

CDK4 and CDK6 regulate ciliogenesis and Hedgehog signaling via their novel substrate SMYD2.
Abstract
Dysregulation of cyclin-dependent kinases 4 and 6 (CDK4/6) by unknown mechanisms is highly prevalent in human disease. In this study, we identify direct cross-talk between CDK4/6 and the epigenome via its previously unidentified substrate, SMYD2, a histone/lysine methyltransferase. CDK4/6 positively regulates the phosphorylation and enzymatic activity of SMYD2, while SMYD2 also positively regulates the expression of CDK4/6. We also identify SMYD2 as an α-tubulin methyltransferase, thus connecting CDK4/6-SMYD2 signaling to microtubule dynamics. In addition, depletion or inhibition of CDK4/6 and SMYD2 resulted in increased cilia assembly by affecting (i) microtubule stability and (ii) the expression of IFT20, further connecting CDK4/6-SMYD2 to ciliogenesis. In clinical settings such as breast cancer and autosomal dominant polycystic kidney disease (ADPKD), targeting the up-regulated CDK4/6 and SMYD2 with inhibitors results in restoration of the primary cilium in tumor and cystic cells, which may normalize cilia-mediated extracellular signals that regulate growth, development, and cellular homeostasis.
INTRODUCTION
Dysregulation of cyclin-dependent kinase 4 (CDK4) and the closely related CDK6 is highly prevalent in human disease such as cancer, and inhibitors against these kinases are currently used to restrict tumor growth (1). During the cell cycle, CDK4 and CDK6, in complexes with D-type cyclins, propel G1 phase quiescent cells into S phase (DNA synthesis phase) of the cell cycle. In contrast to CDK1 and CDK2 that phosphorylate hundreds of cellular protein substrates, CDK4/6 has very few identified substrates (2). Almost all previous studies have focused on the retinoblastoma (Rb) protein family (RB, p107, and p130) as the main substrates of CDK4/6 (3). However, both Rb family proteins and the other few known substrates (4) provide limited insights into the mechanisms of CDK4/6-associated human disease. Thus, it would be informative to identify novel CDK4/6 substrates to understand the mechanisms that underlie CDK4/6-dependent abnormal cell proliferation and cell cycle–based therapies.
The biogenesis and resorption of primary cilia are intimately associated with the cell cycle, as primary cilia form during the G0-G1 phase (quiescence) and resorb before mitosis (5). The primary cilium is a nonmotile sensory and signaling organelle, which is present in many diverse cell types throughout the human body (6). Growth of the primary cilium is initiated in a specific region of the plasma membrane, resulting in the entire ciliary structure being enclosed by the plasma membrane of the cell, while inside the cilium is a microtubule-based cytoskeletal structure called the axoneme, which grows from the basal body (7). The ciliary membrane and the basal body microtubules are connected by transition fibers, which form a transition zone to regulate the entry and exit of molecules to and from the cilia. Primary cilia function as cellular antennae on eukaryotic cells to sense specific signaling cues from the extracellular environment, including mechanosensation, chemosensation, and thermosensation (8). Genetic diseases where the assembly and function of primary cilia are defective, ciliopathies, indicate that primary cilia are tightly associated with normal development in mammals (9). The enzymatic activities of CDK4/6 in the G1 phase to drive quiescent cells into the S phase and cilia formation in the G0-G1 quiescent phase suggest an intrinsic connection between CDK4/6 activity and ciliogenesis during the first gap phase of the cell cycle, the focus of our studies.
Epigenetic chromatin modifications, which are dynamic across the cell cycle, play an essential role in regulating cell cycle progression locally by controlling the expression of individual genes and globally by controlling chromatin condensation and chromosome segregation. Abnormalities in gene expression and protein function related to cell cycle regulation result in dysregulated cell proliferation, as has been characterized in cancer cells and cystic renal epithelial cells (10). In the G1 phase and the G1-S transition of the mammalian cell cycle, many genes are actively transcribed (11). This is accompanied by global posttranslational modifications (PTMs), including acetylation, methylation, phosphorylation, ubiquitination, and sumoylation, on specific histone tails of nucleosomes mediated by histone modifiers (12). During the cell cycle, specific tubulin subunits within microtubules can also be posttranslationally modified, including phosphorylation, detyrosination, polyglutamylation, polyglycylation, and acetylation, to affect specialized microtubule structures, such as centrioles, basal bodies, and primary cilia (13). PTMs on tubulins direct microtubule-based functions at specific cellular locations. Although the enzymatic activities of CDK4/6 are required for G1 to S phase transition, if and how CDK4/6 regulate the histone code and tubulin code in this gap phase is unknown.
We report here that CDK4/6 is required for the roles of SMYD2, a SET (suppressor of variegation, enhancer of zeste, trithorax) and MYND (myeloid-Nervy-DEAF1) domain-containing protein methyltransferase, in the methylation of histones and tubulins, providing evidence for cross-talk between CDK4/6 and the regulation of the epigenome and ciliogenesis during the cell cycle. We show that CDK4 and CDK6 interact with and regulate SMYD2 enzymatic methylation of histone H3 lysine 4 (H3K4) and lysine 36 (H3K36) and methylation of α-tubulin. Methylation occurs on histone H3K4 at the promoters of CDK4 and CDK6 to promote their transcription, whereas methylation occurs on histone H3K36 at the promoter of IFT20 to repress its transcription. CDK4/6-SMYD2 methylates α-tubulin at lysine-394 (TubK394me). Methylated microtubules were located at the basal body and surrounding regions and colocalized with the Golgi to regulate cilia protein trafficking from the Golgi to cilia. Targeting CDK4/6 and SMYD2 with inhibitors selectively decreased the methylation of α-tubulin at lysine-394 and increased the expression of IFT20, which led to the stabilization of microtubules and increased IFT20-mediated ciliary protein transport from the Golgi to cilia. This results in an increase in the number of ciliated cells and cilia length, suggesting two novel modes of action for these clinical agents in regulating ciliogenesis in disease.
RESULTS
CDK4 and CDK6 interact and regulate the phosphorylation of SMYD2
The role of CDK4/6 in cell cycle regulation has been mainly focused on the phosphorylation of its key substrates, Rb family proteins. To better understand CDK4/6-dependent cell proliferation and the mechanisms of cell cycle–based therapy, it is necessary to identify novel CDK4/6 substrate(s) that may have diverse roles in cell cycle regulation. Because CDK4/6 interacts with and phosphorylates Rb, and SMYD2 also interacts with and methylates Rb, this suggests an intrinsic connection between CDK4/6 and SMYD2 either directly or indirectly via Rb. We found that CDK4/6 interacts with SMYD2 in renal cortical tubular epithelial (RCTE) cells by coimmunoprecipitation assay with either CDK4 and CDK6 antibodies or a SMYD2 antibody (Fig. 1, A to C). To determine whether CDK4 and CDK6 directly interact with SMYD2 and the binding domain of SMYD2 with CDK4/6, we generated a series of glutathione S-transferase (GST) fusion proteins containing wild-type human SMYD2 and truncated fragments of SMYD2 (Fig. 1D, top), which were purified from Escherichia coli. We first detected the expression of GST-tagged SMYD2 proteins from various constructs in SDS–polyacrylamide gel electrophoresis (SDS-PAGE) gels stained with Coomassie blue (Fig. 1D). Then, we found that only the fusion proteins containing the (S)ET domain of SMYD2 could pull down CDK4 and CDK6 (Fig. 1D, bottom), indicating that the (S)ET domain was required for the binding of SMYD2 to CDK4 and CDK6. We further found that deletion of the (S)ET domain of SMYD2 interrupted the interaction between SMYD2 and CDK4/6 in human embryonic kidney (HEK) 293T cells transfected with green fluorescent protein (GFP)–tagged SMYD2 constructs (Fig. 1E). These results support a direct interaction between CDK4/6 and SMYD2, implying an enzyme substrate relationship between them.
Fig. 1. CDK4/6 interacts with SMYD2 and regulates its phosphorylation.
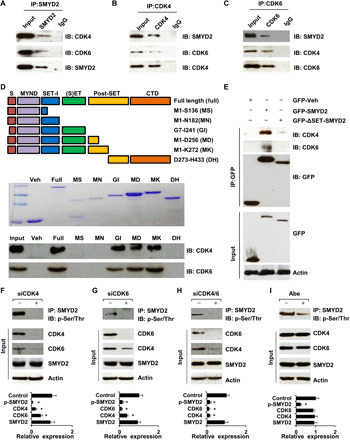
(A to C) Interactions of CDK4 (A) or CDK6 (B) with SMYD2, and SMYD2 with CDK4 or CDK6 (C) in RCTE cells were detected by immunoprecipitation (IP) and immunoblotting (IB). Immunoglobulin G (IgG) was used as a negative control. (D) Schematic of GST-SMYD2 fusion protein constructs (top), which was detected using Coomassie blue staining (middle). GST pull-down assays indicated the interaction of GST-SMYD2 fusion proteins with CDK4 and CDK6 (bottom). (E) GFP-tagged SET domain–deleted SMYD2 cannot pull down CDK4 and CDK6 in HEK293T cells. (F to I) The phosphorylation of SMYD2 was decreased in RCTE cells transfected with siRNAs to CDK4 (F), CDK6 (G), or both (H), and in RCTE cells treated with Abe (10 μM) (I). Knockdown of CDK4 decreased the expression of CDK6, and vice versa for CDK6 in these cells. The quantification analysis (n = 3) of band intensities was shown in the graphs (bottom), in that the density of each protein band was normalized to actin, and then the value was divided by the value of corresponding siRNA and vehicle band density to actin. The value of the control band to actin was set to 1.
