

Significance Statement
In patients with CKD, receptor Notch3 is strongly upregulated. Conversely, in experimental kidney disease models, Notch3 deficiency protects from organ damage. To determine whether Notch3 on immune cells or tissue-resident cells participates in the inflammatory response, animals with bone marrow chimerism were generated. These animal strains do not exhibit phenotypic differences in the absence of disease. However, after unilateral ureteral obstruction, distinct alterations in the immune response and organ fibrosis become apparent. Notch3 receptors expressed by immune cells are of relevance for transmigration into tissue; the receptors expressed by resident kidney cells orchestrate organ fibrosis. These events seem to be separable and distinct.
Keywords: fibrosis, chronic kidney disease, cell signaling, chronic inflammation, cell adhesion
Visual Abstract

Abstract
Background
Kidney injuries that result in chronic inflammation initiate crosstalk between stressed resident cells and infiltrating immune cells. In animal models, whole-body receptor Notch3 deficiency protects from leukocyte infiltration and organ fibrosis. However, the relative contribution of Notch3 expression in tissue versus infiltrating immune cells is unknown.
Methods
Chimeric mice deficient for Notch3 in hematopoietic cells and/or resident tissue cells were generated, and kidney fibrosis and inflammation after unilateral ureteral obstruction (UUO) were analyzed. Adoptive transfer of labeled bone marrow–derived cells validated the results in a murine Leishmania ear infection model. In vitro adhesion assays, integrin activation, and extracellular matrix production were analyzed.
Results
Fibrosis follows UUO, but inflammatory cell infiltration mostly depends upon Notch3 expression in hematopoietic cells, which coincides with an enhanced proinflammatory milieu (e.g., CCL2 and CCL5 upregulation). Notch3 expression on CD45+ leukocytes plays a prominent role in efficient cell transmigration. Functionally, leukocyte adhesion and integrin activation are abrogated in the absence of receptor Notch3. Chimeric animal models also reveal that tubulointerstitial fibrosis develops, even in the absence of prominent leukocyte infiltrates after ureteral obstruction. Deleting Notch3 receptors on resident cells blunts kidney fibrosis, ablates NF-κB signaling, and lessens matrix deposition.
Conclusions
Cell-specific receptor Notch3 signaling independently orchestrates leukocyte infiltration and organ fibrosis. Interference with Notch3 signaling may present a novel therapeutic approach in inflammatory as well as fibrotic diseases.
Chronic inflammation of solid organs often results in destruction of organ architecture and perpetuated fibrosis, with functional deficits and ultimately organ failure.1,2 The list of injuries causing inflammation includes acute and chronic infections, toxic agents, autoimmune diseases, and metabolic alterations (e.g., hyperglycemia and uremia).3–6 Key events after primary insults are stress responses of resident organ cells and leukocyte recruitment. The composition of the infiltrate depends on the timing and severity of the immune response, which encompasses defense mechanisms, such as complement activation, phagocytosis, and antigen presentation.7 Approximately 15% of adults within the United States (37 million people) have CKD.8 Despite the enormity of this problem, therapeutic options are scarce and mostly ineffective. Therefore, an improved understanding of the cellular and molecular mechanisms leading to inflammation and renal fibrosis is essential to develop strategies to combat these processes, because fibrosis-related organ diseases account for almost 50% of all deaths.9–12 Currently, established models of renal fibrosis propose that kidney injury leads to immune cell infiltration, fibroblast activation, extracellular matrix (ECM) deposition, microvascular rarefaction, and tubular atrophy.9 Inflammation is considered the driving force in fibrotic diseases, with an intimate crosstalk between resident and infiltrating cells; therefore, targeting immune cell infiltration is deemed to be a therapeutic option.13–15 However, it is not known whether immune cell recruitment is the culprit of organ fibrosis, or a bystander that modulates reparative processes.1
In CKD, the Notch receptor family plays a key role in both inflammatory responses and organ fibrosis.16–19 Animals deficient in receptors Notch1 and -3 demonstrate similar protective effects on organ fibrosis. Notch3-deficient mice exhibit significantly reduced kidney fibrosis (>50%), fewer myofibroblasts (approximately 50%), and fewer infiltrating immune cells (approximately 30% of total leukocytes) compared with wild-type (WT) mice after ureteral obstruction.18 The inflammatory chemokines CCL2 and CCL5 are reduced by >50% in Notch3-deficient mice.18 Similar findings are reported in Notch3-deficient animals undergoing nephrotoxic serum nephritis and ischemia reperfusion. Thus, genetic ablation of receptor Notch3 protects mice from kidney damage.18–20
However, it is not clear whether this protection is mediated by Notch3 deficiency on immune cells or by a lack of Notch3 expression on tissue-resident cells. Here, we use bone marrow (BM) chimeras to clarify cell type–specific roles of receptor Notch3 and discover that Notch3 receptors are not only relevant for leukocyte transmigration, but are also key molecules for the fibrogenic niche. Unexpectedly, organ fibrosis is not reduced in the absence of prominent leukocyte infiltration and vice versa.
Methods
Animals
Studies were performed using Notch3-knockout animals and their WT littermates in a C57BL/6J (CD45.2) background (kindly provided by Dr. Anne Joutel, Institut National de la Santé et de la Recherche Médicale).21,22 The commercially available CD45.1 mice are C57BL/6J background. The health of the mice corresponded to the Federation of European Laboratory Animal Science Association guidelines. The animals were housed and bred in the Central Animal Facility of the Otto-von-Guericke University Magdeburg Medical Faculty. The mice were kept under specific pathogen-free conditions in individual, ventilated cages (Techniplast, Buguggiate, Italy). All experiments and procedures were conducted in accordance with the German National Guidelines for the Use of Experimental Animals (Animal Protection Act) and were approved by the state of Sachsen-Anhalt (Aktenzeichen UniMD 42502-2-1135 and UniMD 42502-2-1253). All mouse strains were confirmed by PCR genotyping that was performed as previously described.18 Briefly, the mouse tail was lysed with 200 µl direct PCR tail lysis buffer and 50 µg of proteinase K. PCR was performed using primer pairs for WT (forward, CCATGAGGATGCTATCTGTGAC; reverse, CACATTGGCACAAGAATGAGCC) and Notch3 knockout (forward, TCGCCTTCTTGACGAGTTCT; reverse, GCGATGCAATTTCCTCATTT). The PCR cycler performed 35 cycles at 94°C, 60°C, and 72°C (each for 60 seconds). Products were separated on 1% agarose/ethidium bromide gels.
Leishmania major Infection
Leishmania major inoculation was performed as described.23 Briefly, dsRed-expressing parasites were grown at 26°C, for a maximum of five passages, in M119 medium supplemented with 10% heat-inactivated FBS, 0.1 mM adenine, 1 µg/ml biotin, 5 mg/ml hemin, and 2 µg/ml biopterin (all from Sigma-Aldrich, Darmstadt, Germany). For infection, 105 stationary-phase promastigotes were resuspended in 10 µl PBS and injected into the ear dermis.
Experimental Kidney Disease
Unilateral ureter obstruction (UUO) was performed with sex-matched, 12- to 16-week-old mice. The right ureters of WT and knockout mice were ligated for 5 and 14 days. Both kidneys were collected for preparation of cortical RNA, protein lysates, immunohistochemistry, and flow cytometry (FACS). Blood sampling by heart puncture was subjected to complete blood cell counting (ADVIA 120; Bayer Diagnostics Munich, Germany) and plasma specimens were stored at −80°C until further analysis. Contralateral kidneys were harvested for comparative analyses.
Primary Cell Cultures
BM cells were obtained from the femurs of mice as previously described.24 Cells were counted before BM transplantation or homing assays, aliquoted in 1×106 or 50,000 cells, respectively, and stored on ice. Polarization into BM-derived macrophages (BMDMs) was propagated in DMEM (Thermo Fisher, Dreieich, Germany) medium supplemented with 10% FBS (Thermo Fisher), 50 U/ml penicillin (Thermo Fisher), 50 μg/ml streptomycin (Thermo Fisher), and 10 ng/ml macrophage colony-stimulating factor (M-CSF; PeproTech) overnight. The next day, nonadherent cells (1×108 cells/plate) were transferred into a new petri dish and maintained for 6 days in cell culture medium with M-CSF (10 ng/ml) and the following respective cytokines: IL-13 (20 ng/ml) and IL-4 (30 ng/ml) (PeproTech) for the anti-inflammatory response; or IL-6 (20 ng/ml) (PeproTech) for the proinflammatory response. Half of the medium was exchanged daily. Tubular cells were prepared as previously described.24
Preparation of Tubular Cell–Derived ECM Scaffold
Serum-starved tubular cells were treated without or with recombinant Sonic hedgehog (Shh) protein (50 ng/ml; Merck Millipore, Darmstadt, Germany) for 3 days. To establish the Notch3 dependency of Shh-induced ECM scaffold, we added a mouse-specific Notch3 receptor blocking antibody (provided by Genentech) 1 hour before stimulation with Shh. To isolate ECM, cells were detached with EGTA (Sigma-Aldrich) in calcium-free PBS (Thermo Fisher), with shaking at 4°C for 1 hour. The procedure was repeated four times until all cells were removed. The tubular cell–derived ECM scaffold was washed with PBS and collected by scraping with a rubber policeman in PBS for subsequent Western blot analysis.
BM Transplantation
Female (8–10 weeks old) C57BL/6N (n=6) and Notch3−/− (n=6) mice were used as BM donors for age- and sex-matched C57BL/6J (n=12) and Notch3−/− (n=12) recipient mice. Recipients were irradiated with a total dose of 8 Gy divided into two doses (4 Gy each) at an interval of 3 hours. BM cells were isolated from the femur and tibia of donor mice as described.24 At 4 hours after irradiation, BM cells were injected via tail vein. All mice received the antibiotic neomycin sulfate (2 mg/ml) (Merck, Darmstadt, Germany) via drinking water for 2 weeks. BM reconstitution was confirmed after 4 weeks.
Homing Experiments
Successful BM cell homing was visualized by fluorescence cytometry of BM cells. LSK (lineage-negative stem cell antigen 1 positive kit positive) cells were isolated by sorting from BM cells (FACSAria III; BD) and staining with Vybrant DiD (Thermo Fisher) according to the manufacturer’s instructions. Cells (5×104) were injected into irradiated mice, and 16 hours later these mice were euthanized and BM cells and axillary lymph nodes were analyzed for DiD-positive cell numbers by fluorescence cytometry.
Adoptive Transfer of Immune Cells and Quantification of Tissue Infiltration
BM cells were isolated using CD45.1 WT and CD45.2 Notch3−/− mice as donors (see above for isolation procedure) without erythrocyte lysis. Staining of cells was performed by incubation for 10 minutes at 37°C in 5 µM carboxyfluorescein succinimidyl ester (CFSE; Thermo Fisher). After two washes in PBS supplemented with 10% FCS, 2.5×107 CD45.1 WT cells were mixed with 2.5×107 CD45.2 Notch3−/− cells in 300 µl PBS and injected into the tail vein in WT recipient mice. For assessment of cell infiltration, kidney tissue was mechanically crushed and digested in buffer I (RPMI; Thermo Fisher), 0.1% BSA (Sigma-Aldrich), 1 mg/ml collagenase D (Sigma-Aldrich), and 100 µg/ml DNase I (Sigma-Aldrich) for 30 minutes. After repeated mechanical tissue disintegration, the entire well contents were passed through 70- and 40-µm cell strainers. Erythrocytes were lysed and cells were analyzed by flow cytometry. Ears were separated into dorsal and ventral sheets using jagged forceps, digested in RPMI 1640 medium containing collagenase (1 mg/ml) and DNase (50 ng/ml) (both from Sigma-Aldrich) for 45 minutes at 37°C, and then passed through a 70-µm cell strainer.
Flow Cytometry
Single-cell suspensions of tissue samples (kidney and ear) and isolated BMDMs were labeled using antibodies directed against surface receptors/proteins. Fixation and cell membrane permeabilization was performed with the FOXP3 Fix/Perm buffer set (BioLegend, Koblenz, Germany). Nonspecific binding was minimized with 5% mouse serum. Surface markers included the following: CD11b clone M1/70 APC/Cy7, CD3 clone 145-2C11 APC, CD45 clone 30-F11 PE, F4/80 clone BM8 Pacific Blue, Gr1 (Ly6G/Ly6C) clone RB6-8C5 PerCP/Cy5.5, CD45.1 clone A20 PerCP, CD45.2 clone 104 APC, Ly6G clone 1A8 BV421, Ly6C clone HK1.4 PE-Cy7, MHC-II clone M5/114.15.2 BV510, CD29 clone HMbeta1 PECy7, CD18 clone M18/2 FITC, and IL6R clone D7715A7 PECy7 (all from BioLegend). Flow cytometry was performed with a FACS Canto II or Fortessa (BD Immunocytometry Systems). Data collection and analyses were achieved using FlowJo software. Spectral spillover was corrected by creating a compensation matrix generated with the help of single antibody stainings and fluorescence minus one control samples.
