

Significance Statement
Nephrons are derived from nephron progenitor cells. Nephron progenitors are depleted during kidney development, which makes the kidney unable to regenerate nephrons. Therefore, understanding the signaling molecules that regulate nephron progenitor cell generation and maintenance is of great interest for kidney regeneration. Sprouty1 regulates nephron progenitor maintenance by inhibiting Fibroblast growth factor (FGF) signaling. Deletion of Sprouty1 rescues renal agenesis and nephron progenitor depletion in Fgf9/20 loss-of-function kidneys. Deletion of one copy of Fgf8 further decreases FGF signaling, which blocks kidneys’ response to Sprouty1, resulting in failure of nephron progenitor maintenance. This study thus identifies the reciprocal functioning of FGF-Sprouty1 signaling during nephron progenitor development.
Keywords: FGF signal, Spry1, nephron progenitor cells, kidney development
Visual Abstract

Abstract
Background
Nephron progenitor cells (NPCs) give rise to all segments of functional nephrons and are of great interest due to their potential as a source for novel treatment strategies for kidney disease. Fibroblast growth factor (FGF) signaling plays pivotal roles in generating and maintaining NPCs during kidney development, but little is known about the molecule(s) regulating FGF signaling during nephron development. Sprouty 1 (SPRY1) is an antagonist of receptor tyrosine kinases. Although SPRY1 antagonizes Ret-GDNF signaling, which modulates renal branching, its role in NPCs is not known.
Methods
Spry1, Fgf9, and Fgf20 compound mutant animals were used to evaluate kidney phenotypes in mice to understand whether SPRY1 modulates FGF signaling in NPCs and whether FGF8 functions with FGF9 and FGF20 in maintaining NPCs.
Results
Loss of one copy of Spry1 counters effects of the loss of Fgf9 and Fgf20, rescuing bilateral renal agenesis premature NPC differentiation, NPC proliferation, and cell death defects. In the absence of SPRY1, FGF9, and FGF20, another FGF ligand, FGF8, promotes nephrogenesis. Deleting both Fgf8 and Fgf20 results in kidney agenesis, defects in NPC proliferation, and cell death. Deleting one copy of Fgf8 reversed the effect of deleting one copy of Spry1, which rescued the renal agenesis due to loss of Fgf9 and Fgf20.
Conclusions
SPRY1 expressed in NPCs modulates the activity of FGF signaling and regulates NPC stemness. These findings indicate the importance of the balance between positive and negative signals during NPC maintenance.
Receptor tyrosine kinases (RTKs) are activated upon binding of their cognate ligands and regulate many aspects of organogenesis. The intracellular signals initiated by RTK activation determine cellular behavior, such as proliferation, survival, cell fate determination, and morphogenesis. Misregulation of RTK activity leads to the onset and progression of a wide range of disease, such as diabetes, inflammation, bone disorders, atherosclerosis, angiogenesis, and various cancers.1,2 Therefore, tight regulation of the activity of RTK should be guaranteed during development and homeostasis.3
RTK plays a crucial role during kidney development. Glial cell line–derived neurotrophic factor (GDNF) produced in the nephron progenitor cells (NPCs) activates the RET/GFRα1 RTK/coreceptor in the ureteric bud (UB) tips.4 Deletion of Gdnf, Ret, or Gfrα1 during development results in renal agenesis due to failure of UB induction.5–8 UB branching is sensitive to the level of GDNF because deleting one copy of Gdnf or decreasing expression of Gdnf due to loss of Fras1 produces unilateral renal agenesis or hypoplasia.9,10 However, FGF9 and FGF20 activate FGF receptor 1 (FGFR1) and FGFR2 in NPCs and maintain the stemness of NPCs. Loss of FGF20 in human, loss of Fgf9 and Fgf20 in mice, or metanephric mesenchyme-specific deletion of Fgfr1 and Fgfr2 causes bilateral renal agenesis.11–13 In addition, reduction in Fgf9 and Fgf20 levels results in loss of NPCs and premature differentiation, indicating that stemness of NPCs are sensitive to the copy number of Fgf genes.12
Sprouty (SPRY) is a negative feedback regulator of RTK signaling. During kidney development, SPRY negatively regulates activity of the GDNF-RET signal. Either knockout or treatment of Spry1 with antisense oligonucleotides generates supernumerary and dilated UBs.14,15 In addition, ectopic expression of SPRY2 in the developing kidney leads to renal hypoplasia and agenesis.16 Furthermore, deleting Spry1 restores kidneys in the Gdnf (or its expression level) or Ret knockout mice.17–19 When both GDNF-RET and SPRY signaling is depleted, FGF10 promotes UB branching.18 Although these studies point to the importance of SPRY1 in UB branching, the function of SPRY1 in the NPCs in vivo remains to be determined.
Here, we show SPRY1 regulates NPC maintenance by modulating FGF signaling. Both germ line and NPC-specific deletion of Spry1 rescues renal agenesis in Fgf9−/−;Fgf20−/− embryos. We also show that FGF8 is another FGF ligand promoting NPC maintenance. Combinatory gene deletion analyses show that balance of FGFs and Spry1 level is critical to maintain NPCs.
Methods
Mice
Spry1−/+15, Spry1fl/+ (MMRRC:029870),15 Fgf8fl/+,20 Fgf8−/+,21 Fgf9−/+,22 Fgf20−/+,23 Fgf20Cre/+,24 and RosaTdTomato/+ (Jax#007905) mouse lines were used. All mice were on a mixed C57BL/6J and 129X1/SvJ genetic background and maintained in the University of Nebraska Medical Center animal facility according to animal care regulations, and the Animal Care and Use Committee (protocol number 16-005-02-EP).
Histology
For histologic analysis, embryonic day 18.5 (E18.5) kidneys were dissected from embryos, fixed with 4% paraformaldehyde overnight at 4°C, and dehydrated with ethanol gradients. Dehydrated kidneys were embedded in paraffin and sectioned. The paraffin-embedded sections were deparaffinized, hydrated, and stained with hematoxylin and eosin. Stained slides were dehydrated, mounted, and photographed with a Zeiss microscope.
Immunohistochemistry
Embryos (E10.5 and E11.5) were incubated with 30% sucrose solution. For E18.5 samples, kidneys were isolated from embryos and incubated with series of sucrose solutions (10%, 20%, and 30%). Samples were frozen sectioned and kept at −80°C for storage. For antibody staining, sections were washed with PBS with 0.5% Triton X-100 (PBST) for 30 minutes at room temperature (RT) and incubated with blocking solution (5% donkey serum, in PBST) for 1 hour at RT. Sections were incubated with primary antibodies in PBST with 1% donkey serum overnight at 4°C. Sections were washed three times with PBS and incubated with secondary antibodies for 30 minutes at RT. After washing three times with PBS, slides were mounted with Vectorshield (Vector Laboratories). Images were acquired with a Zeiss Axioimage Z2 equipped with ApoTome. Antibodies and reagent used were Six2 (1:500; Proteintech), cytokeratin-8 (1:40; Developmental Studies Hybridoma Bank), biotinylated–dilochos biflorus agglutinin (1:500; Vector Laboratories), FoxD1 (1:200; Santa Cruz), and Pax2 (1:200; Covance). Secondary antibodies conjugated with Alexa Fluor 488 and Alexa Fluor 555 (1:500; Molecular Probes) were used.
Proliferation and Cell Death Analyses
To analyze nephron progenitor proliferation and cell death, frozen sections were prepared from the entire putative kidney field of E10.5 or E11.5 embryos. Alternate sections were subjected to staining for EdU and Six2 (for proliferation) or terminal deoxynucleotidyl transferase–mediated digoxigenin-deoxyuridine nick-end labeling and Pax2 (for cell death). For EdU pulse labeling, pregnant females containing E10.5 and E11.5 embryos were injected with 50 µg/g (body wt) of EdU according to the manufacture’s recommendations. Two hours after injection, the mother mice were euthanized and embryos were collected and processed for frozen sectioning. Staining of EdU was followed by the Click-iT EdU Imaging Kit protocol (C10338; Invitrogen). Terminal deoxynucleotidyl transferase–mediated digoxigenin-deoxyuridine nick-end labeling staining was performed according to the manufacturer’s recommendation (In situ Cell Death Detection Kit; Roche).
In Situ Hybridization
For in situ hybridization of sections, paraffinized slides were hydrated with diethyl pyrocarbonate–treated ethanol series and water, washed with hybridization solution, and incubated overnight with digoxigenin-labeled Spry1 probe. After washing, samples were incubated with anti-digoxigenin antibody conjugated with alkaline phosphatase (Roche) and the color reaction was performed using alkaline phosphate substrate (Roche). For whole-mount in situ hybridization, embryos were dissected in diethyl pyrocarbonate–treated PBS and fixed with 4% paraformaldehyde. After washing, samples were dehydrated with methanol series. Samples were rehydrated, treated with protease, stabilized with hybridization solution, and incubated overnight with digoxigenin-labeled RNA probes. After washing, samples were incubated with anti-digoxigenin antibody and visualized using colorimetric alkaline phosphate substrate. Samples were photographed on a dissecting microscope. Probes used for these studies were Wnt9b,25 Wnt4,26 Pax2,27 Ret,28 Gdnf,5 Etv4,29 and Etv5.29
Statistical Analyses
GraphPad Prism was used to perform a Welch t test or a nonparametric Kruskal–Wallis test with the Dunn test compensating for multiple comparisons where appropriate. A value of P<0.05 was considered statistically significant. For comparative analysis of kidney sizes, kidney perimeter was measured and individual measurements were used for statistical analysis. Three or more animals from at least two independent experiments were examined.