To investigate whether CDK4/6 regulates the phosphorylation of SMYD2 via their interaction, we knocked down CDK4, CDK6, or both CDK4/6 with small interfering RNA (siRNA), as well as inhibited the activity of CDK4/6 with their inhibitor, abemaciclib (Abe), and examined the phosphorylation of SMYD2 in RCTE cells. Because of the lack of an anti–phospho-SMYD2 antibody, we used an anti-SMYD2 antibody to pull down SMYD2 and then the SMYD2 bands were blotted with an anti–phospho-(Ser/Thr) antibody. In all cases, we found decreased phosphorylation of SMYD2 in RCTE cells compared to cells treated with control siRNA and vehicle (H2O) (Fig. 1, F to I). These results suggest that CDK4 and CDK6 are required to phosphorylate SMYD2.
CDK4/6 regulates the enzymatic activity of SMYD2, and SMYD2 also regulates the expression of CDK4 and CDK6
SMYD2, as a histone/lysine methyltransferase, regulates the methylation of H3K4 and H3K36 (14). To investigate whether CDK4/6-mediated phosphorylation of SMYD2 affects its enzymatic activity, we knocked down CDK4/6 or SMYD2, as above, and then examined the methylation of H3K4 and H3K36. We found decreased mono-, di-, and trimethylation of H3K4 and H3K36 compared to cells treated with control siRNA (Fig. 2, A and B). Similar effects of decreased H3K4 and H3K36 methylation were detected in RCTE cells treated with the CDK4/6 inhibitor Abe or the SMYD2 inhibitor AZ505 (Fig. 2, C and D). Unexpectedly, we found that knockdown or inhibition of SMYD2 decreased the mRNA and protein levels of CDK4 and CDK6 (Fig. 2, E and F). Using a chromatin immunoprecipitation (ChIP) assay, we further found that SMYD2 and methylated H3K4 bind to the promoters of CDK4 and CDK6 (Fig. 2, G and H). Overall, these results suggest that (i) CDK4/6 regulates the enzymatic activity of SMYD2 to methylate histones H3K4 and H3K36 via their interaction and (ii) SMYD2-mediated H3K4 methylation regulates the expression of CDK4/6, which forms a positive feedback loop between CDK4/6 expression and SMYD2 activity, connecting CDK4/6 directly with the epigenome.
Fig. 2. Targeting CDK4/6 decreased the methylation of histones mediated by SMYD2 in RCTE cells and SMYD2 also regulated the expression of CDK4 and CDK6.
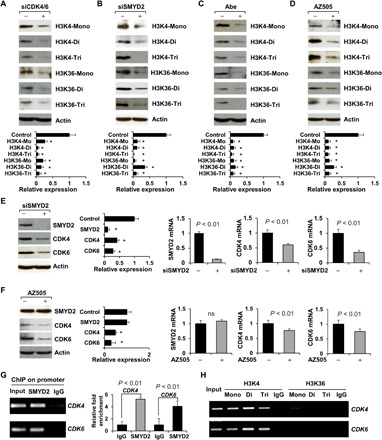
(A to D) Western blot analysis indicated that knockdown of CDK4 and CDK6 with siRNAs (A) or knockdown of SMYD2 with siRNA (B) as well as inhibition of CDK4/6 with Abe (C) or inhibition of SMYD2 with AZ505 (D) decreased the mono-, di-, and trimethylation of histone H3 at lysine 4 (H3K4) and lysine 36 (H3K36) in RCTE cells. The quantification and statistical analysis (n = 3) were shown in the graph (bottom). *P < 0.01 as compared to each control. (E and F) Knockdown (E) or inhibition (F) of SMYD2 decreased the mRNA and protein levels of CDK4 and CDK6 in RCTE cells, examined with qRT-PCR and Western blotting. *P < 0.01 as compared to each control (n = 3). ns, not significant. (G) SMYD2 bound to the promoter of CDK4 and CDK6. ChIP-qPCR analysis was performed with an SMYD2 antibody, or normal rabbit IgG in RCTE cells. (H) ChIP assay was performed with mono-, di-, and trimethylated H3K4 and H3K36 antibodies and normal rabbit IgG in RCTE cells.
Targeting CDK4/6 and SMYD2 decreased mitotic entry of renal epithelial cells
CDK4 and CDK6, in complexes with D-type cyclins, drive G0-G1 quiescent cells into the S phase of the cell cycle (15). To investigate whether targeting CDK4/6 and SMYD2 affects cell cycle progression, we examined mitotic entry in mouse inner medullary collecting duct (mIMCD3) cells treated with either a CDK4/6 (Abe) or SMYD2 inhibitor (AZ505). In both cases, we found that inhibition decreased mitotic entry, as shown by the decrease in mitotic cells stained with p-H3, a mitotic cell marker, in treated compared to control cells (fig. S1, A and B). In addition, we isolated primary renal epithelial cells from kidneys of wild-type mice and mice where Smyd2 was knocked out in kidney distal tubules and collecting ducts using the Smyd2flox/flox:Ksp-Cre mouse (16). We found that depletion of Smyd2 in primary renal epithelial cells decreased mitotic entry of these cells, as shown by decreased p-H3 staining, compared to wild type (fig. S1C). These results suggested that targeting CDK4/6 and SMYD2 decreases mitotic entry of renal epithelial cells.
CDK4/6 and SMYD2 are localized at the basal body of RCTE cells and RPE cells
The decrease of mitosis by targeting CDK4/6 and SMYD2 should block the cell cycle at the quiescent G0-G1 phase, a phase when primary cilia are assembled, so we wondered if they play a role in regulating ciliogenesis. To test this hypothesis, we first detected the localization of CDK4/6 and SMYD2 on cilia, induced with serum starvation, in RCTE and hTERT-RPE1 (RPE) cells. In support of this, we found that CDK4 and CDK6 as well as SMYD2 colocalized with a basal body marker, γ-tubulin, in about 90% of RCTE cells (fig. S1, D and E), but were not on the ciliary axoneme, as detected by α-acetyl-tubulin staining (fig. S1, E and F). In addition, we found that SMYD2 colocalized with CDK6 in RCTE cells (fig. S1G, top). The basal body localization of CDK4 supported a colocalization of CDK4 and SMYD2 in RCTE cells (fig. S1G, bottom). The basal body localization of CDK4/6 and SMYD2 links them to the base of primary cilia in RCTE and RPE cells.
Loss of SMYD2 and CDK4/6 promotes cilia assembly, and overexpression of SMYD2 inhibits ciliogenesis
To further investigate the role of SMYD2 in ciliogenesis, we promoted ciliation in the Smyd2 knockout primary renal epithelial cells by depletion of serum for 48 to 72 hours. We found that knockout of Smyd2 resulted in an increase in ciliated cells (34.8% versus 63.7%) and an increase in cilia length (14.02 ± 3.39 μm versus 7.96 ± 2.82 μm) compared to wild type (Fig. 3A and fig. S2A). Knockout of Smyd2 also resulted in more and longer cilia in tubule lumens in Smyd2flox/flox:Ksp-Cre kidneys compared to wild-type kidneys (fig. S2, B and C). Furthermore, knockdown of Smyd2 with siRNA or inhibition with AZ505 resulted in an increase in cilia length (5.05 ± 2.13 μm to 8.46 ± 3.93 μm and 5.11 ± 1.92 μm to 7.91 ± 2.48 μm, respectively) in mIMCD3 cells compared to a control siRNA and dimethyl sulfoxide (DMSO)–treated cells (Fig. 3, B and C). Consistent with this, knockdown of CDK4/6 with siRNA or CDK4/6 inhibition with Abe also increased cilia length (4.75 ± 2.58 μm to 8.75 ± 2.71 μm and 5.01 ± 1.75 μm to 8.51 ± 2.23 μm, respectively) in mIMCD3 cells compared to a control siRNA and solvent (H2O)–treated cells (Fig. 3, D and E). Overexpression of GFP-tagged SMYD2 (green), which was enriched at the basal body and in the cytosol (fig. S2D), decreased the percentage of ciliated cells and cilia length in HEK293T cells compared to GFP vehicle–transfected cells (fig. S2E). Our results indicate that targeting CDK4/6 and SMYD2 promotes ciliogenesis, whereas overexpression of SMYD2 reduces cilia assembly in renal epithelial cells.
Fig. 3. Depletion of Smyd2 promotes cilia assembly in mouse renal epithelial cells.