Integrin Signaling
Monitoring of activated β1-integrins was performed as described.25 Briefly, BMDMs were stimulated in 25 mM HEPES buffer (pH 7.4), supplemented with 150 mM sodium chloride (NaCl) and magnesium chloride. As positive control for activated β1-integrin, cells were incubated in buffer solution containing 5 mM manganese(II) chloride. Stimulation was performed with IL-6 (15 ng/ml; Peprotech) alone or together with anti–IL-6 blocking antibody (R&D) for the indicated time points. After stimulation, cells were incubated with 9EG7 antibody (CD29 clone 9EG7, antibody detecting active β1-integrin; BD Biosciences, Heidelberg, Germany) and, after washing with modified FACS buffer (25 mM HEPES, pH 7.4, 150 mM NaCl, 5% FCS, 0.1% sodium azide), rat IgG-specific, FITC-conjugated, goat anti-rat antibody (BD Biosciences) was added. Washes with buffer solution removed unbound antibodies.
NF-κB Signaling
Cells were stimulated with 1 µg/cm2 of recombinant tenascin-C (TNC; Merck) for the indicated time points and subsequently washed with ice-cold PBS. Cell lysis was performed with radioimmunoprecipitation assay lysis buffer (50 mM Tris base, 150 mM NaCl, 1 mM EDTA, 1% nonidet P-40, 0.25% sodium deoxycholate) supplemented with cOmplete Mini Protease Inhibitor Cocktail Phospho-Stop (Roche). Lysates were cleared by centrifugation at 14,000 × g. Protein concentrations were determined using Lowry assay.
Cytokine Quantification
In tissue lysates of mechanically homogenized kidneys, cytokine levels of MIP1α, MIG, KC, IL-10, TNFα, CCL5, and CCL2 were determined using a flow cytometry–based bead assay (BD Biosciences) using FACS Canto II.
Immunohistochemistry
Methacarn-fixed, paraffin-embedded tissue sections were used for immunohistochemistry (avidin-biotin complex method). Unmasking of antigens was optimized as follows: for F4/80 staining, sections were incubated with protease 1 (Ventana, Basel, Switzerland) for 10 minutes at 20°C. Endogenous peroxidase activity was inhibited by 3% hydrogen peroxide. After washing, the sections were incubated with blocking solution (20% FCS in 1× Tween 20–PBS) for 1 hour at room temperature and incubated overnight at 4°C with the primary antibody diluted in 1% FCS plus 1× Tween 20–PBS. The biotinylated secondary antibody was incubated for 30 minutes at room temperature, and sections were washed three times in 1× Tween 20–PBS. Avidin-biotin-peroxidase complex (Elite Kit; Vector Laboratories) was used for staining, followed by counterstaining with hematoxylin.
Periodic Acid–Schiff Staining
Sections were deparaffinized and rehydrated. For staining, the sections were incubated with periodic acid solution and Schiff reagent (both from Sigma-Aldrich), followed by an extensive washing step. The counterstain was carried out with hematoxylin solution.
Sirius Red Staining
Deparaffinized sections were incubated with 0.1% Sirius Red in saturated picric acid (Dr. K. Hollborn & Söhne GmbH & Co KG, Leipzig, Germany) and were destained with 0.01 N hydrochloric acid. The dehydrogenation and subsequent embedding were similar to the description above.
Automated Multidimensional Fluorescence Microscopy
Snap-frozen tissue embedded into O.C.T. (Sakura Finetek) was used to perform cryo-sections (10 µm thickness) that were placed on silan-coated slides. Tissue samples were fixed with 2% paraformaldehyde (Santa Cruz), permeabilized with 0.2% Triton X-100, and blocked with 1% BSA (Sigma) in PBS. Multiepitope ligand cartography (MELC) analysis was performed as previously described.24 The antibodies used are listed in Table 1. The appropriate working dilutions, incubation times, and positions within the MELC experiment were validated systematically using conditions suitable to MELC, as described,26 with subsequent data analysis.27
Table 1.
List of primary antibodies
| Primary Antibodies | Catalog No. | Dilution | Company | Dye |
|---|---|---|---|---|
| Immunhistochemistry and immunofluorescence | ||||
| Gli1 (IF) | AF3455 | 1:200 | R&D Systems | |
| Notch3 (IHC) | ab23426 | 1:200 | Abcam | |
| F4/80 (IHC) | MON3099 | 1:100 | Monosan | |
| Western blotting | ||||
| Notch3 | sc-5539 | 1:500 | Santa Cruz | |
| TNC | ab108930 | 1:500 | Abcam | |
| Tubulin | T5168 | 1:1000 | Sigma | |
| Phospho–NF-κB–p65 (S536) | 3033 | 1:1000 | Cell Signaling | |
| Phospho-IκBα (S32) | 2859 | 1:1000 | Cell Signaling | |
| NF-κB–p65 | 6956 | 1:1000 | Cell Signaling | |
| IκBα | 9242 | 1:1000 | Cell Signaling | |
| Vinculin | sc-59803 | 1:1000 | Santa Cruz | |
| CYLD | 8462 | 1:1000 | Cell Signaling | |
| SMA | ab5694 | 1:1000 | Abcam | |
| Phospho-STAT3 (S727) | 9134 | 1:1000 | Cell Signaling | |
| GAPDH | sc-20357 | 1:2000 | Santa Cruz | |
| Arg1 | 610709 | 1:1000 | BD Biosciences | |
| MELC | ||||
| Vimentin | 1:120 | 515635 | Exbio | Alexa Fluor 488 |
| Collagen type 4 | 1:240 | 1340–30 | Southern Biotech | Alexa Fluor 488 |
| SMA | 1:240 | F3777 | Sigma | FITC |
| Propidium iodide | 1:5000 | P4864 | Sigma | |
| Phospho-NF-κB–p65 (S529) | 1:20 | 558421 | BD Biosciences | Alexa Fluor 488 |
| CD45 | 1:400 | 553080 | BD Biosciences | FITC |
| F4/80 | 1:20 | 53–4801 | eBioscience | Alexa Fluor 488 |
| CD4 | 1:20 | 557667 | BD Biosciences | Alexa Fluor 488 |
| CD3ε | 1:60 | 100210 | Biolegend | Alexa Fluor 488 |
IF, immunofluorescence; IHC, immunohistochemistry; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; S, serine.
Gene Array
RNA extraction from kidney tissue lysates and gene array analysis have been described.24 The extraction protocol was as follows: RNA was prepared using a Qiagen (Valencia, CA) RNAeasy Kit. RNA was quantified using a NanoDrop-1000 spectrophotometer, and quality was monitored with the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). The labeling protocol was as follows: cyanine 3–labeled complementary RNA (cRNA) was prepared from 0.5 μg RNA using the One-Color Low RNA Input Linear Amplification PLUS kit (Agilent Technologies), according to the manufacturer’s instructions, followed by RNAeasy column purification (Qiagen). Dye incorporation and cRNA yield were checked using the NanoDrop ND-1000 Spectrophotometer. The hybridization protocol was as follows: cyanine 3–labeled cRNA (1.5 μg; sp act, >10.0 pmol cyanine 3/μg cRNA) was fragmented at 60°C for 30 minutes in a reaction volume of 250 ml containing 1× Agilent fragmentation buffer and 2× Agilent blocking agent, following the manufacturer’s instructions. On completion of the fragmentation reaction, 250 ml of 2× Agilent hybridization buffer was added to the fragmentation mixture and hybridized using the Agilent 4×44k Mouse v2 (design identifier, 026655) for 17 hours at 65°C in a rotating Agilent hybridization oven. After hybridization, the microarrays were washed for 1 minute at room temperature with GE Wash buffer 1 (Agilent Technologies) and for 1 minute with 37°C GE Wash buffer 2 (Agilent Technologies), and then dried immediately by brief centrifugation. For data processing, the scanned images were analyzed with Feature Extraction Software 10.5 (Agilent Technologies) using default parameters to obtain background-subtracted and spatially detrended processed signal intensities.
Proliferation Assay
BMDMs (1×104 cells per well) were seeded in six-well plates. The medium used was complete DMEM with M-CSF (10 ng/ml; PeproTech) for nonpolarized macrophages, M-CSF and IL-6 for proinflammatory macrophages, or M-CSF together with IL-4 and IL-13 for anti-inflammatory macrophages. Half of the medium was exchanged daily. The cell number was quantified manually by counting in Neubauer chambers.
Lysate Preparation and Western Blot Analysis
Kidney tissue was mechanical homogenized in RIPA buffer (50 mM Tris hydrochloride, 150 mM nonidet P-40, 1 mM sodium deoxycholate, 1 mM EDTA, and 1 mM sodium orthovanadate) containing complete protease inhibitor cocktail (Roche) at 4°C for 15 minutes. Protein was quantified using the Bio-Rad protein assay. Heat-denatured protein samples were separated using 10% SDS-PAGE and blotted onto nitrocellulose membranes. The membranes were blocked with 5% dry milk in TBS/Tween 20, and then incubated with primary antibodies diluted in TBS/Tween 20 overnight at 4°C. The applied antibodies are listed in Table 1. As secondary antibodies, horseradish peroxidase–conjugated goat anti-rabbit IgG (Biozol) or goat anti-mouse IgG (Biozol, Germany) serum was added for 30 minutes. Pierce ECL substrate (Thermo Scientific) was used for detection.
Adhesion Assay
For adhesion to culture plates, BMDMs were polarized with M-CSF over 7 days and then detached by addition of trypsin/EDTA. Suspended cells were serum starved for 1 hour in adhesion assay medium (10 mM HEPES, pH 7.4, 137 mM NaCl, 1 mM magnesium chloride, 1 mM calcium chloride, 2.7 mM potassium chloride, 4.5 g/L glucose, 3% BSA [wt/vol]). Thereafter, 2.5×105 cells per well were plated out on cell culture plates (12-well; Greiner Bio-One) in the same medium supplemented with 8% FCS, and 5 mM manganese sulfate, if indicated. Seeded cells were washed and fixed with 4% paraformaldehyde (wt/vol) in PBS, and stained with Alexa Fluor 546 phalloidin and 4′,6-diamidino-2-phenylindole (DAPI). Positive nuclear staining of DAPI represented adherent cells. The whole well was imaged using a 2.5× objective and cell numbers were counted. Human umbilical vein endothelial cells (HUVECs) were grown as described.27 HUVECs were activated with 1 µM PMA for 12 hours before adding freshly isolated BM cells. Notch3 WT cells were labeled with CFSE (0.5 µM). Notch3-knockout cells were stained with 0.1 µM CellTracker Deep Red (Thermo Fisher). After two washes in PBS supplemented with 10% FCS, WT cells were mixed with Notch3−/− cells at a 1:1 ratio and incubated for 1 or 2 hours. Adhesion of cells BM-derived cells to HUVECs was analyzed after detachment from the surface by flow cytometry.
Databases
The Nephroseq database (www.nephroseq.org) was used for analysis and visualization of the expression of the Notch receptor protein family in kidney tissue. The ImmGen database (www.immgen.org) was used for analysis and visualization of Notch receptor family expression among immune cell compartments.
Unique molecular identifier count data from Lake et al.28 was downloaded from Gene Expression Omnibus (GSE121862). Counts were normalized and visualized using Seurat version 3.0.1.29
Statistical Analyses
All results were confirmed in three independent experiments, if not otherwise stated. Data management and statistical analysis was performed using GraphPad Prism software version 7.03 (GraphPad Software, San Diego, CA). Results were calculated and presented as mean±SD. For multiple comparisons, statistical difference was calculated by one-way ANOVA. Post hoc analysis was performed with the Tukey test when ANOVA showed significant differences. P<0.05 was considered statistically significant.
Results
Notch Receptors Are Differentially Expressed in Kidney Disease
Database and single-cell RNA sequence analyses reveal differential Notch receptor expression in healthy and injured kidneys. Both Notch1 and -3 are highly expressed in diseased kidneys, but in different cell types (Figure 1, A–C). Immunohistochemistry reveals a strongly activated Notch3 signaling pathway in diseased human (lupus nephritis, IgA nephritis) and mouse (UUO) kidneys, predominantly in tubular as well as tubulointerstitial cells.18,19 Costaining using the pericyte marker Gli1 together with Notch3 confirms coexpression in pericytes, which is similarly determined by single-cell RNA sequencing (Figure 1, D and E). Furthermore, a distinct expression profile of the Notch receptors in immune cells exists, even in the absence of disease (Figure 1F).
Figure 1.
