

Significance Statement
Genome-wide association studies (GWAS) are a powerful tool to identify genetic variants associated with CKD. However, knowledge of CKD-relevant target tissues and cell types important in the pathogenesis is incomplete. Integrating large-scale kidney function GWAS with gene expression datasets identified kidney and liver as the primary organs for kidney function traits. In the kidney, proximal tubule was the critical cell type for eGFR and urate, as well as for monogenic electrolyte or metabolic disease genes. Podocytes showed enrichment of genes implicated in glomerular disease. Compendia connecting traits, genes, and cell types allow further prioritization of genes in GWAS loci, enabling mechanistic studies.
Keywords: chronic kidney disease, genetics and development, gene expression, proximal tubule
Visual Abstract

Abstract
Background
Genetic variants identified in genome-wide association studies (GWAS) are often not specific enough to reveal complex underlying physiology. By integrating RNA-seq data and GWAS summary statistics, novel computational methods allow unbiased identification of trait-relevant tissues and cell types.
Methods
The CKDGen consortium provided GWAS summary data for eGFR, urinary albumin-creatinine ratio (UACR), BUN, and serum urate. Genotype-Tissue Expression Project (GTEx) RNA-seq data were used to construct the top 10% specifically expressed genes for each of 53 tissues followed by linkage disequilibrium (LD) score–based enrichment testing for each trait. Similar procedures were performed for five kidney single-cell RNA-seq datasets from humans and mice and for a microdissected tubule RNA-seq dataset from rat. Gene set enrichment analyses were also conducted for genes implicated in Mendelian kidney diseases.
Results
Across 53 tissues, genes in kidney function–associated GWAS loci were enriched in kidney (P=9.1E-8 for eGFR; P=1.2E-5 for urate) and liver (P=6.8·10-5 for eGFR). In the kidney, proximal tubule was enriched in humans (P=8.5E-5 for eGFR; P=7.8E-6 for urate) and mice (P=0.0003 for eGFR; P=0.0002 for urate) and confirmed as the primary cell type in microdissected tubules and organoids. Gene set enrichment analysis supported this and showed enrichment of genes implicated in monogenic glomerular diseases in podocytes. A systematic approach generated a comprehensive list of GWAS genes prioritized by cell type–specific expression.
Conclusions
Integration of GWAS statistics of kidney function traits and gene expression data identified relevant tissues and cell types, as a basis for further mechanistic studies to understand GWAS loci.
Genome-wide association studies (GWAS) are a powerful method to identify genetic variants associated with complex traits and diseases, but a systematic translation of associated gene loci into mechanistic insights has been challenging and is still lacking for the majority of loci.1 One reason that has complicated such translational efforts is that target tissues and cell types important in disease pathogenesis are partly unknown, both with respect to the global genetic architecture of complex diseases and traits as well as on the level of the individual gene.
The analytical integration of findings from GWAS of human traits and diseases with information on gene expression represents an attractive option to link two unbiased sources of evidence to connect genetic variation to the respective phenotypes.1–3 Advances in bulk tissue as well as single-cell RNA sequencing (scRNA-seq) and single-nucleus RNA sequencing (snRNA-seq)4–7 together with the establishment of community gene expression profiling efforts, such as the GTEx Project,8,9 and the availability of murine and human kidney single-cell transcriptomes10,11 offer new opportunities to highlight trait-relevant target tissues and cell types in an unbiased manner.
Such opportunities are of high interest in areas where few specific treatments exist and novel therapeutic options are lacking, such as in CKD. CKD affects 10% of the global adult population,12 is a leading cause of death,13 is treated insufficiently and largely symptomatically,14 and lags behind other disciplines with respect to the number of clinical trials.15 In this project, we therefore capitalize on the recent generation of powerful GWAS summary statistics from the CKDGen Consortium for kidney function–related traits—eGFR,16 urinary albumin-creatinine ratio (UACR),17 BUN,16 and serum urate.18
Our objective was to identify relevant target tissues in which the genes associated with these traits are highly expressed, followed by the subsequent identification of relevant kidney cell types for the respective traits as a whole and also at the level of the individual genes in order to pave the way for translational studies.
Methods
GWAS Summary Statistics
GWAS summary statistics of the four kidney function traits eGFR, UACR, BUN, and serum urate16–18 were downloaded from the CKDGen Consortium Data website (https://ckdgen.imbi.uni-freiburg.de). To match the European linkage disequilibrium (LD) score reference in linkage disequilibrium score regression for specifically expressed genes (LDSC-seg), we used the European ancestry–specific GWAS summary statistics. GWAS summary statistics for asthma (ICD10 code J45) from UK Biobank were downloaded from Gene Atlas,19 and those for schizophrenia from the CLOZUK + PGC2 Consortia20 were downloaded from the Walters group data repository (https://walters.psycm.cf.ac.uk/).
Genetic Correlations of GWAS Traits
Pairwise genetic correlations and single-nucleotide polymorphism (SNP)–based heritability of the six GWAS traits were estimated by LDSC21 software with the option “-rg.”22 LD scores were calculated for all SNPs that were part of the HapMap Project Phase 323 using the 1000 Genomes Project Phase 3 European population genotypes as the LD reference.24 The MHC region was excluded from the analysis due to its unusual LD pattern as recommended. A Bonferroni correction was applied to control for multiple testing.
Processing of Gene Expression Datasets
GTEx V7 RNA-sequencing (RNA-seq) data was downloaded as transcripts per million (TPM) from the GTEx Portal8 (https://gtexportal.org/home/). Genes for which fewer than four samples had at least one read count per million in the TPM matrix were filtered out. We used 53 tissues grouped by 30 categories as defined in the provided sample annotation file. Sample sizes of the tissue and tissue categories are available in Supplemental Table 1. Age and sex were extracted from the subject phenotype file and used as covariates in linear models.
Human kidney scRNA-seq data (Young) are available as supplemental data 1 of Young et al.11 This dataset was used as our main dataset and includes single-cell transcriptomes from a number of renal tumor samples as well as samples from healthy fetal, pediatric, adolescent, and adult kidneys. We used the data of 8707 cells in the bar-code group of normal epithelium and vasculature without the bulk of proximal tubule (PT) cells, which originated from five healthy adult kidneys. The authors’ R code (https://github.com/constantAmateur/scKidneyTumors) was adapted to process and normalize the data using the R package Seurat25 v2.3.4. The individual 10× sequencer channel information was corrected for by the batch correction algorithm “ComBat” while preserving zero expression values. Expression data were then normalized by the Seurat function “NormalizeData” with default parameters. Cluster assignment of the cells was taken from Supplemental Table 11.
Mouse kidney scRNA-seq data (Park) were downloaded as an RNA-seq count matrix of 43,745 kidney cells from seven mice from Gene Expression Omnibus10 (GEO; accession no. GSE107585). Data were processed with Seurat25 v2.3.4. Normalization was performed by the function “NormalizeData” with default parameters, and the Seurat function “removeBatchEffect” was used to correct for the batch variable “mouse identifiers.” Cluster assignment information was extracted from the cell identifiers.
Human kidney snRNA-seq data (Lake) were provided by Lake et al.26 This dataset from the Kidney Precision Medicine Project Consortium includes single-nucleus transcriptomes from tumor-free regions of nephrectomies and discarded deceased donor kidneys from 15 different individuals.26 The Seurat object file contained gene expression values of 17,659 single nuclei in 30 cell clusters that had passed quality control.
Human kidney snRNA-seq data (Wu) were downloaded as an RNA-seq count matrix of 4524 single nuclei from a healthy human adult from GEO27 (accession no. GSE118184). Data were processed using Seurat v2.3.4 as described previously.25 Normalization by the “NormalizeData” function was performed with default parameters. Assignment of cells to 17 cell clusters was provided by the authors.
Human kidney organoid scRNA-seq data originated from Wu et al.27 and were downloaded as a count matrix from GEO (accession no. GSE118184). Two subsets of the induced pluripotent stem cell organoid data were used: 18,072 cells with the Morizane protocol and 26,890 cells with the Takasato protocol. Processing of the datasets followed the same procedures as in the snRNA-seq data by Wu et al.27
Rat microdissected tubule scRNA-seq data (Lee) are available from supplemental dataset 1 of Lee et al.28 RNA-seq was used to profile gene expression in each of 14 microdissected renal tubule segments and glomeruli of rat kidneys. We imported data from the worksheet “RPKM_withPAseq,” which includes reads per kilobase of exon model per million mapped reads of 105 tubule samples and 5 glomeruli samples.
Mouse glomerulus scRNA-seq data (Karaiskos) were downloaded from https://shiny.mdc-berlin.de/mgsca/. Karaiskos et al.29 used scRNA-seq of 12,954 mouse glomerular cells to generate a normalized and filtered gene expression matrix with cluster information. We processed the data containing five major cell clusters using Seurat25 v2.3.4 and performed normalization using the same procedures as with the other single-cell datasets.