Results
Loss of Spry1 Partially Restores Kidney Phenotypes Caused by Loss of Fgf9 and Fgf20
To test whether Spry1 regulates Fgf9 and Fgf20 activity during kidney development, we generated Fgf9, Fgf20, and Spry1 triple compound mutants. In this study, we used Fgf20−/− embryos as controls. Previously, we identified that Fgf20−/− embryos produce 15% smaller kidneys than Fgf20−/+ with a decreased number of nephrons.12 Kidney size of Fgf20−/− and Fgf20−/−;Spry1−/+ embryos were comparable at E18.5 (Figure 1, A, B, Q, and R). Histologic morphology appeared normal in both genotypes (Figure 1, G and H). In Fgf9−/+;Fgf20−/− embryos, ten out of 18 kidneys were hypoplastic and one kidney was aplastic (Figure 1, C and Q). Kidney size was decreased to 61.8%±30.1% compared with control (P<0.001) (Figure 1R). Fgf9−/+;Fgf20−/− kidneys were also cystic (Figure 1I). In Fgf9−/+;Fgf20−/−;Spry1−/+ embryos, five out of 18 kidneys were hypoplastic and kidney size was significantly restored compared with Fgf9−/+;Fgf20−/− embryos (87.4%±15.0%, P<0.005; Figure 1, D, Q, and R). Renal cysts were not detected in the Fgf9−/+;Fgf20−/−;Spry1−/+ kidneys (Figure 1J). However, kidney size was not restored compared with controls (Figure 1R). All kidneys in Fgf9−/−;Fgf20−/− embryos were missing (n=8) (Figure 1, E and Q). Interestingly, all Fgf9−/−;Fgf20−/−;Spry1−/+ embryos generated kidneys. Among 14 kidneys analyzed, four kidneys were normal and ten kidneys were hypoplastic (Figure 1, F and Q). Size of the kidneys in Fgf9−/−;Fgf20−/−;Spry1−/+ embryos was restored to that of Fgf9−/+;Fgf20−/− embryos (Figure 1R). Histologic analysis showed that kidney structures were comparable with controls (Figure 1K). Nephron progenitors were repopulated in Fgf9−/+;Fgf20−/−;Spry1−/+ and Fgf9−/−;Fgf20−/−;Spry1−/+ kidneys compared with Fgf9−/+;Fgf20−/− and Fgf9−/−;Fgf20−/−, respectively (Figure 1, L–P′). These data indicate that deleting one copy of Spry1 partially restores kidney size and morphology caused by loss of Fgf9 and Fgf20.
Figure 1.
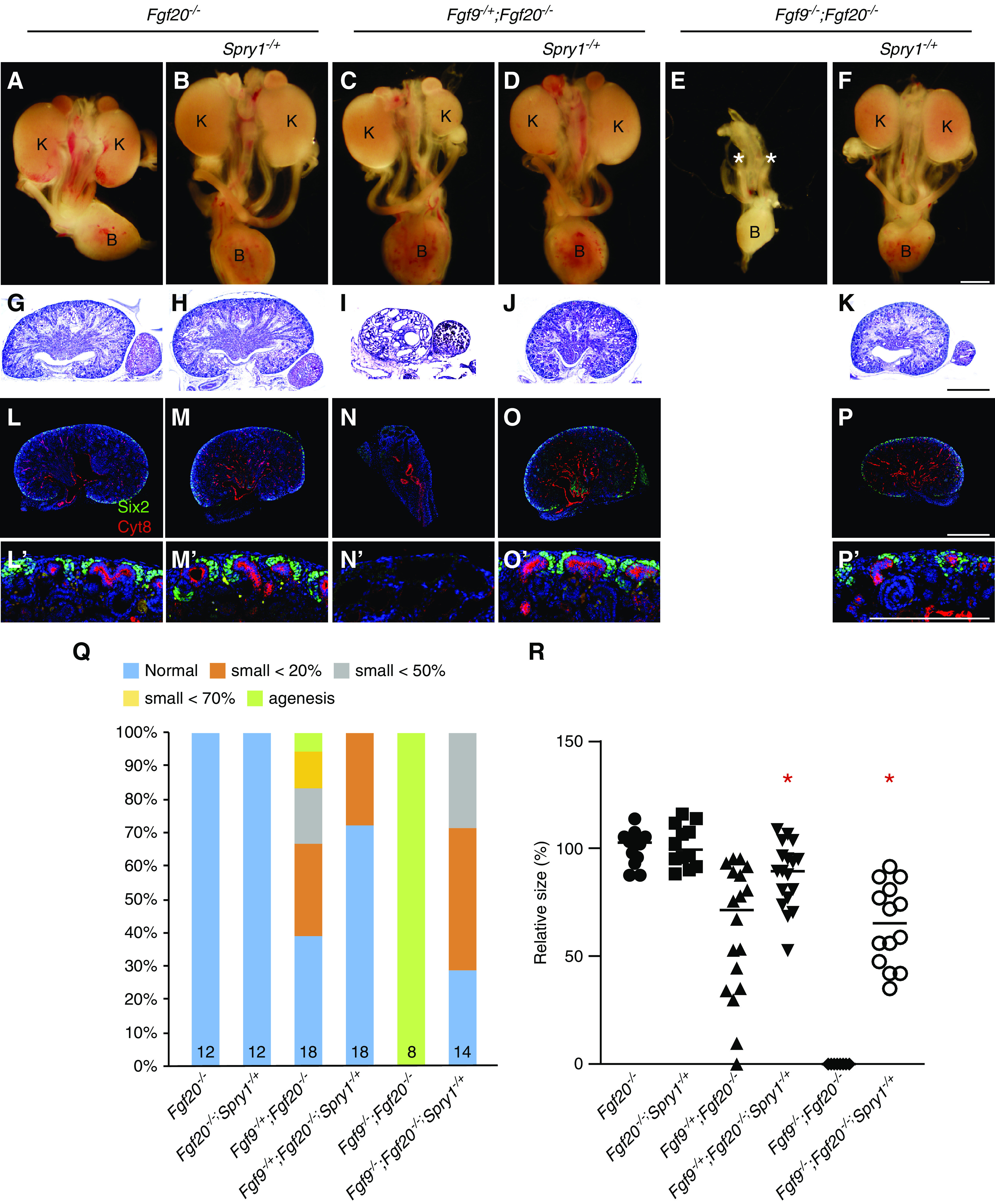
Loss of one copy of Spry1 rescues renal phenotypes caused by loss of Fgf9 and Fgf20. (A–F) Morphology of urogenital system in E18.5 Fgf9, Fgf20, and Spry1 compound mutants. (E) Renal agenesis caused by loss of Fgf9 and Fgf20 was restored by (F) deleting one copy of Spry1. * in (E) indicates loss of kidneys. (G–K) Hematoxylin and eosin staining of E18.5 kidneys of Fgf9, Fgf20, and Spry1 compound mutants. (I) Renal cysts of Fgf9-/+;Fgf20−/− animals disappeared after (J) deleting one copy of Spry1. (L–P) Sections of E18.5 kidneys stained with cytokeratin-8 (Cyt8) and Six2 in E18.5 kidneys of Fgf9, Fgf20, and Spry1 compound mutants. (N) Loss of NPCs caused by deletion of Fgf9 and Fgf20 were restored by (O and P) deleting one copy of Spry1. (L′–P′) High power image of nephrogenic zone. (Q) Percentage of kidney phenotypes and (R) relative kidney size of Fgf9, Fgf20, and Spry1 compound mutants. *P<0.01. Scale bar, 100 µm. K, kidney; B, bladder.
Spry1 is reported to be expressed in both UBs and NPCs during kidney development.14,15 We observed that Spry1 was expressed in both UBs and NPCs at E11.5 and its expression was decreased in NPCs of E13.5 and E14.5 kidneys (Supplemental Figure 1). Spry1 expressed in the UBs negatively regulates GDNF-RET-induced UB branch morphogenesis.17,18 We hypothesized that Spry1 expressed in the NPCs antagonizes FGF9- and FGF20-induced NPC maintenance. To investigate this, we conditionally deleted Spry1 in NPCs using an Fgf20Cre knock-in mouse line, which serves as both a loss of Fgf20 and Cre driver of NPCs.12 Cre recombinase in Fgf20Cre was active only in NPCs and their derivatives (Supplemental Figure 2).
