

scLearn is a metric learning-based framework with measurement and threshold learned automatically for single-cell assignment.
Abstract
Efficient single-cell assignment without prior marker gene annotations is essential for single-cell sequencing data analysis. Current methods, however, have limited effectiveness for distinct single-cell assignment. They failed to achieve a well-generalized performance in different tasks because of the inherent heterogeneity of different single-cell sequencing datasets and different single-cell types. Furthermore, current methods are inefficient to identify novel cell types that are absent in the reference datasets. To this end, we present scLearn, a learning-based framework that automatically infers quantitative measurement/similarity and threshold that can be used for different single-cell assignment tasks, achieving a well-generalized assignment performance on different single-cell types. We evaluated scLearn on a comprehensive set of publicly available benchmark datasets. We proved that scLearn outperformed the comparable existing methods for single-cell assignment from various aspects, demonstrating state-of-the-art effectiveness with a reliable and generalized single-cell type identification and categorizing ability.
INTRODUCTION
Single-cell transcriptomics are now indispensable for revealing the heterogeneity of complex tissues and organisms (1–6); however, it is limited by the heavy reliance on manual annotation and inspection of cell type–specific marker genes, which is time-consuming, labor intensive, and irreproducible. Efficiently assigning cells into proper cell types or states presents to be one of the grand challenges in single-cell data analysis (7).
Recently, cell type assignment strategies without prior marker gene annotations have emerged to solve these issues (8–15). Basically, these strategies use a large amount of labeled datasets as a reference to automatically assign or categorize cell types for the new query cells without prior cell type–specific marker gene information. They also simultaneously label those cells whose cell types are absent in the reference as “unassigned.” Although these strategies have advanced progress in single-cell type identification and categorization, single-cell sequencing data are inherently heterogeneous and noisy (16), which make it challenging for these strategies to perform well on different single-cell sequencing datasets and different single-cell types (8, 15). Basically, the performance of existing methods can be evaluated in two test scenarios, i.e., positive control scenario and negative control scenario [also called rejection task (15)]. In the test of the positive control scenario, the single cells with labeled cell types are collected as the reference, and the coming single cells with the same tissues and cell types as the reference are taken as the query. The goal of this test is to assign the query cells with the proper cell types presented in the reference datasets. On the other hand, in the test of the negative control scenario, the reference and the query are from the completely different tissues and cell types, and the goal of this test is to identify these query cells as unassigned cells. A comprehensive study of the existing single-cell assignment methods indicated that several issues remain: (i) the assignment performance of existing methods is greatly influenced by different cell types and datasets for both positive control scenario and negative control scenario, failing to achieve a well-generalized and robust assignment performance in different cell types, and (ii) the current methods are biased toward one specific test scenario. For example, the recently proposed scmap-cluster (8) method performs well in negative control scenario, while it performs poorly in positive control scenarios, leaving a large percentage of cells incorrectly unassigned (8). On the other hand, scmap-cell (8), a proposed complementary method to scmap-cluster (8), performs totally opposite to scmap-cluster. (iii) The threshold to assign query cell as unassigned is selected empirically, which makes most of the existing tools inefficient to identify novel cell types that are absent in the reference datasets.
In addition, in real single-cell assignment application scenario, we basically have no idea what the query cells look like. Therefore, an assignment method that performs well generalized in both positive control scenario and negative control scenario with robustness to various single-cell sequencing datasets and cell types is highly desired. Traditional strategies have failed to achieve well-generalized single-cell assignments for different tasks, mainly because the measurement/similarity used for cell type assignment is manually designed. Therefore, the performance is substantially influenced by the inherent heterogeneity and complexity of different single-cell sequencing datasets and different single-cell types (17–19). To this end, we present scLearn (https://github.com/bm2-lab/scLearn), a learning-based framework that automatically infers the quantitative measurement/similarity and threshold that can be used for different single-cell assignment tasks to obtain well-generalized performance on different single-cell types. The main contributions of scLearn are as follows: (i) scLearn is robust to different assignment tasks with a well-generalized assignment performance. (ii) scLearn is efficient in the identification of novel cell types that are absent in the reference datasets, and (iii) a multilabel single-cell assignment strategy is proposed in scLearn to assign a single cell to proper time status and cell type simultaneously, proven to be effective for cell development and lineage analysis with additional temporal information (20, 21). We first evaluated scLearn among 30 publicly available benchmark datasets (table S1) (2, 22–39) and indicated that scLearn outperformed the comparable existing methods for single-cell assignment from various aspects, demonstrating state-of-the-art effectiveness with a reliable single-cell type identification and categorizing ability. The effectiveness of scLearn on multilabel single-cell assignment is further validated on publicly available datasets (20, 21), proven to be effective for single-cell data analysis with additional temporal information.
RESULTS
General pipeline of scLearn
scLearn is a learning-based framework designed to intuitively carry out a cell search by measuring the similarity between query cells and each reference cell cluster centroid using measurement and thresholds learned from reference datasets, rather than manually designing the measurement/similarity or empirically selecting the threshold to determine unassigned cells. Basically, scLearn comprises three main steps (Fig. 1 and see Materials and Methods): data preprocessing, model learning, and cell assignment.
Fig. 1. The scLearn workflow.
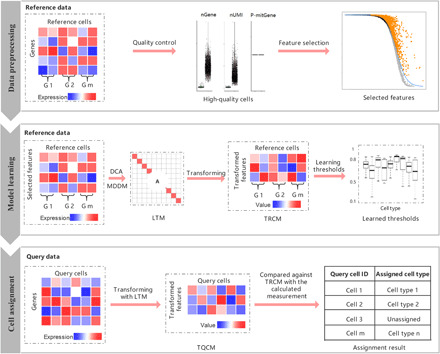
scLearn comprises three steps: data preprocessing, model learning, and cell assignment. (i) In the first step, the main processes comprise routine normalization, cell quality control, rare cell type filtering, and feature selection; nGene, number of genes; nUMI, number of unique molecular identifiers; P-mitGene, percentage of mitochondrial genes; and G, cell group. (ii) In the second step, for single-label single-cell assignment, discriminative component analysis (DCA) is applied to learn the transformation matrix. For multilabel single-cell assignment, MDDM (multilabel dimension reduction via dependence maximization) is applied to learn the transformation matrix. Then, with the learned transformation matrix, the transformed reference cell samples are obtained for the following assignment. The thresholds for labeling a cell as unassigned for each cell type are also automatically learned. LTM, learned transformation matrix, which can be calculated as the optimal transformation matrix for single-label single-cell assignment or by Eq. 6 for multilabel single-cell assignment, respectively (see Materials and Methods); TRCM, transformed reference cell matrix, which can be calculated using Eq. 1 (see Materials and Methods). (iii) In the third step, the transformed query cell samples are obtained on the basis of LTM with an available optional cell quality control procedure. The transformed query samples are compared against the TRCM to derive the measurement fulfilling the cell-type assignment with the rejection task. TQCM, transformed query cell matrix, which can be calculated using Eq. 2 (see Materials and Methods).
