

CRISPR-Cas9 knockin of the HBB gene paves the way for a general therapeutic solution for treating hemoglobinopathies.
Abstract
Leveraging the endogenous homology-directed repair (HDR) pathway, the CRISPR-Cas9 gene-editing system can be applied to knock in a therapeutic gene at a designated site in the genome, offering a general therapeutic solution for treating genetic diseases such as hemoglobinopathies. Here, a combined supramolecular nanoparticle (SMNP)/supramolecular nanosubstrate–mediated delivery (SNSMD) strategy is used to facilitate CRISPR-Cas9 knockin of the hemoglobin beta (HBB) gene into the adeno-associated virus integration site 1 (AAVS1) safe-harbor site of an engineered K562 3.21 cell line harboring the sickle cell disease mutation. Through stepwise treatments of the two SMNP vectors encapsulating a Cas9•single-guide RNA (sgRNA) complex and an HBB/green fluorescent protein (GFP)–encoding plasmid, CRISPR-Cas9 knockin was successfully achieved via HDR. Last, the HBB/GFP-knockin K562 3.21 cells were introduced into mice via intraperitoneal injection to show their in vivo proliferative potential. This proof-of-concept demonstration paves the way for general gene therapeutic solutions for treating hemoglobinopathies.
INTRODUCTION
Hemoglobinopathies are a group of inherited genetic disorders caused by hemoglobin beta (HBB) gene mutations (1). Adult hemoglobin consists of a tetramer of two α-globin and two β-globin subunits (α2β2). Several mutations in the HBB gene have been identified, which are associated with either structural changes or reduced production of β-globin protein, resulting in either altered hemoglobin production or function. These alterations are observed in the two most prevalent forms of β-hemoglobinopathies, i.e., sickle cell disease (SCD) and β-thalassemia (2). For example, SCD is caused by an A-to-T point mutation in the HBB gene, which leads to a substitution of a valine for a glutamic acid at position 6 in the β-globin chain. In addition, more than 400 mutations in the HBB gene have been identified as associated with β-thalassemia. Traditionally, patients with severe hemoglobinopathy phenotypes require lifelong supportive care, which can include frequent red blood cell (RBC) transfusions (3). Allogeneic hematopoietic stem cell (HSC) transplantation (4) represents the only curative therapy for these patients. However, the lack of availability of well-matched donors to avoid transplant-associated comorbidities, such as graft-versus-host disease, remains a critical challenge to achieving the best treatment outcomes. Gene therapies are emerging as promising alternative options for patients lacking an allogenic compatible HSC donor. Lentiviral vectors (LVs) carrying genetic materials encoding for either a corrected HBB gene or modifying regulators of hemoglobin production are now being applied clinically to enable effective autologous gene-modified HSC transplantation strategies (5). However, these LV-based gene addition approaches require correction of long-term repopulating HSCs and are extremely expensive to manufacture. Furthermore, LVs are limited by the size and type of cargo that they can deliver and may integrate semirandomly into the genome, generating aberrant transcripts that may potentially trigger oncogenesis (5). Compared with LV-based gene transduction, genome-editing technologies offer a more practical approach, because they are designed to modify a single, safe genomic target (6).
The CRISPR-Cas9 genome editing system is evolving from an already powerful genetic editing research tool to a promising technology for treating genetic diseases (7). The CRISPR-Cas9 system is composed of two functional components, i.e., the Cas9 endonuclease and an engineered short, single-guide RNA (sgRNA), which form a ribonucleoprotein (RNP) complex, Cas9•sgRNA. On the basis of simple base pairing, the Cas9•sgRNA complex recognizes and cuts at a target site predetermined by the design of the sgRNA, introducing a precise double-stranded break (DSB) (7). Following the formation of a DSB, endogenous DNA repair can occur via either the (i) nonhomologous end joining (NHEJ) or (ii) homology-directed repair (HDR) pathway, offering two classes of therapeutic genome editing approaches. In contrast to the NHEJ pathway, which serves as the foundation of CRISPR-Cas9–mediated gene knockdown and knockout, the HDR pathway enables CRISPR-Cas9–mediated gene correction and knockin, which can integrate a single copy of a therapeutic gene at a predetermined target site via a homologous donor DNA (dDNA) template, offering a more general therapeutic solution for a variety of genetic diseases (7). However, notable challenges remain for developing highly efficient methods for step-by-step delivery of CRISPR-Cas9 genome editing components into diseased cells, such as limited cargo size, different charge properties of CRISPR cargos, low delivery and genome editing efficiencies, and manufacturing challenges (8).
Physical methods, such as electroporation and microinjection, have been used successfully for delivering CRISPR-Cas9 reagents intracellularly via transient disruption of the lipid bilayer of cells (9). However, decreased cell viability and premature differentiation of the engineered stem cell product limit their clinical applications. Viral-based methods remain a popular choice for the delivery of gene-editing machinery, with adeno-associated virus (AAV) being the most promising viral vector (10). However, limitations in packaging capacity (<4.7 kb) (11), high cost, and safety concerns related to immunogenicity associated with AAV remain. Notable research has been devoted to developing less expensive and safer nonviral vectors (9, 12, 13) for delivering CRISPR-Cas9 reagents intracellularly, including lipids, polymers, and nanoparticles. The CRISPR-Cas9 system can be introduced in three forms: DNA (14), mRNA (15), and protein (16). Compared to deliveries of Cas9 DNA and Cas9 mRNA, direct delivery of Cas9•sgRNA has two major advantages: (i) rapid genome editing, as it skips gene transcription and/or translation, and (ii) transient genome editing with reduced off-target effects and toxicity. Because of the large size of the Cas9 protein (~160 kDa), there is a need for more effective delivery vectors (12). Lee et al. (17) developed a delivery vehicle (CRISPR-Gold) composed of gold nanoparticles conjugated to DNA and complexed with cationic endosomal disruptive polymers to deliver CRISPR RNPs and donor template to correct the DNA mutation that causes Duchenne muscular dystrophy (DMD) in mice with reduced off-target effects and reduced muscle fibrosis in mdx mice. Recently, Cheng et al. (18) reported a strategy termed as selective organ targeting (SORT) wherein multiple classes of lipid nanoparticles are systematically engineered to edit extrahepatic tissues exclusively via addition of a supplemental SORT molecule. Lung-, spleen-, and liver-targeted SORT lipid nanoparticles were designed to edit therapeutically relevant cell types selectively including epithelial cells, endothelial cells, B cells, T cells, and hepatocytes. However, most nonviral vector–based CRISPR-Cas9 genome editing systems have been developed for gene knockout (19), knockdown (20), or correction (17) to treat monogenic disorders. There has been relatively limited progress made in gene knockin (21), especially for achieving effective knockin of long-frame therapeutic genes in HSCs, which are notoriously difficult to transfect.