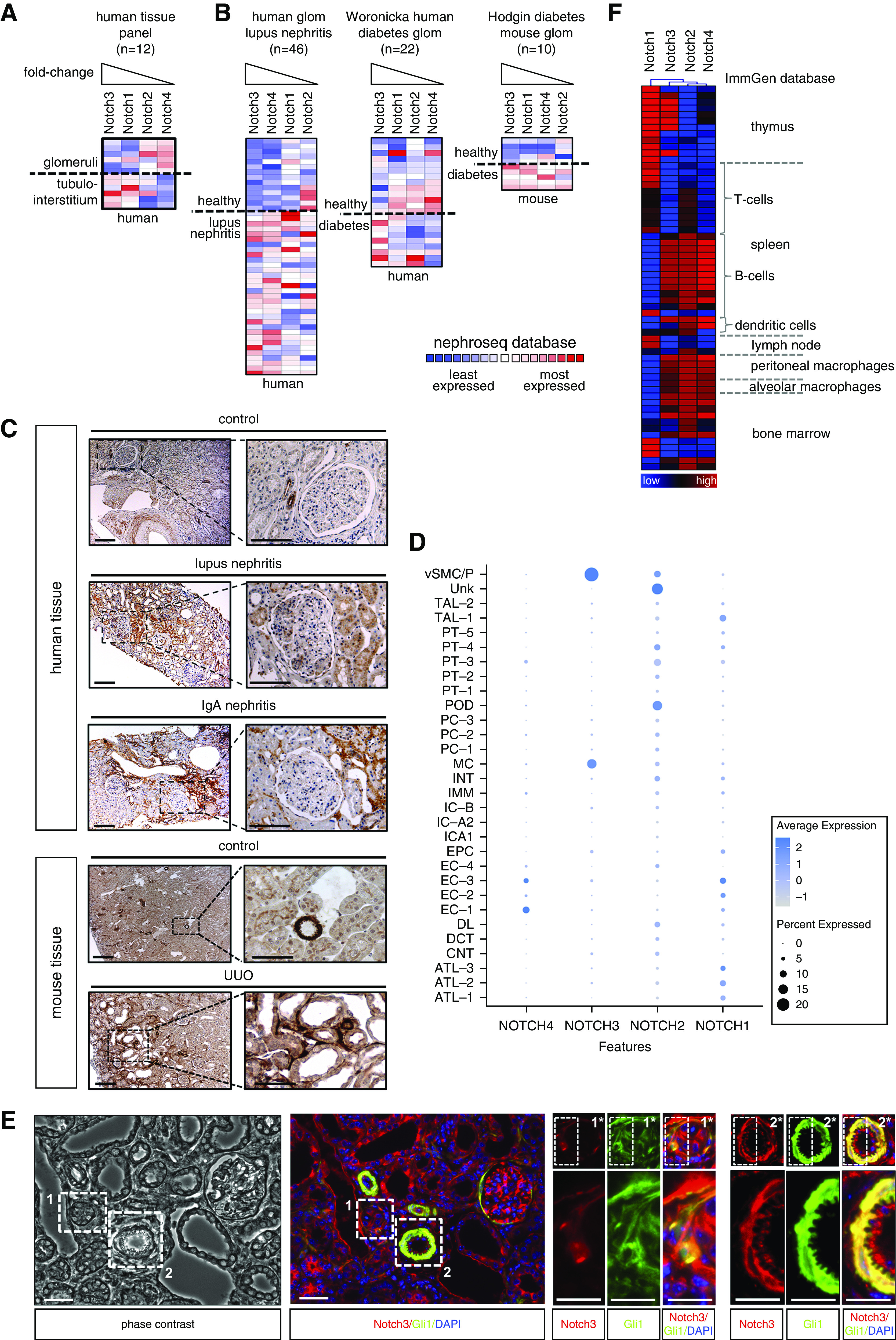
Notch3 receptor expression is upregulated in diseased tissue. Members of the Notch receptor protein family are differentially expressed in (A) healthy glomeruli and the tubulointerstitium. Colors indicate z-scores normalized to depict relative values within rows. The Nephroseq database (www.nephroseq.org) was used for analysis and visualization. (B) Within diseased human and mouse kidney tissue, an upregulation of all members of the Notch family could be observed. Among the four members of the family, Notch3 transcripts were the most strongly regulated in different diseases. (C) Histochemical validation of expression and signaling of Notch3 in diseased human/mouse kidney tissue compared with healthy control tissue. Kidney sections were immunohistochemically stained with specific antibody against the intracellular domain of receptor Notch3. Scale bar, 50 μm. (D) Dot plot of NOTCH1–4 expression in human kidneys using data from Lake et al.28 Dot size indicates the percentage of cells with respective gene expression. The color scale indicates average normalized gene expression. (E) Pericyte marker Gli1 and Notch3 colocalize in vessels and tubulointerstitial cells. Cells positive for Gli1 (green) and Notch3 (red) are seen in vessels and are dispersed in the injured kidneys after UUO in the tubulointerstitium. Scale bars, 100 µm. (Note that the image is rotated 90° clockwise to fit the layout.) (F) Heat-map analysis of Notch receptor family members in immune (cell) compartments, with data obtained from the ImmGen database (www.immgen.org) (blue, low expression; red, high expression). ATL-1/-2/-3, thin ascending limb; CNT, connecting tubule; DCT, distal convoluted tubule; DL, descending limb; EC-1, endothelial cells—glomerular capillaries; EC-2, endothelial cells—AVR; EC-3, endothelial cells—AEA and DVR; EC-4, endothelial cells (unassigned); EPC, epithelial cells (unassigned); glom, glomerular; IC-A2, collecting duct—intercalated cells type A (medulla); IC-A1, collecting duct—intercalated cells type A (cortex); IC-B, collecting duct—intercalated cells type B; IMM, immune cells—macrophages; INT, interstitium; MC, mesangial cells; PC-1, collecting duct—principal cells (cortex); PC-2, collecting duct—principal cells (stressed); PC-3, collecting duct—principal cells (medulla); POD, podocytes; PT-1, proximal tubule epithelial cells (S1); PT-2, proximal tubule epithelial cells (S2); PT-3, proximal tubule epithelial cells (stress/inflammation); PT-4, proximal tubule epithelial cells (fibrinogen+; S3); PT-5, proximal tubule epithelial cells (S3); TAL-1/-2, thick ascending limb; Unk, unknown—novel PT CFH+ subpopulation (S2); vSMC/P, vascular smooth muscle cells and pericytes.
To gain insight into the mechanism(s) by which Notch3 deficiency alters the pattern of kidney damage, we performed gene expression analysis of UUO kidneys. We observe 1463 differentially expressed genes in Notch3 knockouts compared with WT animals, with 353 genes altered by a log2 fold change greater than one (228 down-, 125 upregulated). Gene Ontology and pathway analyses were carried out to determine molecular processes and biologic pathways associated with differentially expressed genes. Pathway analyses reveal reduced inflammatory signaling pathways in knockout animals after UUO (Figure 2). Several of the regulated molecules play key roles in fibrotic and inflammatory processes. To facilitate interpretation of the data, a subset of fibrosis-associated genes, which are known to be upregulated in WT animals after UUO, were selected.30 Immunohistochemistry of TNC and collagens 1 and 3 in diseased kidneys of WT and Notch3-knockout mice confirm these findings (Figure 2, C–E).
Figure 2.
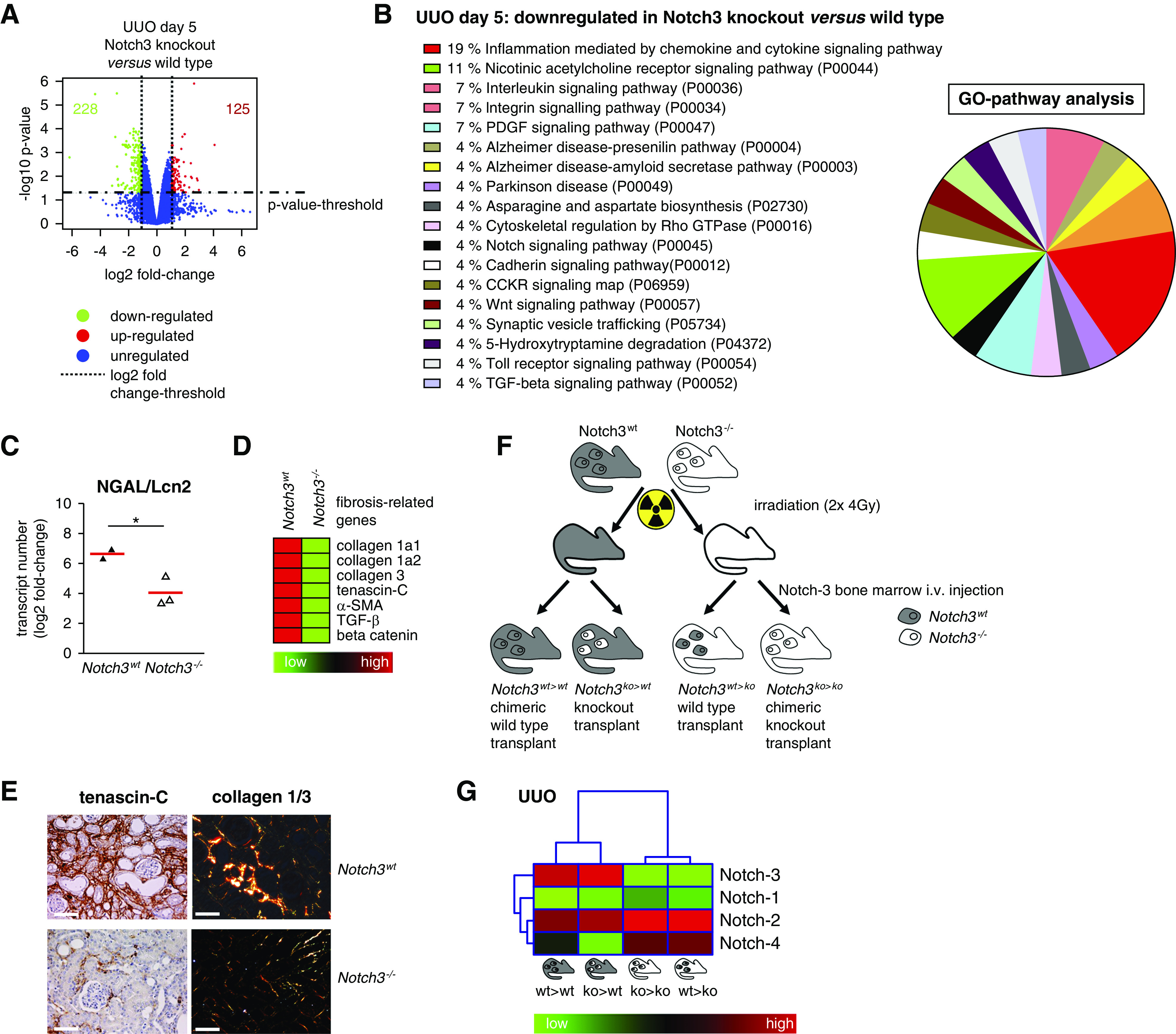
Notch3-knockout animals are protected from UUO-induced disease. (A) Volcano plot of all genes is depicted. The most significantly upregulated genes are shown in red, whereas green represents the significantly downregulated genes. Blue dots are genes that are differentially expressed below the cutoff values of log-fold change and log10 P value. (B) Upregulated genes (fold change ≥1.0 and P>0.05) are classified according to their involvement in different pathways via Panther database. Numbers show the percentage of genes involved in the respective pathways. (C) Quantitative mRNA analysis of the tubular injury marker neutrophil gelatinase–associated lipocalin (NGAL/Lcn2) in kidney tissue of WT and Notch3 receptor–knockout animals revealed that tubular cells are protected from UUO-induced tissue damage in the absence of Notch3 receptor. (D) Heat-map analysis of selected fibrosis-relevant genes after UUO in WT and Notch3-knockout animals (red, upregulated; green, downregulated). (E) Histochemical validation of enhanced TNC and collagen 1/3 expression in WT UUO kidney tissue compared with Notch3-knockout tissue. (F) Scheme of the four chimeric animal models generated and used in the study. BM transplantation was carried out with 8-week-old mice by intravenous (i.v.) injection of 1×106 BM-derived cells 4 hours after irradiation. The images of individual cells refer to hematopoietic (BM-derived) cells; the rest of the mouse body represents nonhematopoietic tissue. The presence of WT Notch3 receptor is indicated by gray, the Notch3 knockout by white. To investigate the contribution of Notch3 expression on BM-derived cells and tissue-resident cells, UUO was performed 4 weeks after transplantation. Kidneys were analyzed on days 5 and 14 after disease induction. (G) Hierarchic clustering and heat-map analysis of Notch1–4 expression in UUO kidneys. GO, Gene Ontology.
