

Significance Statement
Models of autosomal dominant polycystic kidney disease (ADPKD) are possible in nephron progenitor–derived human kidney organoids that form from induced pluripotent stem cells (iPSCs) that have had PKD genes deleted, enabling cyst formation. However, this has not been achieved in the ureteric bud/collecting duct lineage, despite the prevalence of collecting duct cysts in patients. Cysts formed in ureteric bud organoids derived from iPSCs with homozygous deleted PKD1, as well as in ureteric bud organoids generated from heterozygous mutant iPSCs and from a patient with ADPKD who had a heterozygous missense mutation, all upon cAMP stimulation. These PKD1 mutant organoids can model human ADPKD in the collecting duct lineage, complementing existing cell and animal models.
Keywords: ADPKD, collecting ducts, renal development, stem cell
Visual Abstract

Abstract
Background
Autosomal dominant polycystic kidney disease (ADPKD) is the most common hereditary kidney disease leading to renal failure, wherein multiple cysts form in renal tubules and collecting ducts derived from distinct precursors: the nephron progenitor and ureteric bud (UB), respectively. Recent progress in induced pluripotent stem cell (iPSC) biology has enabled cyst formation in nephron progenitor–derived human kidney organoids in which PKD1 or PKD2, the major causative genes for ADPKD, are deleted. However, cysts have not been generated in UB organoids, despite the prevalence of collecting duct cysts in patients with ADPKD.
Methods
CRISPR-Cas9 technology deleted PKD1 in human iPSCs and the cells induced to differentiate along pathways leading to formation of either nephron progenitor or UB organoids. Cyst formation was investigated in both types of kidney organoid derived from PKD1-deleted iPSCs and in UB organoids generated from iPSCs from a patient with ADPKD who had a missense mutation.
Results
Cysts formed in UB organoids with homozygous PKD1 mutations upon cAMP stimulation and, to a lesser extent, in heterozygous mutant organoids. Furthermore, UB organoids generated from iPSCs from a patient with ADPKD who had a heterozygous missense mutation developed cysts upon cAMP stimulation.
Conclusions
Cysts form in PKD1 mutant UB organoids as well as in iPSCs derived from a patient with ADPKD. The organoids provide a robust model of the genesis of ADPKD.
Autosomal dominant polycystic kidney disease (ADPKD) is the most common hereditary kidney disease, affecting approximately one in 1000 individuals.1 The main features of ADPKD are multiple renal cysts that eventually cause renal failure, often accompanied by liver cysts, pancreatic cysts, and cerebral aneurysms. PKD1 and PKD2 are the main causative genes for ADPKD, the former accounting for approximately 85% of cases.2 PKD1 and PKD2 encode polycystin-1 (PC1) and polycystin-2 (PC2) proteins, respectively. PC1 and PC2 are considered to function as a mechanosensor3–6 and a calcium (Ca) channel,7–9 respectively. PC1 and PC2 are detected in the primary cilia of renal epithelial cells,10,11 and possibly form a complex that converts the physical force of urinary flow to Ca influx.9 However, the function of the polycystin complex remains controversial.12
The clinical progression of ADPKD is gradual, and most patients manifest symptoms in adulthood. The majority of patients with ADPKD are heterozygous for PKD1 mutations, initially retaining one copy of the intact allele. It is proposed that this intact allele is lost or mutated during many years of clinical progression, which may explain the late appearance of the disease (two-hit hypothesis).13 In contrast, other studies have shown the presence of microscopic cysts in patients at or before birth, suggesting dosage effects of PC1 proteins (haploinsufficiency) rather than loss of heterozygosity by the two-hit hypothesis.14–16 Currently, the mechanism for ADPKD progression in response to a PKD1 mutation in humans remains unclear.
Mouse models are frequently used to investigate the mechanisms of ADPKD. Conventional Pkd1-null homozygous mice show marked polycystic kidneys during embryogenesis as well as hydrops fetalis likely resulting from cardiac abnormalities, both of which can lead to embryonic lethality.17,18 In contrast, Pkd1 heterozygous mice do not exhibit cysts at birth or renal failure during their lifetime, although microscopic renal cysts are observed at several months after birth.18,19 Thus, there may be species-dependent differences in phenotypic manifestations.
The kidney develops through mutual interactions between nephron progenitors and ureteric buds (UBs), with the former contributing to the renal tubules and glomeruli, and the latter contributing to the collecting duct. Recent advances in stem cell biology have enabled the generation of nephron progenitor–derived kidney organoids (nephron organoids) from pluripotent stem cells including human cells.20–24 Meanwhile, highly efficient gene editing in pluripotent stem cells using the CRISPR-Cas9 system enables disease modeling in organoids.25–27 Nephron organoids lacking PKD1 or PKD2 were shown to form cysts,27–29 and stimulation by cAMP signaling after forskolin administration enhanced cystogenesis in these organoids.28,29 However, most of the cysts in the organoids originated from proximal and/or distal nephron tubules because nephron organoids were used in this research. In contrast, large cysts in patients with ADPKD tended to originate from collecting ducts rather than nephron tubules.30 Indeed, tolvaptan, an antagonist of arginine vasopressin V2 receptor (AVPR2) expressed specifically in collecting ducts, is used for treatment of patients with ADPKD,31 although this treatment alone cannot control or cure the disease. Thus, modeling of cyst formation in the UB/collecting duct in humans would be beneficial for this research field and future treatment. However, conventional nephron organoids lack the UB/collecting duct lineage.32,33
Another issue is that induced pluripotent stem cells (iPSCs) derived from heterozygous patients with ADPKD failed to show more severe cyst formation than control cells.28 Therefore, previous studies mainly used CRISPR-Cas9–mediated mutant iPSCs, and reproducible cyst modeling from patient-derived iPSCs is awaited for allele-dependent mechanistic studies and drug screening.
We previously elucidated the developmental pathway from nascent mesoderm to nephron progenitors in mice, and successfully generated nephron organoids from human iPSCs.20,34,35 More recently, we revealed the distinct developmental pathway to the UB through Wolffian duct progenitors, and established an induction protocol for human UBs.22 Using these two established protocols in this study, we differentially induced PKD1 mutant iPSCs toward UBs and nephron progenitors, and achieved cystogenesis in UB organoids with homozygous and heterozygous mutations, in addition to nephron organoids. In addition, we detected cyst formation in UB organoids with a heterozygous PKD1 missense mutation derived from a patient with ADPKD.
Methods
Construction of Single-Guide RNA and Targeting Vectors
A single-guide RNA (sgRNA) was designed to target exon 15 of the PKD1 gene (target sequence, 5′-GCTCCTCCAACACGACCGTGCGG-3′; PAM sequence, GGG), and cloned into the pHL-H1-ccdB-mEF1a-H-A vector after removal of the ccdB cassette using an In-fusion HD Cloning Kit (Takara Bio) as described previously.26 The sgRNA vector and a Cas9 expression vector (pHL-EF1a-SphcCas9-A) were first transfected into HEK293 cells using Lipofectamine 2000 (Thermo Fisher Scientific) to evaluate their activity. The target region was amplified using Target-fwd and Target-rev primers (Supplemental Table 1). When the resulting fragment was denatured and annealed, a band shift was observed, confirming the formation of a mismatched duplex resulting from small insertion or deletion mutations (indels).36 For the targeting vector, 5′ and 3′ homology arms (0.9 kb and 0.81 kb, respectively) of PKD1 were amplified using genomic DNA from 201B7 iPSCs with the following primers: 5′ arm-fwd and 5′ arm-rev for the 5′ homology arm, and 3′ arm-fwd and 3′ arm-rev for the 3′ homology arm (Supplemental Table 1). After sequence verification, the 5′ homology arm was cloned into the EcoRI site of the HR130PA-1 vector (Systems Biosciences) containing a ppluGFP2 variant37 (precursor of TurboGFP), followed by red fluorescent protein (RFP) and a puromycin-resistance gene (Puro), and the 3′ homology arm was cloned into the BamHI-SphI site of the HR130PA-1 vector.