Polyamidoamine (PAMAM) dendrimer is a kind of hyperbranched polymer with high molecular uniformity, narrow molecular weight distribution, defined size, and an amine terminal surface. These properties enable it to be modified with different numbers of adamantane (Ad) motifs to prepare Ad-grafted PAMAM (Ad-PAMAM) dendrimers. Previously, we demonstrated a convenient and flexible self-assembled synthetic approach for producing supramolecular nanoparticle (SMNP) vectors (22) by mixing three molecular building blocks, i.e., Ad-PAMAM dendrimer, β-cyclodextrin (CD)–grafted branched polyethyleneimine (CD-PEI), and Ad-grafted poly(ethylene glycol) (Ad-PEG). Multivalent molecular recognition between Ad and CD enables modular control over the sizes, surface chemistry, and payloads of SMNP vectors, with a diversity of imaging (22) and therapeutic applications (23). To improve the delivery efficiency of SMNP vectors, we developed a substrate-mediated delivery strategy (24), also known as the supramolecular nanosubstrate–mediated delivery (SNSMD), by which Ad-grafted silicon nanowire substrates (Ad-SiNWS) were used to facilitate the uptake of SMNP vectors into cells. The Ad/CD recognition system drives dynamic assembly and local enrichment of SMNPs onto Ad-SiNWS. Once the cells settle onto the Ad-SiNWS, intimate contact between the cell membrane and the nanowires leads to efficient delivery of SMNP vectors. We envision that the combined utility of SMNP vectors and SNSMD (i.e., a combined SMNP/SNSMD strategy) offers a powerful solution for step-by-step delivery of Cas9•sgRNA and dDNA for highly efficient genome editing in cells, especially for hematologic cells.
Here, we demonstrate that the combined SMNP/SNSMD strategy (Fig. 1) enables the CRISPR-Cas9–mediated knockin of a single-copy HBB gene to the human AAV integration site 1 (AAVS1) safe-harbor locus, providing a safe, effective, cheap, and general curative therapeutic solution for hemoglobinopathies. The K562 3.21 cell line has a known HBB mutation (associated with SCD) (25) and was used as a model system for optimization and feasibility studies. Two different SMNP vectors, i.e., Cas9•sgRNA⊂SMNPs and HBB/green fluorescent protein (GFP)–plasmid⊂SMNPs, were prepared (Fig. 1, B and C) by encapsulating Cas9•sgRNA (targeting the AAVS1 site) and dDNA (HBB/GFP plasmid, detailed gene map in fig. S1) into SMNP vectors via a self-assembled synthetic approach, respectively. Here, CRISPR-Cas9 knockin of the HBB/GFP gene is carried out via two consecutive steps. In step 1, the combined SMNP/SNSMD strategy facilitates cell uptake of Cas9•sgRNA⊂SMNPs, and the internalized and released Cas9•sgRNA specifically recognizes and induces DSB at the AAVS1 site. In step 2, HBB/GFP plasmid⊂SMNPs were added to deliver the HBB/GFP plasmid, and the HDR pathway led to integration of the HBB/GFP gene into the DSB. We examined how the delivery time interval and multiple treatments of these two SMNP vectors affected HBB/GFP knockin efficiency. Under optimized step-by-step delivery conditions, HBB/GFP-knockin K562 3.21 cells were produced, sorted, and expanded. Fluorescence microscopy, polymerase chain reaction (PCR), and Sanger sequencing were used to confirm the successful integration of the 3.7-kb HBB/GFP gene into the AAVS1 site in the cells. Furthermore, the HBB/GFP-knockin K562 3.21 cells were tested with immunofluorescence (IF) analysis and quantitative PCR (qPCR) assays, indicating that the integrated HBB gene was functionally expressed at both protein and mRNA levels, respectively. Last, the HBB/GFP-knockin K562 3.21 cells were introduced into athymic nude mice via intraperitoneal injection to test their ability to proliferate and yield consistent HBB/GFP gene expression in vivo.
Fig. 1. CRISPR-Cas9–mediated knockin of HBB/GFP gene.
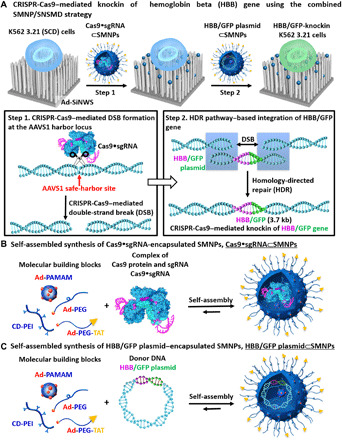
(A) Schematic of the mechanism governing the combined SMNP/SNSMD strategy for CRISPR-Cas9–mediated knockin of HBB/green fluorescent protein (GFP) gene into K562 3.21 (SCD) cells via two consecutive steps. (B) A self-assembled synthetic approach for the preparation of Cas9•sgRNA⊂SMNPs through stoichiometric mixing of Cas9•sgRNA and the four molecular building blocks. (C) A self-assembled synthetic approach for the preparation of HBB/GFP plasmid⊂SMNPs.
RESULTS
Synthesis of EGFP-Cas9•sgRNA⊂SMNPs and analysis of cellular uptake
Our previous experience in using SMNP vectors for coencapsulating a transcription factor protein and a DNA plasmid (26) was used to prepare Cas9•sgRNA⊂SMNPs (Fig. 1B). First, an sgRNA targeting the AAVS1 locus (27) was purchased (Integrated DNA Technologies, Iowa) for preparing Cas9•sgRNA. Cas9•sgRNA⊂SMNPs were prepared through stoichiometric mixing of Cas9•sgRNA and four SMNP molecular building blocks. In search of an optimal formulation of Cas9•sgRNA⊂SMNPs, enhanced GFP (EGFP)–labeled Cas9 protein (EGFP-Cas9, GenCrispr, New Jersey) was used to study uptake into K562 3.21 cells (Fig. 2A). Three batches (15 formulations) of EGFP-Cas9•sgRNA⊂SMNPs (Fig. 2B) were formulated by stepwise modulation of (i) the SMNP/EGFP-Cas9•sgRNA weight ratio (100:1, 100:2, 100:4, 100:6, and 100:8), (ii) SMNP size (100 to 200 nm), and (iii) the coverage of a membrane penetration ligand, transactivator of transcription (TAT) (2 to 10%). Before the cell uptake study, K562 3.21 cells were starved in serum-free RPMI medium for 10 hours to synchronize their cell cycles to G0/G1 phases (28). Approximately 1 × 105 K562 3.21 cells were introduced into each well of eight-well culture plates (10.5 cm2 per well), in which an Ad-SiNWS (2.5 × 3.0 cm2) was immersed in 1.0 ml of RPMI medium. Each formulation of EGFP-Cas9•sgRNA⊂SMNPs (containing 3.0 μg of EGFP-Cas9) was added to one well. After 4′,6-diamidino-2-phenylindole (DAPI) nuclear staining, the K562 cells on Ad-SiNWS were subjected to high-resolution microscopy imaging (fig. S2, A and B). We first examined how the weight ratios [weight % (wt %)] between SMNP vectors and EGFP-Cas9•sgRNA affect cell uptake. The results (Fig. 2B) indicate that a higher percentage of EGFP-positive cells (17%) was achieved at the ratio of 100:4. Using this formulation ratio, we then studied how the sizes of Cas9•sgRNA⊂SMNPs affect cellular uptake. By altering the amount of Ad-PAMAM and CD-PEI in each formulation, we were able to prepare the second batch of EGFP-Cas9•sgRNA⊂SMNPs with five different sizes, ranging from 110 to 200 nm [see dynamic light scattering (DLS) characterization in fig. S2C]. We found that the 120-nm EGFP-Cas9•sgRNA⊂SMNPs led to better delivery performance with 28% EGFP-positive cells. On the basis of this SMNP configuration, we prepared TAT-grafted EGFP-Cas9•sgRNA⊂SMNPs with TAT coverage ranging between 2 and 10%. These studies revealed that EGFP-Cas9•sgRNA⊂SMNPs with 8% TAT coverage exhibited an optimal delivery performance of 35%. We thus identified the optimal synthetic formulation that gave 120-nm 8% TAT–grafted EGFP-Cas9•sgRNA⊂SMNPs, and this formulation was subjected to gene-editing studies. Three control studies, i.e., K562 3.21 cells treated with EGFP-Cas9•sgRNA⊂SMNPs (no Ad-SiNWS), Lipofectamine CRISPRMAX agent (encapsulated EGFP-Cas9•sgRNA), and EGFP-Cas9•sgRNA (without SMNP vectors), were conducted in parallel. The 8% TAT–grafted Cas9•sgRNA⊂SMNPs showed about half the uptake efficiency without nanowires. The EGFP-Cas9-sgRNA delivered with Lipofectamine had a similar efficiency to the SMNPs without nanowires. EGFP-Cas9-sgRNA alone resulted in low efficiency. The markedly compromised EGFP-Cas9 uptake performance highlights the critical roles of the two functional components, i.e., Ad-SiNWS and SMNP vectors, of the combined SMNP/SNSMD strategy. To visualize intracellular distribution and clearance of the SMNPs, we further used CD-PEI-Cy5 instead of CD-PEI, to assemble the Cas9•sgRNA⊂Cy5-SMNPs in the uptake experiment. According to the fluorescent images (fig. S3), it appears that the Cas9•sgRNA⊂Cy5-SMNPs (red) were taken up by the K562 3.21 cells after incubation for 1 day. Thereafter, slow decay of red fluorescence intensity was observed from days 2 to 7, suggesting that the CD-PEI-Cy5 was cleared by the cells. To determine potential off-target effects, we performed PCR and Sanger sequence at five of the top-ranking predicted off-target sites of the sgRNA-AAVS1 via the Cas-OFFinder design tool. The sequencing results showed no detectable off-target events in the top five predicted off-target loci (fig. S4).
