

Sex-specific modulation of a meiotic DNA damage checkpoint limits the requirement for PRDM9 in mammalian fertility.
Abstract
In many mammals, genomic sites for recombination are determined by the histone methyltransferase PRMD9. Some mouse strains lacking PRDM9 are infertile, but instances of fertility or semifertility in the absence of PRDM9 have been reported in mice, canines, and a human female. Such findings raise the question of how the loss of PRDM9 is circumvented to maintain fertility. We show that genetic background and sex-specific modifiers can obviate the requirement for PRDM9 in mice. Specifically, the meiotic DNA damage checkpoint protein CHK2 acts as a modifier allowing female-specific fertility in the absence of PRDM9. We also report that, in the absence of PRDM9, a PRDM9-independent recombination system is compatible with female meiosis and fertility, suggesting sex-specific regulation of meiotic recombination, a finding with implications for speciation.
INTRODUCTION
Meiotic recombination generates genetic diversity and ensures the accuracy of chromosome transmission to the next generation. In many organisms, recombination occurs preferentially at sites in the genome known as recombination hotspots (1). In a subset of mammals, including mice and humans, the positions of hotspots are determined by the specialized histone methyltransferase PRDM9—a meiosis-specific, DNA binding zinc finger protein that uniquely trimethylates both lysines 4 and 36 of histone H3 (2–6). This H3K4me3/H3K36me3 double-positive signature is thought to preferentially facilitate recombination at these sites, to the exclusion of other functional elements (1, 6, 7). In the absence of PRDM9, meiotic double-strand breaks (DSBs) occur in normal numbers but are localized to functional elements enriched for H3K4me3, such as promoters (7). In Prdm9-deficient C57BL/6 mice (henceforth B6.Prdm9−/−), repair of these ectopic DSBs is impaired, leading to prophase I meiotic arrest of both male and female germ cells, with consequent infertility (8). In human males, several point mutations in PRDM9 are associated with nonobstructive azoospermia (9, 10). Together, these observations suggest that PRDM9-dependent recombination is required for successful reproduction, at least in mice and humans.
However, intriguing exceptions have been reported in both mice and a human female. Prdm9-deficient PWD/PhJ (henceforth PWD.Prdm9−/−) male mice have a normal meiotic prophase I, although they are infertile due to low sperm number (PWD.Prdm9−/− females are also infertile) (11). Further, some Prdm9−/− male mice with mixed genetic backgrounds are fertile, suggesting background-dependent genetic modifiers of the phenotype (11). A report of a single fertile PRDM9-null woman shows that PRDM9 can be dispensable for fertility in human females (12). Although Prdm9 is a pseudogene in the canine lineage, both male and female canids reproduce successfully (13, 14). Together, these findings imply that the requirement for PRDM9 in mammalian recombination is complex.
These findings also suggest that a functional Prdm9-independent recombination initiation pathway must exist even in species using PRDM9; elucidating its mechanisms will help decipher the genetic complexity that is likely involved. In male mice, recombination hotspots on autosomes are PRDM9-dependent, but the recombination hotspots within the pseudoautosomal region of the sex chromosomes are activated by a PRDM9-independent recombination initiation pathway (7). This suggests that both PRDM9-dependent and PRDM9-independent recombination pathways are active in tandem during mammalian meiosis and might fulfill functions necessary to bypass reproductive bottlenecks due to dysfunctional or absent PRDM9. Although a PRDM9-independent recombination mechanism exists in male mice, it has been unknown whether a similar pathway functions in females. In this study, we investigated oogenesis and the effect of genetic background to determine (i) whether there is a sex-specific requirement for PRDM9-dependent hotspot activation and (ii) the possible mechanisms allowing organisms to circumvent the loss of PRDM9 and maintain fertility.
RESULTS
Sex-specific requirement for PRDM9-dependent histone methyltransferase activity in mice
We used CRISPR-Cas9 gene editing to create a point mutation in the PR/SET domain of Prdm9 (Glu365Pro, henceforth Prdm9EP) in C57BL/6J (B6) mice (fig. S1, A and B). To determine the effect of this mutation, we performed chromatin immunoprecipitation with massively parallel DNA sequencing (ChIP-seq) for H3K4me3 and the recombinase protein DMC1 (DNA Meiotic Recombinase 1) that binds specifically to the single-stranded ends of resected meiotic DSBs (Fig. 1, A and B, and fig. S1, C to F), on B6 and B6.Prdm9EP/EP spermatocytes. We defined hotspots as PRDM9-dependent H3K4me3 sites in the B6 genetic background based on a previously published study from our lab (15) (see Materials and Methods for details) and compared the number and distribution of H3K4me3 and DMC1 peaks in B6.Prdm9EP/EP spermatocytes to those in wild-type B6 spermatocytes.
Fig. 1. Prdm9EP alters the Prdm9-dependent H3K4me3 and meiotic DSB landscape in spermatocytes.
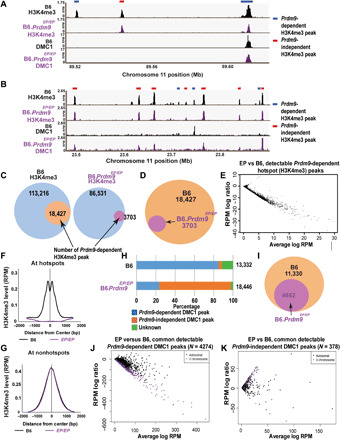
(A and B) Genome browser snapshots of H3K4me3 and DMC1 ChIP-seq peaks in wild-type and Prdm9EP/EP (Glu365Pro mutation) spermatocytes. (A) One strong and one weaker PRDM9-dependent hotspot are shown, together with one PRDM9-independent H3K4me3 peak. (B) Several PRDM9-dependent and PRDM9-independent H3K4me3 peaks are shown. Note the shift in DMC1 peaks to PRDM9-independent H3K4me3 peaks in Prdm9EP/EP spermatocytes. (C) Venn diagrams showing the number of detectable PRDM9-dependent H3K4me3 peaks in wild-type (B6) and Prdm9EP/EP spermatocytes. (D) Venn diagram directly comparing the number of detectable PRDM9-dependent H3K4me3 peaks in wild-type and Prdm9EP/EP spermatocytes. (E) MA plot comparing Prdm9EP/EP versus wild-type signal at PRDM9-dependent H3K4me3 peaks that were detectable in Prdm9EP/EP spermatocytes (n = 3703). (F) Aggregation plot showing normalized average signal intensity [reads per million (RPM)] at known PRDM9-dependent H3K4me3 ChIP-seq peaks (n = 18,838) in wild-type and Prdm9EP/EP spermatocytes. (G) Aggregation plot showing normalized average signal intensity (reads per million, RPM) at common nonhotspot H3K4me3 ChIP-seq peaks (n = 79,043) in wild-type and Prdm9EP/EP spermatocytes. (H) Distribution of DMC1 peaks in wild-type and Prdm9EP/EP spermatocytes. (I) Venn diagram comparing the number of PRDM9-dependent DMC1 peaks in wild-type and Prdm9EP/EP spermatocytes. (J and K) MA plots comparing Prdm9EP/EP versus wild-type DMC1 signal at (J) common PRDM9-dependent DMC1 peaks (n = 4724) and (K) common PRDM9-independent DMC1 peaks (n = 378). Black points represent autosomal peaks, and purple points represent peaks on the X chromosome. EP, Glu365Pro change in amino acid sequence.
We found the Prdm9EP mutation to be a catalytic hypomorph, as it severely reduced but did not entirely abolish the methyltransferase activity of Prdm9 in vivo. The mutation did not affect Prdm9 expression (fig. S1B) but altered the epigenetic landscape at hotspots markedly. In marked contrast to B6 spermatocytes, most hotspots in B6.Prdm9EP/EP spermatocytes lacked a detectable H3K4me3 peak, although residual H3K4me3 enrichment was present at stronger hotspots (Fig. 1, A and B). Quantitatively, there was a fivefold reduction in the number of detectable Prdm9-dependent H3K4me3 peaks in spermatocytes from B6.Prdm9EP/EP males (Fig. 1, C and D), compared to B6 spermatocytes. The intensity of detectable PRDM9-dependent peaks was also sharply reduced relative to B6 (Fig. 1E); the mean normalized read count at these peaks (n = 3703) was reduced more than threefold to 1.29 reads per million (RPM), compared to 4.45 RPM in B6 (P = 1.5 × 10−268, Student’s t test). PRDM9-dependent H3K4me3 signal was so low overall in B6.Prdm9EP/EP spermatocytes that it did not register in aggregation plots of H3K4me3 enrichment at all known PRDM9-dependent sites (Fig. 1F), confirming a significant alteration in PRDM9-dependent methyltransferase activity in B6.Prdm9EP/EP spermatocytes. By contrast, PRDM9-independent H3K4me3 peaks (an internal control) were unaffected (Fig. 1, A, B, and G), showing that the effect of Prdm9EP was restricted to PRDM9-dependent peaks.
The effect of Prdm9EP on the meiotic DSB landscape, as measured by ChIP-seq for DMC1, was equally marked. A quarter of meiotic DSBs occurred at PRDM9-dependent hotspots, while nearly three quarters occurred at PRDM9-independent “default” sites, which are used exclusively in Prdm9-null spermatocytes (7). This was markedly different from B6 spermatocytes, in which about 85% of DMC1 peaks occurred at hotspots and only ~4% occurred at default sites (Fig. 1, A, B, H, and I). These results represented a marked improvement over Prdm9-null spermatocytes, which make no DSBs at hotspots (7). The average intensity of DMC1 signal at hotspots was also reduced in B6.Prdm9EP/EP spermatocytes relative to B6 spermatocytes: The mean normalized read count at common PRDM9-dependent DMC1 peaks (n = 4724) was reduced more than threefold to 12.8 RPM, relative to 41.3 RPM in B6 (P = 1.97 × 10−246, Student’s t test). When DMC1 signals at common peaks between B6 and B6.Prdm9EP/EP were compared directly, we observed a distinct contrast between hotspots and default sites (Fig. 1, J and K). While DMC1 signal tended to be higher at default sites in B6.Prdm9EP/EP spermatocytes relative to B6 spermatocytes, it was much lower at hotspots, especially on the X chromosome (Fig. 1J). This likely implies a lower rate of DSB initiation at hotspots in B6.Prdm9EP/EP, which would be expected given the drastically reduced catalytic activity of PRDM9EP. However, we cannot rule out the possibility that DSB repair is affected as well. In summary, the residual catalytic activity of Prdm9EP is sufficient to induce DSBs at a subset of PRDM9-dependent sites, but not sufficient to rescue meiotic arrest, in spermatocytes.