Kidney size of control (Fgf20Cre/−) and Fgf20Cre/−;Spry1fl/+ embryos were comparable at E18.5 (Figure 2, A, B, G, and H). In Fgf9−/+;Fgf20Cre/− embryos, three out of 16 kidneys were hypoplastic and two kidneys were aplastic, and kidney size was decreased to 75.7%±32.1% compared with control (P<0.05) (Figure 2, C, G, and H). In Fgf9−/+;Fgf20Cre/−;Spry1fl/+ embryos, two out of 18 kidneys were hypoplastic and kidney size was restored to that of control kidneys (Figure 2, D, G, and H). All kidneys in Fgf9−/−;Fgf20Cre/− embryos were missing (n=12) (Figure 2, E and G). In four Fgf9−/−;Fgf20Cre/−;Spry1fl/+ embryos, two kidneys were normal, four kidneys were hypoplastic, and two kidneys were missing (Figure 2, F and G). Size of the kidneys in Fgf9−/−;Fgf20Cre/−;Spry1fl/+ embryos was 56.2%±41.6% compared with controls, which is significantly increased compared with Fgf9−/−;Fgf20Cre/− embryos (P<0.005) (Figure 2H). There was less effective rescue of renal phenotypes in Fgf9−/−;Fgf20Cre/−;Spry1fl/+ compared with Fgf9−/−;Fgf20−/−;Spry1−/+, which could be due to recombination efficiency. Another possibility for why this occurred is due to separate mating to generate Fgf9−/−;Fgf20−/−;Spry1−/+ and Fgf9−/−;Fgf20Cre/−;Spry1fl/+ mice that could cause some genetic background difference to persist. Together, these data indicate that phenotypic rescue of NPC-specific Spry1 deletion recapitulates whole-body deletion of Spry1, thus suggesting SPRY1 in NPCs antagonizes activity of FGF9 and FGF20 to regulate kidney development.
Figure 2.
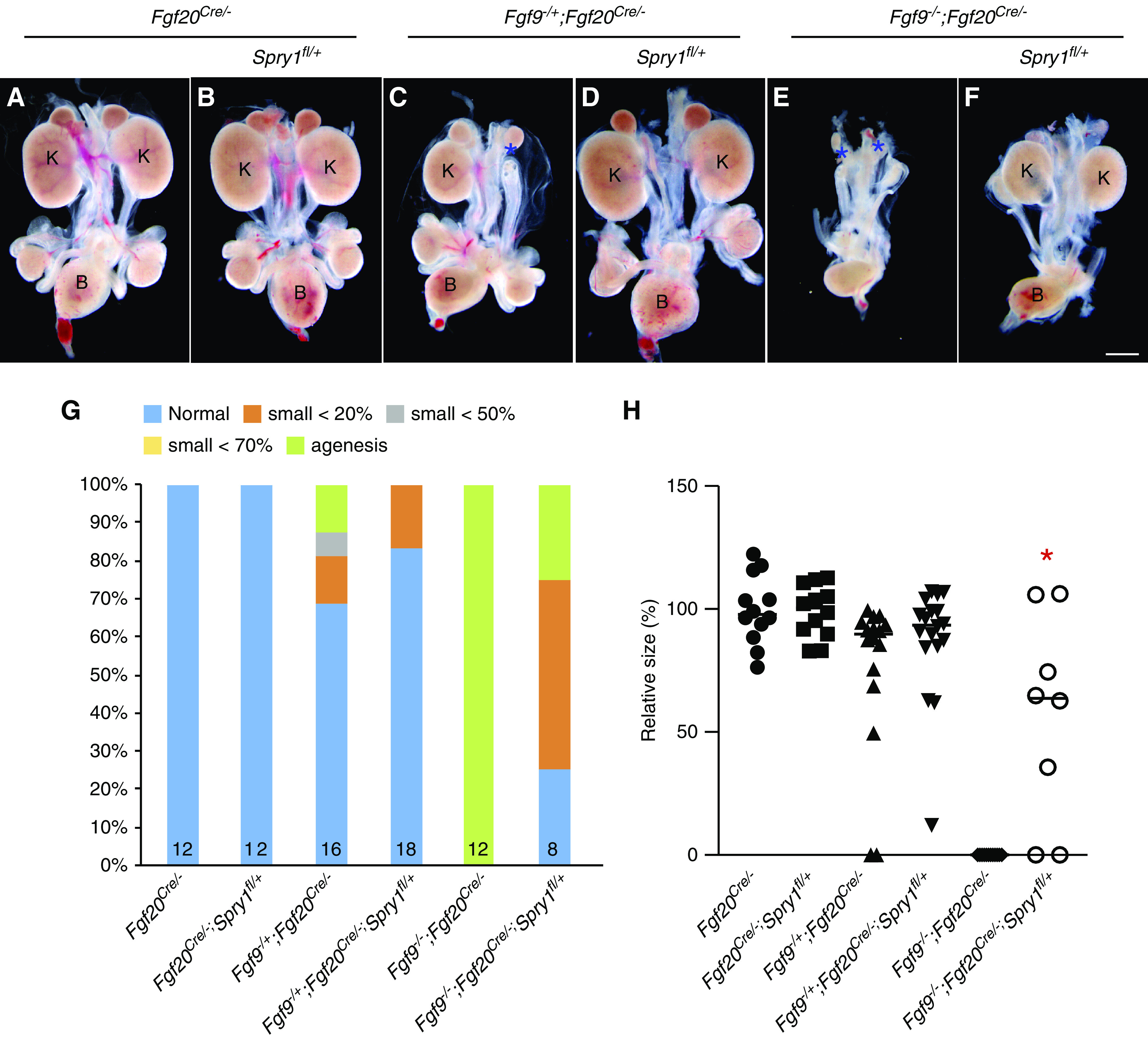
Spry1 in NPCs antagonizes Fgf9- and Fgf20-induced nephrogenesis. (A–F) Morphology of urogenital system in E18.5 nephron progenitor–specific Fgf9, Fgf20, and Spry1 compound mutants. (C and E) Renal agenesis caused by loss of Fgf9 and Fgf20 was restored by (D and F) deleting one copy of Spry1 in nephron progenitors. * in (C and E) indicates loss of kidneys. (G) Percentage of kidney phenotypes and (H) relative kidney size of nephron progenitor–specific Fgf9, Fgf20, and Spry1 compound mutants. *P<0.01. Scale bar, 100 µm. K, kidney; B, bladder.
FGF9 and FGF20 Are Required for Nephron Progenitor Survival and Proliferation
Previously, we identified that loss of Fgf9 and Fgf20 resulted in NPC cell death.12 To further analyze their roles during kidney development, we performed proliferation and cell death analyses of NPCs at the time of NPC induction. At E10.5, the number, proliferation rate, and cell death rate of Six2+ NPCs were comparable to all genotypes (Supplemental Figure 3). At E11.5, the number of NPCs in Fgf9−/−;Fgf20−/− embryos was significantly decreased compared with control (Fgf9−/+;Fgf20−/+) embryos (P<0.01) (Figure 3, A–D and M). The proliferation rate of NPCs in Fgf9−/−;Fgf20−/− embryos was also significantly decreased compared with control embryos (P<0.01) (Figure 3, E–H and N). The cell death rate of NPCs in Fgf9−/+;Fgf20−/− embryos was increased compared with control embryos (P<0.05) (Figure 3, K and O), and cell death rate of NPCs was further increased in Fgf9−/−;Fgf20−/− embryos compared with control (P<0.01) and Fgf9−/+;Fgf20−/− (P<0.05) embryos (Figure 3, L and O).
Figure 3.