Cell cluster labels in each single-cell dataset were taken from the respective publication, with adjustment according to the nomenclature of Chen et al.30 For visualization of expression values as heat maps for GWAS-implicated genes, we transformed the average expression values across cell types to z scores. The transformation was applied to the output of the Seurat function “AverageExpression.” Heat maps were created using the Bioconductor R package ComplexHeatmap.31 We highlighted known monogenic kidney disease genes in heat maps on the basis of their categorization from Rasouly et al.32
Heritability Enrichment by LDSC-seg
We used LDSC-seg33 to define specifically expressed genes in each tissue or kidney cell type. In brief, t statistics for each gene were extracted from a linear model fit of gene expression on tissue or cell-type membership, where in the design matrix, the sample or cell was set to 1 when belonging to the focal tissue or cell type and −1 otherwise.
We used the TPM values for GTEx, normalized expression values of Seurat objects for single-cell datasets, and reads per kilobase of exon model per million mapped reads values for rat tubule data as the dependent variables. In GTEx data, we excluded samples in the same tissue category as the focal tissue when fitting the model (e.g., other brain regions when looking at a specific brain region) and included age and sex as covariates. In the single-cell and rat tubule datasets, each cell type was compared with all other cell types. We then ranked genes by their t statistics and selected the top 10% of genes of each GTEx tissue and kidney cell type to define a set of specifically expressed genes. Sensitivity analyses using the top 5% and 20% to define specifically expressed genes showed robustness of the results to the choice of the 10% threshold. The human homologs of mouse and rat genes were obtained by querying Ensembl Biomart.34
After the top 10% specifically expressed genes were obtained, LD score regression was performed by LDSC21 software for each GWAS summary statistic file in order to test whether trait heritability was enriched in regions surrounding these genes in each evaluated tissue or cell type. Annotation files were first constructed using the specifically expressed genes as input and adding 100 kb on both sides of the transcribed regions. A control annotation file was created for each expression dataset including all genes existing in the dataset. LD scores were then calculated for each annotation. Other parameters were the same as in the LDSC genetic correlation analyses. The magnitude and significance of per-SNP heritability enrichment were then tested for each annotation conditional on all genes’ annotation and the annotations in the baseline model v1.1. Enrichment was defined by the h2 estimate of the individual annotation divided by the proportion of SNPs contained within the annotation. The coefficient P value was calculated from the coefficient (τ) z score.
LDSC-seg analyses were performed for six GWAS traits. Genome-wide correlation analyses demonstrated the four kidney function traits being correlated with each other. Therefore, we defined the significance threshold using a Bonferroni correction for the number of tissues or cell types and the number of trait categories (kidney, asthma, schizophrenia). For example, in the GTEx dataset, the P-value cutoff was 3.1e-4 (0.05 per 53 tissues per three categories). Additionally, we defined a “suggestive” P-value threshold correcting for the number of tissues or cell types only (e.g., GTEx: 9.4e-4=0.05 per 53 tissues).
Gene Set Enrichment of Mendelian Kidney Disease Genes
We used the expression-weighted cell-type enrichment (EWCE) method implemented in the R package EWCE35 to test if the expression of known monogenic kidney disease genes inherited in a Mendelian fashion was enriched in certain kidney cell types. A list of 625 kidney disease genes was on the basis of supplement 3 of Rasouly et al.32 These genes sets were divided into seven subcategories: congenital or developmental, electrolytes or metabolic, glomerular, tubulointerstitial, cancer, genital, and secondary renal diseases. We tested the enrichment using all seven gene sets, although the last three sets do not represent primary Mendelian kidney disease genes. The specificity matrices were generated by the EWCE “generate.celltype.data” function in the human (Young) and mouse (Park) single-cell datasets, as well as two human kidney organoid datasets. Subsequently, EWCE was run with 100,000 bootstrap replicates controlling for transcript length and GC content. We performed multiple-tests correction using the Bonferroni procedure, accounted for the number of cell types and gene sets, and controlled the α at 0.05.
Projecting Cell Types across Four scRNA-Seq Datasets
The “scmapCluster” function in R package scmap36 (v1.4.1) was used to project cell-type annotations across the four single-cell kidney datasets. We started with a similarity threshold of 0.7 and decreased it in steps of 0.1 until the proportion of “unassigned” cells was <0.3 (lowest similarity threshold was 0.3). Sankey plots were created using the “plot” function in scmap. Because the projection was bidirectional, for each pair we plotted only the projection with the lower “unassigned” cells proportion.
Cell-Type Enrichment by Multimarker Analysis of GenoMic Annotation
As a secondary analysis, the R packages EWCE35 and Multimarker Analysis of GenoMic Annotation (MAGMA) Celltyping37 were used to calculate cell-type specificity of gene expression and its association with GWAS traits. MAGMA v1.0.738 was used to calculate gene-level association statistics using a window of 10 kb upstream and 1.5 kb downstream of each gene, accounting for LD, gene length, gene density, sample size, and minor allele count. Because the top 10% of specifically expressed genes were used for the heritability enrichment analyses in LDSC-seg, the “top 10% mode” of MAGMA was used to match these analyses.
Results
This project built on several datasets that have recently been generated and are on the basis of genetic data from up to 1 million individuals. Tables 1 and 2 display an overview of the investigated kidney function traits GWAS and gene expression datasets. Summary statistics from asthma19 and schizophrenia20 as traits that are not known to be connected to kidney function and disease were used as negative controls. Gene expression data of 53 human tissues and cell types were obtained from the GTEx Project (V7),9 as well as from scRNA-seq projects from primary kidney tissue of humans11,26,27 and mice,10,29 kidney organoids,27 and a microdissected tubule RNA-seq dataset from rats.28 The harmonized kidney cell types from human and mouse single-cell datasets are shown in Figure 1.
Table 1.
GWAS datasets used in the analyses
| Source | Phenotype | Sample Size | Reference: PMID |
|---|---|---|---|
| CKDGen | eGFR | 567,460 | 31152163 |
| CKDGen | BUN | 243,031 | 31152163 |
| CKDGen | UACR | 547,361 | 31511532 |
| CKDGen | Urate | 288,666 | 31578528 |
| UKB | Asthma | 28,628 patients; 423,636 controls | 30349118 |
| CLOZUK + PGC2 | Schizophrenia | 40,675 patients; 64,643 controls | 29483656 |
PMID, PubMed ID; UKB, UK Biobank.
Table 2.
Expression datasets used in the analyses
| Name | Species | Dataset Type | Sample Size | Reference: PMID |
|---|---|---|---|---|
| GTEx v7 | Human | Bulk RNA-seq | 11,688 samples, 714 donors | 25954001 |
| Park | Mouse | scRNA-seq | 43,745 cells (7 mice) | 29622724 |
| Young | Human | scRNA-seq | 8707 cells (5 individuals) | 30093597 |
| Lake | Human | snRNA-seq | 17,659 nuclei (15 individuals) | 31249312 |
| Wu | Human | snRNA-seq | 4524 nuclei (1 individual) | 30449713 |
| Lee | Rat | Bulk RNA-seq | 110 samples | 25817355 |
| Karaiskos | Mouse | scRNA-seq | 12,954 cells (8 mice) | 29794128 |
| Organoid (Morizane) | Human | scRNA-seq | 18,072 cells | 30449713 |
| Organoid (Takasato) | Human | scRNA-seq | 26,890 cells | 30449713 |
PMID, PubMed ID.
Figure 1.

Structure of nephron and glomerulus with cell-type labels. Labels colored in black are the cell types in the human single-cell dataset,11 and those colored in gray are the ones in mouse single-cell dataset.10 AVR, ascending vasa recta; B-lymph, B lymphocyte; CD-Trans, collecting duct transient cell; DCT, distal convoluted tubule; DVR, descending vasa recta; Endo, endothelial cell; Fib, fibroblast; GE, glomerular epithelium; IC, intercalated cell; IC-A, intercalated cell type A; IC-B, intercalated cell type B; LOH, loop of Henle; Macro, macrophage; MC, mesangial cell; NE, nephron epithelium; Neutro, neutrophil; NK, natural killer cell; novel, novel cell type; PC, principal cell; Podo, podocyte; T-lymph, T lymphocyte.
As a first step, pairwise genome-wide genetic correlations of the six investigated traits were determined (Methods), and they are summarized in Supplemental Table 2. Significant (P<0.003) genetic correlations were observed for five of six pairs of kidney function traits. eGFR was negatively correlated with BUN (rg=−0.31; P=3.8E-14) and serum urate (rg=−0.21; P=1.3E-9). The correlation with UACR was positive (rg=0.34; P=2.6E-33), which might be caused by UACR containing creatinine as its denominator and urinary albumin values in the numerator being generally low in the mostly population-based studies of the CKDGen Consortium. The observed genetic correlations are consistent with estimates in studies of the individual kidney function traits.16–18 Asthma showed a moderate genetic correlation with serum urate (rg=0.11; P=0.001), and schizophrenia was not correlated with any other trait.