The Prdm9EP mutation also yielded an unanticipated phenotype: sexual dimorphism (Fig. 2, A to C, and fig. S1G). Homozygous B6.Prdm9EP/EP males exhibited the expected meiotic arrest and infertility, phenocopying both the Prdm9-null condition (8) and a recently reported methyltransferase-dead Prdm9 allele (fig. S1G) (16, 17). In contrast to the infertile males, all tested homozygous B6.Prdm9EP/EP females produced at least one—and up to five—litters when mated to B6 males over a period of 6 months (Fig. 2, C and D, and tables S1 and S2). The offspring were grossly normal and healthy, and those tested, males and females, were fertile. Compared to wild-type B6 controls, ovaries from prepubescent B6.Prdm9EP/EP females were smaller but contained all developmental stages including primary follicles (Fig. 2, A and B, and fig. S1G). Quantification of follicles revealed lower numbers of follicles in B6.Prdm9EP/EP ovaries, relative to wild-type and heterozygous ovaries (Fig. 2B). B6.Prdm9EP/EP females are therefore subfertile relative to wild type but escape the unconditional sterility characteristic of Prdm9-null B6 females (8).
Fig. 2. Females homozygous for Prdm9EP are fertile.
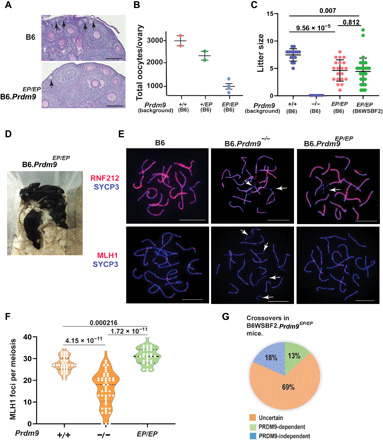
(A) Periodic acid–Schiff (PAS)–stained sections from 3-week postpartum ovaries in wild-type and Prdm9EP/EP mice in the B6 genetic background. Arrows show primary follicles. Scale bars, 100 μm. (B) Oocytes per ovary in wild-type, heterozygous, and Prdm9EP/EP females (error bars, SEM). P values were not calculated as N = 2 is not sufficient for reaching statistical significance. (C) Litter sizes in wild-type, Prdm9−/−, and Prdm9EP/EP female mice (error bars, SEM). P values were calculated using the Mann-Whitney U test with Bonferroni correction for multiple testing. (D) Pups produced by Prdm9EP/EP female mice. (E) Coimmunolabeling detection of RNF212 foci (red, top row), MLH1 (red, bottom row), and SYCP3 (blue) in pachytene oocyte chromatin spreads in wild-type and mutant females. White arrows highlight unsynapsed regions of chromosomes. Scale bars, 10 μm. (F) Violin plot with dots showing numbers of MLH1 foci per meiotic oocyte (error bars, SEM), in wild-type, Prdm9−/−, and Prdm9EP/EP mice. P values were calculated using the Mann-Whitney U test with Bonferroni correction for multiple testing. (G) Diagram showing proportions of PRDM9-dependent and PRDM9-independent crossovers in progeny of B6WSBF2.Prdm9EP/EP females (n = 94). Photo credit: Natalie R. Powers and Tanmoy Bhattacharyya, The Jackson Laboratory.
To determine whether B6.Prdm9EP/EP oocytes exhibit normal cytological features of recombination, we performed immunofluorescence staining on meiotic chromatin spreads from the ovaries of newborn pups (postnatal day 0 or P0), using antibodies against the crossover regulator RNF212 (ring finger protein 212), the crossover marker MLH1 (mutL homolog 1), and the synaptonemal complex (SC) protein SYCP3 (Fig. 2E). In B6. Prdm9EP/EP, ~65% of pachytene oocytes exhibited homologous synapsis of all chromosomes stained with RNF212 and at least one MLH1 focus on each chromosome, in comparison to ~91% of B6 pachytene oocytes (Fig. 2E). In distinct contrast, only ~4% of B6.Prdm9−/− oocytes had all homologous chromosomes synapsed and with at least one MLH1 focus per chromosome (Fig. 2F). Most (~79%) B6.Prdm9−/− pachytene oocytes showed widespread asynapsis and few MLH1 foci on synapsed chromosomes (Fig. 2, E and F, and table S3). The average number of MLH1 foci was significantly higher in B6.Prdm9EP/EP oocytes than in wild-type B6 control oocytes (P = 0.000216, Mann-Whitney U test; Fig. 2, E and F, and table S3). Future work will determine whether this increase in MLH1 count in B6.Prdm9EP/EP oocytes reflects altered crossover rates due to DSBs at a large number of PRDM9-independent sites in B6.Prdm9EP/EP oocytes, providing both PRDM9-dependent and PRDM9-independent DSB sites as candidates for crossover sites, or whether there is some other reason for the increase. Together, these results show that the effects of the Prdm9EP mutation are tolerated better in female meiosis than in male meiosis, with sufficient recombination to ensure at least some viable eggs despite the severely reduced catalytic activity. This finding implies that the requirement for PRDM9-dependent hotspot activation in mammalian reproduction is subject to sex-specific control.
We next investigated whether the absence of an efficient PRDM9-dependent hotspot activation mechanism promotes recombination at PRDM9-independent sites. To determine where crossover sites occur in B6.Prdm9EP/EP females, we used the strategy of outcrossing this mutant to another strain and mapping detectable crossovers in the progeny of Prdm9EP/EP females. We chose WSB/EiJ as the outcross strain because it is highly divergent from the classical laboratory strains but still a member of the Mus musculus (M. m.) domesticus subspecies; it could therefore give us maximum resolution while avoiding fertility issues or unnecessary genetic complexity associated with intersubspecific crosses. We outcrossed B6.Prdm9+/EP mice to WSB/EiJ (WSB) mice, producing male and female B6WSBF1 progeny heterozygous for Prdm9EP. We then intercrossed these F1 mice to produce B6WSBF2.Prdm9EP/EP mice, homozygous for the Prdm9EP mutation. All B6WSBF2.Prdm9EP/EP males were infertile with low testis weight (table S4). We backcrossed three B6WSBF2.Prdm9EP/EP females to wild-type B6 males and genotyped both the F2 females and 20 of their progeny with a genome-wide single-nucleotide polymorphism (SNP) array (18). This allowed us to map some of the crossovers that had occurred in the oocytes giving rise to these progeny. Only maternal crossovers were detectable in these progeny, as their B6 fathers were homozygous at every locus. We limited our focus to the 94 informative crossovers that occurred in the Prdm9EP/EP mothers; crossovers that had occurred in the B6WSBF1 generation were excluded. Of these, 26 occurred in intervals with no known PRDM9-dependent H3K4me3 peak (27.7%) (15). To exclude the possibility that these 26 putative PRDM9-independent crossovers occurred at heretofore unrecognized PRDM9 binding sites on the WSB chromosomes, we searched the B6 and WSB genomic sequences in the crossover regions for PRDM9Dom2 binding motifs (19). Prdm9Dom2 is the endogenous Prdm9 allele in the B6 strain, of which Prdm9EP is a mutant version. Only hotspots defined by PRDM9Dom2 will therefore be bound in Prdm9EP/EP homozygotes. Of these 26 intervals, 17 (18.1% of total) had no apparent PRDM9Dom2 binding motifs in the WSB sequence when compared to the B6 sequence and are considered PRDM9-independent. Of the 94 total crossovers, 12 (12.8%) could be classified as clearly PRDM9-dependent based on very high-resolution crossover intervals containing a known hotspot (Fig. 2G). Notably, the B6WSBF2.Prdm9EP/EP females produced more offspring than B6.Prdm9EP/EP females (average offspring per female per month 4.55 versus 2.69, respectively; table S1), revealing an effect of genetic modifiers from the WSB genetic background or hybrid vigor.
While the small scale and limited resolution of most of the informative crossovers constrain any quantitative conclusions, this analysis does show directly that PRDM9-independent meiotic DSBs yield recombinant chromosomes in mice. These PRDM9-independent crossovers occurred in oocytes that gave rise to grossly normal and healthy animals, further supporting the idea that these PRDM9-independent recombination events support normal chromosome segregation. In addition, the clearly PRDM9-dependent crossovers demonstrate that the residual methyltransferase activity of PRDM9EP is sufficient to promote PRDM9-dependent crossovers in oocytes, as it is for PRDM9-dependent meiotic DSBs in spermatocytes.
In summary, a severe catalytic hypomorph of Prdm9 allows for successful meiosis and fertility in female—but not male—M. m. domesticus mice. Meiotic progression is presumably facilitated by the activation of a normally quiescent PRDM9-independent recombination pathway in response to limited PRDM9 activity.