First, a routine normalization and quality control for single-cell RNA sequencing data is performed. scLearn removes the rare cell types whose cell numbers are less than 10 from the reference datasets. Then, scLearn performs feature selection. Previous works (8, 40) has indicated that M3Drop (40), which is based on a specific dropout rate, obtained a better performance than HVG (high variable gene) (16) method and random selection method in the single-cell assignment. Therefore, we use M3Drop as the feature selection method in scLearn.
Second, scLearn establishes a metric learning-based model to automatically learn the measurement used for cell assignment based on reference cells. In this model, the identification of query cell type is formulated as a single-label single-cell assignment. Discriminative component analysis (DCA) (41) is applied, and a transformation matrix that can be applied to formulate an optimal measurement that naturally fits the relationship between these samples is learned on the basis of the prior sample similarity or dissimilarity (see Materials and Methods). In addition, the assignment of query cell into proper time status and cell type simultaneously is formulated as a multilabel single-cell assignment. In this case, scLearn extended the DCA-based matrix transformation to a multilabel dimension reduction by maximizing the dependence between the original feature space and the associated labels [multilabel dimension reduction via dependence maximization (MDDM) (42)] (see Materials and Methods). For either case, the derived transformation matrix can be multiplied by the original reference data matrix and the query data matrix, respectively, and the learned measurement can be obtained on the basis of the distance/similarity between the transformed data samples. For single-label single-cell assignment, bootstrapping sampling technology is also used in this step to reduce sampling imbalances and to obtain a stable learning-based model. Note that in the single-cell assignment, the accurate identification of novel cell types that are absent in the reference, i.e., assigning these cells as unassigned, is important, while the existing single-cell assignment strategies often fail in this task by adoption of an empirical threshold for unassigned cells, such as a Pearson correlation coefficient of 0.7 or cosine similarity of 0.5 (8), which should differ among distinct datasets with different cell types and annotations. For example, the thresholds of datasets with fine-grained annotation (deep annotation, i.e., cells are categorized in a fine-grained manner) should be larger than those of datasets with coarse-grained annotation (shallow annotation, i.e., cells are categorized in a coarse-grained manner) because the cells in the former datasets are more similar than the cells in the latter datasets. Therefore, one threshold for all datasets and all cell types is not suitable. To this end, in this step, scLearn automatically learns the thresholds of each cell type in each dataset instead of specifying a priori thresholds (see “Model learning” in Fig. 1 and Materials and Methods). Last, according to the measurement and thresholds learned with the learning-based model, scLearn assigns the cell types of the query cells by comparison with the reference datasets (see Materials and Methods).
Evaluating the cell type assignment performance of scLearn with various test scenarios
To evaluate the cell type assignment performance of scLearn, which is formulated as a single-label single-cell assignment, we first validated the rationale of the designed framework of scLearn to ensure that it is inherently suitable for single-cell assignment. For illustration purposes, we used human pancreas cells [Baron_human (22)] as an example plot (Fig. 2). As shown in Fig. 2 (A to D), compared with the commonly used measurements in traditional studies (e.g., Pearson correlation coefficient), the measurements learned by scLearn are much more suitable for the data characteristics, making cells within the same class become more similar (referred to as “intracluster compactness,” see Materials and Methods), while cells with different labels become more dissimilar [referred to as “intercluster complexity,” as described by Abdelaal et al. (15)]. We also tested another two example datasets with different protocols and tissues, i.e., mouse embryo stem cells (31) and mouse retina (figs. S1 and S2) (33). The ability of scLearn to maximize intracluster compactness and intercluster complexity is especially beneficial for single-cell assignment tasks. To further verify the universal performance of scLearn, we calculated the intracluster compactness and intercluster complexity of the clustering results with a manually designed Pearson correlation coefficient and the learned measurement from 30 previously published test datasets (Fig. 2, E and F, and tables S2 and S3).
Fig. 2. Comparison of the clustering results before and after transforming with scLearn.
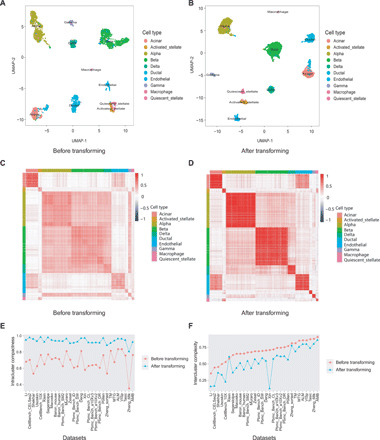
(A) The visualization of clustering results by UMAP (uniform manifold approximation and projection) before DCA-based transforming for the Baron_human (human pancreas) dataset. (B) The visualization of clustering results by UMAP after DCA-based transforming for the Baron_human (human pancreas) dataset. (C) Similarity heatmap calculated with the Pearson correlation coefficient before transforming for the Baron_human (human pancreas) dataset. (D) Similarity heatmap calculated with the Pearson correlation coefficient after transforming for the Baron_human (human pancreas) dataset. (E) Comparison of the intercluster complexity before and after DCA-based transforming by scLearn with all 30 datasets listed in table S1. For datasets with multiple levels of cell type, we used the most fine-grained cell types. (F) Comparison of the intracluster compactness before and after DCA-based transforming by scLearn with all 30 datasets listed in table S1. For datasets with multiple levels of cell type, we used the most fine-grained cell types.
Second, we benchmarked six traditional single-cell assignment strategies, including scmap-cluster (8), scmap-cell (8), scID (9), scPred (10), CHETAH (11), and SVMrejection (12). Note that SVMrejection is an SVM-based single-cell assignment model treating those unmatched query cells to the reference as unassigned, which was also benchmarked in previous studies (8, 15). Abdelaal et al. (15) have proven that SVMrejection achieved a good assignment performance in the previous study. Therefore, although SVMrejection is not designed specifically for single-cell assignment, it is necessary to be included in our benchmark for a comprehensive comparison. Together, these methods are currently the main methods proposed for single-cell type assignment with unassigned option that do not require prior marker gene information. Garnett (43) and CellAssign (44) require prior maker gene information, and SingleR (13) and CellFishing.jl (14) do not offer the unassigned option for cell type identification; therefore, they are not comparable here (15). To be comprehensive and fair, we followed the similar benchmark strategies as those of scmap (8) tested in positive control scenario and negative control scenario and performed a series of comparisons between scLearn and the other six tools on a total of 30 previously published datasets. Note that SVMrejection is not specifically designed for single-cell assignment and does not have a specific feature selection strategy for the current application; therefore, to keep the comparison fair, we applied the same feature selection as that used in scLearn, and the threshold for SVMrejection was set to 0.7 as usual (8, 15), while all the other methods adopted their own default methods and parameters to determine unassigned cells.
