 fragment, which is comparatively benign (Fig. 1) to various proteolytic fragments exhibiting varying degrees of much-reduced toxicity (18). These observations are consistent with studies showing that the physical properties of HTT proteins are strongly influenced by amino acids surrounding the expanded polyQ (60,64). Thus, the degree of polyQ toxicity is highly sensitive to peptide context.
Although expanded polyQ is necessary to promote aggregation, it is not sufficient, as revealed by the lack of aggregates formed by full-length HTT-120Q, by many of the proteolytic fragments (18) and by the HTT118 fragment reported here (Fig. S2). However, since all of the fragments tested here have the first 17 amino acids of HTT which is reportedly central to aggregate formation (65), the influence of HTT-protein context on aggregation suggests that intramolecular interactions influence aggregation behavior. Various studies have demonstrated extensive intramolecular interactions in the N-terminal region of HTT (38,66–69). We suggest that intramolecular interactions may account for how the exon 1 fragment can be an aggressively aggregating cytoplasmic peptide that causes severe pathology, how addition of just 28 additional amino acids of HTT (118) leads to a diffuse cytoplasmic protein that is much less toxic and how the addition of amino acids up to 171 produces a benign but aggregating peptide. These divergent behaviors of modestly different HTT fragments demonstrate how extensive the influence of flanking sequences can be on polyQ behavior.
Further evidence for the influence of flanking sequences on HTT polyQ- peptide behavior comes from appending small peptide tags to the HTT118 and HTT171 fragments. For example, the acidic charged hemagglutinin HA (…YPYDVPDYA*) and uncharged Lumio LUM tags (…GCCPGCCGG*) are generally considered benign when used on a wide variety of proteins. However, the addition of either HA or LUM to the C-terminus of the normally benign HTT171 fragment renders it dramatically more toxic (Fig. 4A). Both tags also affect subcellular behavior (Fig. S2) with the LUM tag causing HTT171 to become diffuse nuclear in trachea (Fig. 4E) and diffuse cytoplasmic in the PG (Fig. 4F) and the HA causing elevated levels of diffuse and aggregated cytoplasmic protein (Fig. 4F).
For example, in the prothoracic gland, the addition of the LUM tag converts the cytoplasmic, aggregated and benign HTT171 fragment to a diffuse and toxic (~92%) fragment (Figs. 4D and F; S2). By comparison, the HA-tagged HTT171 shows elevated levels of cytoplasmic aggregates and a trend towards increased toxicity (~36% vs. 0%) (Fig. 4D). The minimal changes in pathology that accompany the changes in subcellular behavior of tagged HTT171 in trachea suggest that features that affect subcellular localization can be independent of those that affect pathology in a given cell type. This observation, taken together with the fact that ubiquitous expression of the modified fragments elicits major changes in pathology compared to unmodified fragments (Fig. 4A and B), underscores the importance of tissue context in the pathogenic process (Fig. S2).
Based on this work, it is likely that the extra 11 amino acids acquired during cloning (14,16) could render the N171 fragment used in many mammalian studies toxic compared to the behavior of a pure HTT171 fragment (e.g. Fig. 4). This underscores the need for caution when using modified HTT peptides in model studies, since relatively minor innocuous-seeming changes can have dramatic effects on pathology.
Sequestration of full-length suppresses toxicity
Full-length HTT does not form aggregates but can cause tissue pathology both when it occurs as a diffuse nuclear material (e.g. trachea) or when it occurs as a diffuse cytoplasmic material (e.g. in the PG, Fig. 1B and C; S2). Thus, fl-mHTT does not require aggregation to be pathogenic and it can engage targets in either the nucleus or the cytoplasm to cause pathogenicity. The suppression of pathology by incorporation into cytoplasmic co-aggregates of HTT171-120Q indicates that cellular location does not determine toxicity. Rather, the sequestration into aggregates is protective in this case, either by reducing the number of full-length molecules that can contact pathogenic targets in a particular compartment or because sequestration is accompanied by structural changes that inhibit target engagement by full-length HTT.