Genetic background– and sex-limited requirement for PRDM9 in mice
The human case of a fertile PRDM9-null female (12), and the higher fertility in the B6WSBF2.Prdm9EP/EP females, led us to examine the impact of different genetic backgrounds on meiotic recombination and fertility in the absence of Prdm9. To this end, we introgressed the B6.Prdm9-null allele onto the CAST/EiJ and C3H/HeJ inbred backgrounds for five generations (~5% residual heterozygosity, henceforth CAST.Prdm9−/− and C3H.Prdm9−/−, respectively). These strains derive from distinct house mouse subspecies (M. m. castaneus and M. m. domesticus, respectively) that diverged ~0.5 million years ago (20, 21). We chose these strains for the contrast they provide: C3H/HeJ is a classical laboratory strain and closely related to B6, whereas CAST/EiJ is a wild-derived inbred strain of M. m. castaneus, a divergent subspecies. There is no Prdm9-dependent hybrid sterility in B6CASTF1 hybrids, which might influence interpretation of the phenotype (22). Notably, CAST.Prdm9−/− females have functional oocytes and are fertile, producing grossly healthy, fertile offspring, while C3H.Prdm9−/− females, like B6.Prdm9−/− females, are infertile (Fig. 3, A to C, and table S2). To determine the recombination status in P0 CAST.Prdm9−/− oocytes, we immunostained CAST.Prdm9−/− ovarian meiotic spreads with antibodies against MLH1 and SYCP3 (Fig. 3D and table S3). We observed normal synapsis and MLH1 frequency in ~96% of CAST.Prdm9−/− pachytene oocytes, although there was a significant reduction in the average number of MLH1 foci in these oocytes compared to the CAST.Prdm9+/+ control oocytes (P = 0.02149, Mann-Whitney U test; Fig. 3, D and E, and table S3). This might be due to alteration of DSB sites or recombination rate in the absence of PRDM9 in the CAST genetic background. B6CASTF1.Prdm9−/− and C3HCASTF1.Prdm9−/− females were fertile, while B6C3HF1.Prdm9−/− females were infertile (Fig. 3C and table S2), indicating the presence of one or more dominant modifiers in the CAST genetic background that abrogate the requirement for PRDM9 in oocytes for fertility. All Prdm9-deficient males of the genetic backgrounds we tested— CAST.Prdm9−/−, B6.Prdm9−/−, and C3H.Prdm9−/−—exhibited meiotic arrest (fig. S2A) and failed to produce live-born offspring. Although these males are infertile, we did observe round spermatids and elongating spermatids in histological sections of some CAST.Prdm9−/− testes (fig. S2, A to C). This suggests a rescue of meiotic arrest in some spermatocytes, as previously observed in PWD.Prdm9−/− males (11). In conclusion, the requirement for PRDM9 in recombination initiation and fertility in mice is sexually dimorphic and is modulated by background-specific genetic modifiers that lead to fertility despite PRDM9 deficiency.
Fig. 3. CAST/EiJ Prdm9-null females are fertile.
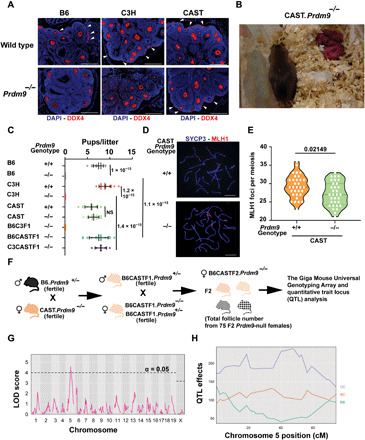
(A) Immunofluorescence staining of histology sections from 3-week postpartum ovaries in wild-type and Prdm9−/− females of different genetic backgrounds. DDX4 (red, also known as MVH) marks oocytes. DNA is stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). Arrowheads indicate primordial follicles. Scale bars, 100 μm. Note the oocyte depletion in Prdm9−/− mice in the B6 and C3H genetic backgrounds, in contrast to survival of oocytes in wild-type and Prdm9−/− mice in the CAST genetic background. (B) Pups produced by CAST.Prdm9−/− female mice. (C) Reproductive productivity of wild-type and Prdm9−/− females in different genetic backgrounds. P values were calculated using the Mann-Whitney U test with Bonferroni correction for multiple testing. NS, not significant. (D) Coimmunolabeling detection of MLH1 foci (red) in pachytene oocyte chromatin spreads, also labeled with antibody against SYCP3 (blue) in wild-type and Prdm9−/− CAST females. Scale bars, 10 μm. (E) Violin plot with dots showing numbers of MLH1 foci per oocyte (error bars, SEM). The genotypes of mice tested are indicated below the graphs. P values were calculated using the Mann-Whitney U test with Bonferroni correction for multiple testing. (F) Scheme of construction of B6CASTF2.Prdm9−/− female cohorts used for SNP array genotyping [Giga Mouse Universal Genotyping Array (GigaMUGA)]. Seventy-five F2 females were generated by crossing B6CASTF1.Prdm9−/− and B6CASTF1.Prdm9+/− females with B6CASTF1.Prdm9+/− males. Eight-week-old B6CASTF2.Prdm9−/− females were phenotyped for oocyte quantity and genotyped. (G) Quantitative trait locus (QTL) mapping, with the total number of ovarian follicles (refer to Materials and Methods for details) per B6CASTF2.Prdm9−/− female as the phenotype. QTL (pink) reached significance on chromosome 5, with a peak at ~100.4 Mb (1.5 LOD drop: 72.9 to 127.65 Mb, mm10). (H) Genotype effects of the chromosome 5 QTL. CAST alleles affect oocyte number positively, as highlighted by CC (violet) and BC (orange) lines, relative to B6 homozygotes (BB, green). CC, homozygous CAST; BB, homozygous B6; BC, heterozygous CAST/B6. Photo credit: Catherine Brunton and Tanmoy Bhattacharyya, The Jackson Laboratory.
To identify the CAST modifier(s) that allow for Prdm9-null female fertility, we generated 75 B6CASTF2.Prdm9−/− female mice to perform quantitative trait locus (QTL) mapping, with the total number of ovarian follicles per female as the phenotype [see Fig. 3 (F and H) and Materials and Methods for details]. We chose this phenotype because the number of oocytes in mature ovaries correlates with the efficacy of meiotic DSB repair and oocyte survival (23). The analysis yielded a significant QTL on chromosome 5, with a peak at ~100.4 Mb [1.5 LOD (logarithm of the odds) score drop: 72.9 to 127.65 Mb, permutation-derived significance threshold calculated at α = 0.05] (Fig. 3, G and H, and fig. S2D). Intriguingly, this QTL contains two critical meiotic genes: ring finger protein 212 (Rnf212 at 108.7 Mb) and checkpoint kinase 2 (Chk2 at 110.8 Mb). The success of the meiotic recombination process depends on the efficient repair of meiotic DSBs and crossover formation, while oocyte survival depends on successful passage through a checkpoint that monitors DNA damage. The roles of RNF212 and CHK2 in DNA damage surveillance in oocytes nominate them as compelling candidate genetic modifiers of PRMD9 (23, 24). RNF212 is a SUMO (Small Ubiquitin-like Modifier) ligase essential for crossover formation and oocyte quality control (24). It has been reported that localization of RNF212 to DSB sites acts as a “memory” of unrepaired DSBs, thus promoting apoptosis of defective oocytes during the diplotene to dictyate meiotic substage transition (24). Loss of Rnf212 leads to persistent DSBs and synapsis defects, and Rnf212 knockout females are infertile (24). The other candidate gene, Chk2, encodes a meiotic checkpoint kinase responsible for DNA damage surveillance in oocytes (23). Ablation of CHK2 prevents oocyte elimination in response to both radiation-induced DNA damage and persistent meiotic DSBs due to genetic mutation of Trip13 (23). Both the Rnf212 and Chk2 genomic sequences are well conserved between B6 and CAST mice (~99% sequence identity; fig. S3, A and B); however, regulatory variants outside the genes themselves may play a critical role. To further elucidate the mechanism behind this QTL, we examined Rnf212 and Chk2 experimentally for their potential roles as genetic modifiers of Prdm9.
Modulation of a meiotic DNA damage checkpoint leads to female-limited fertility in Prdm9-null B6 mice
The first candidate modifier gene we considered was Rnf212. In meiotic spreads, RNF212 protein expression and colocalization patterns were similar between CAST.Prdm9−/− meiotic oocytes and CAST.Prdm9+/+ oocytes (fig. S4). As mentioned above, crossover formation in CAST.Prdm9−/− meiotic oocytes is normal (Fig. 3, D and E), suggesting that the role of RNF212 in crossover formation in CAST.Prdm9−/− remains intact. While the oocyte count of B6.Rnf212−/− females is normal, these mice are infertile due to the absence of crossovers (24). RNF212 deficiency promotes survival of oocytes with genetic and radiation-induced DNA damage, suggesting an additional role for RNF212 as a proapoptotic cell cycle regulator that promotes elimination of defective oocytes (24). In CAST.Prdm9−/− females, delayed activation of RNF212 in this capacity could conceivably promote the survival of oocytes with unrepaired DSBs while still fulfilling its role in crossover formation. Further investigation is needed to understand the exact role of RNF212 in PRDM9-dependent and PRDM9-independent meiotic recombination in CAST oocytes. However, because RNF212 expression and RNF212 colocalization with the SC protein SYCP3 (fig.S4) are similar in Prdm9+/+ and Prdm9−/− CAST oocytes, we did not pursue it further, considering that additional validation would require in-depth sequencing, genetic models, and analysis of functional domains beyond the scope of the present study.
We next turned our attention to Chk2, which encodes a proapoptotic DNA damage surveillance kinase whose ablation is known to allow survival of oocytes with persistent DNA damage (23). We speculated that ablation of Chk2 might also allow for the survival of Prdm9-deficient oocytes. To determine directly whether Prdm9-null oocytes complete meiosis in the face of Chk2 deficiency, we generated Prdm9−/−Chk2−/− double knockout females in the B6 genetic background (henceforth, B6.Prdm9−/−Chk2−/−). Among P0 oocytes from B6.Prdm9−/− single mutants, most exhibited widespread asynapsis and persistent unrepaired DSBs, revealed by pervasive BRCA1 (breast cancer 1 protein), γH2AFX (phosphorylated form of H2A histone family member X), and IHO1 (interactor of HORMAD1 1) signals (figs. S5, A to D, and S6A). Only ~4% of these P0 oocytes had fully synapsed chromosomes stained with RNF212, MLH1, and MLH3 foci (markers of mature crossovers) (Fig. 4, A to D, fig. S5D, and table S3). This phenotype was substantially improved in B6.Prdm9−/−Chk2−/− double-mutant oocytes, with a significant increase in the number of oocytes with fully synapsed chromosomes devoid of BRCA1, γH2AFX, and IHO1 (~26%; figs. S5, E to H, and S6A). We also observed an approximate threefold increase in the number of dictyate oocytes in P0 B6. Prdm9−/−Chk2−/− double-mutant females, compared to B6.Prdm9−/− females (figs. S5, E to H, and S6B). Only a small number of growing follicles survived in B6.Prdm9−/− females (Fig. 5, A and C). We isolated metaphase I (MI) oocytes from young B6.Prdm9−/− and B6.Prdm9−/−Chk2−/− females and noted a significant increase (~25%) in the number of metaphase oocytes with normal chiasmata between the homologous chromosomes in B6.Prdm9−/−Chk2−/− females relative to B6.Prdm9−/− females (Fig. 4, E and F). We conclude that CHK2 eliminated oocytes that failed to repair meiotic DSBs, but oocytes in which recombination was completed were not eliminated and progressed through folliculogenesis. Although ~4% of B6.Prdm9−/− oocytes at P0 had a full complement of crossovers, this is probably not sufficient for fertility, especially given further oocyte elimination after P0 during follicle formation and atresia. We propose that with the removal of Chk2, more Prdm9−/− oocytes escape the meiotic DNA damage checkpoint and subsequently complete DSB repair, resulting in a higher oocyte reserve.
