

Significance Statement
Progressive fibrosis, the underlying pathophysiologic process of CKD, is driven by myofibroblasts and activated profibrotic cells. Treatments targeting these cells for the reversal and amelioration of CKD are lacking. Experimental cytokine therapies administered locally are constrained by low bioavailability and transient activity, but nanoparticles can circumvent this and offer noninvasive kidney-targeted delivery of antifibrotic biologics, such as bone morphogenetic protein 7 (BMP7) and hepatocyte growth factor (HGF)–NK1 (HGF/NK1). The authors used nanoparticles containing plasmid DNA expressing either BMP7 or NGF/NKI enclosed in biocompatible natural materials for intravenous delivery of gene therapy to CKD mouse models. Delivery of BMP7 reversed the progression of fibrosis and regenerated tubules; delivery of HGF/NK1 halted CKD progression by eliminating collagen fiber deposition. Nanoparticle gene therapy shows potential as a noninvasive approach to treat CKD.
Keywords: chronic kidney disease, fibrosis, nanoparticles, gene therapy, regenerative medicine
Visual Abstract

Abstract
Background
Progressive fibrosis is the underlying pathophysiological process of CKD, and targeted prevention or reversal of the profibrotic cell phenotype is an important goal in developing therapeutics for CKD. Nanoparticles offer new ways to deliver antifibrotic therapies to damaged tissues and resident cells to limit manifestation of the profibrotic phenotype.
Methods
We focused on delivering plasmid DNA expressing bone morphogenetic protein 7 (BMP7) or hepatocyte growth factor (HGF)–NK1 (HGF/NK1) by encapsulation within chitosan nanoparticles coated with hyaluronan, to safely administer multifunctional nanoparticles containing the plasmid DNA to the kidneys for localized and sustained expression of antifibrotic factors. We characterized and evaluated nanoparticles in vitro for biocompatibility and antifibrotic function. To assess antifibrotic activity in vivo, we used noninvasive delivery to unilateral ureteral obstruction mouse models of CKD.
Results
Synthesis of hyaluronan-coated chitosan nanoparticles containing plasmid DNA expressing either BMP7 or NGF/NKI resulted in consistently sized nanoparticles, which—following endocytosis driven by CD44+ cells—promoted cellular growth and inhibited fibrotic gene expression in vitro. Intravenous tail injection of these nanoparticles resulted in approximately 40%–45% of gene uptake in kidneys in vivo. The nanoparticles attenuated the development of fibrosis and rescued renal function in unilateral ureteral obstruction mouse models of CKD. Gene delivery of BMP7 reversed the progression of fibrosis and regenerated tubules, whereas delivery of HGF/NK1 halted CKD progression by eliminating collagen fiber deposition.
Conclusions
Nanoparticle delivery of HGF/NK1 conveyed potent antifibrotic and proregenerative effects. Overall, this research provided the proof of concept on which to base future investigations for enhanced targeting and transfection of therapeutic genes to kidney tissues, and an avenue toward treatment of CKD.
CKD is the progressive loss of function cumulating in ESKD and organ failure. Approximately 8%–16% of the world’s population have advanced CKD,1 which has added complications of poor quality of life, high cost and demand for RRT, and risk of cardiovascular disease comorbidity.2,3 Despite multiple etiologies, the underlying pathophysiologic process of CKD is progressive fibrosis mediated by increased tissue myofibroblast cells,4 which can derive from interstitial fibroblasts and mesangial cells. Currently, there are no effective treatments that can prevent or reverse CKD.
Experimental cytokine therapies5 are expensive, with bioactivity and biosafety constrained by susceptibility to rapid breakdown and effects on a multitude of tissues.6–8 Invasive localized administration and repeat dosing can achieve therapeutic effects at target tissues, yet uncontrolled delivery and release are conducive to increased risk of cytotoxicity. Gene therapy offers circumvention of these issues; plasmid DNA (pDNA) synthesis and cloning is inexpensive and, after transient tissue transfection, elevated gene expression results in paracrine production of cytokines to affect the surrounding tissue microenvironment over several days. However, viral carriers have associated risks of immunogenicity,9 whereas lipid-based carriers are unspecific in terms of targeted transfection. Thus, design of noninvasive and nonviral gene carriers to deliver genetic material, in a targeted and specific manner, is a major focus of nanoparticle (NP) biotechnology. Multifunctional polymer-based NPs can influence physiochemical properties of cellular microenvironments, while exhibiting characteristic carrier properties useful in bioimaging and pharmaceutical applications.10–12 Tailoring NPs for kidney retention and to biointerface with key extracellular matrix (ECM) components or cell populations associated with renal fibrosis is becoming increasingly possible due to advances in understanding genomic and proteomic changes during CKD.
Natural polymers are biocompatible, biodegradable, and desirable for therapeutic delivery. Chitosan (CS) is a copolymer of randomly distributed β-1–4–linked d-glucosamine and N-acetyl-d-glucosamine, similar in structure to the N-acetyl-glucosamine and d-glucuronic acid repeats of hyaluronan (HA). CS bioactivity, biointerfacing, and self-assembling advantages are ideal for biomaterial construction.13 Low–mol wt CS (L-CS) NPs with cationic surfaces have increased circulation time and kidney-targeting properties.14–16 Electrostatic accumulation of NPs at the highly negatively charged glomerular basement membrane (GBM)17–19 can help increase the probability of NPs traversing the GBM to affect the cortical interstitium. The addition of anionic coatings (e.g., HA) to decrease surface charge can attenuate random nonspecific protein adsorption and mask CS NPs from macrophage activation and phagocytosis.20,21 Furthermore, HA-coated CS NPs (CS/HA NPs) can help direct therapeutic payloads to cells with abundant HA-binding CD44 receptors, such as epithelial, mesangial, and fibroblast cells,22–25 while conveying multifunctionality through stimulating proliferation26 via the CD44-associated Hippo pathway.27
Previous research identified factors effective in prevention of fibrosis and regulation of the myofibroblast phenotype. Stimulation by bone morphogenetic protein 7 (BMP7) induced internalization and breakdown of the myofibroblast HA glycocalyx (pericellular-coat), which destabilized myofibroblast phenotype and function.22,28 Hepatocyte growth factor (HGF) NK1 variant isoform (HGF/NK1) was suggested to inhibit myofibroblast differentiation while promoting cell migration and proliferation.29 In progressive fibrosis, tissue architecture and ECM composition are compromised. Therefore, as opposed to targeted apoptosis of myofibroblasts, increased uptake of phenotype-influencing therapies may be more conducive to remodeling and reparation of fibrotic ECM. Here, we fabricated biocompatible and biodegradable CS/HA NPs as BMP7 and HGF/NK1 gene carriers (CS/HApDNA NPs) for noninvasive delivery to CKD renal tissues. We hypothesized that CS/HApDNA NPs would actively interact with CD44+ kidney cells; result in the paracrine production of BMP7 or HGF/NK1; attenuate profibrotic phenotype acquisition; promote remodeling of fibrous collagen-rich ECM; and lead to the structural and functional recovery of renal tissue. Specifically, we assessed CS/HApDNA NP characteristics and effect on fibroblast, mesangial cell, and myofibroblast activity in vitro. We then assessed intravenous (iv) delivery of CS/HApDNA NPs, their targeting to kidney tissue, and their capacity to inhibit in vivo models of progressive renal fibrosis.