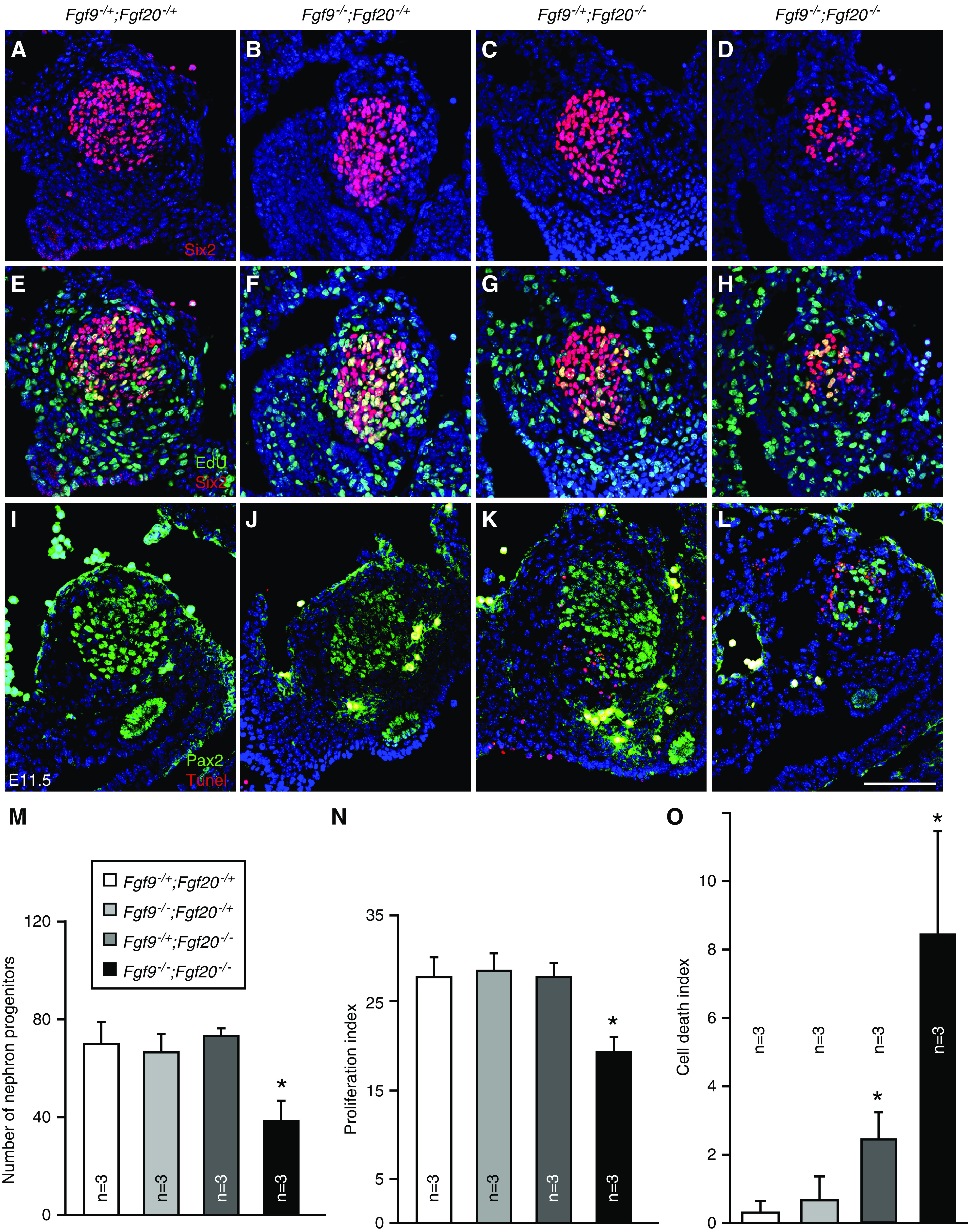
Fgf9 and Fgf20 are required for cell death and proliferation of E11.5 nephron progenitors. (A–D) Six2 staining of E11.5 Fgf9 and Fgf20 compound mutant kidneys. (E–H) Six2 and EdU staining of E11.5 Fgf9 and Fgf20 compound mutant kidneys. (I–L) Pax2 and terminal deoxynucleotidyl transferase–mediated digoxigenin-deoxyuridine nick-end labeling (Tunel) staining of E11.5 Fgf9 and Fgf20 compound mutant kidneys. (M) Quantifying the number of nephron progenitors in Fgf9 and Fgf20 compound mutant kidneys showing the decreased number in the Fgf9 and Fgf20 double mutant. (N) Proliferation index of Fgf9 and Fgf20 compound mutant kidneys showing the decreased nephron progenitor proliferation in Fgf9 and Fgf20 double mutant. (O) Cell death index of Fgf9 and Fgf20 compound mutant kidneys showing the increased NPC death in Fgf9−/+;Fgf20−/− and Fgf9−/−;Fgf20−/− kidneys. Data are shown with mean±SD. *P<0.01. Scale bar, 100 µm.
Fgf9 and Fgf20 Regulate UB Branching
We also investigated whether FGF9 and FGF20 regulate genes required for UB branching. At E10.5, Ret was expressed in UBs and its expression was comparable to all of the genotypes (Supplemental Figure 4, A–D). At E11.5, Ret+ UBs were bifurcated in control and Fgf9−/−;Fgf20−/+ embryos (Supplemental Figure 4, E and F). However, in Fgf9−/+;Fgf20−/− embryos, UBs were not yet bifurcated (Supplemental Figure 4G) and UBs started to regress in Fgf9−/−;Fgf20−/− embryos (Supplemental Figure 4H). Gdnf was highly expressed in NPCs of control and Fgf9−/−;Fgf20−/+ embryos (Supplemental Figure 4, I and J). However, expression of Gdnf was decreased in Fgf9−/+;Fgf20−/− embryos (Supplemental Figure 4K) and diminished in Fgf9−/−;Fgf20−/− embryos (Supplemental Figure 4L). Etv4 and Etv5 are transcription factors regulated by GDNF/RET during UB branching and are responsible for UB branching.18,29–31 Therefore, we tested whether a decrease of Gdnf affects expression of Etv4 and Etv5. At E11.5, the expression of Etv4 and Etv5 was significantly decreased in Fgf9−/+;Fgf20−/− embryos and diminished in Fgf9−/−;Fgf20−/− embryos (Supplemental Figure 4, M–S). Together, these data indicate that FGF20, but not FGF9, is required for UB branching.
Reduction of Spry1 Level Restores Nephron Progenitor Proliferation and Survival in Fgf9 and Fgf20 Double Mutant Kidneys
Next, we investigated whether deleting one copy of Spry1 rescues NPC number, proliferation, and cell death defects caused by loss of Fgf9 and Fgf20. The number of NPCs were comparable in both control (Fgf20−/−) and Fgf20−/−;Spry1−/+ embryos (Figure 4, A, D, and G). The number of NPCs in Fgf9−/+;Fgf20−/−;Spry1−/+ embryos was increased compared with that of Fgf9−/+;Fgf20−/− embryos (P<0.05) (Figure 4, C, D, and G). Also, the number of NPCs in Fgf9−/−;Fgf20−/−;Spry1−/+ embryos was increased compared with that of Fgf9−/−;Fgf20−/− embryos (P<0.001) (Figure 4, E–G). Of note, the number of NPCs in Fgf9−/+;Fgf20−/−;Spry1−/+ and Fgf9−/−;Fgf20−/−;Spry1−/+ embryos was restored close to that of control embryos. Proliferation indexes were comparable to both control and Fgf20−/−;Spry1−/+ embryos. The proliferation index in Fgf9−/+;Fgf20−/−;Spry1−/+ embryos was increased compared with that of Fgf9−/+;Fgf20−/− embryos (P<0.05) (Figure 4, J, K, and N). Also, the proliferation index in Fgf9−/−;Fgf20−/−;Spry1−/+ embryos was increased compared with that of Fgf9−/−;Fgf20−/− embryos (P<0.001) (Figure 4, L–N). Of note, the proliferation indexes in Fgf9−/+;Fgf20−/−;Spry1−/+ and Fgf9−/−;Fgf20−/−;Spry1−/+ embryos was restored close to that of control embryos. Cell death indexes were comparable to both control and Fgf20−/−;Spry1−/+ embryos (Figure 4, O, P, and U). The cell death index in Fgf9−/+;Fgf20−/−;Spry1−/+ embryos was decreased compared with that of Fgf9−/+;Fgf20−/− embryos (P<0.05) (Figure 4, Q, R, and U). Also, the cell death index in Fgf9−/−;Fgf20−/−;Spry1−/+ embryos was decreased compared with that of Fgf9−/−;Fgf20−/− embryos (P<0.001) (Figure 4, S–U). Together, these data indicate that SPRY1 negatively regulates FGF9- and FGF20-dependent NPC survival and cell death.
Figure 4.