Fig. 2. The combined SMNP/SNSMD strategy for delivering EGFP-Cas9•sgRNA or HBB/GFP plasmid into K562 3.21 cells.
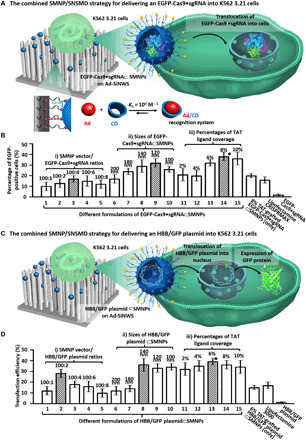
(A) The combined SMNP/SNSMD strategy for delivering EGFP-Cas9•sgRNA into K562 3.21 cells. (B) Three batches (15 formulations) of EGFP-Cas9•sgRNA⊂SMNPs were prepared for delivering EGFP-Cas9•sgRNA into K562 3.21 cells to identify an optimal formulation (marked with an asterisk). (C) The combined SMNP/SNSMD strategy for delivering HBB/GFP plasmid into K562 3.21 cells. (D) Three batches (15 formulations) of HBB/GFP plasmid⊂SMNPs were prepared for GFP transfection studies to identify an optimal formulation (marked with an asterisk).
Synthesis of HBB/GFP plasmid⊂SMNPs and transfection studies
Using a similar screening approach, we identified the optimal formulation for preparing HBB/GFP plasmid⊂SMNPs (Fig. 2C). Three batches (15 formulations) of SMNPs were prepared by stepwise modulation of (i) the SMNP/plasmid weight ratios (100:1, 100:2, 100:4, 100:6, and 100:8), (ii) SMNP size (100 to 200 nm, DLS in fig. S5A), and (iii) the coverage of TAT (2 to 10%). After settling growth-synchronized K562 3.21 cells on Ad-SiNWS in culture plates, individual formulations of the SMNPs (containing 2.0 μg of HBB/GFP plasmid) were added to evaluate transfection of the GFP-encoding transgene. After treatment with SMNPs for 24 hours, fluorescence microscopy was used to quantify the GFP expression in individual cells (fig. S5B). The results revealed (Fig. 2D) that 140-nm HBB/GFP plasmid⊂SMNPs with 6% TAT coverage exhibited an optimal GFP transfection performance of 38%. Three control studies, i.e., K562 3.21 cells treated with HBB/GFP plasmid⊂SMNPs (no Ad-SiNWS), Lipofectamine 3000 agent (encapsulated HBB/GFP plasmid), and HBB/GFP plasmid (without SMNP vectors), were conducted in parallel. This HBB/GFP plasmid⊂SMNP formulation was consequently subjected to time-dependent quantitative imaging at 12, 24, 48, 72, 96, and 120 hours posttreatment (fig. S5C), showing that the highest transfection performance (45%) was observed 48 hours posttreatment, and the GFP signals gradually decayed and diminished completely by 120 hours.
Characterization of SMNPs and Ad-SiNWS
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) were used to characterize Ad-SiNWS, EGFP-Cas9•sgRNA⊂SMNPs, and HBB/GFP plasmid⊂SMNPs, identified above. The SEM and TEM images showed that the diameters and lengths of Ad-SiNWS are ca. 50 to 100 nm and 5 to 10 μm, respectively (Fig. 3, A and D, and fig. S6A). EGFP-Cas9•sgRNA⊂SMNPs (Fig. 3, B and E) and HBB/GFP plasmid⊂SMNPs (Fig. 3, C and F) showed homogeneous spherical morphologies with sizes of 108 ± 37 and 125 ± 43 nm, respectively (fig. S6B). The surface-charge densities of SMNPs were determined by zeta potential measurements in phosphate-buffered saline (PBS) buffer solution, which showed that the zeta potentials of 120-nm 8% TAT–grafted Cas9•sgRNA⊂SMNPs and 140-nm 6% TAT–grafted HBB/GFP plasmid⊂SMNPs were +26 ± 4 and +23 ± 5 mV, respectively (fig. S6C). The assembly of SMNPs onto Ad-SiNWS (Fig. 3G) and the interactions between cells and Ad-SiNWS (Fig. 3, H and I) were also visualized by SEM, supporting the working mechanism of the SMNP/SNSMD strategy. According to stoichiometric calculations (see the Supplementary Materials), we estimate that ca. 130 Cas9•sgRNA complexes and two to three HBB/GFP plasmids were encapsulated into each Cas9•sgRNA⊂SMNP and HBB/GFP plasmid⊂SMNP under the optimal formulation conditions, respectively.
Fig. 3. SEM and TEM images of SMNPs and of Ad-SiNWS.
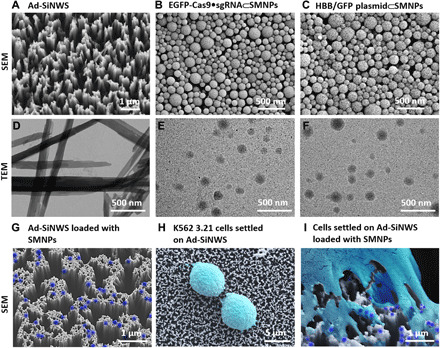
SEM images of (A) Ad-SiNWS, (B) 8% TAT–grafted EGFP-Cas9•sgRNA⊂SMNPs, and (C) 6% TAT–grafted HBB/GFP plasmid⊂SMNPs. TEM images of (D) free nanowires released from Ad-SiNWS, (E) EGFP-Cas9•sgRNA⊂SMNPs, and (F) HBB/GFP plasmid⊂SMNPs. SEM images of (G) EGFP-Cas9•sgRNA⊂SMNPs (colored in dark blue) Ad-SiNWS, (H) K562 3.21 cells (colored in light cyan) settled onto Ad-SiNWS, and (I) a K562 3.21 cell (light cyan) settled on EGFP-Cas9•sgRNA⊂SMNP–grafted (dark blue) Ad-SiNWS.