Test scenario 1: Positive control scenario
The test results of positive control scenario are shown in Fig. 3A and table S4. In the test of positive control scenario, we tested the commonly used pancreas benchmark datasets (table S1) (22–25), each of which was treated as the reference or query data, respectively, resulting in a total of 12 dataset pairs (permutation ) . In addition, seven immune cell benchmark datasets (table S1) (39) were also tested, each of which was treated as the reference or query data, respectively, resulting in a total of 42 dataset pairs (permutation ). In this test scenario, we did not consider the case of negative control, so the reference was processed into cell types that cover the query cell types. Then, we calculate accuracy, i.e., the proportion of the correctly predicted cells among all the query cells, to evaluate the performance of all these methods. The larger accuracy indicates a better performance of the test method. As shown in Fig. 3A, scLearn obtained the best performance among others. In addition, we calculated the SD of all the performance obtained by each method, and scLearn achieved the lowest SD, further proving its robustness and stableness. In summary, in this test scenario, scLearn has obtained the best and the most stable performance among others.
Fig. 3. Benchmark of scLearn with available methods for positive control scenario and negative control scenario.
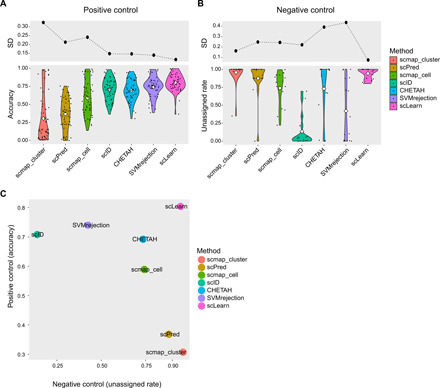
(A) Benchmark in the positive control scenario. The white diamond line represents the mean value. (B) Benchmark in the negative control scenario. The white diamond line represents the mean value. (C) A comprehensive benchmark by integrating the test results from positive control scenario and negative control scenario.
Test scenario 2: Negative control scenario
Although challenging, it is necessary to incorporate an unassigned option in single-cell assignments, which is important in the discovery of novel cell types. In this test scenario (Fig. 3B), the reference cells and the query cells are completely different, and they belong to different cell types. We use unassigned rate i.e., the proportion of the predicted unassigned cells among all query cells, to evaluate the performance of these methods. A higher unassigned rate indicates a better performance. In this study, 16 dataset pairs that were previously used in scmap (8) were tested (table S5). As shown in Fig. 3B, most of the six traditional tools performed poorly in this evaluation. For example, SVMrejection had an unassigned rate that was below 0.5, and scID (9) even obtained the worst unassigned rate of 0.134. Only scLearn and scmap-cluster performed well, and scLearn achieved the most robust and stable results, as indicated by the lowest SD of the performance compared with others. In summary, in the test of negative control scenario, scLearn still obtained a well-generalized performance among others. Note that the datasets used in positive control and negative control scenarios are generated from different protocols. For example, the four pancreas datasets are generated from four different protocols, and the seven peripheral blood mononuclear cell datasets are generated from seven different protocols (see table S1), further proving the robustness of scLearn to different protocols and sequencing platforms.
To further explain the superiority of scLearn, we made a comprehensive comparison by integrating the results of both the two test scenarios (positive control scenario and negative control scenario) (table S6). As shown in Fig. 3C, existing methods are often biased toward one specific test scenario. For example, SVMrejection obtained quite good performance in the positive control scenario, while it performed poorly in the negative control scenario, which is also reported in the previous study (8). On the other hand, scmap-cluster performed well in the negative control scenario, while it performed poorly in the positive control scenario. CHETAH achieved relatively good performance in both two scenarios. scLearn, however, achieved the best and well-generalized performance in both positive control and negative control scenarios.
Test scenario 3: Real application scenario evaluation
In the former test scenarios, scLearn achieved the best cell assignment performance with various testing cases, indicating its suitability for real application scenarios, in which we have basically no idea how the query cells look and the types of assignment tasks the query cells face. To further prove the superiority of scLearn, we simulated a real application scenario in this test. We believe that in the real application scenario, the most likely situation is that some of the query cell types are in the reference (positive controls), while the others are not (negative controls). The former needs to be accurately assigned with correct labels as many as possible, while the latter should be labeled as unassigned as completely as possible. In other words, effective cell assignment is expected to achieve a well-generalized performance on both the positive controls and negative controls, which will benefit categorizing known cell types and discover novel cell types simultaneously.
For the real application scenario (Fig. 4, A to C, and tables S7 and S8), we retained seven common pancreas cell types within the Muraro dataset (23) and Baron dataset (22) as a complete pancreas cell reference. Then, the whole cells in the Baron dataset were used as query cells. For the Baron dataset, besides the above seven common pancreas cell types (positive control), it contained additional seven cell types including “activated stellate cells,” “epsilon cells,” “macrophage cells,” “mast cells,” “quiescent stellate cells,” “Schwann cells,” and “T cells.” These additional cell types were absent in the reference, and they are treated as negative control cell types, which should be predicted as unassigned (negative control). As shown in Fig. 4A and table S7, for the common pancreas cell types, scLearn assigned them correctly with small unassigned rates. For additional cell types, scLearn assigned them with high unassigned rates, while most of the other existing methods failed to keep a balance between these two cases (Fig. 4B). As shown in Fig. 4C and table S8, the calculation of the accuracy (the proportion of correctly assigned cells among the common pancreas cell types) and the specificity (the proportion of correctly predicted unassigned cells among all predicted unassigned cells) further indicated that scLearn obtained the best performance in both measures, proving its well-generalized performance to categorize known cell types and discover novel cell types.
Fig. 4. The test in real application scenario.
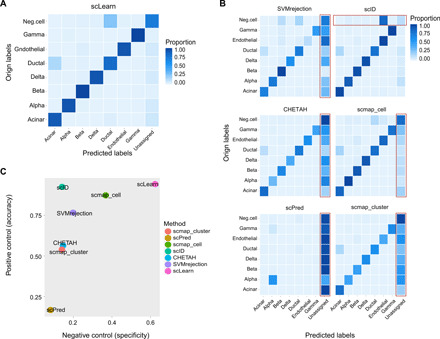
(A) The prediction performance heatmap of confusion matrix for scLearn, with normalization for each row (Orign labels). Neg.cell, negative control cells, including activated stellate cells, epsilon cells, macrophage cells, mast cells, quiescent stellate cells, Schwann cells, and T cells. (B) Prediction performance heatmaps of confusion matrix for the other six methods, including SVMrejection, scID, CHETAH, scmap_cell, scPred, and scmap_cluster, with normalization for each row (Orign labels). The poor prediction results are outlined in the red box. (C) The comprehensive comparison by considering accuracy and specificity simultaneously. The specificity is defined as the proportion of correctly predicted unassigned cells among all predicted unassigned cells.