Methods
Materials
L-CS (mol wt 50,000, 85% deacetylated) was purchased from Haidebei Marine Bioengineering (Jinan, Shandong, China). Medium–mol wt HA (M-HA; mol wt 80,000–100,000) was purchased from Heowns Biochemical Technology (Tianjin, China). BMP7 and HGF/NK1 overexpression plasmids were custom designed in-house and outsourced for synthesis by GenScript (Nanjing, Jiangsu, China). All chemical reagents were purchased from Heowns Biochemical Technology, unless otherwise stated. All tissue culture reagents were from HyClone (Thermo Fisher Scientific, Beijing, China) and plasticware was from BD Biosciences (San Jose, CA), unless otherwise stated. Cells used in experiments were: human lung fibroblasts (HFL1; ATCC CCL-153, Manassas, VA; passages 4–8); human kidney mesangial cells (HKM; passages 4–8) isolated from donor tissue according to previously detailed methodology30 and identified according to previously detailed methodology30,31; and mouse kidney fibroblasts (MKF; passages 2–3) isolated from mouse kidney tissue and identified according to previously detailed methodology.32 TGF-β1 was purchased from R&D Systems (Shanghai, China). All antibodies were purchased from Abcam (Cambridge, UK).
NP Synthesis and Characterization
CS NP cores were synthesized according to optimized protocols from previous literature,33–35 with minor adjustments. Briefly, 1 mg/ml CS was dissolved in 0.1% v/v glacial acetic acid/ddH2O by sonication for 10 minutes. After filtration through a 0.22-µm pore-size filter, the CS solution was adjusted with ddH2O to 0.64 mg/ml. For every 1 ml of 0.64 mg/ml CS solution, 71 µl of 1 mg/ml filtered sodium tripolyphosphate in 0.1% v/v acetic acid/ddH2O containing pDNA (2% w/w of final NP mass yield) was added slowly to the solution under 1000-rpm stirring using a Scilogex OS20-Pro overhead stirrer (Scilogex, Rocky Hill, CT) to achieve ionic gelation. The solution was constantly stirred for an additional 30 minutes at RT before the volume was doubled with the addition of 0.1 M acetate buffer (pH 5), and a sample of CS-encapsulated pDNA NPs (CSpDNA NPs) was taken for further analysis. A volume (1 ml of HA per 1.06 ml of CS NP solution) of 0.1 mg/ml HA in 0.1 M acetate buffer was added to the remaining CS NP solution at a constant rate of 1.5 ml/min, using an elevated peristaltic syringe pump and under a 1500-rpm stirring speed. After the complete addition of the HA solution, stirring was continued for 30 minutes to achieve HA-coated CSpDNA NPs (CS/HApDNA NPs). The NP solution was subjected to dialysis against PBS over 3 days, using dialysis membranes with 10-kDa mol wt cut off. The dialyzed NP solution was subsequently concentrated using Amicon Ultra-4 10K centrifugal spin columns (Merck-Millipore, Billerica, MA) before use in further experimentation. CS, CSpDNA, CS/HA, and CS/HApDNA NPs were resuspended to 5 mg/ml in PBS. Average NP diameter, polydispersity index (PDI), and surface charge (ζ-potential) were assessed by dynamic light scattering (DLS) and using a Zetasizer Nano ZS90 (Malvern Panalytical, Malvern, UK). NPs were adsorbed onto hydrophilic carbon film (Zhongjingkeyi Technology, Beijing, China) and dry NP morphology was assessed by transmission electron microscopy (TEM) (FEI Talos F200C; Thermo Fisher Scientific).
pDNA Release and Protection Assays
CSpDNA and CS/HApDNA NPs were resuspended to 5 mg/ml in PBS. For NP stability assays, pDNA release was measured at the specified time points over a course of 6 months, by measuring the OD of 2 μl of supernatant after centrifugation of the NP solution (15,000 × g, 30 minutes, 4°C). The DNA concentration of the sample was measured using a BioDrop µLITE+ (BioDrop, Cambridge, UK) and expressed as a percentage of the initial encapsulated pDNA concentration. Protection assays were performed by incubating 1 mg/ml NP sample with 10 U of DNase I (Thermo Fisher Scientific), 1 U of chitosanase (Merk-Millipore), 10 µg/ml hyaluronidase (Sigma-Aldrich), or both chitosanase and hyaluronidase, at 37°C for 90 minutes. DNase I was inactivated by heating to 65°C for 10 minutes, before mixing with DNA loading buffer and DNA release/degradation assessment by 1% agarose gel flat-bed electrophoresis. Gels were visualized and photographed using a Tanon 2500 Gel Imaging System (Tanon, Shanghai, China). If the DNA remained encapsulated within NPs, the DNA remained within the loaded wells, because NPs interfered with DNA migration through the gel.
Cell Culture
MKF and HKM cells were cultured in DMEM. HFL1 cells were cultured in Kaighn’s modified Ham’s F-12 medium (F-12K) containing NaHCO3/HEPES (pH 7). Cells were grown to confluence in T75 culture flasks, in the presence of 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells were subcultured using 0.05% trypsin/EDTA at a ratio of 1:3. Cells were maintained at 37°C and 5% CO2, and growth medium was replenished every 3 days until experimentation. Cells were growth-arrested in serum-free medium for 48 hours before experiments, and all experiments were performed under serum-free conditions.