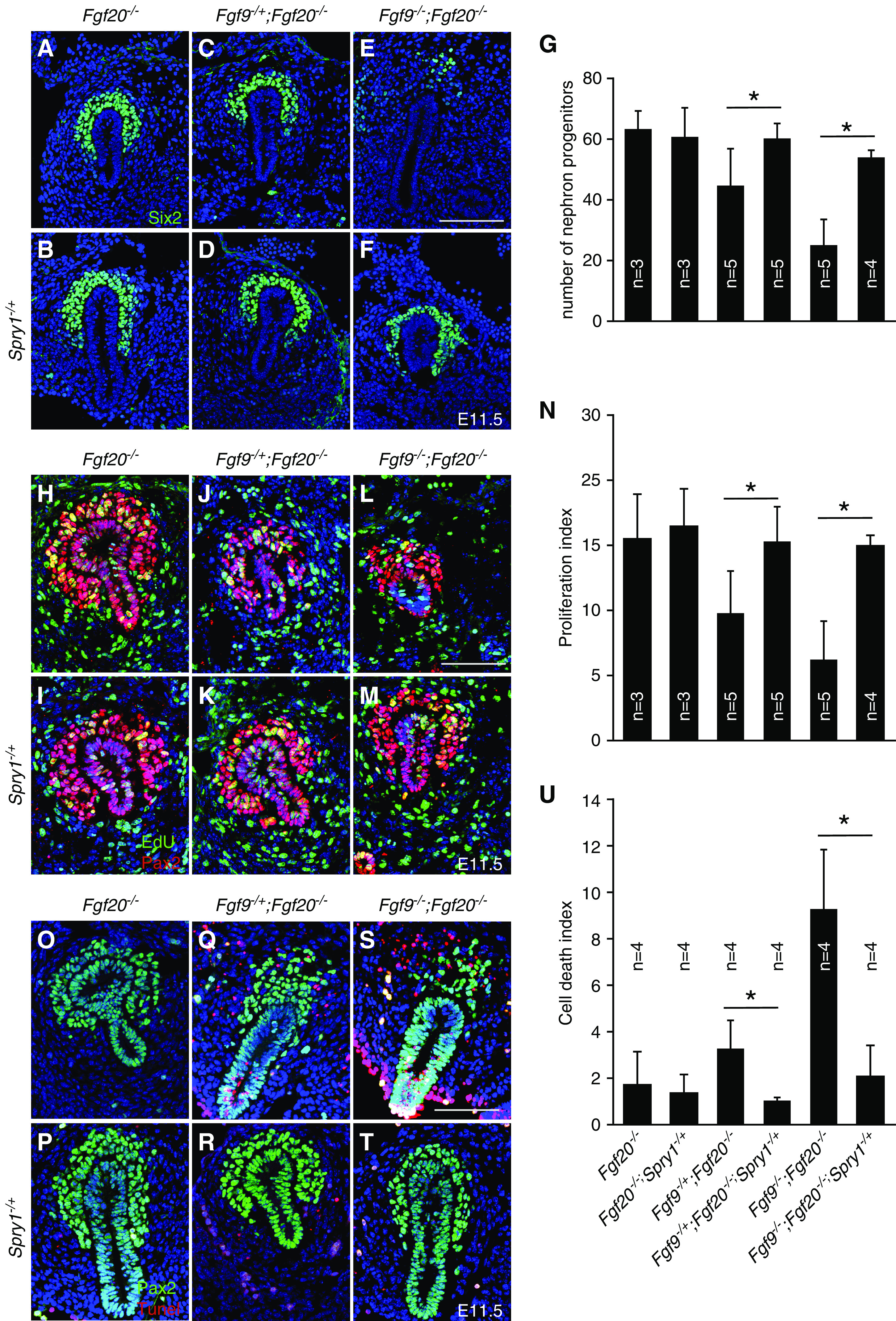
Loss of one copy of Spry1 rescues NPC death and proliferation defects caused by loss of Fgf9 and Fgf20. (A–F) Six2 staining of E11.5 Fgf9, Fgf20, and Spry1 compound mutant kidneys. (G) Quantifying the number of nephron progenitors of Fgf9, Fgf20, and Spry1 compound mutant kidneys, showing that deleting one copy of Spry1 restored loss of nephron progenitors. (H–M) Pax2 and EdU staining of E11.5 Fgf9, Fgf20, and Spry1 compound mutant kidneys. (N) Proliferation index of Fgf9, Fgf20, and Spry1 compound mutant kidneys, showing that deleting one copy of Spry1 restored nephron progenitor proliferation defects. (O–T) Pax2 and terminal deoxynucleotidyl transferase–mediated digoxigenin-deoxyuridine nick-end labeling (Tunel) staining of E11.5 Fgf9, Fgf20, and Spry1 compound mutant kidneys. (U) Cell death index of Fgf9, Fgf20, and Spry1 compound mutant kidneys, showing that deleting one copy of Spry1 rescued NPC death. Data are shown with mean±SD. *P<0.01. Scale bar, 100 µm.
FGF8 Functions Together with FGF20 To Regulate Nephron Progenitor Maintenance
FGF8 is expressed in the NPCs and is required for NPC survival and tubulogenesis at E14.5.21,32–34 Because FGF8 is also required for NPC survival, we hypothesize that FGF8 functions together with FGF9 and FGF20 to maintain NPCs. To investigate this hypothesis, we deleted Fgf8 in the NPC together with Fgf20 using an Fgf20Cre mouse line and analyzed kidneys. Kidneys in Fgf8fl/−;Fgf20Cre/+ and Fgf8fl/+;Fgf20Cre/− embryos were smaller than those of control (Fgf8fl/+;Fgf20Cre/+) embryos (Figure 5, A–C). Fgf8fl/−;Fgf20Cre/− embryos showed bilateral kidney agenesis (Figure 5D). Kidneys of Fgf8fl/−;Fgf20Cre/+ embryos contained no nephrons, which is consistent with previous publications (Figure 5F).21,32 Fgf8fl/+;Fgf20Cre/− kidneys had less nephrons compared with control embryos (Figure 5, E and G). UBs had less branching in both E14.5 Fgf8fl/−;Fgf20Cre/+ and Fgf8fl/−;Fgf20Cre/− kidneys as noted by Wnt9b staining, the marker of UBs (Supplemental Figure 5, A–C).25 Wnt4 expression was diminished in the Fgf8fl/−;Fgf20Cre/+ kidneys, which was consistent with previous publications (Supplemental Figure 5E).21,32 However, Wnt4 expression in the Fgf8fl/−;Fgf20Cre/− kidneys was comparable to control kidneys (Supplemental Figure 5, D and F), indicating that Fgf20 does not regulate Wnt4 expression.
Figure 5.
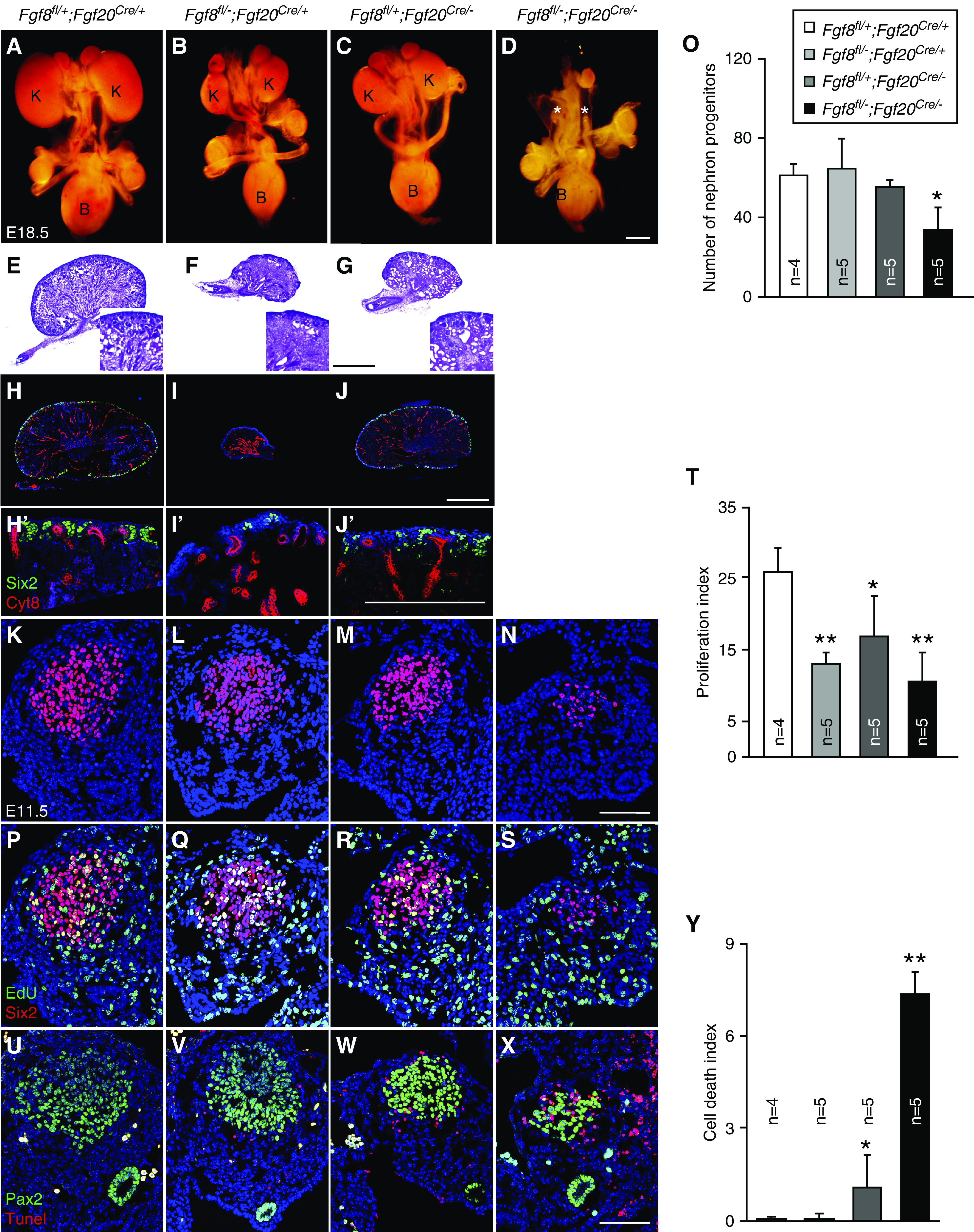
Fgf8 functions redundantly with Fgf20 to maintain nephrogenesis and nephron progenitors. (A–D) Morphology of urogenital system in E18.5 Fgf8 and Fgf20 compound mutants showing bilateral renal agenesis in Fgf8 and Fgf20 double mutant. * in (D) indicates loss of kidneys. (E–G) Hematoxylin and eosin staining of E18.5 Fgf8 and Fgf20 compound mutants, (F) showing loss of nephrons in Fgf8fl/−;Fgf20Cre/+ kidneys. Higher magnification shown in insets. (H–J) Sections of E18.5 kidneys stained with cytokeratin-8 (Cyt8) and Six2 in E18.5 Fgf8 and Fgf20 compound mutants. (H′–J′) High power image of nephrogenic zone from E18.5 Fgf8 and Fgf20 compound mutant kidneys. (K–N) Six2 staining of E11.5 Fgf8 and Fgf20 compound mutant kidneys. (O) Quantifying the number of nephron progenitors of Fgf8 and Fgf20 compound mutants, showing decreased nephron progenitors in Fgf8fl/−;Fgf20Cre/−. (P–S) Six2 and EdU staining of E11.5 Fgf8 and Fgf20 compound mutant kidney slides. (T) Proliferation index of Fgf8 and Fgf20 compound mutants, showing decreased nephron progenitor proliferation in all Fgf8 and Fgf20 compound mutants. (U–X) Pax2 and terminal deoxynucleotidyl transferase–mediated digoxigenin-deoxyuridine nick-end labeling (Tunel) staining of E11.5 Fgf8 and Fgf20 compound mutant kidneys. (U) Cell death index of Fgf8 and Fgf20 compound mutants, showing increased nephron progenitor cell death in Fgf8fl/+;Fgf20Cre/− and Fgf8fl/−;Fgf20Cre/−. Data are shown with mean±SD. *P<0.05, **P<0.01. Scale bar, 500 µm in (D), (G), (J), and (J′), 100 µm in (N) and (X). K, kidney; B, bladder.