Extending scLearn to multilabel single-cell assignment
Temporal information is also essential for single-cell data analysis in certain application scenarios, especially for cell development and lineage analysis (20, 21). In such cases, individual cell commonly requires temporal annotations besides cell type categorization. Every cell can be labeled in two aspects, i.e., cell type and time point. Therefore, assigning or labeling single cells with proper time status during the development besides categorizing the cell type is highly desired. This problem can be formulated as a multilabel single-cell assignment task. Basically, it can be addressed through the following two strategies, i.e., assigning the query cell to individual label type separately (separate assignment) or assigning the query cell with a synthetic label space by combining the two label type into one fine-grained label space (fine-grained assignment). In our study, the later one can be explained as combining the original two label spaces <time point>, <cell type> into one fine-grained label space <time point, cell type>. For separate assignment, we performed the traditional single-cell assignment for each type of labels and then combined the results for performance evaluation. For fine-grained assignment, we first combined the two separate types of labels into one fine-grained label and then performed the traditional single-cell assignment for performance evaluation. Nevertheless, in single-cell data analysis, different label types are often related to each other. For example, in the process of cell development, specific cell types are often enriched in certain time phasing (20). Therefore, for multilabel single-cell assignment, it is important and necessary to incorporate label relationship information between different label types to boost the assignment performance. In our study, we extend scLearn to a multilabel single-cell assignment paradigm by applying a multilabel dimension reduction–based matrix transformation via maximizing the dependence between the original feature space and the associated labels [MDDM (42)], which also considers the correlations between labels (see Materials and Methods).
To verify the superiority of MDDM-based scLearn on multilabel single-cell assignment, we first validated the rationale of the MDDM-based scLearn by comparing the MDDM-based learning and DCA-based learning with a geometric intuition (Fig. 5A). As shown in Fig. 5A, the main advantage of MDDM-based learning is the utilization of the relationship of multilabels, so it can separate cell type and time state simultaneously. Then, we also made a visualization of clustering results by uniform manifold approximation and projection (UMAP) before and after MDDM-based transforming for ESC (embryonic stem cell) (Fig. 5B) (20). As shown in Fig. 5B, the clustering of the real dataset ESC visually proved this advantage. We further compared MDDM-based scLearn with DCA-based scLearn in the following two strategies, i.e., separate assignment and fine-grained assignment on three single-cell RNA sequencing datasets with temporal and cell type information simultaneously (Fig. 5C and table S9) (20, 21). The three multilabel datasets are ESC (20), mouse embryo ventral midbrain cells (mEM) (20), and human prefrontal cortex cells (hPFC; for details, see Materials and Methods) (21). We performed the comparisons with self-projection (8), i.e., 70% cells were randomly selected to train the model, and the remaining 30% cells were used for testing 10 times. The results were evaluated with micro_F1 (42), which is designed specifically for multilabel tasks by calculation of the traditional F1 measurement on the predictions of different labels as a combined one (see Materials and Methods). The higher the micro_F1, the better the performance. As seen in Fig. 5, for multilabel single-cell assignment, MDDM-based scLearn performed much better than DCA-based scLearn with a higher micro_F1 on each strategy, respectively. Overall, MDDM with fine-grained assignment is recommended for multilabel single-cell assignment compared with the others.
Fig. 5. Comparison of DCA-based scLearn and MDDM-based scLearn for multilabel single-cell assignment.
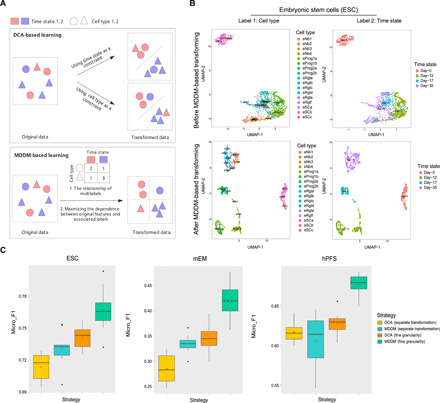
(A) Geometric intuition for DCA-based learning and MDDM-based learning. (B) Visualization of clustering results by UMAP before and after MDDM-based transforming for the ESC (embryonic stem cells) dataset. (C) The micro_F1 of DCA-based scLearn and MDDM-based scLearn under two strategies for three multilabel datasets were presented. mEM, mouse embryo ventral midbrain cells; hPFC, human prefrontal cortex cells.
DISCUSSION
Here, we present a novel learning-based framework, scLearn, for single-cell assignment, in which the measurement and thresholds to determine unassigned cells are both learned from reference datasets instead of a priori empirical settings. We demonstrated the superiority and robustness of scLearn by performing three main evaluations with comparisons to other mainstream cell assignment methods on a total of 30 previously published datasets. Together, scLearn is proven to achieve a well-generalized and robust performance in both positive control scenario and negative control scenario. Moreover, we presented a multilabel single-cell assignment paradigm to extend scLearn to assign a single cell to proper time status and cell type simultaneously. The effectiveness of scLearn on such multilabel single-cell assignment is validated on publicly available datasets as well, proven to be effective for single-cell development and lineage analysis with additional temporal information.
The whole pretrained models for all 30 datasets and for a comprehensive reference single-cell atlas of 20 mouse organs (2) are also built in scLearn (table S10), including commonly used brain cells, immune cells, pancreas cells, embryo stem cells, retina cells, lung cancer cell lines, and all 20 mouse organs with coarse-grained and fine-grained annotation, which can be directly used and can be beneficial for the related single-cell categorizing used by researchers.
In summary, single-cell assignment is difficult and more challenging when applied to real-world scenarios. Although several methods have been presented, most only perform well on a specific test scenario and fail to achieve a well-generalized performance. In real application scenarios, we often need to consider how to label cells as unassigned if their cell types are absent in the reference dataset while, at the same time, assigning known cell types correctly. Facing these challenges, scLearn provides great improvements and contributions to solve these issues by achieving a well-generalized and robust single-cell type identification and categorizing ability.
The temporal information is also important for many single-cell studies such as cell development and lineage analysis (20, 21). Therefore, an efficient multilabel single-cell assignment is highly desired. The multilabel single-cell assignment paradigm presented in scLearn serves as a novel contribution to single-cell data analysis community. This paradigm is not restricted to two labels, and it is extendable to multiple labels. Such paradigm is expected to be beneficial for efficient investigation of the tempo-spatial characteristics of single-cell data by considering the tempo-spatial correlations among the cell microenvironment.
Note that although scLearn has greatly improved the performance of the negative control scenario while simultaneously maintaining a balance of good performance for positive controls, the problem of determining unassigned cells, which is the essential step for discovering novel cell subtypes, requires further improvement. In addition, for rare cell types whose cell number is less than 10, most of the single-cell assignment methods, including the current version of scLearn, are not satisfactory. In the current study, these cell types were excluded because they contain limited information and are unreliable for subsequent assignment. Therefore, efficient cell assignment and detection of rare cell types remain future challenges (7, 45, 46).