NP Transfection Efficiency Testing
Cells were grown to 50% confluence on eight-well chamber slides (Nunc, Thermo Fisher Scientific), before treatment with 10 mg/ml CSFluc/EGFP or CS/HAFluc/EGFP NPs containing various percentages of pDNA (w/w of CS/HA). After 48 hours of treatment time, cells were washed, fixed with 4% paraformaldehyde, and mounted in DAPI Fluoromount-G (Thermo Fisher Scientific). Cells were analyzed and imaged for EGFP expression using a Zeiss Axio Imager Z1 UV fluorescence microscope (Zeiss, Jena, Germany). Cells positive for EGFP expression were counted and the percentage efficiency of transfection was calculated and graphed. Alternatively, bioluminescence intensity (BLI) assays were performed to detect firefly luciferase (Fluc) activity in transfected cells, using an IVIS Lumina imaging system (PerkinElmer, Waltham, MA). Naked pDNA with no carrier was used for negative controls. Lipofectamine LTX (Invitrogen, Thermo Fisher Scientific) transfection was used for positive controls.
Functional Cell Assays
Cells were seeded into 12-well culture plates at a density of 2.5×104 and, when cultures had reached 50% confluence, the culture medium was replaced with fresh serum-free F-12K or serum-free DMEM for 48 hours (growth arrest). Cells were treated with the indicated treatments over 24 hours. Cell Counting Kit-8 (CCK-8; Beyotime, Haimen, China) assays were performed as follows: 10% of CCK-8 reagent was added to the target wells (90% F-12K or 90% DMEM), 1 hour before the indicated time points. CCK-8 utilizes water-soluble tetrazolium salt–SST-8, which is reduced by dehydrogenases of cells, to give soluble orange-colored formazan. The amount of the formazan dye generated by dehydrogenase is directly proportional to the number of cells. Plates were incubated for 1 hour before the culture medium was aliquoted into wells of a 96-well plate. Absorbance was measured at 450-nm wavelength, using a Bio-Rad optical plate reader (Bio-Rad, Hercules, CA). Fresh culture medium was used to replenish the culture plates.
Cells were seeded into 12-well culture plates and allowed to reach 100% confluence. Growth-arrested cell monolayers were subjected to scratch-wound–induced monolayer interruption by scratching a 20-μl pipette tip across the center of each well. The indicated treatments were immediately added to the culture wells and, at 0, 1, 2, and 3 days, plate wells were observed under a light microscope. Images were captured using a digital camera and migration was assessed by measuring the remaining wound area using ImageJ v.1.37c software (National Institutes of Health [NIH], Bethesda, MD).
Reverse Transcription and Quantitative Real-Time PCR
Reverse transcription and real-time quantitative PCR (qRT-PCR) was used to assess the mRNA expression of genes of interest, utilizing custom PCR primers (Supplemental Table 1) synthesized by BGI Genomics (Beijing, China). Cells were grown in six-well culture plates and washed with PBS before lysis and collection in TRIzon Reagent (CWBio, Beijing, China). Tissues were harvested, halved, and rinsed thoroughly with PBS before complete homogenization in TRIzon Reagent (CWBio) using a HerosMole TL-1000 homogenizer (DHS Bio, Beijing, China). Total RNA purification was according to the manufacturer’s protocol. Reverse transcription was done using the TransScript First-Strand cDNA Synthesis SuperMix kit with one-step gDNA removal according to the manufacturer’s protocol (TransGen, Beijing, China), using the random primer method for the initiation of cDNA synthesis. Nuclease-free H2O replaced the RNA sample to serve as a negative control for the reverse transcription reaction. For qRT-PCR, 10 µl of 2× TransStart Top Green qPCR SuperMix was added to each well of a 96-well PCR plate containing 0.4 µl of forward primer, 0.4 µl of reverse primer, 500 ng of RNA, and nuclease-free water to a total volume of 20 µl. The qRT-PCR reaction was completed using a CFX96 Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA). Amplification used the following parameters: 94°C for 30 seconds; followed by 40 cycles of 94°C for 5 seconds, 55°C for 15 seconds, 72°C for 10 seconds; and then a dissociation stage (melt-curve). Simultaneous qRT-PCR reactions were performed for GAPDH or Gapdh as standard reference genes and using nuclease-free water in the place of cDNA as a negative control.
ELISA
The absolute protein quantifications of BMP7 and HGF were measured in cell supernatant, blood serum, and homogenized kidney tissue using commercially available ELISA kits (BMP7, #DBP700; R&D Systems; HGF, #DHG00B; R&D Systems). Assays were performed according to the manufacturer protocols. Cell supernatant was collected every 2 days for analysis. Culture wells were replenished with fresh culture medium containing 0.1% FBS. An equal concentration of supernatant protein (200 µg/ml) was assessed in each ELISA plate well to account for differences in cell numbers. Background detection for DMEM containing 0.1% FBS was performed alongside ELISA assays to account for the presence of cytokines in FBS.
Immunocytochemistry
Cells were grown to 50%–60% confluence in eight-well glass chamber slides (Nunc, Thermo Fisher Scientific), before growth arrest and treatment. Cells were washed with PBS before fixation in 4% paraformaldehyde for 10 minutes at room temperature. Next, cells were permeabilized with 0.1% (v/v) Triton X-100 in PBS for 10 minutes at room temperature. Slides were blocked with 1% goat serum (TBD Science, Tianjin, China) for 30 minutes before washing with 0.1% (w/v) BSA in PBS. Subsequently, slides were incubated with monoclonal mouse anti–α-smooth muscle actin (α-SMA) antibody (1:50; Sigma-Aldrich), diluted in 0.1% BSA-PBS, overnight at 4°C. After further wash steps, slides were incubated with goat anti-mouse IgG CF-488 secondary antibody (1:1000) in 0.1% BSA-PBS for 1 hour at room temperature and under darkness. For F-actin visualization, after fixation and permeabilization, cells were incubated with phalloidin conjugated to FITC (Sigma-Aldrich) in 0.1% BSA-PBS overnight at 4°C. Cells were then mounted in DAPI Fluoromount-G (Thermo Fisher Scientific) and analyzed and imaged using a Zeiss Axio Imager Z1 UV fluorescence microscope (Zeiss).