We next applied LDSC-seg,33 which uses GWAS summary statistics to test for the presence of heritability enrichment of specifically expressed genes across human tissues, to identify kidney function–relevant tissues (Methods, Supplemental Table 3). Significant (P<2.4E-4) enrichment of heritability of eGFR was observed in regions around genes specifically expressed in kidney (P=9.1E-8; 2.2-fold enrichment) and liver (P=6.8E-5; 2.3-fold enrichment) (Figure 2). Significant enrichment in kidney tissue was also observed for BUN (P=1.2E-4; 1.9-fold enrichment) and serum urate (P=1.2E-5; 2.1-fold enrichment). There was no significant enrichment for UACR, but nominally significant heritability enrichment was observed in kidney, liver, and aorta (Figure 2). Heritability of schizophrenia, the negative control, was enriched in regions surrounding genes specifically expressed in multiple brain but no other tissues (Figure 2). Asthma did not show significant enrichment in any tissue.
Figure 2.
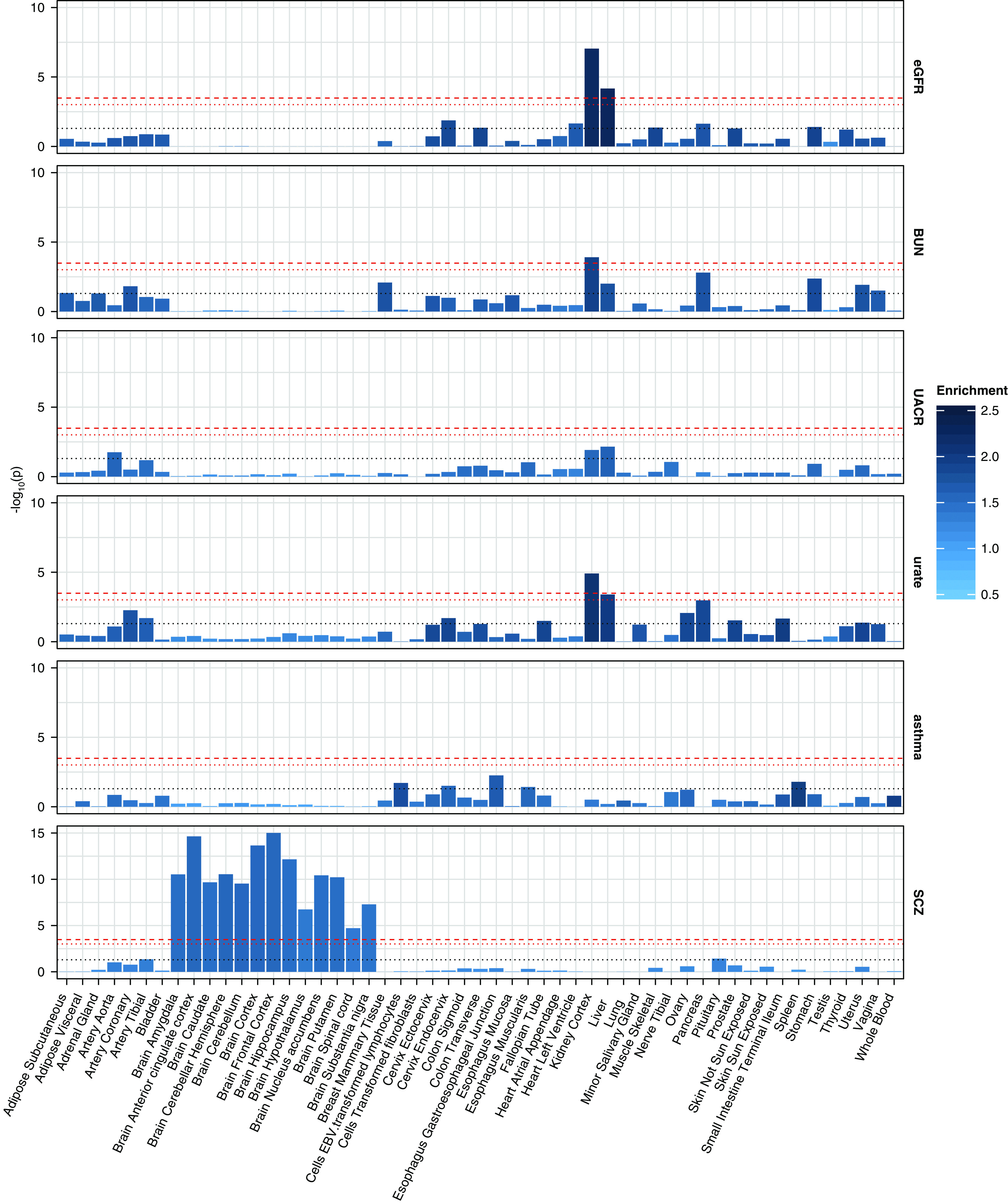
Heritability enrichment for six traits in GTEx tissues. Bar plots of −log10 (P values) for four kidney traits, asthma, and schizophrenia across GTEx tissues. The dashed red lines denote Bonferroni-adjusted significance (P=0.05/53/3=3.1E-4), the dotted red lines denote suggestive significance (P=0.05/53=9.4E-4), and the dotted gray lines denote nominal significance (P=0.05). The color intensity of the bars denotes the enrichment values. SCZ, schizophrenia.
Given the strong enrichment of heritability in the kidney, we next investigated which kidney cell types showed enriched heritability in genomic regions around genes that are highly expressed in specific cell types. Lists of specifically expressed genes as determined by a linear model fit of gene expression on tissue or cell-type membership using both a human scRNA-seq dataset11 and a murine scRNA-seq dataset10 are presented in Supplemental Tables 4 and 5, respectively. The validity of these lists is underscored by the presence of many positive controls, such as NPHS2 and NPHS1 as top genes for podocytes, LRP2 and CUBN for PT cells, and UMOD and several claudins for the loop of Henle. To facilitate linking this information to existing knowledge about monogenic kidney diseases, we annotated the presence and type of monogenic kidney disease32 for each gene and cell type. These annotations also showed cell-type specificity. For example, four of the ten most specifically expressed genes in the human distal tubule are known as monogenic kidney disease genes for electrolyte or metabolic disorders (Supplemental Table 4).
Investigating the four kidney function GWAS for heritability enrichment in the human kidney scRNA-seq dataset,11 we found significant (P<5.2E-4) heritability enrichment for eGFR and urate in a PT cluster PT1 (P=8.5E-5 and P=7.8E-6, respectively). Suggestive (P<2.3E-3) enrichment was also found for another PT cluster, PT2, for eGFR and urate, as well as in distal tubule cells for eGFR (Figure 3). Of note, a similar enrichment P value despite the considerably smaller sample size of the serum urate GWAS (n=288,666) than the eGFR GWAS (n=567,460) may suggest that PT has a more specific role in urate metabolism than in regulation of eGFR. Association statistics for BUN also looked similar, but showed no significantly enriched clusters, at a GWAS summary statistic sample size comparable with serum urate (n=243,031). Conversely, for UACR, the highest heritability enrichment was observed in glomerulus, podocyte, and PT. None of these were significant, but the finding reflects existing physiologic knowledge about albuminuria, with a role of the glomerulus in the filtration of albumin and the role of the PT in its subsequent receptor-mediated reabsorption.
Figure 3.