Immune Cell Infiltration into the Tissue Requires Intact Notch3 Signaling
To specifically address the role of receptor Notch3 on immune versus tissue cells, BM chimeras were generated (Figure 2, F and G, Table 2). Extensive analyses confirm full reconstitution and complete BM chimerism in the animal strains (Supplemental Figures 1–3, Supplemental Table 1). Five days after UUO induction, the immune cell infiltrate was analyzed. The percentage of infiltrating CD45+ cells in injured kidneys is reduced by approximately 70% in Notch3-deficient BM transplanted into WT recipients (Notch3ko>wt) compared with knockout animals receiving WT BM cells (Notch3wt>ko) (Figure 3). Among the CD45+ cells infiltrating the kidney, we observe a 50% reduction of CD3+ T cells, macrophages, and monocytes, and a 70% reduction of neutrophils. These differences are similarly seen in Notch3ko>ko and Notch3ko>wt strains, which contrast to the results seen in Notch3wt>ko and Notch3wt>wt animal strains.
Table 2.
Scheme of BM transplantation
 |
Figure 3.
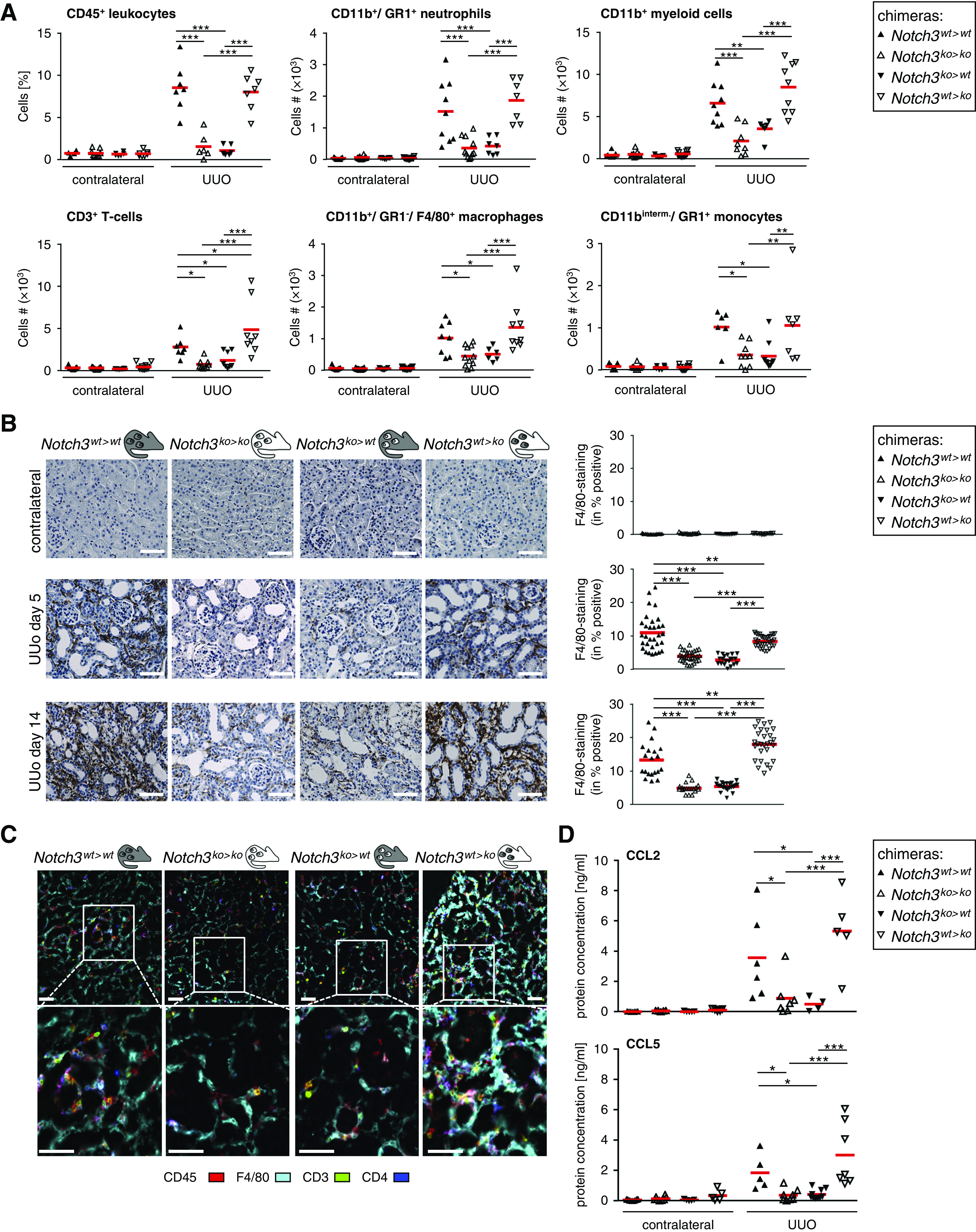
Immune cell infiltration is dependent on Notch3 expression on BM-derived cells. (A) Effect of Notch3 expression on immune cell infiltration after the induction of UUO. Healthy (contralateral) and diseased kidneys of the four types of chimeric mice were harvested after UUO. Kidney tissue was digested with collagenase and the resulting single-cell suspension was stained and characterized by flow cytometry (gating strategy shown in Supplemental Figure 6). All data collection and analyses were performed using FlowJo software (Ashland, OR). Controls were used for gating analyses to distinguish immunopositive from unstained cell populations (n=12 in each group). Cell numbers were calculated from the CD45+ leukocyte population. (B) Immunostaining of infiltrating monocytes/macrophages was performed using the F4/80 antibody. Quantification was performed by assessing the positively stained cortical area. Diagrams show data obtained by computer-based morphometric analysis on days 5 and 14 after induction of UUO in obstructed and contralateral kidneys. Values indicate the relative area of tissue that stained positive. Data represent mean±SD (n=12 in each group). Scale bar, 50 μm. (C) Representative pictures of automated multidimensional fluorescence microscopy (MELC) imaging of diseased kidneys of chimeric mice. Immune cells were stained using CD45 (red), CD3 (green), CD4 (dark blue), and F4/80 (cyan). Mean fluorescence intensities were calculated as described in the Methods section. n=3. Scale bar, 50 μm. (D) Ligated or contralateral kidneys of chimeric mice were homogenized, and levels of CCL2 and CCL5 were determined after ureteral ligation using flow cytometry–based bead assays (n=6 animals per group). *P<0.05; **P<0.005; ***P<0.001.
Next, monocytes/macrophages were immunostained to assess their spatial distribution. After UUO, a strong, predominantly tubulointerstitial cell infiltrate is seen in WT (Notch3wt>wt) and WT BM chimeras (Notch3wt>ko) (Figure 3B), which is blunted by 70% in Notch3ko>ko animals and reduced by 50% in mice receiving Notch3-deficient BM (Notch3ko>wt). Notably, in Notch3wt>ko animals, a similar number of inflammatory cells as in WT animals are recruited, suggesting that receptor Notch3 expression by resident cells is not a prerequisite for cell recruitment.
The composition and spatial organization of cells within the inflamed kidney was visualized by MELC (Figure 3C). After UUO, Notch3wt>wt and Notch3wt>ko mice exhibit strong infiltrates of CD45+ leukocytes, which are primarily F4/80+ macrophages, with a smaller fraction of CD3+ T cells. In contrast, Notch3ko>ko and Notch3ko>wt animal strains have markedly less infiltrating immune cells (approximately 50%). From these results, we conclude that Notch3 expression on circulating immune cells is critical for successful cell transmigration into the damaged kidney tissue.
Given the blunted immune cell infiltration, it is hypothesized that the crosstalk between the damaged kidney cells and circulating immune cells is disturbed. Among the numerous chemokines quantified (Methods), CCL2 and CCL5 are regulated in a Notch3-dependent manner (Figure 3D), suggesting an inability of injured tissue to perform chemokine synthesis.
The absence of infiltrating immune cells in Notch3ko>wt and Notch3ko>ko animal strains is possibly linked with insufficient mobilization of cells from the BM or dysfunctional cell transmigration into tissue. Because the absolute number of circulating leukocytes does not differ in the chimeric animal strains (Supplemental Figure 3D), it is hypothesized that Notch3-deficient immune cells are unable to extravasate from the blood circulation. To test this hypothesis, we performed an adoptive transfer of WT and Notch3-deficient cells using a well-defined infection model.23 CSFE-labeled BM cells (WT, CD45.1; Notch3 knockout, CD45.2) were transferred in equal numbers into L. major–infected WT animals. Labeled WT immune cells are efficiently recruited to the site of infection, whereas the number of Notch3-deficient cells is markedly reduced (20%–40% reduction on days 1 and 2, and 80% on day 5 after adoptive transfer) (Figure 4, A and B). Parallel analysis of the BM on days 1, 2, and 5 confirm that the Notch3-deficient cells are able to home to the BM after adoptive transfer. For monocytes and neutrophils, a Notch3-dependent infiltration into the ear is observed (Figure 4, B and C). In the BM, the reverse effect is seen, because more Notch3 knockout cells are counted (Figure 4, D–F). The results from the ear infection model, together with UUO, suggest that recruitment of immune cells into diseased organs is dependent on Notch3 receptor expression by immune cells. These results suggest that a retention of immune cells within the BM occurs and, at the same time, the adhesion and/or transmigration of circulating cells into the tissue is blunted.
Figure 4.
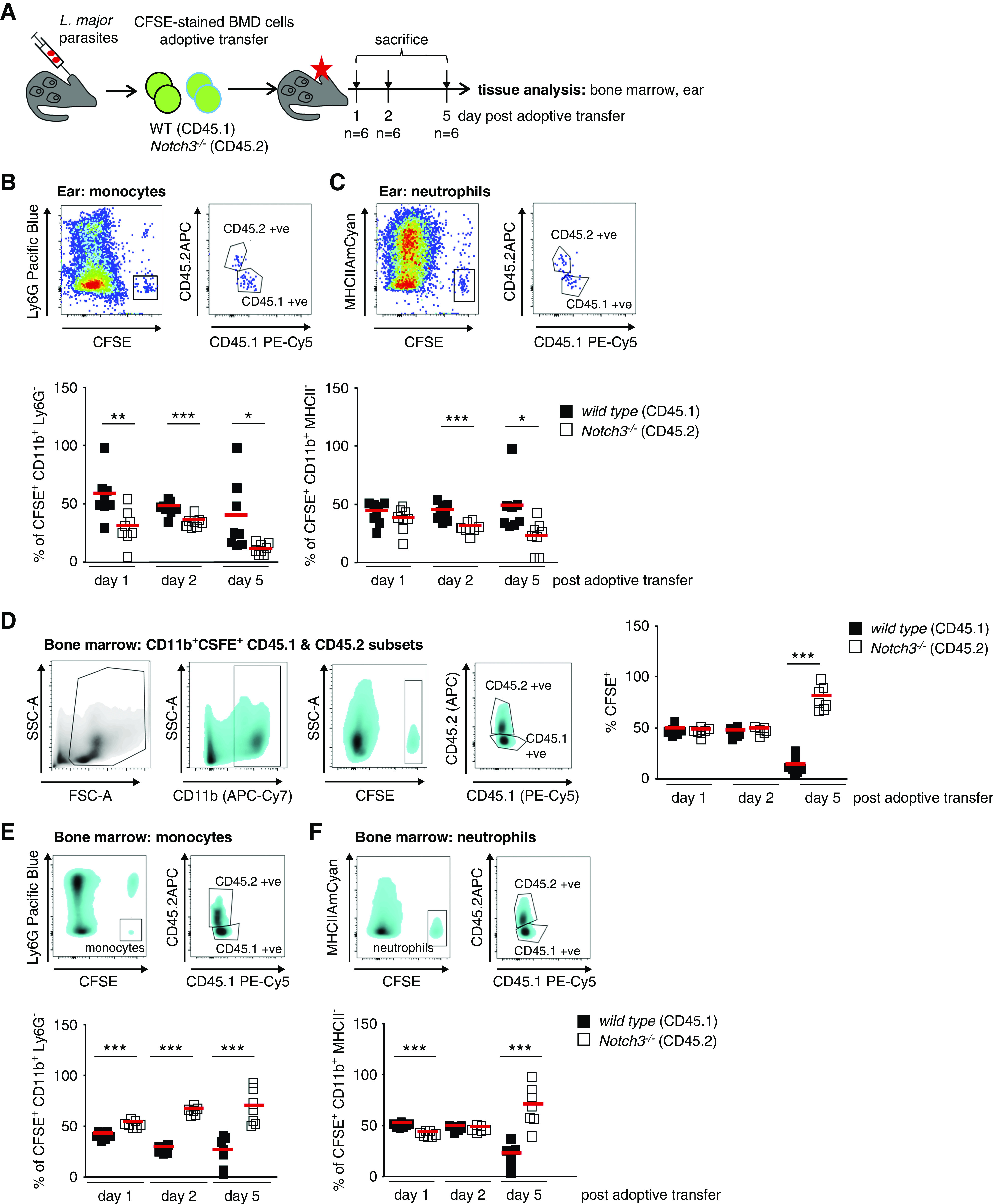
Notch3 receptor expression on immune cells is required for effective recruitment to site of Leishmania infection. (A) Experimental setup of adoptive BM cell transfer. CFSE-stained BM of WT (CD45.1) and Notch3-knockout (CD45.2) mice were adoptively transferred to recipients after 3 weeks of infection with dsRed-expressing L. major parasites in the ear dermis. Infected ear tissues were analyzed on days 1, 2, and 5 after application by flow cytometry. For further tracking of the immune cells, the immune cell composition of the BM was analyzed as well. (B and C) Representative flow cytometry plots of infected ear tissues to identify CSFE-stained WT and Notch3-knockout monocytes and neutrophils. Diagrams show the cell population frequencies quantified by flow cytometry. Mean values are indicated by red line. (D) Flow cytometry analysis of the immune cell composition within the BM revealed an equal contribution of WT and Notch3-knockout adoptively transferred cells at days 1 and 2 after transfer. At day 5, more Notch3-knockout cells were harvested from the BM, whereas there were fewer WT cells. (E and F) Representative flow cytometry plots of the BM to identify CSFE-stained WT and Notch3-knockout monocytes and neutrophils. Diagrams showing the cell populations frequencies measured by flow cytometry. Mean values are indicated by the line. FSC-A, forward scatter area; SSC-A, side scatter area. *P<0.05; **P<0.005; ***P<0.001.