Fig. 4. Chk2 ablation rescues meiosis in Prdm9-null females in the B6 genetic background.
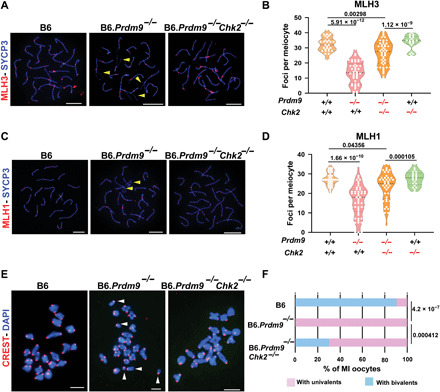
(A and C) Coimmunolabeling detection of MLH3 foci (red, A), MLH1 (red, C), and SYCP3 (blue, both A and C) in pachytene oocyte chromatin spreads from wild-type and mutant females. Yellow arrowheads highlight unsynapsed regions of chromosomes. Scale bars, 10 μm. (B and D) Violin plots with dots showing numbers of MLH3 (B) and MLH1 (D) foci per meiotic oocyte (error bars, SEM). The genotypes of mice tested are indicated below the graphs. P values were calculated using the Mann-Whitney U test with Bonferroni correction for multiple testing. (E and F) Chromosome configuration in meiosis MI analyzed by chromosome spreads from different mutant and control oocytes, pictured in (E) and quantified in (F). In (E), DNA (blue) and kinetochores (red) were detected by DAPI and CREST antiserum, respectively. Scale bars,10 μm. Multiple univalents in a B6.Prdm9−/− oocyte are indicated with white arrowheads. The examples of oocytes from B6 and B6.Prdm9−/−Chk2−/− mice reveal all chromosomes organized in bivalents. P values were calculated using χ2 test.
Fig. 5. Chk2 ablation rescues fertility in Prdm9-null females in the B6 genetic background.
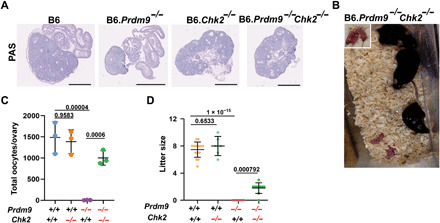
(A) PAS-stained histological sections of 3-week postpartum ovaries from wild-type, single-, and double-mutant females in the B6 genetic background. Scale bars, 200 μm. (B) Pups produced by B6.Prdm9−/−Chk2−/− female mice. (C) Oocyte quantification in mutant and wild-type animals. (D) Litter sizes in mutant and control females. P values were calculated using the Mann-Whitney U test with Bonferroni correction for multiple testing. Photo credit: Tanmoy Bhattacharyya, The Jackson Laboratory.
To test the impact of improved meiotic phenotypes on oocyte number and fertility, we estimated the oocyte number in B6.Prdm9−/−Chk2−/− females and performed fertility tests with wild-type B6 males. In contrast to B6.Prdm9−/− females, B6.Prdm9−/−Chk2−/− females produced at least one litter of grossly healthy, fertile offspring over a 6-month period (Fig. 5, B to D, fig. S6D, and table S2), indicating some functional ovarian reserve. However, there was a high incidence of pregnancy losses and perinatal deaths (fig. S6E), suggesting that oocyte quality is compromised, consistent with the observations of metaphase oocytes in B6.Prdm9−/−Chk2−/− females. The extent of this fertility was much lower than that observed in CAST.Prdm9−/− females, suggesting CAST modifiers in addition to Chk2. In contrast to females, Chk2 deficiency did not rescue fertility in B6.Prdm9−/− males; both single- and double-mutant males were infertile with complete meiotic arrest (fig. S6C). Together, these results demonstrate that, in females, a Prdm9-independent recombination pathway can give rise to the required number of crossovers for production of healthy offspring, probably facilitated by modulation of the Chk2-dependent DNA damage surveillance mechanism.
DISCUSSION
In this study, we find that female meiosis tolerates critically low levels of catalytically active PRDM9, or even no PRDM9 at all in some genetic backgrounds, because a PRDM9-independent recombination pathway compensates successfully. The meiotic DNA damage repair protein CHK2 acts as a modifier; removing it allows completion of meiosis and fertility in females lacking active PRDM9. This is not the case for males; Prdm9EP/EP and Prdm9-null males in the genetic backgrounds examined were infertile. Overall, these results demonstrate that the requirement for functional PRDM9 during meiosis varies substantially by genetic background and by sex and that there are alternative pathways for effective recombination.
As shown by our genetic analyses, elimination of CHK2-dependent DNA damage checkpoint activation can restore female fertility in the absence of PRDM9. CHK2 triggers elimination of oocytes with persistent DNA damage. It is possible that PRDM9-independent DSBs occur later or that their repair is inefficient or prolonged, thus causing DSBs to persist past the timely activation of the CHK2-dependent DNA damage checkpoint, leading to oocyte elimination. Ablation of Chk2 and consequent elimination of checkpoint surveillance may promote survival of oocytes by allowing extra time for PRDM9-independent crossovers to accumulate, permitting completion of meiosis in a sufficient number of oocytes to produce offspring. However, this is not a fully adequate explanation of the modifier effect because, although B6.Prdm9−/−Chk2−/− females were fertile, they were not as productive as CAST.Prdm9−/− females. Thus, while CHK2 is clearly mechanistically involved, at least in B6 females, it alone does not explain the disparity between B6 and CAST female fertility in the absence of PRDM9. Furthermore, the predicted causative CAST variant(s) in Chk2 seem to be dominant and possibly neomorphic, unlike the recessive null mutation that restores fertility in B6.Prdm9−/−Chk2−/− females.
A recent study showing partial restoration of meiosis in PWD. Prdm9−/− males, but not females, speculated that mice with higher crossover rates can alleviate the synapsis defects characteristic of Prdm9-null meiocytes by increasing the probability of synapsis-promoting DSBs (11). PWD males have higher recombination rates than females—unlike most other mouse strains, including B6 and CAST, in which females have higher recombination rates than males (25, 26). The dosage effect of the crossover maturation factor RNF212 on crossover rate (27) suggests that alteration in the level or timing of Rnf212 expression could markedly alter crossover rate. Thus, higher crossover rates in CAST females (or PWD males) in comparison to CAST males (or PWD females) may be modulated by allele-specific genetic regulation or functional variation in RNF212 dosage during pachynema in different genetic backgrounds and sexes. Alternatively, if RNF212 does indeed participate in DNA damage surveillance as a memory of persistent DSBs (24), some allelic variants may be more permissive than others. It is therefore possible that both Chk2 and Rnf212 are modifiers of the PRDM9-independent fertility phenotype or that additional CAST genome variants affect timing or efficacy of DNA repair, the checkpoint, or other features of meiosis that determine whether or not oocytes activate the checkpoint.
Another possible explanation of fertility restoration in CAST females lacking PRDM9 is that repair of PRDM9-independent DSBs is more efficient in the CAST genetic background than in the B6, with fewer oocytes exhibiting enough persistent to activate the DNA damage checkpoint. Ability to repair DSBs may be a limiting factor in B6 mice; the high pre- and perinatal death rate (fig. S6E) and the aneuploidy rate we observed in developing oocytes (Fig. 4, E and F) indicate that, in B6 mice, the PRDM9-independent pathway leads to maturation of defective oocytes. Although similar modification of an infertility phenotype via removal of Chk2 was reported for Trip13-deficient females (23), there are critical differences between causes of infertility in Trip13- and Prdm9-deficient females. In Trip13 mutants, crossovers originate from PRDM9-dependent DSBs, and complete synapsis is achieved (23, 28); in contrast, Prdm9-deficient oocytes exhibit widespread chromosomal asynapsis and form crossovers at ectopic DSBs. Trip13-deficient oocytes are eliminated due to inefficient repair of DSBs in a noncrossover pathway (23). Removal of Chk2 in Trip13 mutants reestablishes ovarian reserve and fertility by allowing oocyte survival and additional time for DSB repair (23); however in Prdm9-deficient oocytes, Chk2 removal must also allow chromosomal synapsis and recombination at non-PRDM9 DSBs. To our knowledge, this is the first evidence that modulation of a DNA damage checkpoint protein can allow survival of oocytes that undergo genetic recombination via a PRDM9-independent pathway.
While checkpoint modulation appears to be a feature allowing oocyte, but not spermatocyte, survival in the absence of PRDM9, at least in certain genetic backgrounds, the question of the genetic and cellular mechanisms behind this dimorphism is undoubtedly more complex. Although recombination is essentially the same process in both sexes, spermatogenesis and oogenesis are fundamentally different cell differentiation programs. Two key differences unique to males that might explain the sexually dimorphic requirement for PRDM9 are meiotic sex chromosome inactivation (MSCI), which is the silencing of most genes on the sex chromosomes from pachynema of meiotic prophase I into spermiogenesis (29), and deployment of Piwi-interacting RNAs (piRNAs) (30, 31). MSCI is triggered by XY asynapsis in spermatocytes and is marked by sequestration of the sex chromosomes in the “sex body” (29). Spermatocytes undergoing PRDM9-deficient meiotic arrest in B6 mice do not form a normal sex body, as unsynapsed autosomes and unrepaired DSBs compromise recruitment of silencing factors to sex chromosomes (8, 17, 32). This could potentially lead to failure of MSCI, triggering meiotic arrest of PRDM9-deficient spermatocytes (8, 32). Inappropriate critical gene silencing on the asynapsed autosomes due to meiotic silencing of unsynapsed chromatin (MSUC) may also contribute to spermatocyte elimination (29). Oocytes, which are not affected by MSUC, may escape these outcomes (29). In mammals, piRNAs are necessary to suppress expression of L1 retrotransposons during spermatogenesis (30, 31). Mutation or deficiency of the piRNA pathway contributes to male-limited sterility (33–35), and it was recently reported that the expression of piRNAs is misregulated in Prdm9-null spermatocytes (36). Thus, it is possible that dysregulation of the piRNA pathway contributes to the postmeiotic problems in Prdm9-null germ cells that undergo partial meiotic arrest, as seen in PWD (11), CAST, and other genetic backgrounds (this report).