Summary
Our studies demonstrate that the behavior of expanded polyQ HTT fragments in a particular cell can be quite sensitive to small changes in peptide composition, and the behavior of a particular peptide can be quite sensitive to the cellular environment (Fig. S2). Thus, expanded polyQ is necessary but not sufficient for pathology, expanded poly Q is necessary but not sufficient to promote aggregate formation and that aggregated polyQ (or diffuse polyQ peptides) is neither necessary nor sufficient to cause pathology.
Materials and Methods
Construction of transgenes
Details of producing transgenes with 120Qs that do not contain extraneous sequences remaining from cloning steps are described in Barbaro et al., 2015 and involved use of paired class IIS restriction enzymes (referred to as the scarless method since it leaves no extraneous bases) and iterative cloning steps to produce a set of ‘clean’ transgenes. Briefly, this process involved initially producing a 171 amino acid expanded HTT construct by the scarless method and then swapping this domain as an EcoRI-XhoI domain into the unexpanded full-length HTT construct. All other fragments were made from this one by PCR. To produce C-terminally tagged transgenes, the reverse PCR primer included the coding sequence for the tag. We added either a 9 amino acid hemagglutinin tag (HA) (…YPYDVPDYA) (51) or a 9 amino acid tetracysteine ‘Lumio’ tag (LUM) (…GCCPGCCGG) (52) to the C-terminus of a ‘true’ 171 fragment (ending with...PRLQLE) or the 118 fragment (ending with...IVAQSV). When complete, fragments were cloned into the transforming vectors with EcoRI and NotI digestion sites that were also generated by the forward and reverse PCR primers, respectively. The vector for targeted transformation, pUASTattB, EF362409 was used to insert constructs into the 51D site (Bishof et al 2006). The same construct that was used to insert fl-HTT-120Q at the 51D site was also used for random insertion using the pUAST vector (Brand and Perrimon, 1993). One line with the insert at 55C6 produces higher levels of lethality than the insert at 51D and was used in some cases to allow a greater range of sensitivity for rescue experiments. The 55C6 insert (referred to as 55C) is within the region in bold underline CATTTAGCAAAAAGCTTGAGGCAATAAGGTGTGTATATATGTACAGTAAATAGAG AGATCAGTCGAAAGAAAAACACTGACGGGTAAACAACACGTATCATGTCGAGCTCTGGAATTG and the orientation of the P-element is 3′P-5′P as related to this sequence.
The shortstop fragment corresponds to the first two exons of Htt. Although initially described as 117 aa (17), it actually contains the first 118 aa (20). It arose by a spontaneous deletion during construction of a genomic YAC clone.
Fly husbandry
Cultures were maintained on a standard cornmeal/sugar/agar medium and kept on a 12-h light cycle. The polyQ expressing flies used in this study were mated with Act>GFP flies w[1118]; M{w[+mC] = Act5C-EGFP.rut3′UTR}ZH-51C/SM6b (Bloomington stock number B#52665) to generate a benign marker for segregation analysis. The resulting HTT polyQ/act > GFP males were crossed to various Gal4 driver females and survival to adulthood (eclosion) was monitored. Drivers used here include the ubiquitous da-Gal4 (B#8641), the INKA cell driver (ecdysis triggering hormone) ETH-Gal4 (B#51892), the prothoracic gland driver, spookier, spok-Gal4 (a kind gift of M. O’Connor, U Minnesota) and the pan-neuronal elav-Gal4 C155 driver (B#458). Progeny bearing no transgene expressed GFP and were used as internal controls. The tracheal driver, btl-Gal4 (btl-Gal4, UAS-GFP (2), (a kind gift of A.S. Ghabrial, (70)), was crossed to generate btl-Gal4/CyO heterozygotes which were then crossed to HTT polyQ flies. Specifically, btl-Gal4, UAS-GFP was crossed to Sco/CyO and btl-Gal4, UAS-GFP/CyO was recovered, thus eliminating the vine mutant from the background (70, 71). For our experiments, btl-Gal4, UAS-GFP/CyO males were crossed to different UAS-Htt female lines. Transgene expression produced by Gal4 drivers is temperature dependent such that levels can be modulated roughly two-fold by rearing cultures between 22.5 and 29° (72, 73).
