

Significance Statement
Most studies investigating the mechanisms of antibody-mediated rejection, a major cause of kidney allograft failure, have focused on characterizing the role of donor-specific antibodies (DSAs), whereas the alloreactive cellular component has been less studied. On the basis of a multidimensional and concomitant profiling of circulating T follicular helper (TFH) cells and B cells, the authors identified highly coordinated responses of circulating TFH cells and activated B cells at phenotypic, functional, and transcriptional levels in patients with antibody-mediated rejection. The levels of circulating TFH cell and B cell activation were predictive of DSA pathogenicity, histologic severity, and allograft loss. This study provides novel mechanistic insights into the cellular and molecular processes underlying antibody-mediated rejection and a rationale for monitoring and therapeutic targeting of circulating TFH cell–B cell interaction during antibody-mediated rejection.
Keywords: acute allograft rejection, lymphocytes, immunology, kidney transplantation, transcriptional profiling, transplant outcomes
Visual Abstract

Abstract
Background
Although antibody-mediated rejection (ABMR) has been long recognized as a leading cause of allograft failure after kidney transplantation, the cellular and molecular processes underlying the induction of deleterious donor-specific antibody (DSA) responses remain poorly understood.
Methods
Using high-dimensional flow cytometry, in vitro assays, and RNA sequencing, we concomitantly investigated the role of T follicular helper (TFH) cells and B cells during ABMR in 105 kidney transplant recipients.
Results
There were 54 patients without DSAs; of those with DSAs, ABMR emerged in 20 patients, but not in 31 patients. We identified proliferating populations of circulating TFH cells and activated B cells emerging in blood of patients undergoing ABMR. Although these circulating TFH cells comprised heterogeneous phenotypes, they were dominated by activated (ICOS+PD-1+) and early memory precursor (CCR7+CD127+) subsets, and were enriched for the transcription factors IRF4 and c-Maf. These circulating TFH cells produced large amounts of IL-21 upon stimulation with donor antigen and induced B cells to differentiate into antibody-secreting cells that produced DSAs. Combined analysis of the matched circulating TFH cell and activated B cell RNA-sequencing profiles identified highly coordinated transcriptional programs in circulating TFH cells and B cells among patients with ABMR, which markedly differed from those of patients who did not develop DSAs or ABMR. The timing of expansion of the distinctive circulating TFH cells and activated B cells paralleled emergence of DSAs in blood, and their magnitude was predictive of IgG3 DSA generation, more severe allograft injury, and higher rate of allograft loss.
Conclusions
Patients undergoing ABMR may benefit from monitoring and therapeutic targeting of TFH cell–B cell interactions.
A significant advancement in the field of kidney transplantation has been the recognition that the alloimmune response mediated by anti-HLA donor-specific antibodies (DSAs) are deleterious.1,2 There is a broad spectrum of allograft injury related to these DSAs. Antibody-mediated rejection (ABMR) is the most severe manifestation of DSA pathogenicity and involves C1q-binding IgG1 and IgG3 DSAs, which convey microvascular inflammation and complement activation in allograft capillaries. In contrast, IgG4 DSAs are associated with delayed and chronic damage.3 Current therapeutic strategies for preventing or reversing ABMR that are designed to deplete B cells and DSAs have had limited success. Thus, there remains a substantial unmet need for new therapeutic solutions to efficiently combat ABMR.4
Given that DSAs are directed against protein antigens, it is postulated that they are generated through T-dependent B cell responses. T follicular helper cells (TFH) are CD4+ T cells specializing in the control of cognate antigen-specific B cell responses.5 They express CXCR5, which permits trafficking to B cell follicles in response to CXCL13. TFH cells promote germinal center (GC) formation by providing critical help to B cells, enabling their proliferation and differentiation into memory B cells and plasma cells that secrete high-affinity antibodies. The TFH compartment comprises a memory pool that recirculates in blood and is largely quiescent in the absence of antigen stimulation.6,7 Because of difficulties in accessing lymphoid tissues in humans, the analysis of circulating TFH (cTFH) has proved valuable in understanding the alterations of TFH response that contribute to human diseases. cTFH correlate with the magnitude of antibody responses during vaccination and are reliable surrogate indicators of GC activity during infections and disease manifestations in autoimmunity.8 cTFH can upregulate CD40L, which reflects an activated state.9 Although activated cTFH do not sustain the expression of Bcl6, these cells represent long-lived memory cells that share functional properties with bona fide GC-TFH, with which they share the expression of ICOS and PD-1 as well as the production of IL-21; the latter is dependent on the transcription factor c-Maf.10 Indeed, c-Maf may also participate in TFH lineage maintenance by inducing sustained expression of CXCR5 when Bcl6 is downregulated as TFH become memory cells.11 cTFH are functionally heterogeneous on the basis of their ability to produce distinctive cytokines: (1) Th2-cTFH and Th17-cTFH, which provide efficient help to generate broadly neutralizing antibodies, and (2) Th1-cTFH, which provides suboptimal help that is selective to memory B cells.12
Although recent studies in animal models have shown that ABMR results from increased TFH activity, little is known about the dynamics and contribution of TFH cells to ABMR in humans.13 We have recently documented the existence of human alloreactive memory cTFH and examined their relationship to early DSA generation post-transplant.14 However, whether these cTFH play a role in triggering and sustaining the ABMR process still needs to be explored. Therefore, we undertook a concomitant profiling of cTFH and B cell compartments using multiple analytic approaches to provide insights into the cellular and molecular processes that occur during ABMR.
In a cohort of 105 kidney recipients, we provide evidence that activated cTFH and B cells that emerge in blood of patients who developed ABMR were highly distinct from those of patients who generated DSAs but did not undergo ABMR, and patients who did not generate DSAs. These distinctive cTFH and B cell responses appeared highly coordinated at phenotypic, transcriptional, and functional levels, indicative of active GC activity. Furthermore, accumulation of activated cTFH and B cells stratified a subset of patients with more severe allograft rejection lesions and worse clinical outcome. Therapeutic interventions targeting cTFH–B cell interaction may provide the greatest benefit to such patients.
Methods
Study Design
This study was performed on samples from patients who underwent kidney transplantation between January 2013 and December 2017 at University of Pittsburgh Medical Center and who were recruited to participate to a biorepository initiative at Thomas E. Starzl Transplantation Institute (STI). All patients signed a written informed consent (institutional review board numbers PRO12030552 and PRO17020318).
A total of 530 patients were screened for the following immunologic events: presence of post-transplant DSA and biopsy-proven ABMR. We identified 51 patients who developed DSA in the first 24 months post-transplant and had available PBMCs, defining two study groups: patients with ABMR (DSA+ABMR+, n=20) and patients without ABMR (DSA+ABMR−, n=31). Fifty four age- and sex-matched patients with no DSA or ABMR (DSA−) in the first 24 months post-transplant were selected to form the third study group. The flow chart of the study design is depicted in Supplemental Figure 1A. Clinical data of the study patients were extracted from the prospective database of the STI. In addition, patients were followed until May 1, 2019 and allograft loss was assessed.
Blood Samples
PBMCs, plasma, and sera were prospectively collected and banked at pretransplant, 1, 3, 6, 12, and 24 months post-transplant, and at the time of clinically indicated kidney allograft biopsies. The presence of DSAs in sera was systematically assessed at these time points. Surveillance protocol allograft biopsies were performed at 3 and 12 months post-transplant (Supplemental Figure 1B).
We analyzed cross-sectional PBMCs, plasma, and serum samples banked from the blood collected at the time of the immunologic event: (1) detection of post-transplant DSA for DSA+ABMR− patients and (2) detection of ABMR in presence of DSA for DSA+ABMR+ patients. In DSA− patients, the blood samples were analyzed at the time of a protocol biopsy and their time points were matched with those from DSA+ABMR− and DSA+ABMR+ patients (Supplemental Figure 1, B and C). Patients for whom cryopreserved PBMC samples were available at time points considered (pretransplant, 1, 3, 6, and 12 months post-transplant) were included in the longitudinal flow cytometry analysis in Figure 7.
Figure 7.
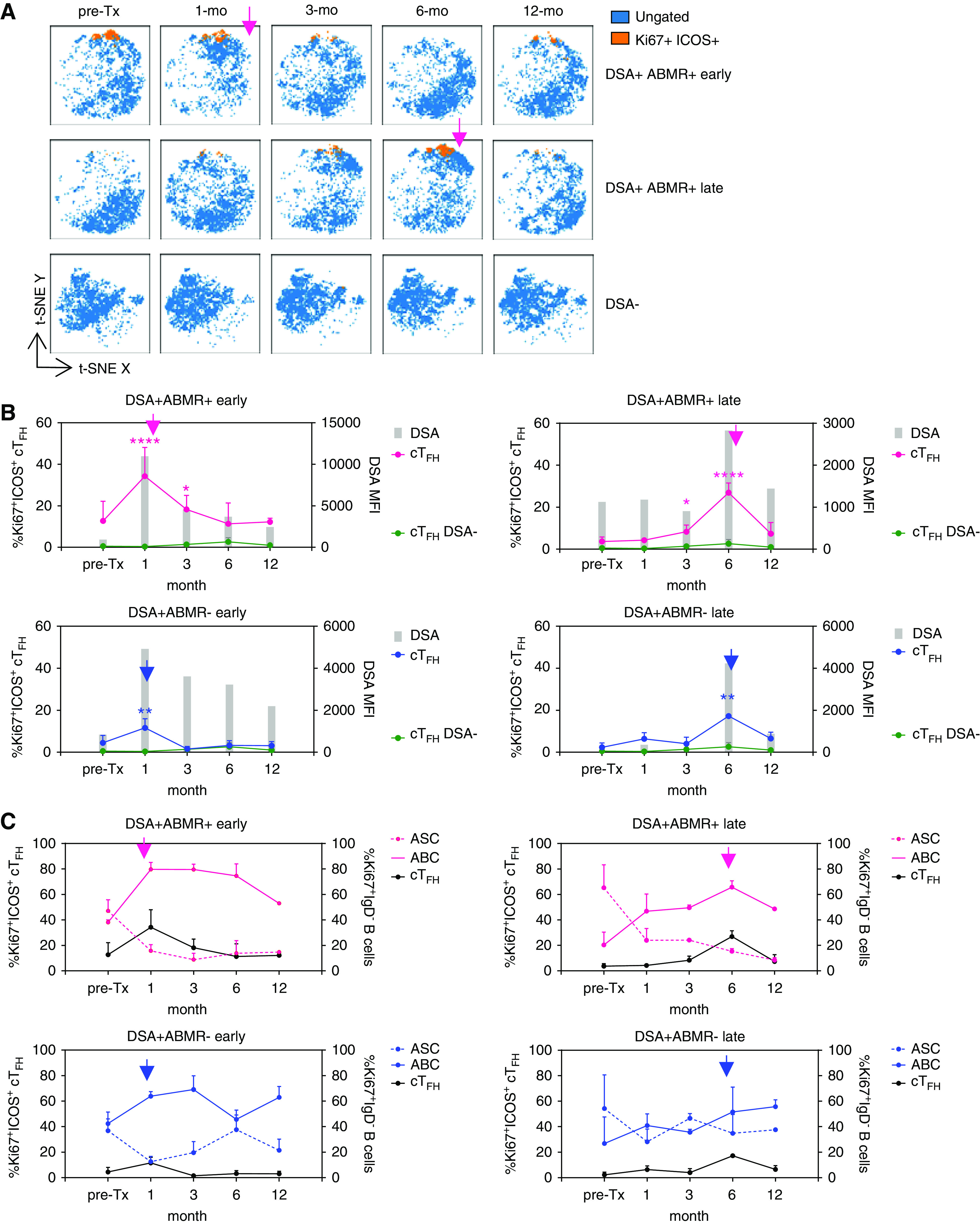
Dynamics of cTFH and ABC responses correlate with timing of ABMR onset. Patients were sampled longitudinally from pretransplant to the indicated intervals. (A) t-SNE projections of cTFH cells from three individual patients, overlaid with those that are Ki67+ICOS+. Each t-SNE map is on the basis of n=3000 cells per time point. Pink arrows indicate the time point of onset of ABMR. (B) Kinetics of emergence of Ki67+ICOS+ cTFH (connecting lines) and DSAs (gray bars) in indicated patient groups: DSA− (n=5), DSA+ABMR− early (n=5), DSA+ABMR− late (n=2), DSA+ABMR+ early (n=2), and DSA+ABMR+ late (n=2). Mixed-effects model for comparison of %Ki67+ICOS+ cTFH between DSA+ABMR+ or DSA+ABMR− groups with the DSA− group. *P<0.05; **P<0.01; ****P<0.001. (C) Kinetics of cTFH, ABCs, and ASCs in patient groups, as in (B). Blue arrows indicate the time of DSA emergence post-transplant and pink arrows indicate the time point of ABMR onset. cTFH, ABC, and ASC data are shown as percentage±SEM and DSA bars represent mean values.