Animal Studies
A total of 46 C57BL/6 mice (male, 6–7 weeks old and average weight 24.95±0.53 g at point of experimentation) were used in this study. Mice were randomly assigned into groups of five for Sham, UUO, CS/HA, CS/HABMP7, and CS/HAHGF/NK1 treatments. Whereas three mice were used for CS/HAFluc/EGFP detection of Fluc expression in major organs after iv NP administration, six mice were used for UUO model generation (two per group: 3, 5, and 7 days post UUO), and 12 mice were used to determine NP biodistribution and biosafety (three per group: Sham, CS/HA, CS/HABMP7, and CS/HAHGF/NK1). Animal experiments were approved by the Animal Experiments Ethical Committee of Nankai University and complied with the Guide for Care and Use of Laboratory Animals. For UUO surgery, mice were anesthetized by isoflurane inhalation. Ventral flank fur was removed using hair-removal cream and the skin was sterilized using ethanol. A small 2-cm incision through the skin and peritoneal cavity was made and the ureter of the animal’s left kidney was tied off using 4–0 silk suture. The peritoneum and skin were then sutured using medical-grade sutures. Animals were kept at 37°C and monitored until they regained consciousness. On the third day post UUO surgery, 25 mg of NP per kilogram of body mass was delivered by iv injection into tail veins. In Sham animals, an equal volume of PBS was delivered. Mice were housed in standard rodent cages with ad libitum access to water and food. At 7 days postsurgery, animals were euthanized by isoflurane inhalation followed by overdose injection of chloral hydrate. Blood samples were collected from the retro-orbital sinus for assessment of blood serum creatinine (SCr) and BUN, according to kit manufacturer protocols (Cayman Chemical, Ann Arbor, MI). Before organ collection, blood was removed by perfusion injections of PBS into the left ventricle of the heart.
Tissue Histopathology and Immunostaining
Kidney tissue was halved and fixed in 10% formalin overnight and then embedded in optimal cutting temperature compound (Tissue Tek; Sakura Finetek, Torrance, CA) for cryosectioning to 4-µm-thick sections. Sections were stained with hematoxylin and eosin (H&E) to observe tissue architecture and tubular atrophy; periodic acid–Schiff (PAS) was used to observe hyaline cast formation. Masson’s trichrome and Sirius Red were used to visualize deposition and the morphometrics of collagen, respectively. Immunostaining was performed to visualize proliferating cell nuclear antigen (PCNA), α-SMA, collagen IV, fibronectin, EGFP, E-cadherin, PDGF receptor-α (PDGFRα), CD68, and CD31 protein expression, following the antibody and appropriate secondary antibody manufacturer’s recommendations (Abcam). Staining was quantified using ImageJ v.1.37c software (NIH). Fully blinded tissue collection and sectioning (three separate researchers), staining and imaging (three separate researchers), and data analysis (two separate researchers) were performed by different researchers throughout the investigation.
Statistical Analyses
For all data with two independent variable groupings, two-way ANOVA was used, followed by Bonferroni’s multiple comparisons and post-hoc Tukey’s test. For in vitro experiments, graphical data are expressed as mean±SEM of three independent experiments; for in vivo experiments, graphical data are expressed as mean±SD of five animals. All data were analyzed using GraphPad Prism v.7 (GraphPad Software, San Diego, CA). Where considered statistically significant, analysis is displayed on figures as: *P≤0.05; **P≤0.01; and ***P≤0.001; or NS if no significance was found.
Results
Characterization of CS/HA NPs
L-CS was chosen to encapsulate pDNA due to cationic kidney-targeting properties14–16 and improved endosomal complex disassembly and escape versus high-mol wt CS.35 Medium-mol wt HA (M-HA) coatings were chosen to exploit increased CD44 binding affinity36 and endocytosis37 (Figure 1A). After optimization of size, PDI, and charge, 50-kDa CS cores (Supplemental Figure 1A) and 1:4 HA coatings (HA to CS ratio; Supplemental Figure 1B) were used to synthesize HA-coated and pDNA-loaded CS NPs (CS/HApDNA NPs). Finalized CS/HApDNA NPs were assessed by DLS and TEM (Figure 1B). TEM showed that dry-state NPs were spherical, with diameters of 45.67±10.16 nm. DLS demonstrated that wet-state CS/HApDNA NP diameters ranged between 200 and 280 nm (Supplemental Figure 2A, Supplemental Table 2), indicating hydrophilic NPs that swelled to approximately five-fold their dry-state size. NP wet/dry size differences were determined suitable to pass glomerular capillary endothelium fenestrae under fluctuating kidney tonicity. Inclusion of 2% w/w pDNA within CS cores resulted in HA coatings that were more densely compacted with further reductions in surface charge. PDI ranged from 0.30 to 0.37 across all NPs synthesized (Supplemental Figure 2B), showing consistent uniformity and reproducibility of the method. Retained positive surface charge (ζ-potential) (Supplemental Figure 2C) of CS/HApDNA NPs suggested that circulation time and glomeruli accumulation would be maintained. CS/HApDNA NPs remained stable for 1 month postsynthesis (Supplemental Figure 3A), preserving approximately 85% and 70% of encapsulated pDNA when kept undisturbed at 4°C and RT, respectively. Incubation in 37°C serum led to approximately 62% of DNA retention after 1 week, suggesting that the presence of additional proteins interfered with polymer stability, an outcome that may be reflective of in vivo circulation. Therefore, NPs were stored at 4°C and used within 1 month. DNA protection assays indicated that DNase I degradation of pDNA was prevented by CS/HA encapsulation (Supplemental Figure 3B). Degradation of HA by hyaluronidase could not release pDNA from CS (Supplemental Figure 3C). Only when hyaluronidase and chitosanase enzymes were used together, could pDNA be released from NPs.
Figure 1.