Generation of Human PKD1 Mutant iPSCs
The 201B7 human iPSC line was maintained on mouse embryonic fibroblasts as previously described.38 The cells were pretreated with 10 μM Y-27632 (#257-00511; Wako) at 1 hour before electroporation and a single-cell suspension was prepared using Dissociation Solution (#RCHETP002; Reprocell) followed by Accutase (Millipore). The sgRNA vector, Cas9 expression vector, and targeting vector (5 μg each) were electroporated into dissociated human iPSCs using a Super Electroporator NEPA21 (Nepagene) with two poring pulses (125 V, 2.5 ms) followed by five transfer pulses (20 V, 50 ms). The electroporated cells were plated onto puromycin-resistant DR4 feeders39 and puromycin was added at 5 days after electroporation. The puromycin concentration was incrementally increased from 0.25 to 0.5 μg/ml by 0.05 μg/ml per day for 5 days. After the puromycin selection, 48 single colonies were picked and screened by PCR with the following primers: Target-fwd and Target-rev, Left-fwd and Left-rev, and Right-fwd and Right-rev (Supplemental Table 1). After the PCR screening, we obtained 11 candidate PKD1+/+ clones, 17 candidate PKD1+/− clones, and 12 PKD1−/− clones. Subsequently, we sequenced the target regions in the PKD1+/+ and PKD1+/− candidate clones to exclude clones with indels on the assumptive wild-type alleles. Finally, we obtained seven PKD1+/+, six PKD1+/−, and 12 PKD1−/− clones. Two clones each for the PKD1+/+, PKD1+/−, and PKD1−/− genotypes (clones 2 and 5, 32 and 41, and 12 and 17, respectively) were adapted to a feeder-free condition,34 and used for subsequent analyses. All six clones, together with their maternal cell line (210B7) used in this study, had almost normal karyotypes except for the long arm of chromosome 10. This long arm contained an additional small chromosomal fragment of unknown origin (46, XX, add[10][q26]), possibly resulting from preferential selection of fast-growing cells. However, this abnormality was considered unlikely to affect the cyst phenotypes, because PKD1 is located on chromosome 16 (16p13.3). Although we confirmed in-frame insertion of the T2A–green fluorescent protein cassette or RFP in the PKD1 mutated alleles, green fluorescent protein or RFP signals in iPSCs and nephron progenitors were not detectable by either microscopy or FACS analysis due to unknown reasons which possibly include silencing of the promoter regions.
Nephron Organoid Induction from PKD1 Mutant iPSCs
The established iPSC clones were induced toward nephron progenitors using a five-step protocol as described.22 At day 13, some of the induced spheres were analyzed by flow cytometry to measure the percentages of ITGA8+/PDGFR− nephron progenitors in the spheres (Supplemental Material, Supplemental Reference). The percentages (31%–32%) were lower than those in our previous report using 201B7 iPSCs (40%–50%),20 possibly because of the multiple passages upon gene manipulation. However, the variation in induction of nephron progenitor cells among the three genotypes was negligible. The remaining spheres were cocultured with mouse embryonic spinal cord at the air-fluid interface to initiate nephron differentiation.40 After 6 days of coculture (day 19), the spheroids were removed from the spinal cord and cultured in Transwell inserts (#3428; Costar) with the medium containing KnockOut Serum Replacement (#10828010; Thermo Fisher Scientific).41 Subsequently, 25 μM forskolin (067-02191; Wako) or 300 nM vasopressin (V9879; Sigma) was added to the medium from day 19 to day 25 (forskolin) or day 31 (vasopressin). Each treatment was repeated at least three times, and two clones per genotype were used in all experiments.
UB Organoid Induction from PKD1 Mutant iPSCs
The iPSC clones were induced toward UBs using a seven-step protocol as described.22 At day 6.25, the percentages of CXCR4+/KIT+ Wolffian duct progenitors in the spheres were analyzed by flow cytometry. At day 12.5, the UB spheres were transferred to 24-well Transwell inserts (#3422; Costar), mounted in 150 μl of branching medium containing 50% Matrigel (#356230; Corning), and cultured in the presence of 500 μl of branching medium without Matrigel. Subsequently, 25 μM forskolin or 300 nM vasopressin was added to the medium from day 18.5 to day 30. Each treatment was repeated at least three times, and two clones per genotype were used in all experiments.
UB Organoid Induction from iPSCs Derived from Patients with ADPKD
The establishment of iPSC lines (CiRA00007 and CiRA00009) derived from patients with ADPKD was reported previously.42 CiRA00007 was derived from patient 4 with a missense mutation in PKD1 (G3818R), and CiRA00009 was derived from patient 6 with a nonsense mutation in PKD1 (Q3895X). These clones had normal karyotypes and their PKD1 mutations were confirmed to be identical to those in the patient fibroblasts by short tandem analysis.42 The patient-derived lines, as well as the control line 201B7,38 were maintained in a feeder-free condition34 and used for induction experiments. The concentrations of BMP4 in the first and second steps of the UB induction protocol were titrated to maximize the percentages of CXCR4+/KIT+ Wolffian duct progenitors. The optimized BMP4 concentrations were 3 ng/ml for the patient 4–derived line and 0 ng/ml for 201B7. Because the percentage of Wolffian progenitors in the patient 6–derived line was <2% at any BMP4 concentration, the line was excluded from further analysis. All experiments using patient-derived iPSCs were performed in accordance with institutional guidelines and approved by the Licensing Committee of Kumamoto University. The names of the ethics committees were the Ethics Committee for Epidemiologic and General Research and the Ethics Committee for Human Genome and Gene Analysis Research at the Faculty of Life Science, Kumamoto University (approval numbers 1453 and 359, respectively). Analysis of patient-derived nephron organoids will be described elsewhere (K.O., manuscript in preparation).
Measurement of Cyst Areas
We evaluated the cyst areas in nephron organoids and UB organoids on bright-field images using ImageJ software.43 When multiple cysts were observed in an organoid, the total area was calculated for that organoid.
Statistical Analyses
Data are presented as mean±SEM. Statistical analyses were performed by a t test for differences between two groups. The Tukey–Kramer test was applied for differences among multiple groups. Differences with values of P<0.05 were considered statistically significant.
Result
Establishment of PKD1 Mutant iPSCs by Genome Editing
To examine the role of PKD1 in human kidney cystogenesis, we inserted a drug-resistance gene cassette into the PKD1 locus of human iPSCs by CRISPR-Cas9–mediated homologous recombination (Figure 1A). Among the 46 exons in the PKD1 gene, we targeted exon 15 encoding the majority of the extracellular PKD repeat domains of PC1 protein (Supplemental Figure 1, A and B). Exon 15 is the largest exon in the PKD1 gene, and has been reported to contain numerous mutations, including frameshift mutations, in patients with ADPKD (http://pkdb.mayo.edu/). Unlike mice, however, humans possess PKD1 pseudogenes,44,45 five of which have similar sequences to those in and around exon 15 (Supplemental Figure 1A). Therefore, we designed an sgRNA that specifically targeted PKD1 exon 15, and not the pseudogenes. We introduced the sgRNA, together with the Cas9 expression vector and targeting vector, into human iPSCs. After puromycin selection, we screened for heterozygous (PKD1+/−) and homozygous (PKD1−/−) mutant clones by PCR (Figure 1B). Southern blotting confirmed that mutations were only introduced into the PKD1 gene, and not the pseudogenes (Figure 1C, Supplemental Figure 1A). Indeed, PKD1−/− clones lacked full-length PC1 protein but expressed truncated proteins, as determined using an antibody against an N-terminal region of PC1 (Figure 1D). In PKD1+/− clones, reduced expression of full-length PC1 protein was observed compared with PKD1+/+ clones (Figure 1D). Thus, we successfully generated heterozygous and homozygous PKD1 mutant iPSC clones with intact PKD1 pseudogenes in humans.
Figure 1.

