

Filia regulates genome integrity and neurogenesis of postnatal hippocampal neural stem/progenitor cells.
Abstract
Endogenous DNA double-strand breaks (DSBs) formation and repair in neural stem/progenitor cells (NSPCs) play fundamental roles in neurogenesis and neurodevelopmental disorders. NSPCs exhibit heterogeneity in terms of lineage fates and neurogenesis activity. Whether NSPCs also have heterogeneous regulations on DSB formation and repair to accommodate region-specific neurogenesis has not been explored. Here, we identified a regional regulator Filia, which is predominantly expressed in mouse hippocampal NSPCs after birth and regulates DNA DSB formation and repair. On one hand, Filia protects stalling replication forks and prevents the replication stress-associated DNA DSB formation. On the other hand, Filia facilitates the homologous recombination–mediated DNA DSB repair. Consequently, Filia−/− mice had impaired hippocampal NSPC proliferation and neurogenesis and were deficient in learning, memory, and mood regulations. Thus, our study provided the first proof of concept demonstrating the region-specific regulations of DSB formation and repair in subtypes of NSPCs.
INTRODUCTION
Tissue stem cells can reconstruct the tissue and are essential for organogenesis, tissue homeostasis, and repair. Genome integrity is crucial for stem cells to maintain their identity and normal functions. Genetic instability may cause cell cycle arrest and apoptosis, induce stem cell differentiation, and skew the stem cell differentiation potential. This will not only decline the quantity and quality of stem cells but also passage the DNA damages to their progenies. Thus, stem cell genomic instability is implicated in many developmental and degenerative disorders, as well as in the stem cell–based tumorigenesis. Neural stem/progenitor cells (NSPCs) undergo active proliferation during fetal neurogenesis. NSPC proliferation and neurogenesis also take place after birth at two specific brain regions including the subventricular zone (SVZ) and the subgranular cell layer (SGL) in the hippocampal dentate gyrus. The causative relations between genomic instability of fetal NSPCs and cortical development failures have been well documented. For instance, induction of genomic instability in Nestin-expressing fetal NSPCs by conditional depletion of ATR (ataxia telangiectasia and Rad3 related) (ATM- and Rad3-related), a central coordinator of DNA replication stress response, caused severe neurodevelopmental failure and animal death at postnatal day 7 (P7) (1). Similarly, specific knockout of TopBP1 [topoisomerase (DNA) II binding protein 1], which activates ATR kinase and ensures genomic stability, in fetal NSPCs resulted in neurodevelopmental defects and animal death at P14 (2). A recent study identified a stem cell–enriched nucleolar protein nucleostemin, which repairs DNA damages in NSPCs by recruiting Rad51 to replication-associated DNA double-strand breaks (DSBs). Conditional loss of nucleostemin in Nestin-expressing fetal NSPCs severely impaired the neurodevelopment and caused newborn death (3). Collectively, these studies demonstrated that severe genomic instability induced by deficiency in replication stress response or DNA DSBs repair in proliferating fetal NSPCs can cause life-threatening impairment in embryonic neurodevelopment.
Beyond the detrimental effect, DSBs, on the other hand, provide opportunities to functionally modify genomic information and to generate genetic diversity. Two major pathways are implicated in DSBs repair: homologous recombination (HR)–mediated pathway and classical nonhomologous end-joining (cNHEJ) pathway. HR repair has low efficiency but high accuracy, whereas cNHEJ ligates two broken ends together with high efficiency but is prone to generate DNA translocation and gene diversification. RAG1 (recombination activating 1)–mediated endogenous DSB formation and their repair by cNHEJ have been shown to play essential roles in V (variable), D (diversity), or J (joining) genes recombination in the development and function of adaptive immune system (4). Similarly, during fetal neurogenesis, endogenous DNA DSBs frequently arise due to DNA replication, transcription activation, and oxidative stress in proliferating NSPCs. Notably, many long genes critical for neurogenesis in NSPCs harbor recurrent DSB clusters (RDCs), which represent replication fragile sites and are prone to generate DSBs under mild replication stress or even at unperturbed conditions (5). Appropriate endogenous DNA DSB formation and repair in NSPCs play fundamental roles in neurogenesis, and aberrations may contribute to the abnormal neurogenesis and brain disorders. In support, a recent study reported the increased DSB formation in NSPCs differentiated from human induced pluripotent stem cells of patients with macrocephalic autism spectrum disorder (6).
NSPCs with distinct spatial or temporal distributions exhibit heterogeneity in terms of lineage fates, neurogenesis activity, and gene expression profiles (7, 8). This raises the question whether different subtypes of NSPCs exhibit heterogeneity with respect to the source of endogenous DNA damages, the repair pathways, and the underlying regulators. However, to the best of our knowledge, no evidence has been obtained to answer this question.
Among the brain regions, the hippocampus is responsible for memory, spatial navigation, and mood regulations. The hippocampus develops relatively late compared to other brain regions. Most hippocampal neurons in rodents are generated in the first postnatal week, and the adult structure of the dentate gyrus is considered to be established by P14. Notably, hippocampal NSPCs in SGL of the dentate gyrus sustain continuous self-renewal and neurogenesis throughout lifetime (9–11), conferring a critical cellular basis for heightened plasticity, learning, memory, and mood regulations in this brain region. On the basis of the properties of hippocampal NSPCs, we reasoned that hippocampal NSPCs may have some unique characters in genomic stability regulations and their genomic alternations could cause behavioral phenotypes and neuropsychiatric disorders.
In this study, we identified a specific regulator Filia [official name Khdc3 (KH domain containing 3, subcortical maternal complex member), also known as Ecat1 (ES cell-associated transcript 1)], which is predominantly expressed in hippocampal NSPCs after birth and persists in adulthood to regulate genomic stability and ensure normal hippocampal neurogenesis and functions. Filia was first identified as a specific gene highly expressed in mouse embryonic stem cells (ESCs). Later studies reported its expression in growing oocytes (12). The protein structure is poorly characterized, and an atypical KH [hnRNP K (heterogeneous nuclear ribonucleoprotein K) homology] domain is predicted at its N terminus. Its cellular and physiological functions were also recently started to be uncovered. For instance, our previous work revealed that oocyte-expressed Filia ensures euploidy of cleavage stage embryos. Depletion of maternal Filia caused preimplantation development arrest or delay and reduced fecundity (13). Most recently, we investigated its functions in mouse ESCs and uncovered its critical roles in safeguarding genomic stability (14, 15). We also detected the expression of Filia in in vitro propagated NSPCs derived from the hippocampus of newborn mice. The expression was markedly induced by exogenous DNA damage insults, suggesting that Filia may play roles in preserving genomic stability of NSPCs. However, Filia−/− mice can live to adulthood (13) and have normal brain size. These intriguing observations prompted us to examine the spatiotemporal expressions and regional functions of Filia in the central nervous system. Our results provided the proof of concept, demonstrating that subtypes of NSPCs exhibit heterogeneity on genomic stability regulation to accommodate region-specific neurogenesis.
RESULTS
Filia is predominantly expressed in hippocampal NSPCs after birth
Filia was previously identified in mouse ESCs and played critical roles in ensuring their genomic stability (14, 15). We wondered whether Filia also safeguarded genomic integrity of NSPCs. Filia proteins were detected in propagated NSPCs derived from the hippocampus of newborn wild-type (WT) mice. The protein expression of Filia was markedly induced by etoposide or hydroxyurea (HU) treatment (Fig. 1A), which causes the DNA DSBs and DNA replication stress, respectively (16). This observation suggested that Filia might play roles in regulating genomic stability of NSPCs.
Fig. 1. Filia is restrictively expressed in hippocampal NSPCs after birth.
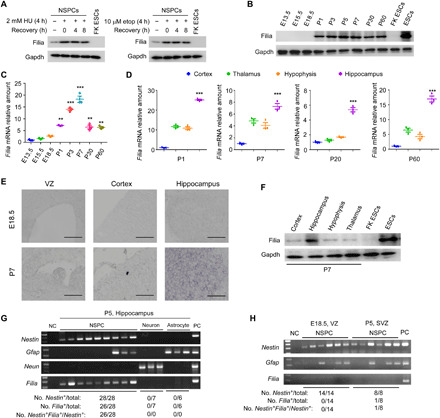
(A) Filia proteins were expressed in cultured mouse NSPCs. HU or etoposide (etop) treatment stimulated Filia expression. Filia knockout ESCs (FK ESCs) were used as negative control. Gapdh, glyceraldehyde-3-phosphate dehydrogenase. (B) Filia expressions in whole-brain tissue collected at different embryonic and postnatal stages. Filia is predominantly expressed after birth. (C) Real-time PCR confirmed the predominant expression of Filia mRNAs after birth in whole-brain tissue. (D) Real-time PCR showed that Filia mRNAs were highly expressed in the hippocampus at P1, P7, P20, and P60 when compared to the other brain regions of the cortex, thalamus, and hypophysis. (E) In situ hybridization verified the prominent expression of Filia mRNAs in the hippocampus after birth. Scale bars, 400 μm. (F) Filia proteins were dominantly detected in the hippocampus when examined at P7. WT ESCs and Filia knockout ESCs were used as positive and negative controls, respectively. (G) PCR detection of Filia mRNA expression in single cells freshly prepared from the hippocampus at P5. (H) Filia mRNA expression was barely detected by PCR in single NSPCs freshly prepared from the VZ at E18.5 or the SVZ at P5. Data were represented as means ± SEM; one-way analysis of variance (ANOVA) with Bonferroni post hoc test, **P < 0.01 and ***P < 0.001.