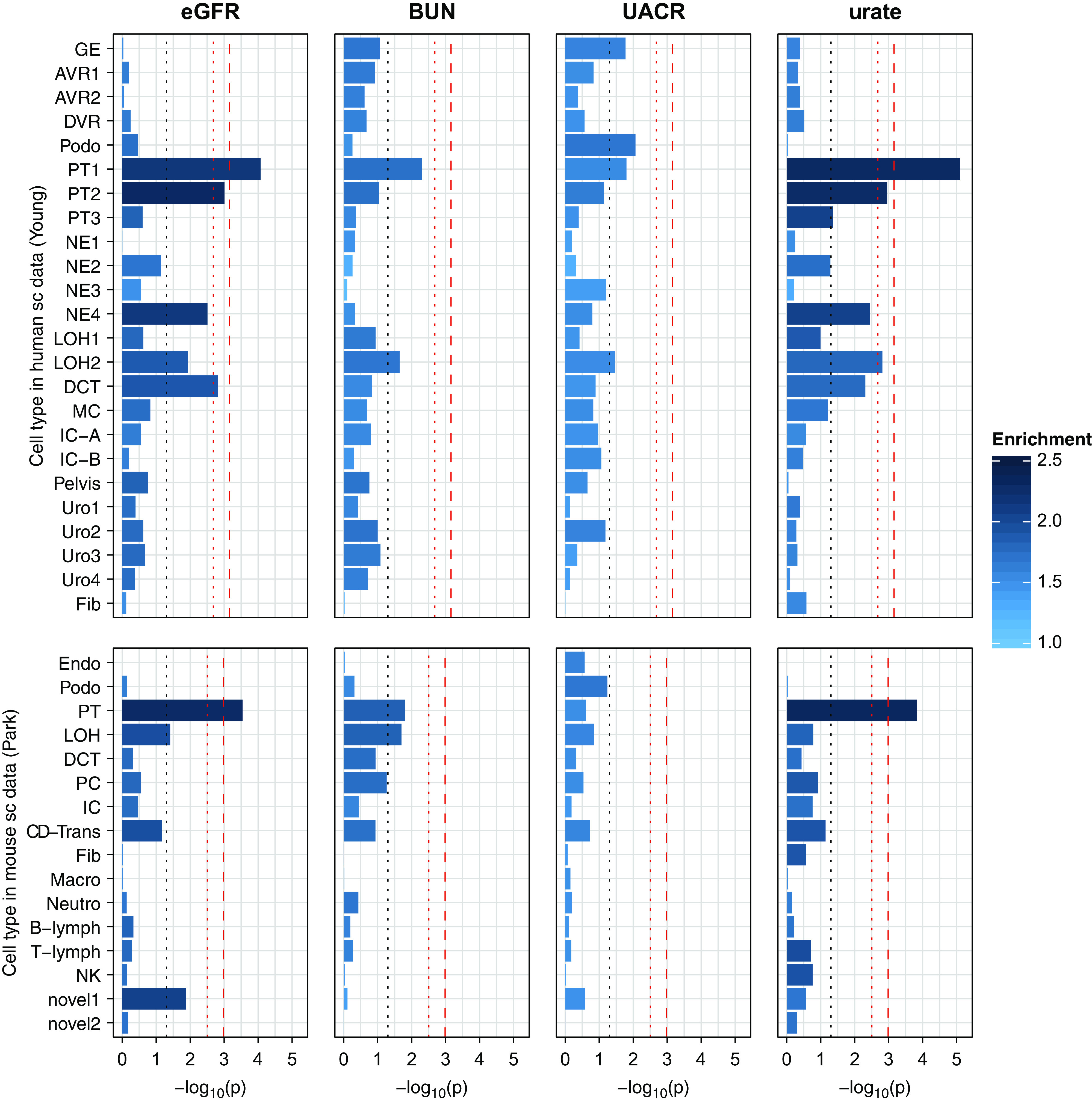
Heritability enrichment for kidney traits in human and mouse single-cell (sc) datasets. Bar plots of −log10 (P values) for four kidney traits across cell types in human and mouse sc datasets. The dashed red lines denote Bonferroni-adjusted significance (P=0.05/24/3=6.9E-4 in human and P=0.05/16/3=1.0E-3 in mouse), the dotted red lines denote suggestive significance (P=0.05/24=2.1E-3 and P=0.05/16=3.1E-3), and the dotted gray lines denote nominal significance (P=0.05). The color intensity of the bars denotes the enrichment values. AVR, ascending vasa recta; B-lymph, B lymphocyte; CD-Trans, collecting duct transient cell; DCT, distal convoluted tubule; DVR, descending vasa recta; Endo, endothelial cell; Fib, fibroblast; GE, glomerular epithelium; IC, intercalated cell; IC-A, intercalated cell type A; IC-B, intercalated cell type B; LOH, loop of Henle; Macro, macrophage; MC, mesangial cell; NE, nephron epithelium; Neutro, neutrophil; NK, natural killer cell; novel, novel cell type; PC, principal cell; Podo, podocyte; T-lymph, T lymphocyte.
Single-cell data from mouse kidney10 implicated the same cell types (Figure 3). Although only the PT met multiple testing–corrected criteria for significant enrichment, the consistency in implicated cell types indicates high homology of important genes across species. LDSC results for all traits are shown in Supplemental Tables 6 (human) and 7 (mouse). The results were robust to the choice of expression dataset: the use of two publicly available snRNA-seq datasets from humans26,27 confirmed very similar patterns of enrichment, with strongest results in the PT (Supplemental Figure 1, Supplemental Tables 8 and 9). In the snRNA-seq dataset by Wu et al.,27 the PT cells were separated into three clusters corresponding to segments 1–3 of the PT. Although the strongest enrichment was in segment 2 (P=1.5E-4) for eGFR, it was segment 1 for urate (P=1.3E-5).
The LDSC-seg results of the microdissected rat tubule data28 and a mouse glomerulus single-cell dataset29 (Supplemental Figure 2, Supplemental Tables 10 and 11) further confirmed these findings. In the rat tubule data, the PT segments showed significant enrichment for eGFR, thus providing evidence that our main findings can be recapitulated on the single functional unit of the kidney. In the mouse glomerulus data, tubule cells were enriched for BUN, eGFR, and urate, whereas their proportion was only 6%, indicating the robustness of the observed PT enrichment to cell cluster size.
No kidney cell types in any of the four single-cell and single-nucleus datasets showed evidence for heritability enrichment on the basis of the negative control schizophrenia. For asthma, a few suggestive enrichment signals were observed, including distal tubule in the human single-cell dataset (P=0.002), T lymphocyte in the mouse single-cell dataset (P=0.003), and macrophage in the human single-nucleus dataset by Wu et al.27 (P=5.9E-4). T lymphocytes and macrophages are important cell types in the etiology of asthma.39,40 However, the heritability enrichment in these cell types does not necessarily suggest that these immune cells in the kidney play important roles in asthma. A more reasonable explanation is that these cells have unique gene expression patterns so that the enrichment can be also detected in the kidney.
To further substantiate these results, we applied a complementary enrichment method37 that uses an MAGMA38-based approach. Different from the LDSC-seg, this method tests the association between gene-level statistics with cell-type specificity. After computing gene-level association statistics and estimating the cell-type specificity of each gene, specificity-based bins were tested for a positive association between the binned fractions and the gene-level association statistics in each cell type for all four kidney function traits in the human and the mouse single-cell datasets (Methods). There was a suggestive association for eGFR with the top 10% of genes specifically expressed in human PT cells (P=0.005 in PT2; P=0.01 in PT1). Their association with serum urate was also statistically significant (P=1E-5 and P=1.7E-3, respectively), further supporting the important and specific role of the PT in urate metabolism. Similar results were observed on the basis of murine gene expression, where PT cell specificity showed a significant association with eGFR and urate association statistics (P=5.6E-5 and P=2.3E-6, respectively). Altogether, these results demonstrate that the significantly enriched cell types can be detected by independent methods. Full MAGMA association results are summarized in Supplemental Figure 3 and Supplemental Tables 12 (human) and 13 (mouse).
GWAS of eGFR, UACR, and serum urate have implicated genes underlying the association signals by functional annotation combined with statistical fine mapping or colocalization analyses.16–18 This process resulted in lists of genes most likely to causally influence the studied traits, which may not be the genes closest to the SNPs with the strongest association signals. We therefore examined the expression patterns of these genes in human and murine kidney cell types. A strong expression signal was observed for many eGFR genes in PT cells in humans (Figure 4) and mice (Supplemental Figure 4), in agreement with the findings from the heritability enrichment analyses. UACR-associated genes were highly expressed in podocytes (Figure 4, Supplemental Figure 4). The benefit of examining gene expression in multiple single-cell datasets from different species is illustrated by the example of CACNA1S, which shows high expression in B lymphocytes in mice but not in B lymphocytes or any other cell types in human kidney. Similar to eGFR genes, urate-associated genes showed a strong expression signal in PT cells (Supplemental Figure 5 for human and Supplemental Figure 6 for mice), consistent with the heritability enrichment findings for urate.
Figure 4.

Cell type–specific expression of eGFR and UACR genes in the human single-cell dataset. Heat map showing expression of genes implicated in eGFR (upper) and UACR (lower) across cell types (x axis) in the human single-cell dataset. The expression values are z-score transformed. Genes known to underlie primary monogenic kidney diseases are colored in blue. AVR, ascending vasa recta; DCT, distal convoluted tubule; DVR, descending vasa recta; Fib, fibroblast; GE, glomerular epithelium; IC-A, intercalated cell type A; IC-B, intercalated cell type B; LOH, loop of Henle; MC, mesangial cell; NE, nephron epithelium; Podo, podocyte.
To help prioritize genes in the eGFR and UACR GWAS loci, we built candidate lists containing all genes within a 500-kb window around each locus’ index SNP. For these genes, we list the cell types (if any) where the gene appears in the top 10% specifically expressed gene list (Supplemental Table 14). For example, rs10846157 is an index SNP identified in GWAS of eGFR16 (P=8.3E-21). Although the closest gene is RERG, using this approach prioritizes the neighboring gene PTPRO. PTPRO is specifically expressed in podocytes and well known as a cause of monogenic steroid-resistant nephrotic syndrome (OMIM #614196). This systematic approach results in a comprehensive list of GWAS genes prioritized by cell type–specific expression.