PKD1 mutant iPSCs are generated by CRISPR-Cas9-mediated homologous recombination. (A) Strategy for targeting the human PKD1 gene. A cassette containing green fluorescent protein (GFP), RFP, and puromycin-resistance gene (Puro) is inserted into exon 15 of PKD1. Gray arrowheads, loxP sites; black hexagons, insulators; black arrowheads, PCR primers used for (B); red box, Southern blot probe on a 5′ region outside the left arm that hybridizes to pseudogenes other than authentic PKD1. (B) PCR screening of homologous recombinants. The primer positions are shown in (A). (C) Southern blot analysis of wild-type (+/+), heterozygous (+/−), and homozygous (−/−) clones. Genomic DNAs were digested with SphI and EcoRI and hybridized with the probe (red box in [A]). Because the pseudogenes have higher copy numbers, their bands are darker than the authentic PKD1 band. (D) Western blots of PC1 proteins in iPSCs. An antibody against an N-terminal region of PC1 was used. The asterisk indicates shorter bands, presumably representing truncated PC1 proteins in the heterozygous (+/−) and homozygous (−/−) clones. B, BamHI; E, EcoRI; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; S, SphI; WT, wild type.
PKD1 Mutant Nephron Organoids Exhibit Cystogenesis upon Forskolin Treatment
We induced the established PKD1 mutant iPSCs toward nephron progenitors and subsequently to nephron organoids, based on our previously reported protocol20,22 (Figure 2A). At day 13, induction of ITGA8+/PDGFRA− nephron progenitors was comparable among the PKD1+/+, PKD1+/−, and PKD1−/− clones (Figure 2B), suggesting the PKD1 mutation did not affect induction of iPSCs into nephron progenitors. At day 25, all clones showed robust nephron formation: glomeruli (WT1+) with proximal (LTL+) and distal (CDH1+) tubules were observed in all organoids of the three genotypes (two clones per genotype) (Figure 2, C and D). However, no cysts were detected, even in PKD1−/− organoids, consistent with previous reports on mouse and human iPSC-derived organoids.29,46
Figure 2.

PKD1 mutant nephron organoids do not show cystogenesis. (A) Outline of the induction protocol for nephron organoids from iPSCs through nephron progenitors. Spinal cord was attached to nephron progenitors at day 13 to initiate nephrogenesis. After removing the spinal cord at day 19, the nephron organoids were cultured up to day 25. (B) Flow cytometry analysis at day 13. The percentages of the ITGA8+/PDGFRA− nephron progenitor fraction were determined from five independent experiments (mean±SEM). (C) Representative bright-field images of nephron organoids at day 25. Scale bars, 200 μm. (D) Representative confocal images of immunostaining at day 25. Nephron segments of the glomeruli (WT1+; red), proximal tubules (LTL+; green), and distal tubules (CDH1+; blue) were formed in all three genotypes. Scale bars, 200 μm (upper panels) and 50 μm (lower panels). DAPI, 4′,6-diamidino-2-phenylindole.
Because stimulation of cAMP signaling was reported to be required for renal cyst formation in nephron organoids in vitro,46 we administered forskolin (an activator of cAMP signaling) to the nephron organoids at day 19 (Figure 3A). Although forskolin treatment caused cyst formation in all three genotypes, cystogenesis was most prominently observed in PKD1−/− organoids (Figure 3B). Indeed, cyst area calculations revealed significantly more severe, although variable, cystogenesis in PKD1−/− organoids compared with PKD1+/+ organoids, although the latter did show mild cyst formation (Figure 3C). In all organoids examined, cysts were mainly formed in LTL+ proximal tubules and WT1+ glomerular parietal epithelial cells (Bowman capsule epithelia), irrespective of the genotypes, whereas cyst formation in CDH1+ distal tubules was less frequent (Figure 3D, Supplemental Figure 2). These observations are consistent with the early-stage phenotype of ADPKD model mice, in which cystogenesis progresses from proximal to distal tubules.47 Primary cilia were detected on the luminal side of all cysts derived from proximal tubules regardless of the PKD1 mutation (Supplemental Figure 3A). Their length and morphology did not appear to differ markedly among the genotypes, but the section staining precluded precise evaluation. Taken together, cyst formation was observed in human nephron organoids with homozygous or heterozygous PKD1 mutations upon cAMP activation.
Figure 3.

PKD1 mutant nephron organoids exhibit cystogenesis upon forskolin treatment. (A) Outline of the induction protocol for nephron organoids with forskolin treatment. After removing the spinal cord at day 19, forskolin was added at a final concentration of 25 μM. (B) Representative bright-field images of nephron organoids at day 25 after treatment with forskolin. Scale bars, 200 μm. (C) Cyst areas in nephron organoids (n=6 per clone). Two clones were analyzed for each genotype. Data are shown as mean±SEM. *P<0.05, **P<0.01. (D) Representative confocal images of immunofluorescence at day 25. LTL+ cysts (presumably derived from proximal tubules; asterisks) and WT1+ cysts (presumably derived from glomerular parietal epithelial cells; arrowheads) were detected in organoids, irrespective of the genotypes. Scale bars, 200 μm (upper panels) and 50 μm (lower panels). DAPI, 4′,6-diamidino-2-phenylindole.
PKD1 Mutant UB Organoids Exhibit Cystogenesis upon Forskolin Treatment
Next, we induced UB organoids from the PKD1 mutant iPSCs (Figure 4A) using our previously reported selective UB induction protocol.22 When the UB organoids were examined on day 6.25 of the induction protocol, the percentages of CXCR4+/KIT+ Wolffian duct progenitors were comparable among the PKD1+/+, PKD1+/−, and PKD1−/− clones (Figure 4B). The spheres were placed in a branching culture condition on day 12.5, and the first branching of the UB tips was observed by day 18.5 (Figure 4C). At this stage, the organoids of all three genotypes expressed marker genes for the ureteric epithelium: RET and WNT11 (tip markers), WNT9B and AQP2 (stalk markers), and GATA3 and CALB1 (general ureteric epithelium markers), as determined by quantitative PCR (Figure 4D). At day 30, when the UB stalks elongated and the tips underwent a few rounds of branching, RET was expressed at the tips, whereas cytokeratin 19 (KRT19) was abundantly expressed in the stalks in the organoids of all three genotypes (Figure 4, C and E). These data indicate that the PKD1 mutation did not affect UB induction and overall tip-stalk patterning, although the stalks of PKD1−/− UBs appeared shorter at day 30 (Supplemental Figure 4), possibly reflecting a mild impairment of cell polarity and/or convergent extension in the stalks.48 Nevertheless, no cyst formation was observed without forskolin treatment even in PKD1−/− UB organoids, consistent with the results in nephron organoids.
Figure 4.

PKD1−/− UB organoids do not show cystogenesis. (A) Outline of the induction protocol for UB/collecting duct organoids from iPSCs. Wolffian duct progenitors were sorted and reaggregated into spheres at day 6.25. At day 12.5, the spheres were transferred to a branching condition and cultured until day 30. (B) Flow cytometry analysis at day 6.25. The percentages of the CXCR4+/KIT+ Wolffian duct progenitor fraction were determined from five independent experiments (mean±SEM). (C) Representative bright-field images of UB organoids at day 18.5 and 30. Scale bars, 200 μm. (D) Quantitative PCR analysis of UB markers at day 18.5 (two clones per genotype in three independent experiments). (E) Representative confocal images of immunofluorescence at day 30. The KRT19− structures in the center of UB organoid were not cysts but occupied by DAPI+ cells. Scale bars, 200 μm (upper panels) and 50 μm (lower panels). DAPI, 4′,6-diamidino-2-phenylindole.