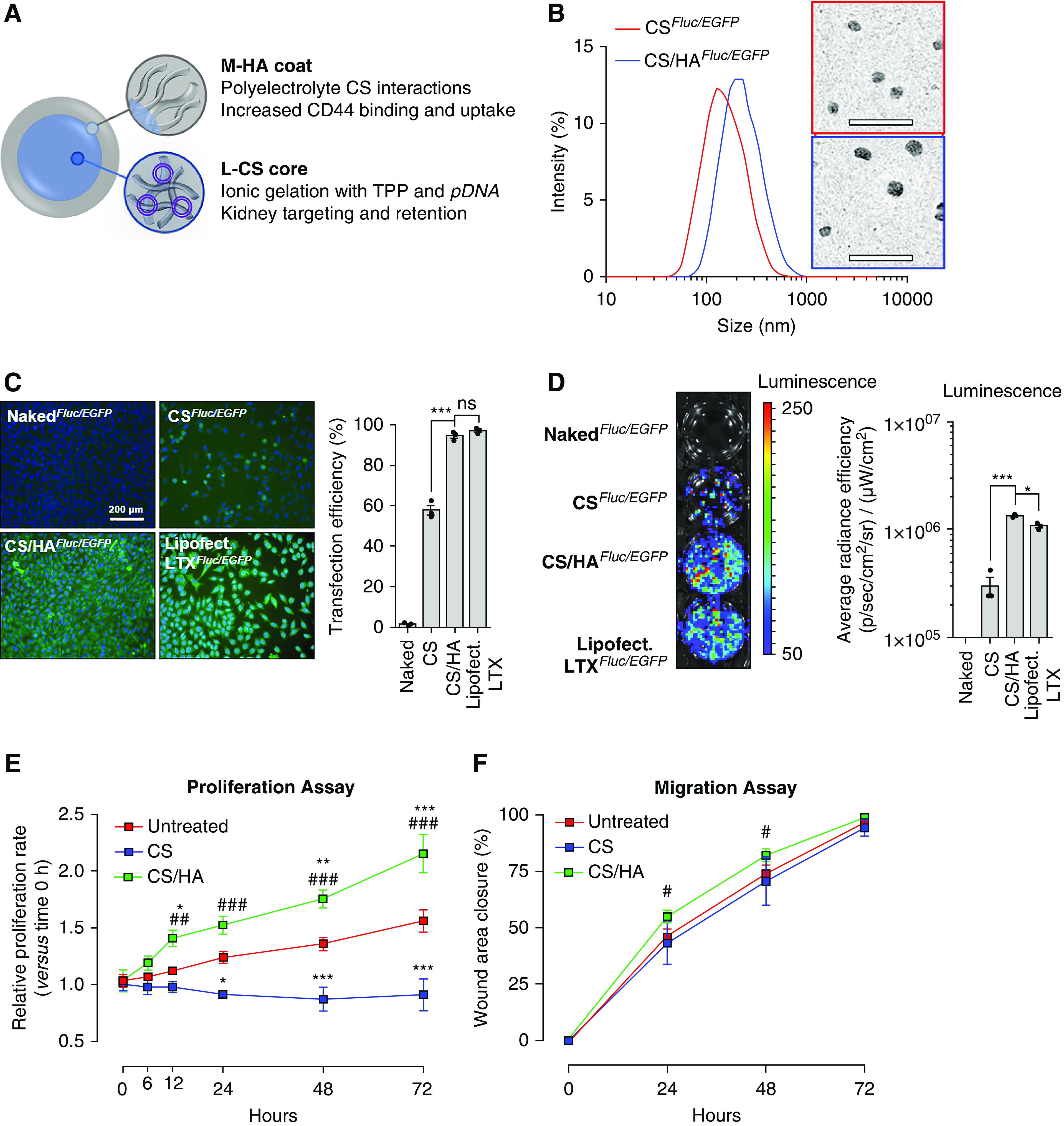
CS/HApDNA NPs have consistent size, protect against DNA release, and have high transfection efficiency. (A) Schematic of L-CS/M-HA NP design and material rationale. (B) DLS of CSFluc/EGFP and CS/HAFluc/EGFP NP size with representative TEM images alongside. Scale bars, 200 nm. (C) Transfection efficiency of equivalent 2% w/w pDNA in 10 µg/ml CS/HA NPs, using naked pDNA; CS NP; CS/HA NP; and Lipofectamine LTX incubations with MKFs, as visualized by fluorescence microscopy of EGFP expression at 48 hours post-treatment. Scale bar, 200 µm; original magnification, ×100. Quantification of percentage of cells transfected is shown alongside. (D) Transfection efficiency as visualized by BLI assay for Fluc activity at 48 hours post-treatment. Quantified luminescence is shown alongside. (E) Proliferation and (F) migration rates of untreated, CS NP–, and CS/HA NP–treated HFL1 cells over the course of 72 hours. All data are representative of three independent experiments; numeric data are displayed as the mean±SEM and statistical analysis is shown as: *P≤0.05; ***P≤0.001.
Transfection Efficiency of CS/HA NPs
MKFs monolayers incubated with various NP concentrations for 48 hours were assessed for cytotoxicity (Supplemental Figure 4A). No cytotoxicity was observed in 0.01–10 µg/ml concentration ranges. Cytotoxicity was observed at 100 µg/ml CS-only NP treatments and at 1000 µg/ml CS/HA NP treatments. Therefore, to avoid cytotoxicity, 10 µg/ml NPs were selected for transfection efficiency evaluation. CS/HAFluc/EGFP NPs loaded with various amounts of pDNA were used to transfect 50%-confluent MKFs (Supplemental Figure 4B). It was observed that 1%–5% w/w pLV-Fluc/EGFP–containing CS/HA NPs resulted in detectable expression of EGFP by fluorescence microscopy. EGFP expression in HFL1 cells was saturated at 2% w/w pLV-Fluc/EGFP loading (Supplemental Figure 4C). Coating with HA resulted in markedly improved transfection efficiency, compared with CS-only NPs, with efficiency rates similar to standardized Lipofectamine LTX transfection (Figure 1C). It was noted that CS-only NPs restricted cell growth, whereas CS/HA NPs increased cell numbers per microscopic field view. BLI assays were used to screen for the more sensitive detection of Fluc activity (Figure 1D), which also suggested that M-HA coatings enhanced transfection efficiency. Increased cell numbers could explain the significantly higher detectable BLI signal in CS/HA-mediated transfection than in Lipofectamine LTX–transfected cells.
We next assessed CD44-dependent CS/HA internalization and transfection. MKFs were transfected with siRNA against Cd44 (siCd44) and knockdown (65%–70% knockdown efficiency) was confirmed by qRT-PCR (Supplemental Figure 5A). Interestingly, CS/HA NPs stimulated the expression of Cd44, but this was inhibited by siCd44 transfection. Fluc expression from CS/HAFluc/EGFP treatments was attenuated by siCd44 pretreatment (Supplemental Figure 5B). Similarly, BLI for Fluc activity was significantly decreased in siCd44-transfected cells (Supplemental Figure 5C). These data confirmed our hypothesis that CS/HA-mediated transfection was predominantly regulated via CD44-dependent internalization.