In NPC maintenance, Six2+ NPCs were decreased in E18.5 kidneys of Fgf8fl/−;Fgf20Cre/+ and Fgf8fl/+;Fgf20Cre/− embryos compared with controls (Figure 5, H–J′). At E11.5, the numbers of NPCs were comparable in Fgf8 and Fgf20 compound heterozygous mutant (control, Fgf8fl/−;Fgf20Cre/+, and Fgf8fl/+;Fgf20Cre/−) embryos (Figure 5, K–M and O). However, the number of NPCs in Fgf8fl/−;Fgf20Cre/− embryos was significantly decreased (P<0.05) compared with control embryos (Figure 5, N and O). Proliferation rates of NPCs in Fgf8fl/−;Fgf20Cre/+, Fgf8fl/+;Fgf20Cre/−, and Fgf8fl/−;Fgf20Cre/− embryos were significantly decreased (P<0.01, P<0.05, P<0.01, respectively) compared with control embryos (Figure 5, P–T). The cell death rate of NPCs in Fgf8fl/−;Fgf20Cre/+ embryos was comparable compared with controls (Figure 5, U, V, and Y). The cell death rate of NPCs in Fgf8fl/+;Fgf20Cre/− embryos was increased (P<0.05) (Figure 5, W and Y). The cell death rate of NPCs was further increased in Fgf8fl/−;Fgf20Cre/− embryos compared with controls (P<0.01) and Fgf8fl/+;Fgf20Cre/− (P<0.01) embryos (Figure 5, X and Y). In addition, similar to Fgf9−/−;Fgf20−/−,12 Pax2 expression in the NPCs was decreased in the Fgf8fl/−;Fgf20Cre/− embryos (Supplemental Figure 6, A–D).
We also analyzed early UB branching. At E11, Ret was expressed in UBs and its expression was comparable to all of the genotypes, suggesting UB induction is not affected (Supplemental Figure 6, E–H). At E11.5, Ret+ UBs were bifurcated in control embryos (Supplemental Figure 6J). However, in Fgf8fl/−;Fgf20Cre/+, Fgf8fl/+;Fgf20Cre/−, and Fgf8fl/−;Fgf20Cre/− embryos, UBs were not yet bifurcated (Supplemental Figure 6, J–L). Gdnf expression was comparable to control and Fgf8fl/−;Fgf20Cre/+ embryos (Supplemental Figure 6, Q and R). However, expression of Gdnf was decreased in Fgf8fl/+;Fgf20Cre/− embryos (Supplemental Figure 6O) and diminished in Fgffl/-; Fgf20Cre/- embryos (Supplemental Figure 6P). In addition, Etv4 and Etv5 were diminished in Fgf8fl/−;Fgf20Cre/− embryos (Supplemental Figure 6, Q–X). Together, these data indicate that Fgf8, similar to Fgf9, functions together with Fgf20 to regulate kidney development.
Spry1 Heterozygous Deletion Does Not Rescue Kidney Defects of Fgf8 and Fgf20 Double Knockout
Next, we questioned whether the loss of Spry1 also rescues renal phenotypes caused by loss of Fgf8 and Fgf20. Therefore, we generated Spry1, Fgf8, and Fgf20 compound mutants and analyzed their kidneys. In Fgf8fl/−;Fgf20Cre/− embryos, among 16 kidneys, two very small kidneys were observed and 14 kidneys were missing (Figure 6, A and C). In Fgf8fl/−;Fgf20Cre/−;Spry1−/+ embryos, two out of ten kidneys were severely hypoplastic and eight kidneys were missing (Figure 6, B and C). In addition, NPC number, proliferation rate, and cell death index were not rescued in the Fgf8fl/−;Fgf20Cre/−;Spry1−/+ embryos compared with Fgf8fl/−;Fgf20Cre/− embryos (Figure 6, D–F).
Figure 6.

Loss of one copy of Spry1 does not rescue renal phenotypes caused by loss of Fgf8 and Fgf20. (A and B) Morphology of urogenital system in E18.5 (A) Fgf8fl/−;Fgf20Cre/− and (B) Fgf8fl/−;Fgf20Cre/−;Spry1−/+ animals. Renal agenesis caused by loss of Fgf8 and Fgf20 was not rescued by loss of one copy of Spry1. * in (A) and (B) indicate loss of kidneys. (C) Percentage of kidney phenotypes of Fgf8fl/−;Fgf20Cre/− and Fgf8fl/−;Fgf20Cre/−;Spry1−/+ animals. (D) Quantifying number of nephron progenitors, (E) proliferation index, and (F) cell death index of Fgf8fl/−;Fgf20Cre/− and Fgf8fl/−;Fgf20Cre/−;Spry1−/+ animals, showing there are no differences between two genotypes. Data are shown with mean±SD. *P<0.01. Scale bar, 100 µm. B, bladder.
SPRY1 Antagonizes FGFs in a Dose-Dependent Manner
We identified that FGF8, FGF9, and FGF20 function together to regulate NPC maintenance. We also identified that deletion of Spry1 did not rescue renal phenotypes caused by loss of both Fgf8 and Fgf20. These results suggest that antagonism of SPRY1 to the FGF signal is limited by the copy number of Fgf genes. To investigate this, we generated Fgf8, Fgf9, Fgf20, and Spry1 compound mutants and analyzed their kidneys. In Fgf8fl/+;Fgf9−/+;Fgf20Cre/− embryos, one out of ten kidneys was normal, six were hypoplastic, and three were missing (Figure 7, A and E). Kidney size was 34.8%±34.3% (P<0.001) compared with Fgf8fl/+;Fgf9−/+;Fgf20Cre/−Spry1−/+ embryos (Figure 7F). In Fgf8fl/+;Fgf9−/+;Fgf20Cre/−Spry1−/+ embryos, all kidneys (ten out of ten) were increased in size compared with Fgf8fl/+;Fgf9−/+;Fgf20Cre/− embryos (Figure 7, B, E, and F). All of the kidneys (four out of four) in Fgf8fl/+;Fgf9−/−;Fgf20Cre/− embryos were missing (Figure 7, C, E, and F). Fgf8fl/+;Fgf9−/−;Fgf20Cre/−;Spry1−/+ embryos also lost all kidneys (Figure 7, C–F). These data indicate the rescue of kidney phenotypes in the loss of one copy of Spry1 is dependent on the dosage of gene copy numbers of Fgfs.
Figure 7.

Antagonism of Spry1 depends on dosage of FGF ligands. (A–D) Morphology of urogenital system in E18.5 Fgf8, Fgf9, Fgf20, and Spry1 compound mutants showing that (B) deleting one copy of Spry1 rescues kidney agenesis in (A) Fgf8fl/+;Fgf9−/+;Fgf20Cre/− but (C and D) not in Fgf8fl/+;Fgf9−/−;Fgf20Cre/−. * in (A), (C), and (D) indicate loss of kidneys. (E) Percentage of kidney phenotypes and (F) relative kidney size of Fgf8, Fgf9, Fgf20, and Spry1 compound mutants, showing that deleting one copy of Spry1 increased kidney size in Fgf8fl/+;Fgf9−/+;Fgf20Cre/− but not in Fgf8fl/+;Fgf9−/−;Fgf20Cre/−. *P<0.01. (G) Model of FGFs and SPRY1 during NPC maintenance. FGF8/9/20 activated FGFR1/2 to promote survival and proliferation of early nephron progenitors putatively through RAS-MAPK/PI3K-AKT. SPRY1 fine-tunes this process by antagonizing FGFR activity. Scale bar, 100 µm. K, kidney; B, bladder.