MATERIALS AND METHODS
Single-cell type assignment benchmark datasets tested for scLearn
The 30 single-cell type assignment benchmark datasets were curated from two parts: One was collected in previous work of scmap (8) (https://hemberg-lab.github.io/scRNA.seq.datasets), and the other one was curated from the following benchmark study (15) (https://doi.org/10.5281/zenodo.3357167). These datasets were converted into Bioconductor SingleCellExperiment (http://bioconductor.org/packages/SingleCellExperiment) class objects. Different datasets were used in different evaluation scenarios, and they are listed in table S1.
Data preprocessing
The first step of scLearn is data preprocessing, which mainly consists of three parts: cell quality control, rare cell type filtering, and feature selection. The quality of the reference datasets is important and must be reliable; thus, scLearn evaluates cell quality based on strict criteria following three considerations: the number of genes detected (default, >500), the number of unique molecular identifiers induced (default, >1500), and the percentage of mitochondrial genes detected (default, <10% among all genes). Only cells satisfying all three criteria are retained to formulate the reference data. All datasets were scaled to 10,000 and normalized with log(counts + 1). Next, scLearn removed rare cell types whose cell number is less than 10 because such cell types are less informative and unreliable for subsequent assignment. Last, to select informative features, we applied M3Drop (40), which was proven to be more suitable than HVG (16, 40) and random selection method for single-cell assignment with a default threshold of 0.05. The features were only selected from the reference datasets, and those absent in the query dataset were all supplemented with zeros.
Model learning for single-cell type assignment
scLearn formulates a metric learning-based framework in which the measurement for cell type assignment is learned from the reference data instead of empirical settings such as traditional Euclidean distance, Pearson correlation coefficient, cosine similarity, Spearman correlation coefficient, etc. Specifically, scLearn first randomly selects some cell samples from each reference class with similar or dissimilar information constraints. Then, with DCA (41), a transformation matrix that leads to an optimal measurement that naturally fits the relationship between these samples is learned according to the similarity or dissimilarity of prior samples.
Specifically, the idea of DCA is to learn the optimal transformation matrix A that leads to the optimal distance measurement by both maximizing the total variance between the discriminative data chunklets and minimizing the total variance of data instances in the same chunklets, in which the chunklets can be formed by the positive constraints (similar).
When the optimal transformation matrix A is solved, the transformed reference cell matrix (TRCM) and transformed query cell matrix (TQCM) can be calculated as follows
| (1) |
| (2) |
where Rselected features is the reference expression matrix with selected features, and Qselected features is the query expression matrix with selected features. A is the optimal transformation matrix. Last, the single-cell type assignment can be fulfilled by calculating the distance/similarity between the samples in TRCM against the reference TQCM. In our study, we adopted Pearson correlation after transforming to calculate the similarity throughout the study, while other measurements, such as cosine and Spearman, were also tested. In general, scLearn is robust to different measurements adopted here (fig. S3). This measurement can be treated as the newly learned measurement from the reference data rather than empirically selected. Note that in this step, scLearn obtains a stable optimal distance measurement by bootstrapping 10 times to reduce sampling imbalances.
Learning the thresholds to determine unassigned cells
One threshold is not suitable for all cell types and datasets. Therefore, scLearn also learns the thresholds for each cell type in each dataset instead of empirically specifying a prior threshold. Specifically, for each cell type of the reference dataset, with a learned TRCM (calculated using Eq. 1), scLearn calculates the cluster centroid, and then the similarities between the cluster centroid and each cell are calculated using the Pearson correlation coefficient. In other words, for each cell type, scLearn obtains its similarity distribution with the learned measurement. Last, scLearn automatically selects the value of the last 1% among the distribution as a threshold for each cell type. The robustness of such cutoff is also tested, as shown in fig. S4.
Query cell assignment
With the learned transformation matrix and thresholds, query cells can be assigned to the reference data. Intuitively, scLearn carries out a search by measuring the similarity between query cells and each reference cluster centroid with the learned measurement and thresholds. First, for the query data, cell quality control is optional for users, and the query data were scaled to 10,000 and normalized with log(counts + 1). Then, the TQCM is obtained using Eq. 2. The similarities between each transformed query cell and the transformed reference cluster centroid are calculated with the Pearson correlation coefficient. Last, the calculated similarity values are compared to corresponding learned thresholds for each reference cell type. If there is no similarity value larger than its corresponding threshold, then the query cell is labeled unassigned. If there is only one similarity value larger than its corresponding threshold, then the query cell belongs to the corresponding cell type with no ambiguity. If there is more than one similarity value larger than their respective corresponding thresholds, then (i) if the difference between the largest similarity value does not exceed 0.05, we consider that this assignment is ambiguous and this query cell is also labeled unassigned because the two values are too similar, and (ii) if the difference between the two largest similarity values exceeds 0.05, this query cell is labeled as the corresponding cell type with the largest similarity value.
Intracluster compactness and intercluster complexity
To evaluate the performance of scLearn, we first validated the ability of scLearn for well-generalized single-cell assignment. We defined the intracluster compactness (Eq. 3) and the intercluster complexity [Eq. 4 as described by Abdelaal et al. (15)] to quantitatively indicate the reference data distribution.
Intracluster compactness represents the degree of intraclass similarity for each cell type in the dataset. The higher the compactness, the greater the intraclass similarity of the cell types in the dataset, and the easier it is to assign query cells. Intuitively, for a dataset, intracluster compactness is the mean of the similarities to the average expression of selected genes for each cell type in the dataset
| (3) |
where n is the cell type number, mi is the cell number of the i-th cell type, corr() is the calculated Pearson correlation coefficient, avgi is the average expression of selected genes for the i-th cell type, and valueij is the j-th cell expression in the i-th cell type.
Intercluster complexity was previously defined by Abdelaal et al. (15), as calculated in Eq. 4, and used to represent the degree of interclass similarity for each cell type in the dataset
| (4) |
where n is the cell type number, avgi and avgj are the average expression of selected genes for the i-th and j-th cell type, respectively, and corr() is the calculated Pearson correlation coefficient.
Here, we used selected genes instead of all of the genes to make the equation more informative. In general, for a dataset, the higher the intercluster compactness and the lower the intercluster complexity, the easier it is to obtain a good assignment performance on the dataset.
The evaluation criteria for single-cell type assignment
The benchmark performance in different test scenarios is evaluated with the following metrics. (i) For positive control scenario, we adopted accuracy, i.e., the proportion of correctly assigned cells among all query cells. A higher accuracy indicates a better performance. (ii) For negative control scenario, because the reference cells and the query cells are completely different, only unassigned rate is applied to evaluate the performance. The unassigned rate is calculated as the proportion of predicted unassigned cells. A higher unassigned rate indicates a better performance. (iii) For real application scenario, the performance in positive controls is evaluated with accuracy, i.e., the proportion of correctly assigned cells among the known cell types. The performance in negative controls is evaluated with unassigned rate, i.e., the proportion of correctly predicted unassigned cells among all predicted unassigned cells. A comprehensive evaluation by considering these two measurements simultaneously is needed in real application scenario.