Effects of CS/HA NP Delivery of BMP7 and HGF/NK1 on Cellular Function
To further investigate the observed increases in cell numbers after CS/HA NP treatments, we assessed cellular proliferation and migration in response to NPs. Treatment with CS NPs resulted in arrest of proliferation over 72 hours, when compared with untreated cells. In contrast, CS/HA NP treatment significantly increased proliferation rates of fibroblasts at all time points, from 12 hours onwards (Figure 1E). However, CS NPs had no influence over migration rates. CS/HA NP treatments resulted in significant, albeit relatively minor, increases in migration rates at 24–48 hours (Figure 1F). In brief, CS-only NPs inhibited cell division but not locomotion, whereas CS/HA NPs positively regulated both functions.
Next, pDNA encoding BMP7 or HGF/NK1 open-reading frames was encapsulated within CS/HA NPs (herein designated CS/HABMP7 and CS/HAHGF/NK1, respectively). Significantly elevated expression of BMP7 (Figure 2A) and HGF/NK1 mRNA (Figure 2B) was confirmed at 48 hours post CS/HA NP delivery to fibroblasts and mesangial cells. Detection of full-length HGF mRNA by qRT-PCR confirmed that HGF/NK1 overexpression was specific (Supplemental Figure 6A). Conditioned medium from cultures was subjected to western blot for total protein expression of BMP7 or HGF/NK1 (Supplemental Figure 6B). Plasmid-loaded CS/HA NPs successfully increased both mRNA and protein expression of BMP7 or HGF/NK1. Empty CS/HA NP treatment resulted in a slight but significant induction of BMP7 mRNA, and this increase was reflected in immunoblots of conditioned medium. Prolonged protein secretion was assessed by ELISA assays. BMP7 production peaked at 6 and 2 days in fibroblasts and mesangial cells, respectively (Figure 2C). BMP7 remained significantly elevated in supernatant for at least 10 days. HGF/NK1 production was assessed by pan-HGF ELISA, exhibiting peak production at 6 and 4 days in fibroblasts and mesangial cells, respectively (Figure 2D). HGF/NK1 expression remained significantly elevated in supernatant for at least 8 days. These data were indicative of the longevity of transient transfection by CS/HA NPs. We next assessed influence of gene delivery on cellular function, first confirming the effects of BMP7 production on pericellular HA glycocalyx thickness (Supplemental Figure 6C). BMP7-transfected myofibroblasts resulted in a reduced glycocalyx thickness, more comparable to resting fibroblasts, whereas HGF/NK1 transfection did not influence HA glycocalyx size. Regarding proliferation, CS/HABMP7 tended to promote earlier increases in cell growth, but rates were not significantly higher than empty CS/HA or CS/HAHGF/NK1 NPs. All NP treatments induced increased rates of proliferation, compared with untreated cells (Supplemental Figure 6D). Migration rates were greatly influenced by CS/HAHGF/NK1 NP treatments, more so than empty and BMP7-loaded NPs (Supplemental Figure 6E). Taken together, these data demonstrated BMP7 promotion of proliferation, HGF/NK1 promotion of migration, and the positive regulation of cell functions by CS/HA NP treatment, further highlighting enhanced biocompatibility conveyed by M-HA–coated NPs.
Figure 2.
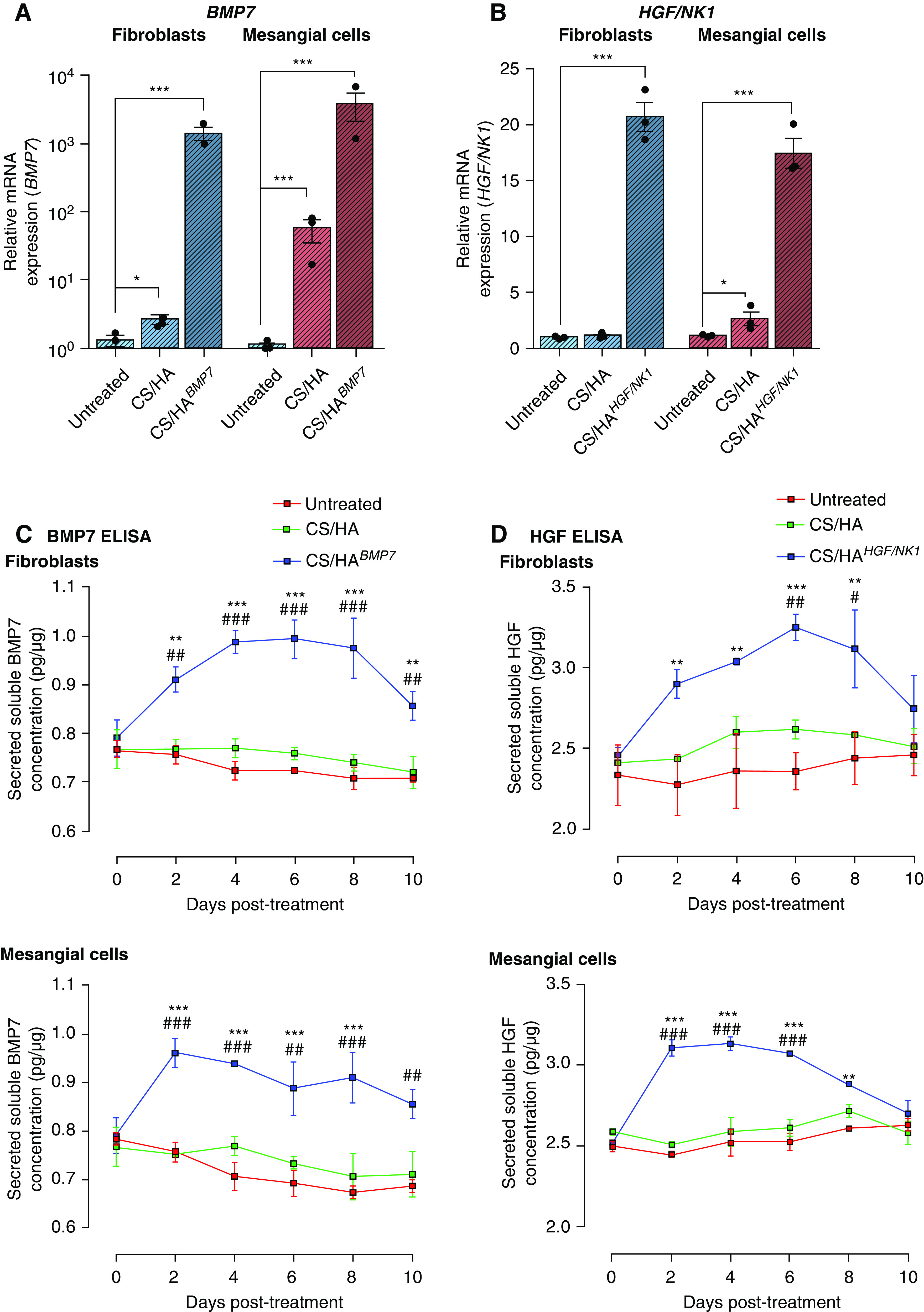
CS/HA, CS/HABMP7, and CS/HAHGF/NK1 NP–mediated transfections result in successful paracrine production over 10 days in fibroblasts and mesangial cells. (A) Successful BMP7 overexpression in CS/HABMP7 NP–treated HFL1 cells (blue bars) and HKM cells (red bars) after 72 hours, as measured by qRT-PCR. (B) Successful HGF/NK1 overexpression in CS/HAHGF/NK1 NP–treated HFL1 cells (blue bars) and HKM cells (red bars) after 72 hours, as measured by qRT-PCR. (C) BMP7 overexpression in CS/HABMP7 NP–treated HFL1 and HKM cells results in elevated production and secretion of BMP7 into the supernatant over 10 days, as measured by BMP7 ELISA assay. (D) HGF/NK1 overexpression in CS/HAHGF/NK1 NP–treated HFL1 and HKM cells results in elevated production and secretion of HGF/NK1 into the supernatant over 10 days, as measured by HGF ELISA assay. All data are representative of three independent experiments; graphed data are displayed as the mean±SEM and statistical significance is shown as: *P≤0.05, **P≤0.01, and ***P≤0.001 versus untreated; #P≤0.05, ##P≤0.01, and ###P≤0.001 versus CS/HA.