Complementary to genes implicated by GWAS, we next asked whether certain gene sets known to cause Mendelian forms of kidney disease32 were expressed at higher levels within a particular cell type than expected by chance, using the EWCE method35 (Methods). To this end, 625 genes previously reported to be connected to Mendelian kidney disease32 were divided into seven subcategories with a focus on four primary kidney disease categories, namely congenital or developmental, electrolytes or metabolic, glomerular, and tubulointerstitial. In the human single-cell gene expression dataset, five enriched cell clusters were identified for genes implicated in monogenic electrolyte or metabolic disease (Figure 5, Supplemental Table 15), including PT, nephron epithelium, and loop of Henle (three of them with P<1E-5). Of these, PT was confirmed in the mouse dataset (Supplemental Figure 7, Supplemental Table 16). For single-gene causes of glomerular diseases, podocyte was highly enriched (P<1E-5 in human and mouse). Fibroblast showed an unexpected enrichment in congenital or developmental diseases in human (P=0.001) but not in mouse (P=0.94). In order to identify cell types related to congenital or developmental disease genes without the presence of nonkidney cell types, we additionally performed EWCE using two kidney organoid datasets from Wu et al.27: 18,072 cells obtained with the Morizane protocol and 26,890 cells with the Takasato protocol. For congenital and developmental disease genes, we detected enrichment in clusters belonging to mesenchymal stem cells in both datasets (Supplemental Figures 8 and 9, Supplemental Tables 17 and 18), which is consistent with existing knowledge about developmental diseases of the kidney. In summary, EWCE analyses correctly identified the effector cell type of genes underlying Mendelian diseases of electrolytes or metabolic as well as glomerular disorders, thereby validating the categorization of disease genes.
Figure 5.
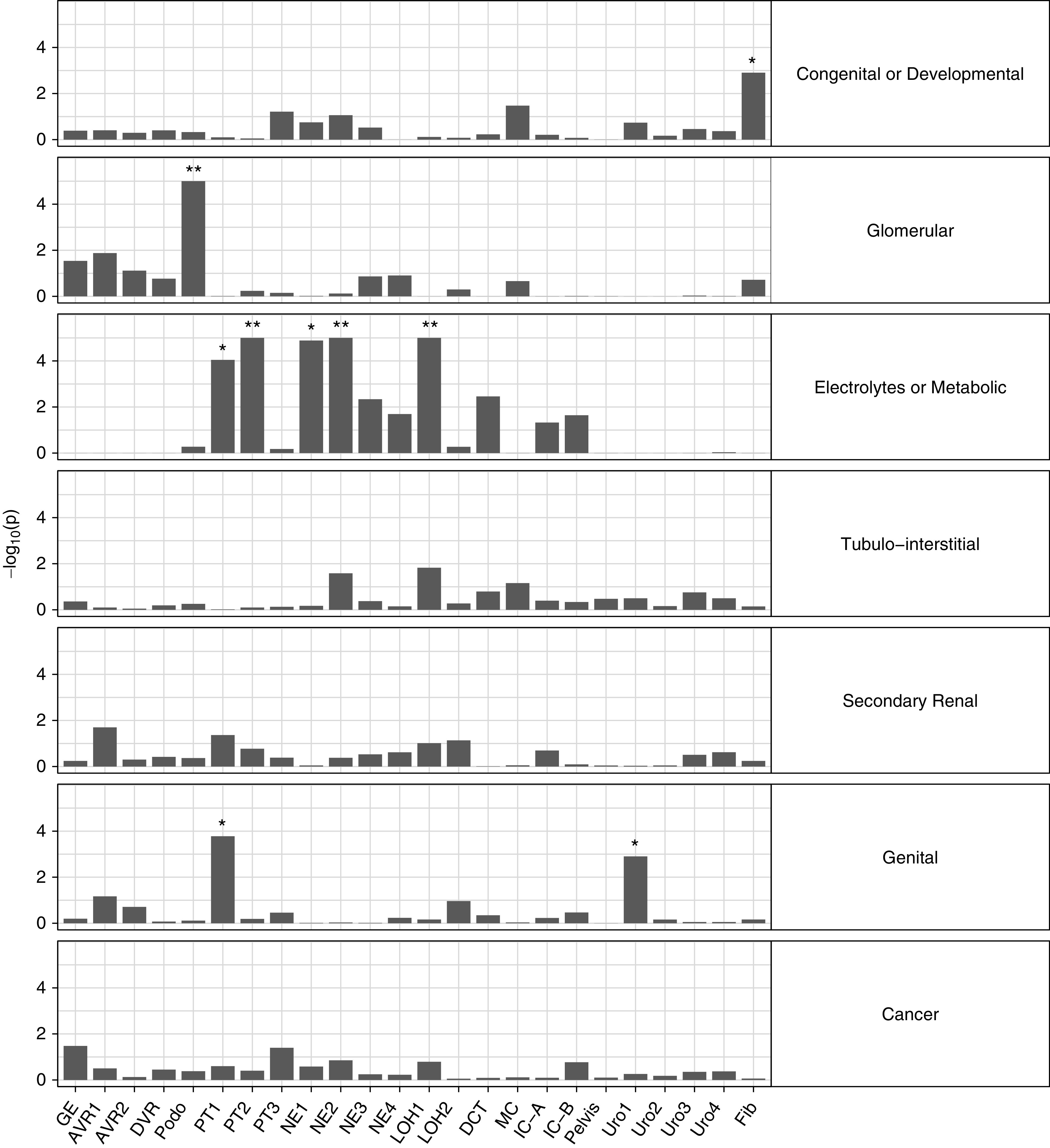
Gene set enrichment analyses for Mendelian kidney genes in the human single-cell dataset. Gene set enrichment analyses for seven Mendelian kidney disease gene sets in the human single-cell dataset. The y axis indicates −log10 (P values). AVR, ascending vasa recta; DCT, distal convoluted tubule; DVR, descending vasa recta; Fib, fibroblast; GE, glomerular epithelium; IC-A, intercalated cell type A; IC-B, intercalated cell type B; LOH, loop of Henle; MC, mesangial cell; NE, nephron epithelium; Podo, podocyte. *Bonferroni significant; **Bonferroni significant and P<1E-5.
The six kidney RNA-seq datasets originated from different species (human, mouse, and rat) and different technologies (scRNA-seq, snRNA-seq, and bulk RNA-seq). In order to investigate to what extent the cell-type assignment was consistent across the four kidney single-cell datasets, we systematically projected cell types from one dataset onto the cell types identified in a different dataset on the basis their gene expression values36 (Methods). As shown in the Sankey plots (Supplemental Figure 10), cell-type annotations from the human and mouse single-cell datasets aligned well. For instance, PT cells in mice overwhelmingly matched to the three PT clusters (PT1, PT2, and PT3) in humans. On the other hand, the human scRNA-seq11 dataset and the snRNA-seq dataset by Wu et al.27 did not match very well using the default similarity threshold of 0.7 (Methods), as reflected by the proportion of “unassigned” cells. Lowering the similarity threshold to 0.3 resulted in a much better match between cell types, such as for PT, collecting duct, and distal tubule. It is worth noting that in this projection, the PT1 cluster in the scRNA-seq dataset predominately matched the segments 1–3 of the PT in the snRNA-seq dataset by Wu et al.27, which was consistent with the observed enrichments for eGFR and urate in the respective dataset. Cell-type matching between human single-nucleus26,27 and mouse single-cell gene expression10 was also improved at a similarity threshold of 0.3. Along the same lines, the projection between two human single-nucleus datasets26,27 did not work well at a threshold of 0.7 but was much improved for almost all cell types using a threshold of 0.4. Notably, there were inconclusive projection results, such as the comparison of a recently generated human single-nucleus gene expression dataset (Lake26) with the mouse10 dataset or with the human single-cell dataset.11 Because of biologic variability, technical biases, and methods used for data processing and cell-type assignment in each single-cell experiment, the comparison of cell types across datasets may sometimes provide different answers. Nevertheless, the comparison illustrates the gain in evaluating several single-cell and single-nucleus datasets to comprehensively address a study question and revealed interesting insights into the comparability of cell-type assignment across datasets.
Discussion
GWAS of kidney function traits have identified hundreds of associated loci.16–18 However, translation of the associated genetic loci into genes, relevant cell types, and ultimately, mechanistic insights is still a challenging task. By leveraging GWAS summary statistics and gene expression data, we identified kidney function trait-related target tissues and kidney cell types. At the tissue level, enriched heritability was found in eGFR, BUN, and serum urate–associated genetic regions highly expressed in the kidney as well as in liver. The enrichment in both kidney and liver may be driven by shared highly expressed genes in both organs. Shared expression patterns may reflect the fact that kidney and liver are both organs with a high proportion of epithelial cells that may share transcription programs. Indeed, Lindgren et al.41 reported that HNF1A and HNF4A are critical transcription factors regulating gene expression in both liver and PT cells. More generally, many solute carriers and enzymes exhibit shared expression in both organs. Thus, kidney function traits showed tissue-specific signatures of trait-associated genes, with unbiased transcriptomics data recapitulating existing biologic knowledge about the physiology underlying the traits of interest.