UB organoids were subsequently treated with forskolin from day 18.5 (Figure 5A), resulting in cyst formation in PKD1−/− organoids (75.0%±22.3% for clone 12; 90%±12.2% for clone 17; n=20 per clone) on day 30 (Figure 5, B and C, Supplemental Figure 5). Although not all epithelia contributed to cyst formation, bubble-like structures were formed in some of the UB stalk regions, reminiscent of the outpocketing phenomenon observed in the initial stages of cystogenesis in vivo.49 In contrast, no cysts were formed in PKD1+/+ organoids, unlike nephron organoids that showed mild cyst formation (see Figure 3, B–D). Some PKD1+/− UB organoids (30.0%±18.7% for clone 32; 25.0%±15.8% for clone 41; n=20 per clone) also exhibited cyst formation, although the cyst areas and frequencies were less than those in PKD1−/− organoids (Figure 5, C and D). Therefore, PKD1-dependent cystogenesis was achieved in human UB organoids upon cAMP activation. Primary cilia were present on the luminal side of the cysts, as seen in nephron organoids (Supplemental Figure 3B).
Figure 5.

PKD1 mutant UB organoids exhibit cystogenesis upon forskolin treatment. (A) Outline of the induction protocol for UB organoids with forskolin treatment. At day 18.5, forskolin was added at a final concentration of 25 μM. (B) Time-course bright-field images of UB organoids upon forskolin treatment. Scale bars, 200 μm. (C) Frequency of cyst formation in UB organoids (two clones per genotype; n=4 per clone in five independent experiments). (D) Cyst areas in UB organoids (n=20 per clone). Data are shown as mean±SEM. *P<0.05, **P<0.01.
Cysts Are Composed of UB Epithelia
We characterized the cysts in UB organoids using molecular markers. At day 24, reflecting an initial stage of cyst formation, the cysts in PKD1−/− organoids expressed general ureteric epithelial markers, CALB1 and DBA, as seen in control organoids (Figure 6A). PAX2 is a transcription factor expressed in the ureteric and nephron lineages, whereas GATA3 is expressed in the UB and distal renal tubules. Our data showed that the cyst epithelia in PKD1−/− organoids, as well as normal epithelia in control organoids, expressed PAX2 and GATA3 at day 24 (Figure 6B). These markers were still detected at day 30 (Supplemental Figure 6, A and B). A ureteric tip marker, RET, was undetectable in the cyst epithelia, whereas a ureteric stalk marker, KRT19, was detected (Figure 6C, Supplemental Figure 6C). These data indicate that cysts in PKD1 mutant UB organoids were indeed derived from UB epithelia, most likely from those in the stalk regions. However, AQP2, which was expressed in the stalk regions of control organoids, was not detectable in the cyst epithelia of mutant organoids (Figure 6D, Supplemental Figure 6D), suggesting that AQP2-negative UB epithelia may have contributed to the cyst formation or failed to differentiate into the more mature AQP2-positive state. Alternatively, AQP2 expression may have been downregulated in the cyst epithelia.
Figure 6.
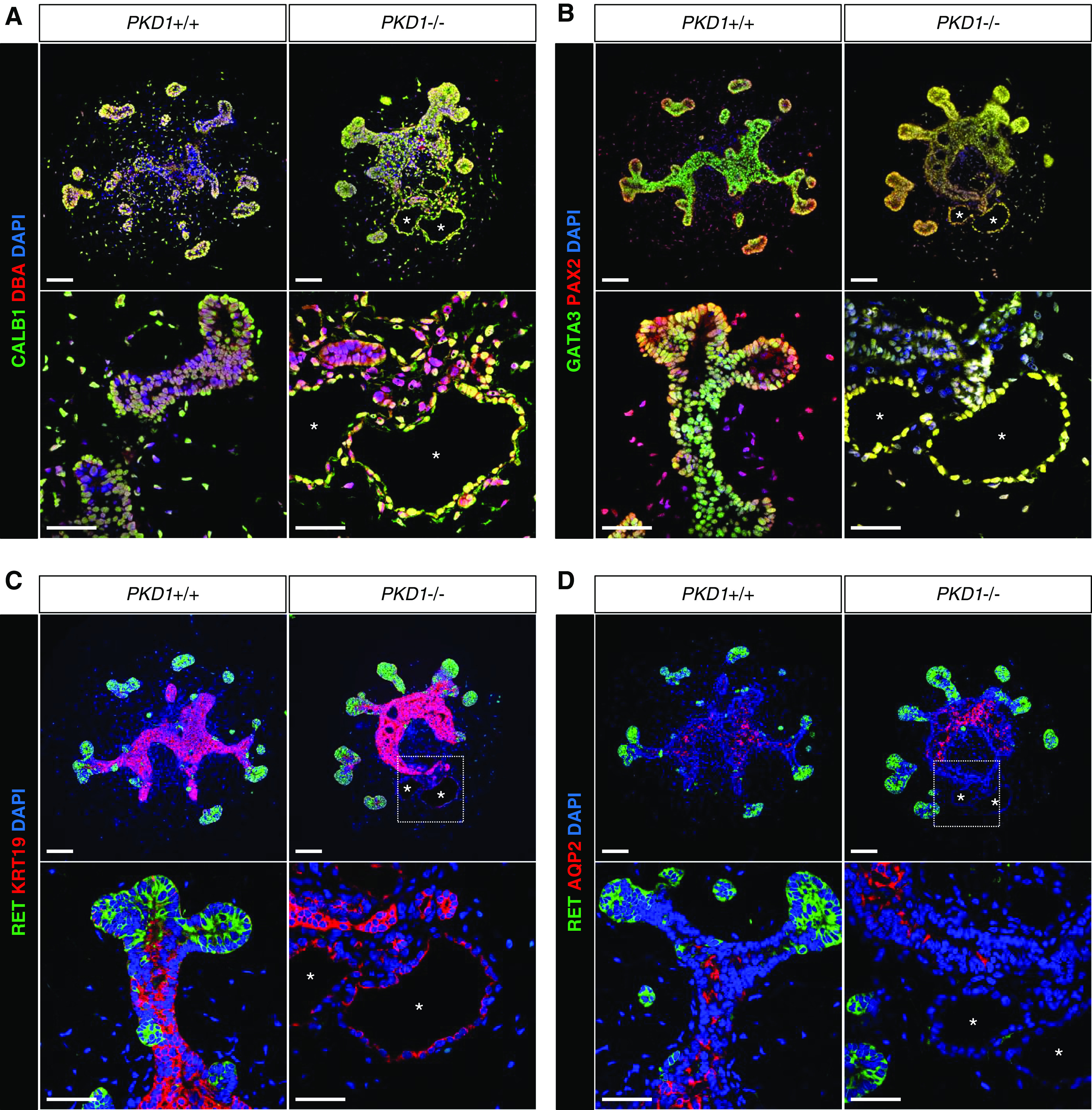
PKD1 mutant cysts express markers for UB epithelia. Confocal images of immunofluorescence in UB organoids at day 24. (A) CALB1 (green) and DBA (red). (B) GATA3 (green) and PAX2 (red). (C) RET (green) and KRT19 (red). (D) RET (green) and AQP2 (red). The asterisks indicate the lumens of cysts. Scale bars, 100 μm (upper panels) and 50 μm (lower panels). DAPI, 4′,6-diamidino-2-phenylindole.
Vasopressin Treatment Induces Cyst Formation in UB Organoids
Through administration of forskolin, we successfully induced cyst formation in both nephron and UB organoids. However, natural ligands that activate cAMP must exist in vivo. Therefore, we examined the expression of vasopressin receptors, because vasopressin is known to induce cAMP in UB-derived collecting ducts and exacerbate cyst formation in patients with ADPKD.50 We found that the arginine vasopressin receptor 1A gene (AVPR1A) was expressed in UB organoids, but not in nephron organoids (Figure 7A). In contrast, AVPR1B and AVPR2 were minimally detected in both types of organoid. These results were consistent with RNA-sequencing data for the human embryonic kidney in vivo51 (Supplemental Figure 7), which detected AVPR1A, but not AVPR1B or AVPR2, at 10–16 weeks of gestation. Immunostaining confirmed that AVPR1A was expressed in the stalk region of UB organoids and did not overlap with RET, a UB tip marker (Figure 7B). The PKD1 mutation did not affect AVPR1A expression (Figure 7, A and B). UB organoids were then treated with vasopressin from day 18.5 to day 30, resulting in cyst formation in PKD1−/− organoids (12.5%±7.2% for clone 12; 12.5%±7.2% for clone 17) but not in PKD1+/− or wild-type organoids (Figure 7, C and D). In contrast, no cysts were formed in nephron organoids, irrespective of the genotypes, upon vasopressin treatment for the same duration (day 19 to day 31; Figure 7, C and D). Therefore, AVPR1A expressed in PKD1−/− UB organoids may be functional and the mutant organoids responded to vasopressin, its hormone ligand, to form cysts, although its efficiency for cystogenesis was not comparable to that of forskolin.