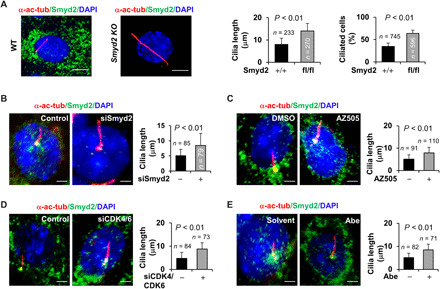
(A) Immunofluorescence staining of primary cilia with α-acetyl-tubulin antibody (α-ac-tubulin) in primary renal epithelial cells isolated from kidneys of wild-type (WT) and Smyd2flox/flox:Ksp-Cre mice, in which Smyd2 was specifically knocked out (KO) by the kidney-specific promoter (Ksp)–driven Cre recombinase in renal epithelial cells and cultured in serum-free medium for 72 hours before subjected to staining. The percentage of ciliated cells and cilia length was measured and statistically analyzed. Scale bar, 5 μm. Error bars represent the SD. N values represent the numbers of cilia (left graph) and cells (right graph), respectively, for each group. (B to E) Knockdown of Smyd2 with siRNA (B) or inhibition of Smyd2 with AZ505 (C) as well as knockdown of CDK4/CDK6 with siRNAs (D) or inhibition of CDK4/CDK6 with Abe (E) in mIMCD3 cells resulted in longer cilia compared to control cells as examined by immunofluorescence with α-acetyl-tubulin and SMYD2 antibodies. Scale bar, 5 μm. Error bars represent the SD. N values represent cilia numbers for each group.
SMYD2 interacts with α- and γ-tubulin and methylates α-tubulin
As major components of primary cilia, the dynamics and therefore stability of microtubules are primarily regulated through PTMs of tubulins (17). However, whether methylation of microtubules affects ciliogenesis remains elusive. SMYD2, as a lysine methyltransferase, methylates both histone and nonhistone substrates (14), but whether it regulates tubulin methylation and stability is unknown. We found that a SMYD2 antibody could pull down both α- and γ-tubulin, and α- and γ-tubulin antibodies could pull down endogenous SMYD2 in RCTE cells (Fig. 4A) and GFP-tagged SMYD2 in HEK293T cells (Fig. 4, B and C). We did not identify the interaction between SMYD2 and β-tubulin in RCTE cells (Fig. 4D). To investigate whether SMYD2 directly interacts with tubulins, we performed GST pull-down assays. We found that GST-tagged Smyd2 pulled down both recombinant human α- and γ-tubulin (Fig. 4E). We further found that the binding domain of SMYD2, which interacts with α- and γ-tubulin, was localized between amino acids 137 to 182, as determined by GST pull-down assays with different GST fusion proteins of SMYD2 (Fig. 4F). Next, to investigate whether the interaction between SMYD2 and α- and γ-tubulin results in their methylation, we performed in vitro methylation assays using recombinant α- and γ-tubulin or histone H3 (control) as substrates. We found that freshly purified SMYD2 was able to methylate tubulins and histone H3 (Fig. 4, G and H). However, SMYD2 methylated α-tubulin with much higher efficiency than γ-tubulin, as determined by the density of methylated bands of α- and γ-tubulin compared to the methylated bands of histone H3 (Fig. 4I). These results suggest that α-tubulin is the main substrate and a novel substrate of SMYD2.
Fig. 4. SMYD2 interacts with α- and γ-tubulin and methylates α-tubulin in vitro.
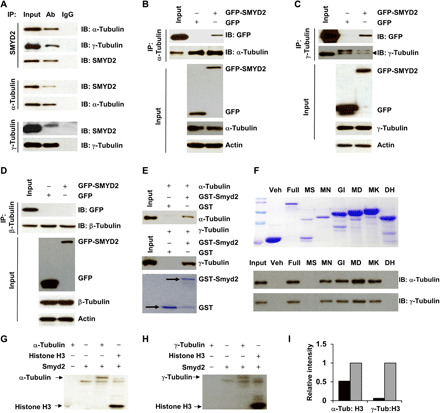
(A) Coimmunoprecipitation of endogenous α- and γ-tubulin with SMYD2 in RCTE cells. IgG was used as a negative control. (B to D) Coimmunoprecipitation of endogenous α-tubulin (B), γ-tubulin (C), and β-tubulin (D) with overexpressed SMYD2 in HEK293T cells. GFP vector–transfected cells were used as a negative control. (E) GST pull-down assays were performed by incubation of GST-SMYD2 fusion protein with 1 μg of recombinant α- or γ-tubulin protein and immunoblotting with an α-tubulin (top) and γ-tubulin antibody (middle). The expression of GST-SMYD2 was detected with Coomassie blue staining (bottom). (F) The expression of GST-SMYD2 constructs was detected using Coomassie blue staining (top). GST pull-down assays of GST-SMYD2 fusion proteins incubated with 1 mg of cell lysate from RCTE cells and immunoblotting using α- and γ-tubulin antibodies (bottom). (G and H) In vitro methylation assay of α-tubulin (G), γ-tubulin (H), and recombinant SMYD2. Histone H3 was used as a positive control in the in vitro methylation assays. (I) Relative intensity of methylated α- and γ-tubulin bands (black box) compared to the intensity of methylated histone H3, which was set to 1 (open box), in the in vitro methylation assays.
SMY2 methylates α-tubulin at lysine-394 (K394) but not at lysine 40 (K40)
To identify potential sites for SMYD2-mediated α-tubulin methylation, we analyzed the amino acid sequence of α-tubulin and found that there are 18 conserved lysine sites in human α-tubulin. Among the 18 lysines, 2, K40 and K394, have been highlighted as particularly important: (i) K40 was reported to be acetylated and methylated by αTAT1 and SETD2, respectively (18); (ii) mutation of K394 abolished the incorporation of α-tubulin into microtubules (19); and (iii) lysines K40 and K394 are highly conserved among species (fig. S3A). To determine whether these two lysine sites are methylated by SMYD2, first, we generated polyclonal antibodies against trimethylated K40 and K394 peptide of α-tubulin, TubK40me3 and TubK394me3 antibodies, respectively. The specificity of these antibodies was characterized by a peptide competition assay as outlined in fig. S3B (top). We found that both TubK40me3 and TubK394me3 antibodies showed strong immunoreactivity with α-tubulin by Western blotting (fig. S3B, left bottom) that was significantly blocked by preincubation of the antibodies with methylated K40 and K394 peptides (fig. S3B, right bottom), but not unmethylated peptides (fig. S3B, middle bottom). We further found that both TubK40me3 and TubK394me3 antibodies could pull down α-tubulin and SMYD2 from RCTE cells (fig. S3C). The TubK40me3 antibody stained mainly within the nucleus (fig. S3D), which was consistent with its reported staining pattern (20), whereas the TubK394me3 antibody stained mainly on one side surrounding the nucleus (fig. S3D), signals that were blocked by preincubation of the antibodies with methylated TubK40me3 peptide or TubK394me3 peptide, respectively (fig. S3D). Our results indicate that the newly generated antibodies are specific for the methylation of α-tubulin at K40 or K394.
To investigate whether SMYD2 regulates the methylation of α-tubulin at K40 and/or K394, we either knocked down SMYD2 with siRNA or inhibited SMYD2 with AZ505 in RCTE cells and found that both of these decreased the methylation of α-tubulin at K394 but not K40 as examined by Western blotting with the TubK394me3 and TubK40me3 antibodies, respectively (Fig. 5, A and B). In addition, overexpression of Flag-tagged SMYD2 increased the methylation of GFP-tagged α-tubulin at K394 but showed no effect on the methylation of GFP-tagged α-tubulin at K40 (Fig. 5C). We also found that overexpression of GFP-tagged SMYD2 increased methylated α-tubulin as stained with the TubK394me3 antibody in HEK293T cells (fig. S3E). Our results indicate that SMYD2 methylates α-tubulin at K394 but not K40.
Fig. 5. SMYD2 methylates α-tubulin at lysine-394 (K394) but not at K40.
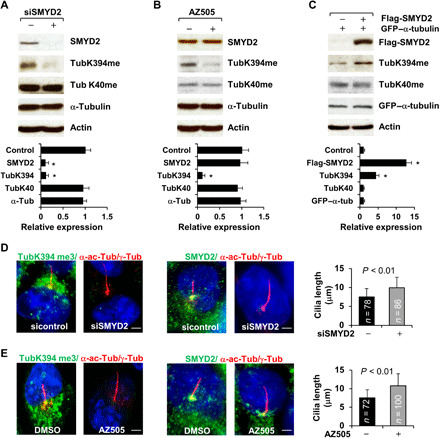
(A and B) Knockdown of SMYD2 with siRNA (A) or inhibition of SMYD2 with AZ505 (B) decreased methylation of α-tubulin at K394 but not at K40 as examined by Western blotting with our generated TubK40me3 and TubK394me3 antibodies in RCTE cells. The quantification and statistical analysis (n = 3) were shown in the graphs (bottom), in which the band density of control siRNA (A) and DMSO (B) was set to 1. (C) Western blot of the methylation of α-tubulin at K40 and at K394 with the TubK40me3 and TubK394me3 antibodies in RCTE cells transfected with Flag-tagged SMYD2 and GFP-tagged α-tubulin. The quantification and statistical analysis (n = 3) were shown in the graph (bottom). (D) Representative images of RCTE cells stained with TubK394me3 (left) or SMYD2 (right) and costained with α-acetyl-tubulin/γ-tubulin (red) antibodies and DAPI (blue). Scale bar, 5 μm. (E) Representative images of RCTE cells stained with TubK394me3 (left, green) or SMYD2 (right, green) and costained with α-acetyl-tubulin/γ-tubulin (red) and DAPI (blue). Scale bar, 5 μm. Error bars represent the SD. N values represent cilia numbers in (D) and (E).