In addition to invoking intriguing meiotic mechanisms, the sexually dimorphic responses to PRDM9 deficiency described here have significant evolutionary implications. Prdm9 is the first and only known mammalian speciation gene (37). Prdm9 interacts with an unknown element on the X chromosome to cause male-limited meiotic arrest and hybrid sterility in F1 hybrids between the females of the M. m. musculus strain PWD/PhJ and the males of certain M. m. domesticus strains, including B6 (37, 38). Sterile (PWDXB6)F1 hybrid males exhibit a significant enrichment of DSBs at PRDM9-independent hotspots and high rates of autosomal asynapsis that trigger pachytene checkpoint activation (38, 39). Chromosome asynapsis is also observed in F1 hybrid females, although the phenotype is markedly less severe than in F1 hybrid males, and females remain fertile (38, 40). Our findings suggest that F1 hybrid females may retain fertility by using a PRDM9-independent DSB repair mechanism to evade checkpoint activation. Our work further nominates Chk2 as a key modifier of these sex and strain differences in the meiotic tolerance for PRDM9-independent DSB repair.
The pattern of male-limited hybrid sterility in the (PWDxB6)F1 model follows Haldane’s rule, which postulates that when only one sex of an interspecies hybrid experiences sterility or inviablity, it is the heterogametic sex (41) . Many hypotheses, largely based on the role of sex chromosomes, have attempted to explain Haldane’s rule, including the faster-male theory, the faster X theory, meiotic drive, and the dominance theory (41). However, the mechanistic basis for sexually dimorphic hybrid sterility remains unclear in most documented cases. The sexually dimorphic requirement for PRDM9 for fertility and the relaxed stringency of DNA damage surveillance in females that we report here provide a possible explanation for Haldane’s rule in the (PWDXB6)F1 hybrid sterility model. Notably, hybrid sterility in this system depends on a genetic interaction between Prdm9 and an X-linked locus affecting DNA DSB repair between homologous chromosomes during meiotic prophase (38, 40). The sex-specific manifestation of (PWDXB6)F1 hybrid sterility may also be driven, at least in part, by sex and genetic differences at an autosomal gene, Chk2.
Finally, Prdm9 is a fundamental evolutionary innovation that is thought to have evolved to direct recombination away from functional elements, safeguarding these regions from recombination-associated mutagenesis (7). As in most cases of evolutionary innovation, however, there are trade-offs. Use of a DNA binding protein to specify regions of recombination requires that sufficient numbers of the cognate binding sites of that protein remain intact. However, PRDM9-dependent hotspots extinguish themselves via gene conversion, leading to gradual erosion of the hotspot-binding motifs recognized by a particular PRDM9 variant and, ultimately, meiotic failure and infertility (42, 43). One mechanism through which populations may avoid this fate is the emergence of new Prdm9 alleles that recognize new suites of hotspots (42, 43).
Prdm9 is highly polymorphic and its DNA binding domain is rapidly evolving via positive selection. Our findings introduce an additional, novel complexity to the dynamic interplay between hotspot erosion and reproductive fitness, i.e., sex differences in the usage or effectiveness of PRDM9-independent pathways may render one sex more resilient to the loss of PRDM9 activity, leading to potential sex-specific fitness effects of hotspot erosion. Further work is required to elucidate the mechanistic details of the sexually dimorphic responses to lack of PRDM9; nevertheless, the theoretical implications raised by this phenomenon present fascinating new insights into the checks and balances that constrain one of the most fundamental evolutionary processes in mammals—genetic recombination.
MATERIALS AND METHODS
Ethics statement
The animal care rules used by The Jackson Laboratory are compatible with the regulations and standards of the U.S. Department of Agriculture and the National Institutes of Health. The protocols used in this study were approved by the Animal Care and Use Committee of The Jackson Laboratory (summary nos. 04008 and 15001). Euthanasia was carried out by cervical dislocation.
Mouse strains
Mice used in this study were acquired from The Jackson Laboratory. The following strains were used: C57BL/6J (stock number 000664), WSB/EiJ (stock number 001145), CAST/EiJ (stock number 000928), C3H/HeJ (stock number 000659), B6;129P2-Prdm9tm1Ymat/J (stock number 010719), C57BL/6J-Prdm9<em2Kpgn>/Kpgn (stock number 28854; Prdm9EP). C57BL/6 N-Atm1Brd Chek2tm1b(EUCOMM)Hmgu/JMmucd (stock number 047090-UCD, also known as Chk2) mice were acquired from the Knockout Mouse Project repository at The Jackson Laboratory. The B6;129P2-Prdm9tm1Ymat/J mice were backcrossed to C57BL/6J for 10 generations before use in this study. C3H-Prdm9−/− and CAST/EiJ-Prdm9−/− congenic mice were generated by backcrossing with B6;129P2-Prdm9tm1Ymat/J mice for five generations. C57BL/6J-Prdm9<em2Kpgn>/Kpgn mice were generated by introducing a point mutation (glu365pro) via CRISPR-Cas9 gene editing, onto the C57BL/6J genetic background (fig. S1A). Gene editing was performed by the Genetic Engineering Technology core at The Jackson Laboratory.
Fertility tests
Control and experimental females (2 to 6 months old) were housed with fertile males for a period of up to 6 months. A female was considered fertile if she gave live birth at least once. Litter size was determined by counting pups on the day of birth. Embryo loss during pregnancy was assessed by comparing the number of live births per pregnancy. Briefly, females were mated to wild-type males and the presence of a copulation plug was checked the next morning and recorded. At 19.5 dpc (days post coitum), pregnant females were expected to deliver. Embryo loss during pregnancy was clear due to sinking of stomach in pregnant females due to reabsorption of embryos by 16.5 dpc.
Tissue fixation, histology, immunofluorescence, and follicle quantification
Whole testes and ovaries were fixed for 24 hours in Bouin’s fixative at room temperature and then washed three times with 70% ethanol at room temperature for 1 hour per wash. Testis cross sections were stained with periodic acid–Schiff (PAS) diastase by standard methods and imaged at ×40 magnification with a NanoZoomer 2.0-HT. Ovaries from 3- and 8-week-old females were fixed and embedded in paraffin, serially sectioned at 5 μm, and stained by hematoxylin and eosin or PAS. Follicle quantification was performed as published previously (23). Briefly, every fifth section was examined for the presence of the following classes of oocytes/follicles: primordial, primary, secondary, and mature. For QTL analysis, total follicle number was used. Statistical differences in follicle number between different genotypes were evaluated using the nonparametric two-tailed Mann-Whitney U test with Bonferroni correction using R.
For immunofluorescence, testes from 2-month-old and ovaries from 3-week-old mice were fixed in 4% paraformaldehyde and sectioned at a thickness of 5 μm, matured overnight, dewaxed, rehydrated, and heated in 10 mM sodium citrate buffer (pH 6.0) for antigen retrieval. Slides with testis sections were incubated with antibody dilution buffer [1.5% bovine serum albumin (BSA), 5% donkey serum, 0.05% Triton X-100 in phosphate-buffered saline (PBS), and 0.2× cocktail of protease inhibitors] for 1 hour at room temperature. Slides were incubated overnight with diluted anti-PRDM9 (1:100) (32), anti-γH2AFX (1:5000; Upstate #07-164), and anti-SYCP3 (1:100; D-1; Santa Cruz #74569) in ADB buffer. Slides with ovary sections were stained using rabbit anti-MVH (mouse Vasa homolog) (1:500; Abcam #27591). Immunofluorescent staining was performed by staining the sections for 2 hours at 37°C, using diluted appropriate secondary antibodies conjugated with Alexa Fluor 488/fluorescein isothiocyanate, Alexa Fluor 594/CY3, and Alexa Fluor 647/CY5 (1:300; Molecular Probes/Invitrogen or Jackson Immunoresearch Laboratories). Stained sections were mounted using ProLong Gold Antifade Mountant with 4′,6-diamidino-2-phenylindole (DAPI) (P36935; Molecular Probes/Invitrogen) at 4°C overnight and imaged after 24 hours using a Leica SP5 confocal microscope and/or Zeiss AxioImager.Z2 epifluorescence microscope. Images were processed and adjusted using Adobe Photoshop (Adobe Systems).
Immunostaining of spread meiocytes
The meiocyte spreads were prepared by using the hypotonic protocol as described previously (38). The nuclei were immunostained using the following primary antibodies: rat polyclonal anti-SYCP3 (44), mouse monoclonal anti-MLH1 (Abcam #14206), mouse monoclonal anti-γH2AFX (Upstate, #05-636), mouse monoclonal anti-SYCP3(D-1) (Santa Cruz #74569), guinea pig polyclonal anti-IHO1 (45), rabbit polyclonal anti-RNF212 (27), rabbit antibody anti-MLH3 (46), and rabbit polyclonal anti-BRCA1(C-20) (Santa Cruz #642); and the following secondary antibodies: goat anti-rabbit immunoglobulin G (IgG)–Alexa Fluor 488 (Molecular Probes, A-11034), goat anti-mouse IgG-Alexa Fluor 568 (Molecular Probes, A-11031), goat anti-rabbit IgG-Alexa Fluor 568 (Molecular Probes, A-11036), goat anti-mouse IgG-Alexa Fluor 350 (Molecular Probes, A-21049), goat anti-mouse IgG-Alexa Fluor 647 (Molecular Probes, A-21236), goat anti-rabbit IgG-Alexa Fluor 647 (Molecular Probes, A-21245), and goat anti–guinea pig IgG-Cy3 (Chemicon, #AP108C). Stained meiocytes were mounted using ProLong Gold Antifade Mountant with DAPI (P36935; Molecular Probes/Invitrogen) at 4°C overnight and imaged after 24 hours using a Zeiss AxioImager.Z2 epifluorescence microscope. Images were processed and adjusted using Adobe Photoshop (Adobe Systems). MLH1 foci were counted using ImageJ software (http://rsbweb.nih.gov/ij/). Statistical differences in MLH1 and MLH3 focus number per meiocyte between different genotypes were evaluated using the nonparametric two-tailed Mann-Whitney U test with Bonferroni correction using R. Meiocytes with partially asynapsed or desynapsed chromosomes were also considered as a data point for the MLH1 and MLH3 analysis.