Benchmarking against existing methods
To demonstrate the superiority of scLearn, we benchmarked six mainstream single-cell assignment strategies, i.e., scmap-cluster (8), scmap-cell (8), scID (9), scPred (10), CHETAH (11), and SVMrejection (12). Garnett (43) and CellAssign (44), which require prior maker gene information, and SingleR (13) and CellFishing.jl (14), which do not offer the unassigned option for cell type identification, therefore are not comparable here (15). We performed all these methods with the same cell quality control and their default parameters and feature selection methods, except for SVMrejection, due to the lack of explicit description of its feature selection procedure. For SVMrejection, a similar M3Drop (40) feature selection as that of scLearn was adopted for a fair comparison, and the threshold to determine unassigned cells of SVMrejection was set to 0.7 as for the others.
Extending scLearn for multilabel single-cell assignment
For multilabel single-cell assignment, scLearn learns the transformation matrix by MDDM (42) instead of DCA (41). In this framework. MDDM attempts to obtain an optimal transformation matrix that can project the original data into a lower-dimensional feature space by maximizing the dependence between the original feature space and the associated class labels and considering the correlations between labels.
Briefly, MDDM comprises three steps: (i) calculating the relationship between different labels, (ii) maximizing the dependence between the feature description and the class labels, and (iii) obtaining the optimal transformation matrix. For convenience, let XD × N denote the feature matrix, where D is the number of features and N is the number of samples. There is a label set Θ, which includes M labels. The proper labels associated with a sample x constitute a subset of Θ, which can be represented as an M-dimensional binary vector y, with 1 indicating that the sample has the corresponding label and 0 otherwise, and then, YM × N is denoted as the label matrix.
First, MDDM calculates the relationship between different labels by constructing the label kernel matrix L, which can be calculated as follows
| (5) |
where yi and yj represent binary label vector of the i-th and j-th sample, respectively.
Then, MDDM tries to maximize the dependence between the feature description and the class labels by applying the Hilbert-Schmidt independence criterion (47) as a measurement of the dependence in the optimization (6) as
| (6) |
where e is an all-one column vector.
The solution to Eq. 6 resulted in the calculation of the eigenvalues λ and eigenvectors p of the matrix XHLHXT. Last, MDDM chooses the first d eigenvectors that enable to construct the optimal transformation matrix AD × d. The thr is between 0 and 1, and the default is set to 99.9% (42).
Benchmark datasets for multilabel single-cell assignment
To verify the superiority of MDDM-based scLearn on multilabel single-cell assignment, three multilabel datasets were curated and tested, i.e., ESC (20), mEM (20), and hPFC (21). The ESC dataset covers different stages of differentiation toward Th+ neurons with a total of 17 cell types and four time points. The mEM dataset covers six developmental stages between embryonic days 11.5 to 18.5 with a total of 26 cell types and six time points. The hPFC dataset covers the developmental stages of gestational weeks 8 to 26 with a total of six cell types and nine time points.
The evaluation criteria for multilabel single-cell assignment
Multilabel single-cell assignment requires suitable evaluation criteria compared with traditional single-label single-cell assignment. Here, we used micro-F1 (42) in our study for multilabel single-cell assignment evaluation. Intuitively, micro-F1 is calculated by averaging the precision and recall of the prediction results. In our multilabel assignment, first we combined the original different label spaces into a fine-grained one and calculated the precision and recall for each fine-grained label. Then, we calculated the averaged precision and averaged recall. Last, we obtained micro-F1 following its routine definition, as listed below
| (7) |
Specifically, two distinct multilabel single-cell assignment strategies exist, i.e., separate assignment and fine-grained assignment, while the evaluation of these two strategies can be treated the same way by combining the original different label spaces into a fine-grained one for the following evaluations. In this case, a combined predicted label is correct only when all types of labels in this combination are correct. As for the determination of unassigned cells, a combined predicted label is unassigned when any type of label in this combination is unassigned.
Reference databases built in scLearn
To facilitate a broad application of scLearn for single-cell assignment, we not only provide the R package of scLearn but also present the pretrained models for the 30 datasets tested in our study, as well as for a comprehensive mouse single-cell RNA sequencing atlas including 20 mouse organs (2) for the direct utilization (table S10).
These reference datasets comprehensively cover commonly used cell type annotations, including brain cells, immune cells, pancreas cells, embryo stem cells, retina cells, lung cancer cell lines, and the whole 20 mouse organs with coarse-grained and fine-grained annotation. They can be directly and successfully applied to the related single-cell assignment by researchers. The R package and the pretrained assignment models can be downloaded from the GitHub website (https://github.com/bm2-lab/scLearn).
Supplementary Material
Acknowledgments
Funding: This work was supported by the National Key Research and Development Program of China (grant nos. 2017YFC0908500 and 2016YFC1303205), the National Natural Science Foundation of China (grant nos. 31970638 and 61572361), the Shanghai Natural Science Foundation Program (grant no. 17ZR1449400), the Shanghai Artificial Intelligence Technology Standard Project (grant no. 19DZ2200900), and the Fundamental Research Funds for the Central Universities. Author contribution: Q.L. conceived the method. Q.L., B.D., C.Z., G.C., and C.T. implemented scLearn. X.C., S.C., S.F., and G.L. processed the single-cell data. Q.L. and B.D. wrote the manuscript with assistance from other authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusion in the paper are present in the paper and/or the Supplementary Materials. The 30 single-cell type assignment benchmark datasets in our study were curated from two parts: One was collected in previous work of scmap (8) (https://hemberg-lab.github.io/scRNA.seq.datasets), and the other one was curated from the following benchmark study (15) (https://doi.org/10.5281/zenodo.3357167). scLearn is developed as an R package available at https://github.com/bm2-lab/scLearn, built in with comprehensive human and mammalian single-cell reference datasets and pretrained models, which can be used directly to facilitate broad applications of single-cell assignment. Additional data related to this paper maybe requested from the authors.