Antifibrotic Gene Delivery In Vitro
TGF-β1 drives myofibroblast differentiation.38–40 To evaluate antifibrotic actions of BMP7 or HGF/NK1 gene delivery in vitro, TGF-β1–stimulated fibroblasts and mesangial cells were treated with or without CS/HApDNA NPs. Both BMP7 and HGF/NK1 gene transfection significantly attenuated TGF-β1–induced fibrosis-associated mRNA: α-SMA, collagen I, and extra domain A (EDA)–fibronectin (Figure 3, A–C). In fibroblasts, collagen III, fibronectin, and TGF-β1 mRNA expression was also inhibited (Supplemental Figure 6F). BMP7 transfection tended to be more effective at inhibiting EDA-fibronectin and TGF-β1 expression, whereas HGF/NK1 transfection tended to be more effective at attenuating the expression of collagens. Immunostaining was performed to visualize stress fiber formation (α-SMA; Figure 3D) and morphologic hallmarks (increased cell size and actin reassembly; Figure 3E), indicative of myofibroblast differentiation. There was clear α-SMA incorporation into stress fibers and enlarged web-like morphologies in TGF-β1 and CS/HA+TGF-β1–treated fibroblasts and mesangial cells. Both BMP7 and HGF/NK1 transfection by CS/HA NPs prevented the expression and visualization of α-SMA. BMP7 reverted cells to spindle-shaped fibroblast- and mesangial-like cells, whereas HGF/NK1-transfected cells maintained enlarged morphologies but without α-SMA expression, a phenomenon suggested to be indicative of promigratory activated fibroblasts or deactivated myofibroblasts.41,42 Western blot analysis assessed total α-SMA protein expression in untreated and TGF-β1–treated cells (Supplemental Figure 6G). TGF-β1 induced α-SMA protein expression eight-fold in fibroblasts and 1.5-fold in mesangial cells, and gene delivery attenuated this fold-induction. Mesangial cell basal expression of α-SMA was evident, a finding explained by increased α-SMA in mesangial cells when cultured in the absence of serum.43 It was noted that culture conditions used in this investigation differed; under culture conditions used here, TGF-β1 was required to induce distinct cytoskeletal stress fiber incorporation of α-SMA. Overall, these data showed that CS/HA-mediated antifibrotic gene delivery prevented TGF-β1–driven myofibroblast differentiation. Before the establishment of animal models for in vivo studies, MKFs were also treated with BMP7- or HGF/NK1-loaded CS/HA NPs. Successful overexpression was confirmed by qRT-PCR (Supplemental Figure 7, A and B) and fibrosis-associated gene expression in human cells was mirrored in mouse cells (Supplemental Figure 7C), as were the inhibitory effects on α-SMA upregulation (Supplemental Figure 7D). Thus, we confirmed that CS/HA NP transfection would convey activity in murine disease models.
Figure 3.

CS/HA-mediated overexpression of BMP7 or HGF/NK1 attenuates in vitro myofibroblast differentiation. Analysis of (A) ACTA2, (B) COL1A1, and (C) EDA-FN1 mRNA expression in HFL1 (blue bars) and HKM (red bars) cells after 72-hour NP treatments, with or without 10 ng/ml TGF-β1. (D) Immunocytochemistry to visualize α-SMA stress fiber formation and (E) F-actin reassembly after 72-hour NP treatments, with or without 10 ng/ml TGF-β1. Scale bars, (D) 20 µm; original magnification, ×400; (E) 50 µm; original magnification, ×200. Images are representative of three independent experiments. Data are shown as the mean±SEM of three separate experiments. Statistical significance is shown as: *P≤0.05; **P≤0.01; ***P≤0.001.