GigaMUGA genotyping and QTL analysis
Giga Mouse Universal Genotyping Array (GigaMUGA) genotyping was carried out by Neogen’s commercial service (18). Invariant SNPs, SNPs with erroneous or missing calls in parental and F1 control samples, SNPs with >10% missing data across all samples, and sites that deviated from Hardy-Weinberg expectations were excluded. Putative genotyping errors were identified as tight double recombinants and recoded as missing data. A total of 54,629 genotypes survived these filters. Cleaned genotypes were then down-sampled to every 10th marker to eliminate a large number of uninformative markers, reduce the impact of genotyping error on map inflation, and expedite the process of map construction. This thinned genotype dataset was then used to construct an empirical genetic linkage map with the est_map call in R/qtl2 (47). Recombination fractions were converted to map distances using the Carter-Falconer mapping function (48) and assuming a 1% residual genotyping error rate.
Single QTL mapping of recombinant B6CASTF2.Prdm9−/− females was performed using the linear mixed model framework implemented in the R/qtl2 package. Conditional genotype probabilities were calculated from the downsized marker dataset without the inclusion of any pseudomarkers. The nonrandom genetic structure among samples was specified via a kinship matrix computed using the leave-one-chromosome-out method. QTL significance thresholds were determined by 1000 permutations of the data. To improve fit to normality, follicle counts were natural log transformed before mapping.
MI spreads
MI spreads and staining were performed as previously described (49). Briefly, pregnant mare’s serum gonadotropin (5 IU) was injected to 4-week-old female mice 48 hours before time of collection of oocytes. Cumulus-oocyte complex or single germinal vesicle oocytes were collected in minimal essential medium (MEM)/polyvinylpyrrolidone (PVP) medium [25 mM Hepes, PVP (3 mg/ml), 2.5 μM milrinone]. After removing cumulus cells, denuded oocytes were matured for MI by incubating in a single drop of CZB (Chatot Ziomek Bavister)/glutamine [81.62 mM NaCl, 4.83 mM KCl, 1.18 mM KH2PO4, 1.18 mM MgSO4 7H2O, 25.12 mM NaHCO3, 1.7 mM CaCl 2H2O, 31.3 mM sodium lactate, 0.27 mM sodium pyruvate, 0.11 mM EDTA, BSA (3 mg/ml), 7 mM taurine, gentamicin (10 μg/ml), phenol red (10 μg/ml), and 1 mM glutamine]. Zona pellucida was removed from matured MI oocytes by using EmbryoMax Acidic Tyrodes solution (Millipore), and then oocytes were placed in spread solution (H2O, 0.16% Triton X-100, 6 mM dithiothreitol, 0.64% PFA). After slides were completely dried, slides were incubated with blocking solution (3% BSA in PBS) for 10 min, at room temperature. Immunostaining was performed by incubating slides in anti-CREST antibody (Anti-Centromere Antibodies derived from human CREST patient serum) (Antibodies Inc., 15-234-0001) diluted in blocking buffer for 3 hours. After washing with blocking buffer, secondary antibody (anti-human Alexa Fluor 647) incubation was performed for 1.5 hours. Slides were mounted in Vectashield with DAPI.
H3K4me3 ChIP-seq
ChIP-seq for H3K4me3 was performed with spermatocytes isolated from 14 dpp (days postpartum) Prdm9EP/EP animals, as previously reported (15), using a commercially available polyclonal α-H3K4me3 antibody (EMD Millipore Cat# 07-473). DNA samples were sequenced on the Illumina NextSeq 500 platform, with 75-bp reads and trimmed for quality using trimmomatic. Sequence data were aligned to the mouse mm10 genome using BWA (Burrows-Wheeler aligner) v0.7.9a, and duplicate reads and reads which failed to align to unique positions in the genome were discarded. This resulted in a total of 26,209,084 and 28,910,782 aligned reads, respectively, for each of two biological replicates. The raw Fastq files were merged and aligned to mm10 by the same procedure for analysis, yielding a total of 45,931,625 aligned reads in the merged sample. Sequence data are available at National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO; www.ncbi.nlm.nih.gov/geo) under accession number GSE144144.
For the wild-type B6 sample, the sequence data used were previously reported by Baker et al. (15). Data are available under GEO accession number GSE52628 (sample accession numbers GSM1273023 and GSM1273024). These data were merged and mapped to mm10 using the above procedure, resulting in 39,973,027 total aligned reads.
H3K4me3 peak calling
H3K4me3 peaks were called using MACS v1.4, default parameters, with a P value cutoff of 0.01, using treatment and control samples. For a control sample, the merged file with the two input B6 biological replicates from Baker et al. (15), mapped to mm10 and described above, was used. To determine overlap between known PRDM9-dependent H3K4me3 peaks and ChIP-seq peak sets, we used BedTools (v2.27.0) intersect with default parameters, except for a requirement for 20% overlap (f = 0.20).
We classified H3K4me3 peaks in spermatocytes as PRDM9-dependent based on a previously published H3K4me3 ChIP-seq experiment from our lab. This experiment compared H3K4me3 peaks in spermatocytes isolated from wild-type B6 males to those from B6.Prdm9Cst knock-in males, in which the DNA binding domain of the highly divergent Prdm9Cst allele replaces that of the endogenous Prdm9Dom2 allele in an otherwise B6 genetic background. PRDM9-dependent H3K4me3 sites are defined as those present in B6 spermatocytes but absent in B6.Prdm9Cst spermatocytes. These PRDM9-dependent H3K4me3 peak locations for B6 were previously described by Baker et al. (15); files with these hotspot locations are available under GEO accession number GSE52628. For the present analysis, their positions were converted to mm10 using the UCSC (University of California, Santa Cruz) Genome Browser tool LiftOver; this file is available under GEO accession number GSE144144.
DMC1 ChIP-seq
Two biological DMC1 ChIP replicates were performed with B6.Prdm9EP/EP males using a previously described method (17, 50). Briefly, testes from two euthanized adult mice (one per replicate) were removed, decapsulated, and cross-linked with 1% paraformaldehyde solution for 10 min. The tissue was homogenized and filtered with a 70-μm cell strainer to obtain germ cells. Cells were washed with lysis buffer 1 [0.25% Triton X-100, 10 mM EDTA, 0.5 mM EGTA, and 10 mM tris-HCl (pH 8.0)] and lysis buffer 2 [0.2 M NaCl, 1 mM EDTA, 0.5 mM EGTA, and 10 mM tris-HCl (pH 8.0] and resuspended in lysis buffer 3 [1% SDS, 10 mM EDTA, and 50 mM tris-HCl (pH 8.0)] with 1× protease inhibitor cocktail. The chromatin was then sheared to ∼1000 bp by sonication and dialyzed against ChIP buffer [0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM tris-HCl, (pH 8.0), and 167 mM NaCl] for at least 6 hours. Ten microliters of chromatin were saved as input. The rest of the chromatin was incubated with an antibody against DMC1 (Santa Cruz, sc-8973) overnight at 4°C. The mixture was then incubated with Protein G Dynabeads (Thermo Fisher Scientific, 10004D) for 4 hours at 4°C. The beads were washed with wash buffer 1 [0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM tris-HCl, (pH 8.0), and 150 mM NaCl], wash buffer 2 [0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM tris-HCl, (pH 8.0), and 500 mM NaCl], wash buffer 3 [0.25 M LiCl, 1% NP-40, 1 mM EDTA, 10mM tris-HCl, (pH 8.0), and 1% deoxycholic acid] and twice with TE (Tris-EDTA) buffer. The chromatin was eluted with dilution buffer [1% SDS and 0.1 M NaHCO3 (pH 9.0)] at 65°C for 30 min and reverse–cross-linked by adding 200 mM NaCl and incubating overnight at 65°C. ChIP and input DNA were purified by the MinElute Reaction Cleanup Kit (Qiagen, 28006). For library preparation, DNA was first end-repaired by incubation with 1× T4 DNA Ligase Reaction Buffer with 0.25 mM deoxynucleotide triphosphates (dNTPs) 3 U of T4 DNA polymerase (NEB, M0203S), 1 U of Klenow Enzyme [New England Biolabs, Inc. (NEB), M0210S], and 10 U of T4 PNK (NEB, B0202S) at 20°C for 30 min, followed by addition of 3′-A overhangs using Klenow Fragment 3′-5′ exo- (NEB, M0212S). After denaturation of DNA at 95°C for 3 min, adapters from the TruSeq Nano DNA LD Library Prep Kit (set A, Illumina, FC-121-4001) were ligated with a Quick Ligation kit (New England Biolabs (NEB), M2200S). The libraries then were amplified using polymerase chain reaction enhancer mix and primer cocktail (Illumina, FC-121-4001) for 12 cycles. For each step, DNA was purified by the MinElute Reaction Cleanup Kit. Libraries were sequenced on the Illumina NextSeq 500 platform, with 75-bp paired-end reads.
DMC1 analysis
Fastq files for paired-end sequenced DMC1 samples were processed with pipelines similar to Brick et al. (51). Briefly, Fastq files for paired-end sequenced DMC1 samples were trimmed using Trimmomatic (v0.32) and subsequently parsed for detection and selection of paired reads having homology at the 5′ and 3′ ends as established by protocols for single strand DNA enrichment. These selected paired-end reads represent the detectable single-stranded DNA reads. The microhomology sequence was removed from the read sequences, and they were subsequently aligned to the mm10 genome using BWA (v0.5.10-tpx). Bam files from this alignment were parsed for detection and subsequent selection of single-ended reads containing true genomic sequence versus fill-in sequence at the microhomologous region. Strandedness could be detected by the alignment flags, and no biases were found between strands. Fastq files were subsequently created containing only the Watson and Crick strand sequences and subsequently processed using nonpaired-end alignment pipelines. Peaks were called using MACS 2.0 with the read extension parameter set to 800 to force pileups.