CRISPR-Cas9 knockin of the HBB/GFP gene in K562 3.21 cells
We then studied the combined SMNP/SNSMD strategy for CRISPR-Cas9 knockin of the HBB/GFP gene in K562 3.21 cells in conjunction with the use of both 120-nm 8% TAT–grafted Cas9•sgRNA⊂SMNPs and 140-nm 6% TAT–grafted HBB/GFP plasmid⊂SMNPs obtained above. Considering the two consecutive steps associated with the CRISPR-Cas9 knockin process (Fig. 1A), we hypothesized that the Cas9•sgRNA and donor HBB/GFP plasmid should arrive in sequence to optimize knockin efficiency. To test this hypothesis, we examined (Fig. 4A) how the delivery time interval (ΔT = 0, 2, 4, 6, 9, or 12 hours) between Cas9•sgRNA⊂SMNPs (containing 3.0 μg of Cas9 protein) and HBB/GFP plasmid⊂SMNPs (containing 2.0 μg of HBB/GFP plasmid) affected the knockin efficiency in growth-synchronized K562 3.21 cells. The treated cells were maintained until termination at day 10, followed by fluorescence microscopy imaging to quantify the GFP signals. Figure 4B compiles serial fluorescent micrographs of the resulting K562 3.21 cells, revealing that the highest HBB/GFP knockin efficiency (12%) was observed at ΔT = 6 hours. Furthermore, to take advantage of the combined SMNP/SNSMD strategy for serial delivery, three rounds of the SMNP treatments that ensure a steady supply of both Cas9•sgRNA and HBB/GFP plasmid over a period of 24 hours were conducted according to the timeline shown in Fig. 4C. Fluorescence microscopy imaging (Fig. 4D) and flow cytometry (fig. S7A) revealed that the three-round SMNP treatments resulted in a higher HBB/GFP knockin efficiency of 21% while showing minimum impact to cell viability and growth (fig. S7B).
Fig. 4. Optimizing the SMNP/SNSMD strategy for CRISPR-Cas9 knockin of the HBB/GFP gene in K562 3.21 cells using both Cas9•sgRNA⊂SMNPs and HBB/GFP plasmid⊂SMNPs.
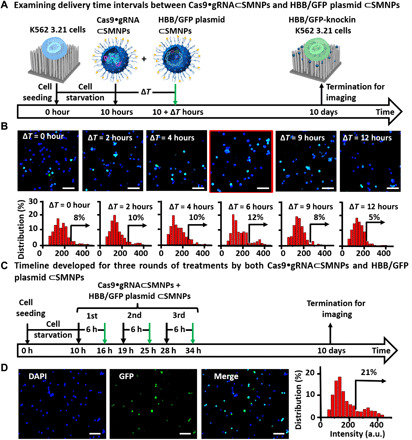
(A) A timeline depicting how different delivery time intervals (ΔT) affect CRISPR-Cas9 knockin performance. (B) Representative fluorescence images and histograms of the K562 3.21 cells obtained for ΔT = 0, 2, 4, 6, 9, or 12 hours. All scale bars are 100 μm. (C) A timeline developed for three rounds of SMNP treatments. (D) Fluorescence images and histograms of the K562 3.21 cells harvested after the three-round SMNP treatments. All scale bars are 100 μm. a.u., arbitrary units.
The studies above prompted us to explore the feasibility of coencapsulating both Cas9•sgRNA and HBB/GFP plasmid into a single SMNP vector to simplify the complicated procedures using the two vectors. On the basis of the previous formulation conditions, we prepared Cas9•sgRNA + HBB/GFP plasmid⊂SMNPs via stoichiometric mixing of the Cas9•sgRNA and HBB/GFP plasmid with the SMNP building blocks (fig. S8A). We also checked the sizes of the coencapsulated SMNPs via SEM and DLS, which showed that the coencapsulated SMNPs have larger sizes and size distributions (240 ± 90 nm; fig. S8, B and C). The surface-charge density of coencapsulated SMNPs was determined by zeta potential measurements in PBS buffer solution, which suggested that the zeta potential was 13 ± 4 mV (fig. S8D). The resulting Cas9•sgRNA + HBB/GFP plasmid⊂SMNPs were then used to study CRISPR-Cas9–mediated knockin (fig. S8E), where we found that the coencapsulated SMNP vector exhibited compromised knockin performance (5%).
The K562 3.21 cells harvested from the three-round SMNP treatments were sorted by flow cytometry to obtain purified HBB/GFP-knockin K562 3.21 cells. Over 20 rounds of culture expansion, these cells displayed consistent GFP signals (Fig. 5A), suggesting their clonal stability. To test the CRISPR-Cas9–mediated knockin (Fig. 5B) of HBB/GFP gene into the AAVS1 site via the HDR pathway, we extracted the genomic DNA from HBB/GFP-knockin K562 3.21 cells, followed by PCR analysis and Sanger sequencing. After PCR amplification, the two characteristic DNA fragments, the 5′ junction (1.1 kb) and the 3′ junction (1.2 kb), signifying the integration of 3.7-kb HBB/GFP into the AAVS1, were detected by electrophoresis (Fig. 5C). Sanger sequencing was used to analyze the DNA fragments at four genome-donor boundaries (colored arrows in Fig. 5, B and D) in both of the 5′ and 3′ junctions, indicating precise integration of the HBB/GFP gene. We further examine whether the integrated HBB gene could functionally express HBB protein. Together with two control cells (untreated K562 3.21 and RBCs from a healthy donor), HBB/GFP-knockin K562 3.21 cells were subjected to IF staining. As shown in Fig. 5E, strong red fluorescence signals (marking IF-stained HBB protein) were observed in both HBB/GFP-knockin K562 3.21 and RBCs. Next, qPCR was performed to quantify HBB mRNA expression (normalized against a housekeeping gene, β-actin) in these cells. The results, shown in Fig. 5F, reveal that increased HBB expression was observed for HBB/GFP-knockin K562 3.21, in contrast to the untreated K562 3.21.
Fig. 5. Analysis of K562 3.21 cells for HBB/GFP gene integration into the AAVS1 site.
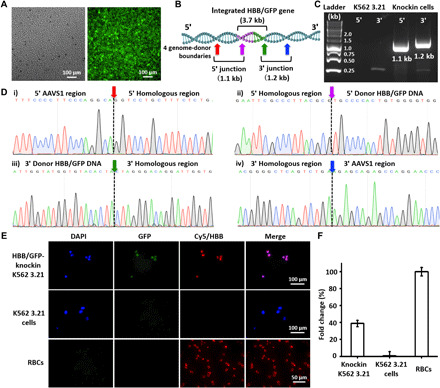
(A) Fluorescence microscopy images of purified HBB/GFP-knockin K562 3.21 cells after 20 rounds of culture expansion. (B) Schematic depicting the integration of HBB/GFP gene into the AAVS1 site, in which the locations of the two HDR junctions and four genome-donor boundaries are labeled. (C) Electrophoretogram shows the two characteristic DNA fragments, i.e., the 5′ junction (1.1 kb) and the 3′ junction (1.2 kb). (D) Sanger sequencing of the four genome-donor boundaries. (E) Representative IF images of HBB/GFP-knockin K562 3.21, K562 3.21, and RBCs. (F) qPCR for quantification of HBB mRNA expression.