Detection and Characterization of DSAs
The presence of anti-HLA antibodies with known reactivity against the donor HLA molecules (DSAs) in sera was systematically assessed at indicated time points described above. Pan-IgG anti-HLA -A, -B, -C, -DRB1/3/4/5, -DQB1, -DQA1, and -DPB1 DSAs were assessed in sera using single-antigen flow bead assays (SAB) (One Lambda Thermo Fisher, Inc.) on a Luminex platform (Luminex Corp.) according to manufacturer’s protocol. DSA-positive sera were tested for the presence of C1q-binding using the C1q-modified SAB assay (One Lambda Thermo Fisher) and IgG subclasses were tested using the SAB assay, substituting PE-conjugated anti-human IgG1, IgG2, IgG3, and IgG4 secondary antibodies for anti-human IgG. Normalized mean fluorescence intensity (MFI) cut-offs for positive results were MFI>1000 for the pan-IgG and IgG subclass assays, and MFI>500 for the C1q assays.3
Kidney Allograft Histology
Renal tissue was fixed in acetic-formol-absolute-alcohol fixative and stained with Masson trichrome and periodic acid–Schiff. Allograft biopsies were scored and graded from 0 to 3; diagnosis of ABMR was histologically defined using the international Banff 2017 criteria15 and was reviewed by an expert clinical transplant pathologist for the following: (1) histologic evidence of acute tissue injury, including one or more of microvascular inflammation (glomerulitis>0 and/or peritubular capillaritis>0), transmural or intimal arteritis >0, acute thrombotic microangiopathy in the absence of any other cause, or acute tubular injury and in the absence of any other apparent cause; and (2) evidence of current antibody interaction with: vascular endothelium with linear C4d staining in peritubular capillaries (C4d>0) or moderate microvascular inflammation (glomerulitis+peritubular capillaritis≥2). Lesions of T cell–mediated rejection (TCMR) were also defined according to Banff criteria. DSA− and DSA+ABMR− patients did not show signs of TCMR or ABMR at the time of sampling, whereas 17 out of 20 DSA+ABMR+ patients displayed concomitant TCMR lesions (Supplemental Table 1). All 20 ABMR cases were acute and C4d-positive, with the exception of one case of chronic active ABMR (transplant glomerulopathy, cg>0) and one case of C4d-negative ABMR.
Spectral Flow Cytometry
Samples were processed in batches to account for the batch effect. Full details of the antibodies used are given in Supplemental Table 7. Briefly, 2–10 million PBMCs were thawed and incubated with a mixture of antibodies diluted in 75% PBS and 25% Brilliant Violet Buffer (BD Biosciences) for 30 minutes at 4°C. Cells were surface-stained in Fc receptor blocking media (10% FCS PBS). Then PBMCs were washed, fixed and permeabilized with fixation/permeabilization buffer (eBioscience) for 40 minutes at 4°C, washed with permeabilization buffer (eBioscience), incubated in the dark for 30 minutes at 4°C with intracellular antibodies, and washed before acquisition on an Aurora spectral flow cytometer (Cytek).
High-Dimensional Flow Cytometry Data Analysis
The flow cytometry data were first curated with FlowJo software (TreeStar Inc.) to exclude debris, dead cells, and doublets, before gating on the cell populations of interest, leaving live events for downstream analyses. Single-cell data were analyzed using Cytobank software.16 All the samples were normalized and analyzed simultaneously. T-distributed stochastic neighbor embedding (t-SNE) analysis makes a pairwise comparison of cellular phenotypes to optimally plot similar cells close to each other and reduces multiple parameters into two dimensions (t-SNE X and t-SNE Y).17 Single cell–based flow cytometry data were normalized, downsampled, and concatenated to create t-SNE maps. We selected all channels except those utilized to gate the cell population of interest, and chose 3000 iterations with a perplexity of 30 and θ of 0.5 to run the t-SNE algorithm. Cell clusters were determined by spanning-tree progression analysis of density-normalized events (SPADE)18 algorithm by using ten target numbers of nodes without downsampled events. The cell clusters identified by SPADE were overlaid on the consensus t-SNE map for visualization and a heatmap was created to identify specific phenotypic patterns.
Donor-Antigen Stimulation for Cytokine Determination by Intracellular Staining and Flow Cytometry
PBMCs were stimulated with donor-cell lysate at 1:5 ratio for 6 hours in presence of Golgi plug (BD Biosciences) and Monensin (eBioscience), at 37°C and 5% CO2.19 Responder cells were washed, surface stained with CD3, CD8, and CXCR5 antibodies for 20 minutes at 4°C, and then washed and fixed with 1% paraformaldehyde (Sigma) for 40 minutes. Subsequently, the cells were permeabilized and incubated with mouse serum (Invitrogen) for 5 minutes at room temperature, followed by CD40L, IL-4, IL-17, IFNγ, and IL-21 antibodies for 30 minutes at room temperature. Cells were washed and acquired on a BD Biosciences Fortessa cytometer.
Cell Sorting
PBMCs were thawed, stained, and sorted on a BD Biosciences FACSAria II cytometer. cTFH cells were sorted as CD19−CD3+CD4+CD45RO+CXCR5+ cells, memory B cells as CD19+CD3−CD38loCD27+ cells, and activated B cells (ABCs) as CD19+CD3−CD38loCD27+CD21lo cells.
Cocultures
Sorted cTFH were cocultured with sorted autologous memory B cells (2×104) at 1:1 ratio with staphylococcal enterotoxin B (SEB) (1 µg/ml; Toxin Technology) in complete RPMI media supplemented with 10% FCS, 100 IU/ml penicillin, 100 mg/ml streptomycin (Life Technologies), 1 M HEPES buffer (Corning), and L-glutamine. After 6 days of coculture, cells were stained with CD4, CD19, CD27, CD38, ICOS, and CD71 antibodies before acquisition on cytometer.
CXCL13 ELISA
CXCL13 was measured in plasma with Human CXCL13 Quantikine ELISA kit (R&D Systems) according to the manufacturer’s protocol.
IgG ELISA
Total IgG production was measured in coculture supernatants with Human IgG Total ELISA kit (eBioscience) and IgG subclasses production with IgG Subclass Human ELISA kit (eBioscience).
RNA Sequencing
Total RNA was extracted from cTFH and ABCs sorted directly into lysis buffer. RNA was isolated using miRNeasy Mini Kit (Qiagen). Complementary DNA synthesis and amplification were performed with a SMARTer Stranded Total RNA-Seq Kit v2 Pico Input Mammalian (Takara). Libraries were sequenced on an Illumina NextSeq 500 using 75-bp paired-end reads. The paired-end reads were checked for quality and adapters using FastQC (version 0.11.7). These quality trimmed reads were later mapped against the Ensembl human reference genome (GRCh38 version 91) using HISAT2 mapper (version 2.1.0). Counts for genes were generated using HT-Seq (version 0.11.2) on the mapped files. EdgeR (version 3.24.1), a bioconductor R (version 3.8) package, was used to analyze differentially expressed genes. Genes differentially expressed (two-fold difference and false discovery rate <0.05) were analyzed using Ingenuity Pathway Analysis (IPA) (Qiagen), with a focus on canonical pathway and upstream regulator analyses. Principal component analysis (PCA) plots were generated on the basis of normalized and scaled fragments per kilobase of transcripts per million read counts using the factoextra and ggplot2 packages in R.
Data Sharing Information
RNA sequencing data is registered with the Gene Expression Omnibus database under accession number GSE145503.
Statistical Analyses
Mean±SD or SEM values and frequencies are provided for the description of the continuous and categorical variables, respectively. The means and proportions were compared using t test and chi-squared test (or the Mann–Whitney U test and Fisher exact test if appropriate, respectively). Multiple groups were analyzed by Kruskal–Wallis test or one-way ANOVA with Tukey post hoc test for adjustment for multiple comparisons. Death-censored allograft survival was assessed using the Kaplan–Meier estimator and compared with the log-rank test. Values of P<0.05 were considered statistically significant, and all tests were two-sided. Analyses were performed using GraphPad Prism version 8, Cytobank (http://www.cytobank.org), Partek Flow (http://www.partek.com/partek-flow/), and R software (https://www.r-project.org/, R Development Core Team, Vienna, Austria).
Results
Clinical Characteristics and Multidimensional Profiling of Transplant Recipients
We enrolled 105 patients who were systematically screened for circulating DSAs and allograft rejection in the first 24 months post-transplant and identified three study groups: patients who did not develop DSAs nor ABMR (DSA−, n=54), those who developed DSAs but did not undergo ABMR (DSA+ABMR−, n=31), and patients who developed DSAs accompanied with ABMR (DSA+ABMR+, n=20) (Supplemental Figure 1A). Their clinical characteristics are shown in Supplemental Table 1. DSA+ABMR− and DSA+ABMR+ had higher rates of retransplantation as compared with DSA− patients, indicating prior exposure to alloantigens (Supplemental Table 1). Accordingly, the majority of these patients developed DSAs, with or without ABMR, in the first 3 months post-transplant (26 out of 31 in the DSA+ABMR− group and 12 out of 20 in the DSA+ABMR+ group), indicative of memory responses (Supplemental Figure 1B).14 We analyzed PBMCs, plasma, and serum from cross-sectional blood samples collected at the time of (1) detection of post-transplant DSAs for DSA+ABMR− patients and (2) detection of ABMR in the presence of DSAs for DSA+ABMR+ patients. For DSA− patients, blood samples were analyzed at matched time points. Using multidimensional profiling, we investigated the phenotypic, transcriptional, and functional states of cTFH and B cells as well as their dynamics in the three study groups of patients (Supplemental Figure 1C, Supplemental Table 2).