Notch3 Deficiency Impairs Integrin Signaling
Adhesion assays demonstrate an inability of Notch3-deficient immune cells to adhere to HUVECs (Figure 5, A and B). To analyze the Notch3-dependent adhesion in more detail, polarized BMDMs were seeded onto cell culture plates. Both WT and Notch3-knockout cells display a comparable level of basal adhesion to plastic. Because adhesion is mediated by integrin activation, we incubated cells with manganese ions (Mn2+) to induce an open “active” integrin conformation.31 After Mn2+ exposure, WT cells exhibit enhanced adhesion, whereas Notch3-knockout cells do not (Figure 5, C–H). The adhesion of IL-6–polarized, Notch3-knockout macrophages is similarly not enhanced after addition of Mn2+, linking absence of the Notch3 receptor with a defect in IL-6 signaling. The adhesion capacities of IL-4/IL-13–polarized WT and Notch3-knockout macrophages after addition of Mn2+ do not differ. Because we do not see differences in the expression levels of β1- and β2-integrins and IL-6 receptor in resting macrophages (Supplemental Figure 4), we hypothesize that receptor Notch3 is involved in IL-6–dependent integrin activation. This is supported by findings with an antibody (9EG7) specific for the active conformation of β1-integrin. BMDMs were stimulated with IL-6 and activation of β1-integrin was monitored. WT cells have a more activated β1-integrin in response to IL-6 or Mn2+ compared with Notch3-knockout macrophages. IL-6 blocking antibody prevents activation of β1-integrin, which serves as a control (Figure 5H).
Figure 5.
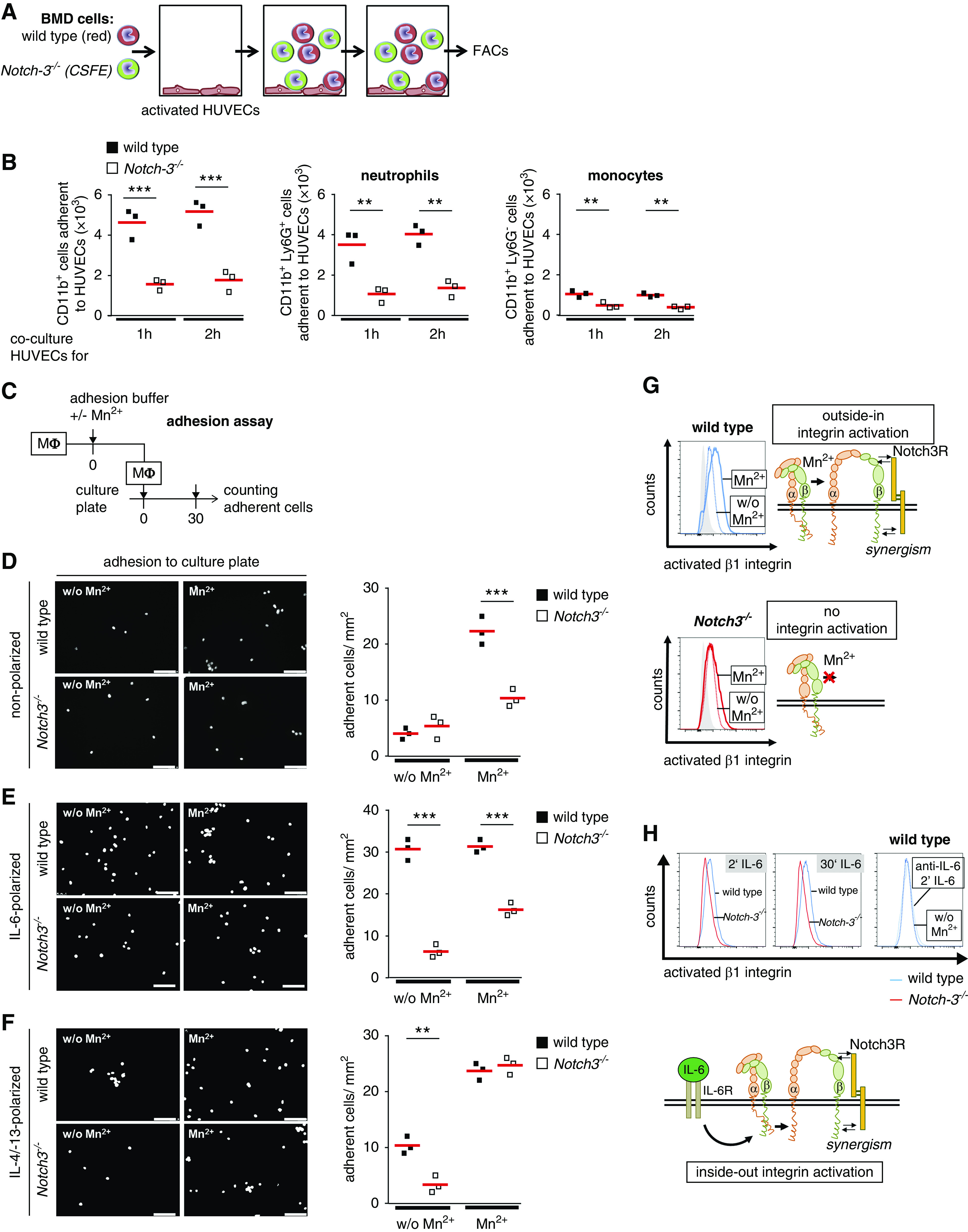
β1-Integrin activation and adhesion of BM-derived (BMD) cells is reduced in the absence of Notch3 expression. (A) Experimental setup of adhesion assay. A mixture of CellTracker Deep Red–stained WT cells and CFSE-stained Notch3-knockout cells were cocultured with PMA-activated HUVECs for 1 or 2 hours. The type and number of bound cells were analyzed by FACS. (B) Diagrams show the numbers of adherent CD11b+ cells as well as neutrophils and monocytes. (C) Experimental setup of adhesion assay. Primary macrophages (Mϕ) were incubated in adhesion buffer. Integrins were activated using Mn2+ ions. Macrophages were added to the culture plate and incubated for 30 minutes. Adherent cells were analyzed after DAPI staining. Representative images and quantitative analysis of (D) adherent nonpolarized cells, (E) IL-6–polarized cells, and (F) IL-4/IL-13–polarized cells. (G) Outside-in activation of β1-integrin was analyzed in the presence (filled line) or absence (dotted line) of Mn2+ in nonpolarized WT (shown in blue) and Notch3-knockout (shown in red) macrophages by FACS using 9EG7 antibody. The gray-filled histograms represent fluorescence in the absence of the primary antibody. (H) For inside-out activation of β1-integrin, nonpolarized macrophages were stimulated with 15 ng/ml IL-6 for the indicated time points and stained for activated β1-integrin using 9EG7 antibody. Specificity was tested using IL-6 blocking antibody (dotted blue line). Experiments were repeated three times each. Notch3R, Notch3 receptor; w/o, without. Scale bar, 100 µm. *P<0.05; **P<0.005; ***P<0.001.
Phenotypic analyses show that both WT and Notch3-deficient cells display no differences in polarization efficiency and proliferation (Supplemental Figure 4, E–H).
Notch3 Deficiency in Kidney Tissue Protects from UUO-Induced Fibrosis
Extensive dilation of collecting ducts, proximal tubules, and distal tubules was present, and tubular cell damage and apoptosis ensue in Notch3wt>wt and Notch3ko>wt animals; however, to a lesser extent in Notch3ko>ko and Notch3wt>ko animals (Figure 6A). The loss of tubular cells is diminished when Notch3 expression is ablated in kidney-resident cells (Figure 6B). Higher numbers of neutrophil gelatinase–associated lipocalin transcript are detected with tubular cell expression of Notch3, supporting a Notch3-dependent damage pattern (Figure 6C). Tubulointerstitial fibrosis with accumulation of collagens 1 and 3 was visualized by Sirius Red staining (Figure 6D, Supplemental Figure 5). A 50% decrease in fibrosis was observed in Notch3ko>ko and Notch3wt>ko mice. MELC analyses revealed that tubulointerstitial cells express more smooth muscle actin (SMA) in animals with resident cells expressing Notch3 (Notch3wt>wt and Notch3ko>wt animals) (Figure 6E). Transcript numbers of fibrosis-related genes, such as collagens 1 and 3, fibronectin-1, TNC, and TGF-β in Notch3wt>ko mice at days 14 after UUO compared with Notch3ko>wt animals are lower (Figure 7A). Furthermore, organ fibrosis in Notch3wt>ko mice is similar to Notch3ko>ko animals, suggesting that tubular Notch3 expression is a driving factor that mitigates the profibrogenic response. This is further supported by the higher transcript numbers of fibrosis-related genes in Notch3ko>wt animals compared with Notch3wt>ko mice (Figure 7A).
Figure 6.