Methylated α-tubulin at lysine-394 is localized at the basal body and the surrounding area, which could be depleted by targeting SMYD2 and CDK4/6
To determine whether basal body–localized SMYD2 affects the methylation of α-tubulin at this region, we costained RCTE cells with the TubK394me3 and γ-tubulin antibodies and found an accumulation of K394-methylated α-tubulin at basal bodies and surrounding areas (fig. S4A). Coimmunostaining with TubK394me3 and α-acetyl-tubulin antibodies as well as α-acetyl-tubulin/γ-tubulin antibodies further supported the basal body localization of K394-methylated α-tubulin and SMYD2; neither K394-methylated α-tubulin nor SMYD2 was detected in the ciliary axoneme (fig. S4, B and C). In addition, we found that knockdown SMYD2 or inhibiting SMYD2 depleted the basal body and cytosol staining of TubK394me3, which was accompanied by increased cilia length in both RCTE (Fig. 5, D and E) and RPE cells (fig. S5, A and B).
Because CDK4/6 interacts with and regulates the phosphorylation and enzymatic activity of SMYD2, we next investigated (i) whether CDK4/6 regulates SMYD2-mediated α-tubulin methylation at K394 and (ii) whether CDK4/6 regulates ciliogenesis in serum-starved cells. Knockdown of CDK4, CDK6, and CDK4/6 with siRNAs, as well as inhibition of CDK4/6 with Abe, (i) decreased the methylation of α-tubulin at K394 (fig. S5, C to F) and (ii) depleted the staining of the TubK394me3 antibody at the basal body and in the cytosol, and increased cilia length in RCTE (fig. S5, G to J) and RPE cells (fig. S5B). However, these treatments did not affect the localization and expression SMYD2 in these cells (fig. S5, C to F). These results indicate that CDK4/6 regulates SMYD2-mediated methylation of α-tubulin at K394 that regulates ciliogenesis.
Methylation of α-tubulin affects the stability of microtubules, which are colocalized with the Golgi
To assess whether CDK4/6-SMYD2–mediated α-tubulin methylation affects the stability of microtubules, we treated RCTE cells with nocodazole to induce microtubule depolymerization and then monitored microtubule reassembly in nocodazole-free culture medium. Treatment with nocodazole for 4 hours completely depolymerized microtubules in most RCTE cells, and cotreatment with the SMYD2 inhibitor AZ505 or the CDK4/6 inhibitor Abe did not block this process (fig. S6A, left). However, when nocodazole-treated RCTE cells were recultured in fresh, prewarmed nocodazole-free medium, the microtubules were mainly reassembled by 30 min in AZ505- or Abe-treated RCTE cells, whereas microtubules were only partially reassembled in control cells within that time (fig. S6, A, right, and B). These results suggest that demethylation of α-tubulin at K394 increases the assembly of microtubules, which might facilitate the trafficking of cilia proteins from the Golgi to the cilia base. In contrast, methylation of α-tubulin by CDK4/6-SMYD2 might retard this trafficking, and this might be accentuated when CDK4/6 and SMYD2 are up-regulated, such as in most cancer cells. To support this notion, we found that the asymmetric localization of TubK394me3 staining on one side of the cell partially overlapped with the Golgi (stained with Giantin), and a similar pattern was also found for SMYD2 staining in RCTE cells (fig. S6C).
To provide direct evidence that methylation of α-tubulin at K394 is involved in cilium assembly and microtubule stability, we generated α-tubulin K394R mutant RCTE cells (see Materials and Methods). We found that our newly generated TubK394me antibody could only detect GFP-tagged wild-type α-tubulin but not GFP-tagged mutant α-tubulin (K394R), as examined by immunostaining and Western blot analysis in RCTE cells (fig. S7, A and B). In addition, we found that mutation of α-tubulin at K394 resulted in an increase of ciliated cells and cilia length compared to RCTE cells expressing wild-type α-tubulin (fig. S7C). In further support of the observation that mutation of α-tubulin at K394 affects the stability of microtubules, we treated wild-type and mutant α-tubulin–expressing RCTE cells with nocodazole to induce microtubule depolymerization and then monitored microtubule reassembly in nocodazole-free culture medium as described above. We found that treatment with nocodazole for 4 hours completely depolymerized microtubules in RCTE cells expressing either GFP-tagged wild-type α-tubulin or GFP-tagged mutant α-tubulin–K394R (fig. S7D, left). However, when nocodazole-treated cells were recultured in fresh, prewarmed nocodazole-free medium, the microtubules were almost completely reassembled within 30 min in α-tubulin K394R mutant–expressing RCTE cells, whereas microtubules were only partially reassembled in wild-type α-tubulin–expressing cells within that same time frame (fig. S7D, right). These results suggest that mutation of α-tubulin at K394 increases the assembly of microtubules and promotes ciliogenesis.
Negative regulation of intraflagellar transport protein IFT20 by CDK4/6-SMYD2 contributes to cilia growth
The partial colocalization of methylated α-tubulin with the Golgi suggests that this modification regulates not only the assembly of microtubules but also perhaps the trafficking of ciliary proteins from the Golgi to cilia, a process that is regulated by an intraflagellar transport protein, IFT20, which is also localized to the Golgi (fig. S6D). We found that the expression of both IFT20 mRNA and protein was up-regulated in SMYD2 knockdown RCTE cells and in cells treated with the SMYD2 inhibitor (Fig. 6, A and B), whereas overexpression of GFP-tagged SMYD2 decreased the expression of IFT20 (Fig. 6C). We further found that SMYD2 and H3K36me3 bound to the promoter of IFT20, as examined by a ChIP assay (Fig. 6D). In addition, we found that siRNA knockdown of CDK4/6 or treatment with Abe increased the mRNA and protein levels of IFT20 (Fig. 6, E and F). We also found that treatment with AZ505 and Abe increased the anterograde trafficking of IFT20 to cilia tips (Fig. 6G). Furthermore, we found increased anterograde trafficking of IFT20 to cilia tips in α-tubulin K394R mutant RCTE cells (fig. S7E). Our results suggest that CDK4/6-SMYD2 negatively regulates not only the expression of IFT20 but also the trafficking of IFT20 from the Golgi to cilia, and inhibiting CDK4/6 and SMYD2 releases this negative regulation to promote cilia assembly.
Fig. 6. Depletion of SMYD2 increases the expression and ciliary trafficking of IFT20.
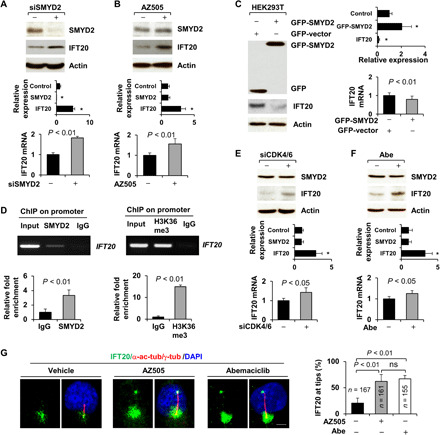
(A and B) Knockdown (A) and inhibition of SMYD2 (B) increased the levels of IFT20 protein (top) and mRNA (bottom) as examined by Western blotting and qRT-PCR in RCTE cells. The quantification and statistical analysis (n = 3) were shown in the graph (bottom), as are also shown in (B), (C), (E), and (F). *P < 0.01 as compared to controls. (C) Overexpression of SMYD2 decreased the levels of IFT20 protein (left) and mRNA (right) as examined by Western blotting and qRT-PCR in HEK293T cells. (D) SMYD2 and H3K36me3 antibodies bound to the promoter of IFT20 in RCTE cells as examined with ChIP assay. (E and F) Knockdown (E) and inhibition of CDK4/6 (F) increased the levels of IFT20 protein (top) and mRNA (bottom) as examined by Western blotting and qRT-PCR in RCTE cells. n = 3. (G) Representative images of RCTE cells stained with IFT20 and α-acetyl-tubulin/γ-tubulin antibodies and DAPI in the presence of AZ505 (middle), Abe (right), and vehicle (left). Scale bar, 5 μm. The statistical analysis was shown in the graph (right). Error bars represent the SD. N values represent cilia numbers.