Expansion of cTFH in Blood of Patients Manifesting Post-Transplant DSAs
We first analyzed CD4+ T cell compartment and distinguished naive (CD45RO−CCR7+) from the memory T conventional (CD45RO+CXCR5−) and cTFH (CD45RO+CXCR5+) subsets. DSA+ABMR+ patients exhibited a significant increase in frequencies of cTFH cells as compared with DSA+ABMR− and DSA− groups (Supplemental Figure 2A). In contrast to naive and T conventional cells, cTFH in DSA+ABMR+ selectively displayed higher expression of PD-1 and ICOS, as well as higher frequencies of PD-1+ICOS+ cTFH, that are hallmarks of activated TFH cells (Supplemental Figure 2, B and C). Thus, activated cTFH are more highly expanded in the context of DSA+ABMR+ compared with DSA+ABMR− responses.
cTFH associated with Post-Transplant DSAs and ABMR Are Heterogeneous and Dominated by Activated and Early Memory Phenotypes
To further delineate the phenotypic alterations in cTFH, we performed high-dimensional t-SNE analyses and created t-SNE maps on the basis of their expression of 18 makers, with samples from all study groups (Figure 1A). We observed considerable differences in cTFH high-dimensional profiles among groups, with specific cells coexpressing ICOS, PD-1, and Ki67 markedly distinguishing DSA+ABMR+ from DSA+ABMR− and DSA− patients (Figure 1B). Analyses by supervised gating further showed that cTFH from DSA+ABMR+ patients displayed higher expression of the memory markers CD127 and CCR7, and the TFH regulators IRF4, c-Maf, and IL-21R. We note that cTFH from DSA+ABMR− contained lower expression of the above markers, and cTFH from DSA− patients expressed higher CD25 (IL-2Ra) and CXCR3, which are known to counteract TFH differentiation (Supplemental Figure 3). We next applied SPADE clustering to refine cTFH cell subpopulations (Figure 1C). Among the ten SPADE clusters, the selective coexpression of ICOS along with PD-1, CD127, and CCR7 appeared to robustly distinguish clusters 4 (Ki67hi) and 8 (Ki67lo) from all other clusters (Figure 1D, Supplemental Table 3). Interestingly, these two clusters were dominant in DSA+ABMR+ patients (Figure 1E). These unsupervised analyses identified Ki67 and ICOS as the most informative markers distinguishing cTFH cells across the patient groups; the frequencies (Figure 1F) and absolute numbers (Supplemental Figure 4A) of Ki67+ICOS+ cTFH were the highest in DSA+ABMR+ patients when compared with DSA+ABMR− and DSA− groups. We note that up to 90% of Ki67+ICOS+ cells coexpressed PD-1 in DSA+ABMR+ patients. Such cells also coexpressed IRF4 and the activation markers CD38 and CD28, but not IL-6R and OX40, and were therefore highly distinctive from Ki67−ICOS− cTFH subset (Supplemental Figure 4B). Thus, during DSA+ABMR+ response, cTFH are enriched for proliferating cells expressing CCR7, CD127, IRF4, and c-Maf, defining an activated and early memory phenotype.
Figure 1.
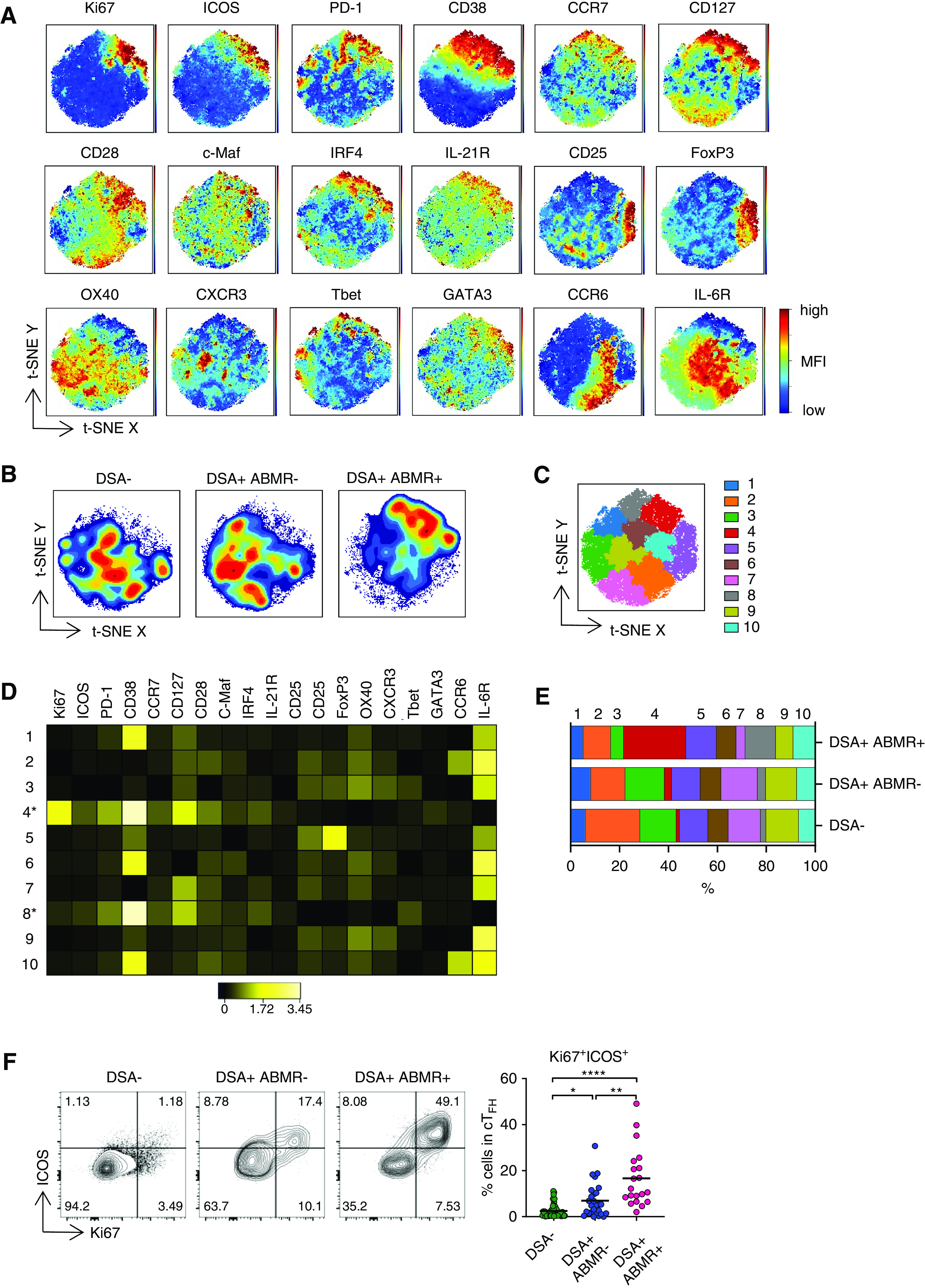
cTFH associated with ABMR are heterogeneous and display activated and early memory features. (A) t-SNE projections were generated using a concatenated file of 56,100 cTFH cells from DSA− (n=20), DSA+ABMR− (n=20), and DSA+ABMR+ (n=20) patients; panels display expression levels of indicated markers (MFI). (B) t-SNE projections of cTFH cell densities in the three patient groups using 18,700 cells from each group, shown in (A). (C) t-SNE map overlaid with 10 cTFH cell clusters delineated by SPADE clustering of the concatenated file, as in (A). (D) Heatmap showing the expression of markers for each cTFH cell cluster according to transformed MFI ratio. Cell clusters 4 and 8 (*) are distinct from others. (E) Stacked bar plot showing cTFH cell cluster distribution on the basis of SPADE clustering as in (C). Clusters 2, 3, 4, 7, 8, and 9 are significantly different in their proportions across the indicated groups. (F) Representative examples of flow cytometry analysis and dot plot of percentages of Ki67+ICOS+ cells in cTFH are displayed for DSA− (n=48), DSA+ABMR− (n=27), and DSA+ABMR+ (n=20) patients. Kruskal–Wallis with Dunn post-test for panel (E and F). *P<0.05; **P<0.01; ****P<0.001. Each dot represents one patient and horizontal lines are mean values.
Activated cTFH Display a GC-Precursor Transcriptional Profile
We next evaluated the transcriptional programs underlying the distinctive phenotypic alterations of cTFH in patients. The transcriptional profiles of purified cTFH from DSA+ABMR− and DSA+ABMR+ were markedly distinguishable from that of DSA− patients (Figure 2A); 927 genes were found to be differentially expressed between DSA+ABMR+ and DSA−, and 652 genes were differentially expressed between DSA+ABMR− and DSA− patients. In addition to the expected upregulation of CXCR5, ICOS, and PDCD1, cTFH from DSA+ABMR+ patients were highly enriched for LEF1 and TCF7, two factors acting upstream of Bcl6 to coordinate differentiation of TFH precursors.20 Consistently, these cTFH also upregulated CD27 and sustained expression of CCR7 at transcript (Figure 2B) and protein (Supplemental Figure 3) levels.21 Additionally, cTFH in DSA+ABMR+ also expressed the highest levels of MAF, IL21R, CD40LG, and SLAMF6, reflecting the multiple signaling molecules involved in TFH differentiation and their function in providing cognate help to B cells (Figure 2B).22 IPA performed on cTFH RNA-sequencing data sets from DSA+ABMR+ patients when compared with DSA− patients confirmed the upregulation of ICOS-L–ICOS and CD28 canonical pathways (Figure 2C, Supplemental Table 4). Moreover, STAT3, which is regulated by IL-21 and coordinates with MAF to mediate TFH differentiation, was predicted by IPA to be a significant activator of the cTFH transcriptional profile of the DSA+ABMR+ group. Conversely, IL-2 was predicted to be downregulated (Supplemental Table 5). Thus, the transcriptional state of cTFH in patients mounting DSA+ABMR+ response reflected that of activated GC-TFH precursors, enriched for multiple receptor and signaling regulators that promote their expansion and differentiation.
Figure 2.