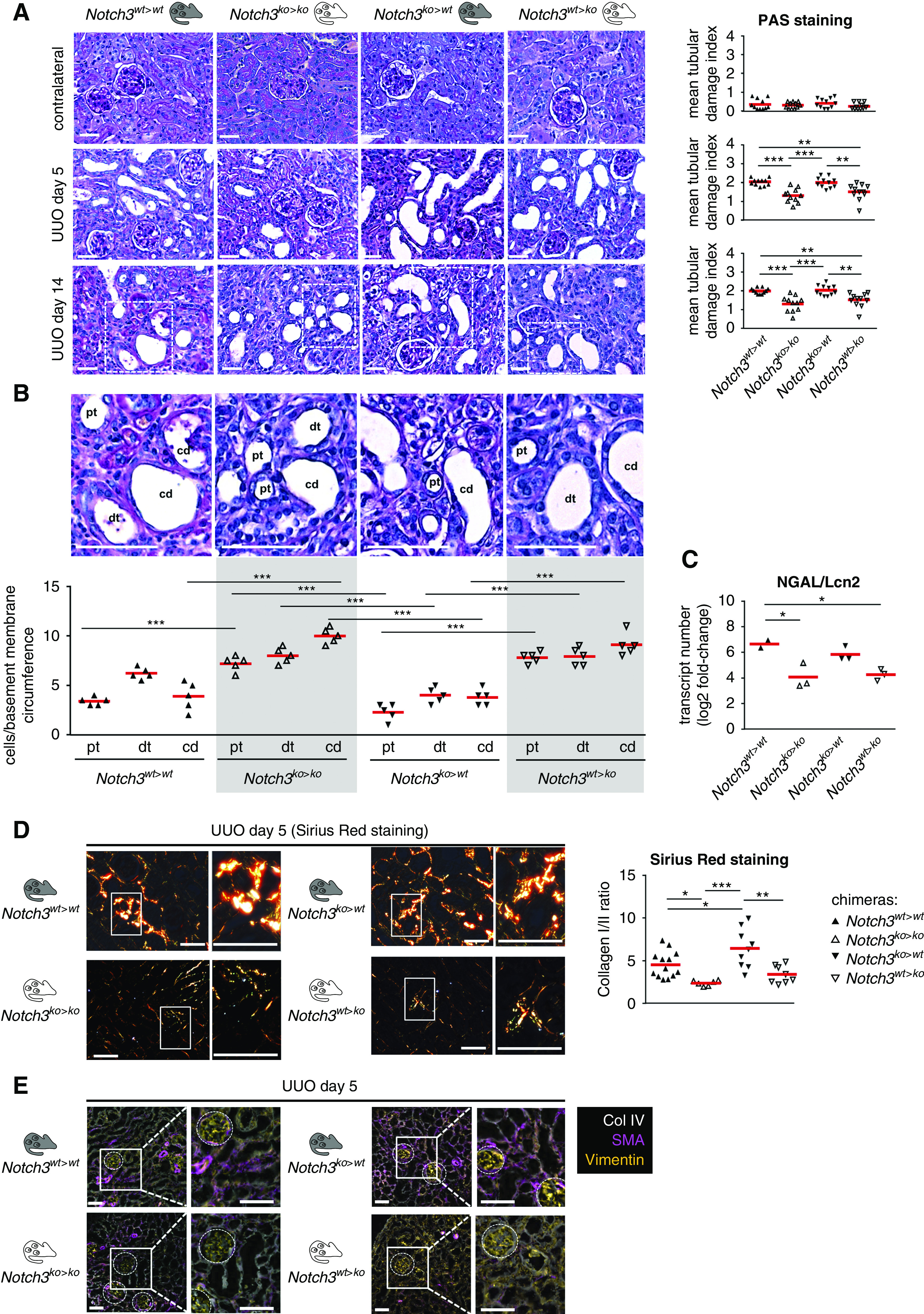
Mice with genetic ablation of Notch3 in tissue-resident cells are protected from tubular epithelial cell damage. (A) In healthy kidneys from the four chimeric animal models, Periodic acid–Schiff (PAS) staining of cortical kidney tissue specimens revealed no apparent dysontogenesis or renal phenotype. Staining of obstructed kidneys at days 5 and 14 of the respective chimeric animals was performed and representative images were taken. The PAS staining showed an increase in tubular damage in WT chimeras and knockout transplants, and less tubular damage in knockout chimeras and WT transplants. Quantification of the results obtained with the tubular histology score, which assesses loss of brush border, tubular dilation, and apoptosis/necrosis of tubular cells. n=12. Scale bar, 50 μm. (B) Analysis of the tubular epithelial attachment to the basement membrane of collecting ducts (cd), proximal tubules (pt), and distal tubules (dt). Tubular cell damage and apoptosis ensues, the loss of tubular cells over the whole nephron was diminished when Notch3 expression is ablated within resident tissue cells. (C) Quantitative mRNA analysis of the tubular injury marker neutrophil gelatinase–associated lipocalin (NGAL/Lcn2) in kidney tissue of WT and Notch3-knockout transplanted mice (Notch3ko>wt) revealed that tubular structure was protected in chimeric Notch3wt>ko mice. (D) Sirius Red staining for collagen deposits showed an increase of collagen deposition in Notch3wt>wt and Notch3ko>wt mice. Representative Sirius Red stainings of obstructed kidneys obtained on day 5 of obstruction are shown. The polarization filter allows us to distinguish collagens 1 (red color) and 3 (green color). Quantification of collagens 1 and 3 and ratio calculation was performed using ImageProPlus software. Scale bar, 50 μm. (E) Representative pictures of automated multidimensional fluorescence microscopy (MELC) imaging of diseased kidneys of chimeric mice. Fibrosis-related proteins were stained using SMA (magenta), collagen 4 (Col IV; white), and vimentin (orange). n=3. Scale bar, 50 μm. *P<0.05; **P<0.005; ***P<0.001.
Figure 7.
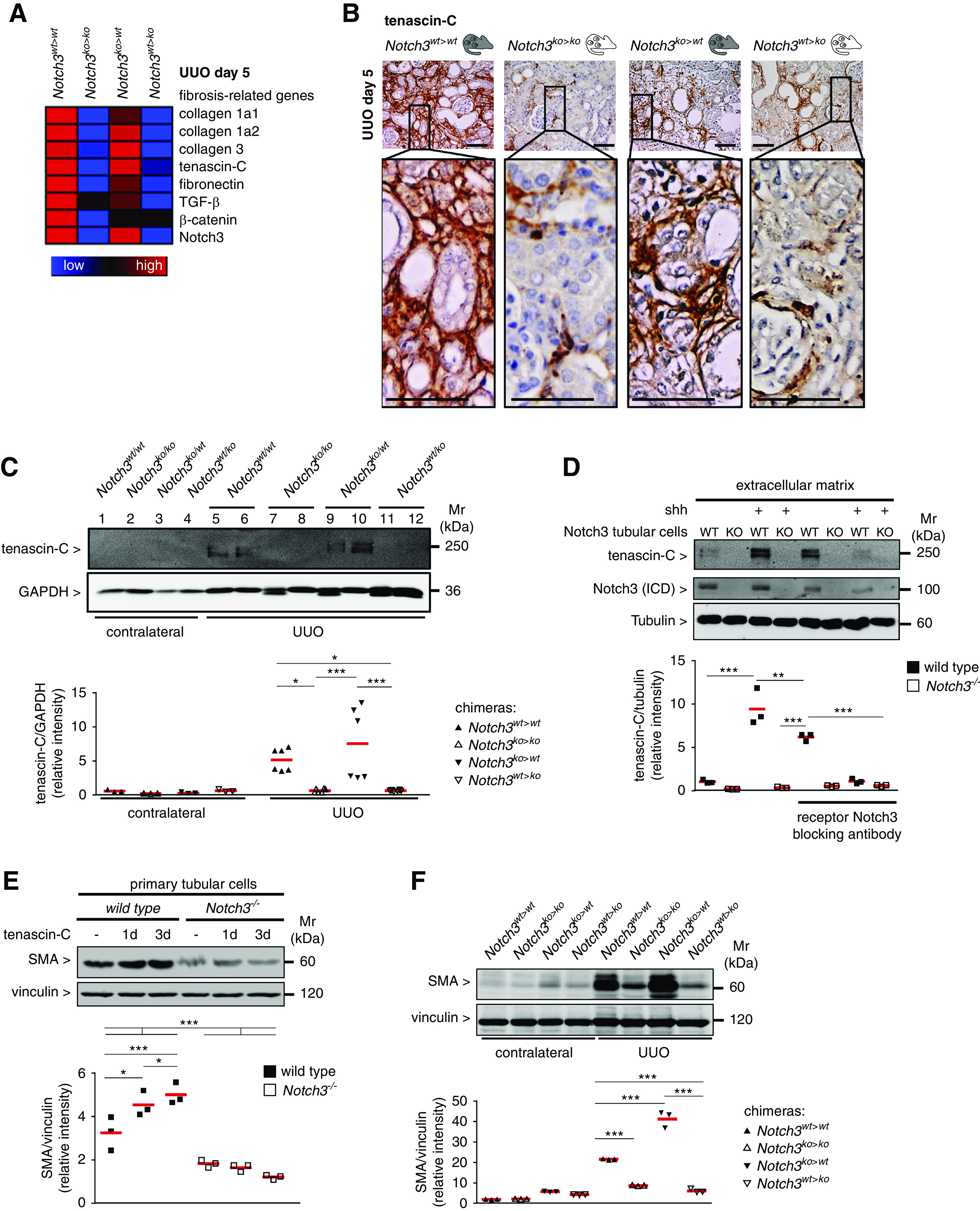
TNC expression in fibrotic kidneys is dependent on Notch3 expression on tissue-resident cells. (A) Heat-map analysis of Notch3 receptor transcripts and selected fibrosis-relevant genes after UUO in Notch3wt>wt, Notch3ko>ko, Notch3ko>wt, and Notch3wt>ko animals (red, upregulated; blue, downregulated). (B) Representative micrographs showing the expression and localization of TNC in kidney tissue after UUO. Scale bar, 50 µm. (C) Western blot analyses of renal expression of TNC protein in contralateral and obstructed kidneys after UUO. (D) Primary tubular cells (Notch3 WT and knockout [KO]) were cultured in a 6-cm dishes and incubated with or without Shh to induce TNC expression. Inhibition of Notch3 signaling was achieved by means of a blocking antibody before stimulation with Shh. After 3 days, cultures were decellularized by addition of EGTA-containing solution, and the remaining ECM scaffold was included in the Western blot analysis for TNC quantification. Cell lysates were tested for Notch3 receptor expression. (E) Western blot analysis of whole-cell lysates from TNC-stimulated primary tubular cells (Notch3 WT and knockout) revealed a time-dependent, TNC-induced expression of SMA in WT cells, which is abolished in Notch3-knockout cells. (F) Western blot analyses demonstrating renal expression of SMA protein in contralateral and obstructed kidneys after UUO. Experiments were each repeated three times. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. ICD, intracellular domain. *P<0.05; **P<0.005; ***P<0.001.
TNC is a component of the fibrogenic niche.32 TNC expression in Notch3ko>wt animals is comparable to Notch3wt>wt animals, whereas for Notch3wt>ko chimera, TNC expression is similar to Notch3ko>ko knockouts (Figure 7, B and C). To test the hypothesis that Notch3 regulates TNC expression, primary tubular cells were incubated with an activator of the Wnt signal pathway, Shh, for 3 days, subjected to decellularization, and then analyzed for the presence of TNC within the ECM. Shh induces TNC expression.32 Indeed, less TNC was detected in Notch3 knockout compared with WT tubular cells. The application of a Notch3 blocking antibody to the medium in WT cells decreased TNC levels similarly to Notch3 knockouts, confirming a Notch3 dependency of TNC expression (Figure 7D). Extracellular TNC stimulates SMA expression in WT tubular cells; however, there was lower expression in Notch3-knockout cells, indicating a TNC-Notch3-SMA axis. Similar results were detected with kidney tissue lysates from chimeric animals. When Notch3 expression was deleted in resident kidney cells, there was also less SMA expression after UUO (Figure 7, E and F). TNC and SMA are both known target genes of NF-κB. From the literature, a close link between Notch3 and NF-κB signaling is described.33 To determine the spatial activation of NF-κB (pp65), MELC analyses were performed in the chimeric animal strains (NF-κB pp65 shown as yellow in Figure 8A). The abundance of nuclear p65 is high in tubular cells from Notch3ko>wt and Notch3wt>wt strains, and low in Notch3wt>ko and Notch3ko>ko animals. Western blotting confirmed these results (Figure 8B). To explore the idea that master regulators of NF-κB signaling are dysregulated in the chosen model, isolated tubular cells were analyzed for the lysine 63 deubiquitinase CYLD (Figure 8C). CYLD is constitutively upregulated in the Notch3-knockout compared with WT cells. Therefore, we propose a model based on this study and published data in which integrin activation and the subsequent activation of NF-κB signaling depends upon Notch3 receptor expression, which is summarized in a scheme shown in Figure 8D.
Figure 8.