In vivo study
To test the proliferative potential of HBB/GFP-knockin K562 3.21 cells in vivo, approximately 1 × 107 HBB/GFP-knockin K562 3.21 cells in 200 μl of Matrigel were injected intraperitoneally in nonirradiated athymic nude mice (n = 3; Fig. 6A). Six weeks after intraperitoneal injection, the mice were euthanized by cervical dislocation under deep anesthesia. To track HBB/GFP-knockin K562 3.21 cells in the xenograft tumors, the IVIS 200 System (PerkinElmer, Waltham, MA) was used to image the entire peritoneal cavity, individual organs, and resected tumors. As shown in Fig. 6B and fig. S9, GFP signals were only observed in the xenograft tumors of the mice injected with HBB/GFP-knockin K562 3.21 cells. Subsequently, the tumor tissues were subjected to PCR analysis (Fig. 6C) and Sanger sequencing (Fig. 6D) to test the stability of the integrated HBB/GFP gene in vivo throughout the repopulating process. Tumor sections were prepared for standard pathology hematoxylin-eosin (H&E) staining and immunohistochemistry (IHC) staining for HBB. Two pathologists reviewed all slides independently, reporting that (i) tumor cells were observed (Fig. 6E) and that (ii) HBB positivity in the cytoplasm of the tumor cells (Fig. 6F) with the RBCs was being used for HBB-positive control (Fig. 6G) and lymphocytes for HBB-negative control (Fig. 6H). Collectively, these experimental data support the fact that the injected HBB/GFP-knockin K562 3.21 cells maintain their ability to proliferate and maintain consistent gene expression in vivo.
Fig. 6. Analysis of the proliferative potential of HBB/GFP-knockin K562 3.21 cells in vivo.
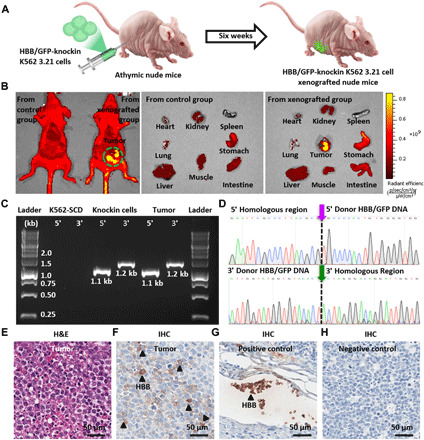
(A) HBB/GFP-knockin K562 3.21 cells were introduced into athymic nude mice via intraperitoneal injection to test the cells’ in vivo growth potential. (B) Representative fluorescence images of mice, organs, and xenograft tumors 6 weeks after intraperitoneal injection. The harvested tumor tissues were subjected to (C) PCR analysis and (D) Sanger sequencing. (E to H) Representative images of hematoxylin-eosin (H&E) staining of the xenograft tumor tissue, immunohistochemistry (IHC) staining for HBB protein expression in the tumor tissue, positive control of IHC staining for HBB, and negative control of IHC staining for HBB, respectively.
DISCUSSION
In the context of hemoglobinopathies, two main CRISPR-Cas9 gene editing strategies have been reported, including (i) knockdown of the BCL11A gene via NHEJ to elevate fetal hemoglobin (HbF) levels and (ii) correction of HBB gene mutations via HDR (29). Genetic variants of the BCL11A gene are found to regulate HbF expression. Knockdown of BCL11A leads to increased HbF levels in the erythroid lineage, which is being applied clinically in gene therapies for hemoglobinopathies (30). However, ubiquitous BCL11A knockdown weakened human RBC enucleation and impaired engraftment of human repopulating HSCs (5). The HDR-based strategy has been investigated for the site-specific correction of the point mutation (A to T) that leads to SCD at the HBB locus in human HSCs (31). However, this strategy can only cure SCD, not β-thalassemia, due to the multiple mutations associated with this disease. Moreover, failed gene correction at the HBB locus can lead to gene disruption by NHEJ, generating a β-thalassemic phenotype instead of correcting the SCD mutation (5). The AAVS1 safe-harbor locus is an ideal site in the human genome for knockin of a new gene, which does not cause alterations of the host genome and is safe for the host cell or organism (32). Hence, knockin of HBB into AAVS1 can provide a safe and general curative therapeutic solution for both SCD and β-thalassemia.
We introduced an effective CRISPR-Cas9–mediated knockin approach using a combined SMNP/SNSMD strategy, which enables step-by-step delivery of Cas9•sgRNA complex and dDNA (HBB/GFP plasmid) encapsulated in two different SMNP vectors. By conducting small-scale combinatorial screenings, optimal formulations were identified for the preparation of TAT-grafted Cas9•sgRNA⊂SMNPs (average sizes = 108 ± 37 nm and 8% TAT coverage) and HBB/GFP plasmid⊂SMNPs (average sizes = 125 ± 43 nm and 6% TAT coverage). Compared with commercially available CRISPR and plasmid delivery reagents (Lipofectamine CRISPRMax and Lipofectamine 3000), the SMNP/SNSMD strategy showed higher performances in cell uptake and GFP transfection. Using this nonviral delivery approach, we demonstrated successful integration of a single-copy HBB/GFP gene into the AAVS1 safe-harbor site of a SCD cell model. Targeted genomic editing by CRISPR-Cas9 can efficiently generate knockout cells via the NHEJ pathway, but the efficiency of gene knockin by HDR is substantially lower (33). Furthermore, increasing the inserted gene size would reduce knockin efficiency (34). Compared with other gene knockin studies (table S1) (34–39), higher knockin efficiency (21%) with longer DNA insertion (3.7-kb HBB/GFP gene) was obtained via optimization of the delivery time interval and multiple treatments of these two SMNP vectors while keeping high cell viability. Physical contact between the cell membranes and the nanowires led to efficient uptake of SMNPs. Meanwhile, the HBB/GFP-knockin K562 3.21 cells maintained their ability to proliferate consistent HBB gene expression in vivo, showing the feasibility of a potential therapeutic solution for hemoglobinopathies.
In summary, this proof-of-concept study highlights (i) the potential of the combined SMNP/SNSMD strategy as an effective delivery platform capable of codelivering Cas9•sgRNA and dDNA into hematologic cells (known to be difficult to transfect) and (ii) the demonstration of an efficient CRISPR-Cas9–mediated knockin of a long-frame DNA sequence (i.e., 3.7-kb HBB/GFP gene) using nonviral vectors. Further research on CD34+ HSCs is currently ongoing. We envision that this approach may enable CRISPR-Cas9–mediated knockin of HBB genes using autologous CD34+ cells, offering a general clinical therapeutic solution for hemoglobinopathies.
MATERIALS AND METHODS
sgRNA synthesis
Briefly, the sequence of sgRNA used to target the AAVS1 locus is ggggccacuagggacaggauGUUUUAGAGCUAGAAAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCUUUUUU, which was synthesized by Integrated DNA Technologies Inc. (Iowa).
Cell culture
The human leukemia K562 3.21 cells were cultured in a humidified atmosphere of 5% CO2/air in RPMI-1640 medium, supplemented with 10% fetal bovine serum, penicillin (100 U/ml), and streptomycin (100 μg/ml). Individual wells of an eight-well plate were inoculated with complete medium containing 10,000 of K562 3.21 cells per milliliter. The plates were incubated at 37°C in a humidified 5% incubator for 18 hours before the experiments.