Transformant lines
yw M{eGFP;RFP.vas-int.Dm}ZH-2A; M{RFP.attP}ZH-51D flies (kind gifts from K. Basler, University of Zurich, (22)) were injected with pUASTattB vectors harboring the different HTT fragments, and transformed flies were recovered based on eye color and crossed with stocks containing CyO in a w[1118] background to generate flies homozygous for the transgene on the second chromosome.
Longevity assays
Triplicate sets of 10–15 flies for each cross were harvested in a 24-h period and were passed to fresh vials every 2 days. Dead flies were counted every 2–3 days.
Survival assays: 10 males and female virgins were allowed to mate in standard food vials for ~ 2 days. Triplicate vials of flies were then passed daily into fresh vials for a span of 4 days. The resulting adult progeny were collected and scored following eclosion.
Adult motor function assays
A fixed number (typically 15) of female flies (HTT-expressing) are transferred to 3 vials placed side-by-side in a rack with vials containing sibling males (non-HTT-expressing). The test flies were then tapped down and allowed to climb every 6 s for ~ 2 min. Video recordings of the climbing flies were analyzed using the Flytracker program with some manual curation (B.A. Barbaro, UCI- https://github.com/brettbarbaro/Flytracker). The average climbing speed in each vial was calculated by subtracting the average position of flies at 2 s from that at 3 s. The results indicate the ratio of climbing speed of females to sibling males.
Immunohistochemistry
The anterior halves of wandering third instar larvae were separated, turned inside out and placed in PBS on ice. These halves were then fixed on shaker with 4% formaldehyde made in PBT (PBS + 0.2% Triton X-100) for 20 min at RT. After fixation, tissues were washed three times with PBT, blocked with 10% BSA in PBT for 2.5 h at RT and probed with primary antibody (s) overnight at 4°C. Tissues were then washed three times, blocked again and incubated with secondary antibody(s) for 1.5 h and washed again. The primary antibodies were rat-Elav-7E8A10 anti-elav (used at 1:200 dilution in PBS; Developmental Studies Hybridoma Bank), VB3130 rabbit anti-N-terminal HTT (used at 1:5000 dilution in PBS; epitope: aa 2-17; VIVA Bioscience) and MAB2170 mouse anti-C-terminal HTT (used at 1:5000 dilution in PBS; epitope: aa 1247 to 1646; Millipore). The secondary antibodies were Alexa Fluor 488 goat anti-rabbit (green), Alexa Fluor 488 goat anti-mouse (green), Alexa Fluor 568 goat anti-rat (red) and Alexa Fluor 568 goat anti-rabbit (red) (all used at 1:250 dilution in PBST, Life Technologies). Required organs were dissected out and mounted in Vectashield with DAPI or were treated with Hoechst and mounted in Vectashield. Images were captured using a Zeiss LSM 700 laser scanning microscope and analyzed using Volocity software.
Statistical analysis
Error bars show standard error of the mean (SEM = standard deviation/square root of n). Statistical significance was established using analysis of variance (ANOVA) with Tukey’s post hoc test on Prism software (GraphPad) (* = P < 0.05, ** = P < 0.01, *** = P < 0.001).
Abbreviations
HD Huntington’s disease polyQ poly glutamine
HTT Huntingtin
fl-mHTT Full-length mutant huntingtin 120Q
HTTex1 HTTexon1
IBs Inclusion bodies
ETH Ecdysis-triggering hormone
LEC Larval epidermal cells
PG Prothoracic gland
HA Hemagglutinin
LUM Lumio
YAC Yeast artificial chromosome
Aggregates and inclusions are used interchangeably to refer to microscopically visible aggregates of HTT.
Toxicity and pathology are used interchangeably to refer to abnormalities such as death and failure to eclose and various developmental abnormalities (e.g. tracheal loss, clefting).
Supplementary Material
Acknowledgements
This work was submitted in partial fulfillment of the Ph.D. degree by B.A. Barbaro. Eric Li conducted and analyzed several of the experiments as part of a science project at Northwood High School, Irvine, CA. The authors gratefully acknowledge the support of the Optical Biology Core facility and the Cancer Center Support grant of the University of California, Irvine (CC grant #CA62203). Stocks obtained from the Bloomington Drosophila Stock Center (NIH P40OD018537) were used in this study.
Funding
This research was supported by the CHDI foundation (HQ-44670) and the National Institute of Health (NS-45283 to JLM).