Figure 7.

Vasopressin treatment induces cyst formation specifically in PKD1 mutant UB organoids. (A) Quantitative PCR analysis for expression of vasopressin receptors in nephron organoids (NP) at day 19 and UB organoids at day 18.5 (n=2 per genotype in three independent experiments). (B) Confocal images of immunofluorescence for RET (green) and AVPR1A (red) in UB organoids at day 18.5. Scale bars, 100 μm (upper panels) and 50 μm (lower panels). (C) Representative bright-field images of nephron (NP) organoids and UB organoids upon vasopressin treatment (NP, from day 19 to day 31; UB, from day 18.5 to day 30). The black arrowhead shows cyst formation in PKD1−/− UB organoids. Scale bars, 200 μm. (D) Frequency of cyst formation upon vasopressin treatment (n=6 per clone in four independent experiments). Data are shown as mean±SEM. **P<0.01. AVP, vasopressin (300 nM).
UB Organoids Generated from Patient-Derived iPSCs Exhibit Cystogenesis upon Forskolin Treatment
Finally, we applied our induction protocol for UB organoids to iPSCs derived from a patient with ADPKD. The patient had a heterozygous missense mutation (PKD1 c.11452G>C; p.Gly3818Arg) and suffered numerous renal cysts, as well as hypertension, intracranial aneurysms, and subarachnoid hemorrhage.42 In the patient’s pedigree, family members with the polycystic kidney phenotype, but not those without the phenotype, had the same point mutation, suggesting that it is likely to be a disease-causing mutation. Identical missense mutations or nonsense mutations at the same position have been reported in other ADPKD families,52,53 supporting the pathogenicity of this particular mutation in our patient. By optimizing the condition in the first step of the UB induction protocol, we successfully induced CXCR4+/KIT+ Wolffian duct progenitors from the patient-derived iPSCs, although the induction efficiency was lower than that for wild-type iPSCs (201B7) (Figure 8A). The UB organoids were moved to a branching culture condition on day 12.5 and treated with forskolin from day 18.5 as described above (see Figure 5A). UB organoids from the patient-derived iPSCs exhibited cyst formation (15.62%±5.41%; n=4), whereas no cysts were formed in wild-type UB organoids (Figure 8, B and C). The poorer tip-stalk patterns in the patient-derived organoids may result from the lower percentages of Wolffian duct progenitors compared with control organoids. The formed cyst epithelia were PAX2+/GATA3+ and RET−/KRT19+ (Figure 8, D and E), consistent with the results for CRISPR-Cas9–mediated PKD1 mutant iPSCs (see Figures 6 and 7). Therefore, we have demonstrated cyst formation from patient-derived iPSCs with a heterozygous mutation in the PKD1 gene, although it is formally necessary to examine clones from multiple patients and to confirm cyst disappearance upon gene correction on the same genetic background (isogenic controls). Taken together with the results shown in Figure 5C, the cystogenesis caused by PKD1 heterozygosity was not limited to the deletions generated by CRISPR-Cas9 technology, but is likely applicable to the PKD1 mutation in the patient.
Figure 8.
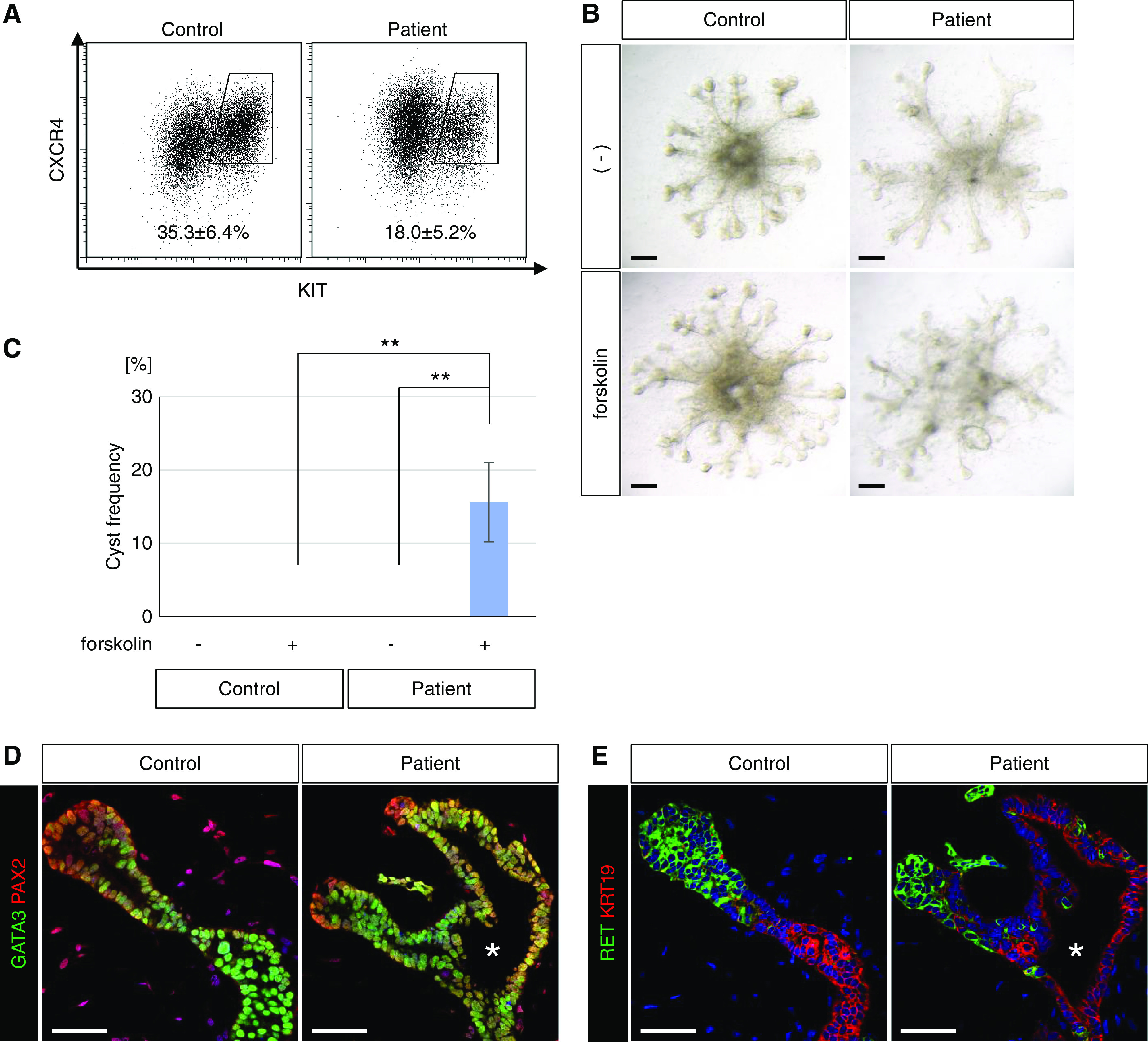
UB organoids generated from iPSCs derived from a patient with ADPKD exhibit cystogenesis upon forskolin treatment. (A) Flow cytometry analysis of UB organoids generated from iPSCs derived from a patient with ADPKD (patient 4) at day 6.25. Wild-type human iPSCs (201B7) were used as controls. The percentages of the CXCR4+/KIT+ Wolffian duct progenitor fraction were determined from three independent induction experiments (mean±SEM). (B) Representative bright-field images of UB organoids upon forskolin treatment from day 18.5 to day 30. Scale bars, 200 μm. (C) Frequency of cyst formation upon forskolin treatment (n=32 in four independent experiments). Data are shown as mean±SEM. **P<0.01. (D and E) Confocal images of immunofluorescence in UB organoids at day 30 with staining for (D) GATA3 (green) and PAX2 (red) or (E) RET (green) and KRT19 (red). The asterisks indicate the lumens of cysts. Scale bars, 50 μm.