Synthesis of EGFP-Cas9•sgRNA⊂SMNPs
Self-assembly was used to prepare the EGFP-Cas9 protein and sgRNA-encapsulated SMNPs (EGFP-Cas9•sgRNA⊂SMNPs). Three batches of EGFP-Cas9•sgRNA⊂SMNPs were formulated by systemically modulating (i) the weight ratios (wt %) between SMNP vector and EGFP-Cas9•sgRNA payload, (ii) sizes of EGFP-Cas9•sgRNA⊂SMNPs, and (iii) the percentages of TAT ligand on SMNP surfaces. The optimized formulation was prepared as follows: A total of 2.0 μl of dimethyl sulfoxide (DMSO) solution containing Ad-PAMAM (7.2 μg) was added into a 50 μl of PBS mixture with EGFP-Cas9 protein (3.0 μg), sgRNA (0.6 μg, molar ratio ≈ 1:1), Ad-PEG (45 μg), CD-PEI (20 μg), and Ad-PEG–TAT (3.6 μg). The resulting mixture was then stirred vigorously to achieve optimal Cas9•sgRNA⊂SMNPs. The mixture was stored at 4°C for 1 hour; afterward, DLS, SEM, and TEM were used to characterize the sizes of EGFP-Cas9•sgRNA⊂SMNPs.
Delivery EGFP-Cas9•sgRNA⊂SMNPs to K562 3.21 cells
Before settling the cells onto Ad-SiMWS, K562 3.21 cells were first starved in serum-free RPMI medium in 75-cm2 cell culture flask for 10 hours to synchronize cells to G0/G1 phases of cell cycle (28) and then centrifuged to remove medium at the rate of 300g for 5 min. Approximately K562 3.21 cells (1 × 105 in 1.0 ml of serum-containing RPMI medium) were introduced into each well of an eight-well plate, where a 2.5 × 3 cm2 Ad-SiNWS was placed. Last, the Cas9•sgRNA⊂SMNPs (containing 3.0 μg of Cas9 protein) in 1.0 ml of serum-containing RPMI medium were added to each well. The cells were coincubated with SMNPs for a certain period. Every 48 hours, 1.0 ml of medium was removed via pipette and then a new 1.0 ml of serum-containing RPMI medium was added to each well. After washing with PBS, the cells in the well were immediately fixed with 2% paraformaldehyde (PFA) and then stained with DAPI. Microscopy-based image cytometry was used to detect the cellular uptake performances of different formulations. After each treatment, the GFP signal was quantified with a fluorescent microscope equipped with a charge-coupled device CCD camera (Nikon TE2000S, Japan).
Delivery Cas9•sgRNA⊂Cy5-SMNPs to K562 3.21 cells
The growth-synchronized K562 3.21 cells (1 × 105 in 1.0 ml of serum-containing RPMI medium) were introduced into each well of an eight-well plate, where a 2.5 × 3 cm2 Ad-SiNWS was placed. The Cas9•sgRNA⊂Cy5-SMNPs (containing 3.0 μg of Cas9 protein) in 1.0 ml of serum-containing RPMI medium were added to each well. The cells were coincubated with SMNPs for a certain period. Every 48 hours, 1.0 ml of medium was removed from each well and replaced with 1.0 ml of serum-containing RPMI medium. After washing with PBS, the cells in the well were immediately fixed with 2% PFA and then stained with DAPI. Microscopy-based image cytometry was used to detect the cellular uptake performances of different formulations. After different treatments, the Cy5 signal was quantified using a fluorescent microscope with a CCD camera (Nikon TE2000S, Japan).
Off-target analysis
Off-target analysis of sgRNA-AAVS1 was performed using the Cas-OFFinder design tool (http://rgenome.net/cas-offinder/). The top five off-target hits with canonical PAM sequence based on our query were as follows: GGGGCaACTAGaGACAGGAaGGG (chromosome 8, three mismatches), GGtGCCACTAGGcACAGGAgCGG (chromosome 8, three mismatches), tGGGCCACTAtGGACAGGAaTGG (chromosome 12, three mismatches), GaGGCCACcAGGGACAGGcTGGG (chromosome 5, three mismatches), and GGGGtacCTAGGGtCAGGATGGG (chromosome 8, four mismatches). The PCR primers for these regions were designed accordingly.
Synthesis of HBB/GFP plasmid⊂SMNPs
A similar self-assembly procedure was applied to prepare the HBB/GFP plasmid⊂SMNPs. Three batches of HBB/GFP plasmid⊂SMNPs were formulated by systematically modulating (i) the weight ratios (wt %) between SMNP vector and EGFP-Cas9•sgRNA payload, (ii) the sizes of HBB/GFP plasmid⊂SMNPs, and (iii) the percentages of TAT ligand on SMNP surfaces. The optimized formulation was synthesized as follows: A total of 2.0 μl of DMSO solution containing Ad-PAMAM (15 μg) was added into a 50 μl of PBS mixture with HBB/GFP plasmid (1.0 μg), Ad-PEG (23 μg), CD-PEI (10 μg), and Ad-PEG-TAT (1.4 μg). The resulting mixture was then stirred vigorously to generate optimal HBB/GFP plasmid⊂SMNPs.
Delivery HBB/GFP plasmid⊂SMNPs to K562 3.21 cells
The growth-synchronized K562 3.21 cells (1 × 105 in 1.0 ml of serum-containing RPMI medium) were introduced into each well of an eight-well plate, where a 2.5 × 3 cm2 Ad-SiNWS was placed. The HBB/GFP plasmid⊂SMNPs (containing 2.0 μg of HBB/GFP plasmid) in 1.0 ml of serum-containing RPMI medium were added to the well. The cells were coincubated with SMNPs for a certain period. After washing with PBS, the cells in each chamber were immediately fixed with 2% PFA and then stained with DAPI. Microscopy-based image cytometry was used to detect the cellular uptake performances of different formulations.
Delivery Cas9•sgRNA⊂SMNPs and HBB/GFP plasmid⊂SMNPs to K562 3.21 cells for gene knockin study
The growth-synchronized K562 3.21 cells (1 × 105 in 1.0 ml of serum-containing RPMI medium) were introduced into each well of an eight-well plate. We first examined how delivery time interval (ΔT = 0, 2, 4, 6, 9, or 12 hours) between Cas9•sgRNA⊂SMNPs (containing 3.0 μg of Cas9 protein in 0.5 ml of serum-containing RPMI medium) and HBB/GFP plasmid⊂SMNPs (containing 2.0 μg of HBB/GFP plasmid in 0.5 ml of serum-containing RPMI medium) affected the knockin efficiency in K562 3.21 cells. The cells were coincubated with two SMNPs for 10 days. Every 2 days, 1.0 ml of medium was removed using a pipette, and then a new 1.0 ml of serum-containing RPMI medium was added to each well. After washing with PBS, the cells in the well were immediately fixed with 2% PFA and then stained with DAPI. Microscopy-based image cytometry was used to measure the delivery performances of different conditions.
Delivery Cas9•sgRNA + HBB/GFP plasmid⊂SMNPs to K562 3.21 cells for gene knockin study
The growth-synchronized K562 3.21 cells (1 × 105 in 1.0 ml of serum-containing RPMI medium) were introduced into each well of an eight-well plate. The Cas9•sgRNA + HBB/GFP plasmid⊂SMNPs (containing 3.0 μg of Cas9 protein and 2.0 μg of HBB/GFP plasmid in 1.0 ml of serum-containing RPMI medium) were added to each well. The cells were coincubated with SMNPs for 10 days. Every 2 days, 1.0 ml of medium was removed using a pipette, and then 1.0 ml of new serum-containing RPMI medium was added to each well. After washing with PBS, the cells in the well were immediately fixed with 2% PFA, stained with DAPI, and then evaluated via microscopy-based image cytometry to evaluate the delivery performance of different conditions.