To understand the physiological roles of Filia in the central nervous system, we first investigated the spatiotemporal expression patterns of Filia in mouse brain. Immunoblotting (Fig. 1B) and quantitative polymerase chain reaction (qPCR) (Fig. 1C) examinations of whole-brain samples collected from WT embryos or mice at different ages consistently showed that Filia expressions remained relatively low at fetal stages [embryonic day 13.5 (E13.5), E15.5, and E18.5] but increased sharply after birth (P1, P3, P7, P30, and P60). We then collected RNA samples from four different brain regions representing the cortex, hippocampus, thalamus, and hypophysis at P1, P7, P20, and P60, respectively. The abundance of Filia mRNAs was highest in the hippocampus at all examined ages (Fig. 1D). In situ hybridization also validated the predominant expression of Filia mRNAs at the hippocampus after birth (Fig. 1E). Consistently, Filia protein expression was highest in the hippocampus region when examined at P7 (Fig. 1F).
To precisely define the identity of Filia-expressing cells in the hippocampus after birth, we analyzed the Filia mRNA expression in single-cell resolution. Single cells were randomly and manually collected from the single-cell suspension freshly prepared from the hippocampus of WT mice at the age of P5. Single-cell complementary DNAs (cDNAs) were prepared by Smart-seq2 and reverse transcription PCR (RT-PCR) examination revealed that Filia transcripts were restrictively detected in Nestin-expressing NSPCs (Fig. 1G). We also performed single-cell analysis to examine Filia expression on NSPCs collected from the ventricular zone (VZ) at E18.5 and the SVZ at P5. Consistently, Filia transcripts were barely detected in Nestin-expressing NSPCs from the VZ and SVZ (Fig. 1H). Together, these data demonstrate that Filia is predominantly and persistently expressed in hippocampal NSPCs after birth and may have restrictive function in the hippocampus.
Loss of Filia increases DNA DSB formation in the hippocampus
Filia protein expression is responsive to exogenous genotoxic insults in cultured NSPCs (Fig. 1A). We reasoned that Filia might participate in the regulation of genomic stability in hippocampal NSPCs under physiological conditions. To this end, we examined the phosphorylation of histone H2AX (H2A.X variant histone) at serine-159 (γH2AX), a surrogate marker of DNA DSBs. Indirect immunofluorescence staining on brain tissue sections showed that higher proportions of NSPCs [proliferating cell nuclear antigen–positive (PCNA+) cells] were positive for γH2AX staining in the hippocampus region of Filia−/− mice when compared to the WT littermates or the other brain regions of Filia−/− mice at P1 and P7 (Fig. 2A). γH2AX level was also notably increased in differentiated cells (PCNA−) in the hippocampus region of Filia−/− mice, suggesting that the DNA DSBs remained in differentiated progenies. This observation is consistent with previous report (17). To further validate the induction of DNA DSBs in the hippocampus, we assessed the DNA damage extent by neutral comet assay, which directly measures the DNA DSBs of individual cells. Single-cell suspensions were freshly prepared from the cortex, hippocampus, thalamus, and hypophysis of Filia−/− and WT littermates at P1, P3, and P7. The hippocampal cells from Filia−/− mice consistently showed longer comet tails than their WT counterparts (Fig. 2B). In contrast, cells from other brain regions had comparable comet tail length between WT and Filia−/− mice (fig. S1A), which was in line with the absence of Filia expression and function in these regions. Filia is persistently expressed in adult hippocampal NSPCs. Concordantly, the elevated DNA DSBs in the hippocampus of Filia−/− mice were detected at the advanced ages of 2 months and 1 year when compared to WT littermates (Fig. 2C). Together, these lines of evidence supported a restrictive function of Filia in the hippocampus.
Fig. 2. Filia depletion causes DNA DSBs in the hippocampus.
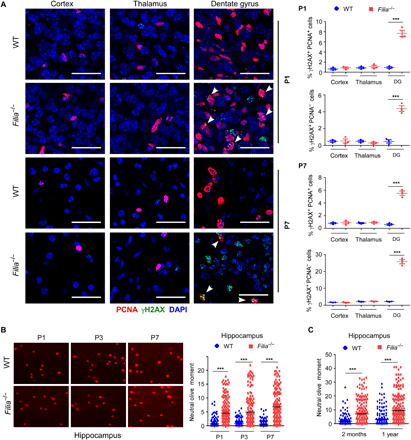
(A) Immunostaining and quantification of brain sections collected at P1 and P7 consistently detected the high level of γH2AX in PCNA+ and PCNA− cells in the dentate gyrus (DG) from Filia−/− mice. Scale bars, 30 μm. Arrowheads, γH2AX+PCNA+ cells. (B) Hippocampal cells were dissociated and performed neutral comet assay. Notably longer comet tails indicating enhanced DNA DSBs were persistently detected in Filia−/− mice at P1, P3, and P7 (n = 3 to 5 mice per group). (C) Filia depletion caused persistent DNA DSBs in hippocampal cells when examined at 2 months and 1 year old by neutral comet assay (n = 3 to 5 mice per group). Data were represented as means ± SEM; two-tailed Student’s t test, ***P < 0.001.
To further corroborate the in vivo results, we also examined the effect of Filia depletion on long-term propagated NSPCs derived from the hippocampus of newborn WT mice. After efficient knockdown of Filia by two independent doxycycline-inducible short hairpin RNAs (shRNAs) (fig. S1B) (15), we consistently observed an increased level of DSBs in cultured NSPCs as measured by neutral comet assay (fig. S1C). This defect was successfully rescued by reexpression of Filia (fig. S1, B and C). Together, these in vivo and in vitro data demonstrate that Filia is required to prevent the endogenous DNA DSBs in NSPCs and their progenies in the hippocampus region.
Loss of Filia impairs proliferation and neurogenesis of hippocampal NSPCs
DNA DSBs can induce cell cycle arrest, apoptosis, or stem cell differentiation, which consequently leads to the reduction of stem cell population (18). We examined whether Filia depletion impaired NSPC proliferation in the hippocampus. Immunostaining with Ki67, a marker of proliferating cells, revealed a notable reduction in the proliferative cells (population of NSPCs) in the dentate gyrus of Filia−/− mice compared to WT littermates at P7, P14, and P60 (fig. S2A). Consistent results were obtained by in vivo 5-bromo-2′-deoxyuridine (BrdU) incorporation assay (Fig. 3, A and B). In line with these observations, we were unable to successfully propagate hippocampal NSPCs in neurospheres from Filia−/− newborn mice (Fig. 3C). The cell proliferation defect was reproducibly detected in cultured NSPCs upon Filia knockdown and was fully rescued by reexpression of Filia (fig. S2B). Although Filia depletion evoked apoptosis in cultured NSPCs (fig. S2C), no obvious cell death was detected by terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) staining in the hippocampus of Filia−/− mice at P1, P7, and P14 (fig. S2D). This suggested that NSPCs with elevated DNA DSBs might lose the stem cell identity instead of undergoing apoptosis in vivo. Similar phenomenon was reported in hematopoietic stem cells (19).
Fig. 3. Filia deletion impairs hippocampal NSPC proliferation and neurogenesis.
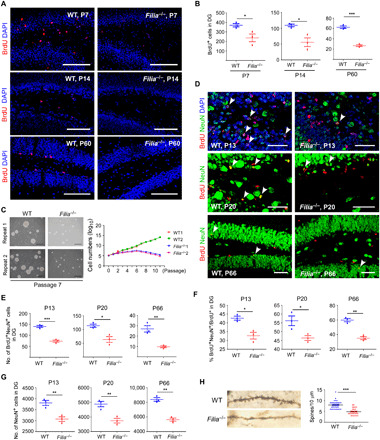
(A) Filia loss compromised hippocampal NSPC proliferation, as monitored by BrdU incorporation when examined at P7, P14, and P60, respectively. Scale bars, 100 μm. (B) Quantification of BrdU+ cells in the dentate gyrus at P7, P14, and P60. (C) NSPCs were derived from the hippocampus of newborn mice and cultured as neurospheres. Left: Photos of neurospheres at passage 7. Right: Growth kinetics of neurospheres from WT and Filia−/− mice. Scale bars, 200 μm. (D) Filia loss impaired the hippocampal neurogenesis in the dentate gyrus. BrdU+NeuN+ cells indicate newborn neurons. Scale bars, 50 μm. (E) Quantification of newborn neurons in the dentate gyrus at P13, P20, and P66. (F) The percentages of NSPCs undergoing neuronal differentiation. (G) Total numbers of neurons in the dentate gyrus. (H) Hippocampal neurons from Filia−/− mice contained much fewer spines than those from WT mice. Left: Morphology of a single neuron from WT and Filia−/− mouse. Right: Spine densities in neurons from WT and Filia−/− mice. Data were represented as means ± SEM; two-tailed Student’s t test, *P < 0.05, **P < 0.01, and ***P < 0.001.
To exclude the possibility that Filia may also directly regulate the proliferation and cell fate determination of NSPCs, we supplied doxycycline to induce Filia knockdown and DNA DSB accumulation in cultured NSPCs. Doxycycline was then withdrawn to allow for the reexpression of Filia. At the earliest time point (day 3 after doxycycline withdrawal) when Filia expression was fully restored (fig. S2E), whereas the DNA damages still persisted (fig. S2F), the proliferation rate and apoptosis of NSPCs were determined. Reexpression of Filia did not rescue the above defects (fig. S2, G and H). These observations suggested that the impaired proliferation of NSPCs in Filia−/− mice may simply be the consequence of DNA DSB accumulation.