Discussion
By applying the two different induction protocols to human iPSCs lacking PKD1, we have demonstrated that both nephron and UB organoids exhibited cyst formation upon forskolin treatment. To the best of our knowledge, this is the first report to show cystogenesis in UB organoids. The UB organoids, but not nephron organoids, responded to vasopressin to form cysts. Furthermore, we reproducibly showed cyst formation from not only heterozygous iPSCs with an artificially truncated PKD1 allele, but also patient-derived heterozygous PKD1 mutant iPSCs. Thus, our UB organoids will serve as useful platforms for modeling of human ADPKD.
Although nephron organoids and UB organoids both exhibited cystogenesis, their responsiveness to forskolin was different. In nephron organoids, even wild-type organoids showed cyst formation, although the severity was much less than that in PKD1 mutant organoids. In contrast, wild-type UB organoids never showed cyst formation, and thus abnormalities were specifically observed in PKD1 mutant organoids. This feature may be related to differences in the nature of the epithelia: UBs were composed of epithelia that were already CDH1+ at day 12.5 in our culture system, whereas proximal renal tubule epithelia were gradually generated and became CDH6+ between days 13 and 19 through mesenchymal conversion of nephron progenitors. Thus, proximal tubules may retain the more nascent characteristics of epithelia. Alternatively, cellular adhesiveness or stiffness of CDH1−/CDH6+ proximal tubule epithelia may be weaker than that of CDH1+ UB epithelia. Although further studies are needed to clarify the differences in epithelial characteristics, cystogenesis specific to PKD1 mutations with no background in wild-type organoids will make the UB organoid system more useful for drug screening to prevent cystogenesis. Because we also demonstrated cyst formation from patient-derived iPSCs, our system will be applicable to various PKD1 alleles observed in patients.
Our data showed that cAMP signaling is required for in vitro cystogenesis in both nephron organoids and UB organoids, consistent with previous reports.29 In an ex vivo culture setting, even Pkd1 mutant mouse embryonic kidneys showed no cyst formation without cAMP stimulation, whereas large cysts were formed in vivo.46 Although cAMP stimulation may not precisely mimic the cyst-forming signals in vivo, cyst formation is considered to be regulated by the balance between cAMP- and PC1/PC2-mediated signals: cAMP signaling enhances cyst formation, whereas Ca signaling mediated by PC1/PC2 channels antagonizes cystogenesis, probably by reducing cAMP levels.54 Sufficient cAMP signal stimulation may not occur in vitro, possibly through a lack of cAMP-stimulating factors or hormones that would be present in vivo.55 One such candidate is vasopressin, which works on its receptors (AVPRs) on the collecting duct cell membrane to increase intracellular cAMP levels.55 Indeed, cyst formation in patients with ADPKD is ameliorated by AVPR2 (V2) antagonists, including tolvaptan. Thus, responsiveness to vasopressin is essential for ADPKD modeling and drug screening. Our UB organoids expressed AVPR1A, but not AVPR2. These expression patterns are consistent with those in the developing human kidney in vivo: AVPR1A is expressed at 10–20 weeks of gestation, whereas AVPR2 expression is initiated later.51 Thus, our UB organoids are likely to represent the first, or at best the second, trimester. This early expression of AVPR1A in the kidney may be specific to humans, because it was not observed in mice (our preliminary data). Although the functions of AVPR1A in human UBs remain unsolved, AVPR1A is expressed by endothelial cells in both humans and mice.56 In contrast to AVPR2 that stimulates cAMP through Gs protein, a different type of G protein, Gq, is coupled to AVPR1A, at least in endothelial cells.57 Such signaling differences may explain the relatively low percentage of cyst formation in UB organoids upon vasopressin treatment, and further maturation accompanying AVPR2 expression may be required to model ADPKD more precisely. Because nephron organoids lack AVPRs, there must be ligands other than vasopressin that can stimulate cystogenesis. Recent single-cell RNA-sequencing analyses of mouse and human kidneys identified many receptors in the nephron, including receptors for parathyroid hormone, somatostatin, growth hormone, and prolactin58 (our unpublished results). Searches for procyst factors will advance research on cystogenesis in the nephron lineage.
Some organoids heterozygous for the PKD1 mutation showed cyst formation within a few weeks of culture, although the severity and frequency of cystogenesis were moderate. Because deletion of the remaining wild-type allele is unlikely to occur within such a short period, the cysts in heterozygotes observed in this study were likely caused by PKD1 haploinsufficiency. Importantly, we also showed cyst formation in UB organoids generated from patient-derived iPSCs heterozygous for a PKD1 mutation after forskolin treatment, although isogenic controls with the corrected gene sequence are formally required for comparison. Thus, heterozygous PKD1 mutant epithelia are likely to be primed for exogenous signals toward cystogenesis, which may underlie the cyst formation in patients with ADPKD. However, our data do not contradict the two-hit hypothesis,13 because patients with ADPKD require a long period (usually many decades) before apparent phenotypic manifestation. Deletion of the remaining wild-type allele (second hit) may occur during this long incubation period with the eventual formation of numerous cysts, which cannot be reproduced in the current organoid model. It is also noteworthy that not all UBs or nephrons exhibited cyst formation, even in the homozygous mutant organoids, which may be equivalent to the two-hit state. These observations alone cannot deny the two-hit hypothesis, because our kidney organoids represent the embryonic stage of development and immaturity of the organoids may cause different degrees of responsiveness to forskolin.
Thus, there remains much room for improvement of our UB organoids. As described above, these organoids were immature and did not express AVPR2, although they responded to vasopressin, possibly through AVPR1A. The addition of exogenous forskolin was required for cyst formation from PKD1 mutant organoids. The cysts were not sufficiently large when compared with those in patients or in animal models in vivo. Therefore, a combination of human kidney organoids with genetically engineered animal models18,59,60 will be necessary to determine the definitive principles underlying this disease. One of the major environmental differences between our organoids and animal models is the absence and presence of urinary flow. Because organ-on-a-chip models have been developed to confer flow onto organoids,61 application of PKD1 mutant organoids to these microdevices will be one possible option to reproduce more physiologic cyst formation.
Taken together, we have demonstrated PKD1-dependent cystogenesis in nephron organoids and UB organoids, with responsiveness to vasopressin in the latter. We further showed cyst formation from patient-derived organoids with a heterozygous PKD1 missense mutation. Our findings, as well as our differential induction protocols and PKD1 mutant iPSCs, will serve as valuable bases for modeling of ADPKD to elucidate the mechanisms underlying the disease and to screen for therapeutic chemicals.
Disclosures
K. Osafune is a founder and member without salary of the scientific advisory board of iPS Portal, Inc., and a founder and chief scientific advisor of RegeNephro Co., Ltd. A. Taguchi has a patent PCT/JP2018/041558 pending. R. Nishinakamura has a patent PCT/JP2018/041558 pending. All remaining authors have nothing to disclose.
Funding
This study was partly supported by the Japan Society for the Promotion of Science KAKENHI grant JP17H06177 (to R. Nishinakamura) and a Japan Agency for Medical Research and Development, Research Center Network for Realization of Regenerative Medicine grant 20bm0804013h0004.
Supplementary Material
Acknowledgments
We thank Tomoko Ohmori and Sayoko Fujimura for technical assistance. We also thank Alison Sherwin, from Edanz Group (https://en-author-services.edanzgroup.com/), for editing a draft of this manuscript.