DNA extraction and PCR
The HBB/GFP-knockin K562 3.21 cells were harvested and then washed with PBS. The genomic DNA was extracted with the commercial QIAamp DNA Mini Kit (Qiagen, Germany) using the manufacturer’s instructions. Then, PCR was conducted to amplify integrated HBB/GFP gene with a S1000TM Thermal Cycler (Bio-Rad) under the following PCR conditions: 95°C for 3 min followed by 35 cycles (95°C for 15 s, 58°C for 15 s, and 72°C for 20 s) and 72°C for 3 min. The PCR products were checked on a 1.5% electrophoresis gel.
The PCR primer sequences are listed as follows: 5′ junction forward primer, 5′-CCGGAACTCTGCCCTCTAAC-3′; 5′ junction reverse primer, 5′-AGTAGGAAAGTCCCATAAGGTCA-3′; 3′ junction forward primer, 5′-AAGCTCATCTGGTCTCCCTTCC-3′; and 3′ junction reverse primer, 5′-TCCTGGGATACCCCGAAGAG-3′.
Quantitative PCR
After adding TRIzol (800 μl), the cells were homogenized, treated with chloroform (160 μl), and centrifuged for 15 min at 4°C. The aqueous phase of the sample was removed by pipette, and 100% isopropanol (400 μl) was added. After being centrifuged for 10 min, the supernatant was removed from the tube, and the pellet was washed with 75% ethanol and centrifuged for 5 min. Afterward, the supernatant was removed, and the pellet was dissolved in deoxyribonuclease- and ribonuclease-free water. RNA (1 μg) was reverse transcribed using the SuperScript III First-Strand Synthesis Kit. qPCR analysis was performed using PowerUp SYBR Green Master Mix (Applied Biosystems, California) with the primers below. Values were normalized against the gene expression of the housekeeping gene β-actin. qPCR primer β-globin forward primer, 5′-CTCATGGCAAGAAAGTGCTCG-3′ and reverse primer, 5′-AATTCTTTGCCAAAGTGATGGG-3′.
IF staining
The living cells were fixed in 4% paraformaldehyde, permeabilized in 0.1% Triton X-100, and blocked in 5% normal bovine serum albumin in PBS. The cells were incubated with an HBB antibody (Abcam, ab214049). After being washed three times with PBS, the cells were incubated with secondary antibodies (donkey anti-rabbit) conjugated with Cy5 (red). DAPI (blue) was used as the nuclear stain. Labeled cells were imaged with a laser-scanning confocal microscope (Olympus). The total amount of retained immunofluorescent material was determined in the green (488 nm) and the red (546 nm) channels.
In vivo studies
All animal studies were conducted under an approved Institutional Animal Care and Use Committee protocol. A total of 1 × 107 HBB/GFP-knockin K562 3.21 cells in 200 μl of Matrigel were injected intraperitoneally in nonirradiated athymic nude female mice (n = 3, 6 to 7 weeks of age; Envigo Company) under brief isoflurane anesthesia. Tumors were staged for 6 weeks. Then, the animals were euthanized and their peritoneal cavities were exposed. For the visualization of HBB/GFP-knockin K562 3.21 cells in organs and tumor, the organs were excised from the peritoneal cavities and placed on a plate with a white background. The IVIS 200 System (PerkinElmer, Waltham, MA) was used to obtain fluorescent images of the whole peritoneal cavity, individual organs, and tumors. Living Image Software version 4.1 (PerkinElmer) was used for GFP fluorescence analysis. To aid in visualization, the GFP signal was pseudocolored by applying a logarithmic grayscale to the whole peritoneal cavity image or by applying reverse logarithmic grayscale to the images of the organs and tumor. Genomic DNA was extracted from the tumor of each mouse by the DNeasy Blood and Tissue Kit (Qiagen GmbH, Germany). The presence of human-specific DNA within the transplanted mice was confirmed by PCR and Sanger sequencing. The IHC experiments were carried out on 4-μm-thick formalin-fixed and paraffin-embedded tissue sections. The sections were prepared for standard pathology H&E staining and IHC staining for HBB using a commercially available detection kit (Dako EnVision Plus-HRP, Dako), according to the manufacturer’s instructions.
Supplementary Material
Acknowledgments
Funding: This research was supported by the NIH (R21EB016270 and DP5OD028181) and gift funding from Dr. H. Ji and Sorrento Therapeutics. This research was also supported by Ministry of Science and Technology (MOST 108-2119-M-010-001, MOST 109-2320-B-075-009), Taipei Veterans General Hospital (V108E-006-5), National Yang-Ming University by the Ministry of Education in Taiwan. Author contributions: P.Y. and S.-J.C. equally contributed to this work. P.Y., S.-J.C., Q.B., and H.-R.T. designed the research. P.Y., S.-J.C., Q.B., J.L., W.L., N.S., R.Y.Z., M.-L.T., and H.I.L. performed the experiments. P.Y., S.-J.C., N.S., R.Y.Z., Y.Z., S.J.J., D.B.K., K.C., P.S.W., S.-H.C., and H.R.T. analyzed the data. P.Y., S.-J.C., Q.B., W.H., and H.-R.T. prepared the manuscript. P.Y., S.-J.C., X.Z., and J.C. drew the figures. M.S., S.J.J., D.B.K., K.C., P.S.W., S.-H.C. revised the manuscript. Z.R., D.L., Z.K., C.Z., and C.-F.L. commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data are available from authors upon request.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/43/eabb7107/DC1
REFERENCES AND NOTES
- 1.Williams T. N., Weatherall D. J., World distribution, population genetics, and health burden of the hemoglobinopathies. Cold Spring Harb. Perspect. Med. 2, a011692 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sirdah M. M., Sievertsen J., Al-Yazji M. S., Tarazi I. S., Al-Haddad R. M., Horstmann R. D., Timmann C., The spectrum of β-thalassemia mutations in Gaza Strip, Palestine. Blood Cells Mol. Dis. 50, 247–251 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Compernolle V., Chou S. T., Tanael S., Savage W., Howard J., Josephson C. D., Odame I., Hogan C., Denomme G., Shehata N., Red blood cell specifications for patients with hemoglobinopathies: A systematic review and guideline. Transfusion 58, 1555–1566 (2018). [DOI] [PubMed] [Google Scholar]
- 4.King A., Shenoy S., Evidence-based focused review of the status of hematopoietic stem cell transplantation as treatment of sickle cell disease and thalassemia. Blood 123, 3089–3094 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Magrin E., Miccio A., Cavazzana M., Lentiviral and genome-editing strategies for the treatment of β-hemoglobinopathies. Blood 134, 1203–1213 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Cavazzana M., Antoniani C., Miccio A., Gene therapy for β-hemoglobinopathies. Mol. Ther. 25, 1142–1154 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doudna J. A., Charpentier E., The new frontier of genome engineering with CRISPR-Cas9. Science 346, 1258096 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Doudna J. A., The promise and challenge of therapeutic genome editing. Nature 578, 229–236 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li L., Hu S., Chen X., Non-viral delivery systems for CRISPR/Cas9-based genome editing: Challenges and opportunities. Biomaterials 171, 207–218 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Senís E., Fatouros C., Große S., Wiedtke E., Niopek D., Mueller A. K., Börner K., Grimm D., CRISPR/Cas9-mediated genome engineering: an adeno-associated viral (AAV) vector toolbox. Biotechnol. J. 9, 1402–1412 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Wu Z., Yang H., Colosi P., Effect of genome size on AAV vector packaging. Mol. Ther. 18, 80–86 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang H.