We further investigated the overall influence of Filia depletion on hippocampal neurogenesis. The proliferating NSPCs were labeled in vivo with BrdU at P7, P14, and P60. Their differentiation into neurons was determined by coimmunostaining with BrdU and NeuN (neuronal nuclei) (marker of neurons) after 6 days of BrdU labeling (examined at P13, P20, and P66, respectively). Much fewer newborn neurons positive for both BrdU and NeuN (BrdU+NeuN+) were detected in the dentate gyrus of Filia−/− mice than in WT littermates (Fig. 3, D and E), indicative of the overall reduced neurogenesis at all examined ages. To better assess the differentiation ability of NSPCs, we calculated the proportions of NSPCs capable of differentiation into neurons. Notably, smaller percentages of BrdU+ NSPCs differentiated into neurons in the dentate gyrus of Filia−/− mice when compared with WT littermates (Fig. 3F). These observations implicated that the loss of Filia compromised the neuronal differentiation of NSPCs. Consistently, fewer neurons (NeuN+ cells) were detected in the dentate gyrus of Filia−/− mice (Fig. 3G). We further examined the morphology of neurons by Golgi staining in the hippocampal CA1 (cornu ammonis 1) region of adult mice at 2 months of age (20). The densities of dendritic spines were notably lower in Filia−/− mice than in WT littermates (Fig. 3H), suggesting a potential alteration in neuron functions.
To corroborate these in vivo observations, we performed in vitro differentiation of cultured NSPCs. Consistently, fewer immature neurons [Tuj1+ (class III beta-tubulin) cells] (fig. S3A) and mature neurons [Map2+ (microtubule-associated protein 2) cells] (fig. S3B) were obtained from cultured NSPCs when Filia was knocked down by doxycycline induction. Moreover, the neurons derived from Filia knockdown NSPCs displayed reduced dendritic length (fig. S3C) and numbers (fig. S3D) than those derived from WT cells. No obvious difference was detected between WT and Filia knockdown NSPCs on their differentiation into astrocytes [Gfap+ (glial fibrillary acidic protein) cells] (fig. S3E). Together, these lines of in vivo and in vitro evidence supported that Filia dysfunction impaired the neuronal differentiation and neurogenesis in the hippocampus of young and adult mice. However, the overall hippocampus size and structure were not notably altered by Filia depletion (fig. S3F). Similarly, Filia−/− mice had normal brain structure (fig. S3F) and weights (fig. S3G).
Loss of Filia has lifetime impact on hippocampus functions
On the basis of the impaired proliferation and neurogenesis of NSPCs, the abnormal neuron morphology, and the persistent DNA DSBs in neural cells in the hippocampus, we hypothesized that Filia−/− mice may have abnormal hippocampus functions. We then performed a series of behavior tests related to hippocampus functions in spatial memory, navigation, and emotions (21). Mice at ages of 2 months and 1 year old were examined. Morris water maze is widely used to assess hippocampus-dependent spatial learning and memory. At 2 months old, both the Filia−/− and WT littermates showed decreased mean escape latency to find the platform during the 9 days of training. However, Filia−/− mice took longer time (Fig. 4A) and traveled more distance (Fig. 4B) in finding the platform beginning at the 7th day. Because Filia−/− and WT mice swam in similar speeds (fig. S4A), these differences reflected the bona fide impairments in spatial learning and memory of Filia−/− mice. Moreover, in the following probe trials in which the escape platform was removed after the 9-day training and the mice were allowed to search for it in a fixed time period, Filia−/− mice again showed fewer platform crossings than WT littermates, no matter the test was performed at 4 or 72 hours after the last training (Fig. 4, C and D). We also repeated the Morris water maze test on 1-year-old mice and obtained the similar results (fig. S4, B to E). These data supported that Filia dysfunction impaired hippocampus-dependent spatial learning and memory.
Fig. 4. Filia loss impairs hippocampus functions in learning, memory, and emotional regulation.
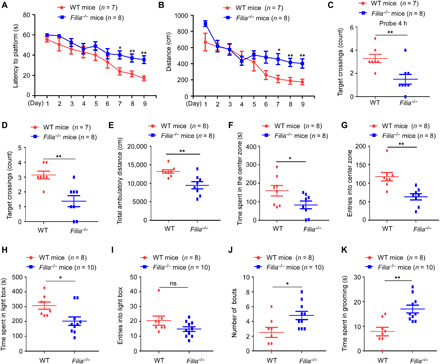
Mice at the age of 2 months old were used in the following tests. Morris water maze test showed that Filia−/− mice spent more time (A) and traveled longer distance (B) in locating the platform at the 7th to 9th days of training. At 4 hours (C) and 72 hours (D) after 9-day probe trials, Filia−/− mice showed fewer platform crossings than WT littermates. In open-field test, Filia−/− mice showed weaker locomotivity (E), spent much less time in the center zone (F), and exhibited fewer entries into the center zone (G). In light-dark box test, Filia−/− mice spent less time in the light box (H) and showed slight difference in the entries into the light box (I). In grooming behavior analysis, Filia−/− mice exhibited more bouts (J) and spent longer time (K) on self-grooming than WT littermates. Data were represented as means ± SEM; two-way ANOVA with Bonferroni post hoc test (A and B) and two-tailed Student’s t test (C to K), *P < 0.05 and **P < 0.01. ns, not significant.
We next investigated whether Filia−/− mice had mood-and-anxiety–related disorders. First, in open-field test designed to probe the rodents’ exploratory behavior and response to novelty, Filia−/− mice displayed reduced locomotion (Fig. 4E and fig. S4F), spent notably less time in the center (Fig. 4F and fig. S4G), and crossed into the center for fewer times (Fig. 4G and fig. S4H) than WT littermates at ages of 2 months and 1 year old. Second, in the light-dark box test used to predict anxiogenic-like activity in mice, Filia−/− mice spent substantially less time in the light box (Fig. 4H), although their entries into the light box did not differ statistically from WT mice (Fig. 4I). Third, by analyzing grooming behavior, we also found that Filia−/− mice exhibited notably more bouts (Fig. 4J) and spent longer time on self-grooming than WT littermates (Fig. 4K). Together, these data support that Filia dysfunction causes lifetime psychiatric disorders and anxiety-like behaviors.
Filia protects the stalled replication forks from collapse and promotes HR repair of DNA DSBs
Previous studies showed that many genes in NSPCs harbor clusters of replication fragile sites, where replication forks stall and are prone to collapse and form RDCs under mild replication stress or even at unperturbed conditions (22, 23). Protection of stalled replication forks at these fragile sites can alleviate the level of endogenous DSBs. Filia localizes on replication forks and protects the stalled fork from collapse in mouse ESCs (15). We asked whether it played similar roles in NSPCs. To this end, we isolated proteins on replication forks of cultured NSPCs by iPOND (isolate proteins on nascent DNA). Immunoblotting analysis of iPOND samples revealed that Filia localized on replication forks in NSPCs under normal and HU-induced replication stress conditions. Moreover, fork stalling induced the robust accumulation of Filia on stalled forks (Fig. 5A). We then assessed the function of Filia on replication forks of NSPCs by DNA fiber assay. Cultured NSPCs were treated with HU to induce replication fork arrest, and the fork restart was evaluated at different time points after HU removal. We found that the fork restart became very inefficient when Filia was depleted (Fig. 5B), indicating that Filia can prevent fork collapse and promote fork restart in NSPCs.
Fig. 5. Filia-ATR axis coordinates stalled fork restart and HR-mediated DNA repair in mouse cultured NSPCs.
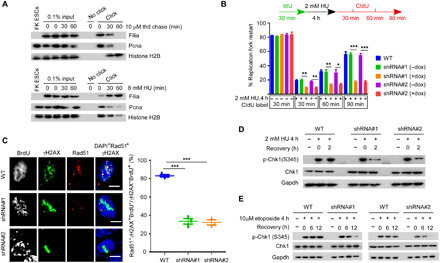
(A) iPOND revealed that Filia is localized at replication forks in cultured mouse NSPCs under unperturbed condition (top) and the amount is robustly stimulated by HU-induced replication stress (bottom). H2B, histone 2B; thd, thymidine. (B) DNA fiber assay showed that doxycycline (dox)–induced Filia knockdown in cultured NSPCs notably attenuated the rates of stalling fork restart (n = 200 fibers from three independent replicates). IdU, 5-iodo-2′-deoxyuridine; CIdU, 5-chloro-2′-deoxyuridine. (C) Cultured NSPCs underwent laser microirradiation. Filia depletion compromised the recruitment of Rad51 to DSB sites labeled with γH2AX at S phase (BrdU+), indicating the impaired HR pathway. Right: Proportions of S phase cells capable of HR repair (three replicates, 50 cells in one replicate). ATR kinase activation, monitored by the phosphorylation of Chk1 at S345, could not be well sustained upon Filia depletion in NSPCs after replication stress (D) or DNA DSBs (E). Scale bars, 5 μm. All data were represented as means ± SEM; two-tailed Student’s t test, *P < 0.05, **P < 0.01, and ***P < 0.001.
DNA DSBs are generally repaired by HR or NHEJ pathway. We also investigated whether Filia participated in HR repair, which is a main repair pathway in proliferating NSPCs (24) and can be monitored by the recruitment of recombinase Rad51 to DNA DSB sites. NSPCs were subject to laser microirradiation, and the recruitment of Rad51 to DSBs was examined after 2 hours of recovery. Rad51 recruitment to DSB sites was considerably suppressed upon Filia knockdown (Fig. 5C), implying an essential role of Filia in HR repair. Consequently, Filia knockdown decreased overall DSB repair efficiency as assessed by neutral comet assay (fig. S5A). Together, these observations suggested that in proliferating hippocampal NSPCs, Filia may protect stalled forks from collapse and promote the HR repair of DSBs, thereby reducing the levels of endogenous DSBs and the genomic alternations.