CDK4/6 and SMYD2 are up-regulated in breast cancer cells and Pkd1 mutant renal epithelial cells, resulting in a decrease of primary cilia formation in these cells
Dysregulation of CDK4 and CDK6 as well as SMYD2 has been reported in breast cancer (21). In addition, Smyd2 is up-regulated in autosomal dominant polycystic kidney disease (ADPKD) mouse kidney tissues and cell lines (14). We found that the expression of CDK4 and SMYD2 is up-regulated in a number of different breast cancer cells, including MCF7, T47D, MDA-MB4231, and MDA-MB468, compared to normal mammary epithelial cells, MCF10A. In contrast, IFT20 is down-regulated in breast cancer cells (Fig. 7A). The expression of CDK6 is only slightly up-regulated in MCF7, T47D, and MDA-MB468 cells but strongly up-regulated in MDA-MB231 cells (Fig. 7A). We also found that the expression of CDK4, CDK6, and SMYD2 was up-regulated and the expression of IFT20 was down-regulated in postnatal Pkd1 homozygous null PN24 cells compared to those in postnatal Pkd1+/−, PH2 cells (Fig. 7B). Up-regulation of CDK4 and SMYD2 was accompanied by a significant decrease of ciliated breast cancer cells; the percentages of cells that were ciliated in MCF7, T47D, MDA-MB231, and MDA-MB468 ranged from 0.5 to 2.4%, whereas it was ~22% in MCF10A cells (fig. S8A). The percentage of ciliated PN24 cells and the cilia length was also decreased compared to Pkd1+/− cells (fig. S8B). However, we found that treatment with the CDK4/6 inhibitor Abe or the SMYD2 inhibitor AZ505 significantly increased the percentages of ciliated breast cancer cells, including in T47D and MDA-MB468 (Fig. 7D), and PN24 cells, as well as increasing cilia length in these cells compared to controls (Fig. 7, E and F). Double knockout of Pkd1 and Smyd2 (Pkd1flox/flox:Smyd2flox/flox:Ksp-Cre mice) resulted in more and longer cilia in kidneys and in primary renal epithelial cells than in Pkd1 single knockout mice or cells (fig. S9, A and B). In testing the role of CDK4/6 and SMYD2 in this process, we found that knockdown of Smyd2 with siRNA and treatment with the SMYD2 inhibitor AZ505 decreased the expression of CDK4 and CDK6 but increased the expression of IFT20 in MDA-MB231 and PN24 cells (fig. S9, C and D). As such, our results indicate that CDK4/6-SMYD2 signaling is involved in the regulation of ciliogenesis in two important disease settings, breast cancer and PKD.
Fig. 7. Primary cilia assembly was decreased in CDK4/6 and SMYD2 up-regulated breast cancer cells and cystic renal epithelial cells, whereas targeting CDK4/6 and SMYD2 restored cilia assembly in these cells.
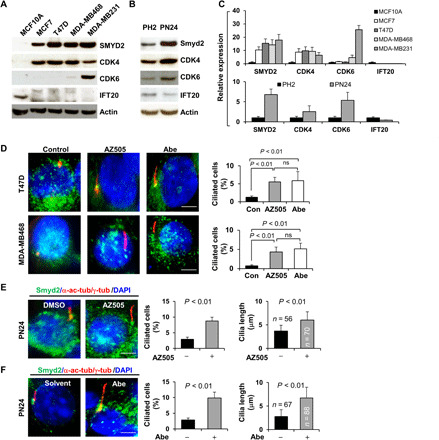
(A) Western blot of SMYD2, CDK4, CDK6, and IFT20 in normal mammary cells MCF10A and breast cancer cells, including MCF7, T47D, MDA-MB468, and MDA-MB231 cells. (B) Western blot of SMYD2, IFT20, CDK4, and CDK6 in PH2 and PN24 cells. (C) The quantification and statistical analysis (n = 3) were shown in the graphs corresponding to Fig. 6A (top) and Fig. 6B (bottom). (D to F) Representative images of T47D (top) and MDA-MB468 cells (bottom) (D) as well as PN24 cells (E and F) stained with SMYD2 (green) and α-acetyl-tubulin/γ-tubulin (red) and costained with DAPI (blue) in the presence of the SMYD2 inhibitor AZ505 (middle) and the CDK4/6 inhibitor Abe as well as vehicle. Scale bar, 5 μm. Statistical analysis of the percentage of ciliated breast cancer cells (n = 200) (P < 0.01) as well as the percentage of ciliated PN24 cells (n = 900) and their cilia lengths are shown in the graph (right). Error bars represent the SD.
Targeting CDK4/6-SMYD2 affects cilia-dependent Hedgehog signaling in RCTE cells and Pkd1 mutant renal epithelial cells
To address whether SMYD2-mediated ciliogenesis affects the functionality of cilia, we investigated the effects of targeting CDK4/6 and SMYD2 on Hedgehog signaling. Hedgehog plays an essential role during vertebrate embryonic development and tumorigenesis (22). Hedgehog signaling can be activated in the presence of Shh [the ligand for Patched 1 (Ptch1) receptor] or the Smoothened agonist, SAG, in a cilia-dependent manner (23). Ptch1 and GLI1 are the main components of Hedgehog signaling involved in signaling transduction, and are also the transcriptional targets of GLI2 and GLI3 during Hedgehog activation (24). We found that treatment with AZ505 or Abe increased the ciliary tip localization of GLI2 and GLI3 in RCTE cells compared to control cells. Treatment with a Smoothened agonist, SAG (500 nM), also resulted in the ciliary tip and axoneme localization of GLI2/3 in RCTE cells, and cotreatment with either AZ505 or Abe further increased the axoneme and ciliary tip localization of GLI2/3 in those cells treated with SAG (Fig. 8A). In addition, we found that treatment with AZ505 or Abe alone did not result in Gli2/Gli3 ciliary localization in Pkd1 mutant PN24 cells, whereas treatment with SAG did result in the ciliary tip and axoneme localization of Gli2 in PN24 cells, and this was further increased when those cells were cotreated with either AZ505 or Abe (Fig. 8B). Furthermore, we found that treatment with SAG induced the up-regulation of Ptch1 and GLI1 and treatment with AZ505 or Abe alone slightly decreased the expression of those proteins in RCTE cells (Fig. 8, C and D), whereas cotreatment with either AZ505 or Abe blocked SAG-induced up-regulation of Ptch1 and GLI1 in those cells (Fig. 8, C and D). We further found that treatment with AZ505 or Abe also blocked SAG-induced up-regulation of Ptch1 and Gli1 in PN24 (Fig. 8, E and F). Our results suggested that targeting CDK4/6-SMYD2 signaling affected not only ciliogenesis but also cilia-dependent Hh signaling activation in RCTE and Pkd1 mutant cells.
Fig. 8. Targeting SMYD2 and CDK4/6 affects cilia-dependent Hedgehog signaling.
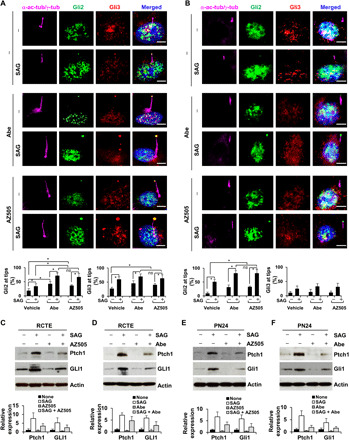
(A and B) Representative images of RCTE (A) and PN24 (B) cells stained with GLI2 (green) and GLI3 (red), and costained with α-acetyl-tubulin/γ-tubulin (purple) antibodies and DAPI (blue). Scale bar, 5 μm. The statistical analysis of the cilia tip localization of GLI2 (left) and GLI3 (right) is shown in the graphs. *P < 0.01. Error bars represent the SD. n = 100 cilia for each group. (C and D) Western blot analysis of Ptch1 and GLI1 in RCTE cells, which were treated with or without SAG and cotreated with or without the SMYD2 inhibitor AZ505 (C) and the CDK4/CDK6 inhibitor Abe (D) in serum-free medium for 48 hours. Statistical analysis of the expression (n = 3) of Ptch1 and GLI1 proteins is shown in the graphs (right). (E and F) Western blot analysis of Ptch1 and Gli1 in PN24 cells, which were treated with or without SAG and cotreated with or without the SMYD2 inhibitor AZ505 (E) and the CDK4/CDK6 inhibitor Abe (F) in serum-free medium for 48 hours. Statistical analysis of the expression (n = 3) of Ptch1 and Gli1 proteins is shown in the graphs (bottom).
DISCUSSION
In this study, we identified a novel substrate of CDK4/6. We found that CDK4 and CDK6 interact with SMYD2, and this interaction results in the phosphorylation and activation of SMYD2, leading to SMYD2 mediating histone H3K4 and H3K36 methylation. We further found that SMYD2 regulates the expression of CDK4 and CDK6 by binding to their promoters together with methylated H3K4, which provides novel evidence of cross-talk between CDK4/6 and the epigenome. SMYD2 also methylates a diverse set of nonhistone proteins, including Rb, p53, STAT3, NF-κB, and HSP90 (14). We identified α-tubulin as a novel nonhistone substrate of SMYD2 (SMYD2 interacts with and methylates α-tubulin at lysine-394) and that CDK4/6 is involved in the regulation of this process. Both CDK4/6 and SMYD2 are localized at the basal body of primary cilia, and we showed that methylated α-tubulins are detected at the basal body and surrounding region using a newly generated antibody (TubK394me3). Methylation of α-tubulin by normal levels of CDK4/6-SMYD2 shows no effect on ciliogenesis in normal human cells, whereas demethylation of α-tubulin, mediated by knockdown of SMYD2 and CDK4/6 or by inhibition of SMYD2 and CDK4/6, demethylates microtubules and blocks ciliary resorption, resulting in an increase in the numbers of ciliated cells and cilia length. In contrast, overexpression of Smyd2 decreases the numbers of ciliated cells and the length of cilia. SMYD2 and its histone substrate H3K36me3 bind to the promoter of the IFT20 gene, which encodes a key IFT protein that regulates the trafficking of ciliary proteins from the Golgi to cilia, to negatively regulate its transcription. Silencing and inhibition of CDK4/6 and SMYD2 increased the expression of IFT20 and the transport of IFT20 to cilia tips, likely increasing the trafficking of ciliary proteins from the Golgi to cilia and promoting cilia assembly. Targeting CDK4/6-SMYD2 with their inhibitors increases the stability or assembly of microtubules and the expression of IFT20, representing two novel ways of regulating the number of ciliated cells and the length of already formed cilia. This study identifies not only a novel substrate of CDK4/6 but also novel roles for CDK4/6 in regulating (i) SMYD2-mediated gene transcription, including itself, during the G0-G1 phase of the cell cycle, and (2) cilia assembly via SMYD2-mediated microtubule methylation and IFT20 expression (fig. S10).