Conflict of Interest Statement
None declared.
References
- 1. Bradford J.W., Li S. and Li X.J. (2010) Polyglutamine toxicity in non-neuronal cells. Cell Res., 20, 400–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lunkes A., Lindenberg K.S., Ben-Haiem L., Weber C., Devys D., Landwehrmeyer G.B., Mandel J.L. and Trottier Y. (2002) Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol. Cell, 10, 259–269. [DOI] [PubMed] [Google Scholar]
- 3. Landles C., Sathasivam K., Weiss A., Woodman B., Moffitt H., Finkbeiner S., Sun B., Gafni J., Ellerby L.M., Trottier Y. et al. (2010) Proteolysis of mutant Huntingtin produces an exon 1 fragment that accumulates as an aggregated protein in neuronal nuclei in Huntington disease. J. Biol. Chem., 285, 8808–8823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sathasivam K., Neueder A., Gipson T.A., Landles C., Benjamin A.C., Bondulich M.K., Smith D.L., Faull R.L., Roos R.A., Howland D. et al. (2013) Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. U. S. A., 110, 2366–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gipson T.A., Neueder A., Wexler N.S., Bates G.P. and Housman D. (2013) Aberrantly spliced HTT, a new player in Huntington's disease pathogenesis. RNA Biol., 10, 1647–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gutekunst C., Li S., Yi H., Mulroy J., Kuemmerle S., Jones R., Rye D., Ferrante R., Hersch S. and Li X. (1999) Nuclear and neuropil aggregates in Huntington's disease: relationship to neuropathology. J. Neurosci., 19, 2522–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim Y.J., Yi Y., Sapp E., Wang Y., Cuiffo B., Kegel K.B., Qin Z.H., Aronin N. and DiFiglia M. (2001) Caspase 3-cleaved N-terminal fragments of wild-type and mutant huntingtin are present in normal and Huntington’s disease brains, associate with membranes, and undergo calpain-dependent proteolysis. Proc. Natl. Acad. Sci. U. S. A., 98, 12784–12789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim Y.J., Sapp E., Cuiffo B.G., Sobin L., Yoder J., Kegel K.B., Qin Z.H., Detloff P., Aronin N. and DiFiglia M. (2006) Lysosomal proteases are involved in generation of N-terminal huntingtin fragments. Neurobiol. Dis., 22, 346–356. [DOI] [PubMed] [Google Scholar]
- 9. Bates G.P., Mangiarini L., Wanker E.E. and Davies S.W. (1998) Polyglutamine expansion and Huntington's disease. Biochem. Soc. Trans., 26, 471–475. [DOI] [PubMed] [Google Scholar]
- 10. Parker J.A., Connolly J.B., Wellington C., Hayden M., Dausset J. and Neri C. (2001) Expanded polyglutamines in Caenorhabditis elegans cause axonal abnormalities and severe dysfunction of PLM mechanosensory neurons without cell death. Proc. Natl. Acad. Sci. U. S. A., 98, 13318–13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sokolov S., Pozniakovsky A., Bocharova N., Knorre D. and Severin F. (2006) Expression of an expanded polyglutamine domain in yeast causes death with apoptotic markers. Biochim. Biophys. Acta, 1757, 660–666. [DOI] [PubMed] [Google Scholar]
- 12. Steffan J.S., Bodai L., Pallos J., Poelman M., McCampbell A., Apostol B.L., Kazantsev A., Schmidt E., Zhu Y.Z., Greenwald M. et al. (2001) Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature, 413, 739–743. [DOI] [PubMed] [Google Scholar]
- 13. DiFiglia M., Sapp E., Chase K.O., Davies S.W., Bates G.P., Vonsattel J.P. and Aronin N. (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science, 277, 1990–1993. [DOI] [PubMed] [Google Scholar]
- 14. Schilling G., Becher M.W., Sharp A.H., Jinnah H.A., Duan K., Kotzuk J.A., Slunt H.H., Ratovitski T., Cooper J.K., Jenkins N.A. et al. (1999) Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum. Mol. Genet., 8, 397–407. [DOI] [PubMed] [Google Scholar]