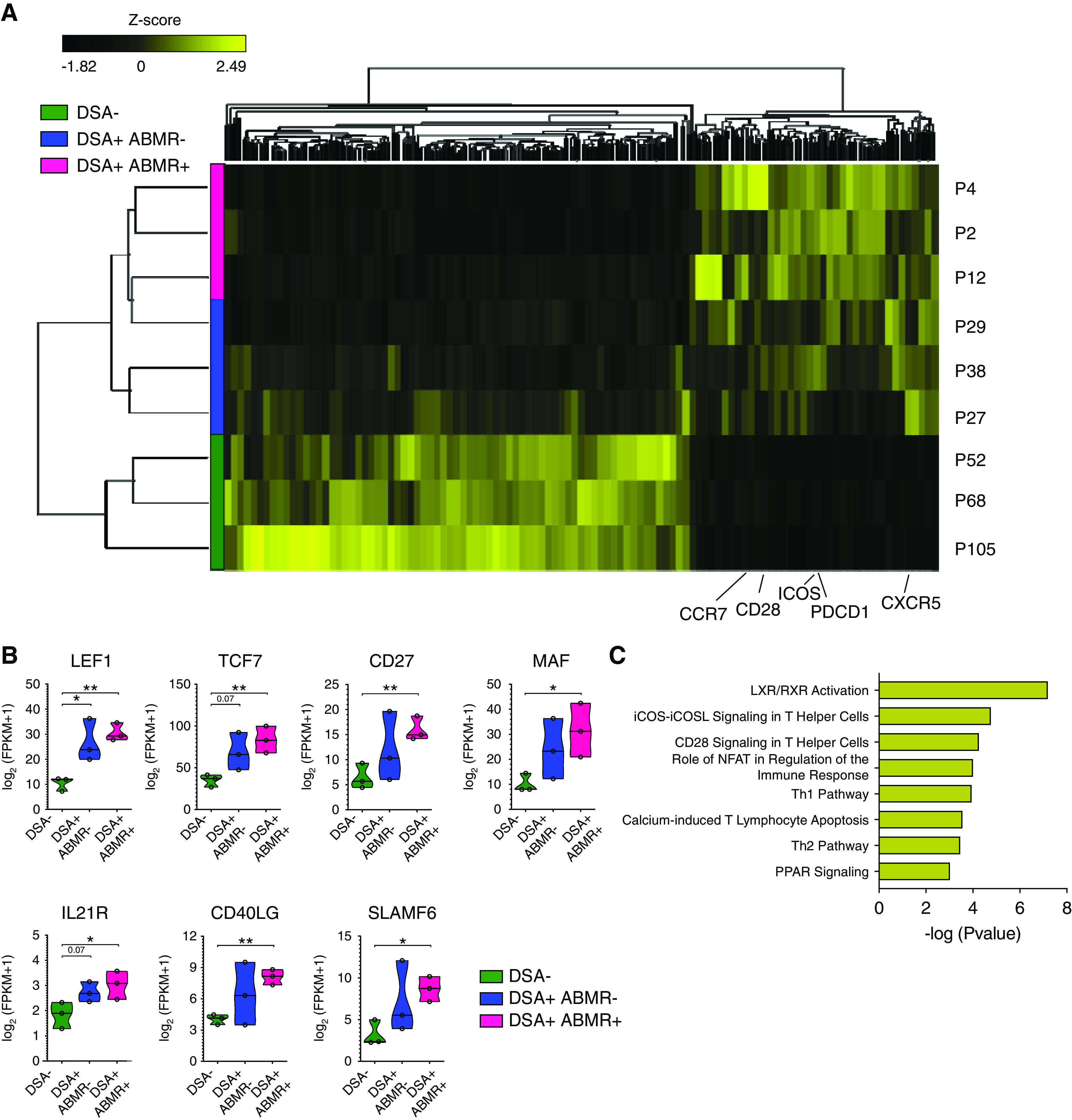
cTFH associated with ABMR display a distinctive GC-precursor transcriptional profile. RNA-sequencing analysis of sorted cTFH was performed in three patient groups: DSA− (n=3), DSA+ABMR− (n=3) and DSA+ABMR+ (n=3). (A) Heatmap generated by hierarchical clustering of genes and the three types of patient samples. Genes used for clustering were differentially expressed (fold change >2, false discovery rate P value <0.05). (B) Violin plots showing the expression levels of displayed genes in cTFH for indicated patient groups. Multiple t test with Holm–Sidak correction. *P<0.05; **P<0.01. (C) Bar plot of significantly upregulated canonical pathways (on the basis of differentially expressed genes between cTFH from the DSA+ABMR+ group versus the DSA− group) aligned by adjusted –log(P value) as predicted by IPA, using Fisher exact test.
Alloreactive cTFH Induce Cognate Memory B Cell Responses and DSAs
We next addressed whether activated cTFH express distinctive cytokines that are critical for shaping B cell responses. We assessed the cTFH cytokine profiles after 6-hour stimulation with donor PBMC lysates in vitro. This enabled the detection of donor-reactive cTFH via their capacity to express intracellular CD40L (Supplemental Figure 5A).14 We observed a significant increase of CD40L+ cTFH (Supplemental Figure 5B), which expressed higher IFNγ in both DSA+ABMR− and DSA+ABMR+ groups. Notably, IL-17 and IL-21 were highest in the latter group (Supplemental Figure 5C).
We next determined the functional effect of cTFH help in promoting the generation of DSAs, using a 6-day coculture system with sorted cTFH and memory B cells stimulated with SEB (Supplemental Figure 6). In presence of memory B cells, cTFH from DSA+ABMR+ patients, in contrast to control patients, significantly upregulated more ICOS and the proliferation marker CD71 (Supplemental Figure 7A). In turn, memory B cells underwent activation and differentiated into antibody-secreting cells (ASCs) (Figure 3A). This was not observed in the absence of cTFH or in the absence of SEB stimulation (Supplemental Figure 7B). Accordingly, the amounts of total IgG, including IgG1 and IgG3 isotypes produced, were most substantially increased in the DSA+ABMR+ group (Figure 3, B and C). Importantly, DSA production was only detected in supernatants from DSA+ABMR+ cocultures (Figure 3D). Thus, cTFH from DSA+ABMR+ patients are enriched for donor-specific cells that are functionally polarized to promote memory B cell activation and the generation of DSAs, including IgG1 and IgG3 isotypes.
Figure 3.
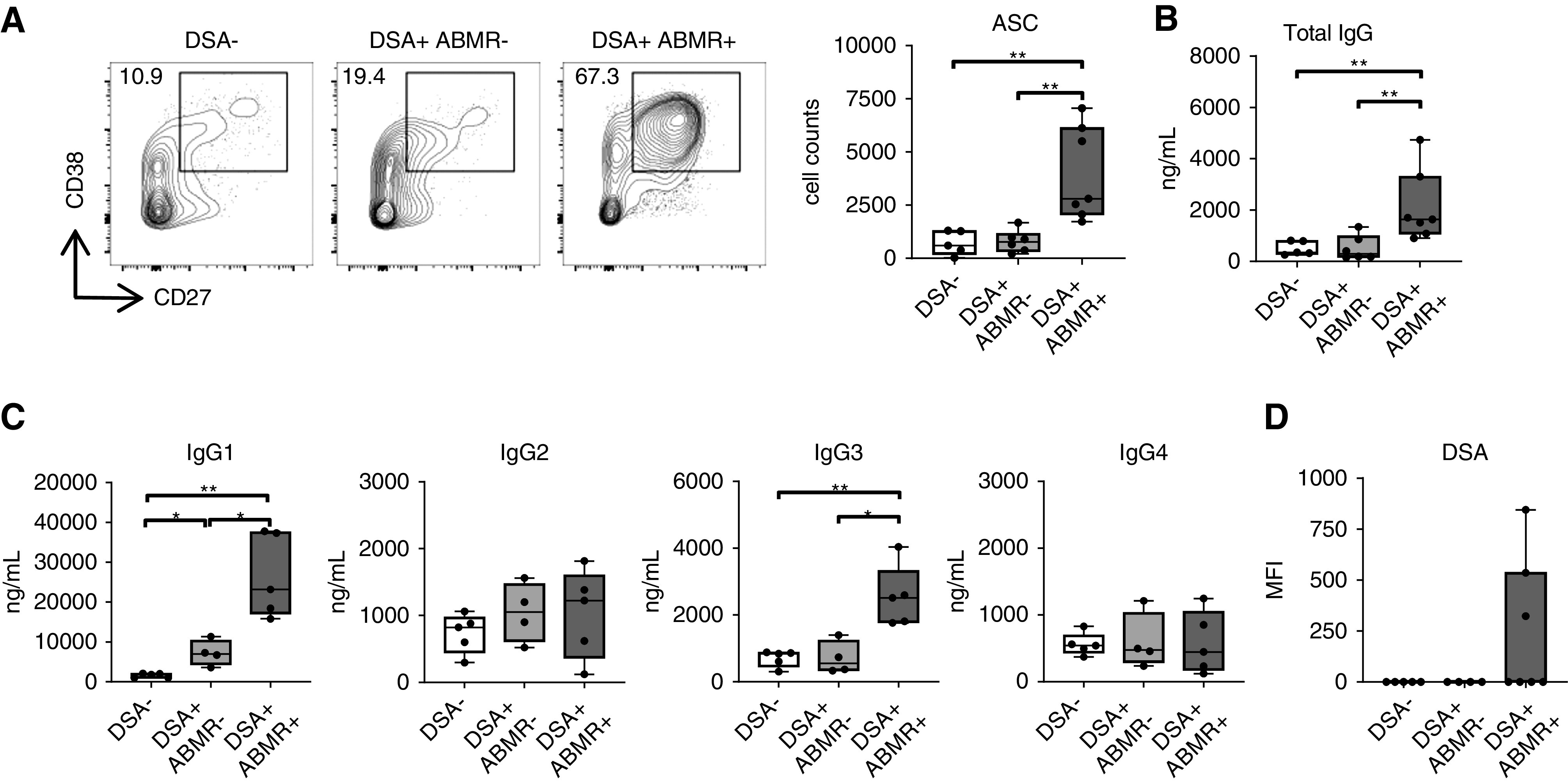
cTFH from ABMR patients promote memory B cells to generate DSAs. Coculture of sorted cTFH with autologous memory B cells in presence of SEB (6 days). (A) Representative examples of individual experiments by flow cytometry analysis and dot plot of CD27+CD38hi ASCs as cell counts in cocultures are displayed for DSA− (n=5), DSA+ABMR− (n=6) and DSA+ABMR+ (n=7) groups. (B and C) Box plots of total IgG and IgG subclasses measured by ELISA in supernatants after 6 days of coculture: (B) DSA− (n=5), DSA+ABMR− (n=6), and DSA+ABMR+ (n=7); and (C) DSA− (n=5), DSA+ABMR− (n=4) and DSA+ABMR+ (n=5). Mann–Whitney U test for (A–C). *P<0.05; **P<0.01. (D) Luminex analysis of DSAs in supernatants after 6 days of coculture for DSA− (n=5), DSA+ABMR− (n=4) and DSA+ABMR+ (n=7) groups.
Activated cTFH Emerge Concomitantly to Proliferating ABCs and ASCs
We next evaluated whether cTFH expansion was coordinated with that of B cells in vivo. We detected a subset of Ki67+ and isotype-switched B cells that were significantly expanded in DSA+ABMR+ patients (Figure 4A). Such cells were also observed, albeit at lower frequencies and numbers (Supplemental Figure 8), in the DSA+ABMR− group. Among Ki67+IgD−CD27+ B cells, we further distinguished CD20+CD38lo ABCs from CD20−CD38hi ASCs.23 ABCs were most highly expanded in DSA+ABMR+ patients, whereas ASCs were increased to a lesser extent when compared with the DSA+ABMR− group (Figure 4B). Importantly, plasma CXCL13 levels, which indicate ongoing GC activity, were significantly elevated in DSA+ABMR+ patients compared with their DSA+ABMR− counterparts, and both Ki67+ICOS+ cTFH and ABCs correlated with CXCL13 levels (Figure 4, C and D).24 Thus, the concomitant emergence in blood of Ki67+ICOS+ cTFH and ABCs are indicative of GC-dependent TFH–B cell interactions in DSA responses, which are exaggerated during ABMR.
Figure 4.