There are extensive connections between the cell cycle machinery and epigenetic chromatin modifications, which are dynamic across the cell cycle to regulate gene expression and cell proliferation. CDK4/6 can be indirectly connected with epigenetic mechanisms via Rb-E2F signaling to regulate cell cycle progression. When a cell enters G1, cyclin D–CDK4/6 phosphorylates Rb at a single phosphorylation site. Then, active mono-phosphorylated Rb causes repression of E2F-targeted genes specifically by recruiting histone deacetylases (HDACs), DNA methyltransferase 1 (DNMT1), and SUV39H1, a histone methyltransferase (HMT) to E2F site–containing promoters, such as cyclin A/E and CDK2 (25). We and others found that Rb can be deacetylated by one of the class III HDACs, SIRT1 (26), and can be methylated by SMYD2 to increase the phosphorylation of Rb, which may be mediated by CDK4/6 (27), leading to increased cystic renal epithelial cell proliferation. In this study, we identify a direct connection between CDK4/6 and the epigenome, in that CDK4/6 directly interacts with and phosphorylates SMYD2 (Fig. 1) to regulate SMYD2-mediated histone H3K4 or H3K36 methylation (Fig. 2), which either promotes or represses gene transcription, respectively. Furthermore, we found that SMYD2 and H3K4 bound to the promoters of CDK4 and CDK6 to increase transcription (Fig. 2, G and H). Our results address how the expression of CDK4/6 and the activity of SMYD2 are regulated and suggest an important feedback loop between CDK4/6 and SMYD2 during cell cycle progression.
The cell division cycle is accompanied by the assembly and resorption/disassembly of primary cilia, which must be regulated. During the cell cycle, cilia are formed in G1 when the mother centriole attaches at the cell cortex. The mother centriole duplicates in S phase to form the daughter centriole (28). Ciliary disassembly occurs in a biphasic manner, with the first wave occurring in G1 and the second wave occurring before mitosis when the cilium is resorbed back into the cell. After the cell divides into its two new cells, the primary cilia are reformed within the cells after the new cells enter G1. However, whether the key cell cycle regulators CDK4 and CDK6 regulate ciliogenesis has not been reported. Our results provide strong evidence that the basal body–localized CDK4/6 and SMYD2 regulate cilia assembly and disassembly, but what are the mechanisms involved in this process?
The ciliary axoneme and rootlet are a microtubule-based cytoskeleton-like structure that originates from the basal body at the proximal end of a cilium. Cilia assembly/disassembly, basal body formation, and ciliary protein trafficking from the Golgi to the basal body involve microtubules. The PTMs of microtubules on specific residues of α- or β-tubulin have been identified (20), and they possibly regulate microtubule dynamics and affect cilia assembly/disassembly (29). It has been reported that deacetylation, glycylation, and glutamylation of α-tubulin at K40 affect cilia disassembly (30). However, little is known about the role of methylation of α-tubulin in microtubule stability and ciliogenesis. In addition, it has been reported that demethylation of arginine residues of axoneme structural components, including radial spoke proteins 1, 2, 5, and 6; tektin; and flagellar-associated protein 172 (FAP172) and FAP250, occurs when Chlamydomonas cells resorb their flagella, and this is regulated by the protein arginine methyl transferase 1 (PRMT1) (31). Furthermore, two CDK-like kinases, FLS1 and FLS2, function in the disassembly of the distal region of the flagellum by controlling the phosphorylation of aurora-like kinase CALK and CrKinesin13, a microtubule depolymerase, and by directly binding with IFT70 (32, 33). Our results that CDK4/6-SMYD2 signaling regulates the methylation of α-tubulin at lysine-394 to destabilize microtubules suggest one mechanism for CDK4/6-SMYD2 regulation of ciliogenesis. However, the mechanism for the asymmetrical distribution of CDK4/6-SMYD2–mediated microtubule methylation and why methylated microtubules are only detected in the basal body but not on the axoneme is still unknown and needs to be further investigated.
Each cell type has a specific optimal cilia length to regulate cellular signaling (34). Therefore, the length of cilia must be regulated to ensure optimal function of the cell. For cilia assembly and elongation, proteins must be selectively imported from the cytoplasm into the cilium and transported from the cell body to the ciliary tip for incorporation in the distal ciliary region, followed by their return to the cell body mediated by IFT complexes. This process serves as an evolutionarily conserved mechanism to regulate the assembly and to maintain the length of all eukaryotic cilia. IFT20 is also localized to the Golgi complex in addition to the basal body and cilia, where all other IFT proteins are found. IFT20 plays a critical role in the delivery or transfer of ciliary membrane proteins from the Golgi complex to the cilium and for the process of ciliary assembly (35). Our results that SMYD2 represses the expression of IFT20 suggest that CDK4/6-SMYD2–mediated repression of IFT20 transcription is another mechanism for CDK4/6-SMYD2 regulation of ciliogenesis. However, our studies leave open the possibility that SMYD2 may also target other ciliary genes, such as other IFTs, to affect cilia biogenesis. How CDK4/6-SMYD2 might regulate the expression of other IFTs needs to be further investigated.
Loss of cilia has been observed in several types of tumors and can lead to aberrant activation of many oncogenic pathways. Recent studies have begun to clarify the key mechanisms that regulate ciliary assembly and disassembly in both normal and tumor cells, allowing possible therapeutic interventions. Dysregulation of CDK4/6 and up-regulation of SMYD2 are reported in breast cancers and cystic kidneys (Fig. 7). As a result, in recent years, small-molecule inhibitors that target CDK4/6 and SMYD2 have been developed. Three of these CDK4/6 inhibitors are currently available for the treatment of metastatic breast cancer (MBC) in combination with aromatase inhibitors or fulvestrant. The results that targeting CDK4/6 and SMYD2 with their inhibitors either alone or in combination increased cilia formation and extension in breast cancer cells and cystic renal epithelial cells (Fig. 7) suggested that dysregulation of CDK4/6 and up-regulation of SMYD2 are potential mechanisms by which cancer and cystic cells lose or shorten their cilia. Because restoration of the primary cilium in tumor cells could promote quiescence and may also normalize cilia-mediated extracellular signals that regulate cell growth, development, and homeostasis, our results also suggest a novel strategy for the treatment of MBC and PKD by using CDK4/6 inhibitors in combination with SMYD2 inhibitors.
In addition, it has been reported that loss of cilia suppresses cyst growth in genetic models of ADPKD, which supports the existence of a cilia-dependent cyst growth–promoting pathway that specifically affects cyst growth (36). That study suggests the possibility that targeting the cilia-dependent pathway can slow cyst growth. One of the cilia-dependent signaling pathways, Hedgehog signaling, is increased in the cystic kidneys of several PKD mouse models, and inhibition of Hedgehog signaling prevented cyst formation in embryonic PKD mouse kidneys treated with adenosine 3′,5′-monophosphate (cAMP) (37). We found that targeting CDK4/6 and SMYD2 with their inhibitors decreased the activation of Hedgehog signaling as seen by an enrichment of Gli2/3 at the axoneme and ciliary tips, and blocked Smoothened agonist SAG-induced up-regulation of Ptch1 and Gli1 in RCTE cells and cystic renal epithelial cells (Fig. 8). These results further support a therapeutic potential for using CDK4/6 inhibitors in combination with SMYD2 inhibitors in the treatment of PKD.
Although our study suggests that CDK4/6-SMYD2 signaling may be a regulatory system that could be exploited for restoring cilia length in disease, we should point out that in contrast to short cilia that were observed in Pkd1 homozygous cystic renal epithelial cells, renal tubular cilia can paradoxically lengthen in several hypomorphic mouse mutants (Nphp3pcy/ko, Nphp9jck/jck, Pkd1RC/RC) (38). The functional consequences of cilia elongation as opposed to cilia shortening are less clear, but we suggest that the loss of fidelity of cilia length control may generally represent a mechanism that permits cystic renal epithelial cell proliferation and cyst growth.