-X., Li M., Lee C. M., Chakraborty S., Kim H.-W., Bao G., Leong K. W., CRISPR/Cas9-based genome editing for disease modeling and therapy: Challenges and opportunities for nonviral delivery. Chem. Rev. 117, 9874–9906 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Rui Y., Wilson D. R., Green J. J., Non-viral delivery to enable genome editing. Trends Biotechnol. 37, 281–293 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li L., Yang Z., Zhu S., He L., Fan W., Tang W., Zou J., Shen Z., Zhang M., Tang L., A rationally designed semiconducting polymer brush for NIR-II imaging-guided light-triggered remote control of CRISPR/Cas9 genome editing. Adv. Mater. 31, 1901187 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Liu J., Chang J., Jiang Y., Meng X., Sun T., Mao L., Xu Q., Wang M., Fast and efficient CRISPR/Cas9 genome editing in vivo enabled by bioreducible lipid and messenger RNA nanoparticles. Adv. Mater. 31, 1902575 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rui Y., Wilson D. R., Choi J., Varanasi M., Sanders K., Karlsson J., Lim M., Green J. J., Carboxylated branched poly (β-amino ester) nanoparticles enable robust cytosolic protein delivery and CRISPR-Cas9 gene editing. Sci. Adv. 5, eaay3255 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee K., Conboy M., Park H. M., Jiang F., Kim H. J., Dewitt M. A., Mackley V. A., Chang K., Rao A., Skinner C., Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat. Biomed. Eng. 1, 889–901 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng Q., Wei T., Farbiak L., Johnson L. T., Dilliard S. A., Siegwart D. J., Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR–Cas gene editing. Nat. Nanotech. 15, 313–320 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee B., Lee K., Panda S., Gonzales-Rojas R., Chong A., Bugay V., Park H. M., Brenner R., Murthy N., Lee H. Y., Nanoparticle delivery of CRISPR into the brain rescues a mouse model of fragile X syndrome from exaggerated repetitive behaviours. Nat. Biomed. Eng. 2, 497–507 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mora-Raimundo P., Lozano D., Manzano M., Vallet-Regí M., Nanoparticles to knockdown osteoporosis-related gene and promote osteogenic marker expression for osteoporosis treatment. ACS Nano 13, 5451–5464 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang H.-X., Song Z., Lao Y.-H., Xu X., Gong J., Cheng D., Chakraborty S., Park J. S., Li M., Huang D., Nonviral gene editing via CRISPR/Cas9 delivery by membrane-disruptive and endosomolytic helical polypeptide. Proc. Natl. Acad. Sci. U.S.A. 115, 4903–4908 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang H., Wang S., Su H., Chen K. J., Armijo A. L., Lin W. Y., Wang Y., Sun J., Kamei K. I., Czernin J., A supramolecular approach for preparation of size-controlled nanoparticles. Angew. Chem. Int. Ed. 48, 4344–4348 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang F., Yang P., Choi J.-s., Antovski P., Zhu Y., Xu X., Kuo T.-H., Lin L.-E., Kim D. N., Huang P.-C., Cross-linked fluorescent supramolecular nanoparticles for intradermal controlled release of antifungal drug—A therapeutic approach for onychomycosis. ACS Nano 12, 6851–6859 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Peng J., Garcia M. A., Choi J.-s., Zhao L., Chen K.-J., Bernstein J. R., Peyda P., Hsiao Y.-S., Liu K. W., Lin W.-Y., Molecular recognition enables nanosubstrate-mediated delivery of gene-encapsulated nanoparticles with high efficiency. ACS Nano 8, 4621–4629 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoban M. D., Lumaquin D., Kuo C. Y., Romero Z., Long J., Ho M., Young C. S., Mojadidi M., Fitz-Gibbon S., Cooper A. R., CRISPR/Cas9-mediated correction of the sickle mutation in human CD34+ cells. Mol. Ther. 24, 1561–1569 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Y., Du J., Choi J. S., Chen K. J., Hou S., Yan M., Lin W. Y., Chen K. S., Ro T., Lipshutz G. S., A high-throughput platform for formulating and screening multifunctional nanoparticles capable of simultaneous delivery of genes and transcription factors. Angew. Chem. Int. Ed. 55, 169–173 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chu V. T., Weber T., Wefers B., Wurst W., Sander S., Rajewsky K., Kühn R., Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 33, 543 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Golding S. E., Rosenberg E., Khalil A., McEwen A., Holmes M., Neill S., Povirk L. F., Valerie K., Double strand break repair by homologous recombination is regulated by cell cycle-independent signaling via ATM in human glioma cells. J. Biol. Chem. 279, 15402–15410 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Lidonnici M. R., Ferrari G., Gene therapy and gene editing strategies for hemoglobinopathies. Blood Cells Mol. Dis. 70, 87–101 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Canver M. C., Smith E. C., Sher F., Pinello L., Sanjana N. E., Shalem O., Chen D. D., Schupp P. G., Vinjamur D. S., Garcia S. P., BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 527, 192–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dever D. P., Bak R. O., Reinisch A., Camarena J., Washington G., Nicolas C. E., Pavel-Dinu M., Saxena N., Wilkens A. B., Mantri S., CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature 539, 384–389 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Papapetrou E. P., Schambach A., Gene insertion into genomic safe harbors for human gene therapy. Mol. Ther. 24, 678–684 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richardson C. D., Ray G. J., DeWitt M. A., Curie G. L., Corn J. E., Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 34, 339–344 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Paix A., Folkmann A., Goldman D. H., Kulaga H., Grzelak M. J., Rasoloson D., Paidemarry S., Green R., Reed R. R., Seydoux G., Precision genome editing using synthesis-dependent repair of Cas9-induced DNA breaks. Proc. Natl. Acad. Sci. U.S.A. 114, E10745–E10754 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang J.-P., Li X.-L., Li G.-H., Chen W., Arakaki C., Botimer G. D., Baylink D., Zhang L., Wen W., Fu Y.-W., Efficient precise knockin with a double cut HDR donor after CRISPR/Cas9-mediated double-stranded DNA cleavage. Genome Biol. 18, 35 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yao X., Wang X., Liu J., Hu X., Shi L., Shen X., Ying W., Sun X., Wang X., Huang P., CRISPR/Cas9–Mediated precise targeted integration in vivo using a double cut donor with short homology arms. EBioMedicine 20, 19–26 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yao X., Wang X., Hu X., Liu Z., Liu J., Zhou H., Shen X., Wei Y., Huang Z., Ying W., Homology-mediated end joining-based targeted integration using CRISPR/Cas9. Cell Res. 27, 801–814 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Song J., Yang D., Xu J., Zhu T., Chen Y. E., Zhang J., RS-1 enhances CRISPR/Cas9-and TALEN-mediated knock-in efficiency. Nat. Commun. 7, 1–7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He X., Tan C., Wang F., Wang Y., Zhou R., Cui D., You W., Zhao H., Ren J., Feng B., Knock-in of large reporter genes in human cells via CRISPR/Cas9-induced homology-dependent and independent DNA repair. Nucleic Acids Res. 44, e85–e85 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/43/eabb7107/DC1