To further understand how Filia coordinates its dual functions in promoting stalled fork restart and HR-mediated DSB repair, we examined the activity of ATR kinase, which is required not only for replication stress response (25) but also for Rad51 assembling into repair foci to facilitate HR repair (26). The involvement of ATR in HR repair was confirmed in cultured NSPCs by blocking ATR activation with specific inhibitor VE-821 (fig. S5, B and C) (27). We found that the persistence of ATR activity, monitored by the phosphorylation of its downstream target Chk1 (checkpoint kinase 1) at serine-345 (pS345) (25), was compromised in Filia knockdown NSPCs in response to HU-induced replication stress (Fig. 5D) or etoposide-induced DNA DSBs (Fig. 5E)(16). In contrast, ATM-Chk2 signaling, which mediates the early response to DNA DSBs and is necessary to initiate both HR and NHEJ repair pathways (28), was intact in Filia-deficient NSPCs (fig. S5D). Thus, Filia-ATR axis coordinates two crucial events, protecting stalled forks from collapse and promoting HR-mediated DSB repair, to suppress the DSB formation and the genomic alternation of proliferating NSPCs in the hippocampus.
Filia loss leads to increased DNA DSBs in many long genes including RDC genes
RDC genes harbor clusters of replication fragile sites that are prone to generate DNA DSBs under mild replication stress or even at unperturbed conditions. On the basis of Filia’s functions in preventing replication stress–associated DNA DSBs, we reasoned that Filia depletion could cause increased DNA DSBs in RDC genes. To test this hypothesis, we performed high-throughput genome-wide translocation sequencing (HTGTS) to map, at the nucleotide-resolution, genome-wide DSBs in cultured WT and Filia−/− NSPCs treated with vesicle dimethyl sulfoxide (DMSO) or aphidicolin (22, 23, 29). Two baits on chromosome 12 (Chr12) and Chr15, respectively, were designed as reported (Fig. 6A). In total, we obtained 107 and 112 DSB hotspots using bait on Chr12 and Chr15, respectively. Among these hotspots, 19 were found in both groups (Fig. 6B and dataset S1). The sizes of DSB hotspots ranged from 30 kb to 3.3 Mb, with a median length of 300 kb (Fig. 6B). The DSB densities were then calculated for the interchromosomal hotspots (hotspots located on chromosomes other than the bait chromosome) and intrachromosomal hotspots (hotspots located on the same chromosome as the bait DSB) for each cell group. Under replication stress induced by aphidicolin treatment, WT NSPCs displayed greater DSB density in both inter- and intrachromosomal hotspots (Fig. 6C). This was consistent with previous report (6) and validated the reliability of our HTGTS results. Under unperturbed culture condition (vesicle DMSO treatment), Filia−/− NSPCs showed notably higher DSB density in either inter- or intrachromosomal hotspots when compared to WT NSPCs (Fig. 6C), indicating that Filia depletion increased the risk of DSB formation in mouse NSPCs.
Fig. 6. Filia loss increases DNA DSBs in RDC genes and alters gene splicing in hippocampal NSPCs.
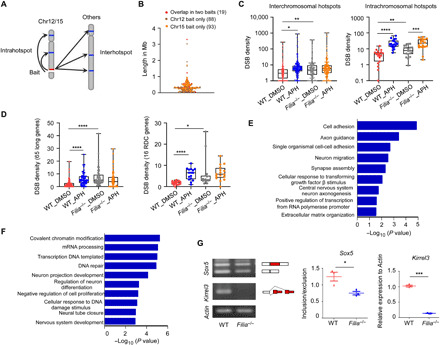
(A) Illustration of HTGTS in mouse NSPCs. Bait was located on Chr12/15. (B) Scatterplot of the sizes of DSB hotspots in megabases. Line represents median length (300 kb). (C) The DSB densities captured by both baits within the inter- and intrachromosomal DSB hotspots. Compared to unperturbed condition (DMSO treatment), aphidicolin (APH) treatment induced greater DSB density in WT NSPCs. Loss of Filia increased the DSB densities under unperturbed condition. (D) The DSB densities of 65 long genes (left) including 16 RDC genes (right) captured by both baits. Note that Filia depletion increased the DSB densities in these genes under unperturbed condition. (E) GO enrichment analyses of 65 long genes whose DSB densities were higher in Filia−/− NSPCs. (F) GO enrichment analyses of genes underlying alternative splicing (AS) in Filia−/− hippocampal NSPCs at P5. (G) Validation of AS of RDC genes Sox5 and Kirrel3 by semiquantitative RT-PCR. Middle and right: Quantifications of isoforms from three replications. Data were represented as means ± SEM; two-tailed Student’s t test, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
In the DSB hotspots, we identified 125 long genes with a size larger than 80 kb (dataset S2). Among them, 65 genes including 16 RDC genes were affected by Filia and displayed increased DSB density in Filia−/− NSPCs under normal culture condition (Fig. 6D and dataset S2). Gene ontology (GO) enrichment analysis revealed that these genes were involved in the regulations of neurogenesis such as cell adhesion, axon guidance, neuron migration, and synapse assembly (Fig. 6E). Together, these data provide compelling evidence, supporting that Filia deletion results in an increase in DSBs in many long genes including RDC genes.
Filia loss leads to robust gene splicing aberration
To further investigate the impacts of Filia depletion on the gene expression and alternative splicing (AS) in hippocampal NSPCs, we used Nestin–green fluorescent protein (GFP) transgenic mice, in which Nestin-expressing NSPCs are labeled with GFP, to isolate NSPCs. GFP-expressing NSPCs were manually collected from single-cell suspensions freshly prepared from the hippocampus of Nestin-GFP/Filia+/+ mice and of Nestin-GFP/Filia−/− mice. RNA sequencing analyses revealed that Filia depletion in hippocampal NSPCs did not cause robust change in overall gene expressions. Only 169 differentially expressed genes (DEGs) were identified (fold change, ≥2; P < 0.05), among which 80 genes were up-regulated and 89 down-regulated in Filia−/− hippocampal NSPCs compared to WT counterparts (fig. S6A and dataset S3). GO enrichment analysis showed that the up-regulated DEGs in Filia−/− NSPCs were related to negative regulation of neuron differentiation and neural precursor cell proliferation (fig. S6B). Examples include Hes1 (30), Hes5 (31), Hey1 (31), and Id2 (32) (fig. S6C). Down-regulated DEGs in Filia−/− NSPCs were enriched in terms of cell-cell adhesion and cell migration (fig. S6B). This was consistent with a recent study in which many genes regulating cell adhesion and migration were decreased in human NSPCs subject to replication stress–induced DNA DSBs (6). Down-regulated genes also play important roles in neurogenesis and brain functions. For instance, Npas4 (33), Cx3cr1 (34), and Ccl3 (35) regulate dendritic spine development. Fscn1 (36) and Osm (37) regulate precursor cell proliferation, neuronal differentiation, and migration (fig. S6C).
We also performed AS analyses. A total of 2258 genes underwent AS in Filia−/− NSPCs compared to WT counterparts (3142 AS events) (adjusted P < 0.05) (dataset S4). GO enrichment analysis showed that these genes were involved in regulating DNA repair, DNA damage response, and neurogenesis (Fig. 6F and dataset S4). Those that play roles in DNA damage response and repair were listed (57 genes; dataset S4), and the AS events of several genes were experimentally validated (fig. S7, A and B). Among these 57 genes, AS events of 13 genes in Filia−/− NSPCs were predicted to cause obvious protein fragment deletions (fig. S7C). For instance, the AS of Trp53bp1 in Filia−/− NSPCs introduced a premature stop codon and led to the loss of normal protein translation and functions (fig. S7D). Trp53bp1 is a key player in DNA damage response and DSB repair (38). Its aberrant splicing in Filia−/− NSPCs may partially contribute to the deficient DSB repairs. We next examined whether the splicing patterns of RDC genes were altered by Filia depletion. Five of 16 RDC genes underwent AS in Filia−/− NSPCs compared to WT cells. The changes in splicing pattern of the Kirrel3 and Sox5 genes were experimentally validated (Fig. 6G). The positions of splicing in Kirrel3 and Sox5 overlapped with the positions of DSBs (fig. S7, E and F), suggesting that the replication stress-induced DNA DSBs at RDC genes might be able to affect splicing. To test this hypothesis, we treated the WT NSPCs with aphidicolin and examined the splicing patterns of Kirrel3 and Sox5. Consistently, aphidicolin treatment and Filia loss led to similar splicing pattern of the two genes (fig. S7G).
Filia ortholog is expressed in rhesus monkey NSPCs and plays conserved roles in guarding genomic integrity
Mouse Filia has ortholog in primates (official symbol KHDC3L), which shares low similarity in amino acid sequence to mouse Filia. To understand whether the primate KHDC3L plays conservative roles in NSPCs and could be relevant to human neurological disorders, we investigated the expression and function of KHDC3L in cultured rhesus monkey NSPCs derived from fetal brain. Similarly, KHDC3L protein was detected in monkey NSPCs, and the expression was induced by etoposide or HU treatment (fig. S8A). We then explored the functions of KHDC3L in monkey NSPCs. Efficient knockdown of KHDC3L via two independent doxycycline-inducible shRNAs (fig. S8B) caused the accumulation of DNA DSBs (fig. S8C), a decrease in proliferation rate (fig. S8D), and an increase in apoptosis (fig. S8E) in monkey NSPCs under normal culture conditions. Notably, KHDC3L played essential roles in stalling fork protection and restart in monkey NSPCs (fig. S8F). Thus, KHDC3L is a critical guardian of genomic stability of primate NSPCs, and its dysfunction could potentially be a relevant mechanism underlying some neurological disorders in humans.