Shohei Kuraoka and Prof. Ryuichi Nishinakamura designed experiments; Shohei Kuraoka performed the experiments and analyses and wrote the original draft; Shunsuke Tanigawa, Atsuhiro Taguchi, and Prof. Hitoshi Nakazato assisted with experiments; Prof. Akitsu Hotta generated the CRISPR-Cas9 vectors; Prof. Kenji Osafune established the patient-derived iPSCs; Akio Kobayashi, Atsuhiro Taguchi, and Prof. Ryuichi Nishinakamura reviewed and edited the manuscript; Akio Kobayashi and Prof. Ryuichi Nishinakamura were responsible for project administration; and Prof. Ryuichi Nishinakamura acquired research funding.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020030378/-/DCSupplemental.
Supplemental Figure 1. Schematic illustration of the PKD1 locus and PC1 protein.
Supplemental Figure 2. The majority of cysts are formed from glomeruli and proximal tubules in nephron organoids.
Supplemental Figure 3. Both nephron and UB organoid cysts express primary cilia marker at the luminal side.
Supplemental Figure 4. PKD1−/− UB organoids exhibit shorter stalks in the absence of forskolin.
Supplemental Figure 5. PKD1 mutant UB organoids exhibit cystogenesis upon forskolin treatment.
Supplemental Figure 6. UB markers are expressed in the cysts of PKD1−/− UB organoids at day 30.
Supplemental Figure 7. AVPR1A is dominantly expressed in the human embryonic kidney.
Supplemental Table 1. Primer sequences.
References
- 1.Yersin C, Bovet P, Wauters JP, Schorderet DF, Pescia G, Paccaud F: Frequency and impact of autosomal dominant polycystic kidney disease in the Seychelles (Indian Ocean). Nephrol Dial Transplant 12: 2069–2074, 1997. [DOI] [PubMed] [Google Scholar]
- 2.Torres VE, Harris PC, Pirson Y: Autosomal dominant polycystic kidney disease. Lancet 369: 1287–1301, 2007. [DOI] [PubMed] [Google Scholar]
- 3.Chauvet V, Tian X, Husson H, Grimm DH, Wang T, Hiesberger T, et al.: Mechanical stimuli induce cleavage and nuclear translocation of the polycystin-1 C terminus [published correction appears in J Clin Invest 115: 788, 2005]. J Clin Invest 114: 1433–1443, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, et al.: Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 33: 129–137, 2003. [DOI] [PubMed] [Google Scholar]
- 5.Patel A, Honoré E: Polycystins and renovascular mechanosensory transduction. Nat Rev Nephrol 6: 530–538, 2010. [DOI] [PubMed] [Google Scholar]
- 6.Kim S, Nie H, Nesin V, Tran U, Outeda P, Bai CX, et al.: The polycystin complex mediates Wnt/Ca(2+) signalling. Nat Cell Biol 18: 752–764, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hanaoka K, Qian F, Boletta A, Bhunia AK, Piontek K, Tsiokas L, et al.: Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature 408: 990–994, 2000. [DOI] [PubMed] [Google Scholar]
- 8.Vassilev PM, Guo L, Chen XZ, Segal Y, Peng JB, Basora N, et al.: Polycystin-2 is a novel cation channel implicated in defective intracellular Ca(2+) homeostasis in polycystic kidney disease. Biochem Biophys Res Commun 282: 341–350, 2001. [DOI] [PubMed] [Google Scholar]
- 9.Su Q, Hu F, Ge X, Lei J, Yu S, Wang T, et al. : Structure of the human PKD1-PKD2 complex. Science 361: eaat9819, 2018 [DOI] [PubMed] [Google Scholar]
- 10.Foggensteiner L, Bevan AP, Thomas R, Coleman N, Boulter C, Bradley J, et al.: Cellular and subcellular distribution of polycystin-2, the protein product of the PKD2 gene. J Am Soc Nephrol 11: 814–827, 2000. [DOI] [PubMed] [Google Scholar]
- 11.Zhou J: Polycystins and primary cilia: Primers for cell cycle progression. Annu Rev Physiol 71: 83–113, 2009. [DOI] [PubMed] [Google Scholar]
- 12.Delling M, Indzhykulian AA, Liu X, Li Y, Xie T, Corey DP, et al.: Primary cilia are not calcium-responsive mechanosensors. Nature 531: 656–660, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pei Y: A “two-hit” model of cystogenesis in autosomal dominant polycystic kidney disease? Trends Mol Med 7: 151–156, 2001. [DOI] [PubMed] [Google Scholar]
- 14.Boyer O, Gagnadoux M-F, Guest G, Biebuyck N, Charbit M, Salomon R, et al.: Prognosis of autosomal dominant polycystic kidney disease diagnosed in utero or at birth. Pediatr Nephrol 22: 380–388, 2007. [DOI] [PubMed] [Google Scholar]
- 15.Reed B, Nobakht E, Dadgar S, Bekheirnia MR, Masoumi A, Belibi F, et al.: Renal ultrasonographic evaluation in children at risk of autosomal dominant polycystic kidney disease. Am J Kidney Dis 56: 50–56, 2010. [DOI] [PubMed] [Google Scholar]
- 16.Grantham JJ, Cook LT, Wetzel LH, Cadnapaphornchai MA, Bae KT: Evidence of extraordinary growth in the progressive enlargement of renal cysts. Clin J Am Soc Nephrol 5: 889–896, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu W, Peissel B, Babakhanlou H, Pavlova A, Geng L, Fan X, et al.: Perinatal lethality with kidney and pancreas defects in mice with a targetted Pkd1 mutation. Nat Genet 17: 179–181, 1997. [DOI] [PubMed] [Google Scholar]
- 18.Lu W, Shen X, Pavlova A, Lakkis M, Ward CJ, Pritchard L, et al.: Comparison of Pkd1-targeted mutants reveals that loss of polycystin-1 causes cystogenesis and bone defects. Hum Mol Genet 10: 2385–2396, 2001. [DOI] [PubMed] [Google Scholar]
- 19.Lu W, Fan X, Basora N, Babakhanlou H, Law T, Rifai N, et al.: Late onset of renal and hepatic cysts in Pkd1-targeted heterozygotes. Nat Genet 21: 160–161, 1999. [DOI] [PubMed] [Google Scholar]
- 20.Taguchi A, Kaku Y, Ohmori T, Sharmin S, Ogawa M, Sasaki H, et al.: Redefining the in vivo origin of metanephric nephron progenitors enables generation of complex kidney structures from pluripotent stem cells. Cell Stem Cell 14: 53–67, 2014. [DOI] [PubMed] [Google Scholar]
- 21.Morizane R, Lam AQ, Freedman BS, Kishi S, Valerius MT, Bonventre JV: Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat Biotechnol 33: 1193–1200, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taguchi A, Nishinakamura R: Higher-order kidney organogenesis from pluripotent stem cells. Cell Stem Cell 21: 730–746.e6, 2017. [DOI] [PubMed] [Google Scholar]
- 23.Mae S-I, Ryosaka M, Toyoda T, Matsuse K, Oshima Y, Tsujimoto H, et al.: Generation of branching ureteric bud tissues from human pluripotent stem cells. Biochem Biophys Res Commun 495: 954–961, 2018. [DOI] [PubMed] [Google Scholar]
- 24.Takasato M, Er PX, Chiu HS, Maier B, Baillie GJ, Ferguson C, et al.: Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature 526: 564–568, 2015. [DOI] [PubMed] [Google Scholar]
- 25.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E: A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li HL, Gee P, Ishida K, Hotta A: Efficient genomic correction methods in human iPS cells using CRISPR-Cas9 system. Methods 101: 27–35, 2016. [DOI] [PubMed] [Google Scholar]
- 27.Freedman BS, Brooks CR, Lam AQ, Fu H, Morizane R, Agrawal V, et al.: Modelling kidney disease with CRISPR-mutant kidney organoids derived from human pluripotent epiblast spheroids. Nat Commun 6: 8715, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cruz NM, Song X, Czerniecki SM, Gulieva RE, Churchill AJ, Kim YK, et al.: Organoid cystogenesis reveals a critical role of microenvironment in human polycystic kidney disease. Nat Mater 16: 1112–1119, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Czerniecki SM, Cruz NM, Harder JL, Menon R, Annis J, Otto EA, et al.: High-throughput screening enhances kidney organoid differentiation from human pluripotent stem cells and enables automated multidimensional phenotyping. Cell Stem Cell 22: 929–940.e4, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Devuyst O, Burrow CR, Smith BL, Agre P, Knepper MA, Wilson PD: Expression of aquaporins-1 and -2 during nephrogenesis and in autosomal dominant polycystic kidney disease. Am J Physiol 271: F169–F183, 1996. [DOI] [PubMed] [Google Scholar]