- 15. Gray M., Shirasaki D.I., Cepeda C., Andre V.M., Wilburn B., Lu X.H., Tao J., Yamazaki I., Li S.H., Sun Y.E. et al. (2008) Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J. Neurosci., 28, 6182–6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schilling G., Klevytska A., Tebbenkamp A.T., Juenemann K., Cooper J., Gonzales V., Slunt H., Poirer M., Ross C.A. and Borchelt D.R. (2007) Characterization of huntingtin pathologic fragments in human Huntington disease, transgenic mice, and cell models. J. Neuropathol. Exp. Neurol., 66, 313–320. [DOI] [PubMed] [Google Scholar]
- 17. Slow E.J., Graham R.K., Osmand A.P., Devon R.S., Lu G., Deng Y., Pearson J., Vaid K., Bissada N., Wetzel R. et al. (2005) Absence of behavioral abnormalities and neurodegeneration in vivo despite widespread neuronal huntingtin inclusions. Proc. Natl. Acad. Sci. U. S. A., 102, 11402–11407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barbaro B.A., Lukacsovich T., Agrawal N., Burke J., Bornemann D.J., Purcell J.M., Worthge S.A., Caricasole A., Weiss A., Song W. et al. (2015) Comparative study of naturally occurring huntingtin fragments in Drosophila points to exon 1 as the most pathogenic species in Huntington's disease. Hum. Mol. Genet., 24, 913–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. O'Brien R., DeGiacomo F., Holcomb J., Bonner A., Ring K.L., Zhang N., Zafar K., Weiss A., Lager B., Schilling B. et al. (2015) Integration-independent transgenic Huntington disease fragment mouse models reveal distinct phenotypes and life span in vivo. J. Biol. Chem., 290, 19287–19306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tebbenkamp A.T., Swing D., Tessarollo L. and Borchelt D.R. (2011) Premature death and neurologic abnormalities in transgenic mice expressing a mutant huntingtin exon-2 fragment. Hum. Mol. Genet., 20, 1633–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schilling G., Jinnah H.A., Gonzales V., Coonfield M.L., Kim Y., Wood J.D., Price D.L., Li X.J., Jenkins N., Copeland N. et al. (2001) Distinct behavioral and neuropathological abnormalities in transgenic mouse models of HD and DRPLA. Neurobiol. Dis., 8, 405–418. [DOI] [PubMed] [Google Scholar]
- 22. Bischof J., Maeda R.K., Hediger M., Karch F. and Basler K. (2007) An optimized transgenesis system for drosophila using germ-line-specific phiC31 integrases. Proc. Natl. Acad. Sci. U. S. A., 104, 3312–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brand A.H. and Perrimon N. (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development, 118, 401–415. [DOI] [PubMed] [Google Scholar]
- 24. Marques Sousa C. and Humbert S. (2013) Huntingtin: here, there, everywhere! J. Huntingtons Dis., 2, 395–403. [DOI] [PubMed] [Google Scholar]
- 25. van der Burg J.M., Bjorkqvist M. and Brundin P. (2009) Beyond the brain: widespread pathology in Huntington's disease. Lancet Neurol., 8, 765–774. [DOI] [PubMed] [Google Scholar]
- 26. Park Y., Filippov V., Gill S.S. and Adams M.E. (2002) Deletion of the ecdysis-triggering hormone gene leads to lethal ecdysis deficiency. Development, 129, 493–503. [DOI] [PubMed] [Google Scholar]
- 27. Ross C.A. and Tabrizi S.J. (2011) Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol., 10, 83–98. [DOI] [PubMed] [Google Scholar]
- 28. Sontag E.M., Vonk W.I. and Frydman J. (2014) Sorting out the trash: the spatial nature of eukaryotic protein quality control. Curr. Opin. Cell Biol., 26, 139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Miller S.B., Ho C.T., Winkler J., Khokhrina M., Neuner A., Mohamed M.Y., Guilbride D.L., Richter K., Lisby M., Schiebel E. et al. (2015) Compartment-specific aggregases direct distinct nuclear and cytoplasmic aggregate deposition. EMBO J., 34, 778–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Saudou F., Finkbeiner S., Devys D. and Greenberg M.E. (1998) Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of Intranuclear inclusions. Cell, 95, 55–66. [DOI] [PubMed] [Google Scholar]