DISCUSSION
DNA DSB formation and repair have been proposed to play important roles in brain development, physiology, and diseases (5, 6, 39). The formation and repair of DSBs are under tight control to ensure the normal neurogenesis and brain functions. Aberrations in the formation and/or repair of DSBs can lead to the neurodevelopmental or neurodegenerative disorders. Unlike in lymphocyte development in which endogenous DSBs are generated by RAG endonuclease (4), no RAG or other endonuclease activity was found to mediate endogenous DSB formation in NSPCs. Instead, many neurogenesis-related genes harbor clusters of replication fragile sites, which form recurrent DNA DSB clusters under mild replication stress or even unstressed conditions. Increased replication stress and DSB formation in NSPCs have been experimentally validated to be associated with macrocephalic autism spectrum disorder (6). Identifying the regulators controlling the replication-associated DSB formation and repair in NSPCs is, therefore, critical to understand the etiology of brain disorders. Within the different brain regions, subtypes of NSPCs show substantial heterogeneities in lineage fates, neurogenesis activity, and gene expression profiles (5, 39). For instance, hippocampal NSPCs have constrained lineage fates but lifetime neurogenesis activity when compared to the fetal cortical NSPCs (7, 8). These differences imply that different subtypes of NSPCs may have distinctive regulations on DSB formation and repair to accommodate region-specific neurogenesis activity. Here, we identified Filia as the first hippocampal NSPC-specific regulator of DSB formation and repair, thereby providing the first proof of concept. Moreover, our study highlights the importance to understand the region-specific regulations of DSB formation and repair in subtypes of NSPCs. In the future, it would be intriguing to systematically investigate the extent and genomic location of endogenous DSBs, the specific regulators of replication-associated DSB formation and repair in fetal cortical and hippocampal NSPCs, respectively.
In hippocampal NSPCs, Filia regulates the extent of DNA DSBs via two approaches. On one hand, Filia localizes at replication forks and protects the stalled forks from collapse, thereby reducing the replication stress–induced DNA DSB formation. On the other hand, Filia facilitates the HR-mediated DNA DSB repair. HR pathway has been shown to play a major role in proliferating NSPCs, whereas cNHEJ is functionally important in postmitotic cells during neural system development. Thus, Filia regulates the overall DSB repair by promoting the HR pathway. Filia depletion induced the robust DNA DSB formation in hippocampal NSPCs. We further mapped the genomic locations of these DSB sites susceptible to Filia loss by HTGTS. Filia depletion increased DSB densities in overall DSB hotspots. Filia depletion affected many long genes that are involved in neurogenesis. For instance, Ctnna2 (40, 41), Dcc (42), Ptk2 (43), and Kirrel3 (44) have been shown to regulate neurogenesis, and their deficiencies cause behavioral abnormalities. DSB formation could potentially alter the protein expression by various means including AS. This provided a causative mechanism for the deficient neurogenesis in Filia−/− mice. It should be noted that in our study, we detected 16 RDC genes showing increased DSB densities in Filia−/− NSPC. The influence of Filia on RDC genes should be underestimated as the HTGTS methodology reports that the most notable recurrent DSB hotspots and the identification of DSB hotspots from fixed baits are restricted by the cellular heterogeneity in genome organization. In addition, Filia depletion caused marked changes in AS of more than 2200 genes. In particular, AS of 57 DNA repair genes was affected. The aberrant splicing of 13 genes in Filia−/− NSPCs was predicted to cause obvious fragment deletions and might be partially responsible for the compromised DSB repair. Filia contains atypical KH domain and may function as a RNA binding protein. Whether Filia can directly regulate RNA splicing requires further investigation.
Similar to hippocampal NSPCs, fetal cortical NSPCs proliferate fast and encounter high risk of replication-associated DNA DSBs. It is intriguing to ask why they do not express Filia. Previous studies proposed that in NSPCs, most RDC genes occupy a single topological domain. This enables the DSBs in an individual RDC gene to undergo end joining via cNHEJ pathway, thereby generating robust gene diversifications (22). On the basis of Filia’s dual roles in preventing replication stress-associated DNA DSB formation and promoting DSB repair by HR, we speculated that forced expression of Filia in fetal cortical NSPCs might reduce the extent of RDC-gene fragility and decrease the density of DSBs in RDCs. In addition, it may also promote repair of DSBs within an individual RDC gene through HR pathway. As a result, ectopic expression of Filia in fetal cortical NSPCs could compromise the diversification of genomic information and neuronal development, complexity, and functions and generate damaging outcome to fetal neurogenesis. In the future, transgenic mice expressing Filia in cortical NSPCs can be generated to test this hypothesis.
Depletion of Filia increased DSB level, impaired hippocampal neurogenesis, and compromised the hippocampus functions in learning, memory, spatial navigation, and mood regulations. The hippocampus is one of the most affected brain regions in Alzheimer’s disease (AD). Recent works in human brains showed that the hippocampal neurogenesis progressively declined as AD advanced, and the impaired neurogenesis was considered as a potentially relevant mechanism underlying memory deficits in AD (9, 11). Genomic instability in hippocampal NSPCs impairs hippocampal neurogenesis and functions. Thus, factors that can cause aberrant DNA DSB accumulation in hippocampal NSPCs are anticipated to speed up the progression of AD. The primate ortholog of Filia (KHDC3L) is expressed in long-term cultured rhesus monkey NSPCs derived from the fetal brain at E91. Our in vitro experiments demonstrated that primate KHDC3L played essential roles in protecting stalled forks and preventing the generation of replication-associated DSBs. Thus, KHDC3L dysfunctions may cause DNA DSBs in primate NSPCs in vivo, leading to the neurodevelopment deficits or hippocampus-associated brain disorders. The variable damaging mutations of KHDC3L have been identified in some female patients with reproductive problems and in human genome Exome Aggregation Consortium database (45–49). However, whether these people had disorders in brain functions, for example, in learning and memory, or psychiatry disorders, was not reported. In the future, more studies are needed to investigate the spatiotemporal expression patterns of KHDC3L in primate NSPCs and the relevance of KHDC3L dysfunctions with neurological disorders.
MATERIALS AND METHODS
Mice
Filia−/− mice were from J. Dean in National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, USA and were maintained on C57BL/6 genetic background. Filia−/− mice were bred with WT C57BL/6 mice to generate heterozygous mutants (Filia+/−), which were then mated to generate Filia−/−, Filia+/−, and Filia+/+ littermates. Filia−/− mice and Filia+/+ littermates (WT controls) were used for experiments. Nestin-GFP transgenic mice (C57BL/6 genetic background) were from Y. Chen in Kunming Institute of Zoology, Chinese Academy of Sciences. Filia−/− and Nestin-GFP mice were crossbred to generate GFP/Filia double gene–edited mice. Genotypes for all animals were confirmed using PCR from genomic tail DNA samples, as described previously (13). Filia and GFP PCR primers are listed in table S1. Animal care and experimental procedures were conducted in compliance with the guidelines of the Animal Care and Use Committee of the Kunming Institute of Zoology, Chinese Academy of Sciences.
Brain section preparation and immunostaining
Mice were transcardially perfused with phosphate-buffered saline (PBS) and 4% paraformaldehyde (PFA); the brains were then fixed in 4% PBS-PFA overnight at 4°C, equilibrated in 15 and 30% sucrose in PBS, and embedded in OCT (optimal cutting temperature compound) or paraffin for sectioning. Hematoxylin-eosin staining was performed in standard procedures (Boster Bio, AR1180).
For TUNEL labeling, brain sections were washed and extracted in 0.3% Triton X-100, and labeling procedures were followed per the manufacturer’s instructions (Roche, 11684795910). To label the in vivo proliferating NSPCs, BrdU was injected at body weight (100 μg/g) 2 hours before mice were euthanized (50). Brain sections were denatured with 2 mol/L HCl in H2O at 37°C for 1 hours, neutralized with 0.1 M borate buffer for 10 min, blocked in 1% bovine serum albumin with 0.3% Triton X-100 for 1 hour at room temperature, and probed with a monoclonal antibody against BrdU overnight at 4°C. To examine the in vivo differentiation of NSPCs, mice at P7, P14, and P60 were injected with BrdU (body weight, 100 μg/g) for three consecutive days. After 4 days of last injection, mice were perfused with 4% PFA, and brain sections were prepared for immunostaining (20 μm per section). Six brain sections representative of the whole hippocampus were used for all stainings (pick 1 section in every 8 sections at P7, pick 1 section in every 10 sections at P14, and pick 1 section in every 12 sections at P60). Antibodies information was listed in table S2.
Golgi staining
Golgi staining was performed as described (20). Briefly, the brains of mice at 2 months old were dissected and washed in Milli-Q water to remove blood from the surface. The brains were immersed in a 1:1 mixture of FD solution A:B (FD Neurotechnologies Inc., FD Rapid Golgistain Kit, PK401C) at room temperature for 14 days, followed by incubation in FD solution C at room temperature and in the dark for 7 days. The brains were then sectioned on a cryostat and mounted on gelatin-coated slides with FD solution C, dried naturally at room temperature. Staining procedures were followed per the manufacturer’s instructions. Spine density was measured on pyramidal neurons that were located in the CA1 region of the dorsal hippocampus.
In situ hybridization
In situ hybridization was performed on brain sections as described (51), with digoxigenin-labeled single-stranded RNA robes for 16 hours at 55°C. After washing twice in 2× saline sodium citrate (SSC) at 55°C, samples were treated with ribonuclease A (10 μg/ml) at 37°C, washed twice again in 0.2× SSC for 30 min, and incubated with BM Purple alkaline phosphatase substrates (Roche, 11442074001). Primers used to synthesize the antisense RNA probes were AGGCGAGCTGAGATTTGGATAT (forward primer) and CTCAGGACACTTCTGGGACAAG (reverse primer).