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/44/eabd0855/DC1
REFERENCES AND NOTES
- 1.Cao J., Spielmann M., Qiu X., Huang X., Ibrahim D. M., Hill A. J., Zhang F., Mundlos S., Christiansen L., Steemers F. J., Trapnell C., Shendure J., The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.The Tabula Muris Consortium; Overall coordination; Logistical coordination; Organ collection and processing; Library preparation and sequencing; Computational data analysis; Cell type annotation; Writing group; Supplemental text writing group; Principal investigators , Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Plass M., Solana J., Wolf F. A., Ayoub S., Misios A., Glažar P., Obermayer B., Theis F. J., Kocks C., Rajewsky N., Cell type atlas and lineage tree of a whole complex animal by single-cell transcriptomics. Science 360, eaaq1723 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Han X., Wang R., Zhou Y., Fei L., Sun H., Lai S., Saadatpour A., Zhou Z., Chen H., Ye F., Huang D., Xu Y., Huang W., Jiang M., Jiang X., Mao J., Chen Y., Lu C., Xie J., Fang Q., Wang Y., Yue R., Li T., Huang H., Orkin S. H., Yuan G.-C., Chen M., Guo G., Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 173, 1307 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Fincher C. T., Wurtzel O., de Hoog T., Kravarik K. M., Reddien P. W., Cell type transcriptome atlas for the planarian Schmidtea mediterranea. Science 360, eaaq1736 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cao J., Packer J. S., Ramani V., Cusanovich D. A., Huynh C., Daza R., Qiu X., Lee C., Furlan S. N., Steemers F. J., Adey A., Waterston R. H., Trapnell C., Shendure J., Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 357, 661–667 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lähnemann D., Köster J., Szczurek E., McCarthy D. J., Hicks S. C., Robinson M. D., Vallejos C. A., Campbell K. R., Beerenwinkel N., Mahfouz A., Pinello L., Skums P., Stamatakis A., Attolini C. S.-O., Aparicio S., Baaijens J., Balvert M., de Barbanson B., Cappuccio A., Corleone G., Dutilh B. E., Florescu M., Guryev V., Holmer R., Jahn K., Lobo T. J., Keizer E. M., Khatri I., Kielbasa S. M., Korbel J. O., Kozlov A. M., Kuo T.-H., Lelieveldt B. P. F., Mandoiu I. I., Marioni J. C., Marschall T., Mölder F., Niknejad A., Raczkowski L., Reinders M., de Ridder J., Saliba A.-E., Somarakis A., Stegle O., Theis F. J., Yang H., Zelikovsky A., McHardy A. C., Raphael B. J., Shah S. P., Schönhuth A., Eleven grand challenges in single-cell data science. Genome Biol. 21, 31 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kiselev V. Y., Yiu A., Hemberg M., scmap: Projection of single-cell RNA-seq data across data sets. Nat. Methods 15, 359–362 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Boufea K., Seth S., Batada N. N., scID uses discriminant analysis to identify transcriptionally equivalent cell types across single-cell RNA-seq data with batch effect. iScience 23, 100914 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alquicira-Hernández J., Sathe A., Ji H. P., Nguyen Q., Powell J. E., scPred: Cell type prediction at single-cell resolution. bioRxiv , 369538 (2018). [Google Scholar]
- 11.de Kanter J. K., Lijnzaad P., Candelli T., Margaritis T., Holstege F. C. P., CHETAH: A selective, hierarchical cell type identification method for single-cell RNA sequencing. Nucleic Acids Res. 47, e95 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ben-Hur A., Horn D., Siegelmann H. T., Vapnik V., Support vector clustering. J. Mach. Learn. Res 2, 125–137 (2001). [Google Scholar]
- 13.Aran D., Looney A. P., Liu L., Wu E., Fong V., Hsu A., Chak S., Naikawadi R. P., Wolters P. J., Abate A. R., Butte A. J., Bhattacharya M., Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 20, 163–172 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sato K., Tsuyuzaki K., Shimizu K., Nikaido I., CellFishing.jl: An ultrafast and scalable cell search method for single-cell RNA sequencing. Genome Biol. 20, 31 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abdelaal T., Michielsen L., Cats D., Hoogduin D., Mei H., Reinders M. J. T., Mahfouz A., A comparison of automatic cell identification methods for single-cell RNA sequencing data. Genome Biol. 20, 194 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brennecke P., Anders S., Kim J. K., Kołodziejczyk A. A., Zhang X., Proserpio V., Baying B., Benes V., Teichmann S. A., Marioni J. C., Heisler M. G., Accounting for technical noise in single-cell RNA-seq experiments. Nat. Methods 10, 1093–1095 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Kim T., Chen I. R., Lin Y., Wang A. Y.-Y., Yang J. Y. H., Yang P., Impact of similarity metrics on single-cell RNA-seq data clustering. Brief. Bioinform. 20, 2316–2326 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Wang B., Zhu J., Pierson E., Ramazzotti D., Batzoglou S., Visualization and analysis of single-cell RNA-seq data by kernel-based similarity learning. Nat. Methods 14, 414–416 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Skinnider M. A., Squair J. W., Foster L. J., Evaluating measures of association for single-cell transcriptomics. Nat. Methods 16, 381–386 (2019). [DOI] [PubMed] [Google Scholar]
- 20.La Manno G., Gyllborg D., Codeluppi S., Nishimura K., Salto C., Zeisel A., Borm L. E., Stott S. R. W., Toledo E. M., Villaescusa J. C., Lönnerberg P., Ryge J., Barker R. A., Arenas E., Linnarsson S., Molecular diversity of midbrain development in mouse, human, and stem cells. Cell 167, 566–580.e19 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhong S., Zhang S., Fan X., Wu Q., Yan L., Dong J., Zhang H., Li L., Sun L., Pan N., Xu X., Tang F., Zhang J., Qiao J., Wang X., A single-cell RNA-seq survey of the developmental landscape of the human prefrontal cortex. Nature 555, 524–528 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Baron M., Veres A., Wolock S. L., Faust A. L., Gaujoux R., Vetere A., Ryu J. H., Wagner B. K., Shen-Orr S. S., Klein A. M., Melton D. A., Yanai I., A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure. Cell Syst. 3, 346–360.e4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muraro M. J., Dharmadhikari G., Grün D., Groen N., Dielen T., Jansen E., van Gurp L., Engelse M. A., Carlotti F., de Koning E. J. P., van Oudenaarden A., A single-cell transcriptome atlas of the human pancreas. Cell Syst. 3, 385–394.e3 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Segerstolpe Å., Palasantza A., Eliasson P., Andersson E.-M., Andréasson A.-C., Sun X., Picelli S., Sabirsh A., Clausen M., Bjursell M. K., Smith D. M., Kasper M., Ämmälä C., Sandberg R., Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab. 24, 593–607 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xin Y., Kim J., Okamoto H., Ni M., Wei Y., Adler C., Murphy A. J., Yancopoulos G. D., Lin C., Gromada J., RNA sequencing of single human islet cells reveals type 2 diabetes genes. Cell Metab. 