This yielded 3,348,889 and 4,119,326 aligned DMC1 reads for the two B6.Prdm9EP/EP replicates, respectively. We merged these two replicates for analysis, yielding 7,468,215 aligned DMC1 reads in the merged file. For comparison to wild type, we used previously reported DMC1 sequence data from wild-type C57BL/6 N males (17). We merged three biological replicates for analysis; these replicates had 2,845,239, 2,938,318, and 5,108,836 aligned DMC1 reads, respectively, yielding a merged file with 10,712,776 aligned DMC1 reads (GEO accession GSE112110; samples SRX3825301, SRX3825302, and SRX3825303).
Peak calling was performed using MACS (v.2.0) with standard treatment (ChIP) and control (input) samples with a false discovery rate value of 0.01. For an input sample, we used previously reported wild-type input data from C57BL/6 N males (17). For analysis, we merged two biological replicates, containing 13,651,687 and 11,604,547 aligned reads, yielding a merged input file with 25,256,234 aligned reads (GEO accession GSE112110; samples SRX3825309 and SRX3825310). This input file was used as the peak calling control for both the wild-type and B6.Prdm9EP/EP merged ChIP files. Our set of PRDM9-independent DMC1 peaks is the set of peaks from Brick et al. (7) in which the authors performed DMC1 ChIP-seq in Prdm9-null mice (GEO accession GSE35498). We mapped this file to mm10 for our analysis; our remapped file is available at GEO accession GSE144144.
MA plots
To generate MA plots [M (log ratio) and A (mean average) Bland–Altman plot], we first counted the number of sequencing reads within each peak using Bedtools (v2.27.0) coverage, using the -counts function with default parameters. We then normalized these counts to RPM mapped reads and used the normalized counts as input for MA plots. The actual plots were generated using the plotMA function of the R package limma (www.r-project.org/), with default parameters.
B6xWSB-Prdm9EP/EP crossover analysis
Wild-type WSB/EiJ males were crossed with B6.Prdm9+/EP heterozygous females to generate B6WSBF1.Prdm9+/EP mice. These F1 mice were crossed together to produce B6WSBF2.Prdm9EP/EP homozygotes. Female B6WSBF2.Prdm9EP/EP homozygotes were then crossed to wild-type C57BL/6J males to generate ((B6xWSB)F2xB6)F3 backcrossed progeny. Three B6xWSBF2.Prdm9EP/EP females and 20 of their ((B6xWSB)F2xB6)F3 offspring were genotyped on the GigaMUGA SNP array. Crossovers were called on the basis of informative SNPs between B6 and WSB, using an in-house R script (available upon request). We defined a crossover as having at least five consecutive informative markers (markers differing in genotype between B6 and WSB) of the appropriate genotype on each side of the event. Crossover calls were confirmed using HaploQA—software developed at The Jackson Laboratory that calls crossovers based on a Hidden Markov Model. Individual chromosomes with more than five crossovers were excluded from analysis. Crossovers present in the mothers were subtracted from those in the offspring. The 94 informative offspring-specific crossover intervals were assessed for overlap with known hotspots—defined as PRDM9-dependent H3K4me3 peaks in the B6 genetic background (n = 18,838) (15)—using bedtools intersect (v2.27.0) with default parameters.
The WSB sequences (ENSEMBL) of crossover intervals with no B6 hotspot were searched for 11-bp PRDM9Dom2 binding motifs using the MEME suite tool FIMO. The PRDM9Dom2 motif table was based on the empirically determined consensus motif (19). The WSB and B6 motif lists were compared, and those WSB intervals with no novel PRDM9Dom2 binding motifs relative to the cognate B6 intervals were noted as such.
Testis extract preparation and Western blotting
Crude testis extract was prepared as reported previously (17) from 12 dpp male mice with the denoted genotypes. Samples were subjected to standard SDS–polyacrylamide gel electrophoresis and Western blotting for detection of PRDM9 (1:1000), using a guinea pig anti-PRDM9 antibody produced in-house (2). Subsequently, the blot was stripped and reprobed with mouse anti–β-tubulin primary antibody (1:10,000, Sigma-Aldrich Cat# T4026). Primary antibodies were detected with horseradish peroxidase (HRP)–conjugated anti–guinea pig (1:5000, Cat# AP193P) and HRP-conjugated anti-mouse secondary antibodies (1:20,000, Bio-Rad Cat# 170-6516), respectively.
Sequence alignment analysis
The DNA sequence for Rnf212 and Chk2 for mm10 reference, CAST/EiJ, and C57BL/6NJ was obtained from published sequence datasets at www.sanger.ac.uk/science/data/mouse-genomes-project and http://useast.ensembl.org/Mus_musculus/Info/Index. The alignment was performed using Blastn. Data were visualized using dot matrix plots.
Quantification and statistical analysis
Statistical tests were performed using GraphPad Prism version 7.0 and R statistical packages.
Supplementary Material
Acknowledgments
This paper is dedicated to the memory of Kenneth Paigen, a visionary, a beloved colleague, and a great mentor. We thank three anonymous reviewers for helpful comments. We thank J. Eppig and all members of the Bolcun-Filas, Handel, Paigen, and Petkov labs for insightful discussions and suggestions. We acknowledge the expertise and contributions of The Jackson Laboratory Scientific Services, including the Histology core, the Microscopy core, Genome Technologies, Genetic Engineering Technology, and Mouse Resources for expertise and help during this project. We thank the Knockout Mouse Project (KOMP2) at The Jackson Laboratory for providing mice. We thank A. Toth, P. Cohen, and N. Hunter for sharing antibody resources. Funding: T.B. was supported in part by a JAX Scholar award (19042802-15-3); N.R.P. was supported in part by NICHD T32 Training Program in Developmental Genetics (T32 HD007065 to The Jackson Laboratory). This work was also supported by grants from the NIH: P01 GM99640 to M.A.H. and K.P., R01 HD093778 to E.B.-F., R01 GM125736 to P.M.P. and P30 CA034196 to The Jackson Laboratory for scientific services. Author contributions: Conceptualization: T.B. and N.R.P; methodology: T.B., N.R.P., B.L.D., and E.B.-F.; investigation: T.B., N.R.P., B.L.D., C.E., R.A.L., and C.B.; writing—original draft: T.B. and N.R.P.; writing—review and editing: T.B., N.R.P., B.L.D., C.E., R.A.L., C.B., K.P., E.B.-F., P.M.P., and. M.A.H; funding acquisition: T.B., N.R.P., B.L.D., K.P., E.B.-F., P.M.P., and M.A.H.; resources: T.B., N.R.P., B.L.D., K.P., E.B.-F., P.M.P., and M.A.H.; supervision: T.B., B.L.D., E.B.-F., P.M.P., and M.A.H. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. All data, code, and materials used in the analysis are available in some form to any researcher or commercially for purposes of reproducing or extending the analysis. H3K4me3 ChIP-seq data are available at NCBI Gene Expression Omnibus (GEO; www.ncbi.nlm.nih.gov/geo) under accession number GSE144144.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/43/eabb6606/DC1
References and Notes
- 1.Paigen K., Petkov P. M., PRDM9 and its role in genetic recombination. Trends Genet. 34, 291–300 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berg I. L., Neumann R., Lam K. W., Sarbajna S., Odenthal-Hesse L., May C. A., Jeffreys A. J., PRDM9 variation strongly influences recombination hot-spot activity and meiotic instability in humans. Nat. Genet. 42, 859–863 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baudat F., Buard J., Grey C., Fledel-Alon A., Ober C., Przeworski M., Coop G., de Massy B., PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 327, 836–840 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parvanov E. D., Petkov P. M., Paigen K., Prdm9 controls activation of mammalian recombination hotspots. Science 327, 835 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Myers S., Bowden R., Tumian A., Bontrop R. E., Freeman C., MacFie T. S., McVean G., Donnelly P., Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science 327, 876–879 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Powers N. R., Parvanov E. D., Baker C. L., Walker M., Petkov P. M., Paigen K., The meiotic recombination activator PRDM9 trimethylates both H3K36 and H3K4 at recombination hotspots in vivo. PLOS Genet. 12, e1006146 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brick K., Smagulova F., Khil P., Camerini-Otero R. D., Petukhova G. V., Genetic recombination is directed away from functional genomic elements in mice. Nature 485, 642–645 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hayashi K., Yoshida K., Matsui Y., A histone H3 methyltransferase controls epigenetic events required for meiotic prophase. Nature 438, 374–378 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Irie S., Tsujimura A., Miyagawa Y., Ueda T., Matsuoka Y., Matsui Y., Okuyama A., Nishimune Y., Tanaka H., Single-nucleotide polymorphisms of the PRDM9 (MEISETZ) gene in patients with nonobstructive azoospermia. J. Androl. 30, 426–431 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Miyamoto T., Koh E., Sakugawa N., Sato H., Hayashi H., Namiki M., Sengoku K., Two single nucleotide polymorphisms in PRDM9 (MEISETZ) gene may be a genetic risk factor for Japanese patients with azoospermia by meiotic arrest. J. Assist. Reprod. Genet. 25, 553–557 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mihola O., Pratto F., Brick K., Linhartova E., Kobets T., Flachs P., Baker C. L., Sedlacek R., Paigen K., Petkov P. M., Camerini-Otero R. D., Trachtulec Z., Histone methyltransferase PRDM9 is not essential for meiosis in male mice. Genome Res. 29, 1078–1086 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Narasimhan V. M., Hunt K. A., Mason D., Baker C. L., Karczewski K. J., Barnes M. R., Barnett A. H., Bates C., Bellary S., Bockett N. A., Giorda K., Griffiths C. J., Hemingway H., Jia Z., Kelly M. A., Khawaja H. A., Lek M., McCarthy S., McEachan R., O’Donnell-Luria A., Paigen K., Parisinos C. A., Sheridan E., Southgate L., Tee L., Thomas M., Xue Y., Schnall-Levin M., Petkov P. M., Tyler-Smith C., Maher E. R., Trembath R. C., MacArthur D. G., Wright J., Durbin R., van Heel D. A., Health and population effects of rare gene knockouts in adult humans with related parents. Science 352, 474–477 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Auton A., Rui Li Y., Kidd J., Oliveira K., Nadel J., Holloway J. K., Hayward J. J., Cohen P. E., Greally J. M., Wang J., Bustamante C. D., Boyko A. R., Genetic recombination is targeted towards gene promoter regions in dogs. PLOS Genet. 