Cell culture
Mouse NSPCs were derived from WT and Filia−/− mice at P5 as described (52). Cells were plated at 100,000 cells/ml in 35-mm plate in complete medium (STEMCELL Technologies, 05702) supplemented with rhEGF (recombinant human epidermal growth factor) (20 ng/ml; STEMCELL Technologies, 02633), basic fibroblast growth factor (bFGF) (10 ng/ml; STEMCELL Technologies, 02634), and heparin (2 μg/ml; STEMCELL Technologies, 07980) and passaged every 5 days. After cells were expanded, neurospheres were dissociated and passaged (recorded as passage 0).
Monkey NSPCs were provided by B. Su in Kunming Institute of Zoology, Chinese Academy of Sciences and maintained in Neurobasal medium (Gibco, 21103-049) supplemented with 1× B-27 (Gibco, 17504044), 1× N-2 (Gibco, 17502048), 1% NEAA (non-essential amino acids) (Gibco, 11140050), 1% GlutaMAX (Gibco, 21103049), 3 μM CHIR99021 (Selleckchem, S2924), 5 μM SB431542 (Cellagen Technology, C72435), bFGF (10 ng/ml; Gibco, PHG0261), and hrLIF (human recombinant leukemia inhibitory factor) (1000 U/ml; Millipore, LIF1050).
shRNA knockdown
shRNA knockdown was conducted using pTRIPZ lentiviral tet-on inducible shRNAmir system as described (15). Filia shRNA and KHDC3L shRNA sequences were listed in table S1. shRNAmir expression vectors (Open Biosystems) were cotransfected with packaging plasmids (psPAX2 and pMD2.G) in 293 T cells to package viruses. After 72 hours of viral infection, WT mouse NSPCs and monkey NSPCs were treated with puromycin (1 μg/ml) to select infected cells. To verify the knockdown efficiency, cells were treated with doxycycline (2 μg/ml) for 72 hours before harvesting for immunoblotting.
In vitro NSPC proliferation and differentiation assays
To examine the proliferation, cultured NSPCs were treated with 10 μM BrdU for 1 hour to label dividing cells (53). In differentiation assay, NSPCs were cultured for 5 days in basal medium (STEMCELL Technologies, 05700) supplemented with differentiation medium (STEMCELL Technologies, 05703) for differentiation. Immunocytochemistry staining was carried out as previously described (14). Antibodies information was shown in table S2. The numbers of BrdU+, Tuj1+, Gfap+, and Map2+ cells were quantified by fluorescence-activated cell sorting. Image-Pro Plus 6.0 was used to quantify dendritic length (21 neurons for all groups) and the numbers of dendrites (50 neurons for all groups). All experiments were repeated in triplicates.
Neutral comet assay
The neutral comet assay was performed as described (54). Comets were analyzed by Komet 7 comet assay software (Andor Technology). At least 100 cells were counted per group. All experiments were performed in three replicates.
Isolate proteins on nascent DNA
iPOND was performed as described (15, 55). NSPCs were arrested in S phase by treating with 2 mM thymidine (Sigma-Aldrich, T1895) for 18 hours, followed by release into thymidine-free medium for 2.5 hours. Cells were then incubated with 10 mM 5-ethynyl-2′-deoxyuridine (EdU) (Life Technologies, A10044) for 10 min. Following EdU labeling, cells were treated with or without HU or treated with 10 μM thymidine for a chase. Cells were then fixed, and the rest of procedures were performed as described.
DNA fiber assay
The DNA fiber assay was performed as described (15, 56). Cells were labeled with 50 μM 5-iodo-2′-deoxyuridine (IdU; Sigma-Aldrich, I7125) for 30 min before washing. Cells were then treated with or without HU (Sigma-Aldrich, H8627) for 4 hours. Following wash, cells were labeled with 50 μM 5-chloro-2′-deoxyuridine (CIdU; Sigma-Aldrich, C6891) for 30, 60, and 90 min, respectively. Cells were collected and suspended at a concentration of 103/μl, and 2.5 μl of cell suspension was used to prepare for the DNA fibers. Immunofluorescent staining was performed using IdU and CIdU antibodies. DNA fibers were analyzed using Olympus FV1000 confocal microscope. The lengths (1 μm = 2.59 kb) of DNA fibers were measured with the ImageJ software. At least 200 DNA fibers were examined for each sample, and each experiment was independently repeated three times.
Quantitative polymerase chain reaction
Total RNA was extracted from brain using the TRIzol reagent (TIANGEN). One microgram of total RNA was reverse-transcribed using PrimeScript RT reagent kit (Takara Bio, RR047A). qPCR was performed using the SYBR Green Premix Ex Taq II (Takara Bio, RR820A). Actin gene was used as the internal control. Primers were listed in table S1.
Immunoblotting
Protein extracts were prepared using radioimmunoprecipitation assay lysis buffer (Beyotime, P0013B). Proteins were separated through a 4 to 12% SDS–polyacrylamide gel electrophoresis and transferred onto nitrocellulose membrane (Roche, 03010040001). After blocked by blocking reagent (Roche, 11096176001) for 1 hour, the membrane was incubated with primary antibodies overnight at 4°C, followed by secondary antibody incubation. Images were captured using a ProteinSimple FluorChem system. Filia antibody was from the previous study (14). Other commercial antibody information was listed in table S2.
Laser microirradiation
Cells were cultured on coverslips and were microirradiated with a 405-nm pulse laser for 10 s using Olympus FV1000 confocal microscope. Cells were then cultured for another 120 min, followed by fixation and immunostaining.
Generation of bait DNA DSB in mouse NSPCs for HTGTS
Bait DSB induction was achieved with a Cas9:sgRNA (single guide RNA) approach (57). sgRNA sequences were listed in table S1, and Cas9:sgRNA expression vectors were constructed as described (58). Briefly, mouse NSPCs were derived from WT and Filia−/− mice at P5. Cells were expanded in neurospheres for several passages. To induce bait DSB, 1 × 105 NSPCs were transfected with 1 μg of Cas9:sgRNA vectors using the Neon Transfection System Kit (Invitrogen, MPK1096). To induce replication stress, NSPCs were treated with 0.5 μM aphidicolin (Sigma-Aldrich, 89458) for 72 hours, followed by additional 24 hours of 0.25 μM aphidicolin treatment. Control NSPCs were treated with vesicle DMSO (1:59 dilution) for 72 hours, followed by additional 24 hours in 1:118 diluted DMSO.
HTGTS and data analyses
HTGTS was performed as described (29). Briefly, 20 μg of genomic DNA was sonicated and subjected to linear amplification–mediated PCR for bait-prey junction amplification. After streptavidin magnetic beads enrichment and adapter ligation, the single-strand DNA fragments are amplified with barcode primers (table S1) for sequencing. Sequencing was performed on the Illumina X Ten platform with 150–base pair (bp) paired-end reads.
Reads were mapped to mm9 genome by Bowtie2 through the HTGTS pipeline (https://github.com/robinmeyers/transloc_pipeline). The bait DSB resections events (junctions fail into ±1 Mb from the bait DSB) and Cas9-sgRNA off-targets events (junctions fail into ±50 bp of the cryptic sgRNA targets sites) were removed. Reads from the same bait are merged and called peaks with SICER using the following parameters: species-mm9; redundancy_threshold-5; window_size-30000; fragment_size-1; effective_genome_fraction-0.74; gap_size-90000; false_discovery_rate-0.1. Peaks with SICER scores of >15 for Chr12 bait and >20 for Chr15 bait were classed as DSB hotspots.
RNA sequencing and data analyses
The hippocampus of Nestin-GFP mice with (n = 4) or without Filia (n = 4) at P5 was dissociated into single cells by collagenase (1 μg/ml). About 200 GFP+ NSPCs were manually collected and lysed in lysis buffer. The full-length polyadenylate-tailed RNA was reverse-transcribed and amplified with 21 PCR cycles to increase the cDNA amount. Sequencing was performed using the Illumina X Ten platform with 150-bp paired-end reads. Two replicates from independent biological samples were analyzed.
Clean reads were mapped to mouse genome (mm10) with TopHat software (version 2.0.8). Gene expression levels were quantified by fragment per kilobase of transcript per million fragments mapped with Cufflinks software (version 2.1.1). DEGs were identified with Cuffdiff software (version 2.1.1). The heatmap was drawn with function heatmap2 of the “gplots” in R package. GO analysis was performed with DAVID using default parameters.
AS was analyzed by a Java program named ASD (AS detector) with default settings (59). Briefly, ASD requires a bam file generated from reads mapping for the WT and Filia−/− samples as input. The mm10 annotation file was used as a reference. Visualization of spliced exons between WT and Filia−/− samples was made by the integrative genomics viewer tool. To validate the AS events, the primers were designed to detect isoforms (table S1). β-Actin was used for normalization.
Single-cell cDNA amplification and RT-PCR
The hippocampus of WT mice at P5 (n = 5) were dissociated into single cells by collagenase (1 μg/ml). The VZ and SVZ of WT mice at E18.5 and P5 (n = 4) were also dissociated into single cells. Single cells were then randomly picked, and cDNAs were amplified by Smart-seq2 (60). Gene expressions were examined by semi-qPCR. Cycling parameters were 95°C for 5 min, followed by 35 or 40 cycles of 95°C for 30 s, 60°C for 30 s, and 72°C for 30 s, final extension for 7 min at 72°C. The PCR primers were listed in table S1.
Behavior tests
All behavioral tests were performed using male mice from littermates. All experiments and data analyses were performed in a blinded manner.
Morris water maze
A circular water tank (diameter, 120 cm) was filled with water, and the water was made opaque with nontoxic white beads. A round platform was hidden 1 cm beneath the surface of the water at the center of a given quadrant of the water tank. Each mouse was subjected to four trials for successive days. The mice that found platform within 60 s were placed on the platform for 15 s after every trial. Training was discontinued when one of the groups succeeded in locating the platform within 10 s. At 24 and 72 hours after the last training, a probe test was carried out by exposing the mice to the pool for 60 s without the platform. The mice were video-tracked using the SMART 3.0 software (Panlab Harvard, MA, USA).