24, 608–615 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Deng Q., Ramsköld D., Reinius B., Sandberg R., Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 343, 193–196 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Pollen A. A., Nowakowski T. J., Shuga J., Wang X., Leyrat A. A., Lui J. H., Li N., Szpankowski L., Fowler B., Chen P., Ramalingam N., Sun G., Thu M., Norris M., Lebofsky R., Toppani D., Kemp D. W. II, Wong M., Clerkson B., Jones B. N., Wu S., Knutsson L., Alvarado B., Wang J., Weaver L. S., May A. P., Jones R. C., Unger M. A., Kriegstein A. R., West J. A. A., Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat. Biotechnol. 32, 1053–1058 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H., Courtois E. T., Sengupta D., Tan Y., Chen K. H., Goh J. J. L., Kong S. L., Chua C., Hon L. K., Tan W. S., Wong M., Choi P. J., Wee L. J. K., Hillmer A. M., Tan I. B., Robson P., Prabhakar S., Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 49, 708–718 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Usoskin D., Furlan A., Islam S., Abdo H., Lönnerberg P., Lou D., Hjerling-Leffler J., Haeggström J., Kharchenko O., Kharchenko P. V., Linnarsson S., Ernfors P., Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci. 18, 145–153 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Tasic B., Menon V., Nguyen T. N., Kim T. K., Jarsky T., Yao Z., Levi B., Gray L. T., Sorensen S. A., Dolbeare T., Bertagnolli D., Goldy J., Shapovalova N., Parry S., Lee C., Smith K., Bernard A., Madisen L., Sunkin S. M., Hawrylycz M., Koch C., Zeng H., Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat. Neurosci. 19, 335–346 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klein A. M., Mazutis L., Akartuna I., Tallapragada N., Veres A., Li V., Peshkin L., Weitz D. A., Kirschner M. W., Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zeisel A., Muñoz-Manchado A. B., Codeluppi S., Lönnerberg P., Manno G. L., Juréus A., Marques S., Munguba H., He L., Betsholtz C., Rolny C., Castelo-Branco G., Hjerling-Leffler J., Linnarsson S., Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Shekhar K., Lapan S. W., Whitney I. E., Tran N. M., Macosko E. Z., Kowalczyk M., Adiconis X., Levin J. Z., Nemesh J., Goldman M., McCarroll S. A., Cepko C. L., Regev A., Sanes J. R., Comprehensive classification of retinal bipolar neurons by single-cell transcriptomics. Cell 166, 1308–1323.e30 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Macosko E. Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A. R., Kamitaki N., Martersteck E. M., Trombetta J. J., Weitz D. A., Sanes J. R., Shalek A. K., Regev A., McCarroll S. A., Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tian L., Dong X., Freytag S., Cao K.-A. L., Su S., Abadi A. J., Amann-Zalcenstein D., Weber T. S., Seidi A., Jabbari J. S., Naik S. H., Ritchie M. E., Benchmarking single cell RNA-sequencing analysis pipelines using mixture control experiments. Nat. Methods 16, 479–487 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Tasic B., Yao Z., Graybuck L. T., Smith K. A., Nguyen T. N., Bertagnolli D., Goldy J., Garren E., Economo M. N., Viswanathan S., Penn O., Bakken T., Menon V., Miller J., Fong O., Hirokawa K. E., Lathia K., Rimorin C., Tieu M., Larsen R., Casper T., Barkan E., Kroll M., Parry S., Shapovalova N. V., Hirschstein D., Pendergraft J., Sullivan H. A., Kim T. K., Szafer A., Dee N., Groblewski P., Wickersham I., Cetin A., Harris J. A., Levi B. P., Sunkin S. M., Madisen L., Daigle T. L., Looger L., Bernard A., Phillips J., Lein E., Hawrylycz M., Svoboda K., Jones A. R., Koch C., Zeng H., Shared and distinct transcriptomic cell types across neocortical areas. Nature 563, 72–78 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng G. X. Y., Terry J. M., Belgrader P., Ryvkin P., Bent Z. W., Wilson R., Ziraldo S. B., Wheeler T. D., Mc Dermott G. P., Zhu J., Gregory M. T., Shuga J., Montesclaros L., Underwood J. G., Masquelier D. A., Nishimura S. Y., Schnall-Levin M., Wyatt P. W., Hindson C. M., Bharadwaj R., Wong A., Ness K. D., Beppu L. W., Deeg H. J., Farland C. M., Loeb K. R., Valente W. J., Ericson N. G., Stevens E. A., Radich J. P., Mikkelsen T. S., Hindson B. J., Bielas J. H., Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hodge R. D., Bakken T. E., Miller J. A., Smith K. A., Barkan E. R., Graybuck L. T., Close J. L., Long B., Johansen N., Penn O., Yao Z., Eggermont J., Höllt T., Levi B. P., Shehata S. I., Aevermann B., Beller A., Bertagnolli D., Brouner K., Casper T., Cobbs C., Dalley R., Dee N., Ding S.-L., Ellenbogen R. G., Fong O., Garren E., Goldy J., Gwinn R. P., Hirschstein D., Keene C. D., Keshk M., Ko A. L., Lathia K., Mahfouz A., Maltzer Z., Graw M. M., Nguyen T. N., Nyhus J., Ojemann J. G., Oldre A., Parry S., Reynolds S., Rimorin C., Shapovalova N. V., Somasundaram S., Szafer A., Thomsen E. R., Tieu M., Quon G., Scheuermann R. H., Yuste R., Sunkin S. M., Lelieveldt B., Feng D., Ng L., Bernard A., Hawrylycz M., Phillips J. W., Tasic B., Zeng H., Jones A. R., Koch C., Lein E. S., Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ding J., Adiconis X., Simmons S. K., Kowalczyk M. S., Hession C. C., Marjanovic N. D., Hughes T. K., Wadsworth M. H., Burks T., Nguyen L. T., Kwon J. Y. H., Barak B., Ge W., Kedaigle A. J., Carroll S., Li S., Hacohen N., Rozenblatt-Rosen O., Shalek A. K., Villani A.-C., Regev A., Levin J. Z., Systematic comparative analysis of single cell RNA-sequencing methods. bioRxiv , 632216 (2019). [Google Scholar]
- 40.Andrews T. S., Hemberg M., M3Drop: Dropout-based feature selection for scRNASeq. Bioinformatics 35, 2865–2867 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.S. C. H. Hoi, W. Liu, M. R. Lyu, W.-Y. Ma, Learning distance metrics with contextual constraints for image retrieval, 2006 IEEE Computer Society Conference on Computer Vision and Pattern Recognition (CVPR’06) (New York, NY, USA, 2006), pp. 2072–2078. [Google Scholar]
- 42.Zhang Y., Zhou Z.-H., Multilabel dimensionality reduction via dependence maximization. ACM Trans. Knowl. Discov. Data 4, 1–21 (2010). [Google Scholar]
- 43.Pliner H. A., Shendure J., Trapnell C., Supervised classification enables rapid annotation of cell atlases. Nat. Methods 16, 983–986 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang A. W., O’Flanagan C., Chavez E. A., Lim J. L. P., Ceglia N., Pherson A. M., Wiens M., Walters P., Chan T., Hewitson B., Lai D., Mottok A., Sarkozy C., Chong L., Aoki T., Wang X., Weng A. P., McAlpine J. N., Aparicio S., Steidl C., Campbell K. R., Shah S. P., Probabilistic cell-type assignment of single-cell RNA-seq for tumor microenvironment profiling. Nat. Methods 16, 1007–1015 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsoucas D., Yuan G.-C., GiniClust2: A cluster-aware, weighted ensemble clustering method for cell-type detection. Genome Biol. 19, 58 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dong R., Yuan G.-C., GiniClust3: A fast and memory-efficient tool for rare cell type identification. bioRxiv , 788554 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.A. Gretton, O. Bousquet, A. Smola, B. Schölkopf, in Algorithmic Learning Theory, 16th International Conference, ALT 2005, Singapore, October 8–11, 2005, Proceedings (2005). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/44/eabd0855/DC1