- 31. Arrasate M. and Finkbeiner S. (2012) Protein aggregates in Huntington's disease. Exp. Neurol., 238, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Benn C.L., Landles C., Li H., Strand A.D., Woodman B., Sathasivam K., Li S.H., Ghazi-Noori S., Hockly E., Faruque S.M. et al. (2005) Contribution of nuclear and extranuclear polyQ to neurological phenotypes in mouse models of Huntington's disease. Hum. Mol. Genet., 14, 3065–3078. [DOI] [PubMed] [Google Scholar]
- 33. Peters M.F., Nucifora F.C. Jr., Kushi J., Seaman H.C., Cooper J.K., Herring W.J., Dawson V.L., Dawson T.M. and Ross C.A. (1999) Nuclear targeting of mutant Huntingtin increases toxicity. Mol. Cell. Neurosci., 14, 121–128. [DOI] [PubMed] [Google Scholar]
- 34. Cho K.H., Daubnerova I., Park Y., Zitnan D. and Adams M.E. (2014) Secretory competence in a gateway endocrine cell conferred by the nuclear receptor betaFTZ-F1 enables stage-specific ecdysone responses throughout development in drosophila. Dev. Biol., 385, 253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McEwen D.G. and Peifer M. (2005) Puckered, a drosophila MAPK phosphatase, ensures cell viability by antagonizing JNK-induced apoptosis. Development, 132, 3935–3946. [DOI] [PubMed] [Google Scholar]
- 36. Sekyrova P., Bohmann D., Jindra M. and Uhlirova M. (2010) Interaction between drosophila bZIP proteins Atf3 and Jun prevents replacement of epithelial cells during metamorphosis. Development, 137, 141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ansaloni A., Wang Z.M., Jeong J.S., Ruggeri F.S., Dietler G. and Lashuel H.A. (2014) One-pot semisynthesis of exon 1 of the Huntingtin protein: new tools for elucidating the role of posttranslational modifications in the pathogenesis of Huntington's disease. Angew. Chem. Int. Ed. Engl., 53, 1928–1933. [DOI] [PubMed] [Google Scholar]
- 38. El-Daher M.T., Hangen E., Bruyere J., Poizat G., Al-Ramahi I., Pardo R., Bourg N., Souquere S., Mayet C., Pierron G. et al. (2015) Huntingtin proteolysis releases non-polyQ fragments that cause toxicity through dynamin 1 dysregulation. EMBO J., 34, 2255–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ochaba J., Lukacsovich T., Csikos G., Zheng S., Margulis J., Salazar L., Mao K., Lau A.L., Yeung S.Y., Humbert S. et al. (2014) Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc. Natl. Acad. Sci. U. S. A., 111, 16889–16894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pavel M., Imarisio S., Menzies F.M., Jimenez-Sanchez M., Siddiqi F.H., Wu X., Renna M., O'Kane C.J., Crowther D.C. and Rubinsztein D.C. (2016) CCT complex restricts neuropathogenic protein aggregation via autophagy. Nat. Commun., 7, 13821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Steffan J.S. (2010) Does Huntingtin play a role in selective macroautophagy? Cell Cycle, 9, 3401–3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tam S., Spiess C., Auyeung W., Joachimiak L., Chen B., Poirier M.A. and Frydman J. (2009) The chaperonin TRiC blocks a huntingtin sequence element that promotes the conformational switch to aggregation. Nat. Struct. Mol. Biol., 16, 1279–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ratovitski T., Gucek M., Jiang H., Chighladze E., Waldron E., D'Ambola J., Hou Z., Liang Y., Poirier M.A., Hirschhorn R.R. et al. (2009) Mutant huntingtin N-terminal fragments of specific size mediate aggregation and toxicity in neuronal cells. J. Biol. Chem., 284, 10855–10867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hackam A.S., Singaraja R., Wellington C.L., Metzler M., McCutcheon K., Zhang T., Kalchman M. and Hayden M.R. (1998) The influence of huntingtin protein size on nuclear localization and cellular toxicity. J. Cell Biol., 141, 1097–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Scherzinger E., Lurz R., Turmaine M., Mangiarini L., Hollenbach B., Hasenbank R., Bates G.P., Davies S.W., Lehrach H. and Wanker E.E. (1997) Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell, 90, 549–558. [DOI] [PubMed] [Google Scholar]