Open-field test
The open field was made of a square black box (40 cm by 40 cm by 40 cm) with a center area in size of 10 cm by 10 cm. Each mouse was placed in the box for 60 min. Overall activity in the box, the times spent in the center area, and entries to center area were measured and tracked by the SMART 3.0 software (Panlab Harvard, MA, USA).
Light-dark box test
An apparatus (45 cm by 27 cm by 27 cm) consisting of a black chamber (18 cm by 27 cm) and a light chamber (27 cm by 27 cm) was used for the light-dark exploration test. Mice were placed into the dark box and allowed to move between the light box and dark box for 30 min. The total number of entries and the time spent in light box were analyzed.
Self-grooming test
Mice were placed in a new plexiglass cage with fresh bedding without nesting or cardboard material. Self-grooming behavior was recorded for 10 min. Cumulative time spent on grooming and the numbers of grooming bouts were scored for each mouse.
Statistical analysis
In the comparisons between Filia−/− and WT mice, statistical analysis was performed by two-tailed Student’s t test, one-way analysis of variance (ANOVA) with Bonferroni post hoc test, or two-way ANOVA with Bonferroni post hoc test. In all scattergrams, values were shown as means ± SEM. All statistical analyses were performed using Prism (version 5.0/8.0) software. We assayed at least three mice for each genotype and stage.
Supplementary Material
Acknowledgments
We thank R.-c. Luo, L.-y. Su, X. Zhou, P.-c. Ma, L. Xu, and Y.-p. Tang for technical helps; B. Su for providing monkey NSPCs; and N.-y. Sheng and Y.-g. Yao for manuscript reading and comments. Funding: This work was supported by the National Key Research and Development Program of China, Stem Cell and Translational Research (2016YFA0100300), the Chinese NSFC grant to P.Z. (31930027), the exchange program of State Key Laboratory of Genetic Resources and Evolution, Kunming Institute of Zoology, Chinese Academy of Sciences (GREKF20-15 to F.M.), and the Basic Research Project of Yunnan Province to L.W. (2018FB056). Author contributions: P.Z., Jingzheng Li, and B.Z. conceived the project. Jiali Li performed most of the experiments. Y.S. performed HTGTS and data analyses. L.W. performed the RNA sequencing analysis. C.S. performed the single-cell cDNA amplification. C.L. and M.T. participated in mice behavioral experiments. S.L. and Jiali Li helped in in situ hybridization experiment. F.-L.M. supervised HTGTS works. P.Z. supervised the study and wrote the manuscript. Competing interests: The authors declare that they have no competing interest. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/44/eaba0682/DC1
REFERENCES AND NOTES
- 1.Lee Y., Shull E. R., Frappart P.-O., Katyal S., Enriquez-Rios V., Zhao J., Russell H. R., Brown E. J., McKinnon P. J., ATR maintains select progenitors during nervous system development. EMBO J. 31, 1177–1189 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee Y., Katyal S., Downing S. M., Zhao J., Russell H. R., McKinnon P. J., Neurogenesis requires TopBP1 to prevent catastrophic replicative DNA damage in early progenitors. Nat. Neurosci. 15, 819–826 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meng L., Lin T., Peng G., Hsu J. K., Lee S., Lin S.-Y., Tsai R. Y., Nucleostemin deletion reveals an essential mechanism that maintains the genomic stability of stem and progenitor cells. Proc. Natl. Acad. Sci. U.S.A. 110, 11415–11420 (201X). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delmonte O. M., Schuetz C., Notarangelo L. D., RAG deficiency: Two genes, many diseases. J. Clin. Immunol. 38, 646–655 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alt F. W., Schwer B., DNA double-strand breaks as drivers of neural genomic change, function, and disease. DNA Repair 71, 158–163 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang M., Wei P.-C., Lim C. K., Gallina I. S., Marshall S., Marchetto M. C., Alt F. W., Gage F. H., Increased neural progenitor proliferation in a hiPSC model of autism induces replication stress-associated genome instability. Cell Stem Cell 26, 221–233.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ming G.-L., Song H., Adult neurogenesis in the mammalian brain: Significant answers and significant questions. Neuron 70, 687–702 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Merkle F. T., Mirzadeh Z., Alvarez-Buylla A., Mosaic organization of neural stem cells in the adult brain. Science 317, 381–384 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Moreno-Jiménez E. P., Flor-García M., Terreros-Roncal J., Rábano A., Cafini F., Pallas-Bazarra N., Ávila J., Llorens-Martín M., Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 25, 554–560 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Boldrini M., Fulmore C. A., Tartt A. N., Simeon L. R., Pavlova I., Poposka V., Rosoklija G. B., Stankov A., Arango V., Dwork A. J., Hen R., Mann J. J., Human hippocampal neurogenesis persists throughout aging. Cell Stem Cell 22, 589–599.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tobin M. K., Musaraca K., Disouky A., Shetti A., Bheri A., Honer W. G., Kim N., Dawe R. J., Bennett D. A., Arfanakis K., Lazarov O., Human hippocampal neurogenesis persists in aged adults and Alzheimer’s disease patients. Cell Stem Cell 24, 974–982.e3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohsugi M., Zheng P., Baibakov B., Li L., Dean J., Maternally derived FILIA-MATER complex localizes asymmetrically in cleavage-stage mouse embryos. Development 135, 259–269 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Zheng P., Dean J., Role of Filia, a maternal effect gene, in maintaining euploidy during cleavage-stage mouse embryogenesis. Proc. Natl. Acad. Sci. U.S.A. 106, 7473–7478 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao B., Zhang W.-D., Duan Y.-L., Lu Y.-Q., Cun Y.-X., Li C.-H., Guo K., Nie W.-H., Li L., Zhang R., Zheng P., Filia is an ESC-specific regulator of DNA damage response and safeguards genomic stability. Cell Stem Cell 16, 684–698 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao B., Zhang W., Cun Y., Li J., Liu Y., Gao J., Zhu H., Zhou H., Zhang R., Zheng P., Mouse embryonic stem cells have increased capacity for replication fork restart driven by the specific Filia-Floped protein complex. Cell Res. 28, 69–89 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagy Z., Soutoglou E., DNA repair: Easy to visualize, difficult to elucidate. Trends Cell Biol. 19, 617–629 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Onishi K., Uyeda A., Shida M., Hirayama T., Yagi T., Yamamoto N., Sugo N., Genome stability by DNA polymerase β in neural progenitors contributes to neuronal differentiation in cortical development. J. Neurosci. 37, 8444–8458 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mandal P. K., Blanpain C., Rossi D. J., DNA damage response in adult stem cells: Pathways and consequences. Nat. Rev. Mol. Cell Biol. 12, 198–202 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Mandal P. K., Rossi D. J., DNA-damage-induced differentiation in hematopoietic stem cells. Cell 148, 847–848 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Su L.-Y., Luo R., Liu Q., Su J.-R., Yang L.-X., Ding Y.-Q., Xu L., Yao Y.-G., Atg5- and Atg7-dependent autophagy in dopaminergic neurons regulates cellular and behavioral responses to morphine. Autophagy 13, 1496–1511 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eichenbaum H., Hippocampus: Cognitive processes and neural representations that underlie declarative memory. Neuron 44, 109–120 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Wei P.-C., Chang A. N., Kao J., Du Z., Meyers R. M., Alt F. W., Schwer B., Long neural genes harbor recurrent DNA break clusters in neural stem/progenitor cells. Cell 164, 644–655 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wei P.-C., Lee C.-S., Du Z., Schwer B., Zhang Y., Kao J., Zurita J., Alt F. W., Three classes of recurrent DNA break clusters in brain progenitors identified by 3D proximity-based break joining assay. Proc. Natl. Acad. Sci. U.S.A. 115, 1919–1924 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Orii K. E., Lee Y., Kondo N., McKinnon P. J., Selective utilization of nonhomologous end-joining and homologous recombination DNA repair pathways during nervous system development. Proc. Natl. Acad. Sci. U.S.A. 103, 10017–10022 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cimprich K. A., Cortez D., ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 9, 616–627 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sørensen C. S., Hansen L. T., Dziegielewski J., Syljuåsen R. G., Lundin C., Bartek J., Helleday T., The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol. 7, 195–201 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Reaper P. M., Griffiths M. R., Long J. M., Charrier J.-D., Maccormick S., Charlton P. A., Golec J. M., Pollard J. R., Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 7, 428–430 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Maréchal A., Zou L., DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 5, a012716 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu J., Meyers R. M., Dong J., Panchakshari R. A., Alt F. W., Frock R. L., Detecting DNA double-stranded breaks in mammalian genomes by linear amplification–mediated high-throughput genome-wide translocation sequencing. Nat. Protoc. 