Discussion
SPRY was originally identified to regulate development of Drosophila trachea through antagonizing branchless and breathless genes, which are orthologs of mammalian FGF and FGFR, respectively.35 The mammalian SPRYs antagonize FGF signaling in many developing organs, including lung, mandible, external genitalia, long bone, auditory, and tooth.36–41 However, in kidney development, SPRY1 seems to function mainly as an antagonist of Ret-GDNF-dependent UB branching.15,17 Of note, Brown et al.42 showed that ectopic expression of SPRY1 in NPCs resulted in loss of NPCs due to NPC cell death, suggesting a possible role of SPRY1 in NPCs. Indeed, we present data indicating that loss of one copy of Spry1 partially rescued renal phenotypes due to loss of Fgf9 and Fgf20 during NPC maintenance. NPC cell death, proliferation defect, and premature depletion were rescued. NPC-specific Spry1 deletion also rescued the Fgf9 and Fgf20 loss-of-function deletion, indicating antagonistic function of Spry1 is in a cell-autonomous manner within NPCs.
FGF8 also functions together with FGF20 to maintain NPCs. At E11.5, Fgf8 is expressed in the NPCs and its expression is restricted to pretubular aggregate (PTA) and renal vesicle as the embryo develops.21,32 Previous studies indicate Fgf8 is required for NPC and PTA survival at later stages of development (E14.5 and later) but is dispensable for NPC proliferation.21,32 Interestingly, in this study, we identified that at an early developmental stage (E11.5), proliferation of NPCs was decreased in all Fgf8, Fgf20 compound mutants (Fgf8fl/−;Fgf20Cre/+, Fgf8fl/+;Fgf20Cre/−, and Fgf8fl/−;Fgf20Cre/−). In addition, NPC cell death was most pronounced in the Fgf8 and Fgf20 double mutant kidneys. Fgf20 does not seem to function together with Fgf8 to maintain PTA, because a PTA marker (Wnt4) was not changed in the Fgf8fl/+;Fgf20Cre/− kidneys. Different from Fgf9 and Fgf20, deleting one copy of Spry1 did not rescue renal phenotypes caused by loss of Fgf8 and Fgf20. No kidneys developed in Fgf8, Fgf20, and Spry1 compound mutants (Fgf8fl/−;Fgf20Cre/−;Spry1−/+). Furthermore, there is no rescue of NPC proliferation and cell death occurs. FGF8, but not FGF9 or FGF20, binds to FGFR-like 1 (FGFRL1).43 FGFRL1 does not contain an intracellular protein tyrosine kinase domain and it is thought to function as a decoy receptor.44 FGFRL1 also interacts with SPRY family members, including SPRED and SPRY1.45 Different from other FGFRs, the SPRY family increases the retention time of FGFRL1 at the plasma membrane, which enhances its chance to bind to its ligands.45 Failure to rescue the renal defects in the Fgf8/20 double mutant may be due to interaction of FGF8 with FGFR1L. Nonetheless, antagonism of SPRY1 to FGFs is dose dependent because loss of one copy of Spry1 rescues renal agenesis in Fgf8fl/+;Fgf9−/+;Fgf20Cre/− but not in Fgf8fl/+;Fgf9−/−;Fgf20Cre/−. Spry1 expression level in the NPCs decreased as the kidneys developed (Supplemental Figure 1) and was undetectable at E18.5 based on a published single-cell RNA sequencing data set.46 In addition, NPC proliferation and cell death defects caused by loss of FGF signaling during early kidney development were rescued by deleting one copy of Spry1. Based on this, we propose that SPRY1 fine-tunes FGF-dependent NPC maintenance during early kidney development (Figure 5G). FGF8, FGF9, and FGF20 bind to FGFR1 and FGFR2 in the NPCs to activate downstream signaling pathways, including RAS-MAPK and PI3K-AKT, which supports NPC proliferation, survival, and stemness.42,47 Upon activation of FGF signaling, Spry1 is upregulated and fine-tunes FGF signaling by antagonizing the RAS-MAPK and PI3K-AKT signaling cascade.48
Another interesting observation is that expression of Gdnf, Etv4, and Etv5 was lost in both Fgf9/20 and Fgf8/20 double mutants. Loss of Etv4 and Etv5 expression would be due to loss of Gdnf expression in the NPCs. Loss of Gdnf expression would be explained partly by NPC cell death. Additional mechanisms regulating FGF signaling that are dependent on GDNF expression should exist. The FGFR/FRS2a/ERK/CREB signaling cascade activates GDNF expression in C6 glioma cells and astrocytes.49 It would be interesting if it were also the case during kidney development. Because GDNF is a critical activator of RET for UB branching morphogenesis and ETV4 and ETV5 are downstream effectors of RET for UB branching,29 it would be interesting to investigate whether lowering the FGF signal in both NPCs and UBs causes renal defects. FGF10 functions to promote UB branching in the absence of Ret-GDNF/SPRY1.18 However, Fgf10 null embryos have a mild effect of UB branching generating small kidneys.50 Deleting both Fgf10 and Fgf20 would give insight for the combination of FGF function in both NPCs and UBs.
In conclusion, fine-tuning of FGF signaling is required for proper NPC maintenance and number of nephrons. This information is important in the fine-tuning of the FGF signal during kidney development.
Disclosures
All authors have nothing to disclose.
Funding
This work was supported by National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases grant P30 DK074038 (to S. Huh).
Supplementary Material
Acknowledgments
We thank Dr. Gail Marin for the Fgf8fl, Fgf8−, and Spry1− mice. We are grateful to Dr. Kameswaran Surendran (Sanford Research) for helpful discussion.
Ms. Ligyeom Ha and Dr. Sung-Ho Huh performed experiments; Ms. Ligyeom Ha, Dr. Sung-Ho Huh, and Dr. Hee-Seong Jang analyzed data; and Dr. Sung-Ho Huh designed experiments and wrote the paper.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020040401/-/DCSupplemental.
Supplemental Figure 1. In situ hybridization of Spry1.
Supplemental Figure 2. Lineage analysis of Fgf20Cre.
Supplemental Figure 3. Fgf9 and Fgf20 are not required for cell death and proliferation of E10.5 nephron progenitors.
Supplemental Figure 4. Fgf9 and Fgf20 regulate genes required for renal branching.
Supplemental Figure 5. Fgf20 does not affect renal phenotypes caused by loss of Fgf8.
Supplemental Figure 6. Fgf8 and Fgf20 regulate genes required for renal branching.
References
- 1.Lemmon MA, Schlessinger J: Cell signaling by receptor tyrosine kinases. Cell 141: 1117–1134, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li E, Hristova K: Role of receptor tyrosine kinase transmembrane domains in cell signaling and human pathologies. Biochemistry 45: 6241–6251, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neben CL, Lo M, Jura N, Klein OD: Feedback regulation of RTK signaling in development. Dev Biol 447: 71–89, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Volovelsky O, Kopan R: Making new kidneys: On the road from science fiction to science fact. Curr Opin Organ Transplant 21: 574–580, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moore MW, Klein RD, Fariñas I, Sauer H, Armanini M, Phillips H, et al. : Renal and neuronal abnormalities in mice lacking GDNF. Nature 382: 76–79, 1996. [DOI] [PubMed] [Google Scholar]
- 6.Pichel JG, Shen L, Sheng HZ, Granholm A-C, Drago J, Grinberg A, et al. : Defects in enteric innervation and kidney development in mice lacking GDNF. Nature 382: 73–76, 1996. [DOI] [PubMed] [Google Scholar]
- 7.Sánchez MP, Silos-Santiago I, Frisén J, He B, Lira SA, Barbacid M: Renal agenesis and the absence of enteric neurons in mice lacking GDNF. Nature 382: 70–73, 1996. [DOI] [PubMed] [Google Scholar]
- 8.Cacalano G, Fariñas I, Wang LC, Hagler K, Forgie A, Moore M, et al. : GFRalpha1 is an essential receptor component for GDNF in the developing nervous system and kidney. Neuron 21: 53–62, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pitera JE, Scambler PJ, Woolf AS: Fras1, a basement membrane-associated protein mutated in Fraser syndrome, mediates both the initiation of the mammalian kidney and the integrity of renal glomeruli. Hum Mol Genet 17: 3953–3964, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cullen-McEwen LA, Drago J, Bertram JF: Nephron endowment in glial cell line-derived neurotrophic factor (GDNF) heterozygous mice. Kidney Int 60: 31–36, 2001. [DOI] [PubMed] [Google Scholar]
- 11.Poladia DP, Kish K, Kutay B, Hains D, Kegg H, Zhao H, et al. : Role of fibroblast growth factor receptors 1 and 2 in the metanephric mesenchyme. Dev Biol 291: 325–339, 2006. [DOI] [PubMed] [Google Scholar]
- 12.Barak H, Huh S-H, Chen S, Jeanpierre C, Martinovic J, Parisot M, et al. : FGF9 and FGF20 maintain the stemness of nephron progenitors in mice and man. Dev Cell 22: 1191–1207, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sims-Lucas S, Cusack B, Baust J, Eswarakumar VP, Masatoshi H, Takeuchi A, et al. : Fgfr1 and the IIIc isoform of Fgfr2 play critical roles in the metanephric mesenchyme mediating early inductive events in kidney development. Dev Dyn 240: 240–249, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gross I, Morrison DJ, Hyink DP, Georgas K, English MA, Mericskay M, et al. : The receptor tyrosine kinase regulator Sprouty1 is a target of the tumor suppressor WT1 and important for kidney development. J Biol Chem 278: 41420–41430, 2003. [DOI] [PubMed] [Google Scholar]