Within the kidney, PT cells were implicated as the relevant cell type for eGFR and serum urate. Moreover, gene set enrichment analyses showed that known single-gene causes of glomerular diseases were highly enriched in podocytes, and electrolyte or metabolic disease genes were enriched in PT cells.
One example for target cell-type prioritization of a GWAS-implicated gene is KNG1. Our analyses show that this gene is highly and specifically expressed in the loop of Henle and was in fact the most specifically expressed gene in this cell type in the human dataset. The index SNP rs11919484 identified in GWAS of eGFR16 is upstream of KNG1. KNG1 encodes for high-molecular weight kininogen, which is part of the kallikrein-kinin system. This system is the target of multiple pharmacologic interventions to lower BP, underscoring the importance of the implicated cell types in BP control. Kininogen is cleaved to bradykinin, which in turn, is linked to kidney function and influences BP, natriuresis, and diuresis via the renin-angiotensin-aldosterone system.42 Another interesting example is TCF21, which is highly specifically expressed in podocytes. The eGFR GWAS index SNP rs3822939 maps close to the gene EYA4, but our analyses prioritize TCF21. Although this gene has not been found to be associated with monogenic kidney disease, it has been shown to be important for kidney development in knockout mice.43 Hypomorphic variants could result in reduced nephron number and/or function, providing a potential link to low eGFR.
Heritability enrichments from human and murine scRNA-seq datasets showed very good agreement across species. Furthermore, two additional human snRNA-seq datasets as well as a kidney tubule segments RNA-seq dataset from rat demonstrated consistent results. Moreover, the findings were confirmed by a second independent enrichment method using linear regression models to correlate gene expression cell-type specificity and gene-level kidney function trait associations. Lastly, the Bonferroni correction we applied was overconservative because many of the cell types within the datasets were related to each other. In total, the tissue- and cell type–specific enrichments that we observed were robust to the choice of species, datasets, and methods, and therefore, they likely represent true positive findings.
Many scRNA-seq datasets across organs from humans and mice have become publicly available over the last few years. Together with genome-wide GWAS summary statistics, they allow for the investigation of cell type–specific expression of implicated genes and eventually, for identifying relevant cell types underlying the studied traits.33,44–48 For kidney traits, Park et al.10 first reported that CKD-associated genes are enriched in PTs by using a z-score enrichment approach. Qiu et al.49 generated tubular and glomerular eQTL data to identify novel genes for CKD and found enrichment of these genes for PT expression. In two recent kidney function GWAS, the expression of implicated genes was profiled across kidney cell types.50,51 The first study used a murine dataset; the second used a dataset derived from a single human kidney. Differences in prioritized genes in this study and previously published studies may be attribute to the better-powered GWAS summary statistics in this study, as well as from differences in causal gene assignment, species, and technical and analytical variability. Still, the results were largely consistent.
Watanabe et al.52 collected 43 publicly available scRNA-seq datasets from humans and mice and tested association of cell types with 26 well powered GWAS using a cell-type specificity regression workflow similar to the one used here. In their study, CKD was included as the only kidney function–related trait, and despite the inclusion of kidney cells, no significant enrichment in any cell type was detected. One possible reason is that the included kidney datasets were from the Mouse Cell Atlas53 and Tabula Muris Consortium,54 and kidney cells constituted only a small proportion of both datasets. Also, the included GWAS of CKD55 was not well powered. Thus, the overall power to detect associations may have been limited.
A very recent GWAS of 38 blood and urine biomarkers in 358,072 participants of the UK Biobank (Sinnott-Armstrong N, et al. [2019] Genetics of 38 blood and urine biomarkers in the UK Biobank. bioRxiv:660506) identified 1857 associated loci. Included kidney biomarkers were creatinine, cystatin C, UACR, urate, and eGFR. Heritability enrichment was tested on the basis of marker gene lists in specific cell types of kidney, liver, and pancreas. In line with our results, enrichments were found for kidney traits in podocytes and tubule cells. Our study focuses on kidney function traits using additional methods as well as several scRNA-seq datasets across organisms and from primary tissue, microdissected tissues, and organoids.
For UACR, we were able to prioritize kidney tissue of 52 other GTEx tissues. Within the kidney, podocytes and PT cells were nominally enriched for UACR. Among the individual genes prioritized as most likely to be causal in the original publication,17 PRKCI was found to be highly and specifically expressed in podocytes. The gene product, a serin/threonine protein kinase, is important in actin cytoskeletal regulation in podocytes.56 A podocyte-specific deletion in mice resulted in nephrotic syndrome,57 and knockdown of PRKCI orthologs in Drosophila nephrocytes resulted in disrupted architecture of slit diaphragm orthologs.17 Furthermore, CUBN was the first gene reported in GWAS of UACR,58 and it was found to be expressed in PT, where the encoded protein is important for the endocytosis of filtered albumin.59 Both Cubulin-knockout mice60 and patients with rare disruptive mutations in the gene61 exhibit proteinuria.
Our findings on the basis of the integration of unbiased data sources are consistent with current biologic knowledge by highlighting the well known role of the PT in urate transport62 and of glomerulum and particularly, podocytes in albuminuria.63 The ability to implicate relevant cell types at a single-gene level is illustrated by the examples above and may help to detect the cell-type origin and to inform mechanistic studies for newly discovered kidney disease risk genes with little preexisting knowledge.
The strengths of our study are in the unbiased input datasets and their examination from different angles: the well powered and very recent GWAS summary statistics of different important kidney traits on the one hand and bulk as well as single-cell gene expression data across species and sequencing technologies on the other hand. In addition, we used two different enrichment methods and incorporated negative controls. We found consistent results supported by existing biologic knowledge and provide comprehensive lists as a resource to the community that can aid post-GWAS follow-up experiments. The restriction to GWAS of European ancestry individuals limits the extrapolation of our findings to groups of non-European ancestry. Differences in cell cluster assignment between the scRNA-seq and snRNA-seq datasets might limit the comparability of cell-type enrichment, but the projection of cell-type annotations across datasets showed good agreement. Furthermore, the power to detect enrichment in rare kidney cell types is limited. This will be alleviated by the future availability of large-scale single-cell data from global initiatives, such as the Human Cell Atlas.64 Although it has been shown that increasing the sample size of sequenced cells increases the significance of the enrichment on average,33 we did not find evidence suggesting that cluster size acted as a confounding factor. Although PT was the dominant cell type in the mouse data,10 the human data that we used11 were from a subcompartment that excluded the mass of proximal tubular cells. We cannot, however, exclude that true enrichment could be missed because of limited power for rare cell types. Lastly, the association of fibroblasts detected with congenital and developmental diseases in one of the human scRNA-seq datasets might be the result of misclassification of these cells. Independent experiments with two kidney organoid datasets lacking fibroblasts showed biologically plausible enrichment in mesenchymal stem cells instead.
The presented heritability enrichment analyses assume a predominant genetic contribution from tissue- or cell type–specific genes. However, it is also conceivable that transcripts not confined to a specific tissue or cell type contribute to the traits of interest. Indeed, colocalization analyses of GWAS and eQTLs support the presence of both signals that colocalize very specifically only with tubular or glomerular eQTLs and signals that show colocalization in many tissues and organs. Lastly, epigenetic modifications are known to contribute to cell type–specific expression. The integrative analyses of epigenetic data are likely to become informative in the future after well powered and—importantly—comparable epigenetic datasets across a wide range of human tissues with matching gene expression data become available.
In summary, the integration of GWAS summary statistics and gene expression data systematically linked genes in kidney function–associated GWAS loci to specific tissues and cell types. These findings will help experimental studies subsequent to GWAS by prioritizing the potentially relevant cell type for each trait-associated gene.
Disclosures
All authors have nothing to disclose.
Funding
The work of A. Köttgen was supported by Deutsche Forschungsgemeinschaft grants KO 3598/3-1 and KO 3598/5-1. The work of A. Köttgen and P. Schlosser was supported by Deutsche Forschungsgemeinschaft grant CRC 992 (to A. Köttgen). The work of A. Köttgen and M. Wuttke was supported by Deutsche Forschungsgemeinschaft grants CRC 1140 (to A. Köttgen) and 246781735 (to A. Köttgen). Y. Li was supported by Deutsche Forschungsgemeinschaft grant KO 3598/4-1 (to A. Köttgen).
Supplementary Material
Acknowledgments
We thank the CKDGen Consortium for making GWAS results publicly available, and our thanks also go to CKDGen Consortium collaborators, all study investigators, personnel, and patients.