9, e1003984 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Munoz-Fuentes V., Di Rienzo A., Vila C., Prdm9, a major determinant of meiotic recombination hotspots, is not functional in dogs and their wild relatives, wolves and coyotes. PLOS ONE 6, e25498 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baker C. L., Walker M., Kajita S., Petkov P. M., Paigen K., PRDM9 binding organizes hotspot nucleosomes and limits Holliday junction migration. Genome Res. 24, 724–732 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diagouraga B., Clement J. A. J., Duret L., Kadlec J., de Massy B., Baudat F., PRDM9 methyltransferase activity is essential for meiotic DNA double-strand break formation at its binding sites. Mol. Cell 69, 853–865.e6 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Bhattacharyya T., Walker M., Powers N. R., Brunton C., Fine A. D., Petkov P. M., Handel M. A., Prdm9 and meiotic cohesin proteins cooperatively promote DNA double-strand break formation in mammalian spermatocytes. Curr. Biol. 29, 1002–1018.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morgan A. P., Fu C. P., Kao C. Y., Welsh C. E., Didion J. P., Yadgary L., Hyacinth L., Ferris M. T., Bell T. A., Miller D. R., Giusti-Rodriguez P., Nonneman R. J., Cook K. D., Whitmire J. K., Gralinski L. E., Keller M., Attie A. D., Churchill G. A., Petkov P., Sullivan P. F., Brennan J. R., McMillan L., Pardo-Manuel de Villena F., The mouse universal genotyping array: From substrains to subspecies. G3 6, 263–279 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walker M., Billings T., Baker C. L., Powers N., Tian H., Saxl R. L., Choi K., Hibbs M. A., Carter G. W., Handel M. A., Paigen K., Petkov P. M., Affinity-seq detects genome-wide PRDM9 binding sites and reveals the impact of prior chromatin modifications on mammalian recombination hotspot usage. Epigenetics Chromatin 8, 31 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keane T. M., Goodstadt L., Danecek P., White M. A., Wong K., Yalcin B., Heger A., Agam A., Slater G., Goodson M., Furlotte N. A., Eskin E., Nellaker C., Whitley H., Cleak J., Janowitz D., Hernandez-Pliego P., Edwards A., Belgard T. G., Oliver P. L., McIntyre R. E., Bhomra A., Nicod J., Gan X., Yuan W., van der Weyden L., Steward C. A., Bala S., Stalker J., Mott R., Durbin R., Jackson I. J., Czechanski A., Guerra-Assuncao J. A., Donahue L. R., Reinholdt L. G., Payseur B. A., Ponting C. P., Birney E., Flint J., Adams D. J., Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477, 289–294 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geraldes A., Basset P., Gibson B., Smith K. L., Harr B., Yu H. T., Bulatova N., Ziv Y., Nachman M. W., Inferring the history of speciation in house mice from autosomal, X-linked, Y-linked and mitochondrial genes. Mol. Ecol. 17, 5349–5363 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.White M. A., Stubbings M., Dumont B. L., Payseur B. A., Genetics and evolution of hybrid male sterility in house mice. Genetics 191, 917–934 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bolcun-Filas E., Rinaldi V. D., White M. E., Schimenti J. C., Reversal of female infertility by Chk2 ablation reveals the oocyte DNA damage checkpoint pathway. Science 343, 533–536 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qiao H., Rao H., Yun Y., Sandhu S., Fong J. H., Sapre M., Nguyen M., Tham A., Van B. W., Chng T. Y. H., Lee A., Hunter N., Impeding DNA break repair enables oocyte quality control. Mol. Cell 72, 211–221.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dumont B. L., Payseur B. A., Genetic analysis of genome-scale recombination rate evolution in house mice. PLOS Genet. 7, e1002116 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balcova M., Faltusova B., Gergelits V., Bhattacharyya T., Mihola O., Trachtulec Z., Knopf C., Fotopulosova V., Chvatalova I., Gregorova S., Forejt J., Hybrid sterility locus on chromosome X controls meiotic recombination rate in mouse. PLOS Genet. 12, e1005906 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reynolds A., Qiao H., Yang Y., Chen J. K., Jackson N., Biswas K., Holloway J. K., Baudat F., de Massy B., Wang J., Hoog C., Cohen P. E., Hunter N., RNF212 is a dosage-sensitive regulator of crossing-over during mammalian meiosis. Nat. Genet. 45, 269–278 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li X., Schimenti J. C., Mouse pachytene checkpoint 2 (Trip13) is required for completing meiotic recombination but not synapsis. PLOS Genet. 3, e130 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turner J. M., Meiotic silencing in mammals. Annu. Rev. Genet. 49, 395–412 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Czech B., Munafo M., Ciabrelli F., Eastwood E. L., Fabry M. H., Kneuss E., Hannon G. J., piRNA-guided genome defense: From biogenesis to silencing. Annu. Rev. Genet. 52, 131–157 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ernst C., Odom D. T., Kutter C., The emergence of piRNAs against transposon invasion to preserve mammalian genome integrity. Nat. Commun. 8, 1411 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun F., Fujiwara Y., Reinholdt L. G., Hu J., Saxl R. L., Baker C. L., Petkov P. M., Paigen K., Handel M. A., Nuclear localization of PRDM9 and its role in meiotic chromatin modifications and homologous synapsis. Chromosoma 124, 397–415 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuramochi-Miyagawa S., Watanabe T., Gotoh K., Totoki Y., Toyoda A., Ikawa M., Asada N., Kojima K., Yamaguchi Y., Ijiri T. W., Hata K., Li E., Matsuda Y., Kimura T., Okabe M., Sakaki Y., Sasaki H., Nakano T., DNA methylation of retrotransposon genes is regulated by Piwi family members MILI and MIWI2 in murine fetal testes. Genes Dev. 22, 908–917 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carmell M. A., Girard A., van de Kant H. J., Bourc’his D., Bestor T. H., de Rooij D. G., Hannon G. J., MIWI2 is essential for spermatogenesis and repression of transposons in the mouse male germline. Dev. Cell 12, 503–514 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Deng W., Lin H., miwi, a murine homolog of piwi, encodes a cytoplasmic protein essential for spermatogenesis. Dev. Cell 2, 819–830 (2002). [DOI] [PubMed] [Google Scholar]
- 36.Fine A. D., Ball R. L., Fujiwara Y., Handel M. A., Carter G. W., Uncoupling of transcriptomic and cytological differentiation in mouse spermatocytes with impaired meiosis. Mol. Biol. Cell 30, 717–728 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mihola O., Trachtulec Z., Vlcek C., Schimenti J. C., Forejt J., A mouse speciation gene encodes a meiotic histone H3 methyltransferase. Science 323, 373–375 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Bhattacharyya T., Reifova R., Gregorova S., Simecek P., Gergelits V., Mistrik M., Martincova I., Pialek J., Forejt J., X chromosome control of meiotic chromosome synapsis in mouse inter-subspecific hybrids. PLOS Genet. 10, e1004088 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gregorova S., Gergelits V., Chvatalova I., Bhattacharyya T., Valiskova B., Fotopulosova V., Jansa P., Wiatrowska D., Forejt J., Modulation of Prdm9-controlled meiotic chromosome asynapsis overrides hybrid sterility in mice. eLife 7, e34282 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bhattacharyya T., Gregorova S., Mihola O., Anger M., Sebestova J., Denny P., Simecek P., Forejt J., Mechanistic basis of infertility of mouse intersubspecific hybrids. Proc. Natl. Acad. Sci. U.S.A. 110, E468–E477 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.A. Coyne, H. A. Orr, Speciation (Sinauer Associates, 2004), pp. 545. [Google Scholar]
- 42.Baker C. L., Kajita S., Walker M., Saxl R. L., Raghupathy N., Choi K., Petkov P. M., Paigen K., PRDM9 drives evolutionary erosion of hotspots in Mus musculus through haplotype-specific initiation of meiotic recombination. PLOS Genet. 11, e1004916 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tiemann-Boege I., Schwarz T., Striedner Y., Heissl A., The consequences of sequence erosion in the evolution of recombination hotspots. Philos. Trans. R. Soc. Lond. B Biol. Sci. 372, 20160462 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eaker S., Pyle A., Cobb J., Handel M. A., Evidence for meiotic spindle checkpoint from analysis of spermatocytes from Robertsonian-chromosome heterozygous mice. J. Cell Sci. 114, 2953–2965 (2001). [DOI] [PubMed] [Google Scholar]
- 45.Stanzione M., Baumann M., Papanikos F., Dereli I., Lange J., Ramlal A., Trankner D., Shibuya H., de Massy B., Watanabe Y., Jasin M., Keeney S., Toth A., Meiotic DNA break formation requires the unsynapsed chromosome axis-binding protein IHO1 (CCDC36) in mice. Nat. Cell Biol. 18, 1208–1220 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Holloway J. K., Sun X., Yokoo R., Villeneuve A. M., Cohen P. E., Mammalian CNTD1 is critical for meiotic crossover maturation and deselection of excess precrossover sites. J. Cell Biol. 205, 633–641 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Broman K. W., Gatti D. M., Simecek P., Furlotte N. A., Prins P., Sen S., Yandell B. S., Churchill G. A., R/qtl2: Software for mapping quantitative trait loci with high-dimensional data and multiparent populations. Genetics 211, 495–502 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Broman K. W., Rowe L. B., Churchill G. A., Paigen K., Crossover interference in the mouse. Genetics 160, 1123–1131 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schindler K., Schultz R. M., The CDC14A phosphatase regulates oocyte maturation in mouse. Cell Cycle 8, 1090–1098 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Khil P. P., Smagulova F., Brick K. M., Camerini-Otero R. D., Petukhova G. V., Sensitive mapping of recombination hotspots using sequencing-based detection of ssDNA. Genome Res. 22, 957–965 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brick K., Thibault-Sennett S., Smagulova F., Lam K. G., Pu Y., Pratto F., Camerini-Otero R. D., Petukhova G. V., Extensive sex differences at the initiation of genetic recombination. Nature 561, 338–342 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/43/eabb6606/DC1