MATERIALS AND METHODS
Cell culture and reagents
Murine IMCD3 cells were maintained at 37°C in 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum. PH2 and PN24 cells (provided by S. Somlo through the George M. O’Brien Kidney Center, Yale University, New Haven, Connecticut, USA) were cultured as described previously (39). Mouse primary renal epithelial cells were isolated from 2-week-old male Smyd2 knockout and wild-type C57BL/6 mouse kidneys. All mice were sacrificed, and kidneys were immediately harvested and washed in sterile ice-cold and 95% O2/5% CO2 equilibrated Krebs-Henseleit saline (KHS; pH 7.4) containing the following: 119 mM NaCl, 4.7 mM KCl, 1.9 mM CaCl2, 1.2 mM KH2PO4, 1.2 mM MgSO4•7H2O, and 25 mM NaHCO3. Each kidney was decapsulated and bisected, and the inner medullary portion was excised. The remaining cortical and outer medullary regions were pulverized and mixed in a solution of 30 ml of KHS containing collagenase (1 mg/ml) (type I; Invitrogen, Carlsbad, CA, USA) for every 1 g of minced kidney. The solution was bubbled with 95% O2/5% CO2 during incubation for 30 min at 37°C. The solution was filtered through a 100-μm mesh sieve (Thermo Fisher Scientific, Houston, TX, USA).
AZ505 was purchased from MedChem Express and dissolved in DMSO (Sigma-Aldrich) at a stock solution of 20 μM. Abe was purchased from Cayman Chemical and dissolved in sterilized water at a stock solution of 5 μM. All the stock solutions were stored at −20°C.
Plasmids
The GFP-tagged wild-type α-tubulin plasmid was purchased from Addgene (pIRESneo-EGFP-alpha Tubulin, Addgene). The GFP-tagged α-tubulin–K394R construct was made according to the instructions for the Site-Directed Mutagenesis Kit (Agilent). The pLKO.1 short hairpin RNA (shRNA) and its vector pLKO.1 were purchased from Sigma-Aldrich. The GFP-tagged SMYD2 plasmid was constructed by cloning full length of Smyd2 into the pAcGFP-C1 vector (Clontech). After 24 to 36 hours, the cells were harvested for further analysis.
Generation of GFP-tagged wild-type and mutant α-tubulin K394R cell lines
The pLKO.1-shRNAs targeting the 3′ untranslated region (UTR) of human α-tubulin (including TUBA1A, TUBA1B, and TUBA1C, which contain the K394 sites) and its vector pLKO.1 were packaged in HEK293T cells according to the protocol for producing lentiviral particles. The harvested lentiviral particles were applied to infect RCTE cells, which were transfected with GFP-tagged wild-type α-tubulin and GFP–α-tubulin–K394 mutant plasmids 24 hours before infection. The 3′UTR is considered a signature of a gene that is unique among different isoforms. The shRNA against the 3′UTR of endogenous α-tubulin should not target the exogenous copy of α-tubulin that lacks a 3′UTR. Twenty-four hours after infection, cells with endogenous α-tubulin knockdown were selected with fresh medium containing puromycin (1 μg/ml), which is included in the shRNA constructs that target 3′UTR of α-tubulin. The selected cells that expressed the GFP-tagged mutant α-tubulin or wild-type α-tubulin (control) could be directly viewed by microscopy.
Western blot analysis and immunoprecipitation
Cell pellets were collected and resuspended in lysis buffer [20 mM tris-HCl (pH 7.4), 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1 mM Na3VO4, 25 mM β-glycerol-phosphate, 0.1 mM phenylmethylsulfonyl fluoride (PMSF), Roche complete protease inhibitor set, and Sigma-Aldrich phosphatase inhibitor set]. The resuspended cell pellet was vortexed for 20 s and then incubated on ice for 30 min and centrifuged at 20,000g for 30 min. The supernatants were collected for Western blot analysis or immunoprecipitation.
For immunoprecipitation, anti-SMYD2, anti–α-tubulin, anti-CDK4, or anti-CDK6 antibodies and their isotype control antibodies were coupled to protein A agarose bead (Pierce) in phosphate-buffered saline (PBS) containing bovine serum albumin (5 mg/ml) (Sigma-Aldrich) for 6 hours at 4°C on a rotating platform. The cell lysates were then incubated with the beads coupled with antibodies overnight at 4°C. The next day, beads were washed with lysis buffer containing an additional 300 mM NaCl, and the immune complexes were eluted off the beads using loading buffer with boiling for 5 min and then subjected to Western blot analysis.
Immunofluorescence staining
Paraffin-embedded sections (5 μm) were subjected to staining. For Smyd2 staining, a polyclonal rabbit anti-Smyd2 antibody (GeneTex; 1:100 dilution) and an Alexa Fluor 488–conjugated donkey anti-rabbit immunoglobulin G (IgG) (H+L) secondary antibody (Thermo Fisher Scientific, 1:1000) were used. Kidney sections were counterstained by 4′,6-diamidino-2-phenylindole (DAPI). Images were analyzed with a Nikon TI2-E microscope.
Generation of anti-TubK40me3 and anti-TubK394me3 antibodies
The TubK40me3 antibody was generated in rabbit using trimethylated K40 peptide [Ac-GQMPSD(KMe3)TIGGGDC-amide] conjugated to keyhole limpet hemocyanin (KLH) as an immunogen, and the TubK394me3 antibody was generated in rabbit using trimethylated K394 peptide (Ac-WARLDH(KMe3)FDLMYAKC-amide) conjugated to KLH as an immunogen (Covance). Both antibodies were purified using serial columns coupled with unmethylated and trimethylated K40 or K394 peptides, respectively.
GST pull-down assays
GST-Smyd2 fusion protein–expressing constructs were generated in a pGEX-6p-1 (GE Healthcare) vector by cloning fragments of human Smyd2 complementary DNA (cDNA) (GenScript) into the Bam HI and Xho I sites via polymerase chain reaction (PCR) using the In-Fusion HD Cloning Kit (Clontech). GST-Smyd2 constructs were transformed to BL21 E. coli and induced using 0.2 mM isopropyl-β-d-thiogalactopyranoside (IPTG). Cells were collected by centrifugation and digested with lysozyme (0.5 mg/ml) (Thermo Fisher Scientific) and 250 U of benzonase (Millipore) on ice for 30 min in PBST (phosphate-buffered saline, 0.1% Tween 20) buffer. The cells were subsequently sonicated for 20 s on, 2 min off for three times to collect the soluble fractions. The GST-Smyd2 fusion proteins were purified using glutathione-agarose beads (Pierce), and expression of the constructs was analyzed on SDS-PAGE gels stained with Coomassie blue. In parallel, the purified GST-Smyd2 fusion proteins were incubated with 1 μg of recombinant α-tubulin (TUBA1A) protein (Origene). Following washing using PBST, the protein complexes were run on SDS-PAGE gels. Immunoblot analyses were performed using a mouse anti–α-tubulin antibody (Santa Cruz Biotechnology).
In vitro methylation assay
We made constructs for GST-SMYD2 and its SET domain deletion mutant, induced expression in the presence of IPTG, and purified the proteins by glutathione agarose beads. In vitro methylation assays were performed using 1 to 2 μg of recombinant proteins incubated with 1 μg of recombinant GST-SMYD2 and 2 μCi of 3H-AdoMet (PerkinElmer) in buffer containing 50 mM tris-HCl (pH 8.0), 10% glycerol, 20 mM KCl, 5 mM MgCl2, and 1 mM PMSF at room temperature overnight. The reaction mixture was resolved by SDS-PAGE followed by autoradiography.
Quantitative reverse transcription PCR
Total RNA was extracted using the RNeasy Plus Mini Kit (Qiagen). Total RNA (1 μg) was used for reverse transcription (RT) reactions in a 20-μl reaction to synthesize cDNA using the iScript cDNA Synthesis Kit (Bio-Rad). RNA expression profiles were analyzed by real-time PCR using iTaq SYBR Green Supermix with ROX (Bio-Rad) in an iCycler iQ real-time PCR detection system. The complete reactions were subjected to the following program of thermal cycling: 40 cycles of 10 s at 95°C and 20 s at 60°C. A melting curve was run after the PCR cycles, followed by a cooling step. Each sample was run in triplicate in each experiment, and each experiment was repeated three times. Expression levels of target genes were normalized to the expression level of actin.
RNA interference
The RNA oligonucleotides that specifically targeted human or mouse Smyd2, CDK4, and CDK6 were purchased from Santa Cruz Biotechnology Inc. The RNA oligonucleotides were transfected with DharmaFECT siRNA transfection reagent (Dharmacon). Twenty-four hours and 48 hours after transfection, cells were harvested and analyzed by Western blotting.
ChIP assay
ChIP assays were performed according to the published protocol (40). Chromatin DNA was subjected to immunoprecipitation with anti-SMYD2, anti–H3K4-monomethylation, anti–H3K4-dimethylation, and anti–H3K4-trimethylation antibodies, or anti–H3K36-monomethylation, anti–H3K36-dimethylation, and anti–H3K36-trimethylation antibodies, or normal rabbit IgG. After the DNA-protein cross-links were reversed, the recovered DNA was analyzed by PCR to detect the binding of SMYD2 and methylated H3K4 or H3K36 on the promoters of human CDK4, CDK6, and IFT20, respectively.
Transfection assays
HEK293T cells were cultured to confluence in DMEM containing 10% fetal calf serum (FCS; FetalClone III, Clontech). The control pACGFPC1 vector or SMYD2 constructs were transfected into cells using the Lipofectamine 2000 Kit (Invitrogen, Carlsbad, CA) according to the manufacturer’s recommendations. Transfected cells were maintained at 37°C for 24 or 48 hours. Cells were harvested and analyzed by Western blotting and quantitative RT-PCR (qRT-PCR).