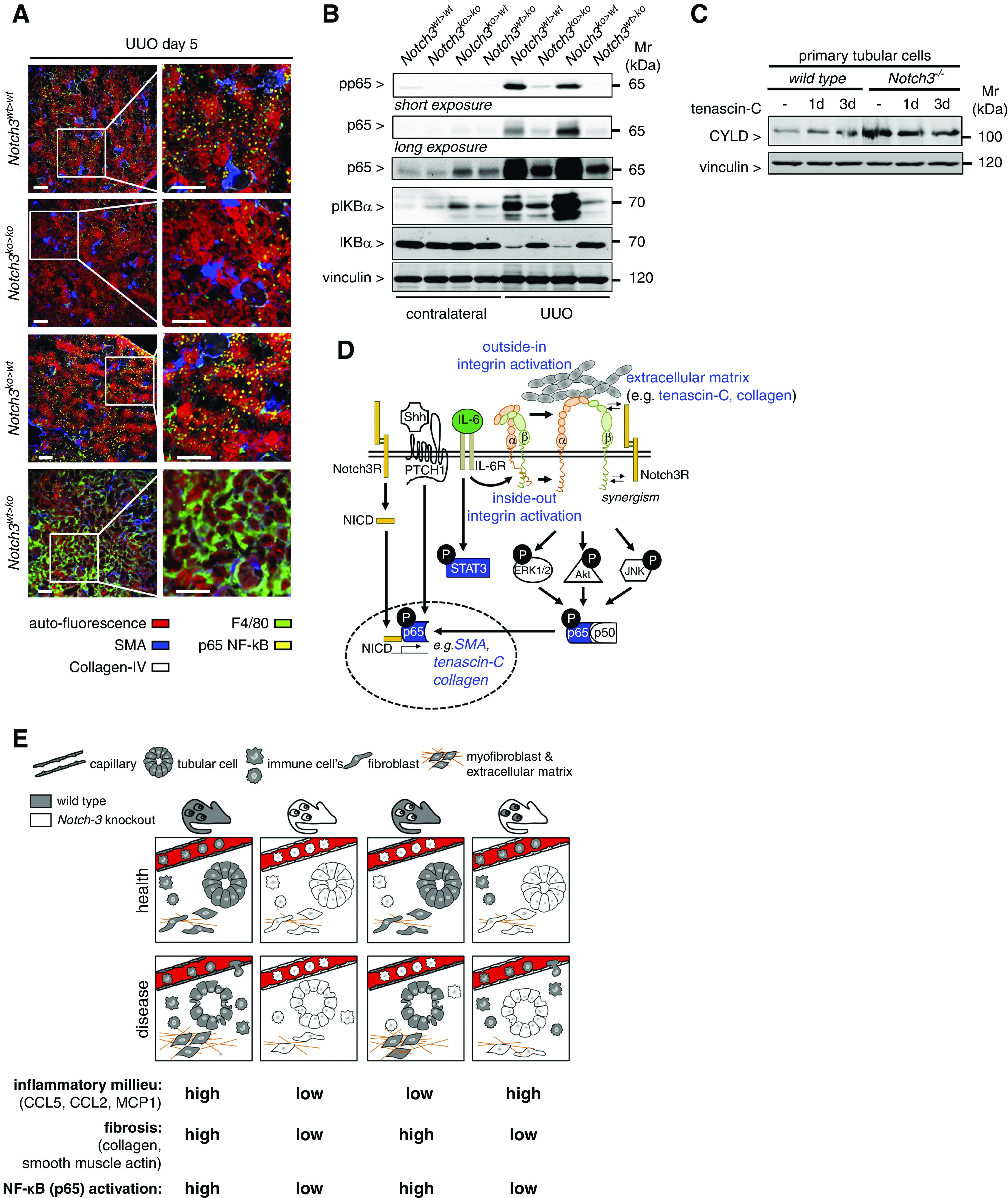
NF-κB (p65) activation in diseased kidneys is dependent on Notch3 expression on tissue-resident cells. (A) Representative images obtained by automated multidimensional fluorescence microscopy (MELC) visualize the spatial distribution of NF-κB–positive cells in diseased kidneys from the four different chimeric animal strains (NF-κB p65 is stained in yellow). F4/80 in green identifies immune cells. Collagen type IV (white) and SMA (blue) allow for discrimination of tissue structures, tubular cells exhibit a red autofluorescence signal. n=3. Scale bars, 50 μm. (B) Western blot analyses were performed with kidney tissue lysates. Antibodies specific for NF-κB (p65), phospho (Ser536)–NF-κB (p65) (with long exposure to visualize the signals in the contralateral kidneys), total IκBα, and phospho (Ser32)–IκBα were applied. (C) Western blot analysis of whole-cell lysates from TNC-stimulated primary tubular cells (WT or Notch3 knockout) revealed an upregulation of the NF-κB inhibitor CYLD in Notch3-knockout cells. Experiments were repeated three times. (D) Schematic illustrating the crosstalk of receptor Notch3 in combination with Shh and integrin signaling leading to the accumulation of ECM. All data obtained within this study are highlighted in blue. Known interactions from the literature are indicated as black lines with white boxes. (E) Cartoon that depicts key findings of the study.
Taken together, the results indicate cell-specific functions of Notch3 receptor. In circulating immune cells, Notch3-dependent integrin activation mediates transmigration and tissue infiltration. In tissue-resident cells, Notch3-dependent signaling enhances matrix synthesis and cell phenotypes that propagate organ fibrosis (Figure 8E).
Discussion
In the pathogenesis of organ fibrosis, tissue injury with subsequent immune cell infiltration, phenotypic alterations toward myofibroblasts, and excess ECM deposition are thought to be intimately linked.34 In the kidney, the origin of myofibroblasts has been deciphered by cell lineage tracking35; however, a causal link between immune cell infiltration and the fibrotic response has yet to be demonstrated. Some reports show that infiltrating hematopoietic cells are a source of collagen type 1 synthesis in the kidney.36 Cell lineage tracing studies confirm that myofibroblasts originate from proliferating tissue-resident fibroblasts and infiltrating BM-derived cells.35,37 Our findings with chimeric animal strains suggest that, in the fibrotic response, Notch3 receptor signaling participates in both immune and resident kidney cells.18,19,38 We hypothesized that Notch3 receptor signaling is required for immune cell recruitment, and that the subsequent organ fibrosis is incited by a prevailing inflammatory tissue milieu. The experimental approach using BM chimeras with animals expressing Notch3 either in tissue or in BM-derived immune cells allowed us to discriminate the relative contribution of Notch3 receptor in each population. Extensive control experiments exclude a participation of Notch3 receptor in BM reconstitution, a prerequisite for meaningful results. The model of UUO is well defined and not immune-mediated, with a clear starting point of tubular damage.39 After UUO, Notch3-deficient tubular cells are protected from immediate damage and remain adherent to the basement membrane, despite a proinflammatory tissue milieu (Supplemental Figure 7). Intriguingly, in the absence of Notch3 receptor in resident kidney cells, organ fibrosis is blunted, although there is marked immune cell infiltrate. Conversely, the Notch3ko>wt chimeras suggest that Notch3 receptor signaling is a prerequisite for most immune cells to transmigrate into the kidney tissue. Thus, dichotomous functions become apparent for Notch3 receptor: one on immune cells, and the other on tissue-resident cells.
In macrophages, Notch signals control cell maturation.40 Macrophages are known to propagate atherogenesis.41 Furthermore, inhibition of Notch3 signaling markedly alleviates microvascular pathologies in diabetes.42 Our results indicate that immune cells lacking Notch3 receptor have an adherence defect, which renders these cells incapable of transmigration into the tissue. Integrin conformational changes are disturbed in the absence of Notch3 signaling. Our findings are supported by studies describing the role of Notch receptors and their ligands in leukocyte adhesion to the endothelium.43–45 We observe Notch3-dependent activation of β1-integrin and monocyte adhesion to endothelial cells, whereas integrin expression per se is not altered with Notch3 deletion. From these results, we conclude that Notch3 receptor is part of the armamentarium of inflammatory cells to perform site-directed transmigration via integrin signaling.
Diminished kidney fibrosis in chimeric animals that have a deletion of Notch3 in resident kidney cells with abundant infiltrates of immune cells is intriguing and may pinpoint to other immune cell functions, such as resolution of the inflammatory response. These may include phagocytosis of cell debris and confinement of inflammation through anti-inflammatory cytokines, with subsequent tissue regeneration.46,47 A verdict on the respective roles that the still-infiltrating cells play is, however, not possible with our results.24,48,49 Furthermore, the absolute number of infiltrating immune cells does not necessarily allow us to form conclusions about their relative effects on tissue fibrosis.36,50
ECM deposition is downregulated in Notch3-knockout mice.18 We identify tissue-resident cells as the drivers of fibrosis. We propose that Notch3 receptor signaling is central in the formation of the fibrotic niche. This depends on a cellular microenvironment composed of a specialized ECM network, secreted factors, and specialized cell types.32,51,52 One critical component of the fibrotic niche is TNC.53 Our findings show that the production of TNC-containing ECM depends on Notch3 expression in resident kidney cells. A crosstalk of Shh/Wnt-induced ECM synthesis and Notch3 signaling exists.32,54 We envision that the activation of Notch3 signaling by ECM resembles that of the membrane-anchored ligands Delta and Jagged, which upregulate TNC expression. Recently, it has been shown that tubular epithelial cell damage triggers the expression and secretion of fibrotic cues (e.g., Shh, Wnt ligands, TGF-β).32 These may stimulate myofibroblasts to produce and secrete TNC and other ECM components, with TNC acting as a ligand for integrins. Our findings corroborate a permissive effect of Notch3 expression on β1-integrin activation. Besides TNC, a strong upregulation of collagens type 1 and 3 and SMA is present when resident kidney cells express Notch3 receptor. Thus, a prominent role is executed by Notch3 signaling in the fibrotic response, similarly to SMA.55
Mechanistically, NF-κB is part of the aforementioned signaling pathways. Recent studies report that the activation of the NF-κB pathway in kidney disease is abrogated in the absence of receptor Notch3.19,56 As one example, Notch3 receptor and p65/NF-κB orchestrate T cell function.57,58 It has been shown that nuclear translocation of p65/NF-κB in tissue-resident cells is reduced when Notch3 receptor is absent.
Taken together, immune cell infiltration and tissue fibrosis are distinct and separable events that are largely regulated via Notch3 receptor: (1) Notch3 expression on immune cells propagates cell adhesion function and transmigration, whereas (2) Notch3 expression on resident kidney cells participates in the induction of organ fibrosis. Our data indicate that, besides immune cells, different kidney-resident cells express Notch3 receptor, e.g., tubular and tububointerstitial cells, pericytes, and mesangial cells. It will be of interest to decipher disease-specific expression patterns and cell-specific roles of receptor signaling in future studies.59 Notably, specific Notch3 blocking antibodies have been developed and entered into clinical trials.60 Given that Notch3-knockout animals are not limited in their life span,21,22 our study results encourage further research with focus on cell-specific Notch3 blocking compounds.
Limitations of this Study
Our findings on Notch3-dependent effects have been obtained in a single experimental model that primarily relies on tubular cell damage with a subsequent inflammatory response. A generalization on other modes of kidney damage is not possible and further results are needed to specify how Notch3 receptor alters immune cell properties in disease.
Disclosures
C. Siebel has a patent “Anti-jagged antibodies and methods of use” (WO 2014/028446) issued, and a patent “Methods of treating cancer using notch antagonists” (US20120328608) issued. All remaining authors have nothing to disclose.
Funding
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under project identifier 97850925 - SFB 854 (to P. Mertens [project A1], B. Isermann [project B26N], F. Heidel [project A20], and A. Müller [Z01, B31N]); project identifier GRK 2408 (to B. Isermann [project 7] and to P. Mertens [project 8]); CRC854 (to L. Philipsen [project Z01]); and grants ME-1365/7-2 and ME-1365/9-2 (to P. Mertens). J. Lindquist was supported by DFG grant LI-1031/4-1. F. Heidel was supported in part by a DFG grant HE-6233/4-1 and by the Thüringian Ministry for Research, Thüringian state program ProExzellenz (RegenerAging, FSU-I-03/14). S. Djudjaj was funded by the Excellence Initiative of the German federal and state governments grant ERS, OPSF470. A. Müller received funding from the European Union’s H2020 European Research Council research and innovation program (StG ImmProDynamics, grant number 714233) and the DFG grant MU3744/4-1. P. Mertens reports receiving federal state Saxony-Anhalt and the European Structural and Investment Funds (ESF, 2014–2020), under project number ZS/2016/08/80645, during the conduct of the study.
Supplementary Material
Acknowledgments
We are grateful to Mrs. Huß, Königsmark, and Schuppe for excellent technical assistance, and to Dr. S. Kliche (Institute of Molecular and Clinical Immunology, Otto-von-Guericke University Magdeburg) for helpful discussion.
Dr. Sabine Brandt, Dr. Tobias Ballhause, Dr. Anja Bernhardt, Dr. Annika Becker, Dr. Delia L. Salaru, Dr. Hien Le-Deffge, Dr. Alexander Fehr, Dr. Yan Fu, Dr. Lars Philipsen, Dr. Sonja Djudjaj, Dr. Andreas Müller, Dr. Rafael Kramann, Dr. Mahmoud Ibrahim, Dr. Robert Geffers, Dr. Berend Isermann, Dr. Florian Heidel, and Dr. Jonathan A. Lindquist performed experiments and analyzed data; Dr. Chris Siebel provided essential reagents; Dr. Sabine Brandt, Dr. Jonathan A. Lindquist, and Dr. Peter R. Mertens wrote the manuscript; and Dr. Peter R. Mertens conceived the study and supervised the work.
Dr. Rafael Kramann reports receiving grants from Chugai Pharma/Roche, outside the submitted work.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2019121289/-/DCSupplemental.
Supplemental Figure 1. Complete blood counts with immune cell composition as well as kidney function in Notch3 knockout and wild type mice.
Supplemental Figure 2. Flow cytometry on bone marrow chimerism from transplanted mice.
Supplemental Figure 3. Confirmation of successful homing, reconstitution, as well as chimerism in bone marrow transplanted mice.
Supplemental Figure 4. Analysis of polarization efficiency, cell proliferation, IL-6 receptor and integrin expression of wild type and Notch3 knockout bone marrow-derived macrophages.
Supplemental Figure 5. Sirius Red staining performed with obstructed kidneys on days 14.
Supplemental Figure 6. Flow cytometric analysis of kidney infiltrating immune cells.
Supplemental Figure 7. Gene array analysis on IL-6 transcript numbers.
Supplemental Table 1. Whole blood counts of wild type, Notch3 knockout and chimeric animals.