- 15.Basson MA, Akbulut S, Watson-Johnson J, Simon R, Carroll TJ, Shakya R, et al. : Sprouty1 is a critical regulator of GDNF/RET-mediated kidney induction. Dev Cell 8: 229–239, 2005. [DOI] [PubMed] [Google Scholar]
- 16.Chi L, Zhang S, Lin Y, Prunskaite-Hyyryläinen R, Vuolteenaho R, Itäranta P, et al. : Sprouty proteins regulate ureteric branching by coordinating reciprocal epithelial Wnt11, mesenchymal Gdnf and stromal Fgf7 signalling during kidney development. Development 131: 3345–3356, 2004. [DOI] [PubMed] [Google Scholar]
- 17.Basson MA, Watson-Johnson J, Shakya R, Akbulut S, Hyink D, Costantini FD, et al. : Branching morphogenesis of the ureteric epithelium during kidney development is coordinated by the opposing functions of GDNF and Sprouty1. Dev Biol 299: 466–477, 2006. [DOI] [PubMed] [Google Scholar]
- 18.Michos O, Cebrian C, Hyink D, Grieshammer U, Williams L, D’Agati V, et al. : Kidney development in the absence of Gdnf and Spry1 requires Fgf10. PLoS Genet 6: e1000809, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pitera JE, Woolf AS, Basson MA, Scambler PJ: Sprouty1 haploinsufficiency prevents renal agenesis in a model of Fraser syndrome. J Am Soc Nephrol 23: 1790–1796, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meyers EN, Lewandoski M, Martin GR: An Fgf8 mutant allelic series generated by Cre- and Flp-mediated recombination. Nat Genet 18: 136–141, 1998. [DOI] [PubMed] [Google Scholar]
- 21.Grieshammer U, Cebrián C, Ilagan R, Meyers E, Herzlinger D, Martin GR: FGF8 is required for cell survival at distinct stages of nephrogenesis and for regulation of gene expression in nascent nephrons. Development 132: 3847–3857, 2005. [DOI] [PubMed] [Google Scholar]
- 22.Colvin JS, Green RP, Schmahl J, Capel B, Ornitz DM: Male-to-female sex reversal in mice lacking fibroblast growth factor 9. Cell 104: 875–889, 2001. [DOI] [PubMed] [Google Scholar]
- 23.Huh SH, Jones J, Warchol ME, Ornitz DM: Differentiation of the lateral compartment of the cochlea requires a temporally restricted FGF20 signal. PLoS Biol 10: e1001231, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huh S-H, Warchol ME, Ornitz DM: Cochlear progenitor number is controlled through mesenchymal FGF receptor signaling. eLife 4: e05921, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carroll TJ, Park J-S, Hayashi S, Majumdar A, McMahon AP: Wnt9b plays a central role in the regulation of mesenchymal to epithelial transitions underlying organogenesis of the mammalian urogenital system. Dev Cell 9: 283–292, 2005. [DOI] [PubMed] [Google Scholar]
- 26.Stark K, Vainio S, Vassileva G, McMahon AP: Epithelial transformation of metanephric mesenchyme in the developing kidney regulated by Wnt-4. Nature 372: 679–683, 1994. [DOI] [PubMed] [Google Scholar]
- 27.Dressler GR, Deutsch U, Chowdhury K, Nornes HO, Gruss P: Pax2, a new murine paired-box-containing gene and its expression in the developing excretory system. Development 109: 787–795, 1990. [DOI] [PubMed] [Google Scholar]
- 28.Pachnis V, Mankoo B, Costantini F: Expression of the c-ret proto-oncogene during mouse embryogenesis. Development 119: 1005–1017, 1993. [DOI] [PubMed] [Google Scholar]
- 29.Lu BC, Cebrian C, Chi X, Kuure S, Kuo R, Bates CM, et al. : Etv4 and Etv5 are required downstream of GDNF and Ret for kidney branching morphogenesis. Nat Genet 41: 1295–1302, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuure S, Chi X, Lu B, Costantini F: The transcription factors Etv4 and Etv5 mediate formation of the ureteric bud tip domain during kidney development. Development 137: 1975–1979, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Riccio P, Cebrian C, Zong H, Hippenmeyer S, Costantini F: Ret and Etv4 promote directed movements of progenitor cells during renal branching morphogenesis [published correction appears in PLoS Biol 14: e1002488, 2016]. PLoS Biol 14: e1002382, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perantoni AO, Timofeeva O, Naillat F, Richman C, Pajni-Underwood S, Wilson C, et al. : Inactivation of FGF8 in early mesoderm reveals an essential role in kidney development. Development 132: 3859–3871, 2005. [DOI] [PubMed] [Google Scholar]
- 33.Atsuta Y, Takahashi Y: FGF8 coordinates tissue elongation and cell epithelialization during early kidney tubulogenesis. Development 142: 2329–2337, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Attia L, Schneider J, Yelin R, Schultheiss TM: Collective cell migration of the nephric duct requires FGF signaling. Dev Dyn 244: 157–167, 2015. [DOI] [PubMed] [Google Scholar]
- 35.Hacohen N, Kramer S, Sutherland D, Hiromi Y, Krasnow MA: Sprouty encodes a novel antagonist of FGF signaling that patterns apical branching of the Drosophila airways. Cell 92: 253–263, 1998. [DOI] [PubMed] [Google Scholar]
- 36.Knosp WM, Knox SM, Lombaert IMA, Haddox CL, Patel VN, Hoffman MP: Submandibular parasympathetic gangliogenesis requires sprouty-dependent Wnt signals from epithelial progenitors. Dev Cell 32: 667–677, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Metzger RJ, Klein OD, Martin GR, Krasnow MA: The branching programme of mouse lung development. Nature 453: 745–750, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ching ST, Cunha GR, Baskin LS, Basson MA, Klein OD: Coordinated activity of Spry1 and Spry2 is required for normal development of the external genitalia. Dev Biol 386: 1–11, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Minowada G, Jarvis LA, Chi CL, Neubüser A, Sun X, Hacohen N, et al. : Vertebrate Sprouty genes are induced by FGF signaling and can cause chondrodysplasia when overexpressed. Development 126: 4465–4475, 1999. [DOI] [PubMed] [Google Scholar]
- 40.Shim K, Minowada G, Coling DE, Martin GR: Sprouty2, a mouse deafness gene, regulates cell fate decisions in the auditory sensory epithelium by antagonizing FGF signaling. Dev Cell 8: 553–564, 2005. [DOI] [PubMed] [Google Scholar]
- 41.Klein OD, Minowada G, Peterkova R, Kangas A, Yu BD, Lesot H, et al. : Sprouty genes control diastema tooth development via bidirectional antagonism of epithelial-mesenchymal FGF signaling. Dev Cell 11: 181–190, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brown AC, Adams D, de Caestecker M, Yang X, Friesel R, Oxburgh L: FGF/EGF signaling regulates the renewal of early nephron progenitors during embryonic development. Development 138: 5099–5112, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Steinberg F, Zhuang L, Beyeler M, Kälin RE, Mullis PE, Brändli AW, et al. : The FGFRL1 receptor is shed from cell membranes, binds fibroblast growth factors (FGFs), and antagonizes FGF signaling in Xenopus embryos. J Biol Chem 285: 2193–2202, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sleeman M, Fraser J, McDonald M, Yuan S, White D, Grandison P, et al. : Identification of a new fibroblast growth factor receptor, FGFR5. Gene 271: 171–182, 2001. [DOI] [PubMed] [Google Scholar]
- 45.Zhuang L, Villiger P, Trueb B: Interaction of the receptor FGFRL1 with the negative regulator Spred1. Cell Signal 23: 1496–1504, 2011. [DOI] [PubMed] [Google Scholar]
- 46.Combes AN, Phipson B, Lawlor KT, Dorison A, Patrick R, Zappia L, et al. : Single cell analysis of the developing mouse kidney provides deeper insight into marker gene expression and ligand-receptor crosstalk [published correction appears in Development 146: dev182162, 2019]. Development 146: dev178673, 2019. [DOI] [PubMed] [Google Scholar]
- 47.Ihermann-Hella A, Hirashima T, Kupari J, Kurtzeborn K, Li H, Kwon HN, et al. : Dynamic MAPK/ERK activity sustains nephron progenitors through niche regulation and primes precursors for differentiation. Stem Cell Reports 11: 912–928, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ornitz DM, Itoh N: The fibroblast growth factor signaling pathway. Wiley Interdiscip Rev Dev Biol 4: 215–266, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hisaoka K, Tsuchioka M, Yano R, Maeda N, Kajitani N, Morioka N, et al. : Tricyclic antidepressant amitriptyline activates fibroblast growth factor receptor signaling in glial cells: Involvement in glial cell line-derived neurotrophic factor production. J Biol Chem 286: 21118–21128, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ohuchi H, Hori Y, Yamasaki M, Harada H, Sekine K, Kato S, et al. : FGF10 acts as a major ligand for FGF receptor 2 IIIb in mouse multi-organ development. Biochem Biophys Res Commun 277: 643–649, 2000. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.