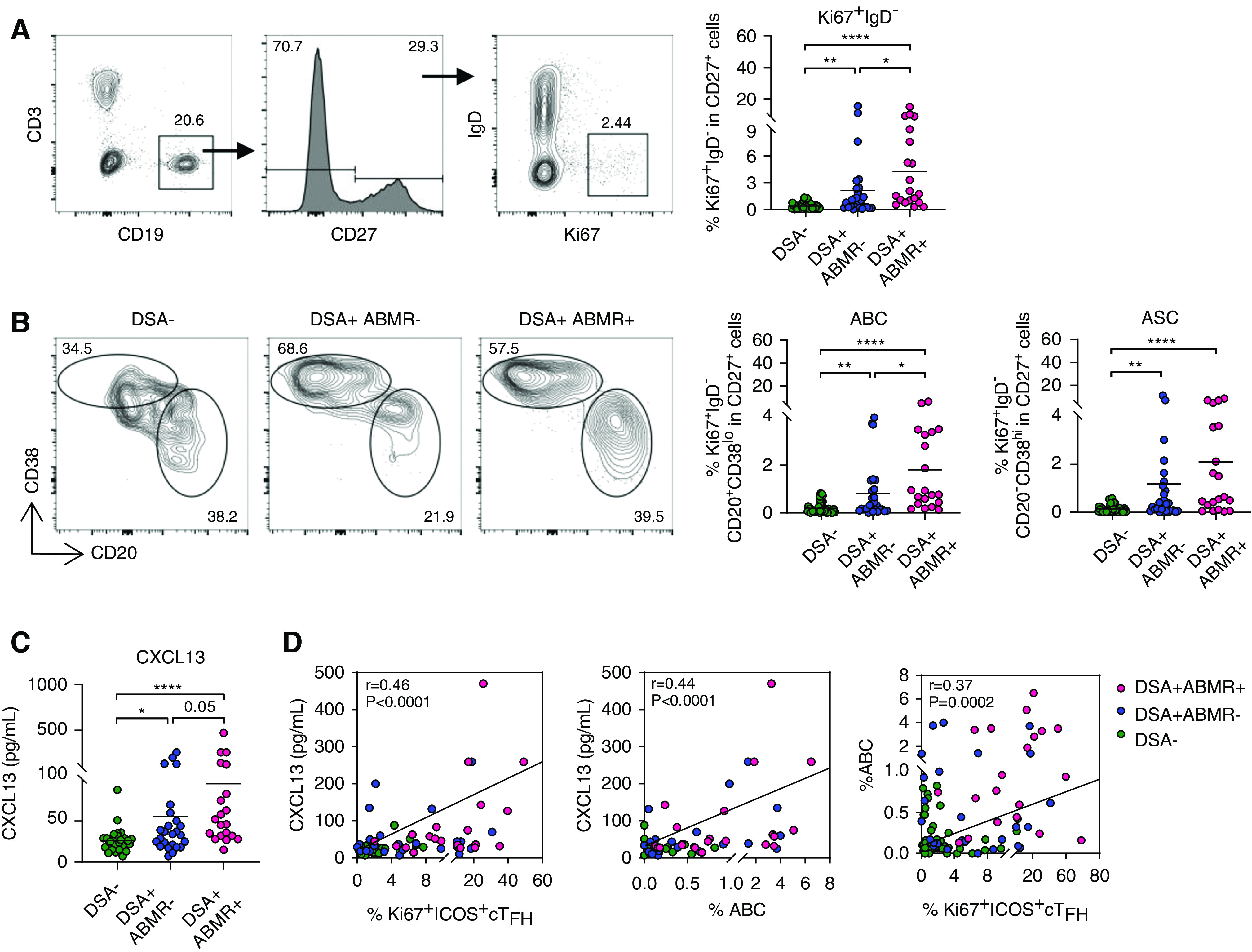
Concomitant emergence of activated cTFH and B cells in blood is reflective of increased GC activity.ctivated B cells emerge concomitantly in blood and reflect active GC responses. Flow cytometry analysis of blood B cells in patients analyzed for cTFH in Figure 1F. (A) Representative examples of flow cytometry analysis and dot plots of percentages of Ki67+IgD− among CD27+ cells are displayed for DSA− (n=48), DSA+ABMR− (n=27) and DSA+ABMR+ (n=20) groups. (B) Representative flow cytometry analysis and percentages of Ki67+IgD−CD20+CD38lo ABCs and Ki67+IgD−CD20−CD38hi ASCs among CD27+ cells are displayed for DSA− (n=48), DSA+ABMR− (n=27) and DSA+ABMR+ (n=20) groups. (C) Plasma CXCL13 levels measured by ELISA for DSA− (n=28), DSA+ABMR− (n=25), and DSA+ABMR+ (n=20) groups. (D) Spearman correlation analysis of Ki67+ICOS+ cTFH and ABCs with plasma CXCL13 levels for DSA− (n=28), DSA+ABMR− (n=25), and DSA+ABMR+ (n=20) groups. Mann-Whitney U test was used for (A–C). *P<0.05; **P<0.01; ****P<0.001. Each dot represents one patient.
Skewed cTFH Polarization Correlates with DSA Pathogenicities
Class-switched and high-affinity antibodies are the output of efficient TFH–B cell interactions in GCs. We therefore measured the quality of circulating DSAs in sera of patients and found that DSA+ABMR+ patients harbored significantly more DSAs of multiple specificities (HLA class 1 plus 2) and IgG subclasses (Supplemental Table 6). Also, higher DSA levels correlated with increased Ki67+ICOS+ cTFH and ABCs, further indicating that mutual expansion of these memory cells tightly reflect the strength of the donor-specific response (Supplemental Figure 9, A and B). In contrast, we observed a higher rate of transient DSAs in DSA+ABMR− patients (Supplemental Table 6).
IgG subclasses and C1q-binding capacity confer different pathogenicities to DSAs during ABMR.3 Accordingly, higher proportions of DSA+ABMR+ patients displayed IgG1, IgG3, and C1q-reactive DSAs compared with DSA+ABMR− patients (Figure 5A). The proportion of patients with IgG2 DSA did not differ and IgG4 DSAs were not detected. The elaboration of more diverse DSA isotypes may be explained by skewed polarization of cTFH subsets with differential capacity to regulate B cell responses.12 On the basis of CXCR3 and CCR6, we identified four subsets within Ki67+ICOS+ cTFH, designated as Th1, Th1/17, Th2, and Th17 (Supplemental Figure 10A). We found a selective increase in cTFH Th2 and Th17 in DSA+ABMR+ patients (Supplemental Figure 10B) that paralleled the elevated IgG1, IgG3, and C1q DSAs (Figure 5B, right panel). Interestingly, in DSA+ABMR− patients, only those who developed IgG1 and C1q DSAs also displayed significantly higher frequencies of cTFH Th2 and cTFH Th17 (Figure 5B, left panel). Consistently, GATA3 and RORgt expression on Ki67+ICOS+ cTFH were most elevated in DSA+ABMR+ patients (not shown). Thus, programming of cTFH into Th2 and Th17 effectors may account for the accumulation of pathogenic DSAs in patients.
Figure 5.
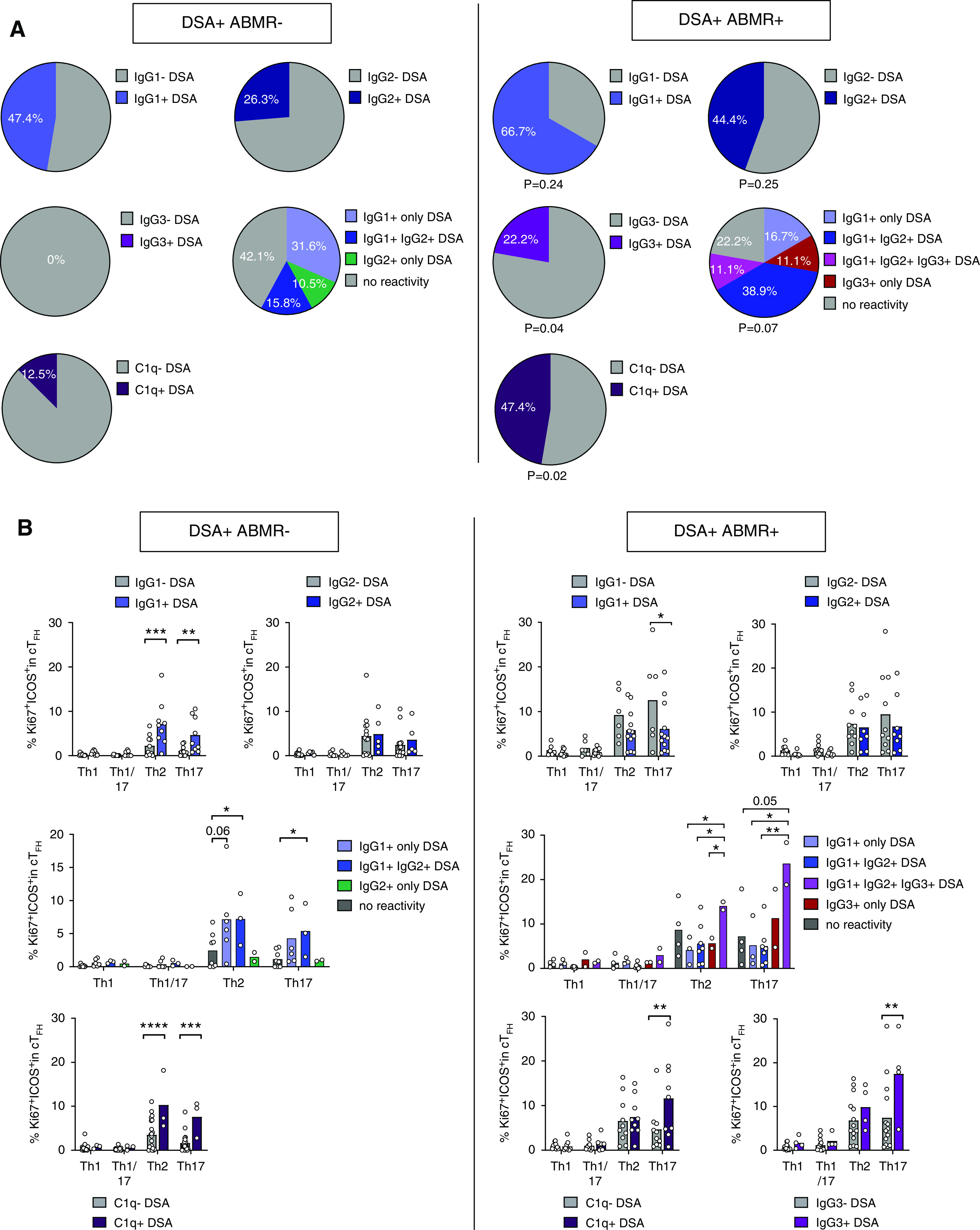
Altered cTFH polarization correlates with differential DSA pathogenicities. (A) Proportions of patients that were positive for the indicated DSA IgG isotypes in the two patient groups are displayed for DSA+ABMR− (n=19, left panel) and DSA+ABMR+ (n=18, right panel). C1q-binding analysis was performed using DSA+ABMR− (n=24, left panel) and DSA+ABMR+ (n=19, right panel) patient serum samples. P values of chi-squared test for the comparison between patient groups are shown. (B) Flow cytometry analysis of percentages of Ki67+ICOS+ Th1, Th1/17, Th2, and Th17 cells among cTFH from DSA+ABMR− and DSA+ABMR+ patients are shown. Sample size as in (A). Kruskal–Wallis test with Dunn post-test. *P<0.05; **P<0.01; ****P<0.001. Each dot represents one patient and horizontal lines are mean values.