References
- 1.Rockey DC, Bell PD, Hill JA: Fibrosis—a common pathway to organ injury and failure. N Engl J Med 372: 1138–1149, 2015 [DOI] [PubMed] [Google Scholar]
- 2.Kramann R, Schneider RK, DiRocco DP, Machado F, Fleig S, Bondzie PA, et al.: Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 16: 51–66, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li L, Kang H, Zhang Q, D’Agati VD, Al-Awqati Q, Lin F: FoxO3 activation in hypoxic tubules prevents chronic kidney disease. J Clin Invest 129: 2374–2389, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petejova N, Martinek A, Zadrazil J, Teplan V: Acute toxic kidney injury. Ren Fail 41: 576–594, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu J, Kumar S, Dolzhenko E, Alvarado GF, Guo J, Lu C, et al.: Molecular characterization of the transition from acute to chronic kidney injury following ischemia/reperfusion. JCI Insight 2: e94716, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Breitkopf DM, Jankowski V, Ohl K, Hermann J, Hermert D, Tenbrock K, et al.: The YB-1:Notch-3 axis modulates immune cell responses and organ damage in systemic lupus erythematosus. Kidney Int 97: 289–303, 2020 [DOI] [PubMed] [Google Scholar]
- 7.Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J, et al.: Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 9: 7204–7218, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Centers for Disease Control and Prevention: Chronic kidney disease in the United States, 2019, Atlanta, GA, US Department of Health and Human Services, Centers for Disease Control and Prevention, 2019 [Google Scholar]
- 9.Liu Y: Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol 7: 684–696, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Djudjaj S, Boor P: Cellular and molecular mechanisms of kidney fibrosis. Mol Aspects Med 65: 16–36, 2019 [DOI] [PubMed] [Google Scholar]
- 11.Kramann R, Fleig SV, Schneider RK, Fabian SL, DiRocco DP, Maarouf O, et al.: Pharmacological GLI2 inhibition prevents myofibroblast cell-cycle progression and reduces kidney fibrosis. J Clin Invest 125: 2935–2951, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bielesz B, Sirin Y, Si H, Niranjan T, Gruenwald A, Ahn S, et al.: Epithelial Notch signaling regulates interstitial fibrosis development in the kidneys of mice and humans. J Clin Invest 120: 4040–4054, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nathan C, Ding A: Nonresolving inflammation. Cell 140: 871–882, 2010 [DOI] [PubMed] [Google Scholar]
- 14.Schroder K, Tschopp J: The inflammasomes. Cell 140: 821–832, 2010 [DOI] [PubMed] [Google Scholar]
- 15.Lech M, Anders HJ: Macrophages and fibrosis: How resident and infiltrating mononuclear phagocytes orchestrate all phases of tissue injury and repair. Biochim Biophys Acta 1832: 989–997, 2013 [DOI] [PubMed] [Google Scholar]
- 16.Sirin Y, Susztak K: Notch in the kidney: Development and disease. J Pathol 226: 394–403, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sweetwyne MT, Tao J, Susztak K: Kick it up a notch: Notch signaling and kidney fibrosis. Kidney Int Suppl (2011) 4: 91–96, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Djudjaj S, Chatziantoniou C, Raffetseder U, Guerrot D, Dussaule JC, Boor P, et al.: Notch-3 receptor activation drives inflammation and fibrosis following tubulointerstitial kidney injury. J Pathol 228: 286–299, 2012 [DOI] [PubMed] [Google Scholar]
- 19.El Machhour F, Keuylian Z, Kavvadas P, Dussaule JC, Chatziantoniou C: Activation of Notch3 in glomeruli promotes the development of rapidly progressive renal disease. J Am Soc Nephrol 26: 1561–1575, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kavvadas P, Keuylian Z, Prakoura N, Placier S, Dorison A, Chadjichristos CE, et al.: Notch3 orchestrates epithelial and inflammatory responses to promote acute kidney injury. Kidney Int 94: 126–138, 2018 [DOI] [PubMed] [Google Scholar]
- 21.Boulos N, Helle F, Dussaule JC, Placier S, Milliez P, Djudjaj S, et al.: Notch3 is essential for regulation of the renal vascular tone. Hypertension 57: 1176–1182, 2011 [DOI] [PubMed] [Google Scholar]
- 22.Krebs LT, Xue Y, Norton CR, Sundberg JP, Beatus P, Lendahl U, et al.: Characterization of Notch3-deficient mice: Normal embryonic development and absence of genetic interactions with a Notch1 mutation. Genesis 37: 139–143, 2003 [DOI] [PubMed] [Google Scholar]
- 23.Olekhnovitch R, Ryffel B, Müller AJ, Bousso P: Collective nitric oxide production provides tissue-wide immunity during Leishmania infection. J Clin Invest 124: 1711–1722, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bernhardt A, Fehr A, Brandt S, Jerchel S, Ballhause TM, Philipsen L, et al.: Inflammatory cell infiltration and resolution of kidney inflammation is orchestrated by the cold-shock protein Y-box binding protein-1. Kidney Int 92: 1157–1177, 2017 [DOI] [PubMed] [Google Scholar]
- 25.Clahsen T, Schaper F: Interleukin-6 acts in the fashion of a classical chemokine on monocytic cells by inducing integrin activation, cell adhesion, actin polymerization, chemotaxis, and transmigration. J Leukoc Biol 84: 1521–1529, 2008 [DOI] [PubMed] [Google Scholar]
- 26.Schubert W, Bonnekoh B, Pommer AJ, Philipsen L, Böckelmann R, Malykh Y, et al.: Analyzing proteome topology and function by automated multidimensional fluorescence microscopy. Nat Biotechnol 24: 1270–1278, 2006 [DOI] [PubMed] [Google Scholar]
- 27.Edelmann B, Gupta N, Schnoeder TM, Oelschlegel AM, Shahzad K, Goldschmidt J, et al.: JAK2-V617F promotes venous thrombosis through β1/β2 integrin activation. J Clin Invest 128: 4359–4371, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lake BB, Chen S, Hoshi M, Plongthongkum N, Salamon D, Knoten A, et al.: A single-nucleus RNA-sequencing pipeline to decipher the molecular anatomy and pathophysiology of human kidneys. Nat Commun 10: 2832, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, et al. : Comprehensive integration of single-cell data. Cell 177: 1888–1902.e21, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arvaniti E, Moulos P, Vakrakou A, Chatziantoniou C, Chadjichristos C, Kavvadas P, et al.: Whole-transcriptome analysis of UUO mouse model of renal fibrosis reveals new molecular players in kidney diseases. Sci Rep 6: 26235, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mould AP, Akiyama SK, Humphries MJ: Regulation of integrin alpha 5 beta 1-fibronectin interactions by divalent cations. Evidence for distinct classes of binding sites for Mn2+, Mg2+, and Ca2+. J Biol Chem 270: 26270–26277, 1995 [DOI] [PubMed] [Google Scholar]
- 32.Fu H, Tian Y, Zhou L, Zhou D, Tan RJ, Stolz DB, et al.: Tenascin-C is a major component of the fibrogenic niche in kidney fibrosis. J Am Soc Nephrol 28: 785–801, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song N, Thaiss F, Guo L: NFκB and kidney injury. Front Immunol 10: 815, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Humphreys BD: Mechanisms of renal fibrosis. Annu Rev Physiol 80: 309–326, 2018 [DOI] [PubMed] [Google Scholar]
- 35.LeBleu VS, Taduri G, O’Connell J, Teng Y, Cooke VG, Woda C, et al.: Origin and function of myofibroblasts in kidney fibrosis. Nat Med 19: 1047–1053, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buchtler S, Grill A, Hofmarksrichter S, Stöckert P, Schiechl-Brachner G, Rodriguez Gomez M, et al.: Cellular origin and functional relevance of collagen I production in the kidney. J Am Soc Nephrol 29: 1859–1873, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kramann R, DiRocco DP, Humphreys BD: Understanding the origin, activation and regulation of matrix-producing myofibroblasts for treatment of fibrotic disease. J Pathol 231: 273–289, 2013 [DOI] [PubMed] [Google Scholar]
- 38.Huang S, Park J, Qiu C, Chung KW, Li SY, Sirin Y, et al.: Jagged1/Notch2 controls kidney fibrosis via Tfam-mediated metabolic reprogramming. PLoS Biol 16: e2005233, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martínez-Klimova E, Aparicio-Trejo OE, Tapia E, Pedraza-Chaverri J: Unilateral ureteral obstruction as a model to investigate fibrosis-attenuating treatments. Biomolecules 9: 141, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krishnasamy K, Limbourg A, Kapanadze T, Gamrekelashvili J, Beger C, Häger C, et al.: Blood vessel control of macrophage maturation promotes arteriogenesis in ischemia. Nat Commun 8: 952, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ewert L, Fischer A, Brandt S, Scurt FG, Philipsen L, Müller AJ, et al.: Cold shock Y-box binding protein-1 acetylation status in monocytes is associated with systemic inflammation and vascular damage. Atherosclerosis 278: 156–165, 2018 [DOI] [PubMed] [Google Scholar]
- 42.Wimmer RA, Leopoldi A, Aichinger M, Wick N, Hantusch B, Novatchkova M, et al.: Human blood vessel organoids as a model of diabetic vasculopathy. Nature 565: 505–510, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Colombo M, Mirandola L, Platonova N, Apicella L, Basile A, Figueroa AJ, et al.: Notch-directed microenvironment reprogramming in myeloma: A single path to multiple outcomes. Leukemia 27: 1009–1018, 2013 [DOI] [PubMed] [Google Scholar]
- 44.Briot A, Bouloumié A, Iruela-Arispe ML: Notch, lipids, and endothelial cells. Curr Opin Lipidol 27: 513–520, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murata A, Okuyama K, Sakano S, Kajiki M, Hirata T, Yagita H, et al.: A Notch ligand, Delta-like 1 functions as an adhesion molecule for mast cells. J Immunol 185: 3905–3912, 2010 [DOI] [PubMed] [Google Scholar]
- 46.Sheikh Z, Brooks PJ, Barzilay O, Fine N, Glogauer M: Macrophages, foreign body giant cells and their response to implantable biomaterials. Materials (Basel) 8: 5671–5701, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Borthwick LA, Wynn TA, Fisher AJ: Cytokine mediated tissue fibrosis. Biochim Biophys Acta 1832: 1049–1060, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Castaño AP, Lin SL, Surowy T, Nowlin BT, Turlapati SA, Patel T, et al.: Serum amyloid P inhibits fibrosis through Fc gamma R-dependent monocyte-macrophage regulation in vivo. Sci Transl Med 1: 5ra13, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huen SC, Cantley LG: Macrophage-mediated injury and repair after ischemic kidney injury. Pediatr Nephrol 30: 199–209, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lever JM, Hull TD, Boddu R, Pepin ME, Black LM, Adedoyin OO, et al.: Resident macrophages reprogram toward a developmental state after acute kidney injury. JCI Insight 4: e125503, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.von Holst A: Tenascin C in stem cell niches: Redundant, permissive or instructive? Cells Tissues Organs 188: 170–177, 2008 [DOI] [PubMed] [Google Scholar]
- 52.Wong GS, Rustgi AK: Matricellular proteins: Priming the tumour microenvironment for cancer development and metastasis. Br J Cancer 108: 755–761, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Midwood KS, Chiquet M, Tucker RP, Orend G: Tenascin-C at a glance. J Cell Sci 129: 4321–4327, 2016 [DOI] [PubMed] [Google Scholar]
- 54.Sivasankaran B, Degen M, Ghaffari A, Hegi ME, Hamou MF, Ionescu MC, et al.: Tenascin-C is a novel RBPJkappa-induced target gene for Notch signaling in gliomas. Cancer Res 69: 458–465, 2009 [DOI] [PubMed] [Google Scholar]
- 55.Noseda M, Fu Y, Niessen K, Wong F, Chang L, McLean G, et al.: Smooth muscle alpha-actin is a direct target of Notch/CSL. Circ Res 98: 1468–1470, 2006 [DOI] [PubMed] [Google Scholar]
- 56.Sakai N, Wada T, Furuichi K, Iwata Y, Yoshimoto K, Kitagawa K, et al.: p38 MAPK phosphorylation and NF-kappa B activation in human crescentic glomerulonephritis. Nephrol Dial Transplant 17: 998–1004, 2002 [DOI] [PubMed] [Google Scholar]
- 57.Barbarulo A, Grazioli P, Campese AF, Bellavia D, Di Mario G, Pelullo M, et al.: Notch3 and canonical NF-kappaB signaling pathways cooperatively regulate Foxp3 transcription. J Immunol 186: 6199–6206, 2011 [DOI] [PubMed] [Google Scholar]
- 58.Kumar V, Palermo R, Talora C, Campese AF, Checquolo S, Bellavia D, et al.: Notch and NF-kB signaling pathways regulate miR-223/FBXW7 axis in T-cell acute lymphoblastic leukemia. Leukemia 28: 2324–2335, 2014 [DOI] [PubMed] [Google Scholar]
- 59.Park J, Shrestha R, Qiu C, Kondo A, Huang S, Werth M, et al.: Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 360: 758–763, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Olsauskas-Kuprys R, Zlobin A, Osipo C: Gamma secretase inhibitors of Notch signaling. Onco Targets Ther 6: 943–955, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.