Prof. Anna Köttgen, Dr. Yong Li, and Dr. Matthias Wuttke designed research; Dr. Cristian Pattaro, Dr. Pascal Schlosser, Dr. Alexander Teumer, and Dr. Adrienne Tin provided essential data; Dr. Stefan Haug, Dr. Yong Li, and Dr. Matthias Wuttke performed data analysis; Prof. Anna Köttgen, Dr. Yong Li, and Dr. Matthias Wuttke wrote the manuscript; and all authors read and approved the final manuscript.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020010051/-/DCSupplemental.
Supplemental Figure 1. Heritability enrichment results of four kidney traits, asthma, and schizophrenia in the human snRNA-seq dataset Lake.
Supplemental Figure 2. Heritability enrichment results of four kidney traits in the rat tubule dataset Lee and the mouse glomerulus scRNA-seq dataset Karaiskos.
Supplemental Figure 3. MAGMA cell-type association results of four kidney traits in human and mouse scRNA-seq datasets.
Supplemental Figure 4. Cell type–specific expression of eGFR- and UACR-associated genes most likely to be causal in the mouse scRNA-seq dataset.
Supplemental Figure 5. Cell type–specific expression of serum urate–associated genes most likely to be causal in the human scRNA-seq dataset.
Supplemental Figure 6. Cell type–specific expression of genes implicated in serum urate in the mouse scRNA-seq dataset.
Supplemental Figure 7. Gene sets enrichment results of Mendelian kidney disease genes in the mouse scRNA-seq dataset.
Supplemental Figure 8. Gene sets enrichment results of Mendelian kidney disease genes in the human organoid scRNA-seq dataset Morizane protocol.
Supplemental Figure 9. Gene sets enrichment results of Mendelian kidney disease genes in the human organoid scRNA-seq dataset Takasato protocol.
Supplemental Figure 10. Projection of assigned cell-type clusters across four single-cell and single-nucleus datasets by scmap.
Supplemental Table 1. Sample sizes of GTEx tissue and tissue categories.
Supplemental Table 2. Genetic correlations and heritability of six GWAS traits.
Supplemental Table 3. LDSC-seg results of six traits in GTEx tissues.
Supplemental Table 4. Specifically expressed genes in the human scRNA-seq dataset (Young).
Supplemental Table 5. Specifically expressed genes in the mouse scRNA-seq dataset (Park).
Supplemental Table 6. LDSC-seg results of six traits in the human scRNA-seq dataset (Young).
Supplemental Table 7. LDSC-seg results of six traits in the mouse scRNA-seq dataset (Park).
Supplemental Table 8. LDSC-seg results of six traits in the human snRNA-seq dataset (Lake).
Supplemental Table 9. LDSC-seg results of six traits in the human snRNA-seq dataset (Wu).
Supplemental Table 10. LDSC-seg results of four kidney traits in the microdissected rat tubule RNA-seq dataset (Lee).
Supplemental Table 11. LDSC-seg results of four kidney traits in the mouse glomerulus scRNA-seq data (Karaiskos).
Supplemental Table 12. MAGMA association results of kidney function traits in the human scRNA-seq dataset (Young).
Supplemental Table 13. MAGMA association results of kidney function traits in the mouse scRNA-seq dataset (Park).
Supplemental Table 14. Prioritization of genes in GWAS loci using expression specificity results.
Supplemental Table 15. Gene set enrichment results of kidney disease genes in the human scRNA-seq dataset (Young).
Supplemental Table 16. Gene set enrichment results of kidney disease genes in the mouse scRNA-seq dataset (Park).
Supplemental Table 17. Gene set enrichment results of kidney disease genes in the human organoid scRNA-seq dataset Morizane protocol.
Supplemental Table 18. Gene set enrichment results of kidney disease genes in the human organoid scRNA-seq dataset Takasato protocol.
References
- 1.Albert FW, Kruglyak L: The role of regulatory variation in complex traits and disease. Nat Rev Genet 16: 197–212, 2015. [DOI] [PubMed] [Google Scholar]
- 2.Tam V, Patel N, Turcotte M, Bossé Y, Paré G, Meyre D: Benefits and limitations of genome-wide association studies. Nat Rev Genet 20: 467–484, 2019. [DOI] [PubMed] [Google Scholar]
- 3.Hekselman I, Yeger-Lotem E: Mechanisms of tissue and cell-type specificity in heritable traits and diseases. Nat Rev Genet 21: 137–150, 2020. [DOI] [PubMed] [Google Scholar]
- 4.Saliba A-E, Westermann AJ, Gorski SA, Vogel J: Single-cell RNA-seq: Advances and future challenges. Nucleic Acids Res 42: 8845–8860, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jaitin DA, Kenigsberg E, Keren-Shaul H, Elefant N, Paul F, Zaretsky I, et al. : Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 343: 776–779, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zeisel A, Muñoz-Manchado AB, Codeluppi S, Lönnerberg P, La Manno G, Juréus A, et al. : Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347: 1138–1142, 2015. [DOI] [PubMed] [Google Scholar]
- 7.Park J, Liu CL, Kim J, Susztak K: Understanding the kidney one cell at a time. Kidney Int 96: 862–870, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.GTEx Consortium : The Genotype-Tissue Expression (GTEx) project. Nat Genet 45: 580–585, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.GTEx Consortium : Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 348: 648–660, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park J, Shrestha R, Qiu C, Kondo A, Huang S, Werth M, et al. : Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 360: 758–763, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Young MD, Mitchell TJ, Vieira Braga FA, Tran MGB, Stewart BJ, Ferdinand JR, et al. : Single-cell transcriptomes from human kidneys reveal the cellular identity of renal tumors. Science 361: 594–599, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eckardt K-U, Coresh J, Devuyst O, Johnson RJ, Köttgen A, Levey AS, et al. : Evolving importance of kidney disease: From subspecialty to global health burden. Lancet 382: 158–169, 2013. [DOI] [PubMed] [Google Scholar]
- 13.GBD 2016 Causes of Death Collaborators : Global, regional, and national age-sex specific mortality for 264 causes of death, 1980-2016: A systematic analysis for the global burden of disease study 2016 [published correction appears in Lancet 390: e38, 2017]. Lancet 390: 1151–1210, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levin A, Tonelli M, Bonventre J, Coresh J, Donner JA, Fogo AB, et al. ; ISN Global Kidney Health Summit participants : Global kidney health 2017 and beyond: A roadmap for closing gaps in care, research, and policy. Lancet 390: 1888–1917, 2017. [DOI] [PubMed] [Google Scholar]
- 15.Inrig JK, Califf RM, Tasneem A, Vegunta RK, Molina C, Stanifer JW, et al. : The landscape of clinical trials in nephrology: A systematic review of. Am J Kidney Dis 63: 771–780, 2014. Available at: Clinicaltrials.gov [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wuttke M, Li Y, Li M, Sieber KB, Feitosa MF, Gorski M, et al. ; Lifelines Cohort Study; V. A. Million Veteran Program : A catalog of genetic loci associated with kidney function from analyses of a million individuals. Nat Genet 51: 957–972, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Teumer A, Li Y, Ghasemi S, Prins BP, Wuttke M, Hermle T, et al. : Genome-wide association meta-analyses and fine-mapping elucidate pathways influencing albuminuria. Nat Commun 10: 4130, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tin A, Marten J, Halperin Kuhns VL, Li Y, Wuttke M, Kirsten H, et al. ; German Chronic Kidney Disease Study; Lifelines Cohort Study; V. A. Million Veteran Program : Target genes, variants, tissues and transcriptional pathways influencing human serum urate levels. Nat Genet 51: 1459–1474, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Canela-Xandri O, Rawlik K, Tenesa A: An atlas of genetic associations in UK Biobank. Nat Genet 50: 1593–1599, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pardiñas AF, Holmans P, Pocklington AJ, Escott-Price V, Ripke S, Carrera N, et al. ; GERAD1 Consortium; CRESTAR Consortium : Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat Genet 50: 381–389, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bulik-Sullivan BK, Loh PR, Finucane HK, Ripke S, Yang J, Patterson N, et al. ; Schizophrenia Working Group of the Psychiatric Genomics Consortium : LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 47: 291–295, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, et al. ; ReproGen Consortium; Psychiatric Genomics Consortium; Genetic Consortium for Anorexia Nervosa of the Wellcome Trust Case Control Consortium 3 : An atlas of genetic correlations across human diseases and traits. Nat Genet 47: 1236–1241, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.International HapMap Consortium : The international HapMap project. Nature 426: 789–796, 2003. [DOI] [PubMed] [Google Scholar]