Coordinated Transcriptional Programs in cTFH and ABCs
To gain insights into the molecular processes underlying the proliferative B cell responses, we undertook RNA-sequencing analysis of sorted ABCs (Supplemental Figure 11A) from the same patients’ samples and same time points as those of sorted cTFH (Figure 2A). As in cTFH, this revealed distinctive gene expression patterns in ABCs from DSA+ABMR+ patients compared with control patients. We then performed combined PCA of the matched cTFH and ABC RNA-sequencing data sets. As expected, PC1 separated cTFH from ABCs on the basis of cellular identity (T versus B cells) (Supplemental Figure 11B). However, PC2 and PC3 clustered cTFH with their corresponding ABCs and clearly segregated the three patient groups (Figure 6A). Covarying genes that enabled this segregation are displayed in Figure 6B. These include genes selected among the top 60 genes driving PC2 and PC3 (Supplemental Figure 11C), with overlaying of additional biologically relevant genes.
Figure 6.
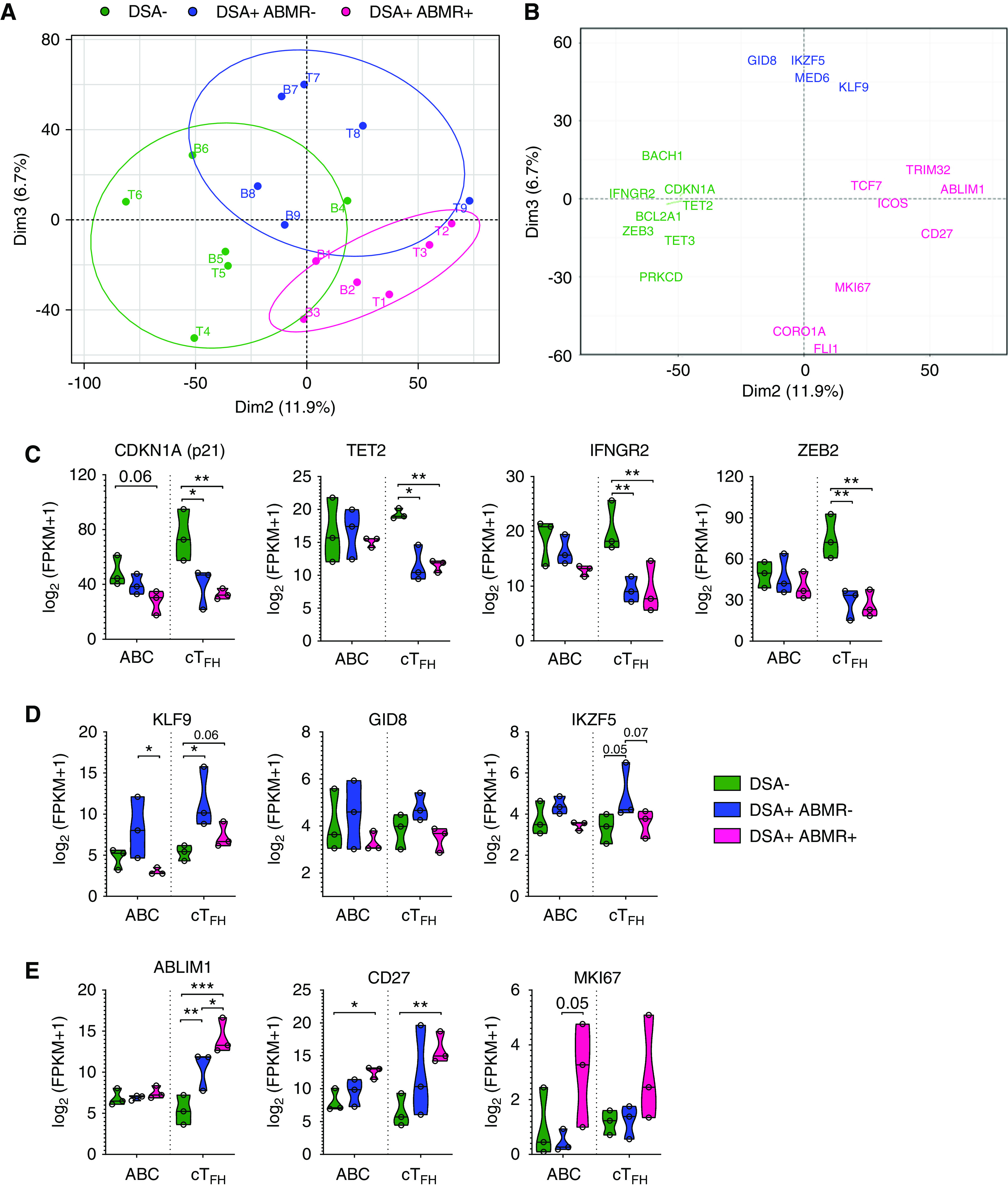
Coordinated transcriptional programs in cTFH and ABCs of patients with ABMR. (A) PCA of the cTFH and ABC RNA-sequencing patterns for each patient group (n=3 patients per group) are depicted. PC2 and PC3 delineate the cTFH (T1–9) and ABCs (B1–9) from DSA− (green), DSA+ABMR− (blue), and DSA+ABMR+ (pink) patients as separate clusters. (B) Genes that drive PC2 and PC3 (selected from the top 60 genes contributing to each PC, along with biologically relevant others) are displayed. (C–E) Violin plots of expression levels of indicated genes that covary in cTFH and ABCs in the three patient groups. Multiple t test with Holm–Sidak correction for (C–E). *P<0.05; **P<0.01; ***P<0.001. Each dot represents one patient.
Specifically, CDKN1A, an inhibitor of cell cycle progression as well as TET2 and IFNGR2, which counteract GC-TFH and B cell differentiation respectively, displayed increased expression in cTFH and ABCs from DSA− patients, consistent with quiescent status (Figure 6C).25 In DSA+ABMR− patients, KLF9 was found increased in ABCs, whereas GID8, another component of β-catenin/Wnt signaling, was upregulated in cTFH (Figure 6D).26,27 Consistent with proliferating states, MKI67 was found selectively and simultaneously upregulated in both cTFH and ABCs from DSA+ABMR+ patients, as were CD27 and the actin-binding regulator ABLIM1 (Figure 6E). Altogether, this demonstrates extensively coordinated cTFH and ABC transcriptional programs underlying each clinical state, including ABMR.
cTFH and ABC Responses Are Dynamic and Peak at the Time of ABMR
Early ABMR (before 3 months) post-transplant is known to involve preformed memory response, whereas late ABMR (after 3 months) is considered to be mediated by de novo DSAs.28 We therefore asked whether early ABMR involved preformed memory cTFH and B cells, and how they differed from that of late ABMR. Ki67+ICOS+ cTFH were readily detectable pretransplant in early DSA+ABMR+ patients, expanded at 1 month post-transplant when ABMR occurred, and remained elevated throughout follow-up (Figure 7A top panel, Figure 7B). Conversely, in late DSA+ABMR+ patients, Ki67+ICOS+ cTFH were minimal at pretransplant and in the first months, and peaked at 6 months post-transplant, when ABMR occurred (Figure 7A middle panel, Figure 7B). Importantly, cTFH dynamics tightly paralleled DSA levels in sera (Figure 7B). Interestingly, the kinetics of ABCs, but not ASCs, paralleled that of Ki67+ICOS+ cTFH in DSA+ABMR+ patients (Figure 7C). Although at lower levels, similar kinetics of Ki67+ICOS+ cTFH (but not of ABCs) were observed in DSA+ABMR− patients, coinciding with the detection of early (1 month) or late (6 month) DSAs (Figure 7, B and C). Thus, the timing of expansion of Ki67+ICOS+ cTFH and ABCs parallels DSA appearance and their magnitude is associated with the onset of ABMR.
Accumulation of cTFH and ABCs Is associated with the Severity of ABMR Manifestations
We have shown that frequencies of Ki67+ICOS+ cTFH markedly differed across patient groups. Using the median frequency of Ki67+ICOS+ cTFH in the DSA+ABMR+ group (Figure 1F), we stratified both DSA+ABMR− and DSA+ABMR+ patients into low (<11.4%) and high Ki67+ICOS+ cTFH (>11.4%) subgroups. On the basis of this stratification, we identified six out of 27 DSA+ABMR− patients and ten out of 20 DSA+ABMR+ patients that manifested high Ki67+ICOS+ cTFH. The latter ten patients displayed the highest enrichment in the CCR7- and CD127-expressing cluster 4 of cTFH compartment (Supplemental Figure 12, A–C) as well as the highest frequencies of ABCs and ASCs (Figure 8A). Importantly, these patients harbored DSA of higher levels, which included proinflammatory IgG3, and manifested significantly higher intimal arteritis, tubulointerstitial inflammation lesions, and thus a more severe histological form of ABMR (Figure 8, B and C). Finally, these DSA+ABMR+ patients displayed the lowest allograft survival rate (Figure 8D). Thus, the magnitude of proliferating cTFH with a precursor phenotype and ABC responses are associated with the onset and severity of ABMR.
Figure 8.