11, 853–871 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baek J. H., Hatakeyama J., Sakamoto S., Ohtsuka T., Kageyama R., Persistent and high levels of Hes1 expression regulate boundary formation in the developing central nervous system. Development 133, 2467–2476 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Sakamoto M., Hirata H., Ohtsuka T., Bessho Y., Kageyama R., The basic helix-loop-helix genes Hesr1/Hey1 and Hesr2/Hey2 regulate maintenance of neural precursor cells in the brain. J. Biol. Chem. 278, 44808–44815 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Chen X.-S., Zhang Y.-H., Cai Q.-Y., Yao Z.-X., ID2: A negative transcription factor regulating oligodendroglia differentiation. J. Neurosci. Res. 90, 925–932 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Ramamoorthi K., Fropf R., Belfort G. M., Fitzmaurice H. L., McKinney R. M., Neve R. L., Otto T., Lin Y., Npas4 regulates a transcriptional program in CA3 required for contextual memory formation. Science 334, 1669–1675 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rogers J. T., Morganti J. M., Bachstetter A. D., Hudson C. E., Peters M. M., Grimmig B. A., Weeber E. J., Bickford P. C., Gemma C., CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J. Neurosci. 31, 16241–16250 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marciniak E., Faivre E., Dutar P., Alves Pires C., Demeyer D., Caillierez R., Laloux C., Buée L., Blum D., Humez S., The Chemokine MIP-1α/CCL3 impairs mouse hippocampal synaptic transmission, plasticity and memory. Sci. Rep. 5, 15862 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sonego M., Gajendra S., Parsons M., Ma Y., Hobbs C., Zentar M. P., Williams G., Machesky L. M., Doherty P., Lalli G., Fascin regulates the migration of subventricular zone-derived neuroblasts in the postnatal brain. J. Neurosci. 33, 12171–12185 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beatus P., Jhaveri D. J., Walker T. L., Lucas P. G., Rietze R. L., Cooper H. M., Morikawa Y., Bartlett P. F., Oncostatin M regulates neural precursor activity in the adult brain. Dev. Neurobiol. 71, 619–633 (2011). [DOI] [PubMed] [Google Scholar]
- 38.FitzGerald J. E., Grenon M., Lowndes N. F., 53BP1: Function and mechanisms of focal recruitment. Biochem. Soc. Trans. 37, 897–904 (2009). [DOI] [PubMed] [Google Scholar]
- 39.McConnell M. J., Moran J. V., Abyzov A., Akbarian S., Bae T., Cortes-Ciriano I., Erwin J. A., Fasching L., Flasch D. A., Freed D., Ganz J., Jaffe A. E., Kwan K. Y., Kwon M., Lodato M. A., Mills R. E., Paquola A. C. M., Rodin R. E., Rosenbluh C., Sestan N., Sherman M. A., Shin J. H., Song S., Straub R. E., Thorpe J., Weinberger D. R., Urban A. E., Zhou B., Gage F. H., Lehner T., Senthil G., Walsh C. A., Chess A., Courchesne E., Gleeson J. G., Kidd J. M., Park P. J., Pevsner J., Vaccarino F. M.; Brain Somatic Mosaicism Network , Intersection of diverse neuronal genomes and neuropsychiatric disease: The brain somatic mosaicism network. Science 356, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park C., Falls W., Finger J. H., Longo-Guess C. M., Ackerman S. L., Deletion in Catna2, encoding αN-catenin, causes cerebellar and hippocampal lamination defects and impaired startle modulation. Nat. Genet. 31, 279–284 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Chu T. T., Liu Y., An integrated genomic analysis of gene-function correlation on schizophrenia susceptibility genes. J. Hum. Genet. 55, 285–292 (2010). [DOI] [PubMed] [Google Scholar]
- 42.Horn K. E., Glasgow S. D., Gobert D., Bull S.-J., Luk T., Girgis J., Tremblay M.-E., McEachern D., Bouchard J.-F., Haber M., Hamel E., Krimpenfort P., Murai K. K., Berns A., Doucet G., Chapman C. A., Ruthazer E. S., Kennedy T. E., DCC expression by neurons regulates synaptic plasticity in the adult brain. Cell Rep. 3, 173–185 (2013). [DOI] [PubMed] [Google Scholar]
- 43.Monje F. J., Kim E.-J., Pollak D. D., Cabatic M., Li L., Baston A., Lubec G., Focal adhesion kinase regulates neuronal growth, synaptic plasticity and hippocampus-dependent spatial learning and memory. Neurosignals 20, 1–14 (2012). [DOI] [PubMed] [Google Scholar]
- 44.Talkowski M. E., Rosenfeld J. A., Blumenthal I., Pillalamarri V., Chiang C., Heilbut A., Ernst C., Hanscom C., Rossin E., Lindgren A. M., Pereira S., Ruderfer D., Kirby A., Ripke S., Harris D. J., Lee J.-H., Ha K., Kim H.-G., Solomon B. D., Gropman A. L., Lucente D., Sims K., Ohsumi T. K., Borowsky M. L., Loranger S., Quade B., Lage K., Miles J., Wu B.-L., Shen Y. P., Neale B., Shaffer L. G., Daly M. J., Morton C. C., Gusella J. F., Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell 149, 525–537 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lek M., Karczewski K. J., Minikel E. V., Samocha K. E., Banks E., Fennell T., O’Donnell-Luria A. H., Ware J. S., Hill A. J., Cummings B. B., Tukiainen T., Birnbaum D. P., Kosmicki J. A., Duncan L. E., Estrada K., Zhao F., Zou J., Pierce-Hoffman E., Berghout J., Cooper D. N., Deflaux N., DePristo M., Do R., Flannick J., Fromer M., Gauthier L., Goldstein J., Gupta N., Howrigan D., Kiezun A., Kurki M. I., Moonshine A. L., Natarajan P., Orozco L., Peloso G. M., Poplin R., Rivas M. A., Ruano-Rubio V., Rose S. A., Ruderfer D. M., Shakir K., Stenson P. D., Stevens C., Thomas B. P., Tiao G., Tusie-Luna M. T., Weisburd B., Won H. H., Yu D., Altshuler D. M., Ardissino D., Boehnke M., Danesh J., Donnelly S., Elosua R., Florez J. C., Gabriel S. B., Getz G., Glatt S. J., Hultman C. M., Kathiresan S., Laakso M., McCarroll S., McCarthy M. I., McGovern D., McPherson R., Neale B. M., Palotie A., Purcell S. M., Saleheen D., Scharf J. M., Sklar P., Sullivan P. F., Tuomilehto J., Tsuang M. T., Watkins H. C., Wilson J. G., Daly M. J., MacArthur D. G.; Exome Aggregation Consortium , Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parry D. A., Logan C. V., Hayward B. E., Shires M., Landolsi H., Diggle C., Carr I., Rittore C., Touitou I., Philibert L., Fisher R. A., Fallahian M., Huntriss J. D., Picton H. M., Malik S., Taylor G. R., Johnson C. A., Bonthron D. T., Sheridan E. G., Mutations causing familial biparental hydatidiform mole implicate c6orf221 as a possible regulator of genomic imprinting in the human oocyte. Am. J. Hum. Genet. 89, 451–458 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reddy R., Akoury E., Phuong Nguyen N. M., Abdul-Rahman O. A., Dery C., Gupta N., Daley W. P., Ao A., Landolsi H., Ann Fisher R., Touitou I., Slim R., Report of four new patients with protein-truncating mutations in C6orf221/KHDC3L and colocalization with NLRP7. Eur. J. Hum. Genet. 21, 957–964 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rezaei M., Nguyen N. M., Foroughinia L., Dash P., Ahmadpour F., Verma I. C., Slim R., Fardaei M., Two novel mutations in the KHDC3L gene in Asian patients with recurrent hydatidiform mole. Hum. Genome Var. 3, 16027 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang W., Chen Z., Zhang D., Zhao B., Liu L., Xie Z., Yao Y., Zheng P., KHDC3L mutation causes recurrent pregnancy loss by inducing genomic instability of human early embryonic cells. PLOS Biol. 17, e3000468 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Favaro R., Valotta M., Ferri A. L., Latorre E., Mariani J., Giachino C., Lancini C., Tosetti V., Ottolenghi S., Taylor V., Nicolis S. K., Hippocampal development and neural stem cell maintenance require Sox2-dependent regulation of Shh. Nat. Neurosci. 12, 1248–1256 (2009). [DOI] [PubMed] [Google Scholar]
- 51.Conlon R. A., Herrmann B. G., Detection of messenger RNA by in situ hybridization to postimplantation embryo whole mounts. Methods Enzymol. 225, 373–383 (1993). [DOI] [PubMed] [Google Scholar]
- 52.Kempermann G., Jessberger S., Steiner B., Kronenberg G., Milestones of neuronal development in the adult hippocampus. Trends Neurosci. 27, 447–452 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Yang C.-P., Gilley J. A., Zhang G., Kernie S. G., ApoE is required for maintenance of the dentate gyrus neural progenitor pool. Development 138, 4351–4362 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Swain U., Subba Rao K., Study of DNA damage via the comet assay and base excision repair activities in rat brain neurons and astrocytes during aging. Mech. Ageing Dev. 132, 374–381 (2011). [DOI] [PubMed] [Google Scholar]
- 55.Sirbu B. M., Couch F. B., Cortez D., Monitoring the spatiotemporal dynamics of proteins at replication forks and in assembled chromatin using isolation of proteins on nascent DNA. Nat. Protoc. 7, 594–605 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Técher H., Koundrioukoff S., Azar D., Wilhelm T., Carignon S., Brison O., Debatisse M., Le Tallec B., Replication dynamics: Biases and robustness of DNA fiber analysis. J. Mol. Biol. 425, 4845–4855 (2013). [DOI] [PubMed] [Google Scholar]
- 57.Frock R. L., Hu J., Meyers R. M., Ho Y.-J., Kii E., Alt F. W., Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat. Biotechnol. 33, 179–186 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cong L., Ran F. A., Cox D., Lin S. L., Barretto R., Habib N., Hsu P. D., Wu X. B., Jiang W. Y., Marraffini L. A., Zhang F., Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou X., Wu W., Li H., Cheng Y., Wei N., Zong J., Feng X., Xie Z., Chen D., Manley J. L., Wang H., Feng Y., Transcriptome analysis of alternative splicing events regulated by SRSF10 reveals position-dependent splicing modulation. Nucleic Acids Res. 42, 4019–4030 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Picelli S., Faridani O. R., Björklund Å. K., Winberg G., Sagasser S., Sandberg R., Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 9, 171–181 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/44/eaba0682/DC1