- 31.Baur BP, Meaney CJ: Review of tolvaptan for autosomal dominant polycystic kidney disease. Pharmacotherapy 34: 605–616, 2014. [DOI] [PubMed] [Google Scholar]
- 32.Wu H, Uchimura K, Donnelly EL, Kirita Y, Morris SA, Humphreys BD: Comparative analysis and refinement of human PSC-derived kidney organoid differentiation with single-cell transcriptomics. Cell Stem Cell 23: 869–881.e8, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Combes AN, Zappia L, Er PX, Oshlack A, Little MH: Single-cell analysis reveals congruence between kidney organoids and human fetal kidney. Genome Med 11: 3, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharmin S, Taguchi A, Kaku Y, Yoshimura Y, Ohmori T, Sakuma T, et al.: Human induced pluripotent stem cell-derived podocytes mature into vascularized glomeruli upon experimental transplantation. J Am Soc Nephrol 27: 1778–1791, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshimura Y, Taguchi A, Tanigawa S, Yatsuda J, Kamba T, Takahashi S, et al.: Manipulation of nephron-patterning signals enables selective induction of podocytes from human pluripotent stem cells. J Am Soc Nephrol 30: 304–321, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakagawa Y, Yamamoto T, Suzuki K, Araki K, Takeda N, Ohmuraya M, et al.: Screening methods to identify TALEN-mediated knockout mice. Exp Anim 63: 79–84, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evdokimov AG, Pokross ME, Egorov NS, Zaraisky AG, Yampolsky IV, Merzlyak EM, et al.: Structural basis for the fast maturation of Arthropoda green fluorescent protein. EMBO Rep 7: 1006–1012, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al.: Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131: 861–872, 2007. [DOI] [PubMed] [Google Scholar]
- 39.Tucker KL, Wang Y, Dausman J, Jaenisch R: A transgenic mouse strain expressing four drug-selectable marker genes. Nucleic Acids Res 25: 3745–3746, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoshimura Y, Taguchi A, Nishinakamura R: Generation of a three-dimensional kidney structure from pluripotent stem cells In: Organ Regeneration: 3D Stem Cell Culture & Manipulation, edited by Tsuji T, New York, Springer, 2017, pp 179–193 [DOI] [PubMed] [Google Scholar]
- 41.Li Z, Araoka T, Wu J, Liao H-K, Li M, Lazo M, et al.: 3D culture supports long-term expansion of mouse and human nephrogenic progenitors. Cell Stem Cell 19: 516–529, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ameku T, Taura D, Sone M, Numata T, Nakamura M, Shiota F, et al.: Identification of MMP1 as a novel risk factor for intracranial aneurysms in ADPKD using iPSC models. Sci Rep 6: 30013, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schneider CA, Rasband WS, Eliceiri KW: NIH image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roelfsema JH, Spruit L, Saris JJ, Chang P, Pirson Y, van Ommen G-J, et al.: Mutation detection in the repeated part of the PKD1 gene. Am J Hum Genet 61: 1044–1052, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kirsch S, Pasantes J, Wolf A, Bogdanova N, Münch C, Markoff A, et al.: Chromosomal evolution of the PKD1 gene family in primates [published correction appears in BMC Evol Biol 9: 14, 2009]. BMC Evol Biol 8: 263, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pinto CS, Raman A, Reif GA, Magenheimer BS, White C, Calvet JP, et al.: Phosphodiesterase isoform regulation of cell proliferation and fluid secretion in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 27: 1124–1134, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ahrabi AK, Jouret F, Marbaix E, Delporte C, Horie S, Mulroy S, et al.: Glomerular and proximal tubule cysts as early manifestations of Pkd1 deletion. Nephrol Dial Transplant 25: 1067–1078, 2010. [DOI] [PubMed] [Google Scholar]
- 48.Karner CM, Chirumamilla R, Aoki S, Igarashi P, Wallingford JB, Carroll TJ: Wnt9b signaling regulates planar cell polarity and kidney tubule morphogenesis. Nat Genet 41: 793–799, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sutters M, Germino GG: Autosomal dominant polycystic kidney disease: Molecular genetics and pathophysiology. J Lab Clin Med 141: 91–101, 2003. [DOI] [PubMed] [Google Scholar]
- 50.Boertien WE, Meijer E, Li J, Bost JE, Struck J, Flessner MF, et al. ; Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease CRISP : Relationship of copeptin, a surrogate marker for arginine vasopressin, with change in total kidney volume and GFR decline in autosomal dominant polycystic kidney disease: Results from the CRISP cohort. Am J Kidney Dis 61: 420–429, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Szabo L, Morey R, Palpant NJ, Wang PL, Afari N, Jiang C, et al.: Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development [published correction appears in Genome Biol 17: 263, 2016]. Genome Biol 16: 126, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heyer CM, Sundsbak JL, Abebe KZ, Chapman AB, Torres VE, Grantham JJ, et al. ; HALT PKD and CRISP Investigators : Predicted mutation strength of nontruncating PKD1 mutations aids genotype-phenotype correlations in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 27: 2872–2884, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peral B, Ong AC, San Millán JL, Gamble V, Rees L, Harris PC: A stable, nonsense mutation associated with a case of infantile onset polycystic kidney disease 1 (PKD1). Hum Mol Genet 5: 539–542, 1996. [DOI] [PubMed] [Google Scholar]
- 54.Di Mise A, Tamma G, Ranieri M, Centrone M, van den Heuvel L, Mekahli D, et al. : Activation of calcium-sensing receptor increases intracellular calcium and decreases cAMP and mTOR in PKD1 deficient cells. Sci Rep 8: 5704, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Belibi FA, Reif G, Wallace DP, Yamaguchi T, Olsen L, Li H, et al.: Cyclic AMP promotes growth and secretion in human polycystic kidney epithelial cells. Kidney Int 66: 964–973, 2004. [DOI] [PubMed] [Google Scholar]
- 56.Treschan TA, Peters J: The vasopressin system: Physiology and clinical strategies. Anesthesiology 105: 599–612; quiz 639–640, 2006 [DOI] [PubMed] [Google Scholar]
- 57.Liu J, Wess J: Different single receptor domains determine the distinct G protein coupling profiles of members of the vasopressin receptor family. J Biol Chem 271: 8772–8778, 1996. [DOI] [PubMed] [Google Scholar]
- 58.Ransick A, Lindström NO, Liu J, Zhu Q, Guo J-J, Alvarado GF, et al.: Single-cell profiling reveals sex, lineage, and regional diversity in the mouse kidney. Dev Cell 51: 399–413.e7, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mangos S, Lam PY, Zhao A, Liu Y, Mudumana S, Vasilyev A, et al.: The ADPKD genes pkd1a/b and pkd2 regulate extracellular matrix formation. Dis Model Mech 3: 354–365, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tsukiyama T, Kobayashi K, Nakaya M, Iwatani C, Seita Y, Tsuchiya H, et al.: Monkeys mutant for PKD1 recapitulate human autosomal dominant polycystic kidney disease. Nat Commun 10: 5517, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Homan KA, Gupta N, Kroll KT, Kolesky DB, Skylar-Scott M, Miyoshi T, et al.: Flow-enhanced vascularization and maturation of kidney organoids in vitro. Nat Methods 16: 255–262, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.