- 46. Vieweg S., Ansaloni A., Wang Z.M., Warner J.B. and Lashuel H.A. (2016) An Intein-based strategy for the production of tag-free Huntingtin exon 1 proteins enables new insights into the Polyglutamine dependence of Httex1 aggregation and fibril formation. J. Biol. Chem., 291, 12074–12086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cooper J.K., Schilling G., Peters M.F., Herring W.J., Sharp A.H., Kaminsky Z., Masone J., Khan F.A., Delanoy M., Borchelt D.R. et al. (1998) Truncated N-terminal fragments of huntingtin with expanded glutamine repeats form nuclear and cytoplasmic aggregates in cell culture. Hum. Mol. Genet., 7, 783–790. [DOI] [PubMed] [Google Scholar]
- 48. de Almeida L.P., Ross C.A., Zala D., Aebischer P. and Deglon N. (2002) Lentiviral-mediated delivery of mutant huntingtin in the striatum of rats induces a selective neuropathology modulated by polyglutamine repeat size, huntingtin expression levels, and protein length. J. Neurosci., 22, 3473–3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Palfi S., Brouillet E., Jarraya B., Bloch J., Jan C., Shin M., Conde F., Li X.J., Aebischer P., Hantraye P. et al. (2007) Expression of mutated huntingtin fragment in the putamen is sufficient to produce abnormal movement in non-human primates. Mol. Ther., 15, 1444–1451. [DOI] [PubMed] [Google Scholar]
- 50. Juenemann K., Weisse C., Reichmann D., Kaether C., Calkhoven C.F. and Schilling G. (2011) Modulation of mutant huntingtin N-terminal cleavage and its effect on aggregation and cell death. Neurotox. Res., 20, 120–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Forsburg S.L. and Sherman D.A. (1997) General purpose tagging vectors for fission yeast. Gene, 191, 191–195. [DOI] [PubMed] [Google Scholar]
- 52. Griffin B.A., Adams S.R. and Tsien R.Y. (1998) Specific covalent labeling of recombinant protein molecules inside live cells. Science, 281, 269–272. [DOI] [PubMed] [Google Scholar]
- 53. Palidwor G.A., Shcherbinin S., Huska M.R., Rasko T., Stelzl U., Arumughan A., Foulle R., Porras P., Sanchez-Pulido L., Wanker E.E. et al. (2009) Detection of alpha-rod protein repeats using a neural network and application to huntingtin. PLoS Comput. Biol., 5, e1000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ratovitski T., Nakamura M., D'Ambola J., Chighladze E., Liang Y., Wang W., Graham R., Hayden M.R., Borchelt D.R., Hirschhorn R.R. et al. (2007) N-terminal proteolysis of full-length mutant huntingtin in an inducible PC12 cell model of Huntington's disease. Cell Cycle, 6, 2970–2981. [DOI] [PubMed] [Google Scholar]
- 55. Wellington C., Singaraja R., Ellerby L., Savill J., Roy S., Leavitt B., Cattaneo E., Hackam A., Sharp A., Thornberry N. et al. (2000) Inhibiting caspase cleavage of huntingtin reduces toxicity and aggregate formation in neuronal and nonneuronal cells. J. Biol. Chem., 275, 19831–19838. [DOI] [PubMed] [Google Scholar]
- 56. Wang J., Gines S., MacDonald M.E. and Gusella J.F. (2005) Reversal of a full-length mutant huntingtin neuronal cell phenotype by chemical inhibitors of polyglutamine-mediated aggregation. BMC Neurosci., 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Evers M.M., Tran H.D., Zalachoras I., Meijer O.C., den Dunnen J.T., van Ommen G.J., Aartsma-Rus A. and van Roon-Mom W.M. (2014) Preventing formation of toxic N-terminal huntingtin fragments through antisense oligonucleotide-mediated protein modification. Nucleic Acid Ther., 24, 4-12. [DOI] [PubMed] [Google Scholar]
- 58. Davies S.W., Turmaine M., Cozens B.A., DiFiglia M., Sharp A.H., Ross C.A., Scherzinger E., Wanker E.E., Mangiarini L. and Bates G.P. (1997) Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell, 90, 537–548. [DOI] [PubMed] [Google Scholar]