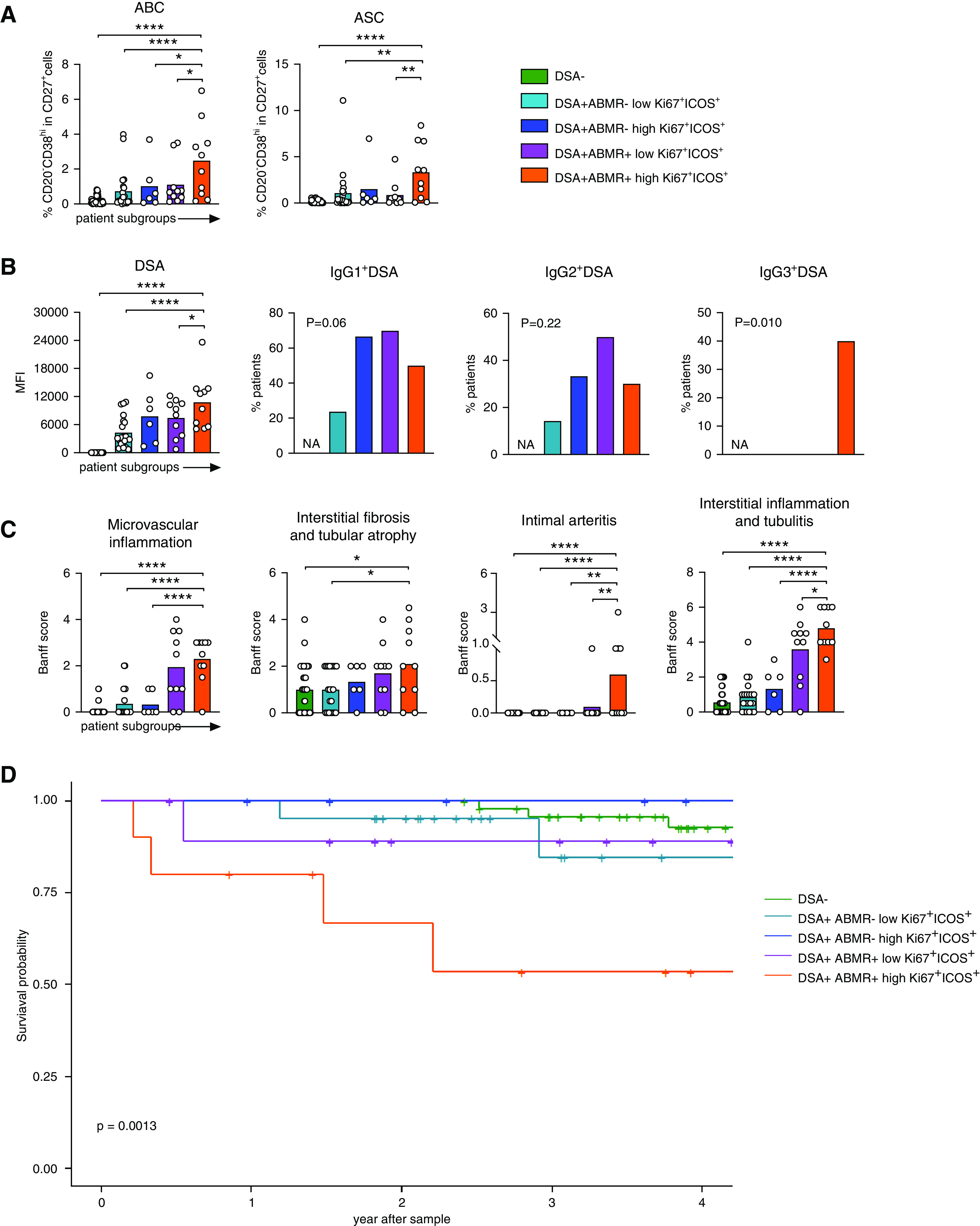
High frequencies of activated cTFH are associated with increased B cell and DSA responses, more allograft injury, and decreased allograft survival. DSA+ABMR− and DSA+ABMR+ patients were stratified into subgroups on the basis of the median percentage of Ki67+ICOS+ cTFH <11.4% (low) and >11.4% (high) in the DSA+ABMR+ group, as shown in Figure 1F. Data shown from indicated patient subgroups: DSA+ABMR− low (n=21), DSA+ABMR− high (n=6), DSA+ABMR+ low (n=10), DSA+ABMR+ high (n=10), and DSA− (n=48) are displayed. (A and B) Dot plots of percentages of ABCs and ASCs by flow cytometry, levels of DSAs, and proportions of patients that were positive for IgG1, IgG2, and IgG3 DSA by Luminex analysis, evaluated at the time of flow cytometry analysis of the cTFH, are displayed for DSA+ABMR− low (n=21), DSA+ABMR− high (n=6), DSA+ABMR+ low (n=10), DSA+ABMR+ high (n=10), and DSA− (n=48) patients. (C) Histologic Banff scores of kidney allograft lesions evaluated at the time of flow cytometry analysis of the cTFH. Microvascular inflammation indicated by glomerulitis+peritubular capillaritis Banff score and interstitial fibrosis and tubular atrophy indicated by IF/TA Banff score. One-way ANOVA with Dunnett post-test for (A–C). *P<0.05; **P<0.01; ****P<0.001. Each dot represents one patient and horizontal lines of bars are mean values. (D) Kaplan–Meier of allograft survival rate in each subgroup of patients; sample size as in (A). Log-rank test.
Discussion
The role of TFH cells in the generation of DSAs has been demonstrated in mouse and primate models of kidney transplantation.13,29,30 Our findings are consistent with animal models and reveal novel features of cTFH phenotypic and molecular states during the generation of DSAs and ABMR in humans. Although previous studies have found an association between increased frequencies of PD-1+ICOS+ or PD-1hiCCR7lo cTFH and DSA development, their relationship to ABMR was not explored.31–34 Interestingly, activated PD-1hiCCR7lo cTFH are thought to represent effector memory cells that can rapidly reacquire GC-TFH effector functions upon secondary activation.35 Here, we identified a novel subset of Ki67+ICOS+ cTFH expressing high levels of CCR7 and CD127, which indicates that these cells are less differentiated than their PD-1hiCCR7lo counterparts. Interestingly, CCR7+CD127+ cells have been suggested to represent early TFH fate-committed cells residing outside of GCs and transitioning into mature GC-TFH.36 Consistent with this possibility, we found that cTFH during DSA responses (with or without ABMR) expressed LEF1 and TCF7, two transcription factors required for optimal TFH responses before GC formation.20,37 These cTFH also displayed a transcriptional program reminiscent of that of GC-TFH, including the expression of MAF, IL21R, CD40LG, and SLAMF6. Furthermore, the upregulation of IRF4 in these cTFH, a key transcription factor promoting T cell differentiation toward TFH lineage by inducing CXCR5, ICOS, and IL-21, strongly supports our conclusion that these cells represent GC-TFH precursors.38 Consistent with the above molecular features, these cTFH provided potent help to autologous B cells and induced their differentiation into ASCs that produced DSAs.
Although it is well established that DSA+ABMR+ patients harbor high levels of DSAs, predominantly of IgG3 isotype, with increased C1q-binding, little is known about the underlying TFH–B cell responses that distinguish such patients from their DSA+ABMR− counterparts. Our results suggest that enrichment in precursor Ki67+ICOS+ cTFH not only distinguishes the two types of patients, but also their states of cytokine polarization. DSA+ABMR+ patients exhibited an expansion of Th17-cTFH, which are known to have higher capacities to provide B cell help and display the most pathogenic potential in other inflammatory diseases, such as autoimmunity. This is consistent with the capacity of IL-17, in addition to IL-21, to promote class-switch recombination toward proinflammatory IgG3 isotypes.39
Concomitant with cTFH expansion, we identified proliferating ABCs in blood of patients developing DSAs and ABMR. These cognate cTFH and ABC responses were extensively coordinated, dynamic, and peaked at the time of rejection. Importantly, these vigorous responses were functionally important as they manifested in vitro in the generation of DSAs. This was likely because of increased numbers of donor-specific cells in the cTFH and memory B cell compartments in patients with ABMR. The stronger proliferative cTFH–B cell responses in vivo were also likely driven by enrichment in donor-specific cells, as they manifested in more pathogenic DSAs generated and increased histologic lesions. As activated cTFH could be detected months before rejection, this encourages future studies to perform systematic cellular monitoring. However, the predictive value of Ki67+ICOS+ and CCR7+CD127+ cTFH needs further validation in independent and larger cohorts of patients before they can be considered as valuable noninvasive biomarkers. In addition, quantifying T follicular regulatory cells and assessing their functional potential to regulate the cTFH and B cell responses in the context of ABMR would be of interest in future studies.
Our integrative PCA of cTFH and ABCs robustly distinguished the patient clinical states according to their transcriptional profiles. From a genomic standpoint, this reflects fundamental aspects of TFH and B cell transcriptional programs. First, the two cell types share the transcriptional determinants IRF4 and Bcl6, which could program similar states of gene expression reflected in shared receptors such as CXCR5 and IL-21R.40 Second, extensive and iterated interactions between cognate TFH and B cells, in GCs, results in mutually coordinated gene expression via their antigen receptors and other signaling systems. The upregulation of CD27 and ABLIM1 in cTFH and ABCs from DSA+ABMR+ patients was reflective of signaling and cytoskeleton modules that could function to promote memory and motility responses, respectively.41,42 In contrast, in DSA+ABMR− patients, cTFH and ABCs appeared to upregulate KLF9 and GID8, two components of the β-catenin/Wnt pathway, which prevents excessive lymphocyte proliferation and favors Th2 over Th17 polarization.43 Altogether, our integrative analyses of cTFH and ABCs at cellular and molecular levels establish the concept that these cells are reflective of a robust coordinated alloreactive response occurring in GCs.
Targeting cTFH–B cell interactions by belatacept has shown promising results in preventing the generation of DSAs and development of ABMR.44 On the basis of our data, we propose that other costimulatory molecules, such as ICOS, CD28, and CD40L, are also potential therapeutic targets. Of note, the abundant expression of CD28 on cTFH during ABMR, in contrast to the downmodulation of OX40, suggest that anti-CD28, but not OX40 antagonists, could efficiently suppress the deleterious cTFH–B cell responses.45,46 Importantly, combined anti-CD40 with belatacept could synergize to suppress cTFH–B cell response upon their detection in blood, and therefore be able to reverse ongoing DSA generation and ABMR.47
Disclosures
All authors have nothing to disclose.
Funding
This work was supported by National Institute of Health grants R21-AI116746 (to D. Metes), R01-AI130010 (to D. Metes), and 5T32AI074490-12 (to K. Louis) and the University of Pittsburgh’s Human Immunology Program at the Thomas E. Starzl Transplantation Institute.
Supplementary Material
Acknowledgments
We thank Tiange Shi and Avantika Srivastava for the help with flow cytometry data analysis, and Dr. Hariharan Sudaram, Michelle Lucas, Beth Elinoff, and David McMichael for help collecting clinical information.
Dr. Kevin Louis and Dr. Diana Metes designed the study. Dr. Kevin Louis, Dr. Camila Macedo, Marilyn Marrari, and Dr. Paul Fadakar performed the experiments. Dr. Kevin Louis, Dr. Elodie Bailly, Louis Lau, Dr. Bala Ramaswami, Dr. Douglas Landsittel, Dr. Uma Chandran, Alexandre Chang, Dr. Geetha Chalasani, and Dr. Masaki Yamada analyzed the data. Dr. Camila Macedo and Dr. Elodie Bailly provided clinical data. Dr. Parmjeet Randhawa provided the pathology data, Dr. Adriana Zeevi provided the tissue typing data and analyzed the donor-specific antibody results. Dr. Kevin Louis, Dr. Diana Metes, Dr. Harinder Singh, and Dr. Carmen Lefaucheur drafted and revised the paper. All authors approved the final version of the manuscript.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020030320/-/DCSupplemental.
Supplemental Figure 1. Study outline and cross-sectional blood samples of kidney transplant patients.
Supplemental Figure 2. Flow cytometry analyses of blood CD4+ T cells.
Supplemental Figure 3. Biaxial flow cytometry analyses of cTFH.
Supplemental Figure 4. Biaxial flow cytometry analyses of Ki67+ICOS+ compared with Ki67−ICOS− cTFH.
Supplemental Figure 5. Flow cytometry analyses of cTFH cytokine production in response to donor-antigen stimulation.
Supplemental Figure 6. Gating strategy for sorting cTFH, memory B cells and ABCs.
Supplemental Figure 7. Coculture analyses of cTFH with autologous memory B cells.
Supplemental Figure 8. Flow cytometry analyses of ABCs and ASCs.
Supplemental Figure 9. Correlation of cTFH, ABCs with DSAs.
Supplemental Figure 10. Flow cytometry analyses of cTFH polarization.
Supplemental Figure 11. Transcriptional profiling of cTFH and ABCs by hierarchical clustering and PCA.
Supplemental Figure 12. High-dimensional flow cytometry and t-SNE analyses of cTFH in individual patients.
Supplemental Table 1. Patient demographics.
Supplemental Table 2. Data and assay table.
Supplemental Table 3. Phenotypic patterns of cTFH cell clusters.
Supplemental Table 4. Affected canonical pathways in cTFH from DSA+ABMR+ versus DSA− patients predicted by IPA.
Supplemental Table 5. Upstream regulators of differentially expressed genes in cTFH from DSA+ABMR+ versus DSA− patients predicted by IPA.
Supplemental Table 6. DSA characteristics.
Supplemental Table 7. Antibodies.