- 24.Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, et al. ; 1000 Genomes Project Consortium : An integrated map of genetic variation from 1,092 human genomes. Nature 491: 56–65, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R: Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36: 411–420, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lake BB, Chen S, Hoshi M, Plongthongkum N, Salamon D, Knoten A, et al. : A single-nucleus RNA-sequencing pipeline to decipher the molecular anatomy and pathophysiology of human kidneys. Nat Commun 10: 2832, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu H, Uchimura K, Donnelly EL, Kirita Y, Morris SA, Humphreys BD: Comparative analysis and refinement of human PSC-derived kidney organoid differentiation with single-cell transcriptomics. Cell Stem Cell 23: 869–881.e8, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JW, Chou C-L, Knepper MA: Deep sequencing in microdissected renal tubules identifies nephron segment-specific transcriptomes. J Am Soc Nephrol 26: 2669–2677, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karaiskos N, Rahmatollahi M, Boltengagen A, Liu H, Hoehne M, Rinschen M, et al. : A single-cell transcriptome atlas of the mouse glomerulus. J Am Soc Nephrol 29: 2060–2068, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen L, Clark JZ, Nelson JW, Kaissling B, Ellison DH, Knepper MA: Renal-tubule epithelial cell nomenclature for single-cell RNA-sequencing studies. J Am Soc Nephrol 30: 1358–1364, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gu Z, Eils R, Schlesner M: Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32: 2847–2849, 2016. [DOI] [PubMed] [Google Scholar]
- 32.Rasouly HM, Groopman EE, Heyman-Kantor R, Fasel DA, Mitrotti A, Westland R, et al. : The burden of candidate pathogenic variants for kidney and genitourinary disorders emerging from exome sequencing. Ann Intern Med 170: 11–21, 2019. [DOI] [PubMed] [Google Scholar]
- 33.Finucane HK, Reshef YA, Anttila V, Slowikowski K, Gusev A, Byrnes A, et al. ; Brainstorm Consortium : Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat Genet 50: 621–629, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kinsella RJ, Kähäri A, Haider S, Zamora J, Proctor G, Spudich G, et al. : Ensembl BioMarts: A hub for data retrieval across taxonomic space. Database (Oxford) 2011: bar030, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skene NG, Grant SGN: Identification of vulnerable cell types in major brain disorders using single cell transcriptomes and expression weighted cell type enrichment. Front Neurosci 10: 16, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kiselev VY, Yiu A, Hemberg M: scmap: Projection of single-cell RNA-seq data across data sets. Nat Methods 15: 359–362, 2018. [DOI] [PubMed] [Google Scholar]
- 37.Skene NG, Bryois J, Bakken TE, Breen G, Crowley JJ, Gaspar HA, et al. ; Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium : Genetic identification of brain cell types underlying schizophrenia. Nat Genet 50: 825–833, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Leeuw CA, Mooij JM, Heskes T, Posthuma D: MAGMA: Generalized gene-set analysis of GWAS data. PLOS Comput Biol 11: e1004219, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Caminati M, Pham DL, Bagnasco D, Canonica GW: Type 2 immunity in asthma. World Allergy Organ J 11: 13, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bentley AM, Menz G, Storz C, Robinson DS, Bradley B, Jeffery PK, et al. : Identification of T lymphocytes, macrophages, and activated eosinophils in the bronchial mucosa in intrinsic asthma. Relationship to symptoms and bronchial responsiveness. Am Rev Respir Dis 146: 500–506, 1992. [DOI] [PubMed] [Google Scholar]
- 41.Lindgren D, Eriksson P, Krawczyk K, Nilsson H, Hansson J, Veerla S, et al. : Cell-type-specific gene programs of the normal human nephron define kidney cancer subtypes. Cell Rep 20: 1476–1489, 2017. [DOI] [PubMed] [Google Scholar]
- 42.Moreau ME, Garbacki N, Molinaro G, Brown NJ, Marceau F, Adam A: The kallikrein-kinin system: Current and future pharmacological targets. J Pharmacol Sci 99: 6–38, 2005. [DOI] [PubMed] [Google Scholar]
- 43.Ide S, Finer G, Maezawa Y, Onay T, Souma T, Scott R, et al. : Transcription factor 21 is required for branching morphogenesis and regulates the gdnf-Axis in kidney development. J Am Soc Nephrol 29: 2795–2808, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Calderon D, Bhaskar A, Knowles DA, Golan D, Raj T, Fu AQ, et al. : Inferring relevant cell types for complex traits by using single-cell gene expression. Am J Hum Genet 101: 686–699, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zeisel A, Hochgerner H, Lönnerberg P, Johnsson A, Memic F, van der Zwan J, et al. : Molecular architecture of the mouse nervous system. Cell 174: 999–1014.e22, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Giri A, Hellwege JN, Keaton JM, Park J, Qiu C, Warren HR, et al. ; Understanding Society Scientific Group; International Consortium for Blood Pressure; Blood Pressure-International Consortium of Exome Chip Studies; Million Veteran Program : Trans-ethnic association study of blood pressure determinants in over 750,000 individuals. Nat Genet 51: 51–62, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sakornsakolpat P, Prokopenko D, Lamontagne M, Reeve NF, Guyatt AL, Jackson VE, et al. ; SpiroMeta Consortium; International COPD Genetics Consortium : Genetic landscape of chronic obstructive pulmonary disease identifies heterogeneous cell-type and phenotype associations. Nat Genet 51: 494–505, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Menon M, Mohammadi S, Davila-Velderrain J, Goods BA, Cadwell TD, Xing Y, et al. : Single-cell transcriptomic atlas of the human retina identifies cell types associated with age-related macular degeneration. Nat Commun 10: 4902, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Qiu C, Huang S, Park J, Park Y, Ko YA, Seasock MJ, et al. : Renal compartment-specific genetic variation analyses identify new pathways in chronic kidney disease. Nat Med 24: 1721–1731, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hellwege JN, Velez Edwards DR, Giri A, Qiu C, Park J, Torstenson ES, et al. : Mapping eGFR loci to the renal transcriptome and phenome in the VA Million Veteran Program. Nat Commun 10: 3842, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morris AP, Le TH, Wu H, Akbarov A, van der Most PJ, Hemani G, et al. : Trans-ethnic kidney function association study reveals putative causal genes and effects on kidney-specific disease aetiologies. Nat Commun 10: 29, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Watanabe K, Umićević Mirkov M, de Leeuw CA, van den Heuvel MP, Posthuma D: Genetic mapping of cell type specificity for complex traits. Nat Commun 10: 3222, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Han X, Wang R, Zhou Y, Fei L, Sun H, Lai S, et al. : Mapping the mouse cell atlas by Microwell-seq [published correction appears in Cell 173: 1307, 2018]. Cell 172: 1091–1107.e17, 2018. [DOI] [PubMed] [Google Scholar]
- 54.Tabula Muris Consortium; Overall coordination; Logistical coordination; Organ collection and processing; Library preparation and sequencing; Computational data analysis; Cell type annotation; Writing group; Supplemental text writing group; Principal investigators : Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562: 367–372, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pattaro C, Teumer A, Gorski M, Chu AY, Li M, Mijatovic V, et al. ; ICBP Consortium; AGEN Consortium; CARDIOGRAM; CHARGe-Heart Failure Group; ECHOGen Consortium : Genetic associations at 53 loci highlight cell types and biological pathways relevant for kidney function. Nat Commun 7: 10023, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Worthmann K, Leitges M, Teng B, Sestu M, Tossidou I, Samson T, et al. : Def-6, a novel regulator of small GTPases in podocytes, acts downstream of atypical protein kinase C (aPKC) λ/ι. Am J Pathol 183: 1945–1959, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huber TB, Hartleben B, Winkelmann K, Schneider L, Becker JU, Leitges M, et al. : Loss of podocyte aPKClambda/iota causes polarity defects and nephrotic syndrome. J Am Soc Nephrol 20: 798–806, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Böger CA, Chen MH, Tin A, Olden M, Köttgen A, de Boer IH, et al. ; CKDGen Consortium : CUBN is a gene locus for albuminuria. J Am Soc Nephrol 22: 555–570, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nielsen R, Christensen EI, Birn H: Megalin and cubilin in proximal tubule protein reabsorption: From experimental models to human disease. Kidney Int 89: 58–67, 2016. [DOI] [PubMed] [Google Scholar]
- 60.Weyer K, Storm T, Shan J, Vainio S, Kozyraki R, Verroust PJ, et al. : Mouse model of proximal tubule endocytic dysfunction. Nephrol Dial Transplant 26: 3446–3451, 2011 [DOI] [PubMed] [Google Scholar]
- 61.Nykjaer A, Fyfe JC, Kozyraki R, Leheste JR, Jacobsen C, Nielsen MS, et al. : Cubilin dysfunction causes abnormal metabolism of the steroid hormone 25(OH) vitamin D(3). Proc Natl Acad Sci U S A 98: 13895–13900, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kahn AM, Weinman EJ: Urate transport in the proximal tubule: In vivo and vesicle studies. Am J Physiol 249: F789–F798, 1985. [DOI] [PubMed] [Google Scholar]
- 63.Nagata M: Podocyte injury and its consequences. Kidney Int 89: 1221–1230, 2016. [DOI] [PubMed] [Google Scholar]
- 64.Regev A, Teichmann SA, Lander ES, Amit I, Benoist C, Birney E, et al. ; Human Cell Atlas Meeting Participants : The human cell atlas. eLife 6: e27041, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.