- 59. Kar K., Jayaraman M., Sahoo B., Kodali R. and Wetzel R. (2011) Critical nucleus size for disease-related polyglutamine aggregation is repeat-length dependent. Nat. Struct. Mol. Biol., 18, 328–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sahoo B., Arduini I., Drombosky K.W., Kodali R., Sanders L.H., Greenamyre J.T. and Wetzel R. (2016) Folding landscape of mutant Huntingtin Exon1: diffusible multimers, oligomers and fibrils, and no detectable monomer. PLoS One, 11, e0155747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Marsh J.L., Walker H., Theisen H., Zhu Y.Z., Fielder T., Purcell J. and Thompson L.M. (2000) Expanded polyglutamine peptides alone are intrinsically cytotoxic and cause neurodegeneration in drosophila. Hum. Mol. Genet., 9, 13–25. [DOI] [PubMed] [Google Scholar]
- 62. Raspe M., Gillis J., Krol H., Krom S., Bosch K., van Veen H. and Reits E. (2009) Mimicking proteasomal release of polyglutamine peptides initiates aggregation and toxicity. J. Cell Sci., 122, 3262–3271. [DOI] [PubMed] [Google Scholar]
- 63. Gillis J., Schipper-Krom S., Juenemann K., Gruber A., Coolen S., van den Nieuwendijk R., van Veen H., Overkleeft H., Goedhart J., Kampinga H.H. et al. (2013) The DNAJB6 and DNAJB8 protein chaperones prevent intracellular aggregation of polyglutamine peptides. J. Biol. Chem., 288, 17225–17237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chen M. and Wolynes P.G. (2017) Aggregation landscapes of Huntingtin exon 1 protein fragments and the critical repeat length for the onset of Huntington's disease. Proc. Natl. Acad. Sci. U. S. A., 114, 4406–4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Thakur A.K., Jayaraman M., Mishra R., Thakur M., Chellgren V.M., Byeon I.J., Anjum D.H., Kodali R., Creamer T.P., Conway J.F. et al. (2009) Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat. Struct. Mol. Biol., 16, 380–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Caron N.S., Desmond C.R., Xia J. and Truant R. (2013) Polyglutamine domain flexibility mediates the proximity between flanking sequences in huntingtin. Proc. Natl. Acad. Sci. U. S. A., 110, 14610–14615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Caron N.S., Hung C.L., Atwal R.S. and Truant R. (2014) Live cell imaging and biophotonic methods reveal two types of mutant huntingtin inclusions. Hum. Mol. Genet., 23, 2324–2338. [DOI] [PubMed] [Google Scholar]
- 68. Giorgini F. (2013) A flexible polyglutamine hinge opens new doors for understanding huntingtin function. Proc. Natl. Acad. Sci. U. S. A., 110, 14516–14517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Vijayvargia R., Epand R., Leitner A., Jung T.Y., Shin B., Jung R., Lloret A., Singh Atwal R., Lee H., Lee J.M. et al. (2016) Huntingtin's spherical solenoid structure enables polyglutamine tract-dependent modulation of its structure and function. Elife, 5, e11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ghabrial A.S., Levi B.P. and Krasnow M.A. (2011) A systematic screen for tube morphogenesis and branching genes in the drosophila tracheal system. PLoS Genet., 7, e1002087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Syed A., Lukacsovich T., Pomeroy M., Bardwell A.J., Decker G.T., Waymire K.G., Purcell J., Huang W., Gui J., Padilla E.M. et al. (2019) Miles to go (mtgo) encodes FNDC3 proteins that interact with the chaperonin subunit CCT3 and are required for NMJ branching and growth in drosophila. Dev. Biol., 445, 37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Duffy J.B. (2002) GAL4 system in drosophila: a fly geneticist's Swiss army knife. Genesis, 34, 1–15. [DOI] [PubMed] [Google Scholar]
- 73. Song W., Smith M.R., Syed A., Lukacsovich T., Barbaro B.A., Purcell J., Bornemann D.J., Burke J. and Marsh J.L. (2013) Morphometric analysis of Huntington's disease neurodegeneration in drosophila. Methods Mol. Biol., 1017, 41–57. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.