References
- 1.Loupy A, Lefaucheur C: Antibody-mediated rejection of solid-organ allografts. N Engl J Med 379: 1150–1160, 2018. [DOI] [PubMed] [Google Scholar]
- 2.Louis K, Hertig A, Taupin J-L, Buob D, Jamme M, Brocheriou I, et al.: Markers of graft microvascular endothelial injury may identify harmful donor-specific anti-HLA antibodies and predict kidney allograft loss. Am J Transplant 19: 2434–2445, 2019. [DOI] [PubMed] [Google Scholar]
- 3.Lefaucheur C, Viglietti D, Bentlejewski C, Duong van Huyen J-P, Vernerey D, Aubert O, et al.: IgG donor-specific anti-human HLA antibody subclasses and kidney allograft antibody-mediated injury. J Am Soc Nephrol 27: 293–304, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Valenzuela NM, Reed EF: Antibody-mediated rejection across solid organ transplants: Manifestations, mechanisms, and therapies. J Clin Invest 127: 2492–2504, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crotty S: T follicular helper cell differentiation, function, and roles in disease. Immunity 41: 529–542, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Asrir A, Aloulou M, Gador M, Pérals C, Fazilleau N: Interconnected subsets of memory follicular helper T cells have different effector functions. Nat Commun 8: 847, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vella LA, Buggert M, Manne S, Herati RS, Sayin I, Kuri-Cervantes L, et al.: T follicular helper cells in human efferent lymph retain lymphoid characteristics. J Clin Invest 129: 3185–3200, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmitt N, Bentebibel S-E, Ueno H: Phenotype and functions of memory Tfh cells in human blood. Trends Immunol 35: 436–442, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chevalier N, Jarrossay D, Ho E, Avery DT, Ma CS, Yu D, et al.: CXCR5 expressing human central memory CD4 T cells and their relevance for humoral immune responses. J Immunol 186: 5556–5568, 2011. [DOI] [PubMed] [Google Scholar]
- 10.Bauquet AT, Jin H, Paterson AM, Mitsdoerffer M, Ho I-C, Sharpe AH, et al.: The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and TH-17 cells. Nat Immunol 10: 167–175, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kroenke MA, Eto D, Locci M, Cho M, Davidson T, Haddad EK, et al.: Bcl6 and Maf cooperate to instruct human follicular helper CD4 T cell differentiation. J Immunol 188: 3734–3744, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morita R, Schmitt N, Bentebibel S-E, Ranganathan R, Bourdery L, Zurawski G, et al.: Human blood CXCR5+CD4+ T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity 34: 108–121, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chhabra M, Alsughayyir J, Qureshi MS, Mallik M, Ali JM, Gamper I, et al.: Germinal center alloantibody responses mediate progression of chronic allograft injury. Front Immunol 9: 3038, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Macedo C, Hadi K, Walters J, Elinoff B, Marrari M, Zeevi A, et al.: Impact of induction therapy on circulating T follicular helper cells and subsequent donor-specific antibody formation after kidney transplant. Kidney Int Rep 4: 455–469, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haas M, Loupy A, Lefaucheur C, Roufosse C, Glotz D, Seron D, et al.: The Banff 2017 Kidney Meeting Report: Revised diagnostic criteria for chronic active T cell-mediated rejection, antibody-mediated rejection, and prospects for integrative endpoints for next-generation clinical trials. Am J Transplant 18: 293–307, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kotecha N, Krutzik PO, Irish JM: Web-based analysis and publication of flow cytometry experiments. Curr Protoc Cytom Chapter 10: Unit10.17, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van der Maaten L, Hinton G: Visualizing Data using t-SNE. J Mach Learn Res 9: 2579–2605, 2008 [Google Scholar]
- 18.Qiu P, Simonds EF, Bendall SC, Gibbs KD Jr., Bruggner RV, Linderman MD, et al.: Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat Biotechnol 29: 886–891, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Korin YD, Lee C, Gjertson DW, Wilkinson AH, Pham T-P, Danovitch GM, et al.: A novel flow assay for the detection of cytokine secreting alloreactive T cells: Application to immune monitoring. Hum Immunol 66: 1110–1124, 2005. [DOI] [PubMed] [Google Scholar]
- 20.Choi YS, Gullicksrud JA, Xing S, Zeng Z, Shan Q, Li F, et al.: LEF-1 and TCF-1 orchestrate T(FH) differentiation by regulating differentiation circuits upstream of the transcriptional repressor Bcl6. Nat Immunol 16: 980–990, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu T, Ji Y, Moseman EA, Xu HC, Manglani M, Kirby M, et al.: The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci Immunol 1: eaai8593, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Locci M, Havenar-Daughton C, Landais E, Wu J, Kroenke MA, Arlehamn CL, et al.; International AIDS Vaccine Initiative Protocol C Principal Investigators: Human circulating PD-1+CXCR3-CXCR5+ memory Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity 39: 758–769, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ellebedy AH, Jackson KJL, Kissick HT, Nakaya HI, Davis CW, Roskin KM, et al.: Defining antigen-specific plasmablast and memory B cell subsets in human blood after viral infection or vaccination. Nat Immunol 17: 1226–1234, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Havenar-Daughton C, Lindqvist M, Heit A, Wu JE, Reiss SM, Kendric K, et al.; IAVI Protocol C Principal Investigators: CXCL13 is a plasma biomarker of germinal center activity. Proc Natl Acad Sci U S A 113: 2702–2707, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishizawa S, Sakata-Yanagimoto M, Hattori K, Muto H, Nguyen T, Izutsu K, et al.: BCL6 locus is hypermethylated in angioimmunoblastic T-cell lymphoma. Int J Hematol 105: 465–469, 2017. [DOI] [PubMed] [Google Scholar]
- 26.Good KL, Tangye SG: Decreased expression of Kruppel-like factors in memory B cells induces the rapid response typical of secondary antibody responses. Proc Natl Acad Sci U S A 104: 13420–13425, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Loosdregt J, Coffer PJ: The role of WNT signaling in mature T cells: T cell factor is coming home. J Immunol 201: 2193–2200, 2018. [DOI] [PubMed] [Google Scholar]
- 28.Tambur AR, Campbell P, Claas FH, Feng S, Gebel HM, Jackson AM, et al.: Sensitization in transplantation: Assessment of risk (STAR) 2017 working group meeting report. Am J Transplant 18: 1604–1614, 2018. [DOI] [PubMed] [Google Scholar]
- 29.Alsughayyir J, Chhabra M, Qureshi MS, Mallik M, Ali JM, Gamper I, et al.: Relative frequencies of alloantigen-specific helper CD4 T cells and B cells determine mode of antibody-mediated allograft rejection. Front Immunol 9: 3039, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burghuber CK, Kwun J, Page EJ, Manook M, Gibby AC, Leopardi FV, et al.: Antibody-mediated rejection in sensitized nonhuman primates: Modeling human biology. Am J Transplant 16: 1726–1738, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Besouw NM, Mendoza Rojas A, Baan CC: The role of follicular T helper cells in the humoral alloimmune response after clinical organ transplantation. HLA 94: 407–414, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cano-Romero FL, Laguna Goya R, Utrero-Rico A, Gómez-Massa E, Arroyo-Sánchez D, Suárez-Fernández P, et al.: Longitudinal profile of circulating T follicular helper lymphocytes parallels anti-HLA sensitization in renal transplant recipients. Am J Transplant 19: 89–97, 2019. [DOI] [PubMed] [Google Scholar]
- 33.Danger R, Chesneau M, Delbos F, Le Bot S, Kerleau C, Chenouard A, et al.: CXCR5+PD1+ICOS+ circulating T follicular helpers are associated with de novo donor-specific antibodies after renal transplantation. Front Immunol 10: 2071, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Graav GN, Dieterich M, Hesselink DA, Boer K, Clahsen-van Groningen MC, Kraaijeveld R, et al.: Follicular T helper cells and humoral reactivity in kidney transplant patients. Clin Exp Immunol 180: 329–340, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He J, Tsai LM, Leong YA, Hu X, Ma CS, Chevalier N, et al.: Circulating precursor CCR7(lo)PD-1(hi) CXCR5+ CD4+ T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity 39: 770–781, 2013. [DOI] [PubMed] [Google Scholar]
- 36.Bentebibel S-E, Schmitt N, Banchereau J, Ueno H: Human tonsil B-cell lymphoma 6 (BCL6)-expressing CD4+ T-cell subset specialized for B-cell help outside germinal centers. Proc Natl Acad Sci U S A 108: E488–E497, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu T, Shin HM, Moseman EA, Ji Y, Huang B, Harly C, et al.: TCF1 is required for the T follicular helper cell response to viral infection. Cell Rep 12: 2099–2110, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmitt N, Liu Y, Bentebibel S-E, Ueno H: Molecular mechanisms regulating T helper 1 versus T follicular helper cell differentiation in humans. Cell Rep 16: 1082–1095, 2016. [DOI] [PubMed] [Google Scholar]
- 39.Mitsdoerffer M, Lee Y, Jäger A, Kim H-J, Korn T, Kolls JK, et al.: Proinflammatory T helper type 17 cells are effective B-cell helpers. Proc Natl Acad Sci U S A 107: 14292–14297, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ochiai K, Maienschein-Cline M, Simonetti G, Chen J, Rosenthal R, Brink R, et al.: Transcriptional regulation of germinal center B and plasma cell fates by dynamical control of IRF4. Immunity 38: 918–929, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Narahara H, Sakai E, Yamaguchi Y, Narahara S, Iwatake M, Okamoto K, et al.: Actin binding LIM 1 (abLIM1) negatively controls osteoclastogenesis by regulating cell migration and fusion. J Cell Physiol 234: 486–499, 2018. [DOI] [PubMed] [Google Scholar]
- 42.Xiao Y, Hendriks J, Langerak P, Jacobs H, Borst J: CD27 is acquired by primed B cells at the centroblast stage and promotes germinal center formation. J Immunol 172: 7432–7441, 2004. [DOI] [PubMed] [Google Scholar]
- 43.Reya T, O’Riordan M, Okamura R, Devaney E, Willert K, Nusse R, et al.: Wnt signaling regulates B lymphocyte proliferation through a LEF-1 dependent mechanism. Immunity 13: 15–24, 2000. [DOI] [PubMed] [Google Scholar]
- 44.Leibler C, Matignon M, Moktefi A, Samson C, Zarour A, Malard S, et al.: Belatacept in renal transplant recipient with mild immunologic risk factor: A pilot prospective study (BELACOR). Am J Transplant 19: 894–906, 2019. [DOI] [PubMed] [Google Scholar]
- 45.La Muraglia GM 2nd, Wagener ME, Ford ML, Badell IR: Circulating T follicular helper cells are a biomarker of humoral alloreactivity and predict donor-specific antibody formation after transplantation. Am J Transplant 20: 75–87, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ville S, Poirier N, Branchereau J, Charpy V, Pengam S, Nerriere-Daguin V, et al.: Anti-CD28 antibody and belatacept exert differential effects on mechanisms of renal allograft rejection. J Am Soc Nephrol 27: 3577–3588, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burghuber CK, Manook M, Ezekian B, Gibby AC, Leopardi FV, Song M, et al.: Dual targeting: Combining costimulation blockade and bortezomib to permit kidney transplantation in sensitized recipients. Am J Transplant 19: 724–736, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.