Mouse strains
Smyd2flox/flox mice were provided by J. Sage (Department of Pediatrics and Genetics, Stanford University Medical Center, Stanford, CA). The Smyd2flox/flox mice developed normally in size and behavior and were fertile. All animal protocols were approved and conducted in accordance with Laboratory Animal Resources of Mayo Clinic and Institutional Animal Care and Use Committee regulations (protocol no. A00003756-18).
Statistics
All data are presented as means ± SEM. All statistical analyses were performed using SPSS Statistics 22 software. P values were calculated by two-tailed unpaired Student’s t test and one-way analysis of variance (ANOVA), and P < 0.05 was considered significant.
Supplementary Material
Acknowledgments
We are grateful to S. Somlo for providing cell lines PH2 and PN24 through the George M O’Brien Kidney Center at Yale University (NIH P30 DK079310). Funding: X.L. acknowledges support from NIH grants NIH R01 DK084097, NIH R01 DK126662, and NIH P30 DK106912 and from the PKD Foundation research grant. P.C.H. acknowledges support from NIH R01 DK058816, DK059597, and NIH P30 DK090728. J.P.C. acknowledges support from the PKD Foundation and the Kansas Research and Translation Core Center (P30 DK106912). Author contributions: L.X.L. performed most experiments and data analysis. J.X.Z., X.W., and H.Z. performed some of the experiments and data analysis. P.C.H. and J.P.C. assisted in data analysis and manuscript preparation. X.L. supervised the whole project, data analysis, and manuscript writing. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/44/eabb3154/DC1
REFERENCES AND NOTES
- 1.Sherr C. J., Beach D., Shapiro G. I., Targeting CDK4 and CDK6: From discovery to therapy. Cancer Discov. 6, 353–367 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hochegger H., Takeda S., Hunt T., Cyclin-dependent kinases and cell-cycle transitions: Does one fit all? Nat. Rev. Mol. Cell Biol. 9, 910–916 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Ewen M. E., Sluss H. K., Sherr C. J., Matsushime H., Kato J. Y., Livingston D. M., Functional interactions of the retinoblastoma protein with mammalian D-type cyclins. Cell 73, 487–497 (1993). [DOI] [PubMed] [Google Scholar]
- 4.Anders L., Ke N., Hydbring P., Choi Y. J., Widlund H. R., Chick J. M., Zhai H., Vidal M., Gygi S. P., Braun P., Sicinski P., A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell 20, 620–634 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Plotnikova O. V., Pugacheva E. N., Golemis E. A., Primary cilia and the cell cycle. Methods Cell Biol. 94, 137–160 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sorokin S., Centrioles and the formation of rudimentary cilia by fibroblasts and smooth muscle cells. J. Cell Biol. 15, 363–377 (1962). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nigg E. A., Stearns T., The centrosome cycle: Centriole biogenesis, duplication and inherent asymmetries. Nat. Cell Biol. 13, 1154–1160 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singla V., Reiter J. F., The primary cilium as the cell’s antenna: Signaling at a sensory organelle. Science 313, 629–633 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Brueckner M., Heterotaxia, congenital heart disease, and primary ciliary dyskinesia. Circulation 115, 2793–2795 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Harris P. C., Torres V. E., Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J. Clin. Invest. 124, 2315–2324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang H. S., Dean D. C., Rb-mediated chromatin structure regulation and transcriptional repression. Oncogene 20, 3134–3138 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Jenuwein T., Allis C. D., Translating the histone code. Science 293, 1074–1080 (2001). [DOI] [PubMed] [Google Scholar]
- 13.Janke C., The tubulin code: Molecular components, readout mechanisms, and functions. J. Cell Biol. 206, 461–472 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li L. X., Fan L. X., Zhou J. X., Grantham J. J., Calvet J. P., Sage J., Li X., Lysine methyltransferase SMYD2 promotes cyst growth in autosomal dominant polycystic kidney disease. J. Clin. Invest. 127, 2751–2764 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neganova I., Lako M., G1 to S phase cell cycle transition in somatic and embryonic stem cells. J. Anat. 213, 30–44 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shao X. L., Somlo S., Igarashi P., Epithelial-specific Cre/lox recombination in the developing kidney and genitourinary tract. J. Am. Soc. Nephrol. 13, 1837–1846 (2002). [DOI] [PubMed] [Google Scholar]
- 17.Magiera M. M., Janke C., Post-translational modifications of tubulin. Curr. Biol. 24, R351–R354 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Jenkins B. V., Saunders H. A. J., Record H. L., Johnson-Schlitz D. M., Wildonger J., Effects of mutating α-tubulin lysine 40 on sensory dendrite development. J. Cell Sci. 130, 4120–4131 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu N., Xiong Y., Ren Y., Zhang L., He X., Wang X., Liu M., Li D., Shui W., Zhou J., Proteomic profiling and functional characterization of multiple post-translational modifications of tubulin. J. Proteome Res. 14, 3292–3304 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Park I. Y., Powell R. T., Tripathi D. N., Dere R., Ho T. H., Blasius T. L., Chiang Y.-C., Davis I. J., Fahey C. C., Hacker K. E., Verhey K. J., Bedford M. T., Jonasch E., Rathmell W. K., Walker C. L., Dual chromatin and cytoskeletal remodeling by SETD2. Cell 166, 950–962 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dean J. L., Thangavel C., McClendon A. K., Reed C. A., Knudsen E. S., Therapeutic CDK4/6 inhibition in breast cancer: Key mechanisms of response and failure. Oncogene 29, 4018–4032 (2010). [DOI] [PubMed] [Google Scholar]
- 22.Jiang J., Hui C. C., Hedgehog signaling in development and cancer. Dev. Cell 15, 801–812 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rohatgi R., Milenkovic L., Scott M. P., Patched1 regulates hedgehog signaling at the primary cilium. Science 317, 372–376 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Cohen M., Kicheva A., Ribeiro A., Blassberg R., Page K. M., Barnes C. P., Briscoe J., Ptch1 and Gli regulate Shh signalling dynamics via multiple mechanisms. Nat. Commun. 6, 6709 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takahashi Y., Rayman J. B., Dynlacht B. D., Analysis of promoter binding by the E2F and pRB families in vivo: Distinct E2F proteins mediate activation and repression. Genes Dev. 14, 804–816 (2000). [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou X., Fan L. X., Sweeney W. E. Jr., Denu J. M., Avner E. D., Li X., Sirtuin 1 inhibition delays cyst formation in autosomal-dominant polycystic kidney disease. J. Clin. Invest. 123, 3084–3098 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lundberg A. S., Weinberg R. A., Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol. Cell. Biol. 18, 753–761 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Firat-Karalar E. N., Stearns T., The centriole duplication cycle. Philos. Trans. R Soc. Lond. B Biol. 369, 20130460 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mirvis M., Stearns T., Nelson W. J., Cilium structure, assembly, and disassembly regulated by the cytoskeleton. Biochem. J. 475, 2329–2353 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pugacheva E. N., Jablonski S. A., Hartman T. R., Henske E. P., Golemis E. A., HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell 129, 1351–1363 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mizuno K., Sloboda R. D., Protein arginine methyltransferases interact with intraflagellar transport particles and change location during flagellar growth and resorption. Mol. Biol. Cell 28, 1208–1222 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu Z., Liang Y., He W., Pan J., Cilia disassembly with two distinct phases of regulation. Cell Rep. 10, 1803–1810 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Zhao Q., Li S., Shao S., Wang Z., Pan J., FLS2 is a CDK-like kinase that directly binds IFT70 and is required for proper ciliary disassembly in Chlamydomonas. PLOS Genet. 16, e1008561 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keeling J., Tsiokas L., Maskey D., Cellular mechanisms of ciliary length control. Cell 5, 6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Follit J. A., Tuft R. A., Fogarty K. E., Pazour G. J., The intraflagellar transport protein IFT20 is associated with the Golgi complex and is required for cilia assembly. Mol. Biol. Cell 17, 3781–3792 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma M., Tian X., Igarashi P., Pazour G. J., Somlo S., Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat. Genet. 45, 1004–1012 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Silva L. M., Jacobs D. T., Allard B. A., Fields T. A., Sharma M., Wallace D. P., Tran P. V., Inhibition of Hedgehog signaling suppresses proliferation and microcyst formation of human autosomal dominant polycystic kidney disease cells. Sci. Rep. 8, 4985 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hopp K., Ward C. J., Hommerding C. J., Nasr S. H., Tuan H.-F., Gainullin V. G., Rossetti S., Torres V. E., Harris P. C., Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J. Clin. Invest. 122, 4257–4273 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shibazaki S., Yu Z., Nishio S., Tian X., Thomson R. B., Mitobe M., Louvi A., Velazquez H., Ishibe S., Cantley L. G., Igarashi P., Somlo S., Cyst formation and activation of the extracellular regulated kinase pathway after kidney specific inactivation of Pkd1. Hum. Mol. Genet. 17, 1505–1516 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmidt D., Wilson M. D., Spyrou C., Brown G. D., Hadfield J., Odom D. T., ChIP-seq: Using high-throughput sequencing to discover protein-DNA interactions. Methods 48, 240–248 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/44/eabb3154/DC1