Prevention of Renal Pathology
Mice received iv tail injections of CS/HAFluc/EGFP NPs, and administration to major organs was assessed by Fluc expression after 3 days (Figure 4A). The results indicated that Fluc transfection had mainly occurred in the kidneys and liver, suggesting NP accumulation within these tissues. A smaller amount of Fluc expression was detected within the spleen, and, in one mouse, low-level Fluc expression was detected in lung tissue. Upon closer examination of kidney tissues from these mice, we observed that EGFP protein expression was predominantly detected in cells in proximity to the glomeruli (magenta arrows) and a number of cells in the cortical interstitium or tubules (cyan arrows) (Supplemental Figure 8), suggesting that successful transfection and protein production had occurred. In light of these findings, we established UUO injury models. According to our model optimization, by day 3 postsurgery fibrosis- and kidney injury–associated genes had elevated expression in UUO kidneys, compared with control kidneys (Supplemental Figure 9A). Tissue damage and collagen deposition indicated the onset of fibrogenesis at day 3, and this was extensive by day 7 (Supplemental Figure 9B). Therefore, we chose to administer NP treatments on day 3 post UUO and assessed kidney tissue at day 7 post UUO surgery (Figure 4B). Closer examination of cell types positive for transfection showed that EGFP+ cells were also E-cadherin+ in tubules and PDGFRα+ surrounding glomeruli and in the cortical interstitium (white arrows; Figure 4C), whereas CD68+ macrophages and CD31+ endothelial cells were negative for EGFP expression (Supplemental Figure 10A), suggesting that the predominant recipients of CS/HApDNA NPs were tubule epithelial cells, mesangial cells, and interstitial fibroblasts. We next confirmed that BMP7 and HGF/NK1 had been successfully overexpressed in unligated control and UUO kidneys, after CS/HApDNA iv injection at both the mRNA (Figure 4, D and E) and protein levels (Figure 4F, Supplemental Figure 10B). Both BMP7 and HGF/NK1 had expression patterns closely resembling the previously observed EGFP expression, and tissue-wide protein expression analysis indicated that protein expression was predominantly localized to the cortex surrounding glomeruli (localization indicated by * on histograms), with minimal expression within the medulla regions (Figure 4G). In addition, assessment of blood serum expression of BMP7 and HGF by ELISA indicated that CS/HApDNA NP treatments resulted in circulating expression readings comparable to Sham control groups (Supplemental Figure 10C).
Figure 4.
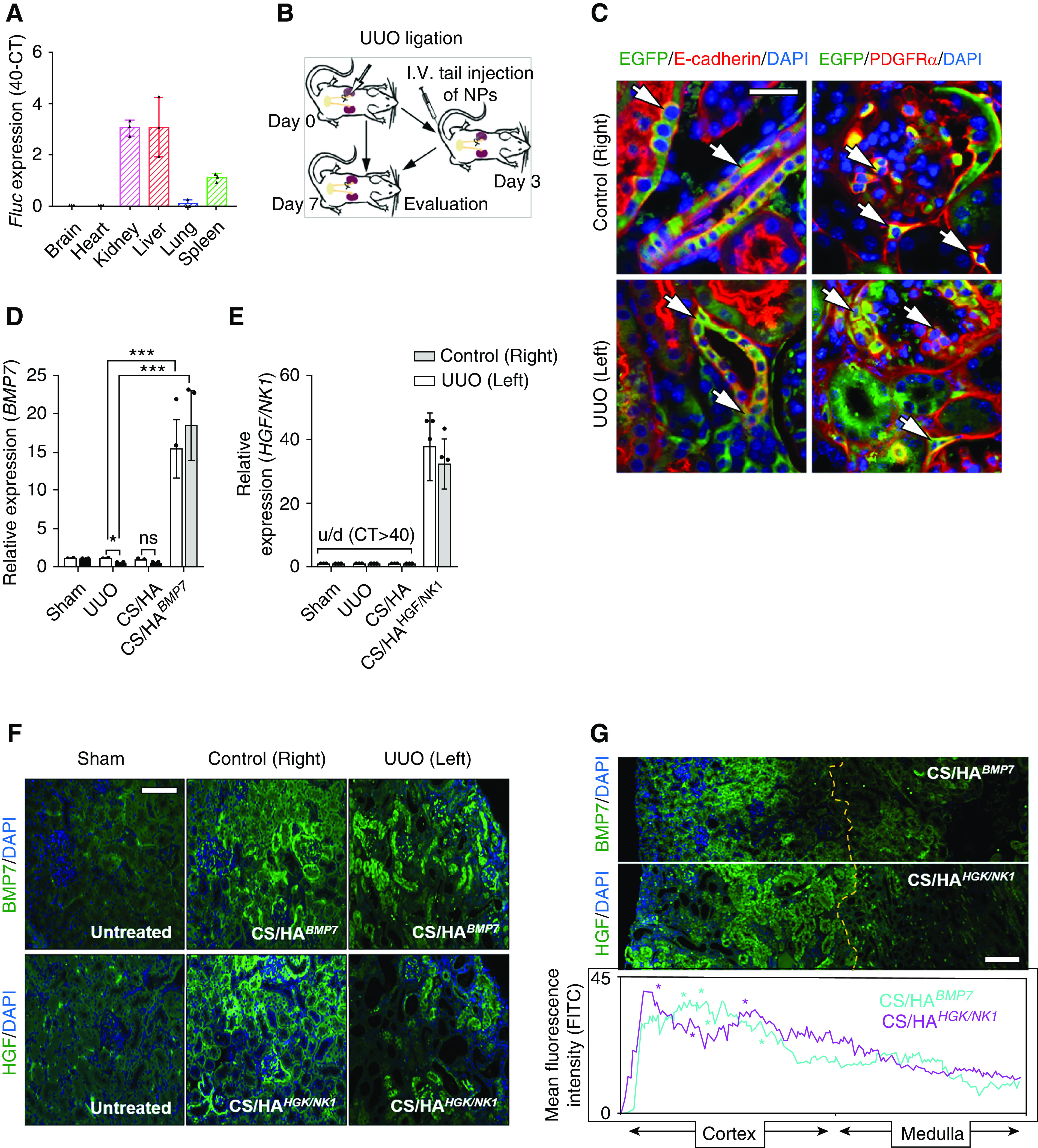
CS/HApDNA NPs successfully target and transfect kidney tissue. (A) Mice received iv tail injections of CS/HAFluc/EGFP NPs and, after 3 days, major organs were assessed for Fluc mRNA expression by qRT-PCR. Data are displayed as 40-CTFluc (n=3). (B) Schematic depicting animal model generation and treatment time; mice received iv injections 3 days after UUO surgery and kidney tissue was evaluated 7 days after UUO surgery. (C) The kidneys of CS/HAFluc/EGFP-injected mice were assessed for EGFP expression using anti-EGFP antibody followed by costaining for E-cadherin (tubule epithelial cells) or PDGFRα (mesangial and fibroblast cells). White arrows indicate EGFP+ cells also expressing costained markers (n=3). Original magnification, ×200; scale bar, 50 µm. (D) BMP7 and (E) HGF/NK1 expression detected in control (right) and UUO (left) kidneys on day 7 post UUO surgery (4 days post CS/HABMP7 or CS/HAHGF/NK1 NP iv injection). BMP7 primers were designed to amplify human and mouse mRNA, whereas HGF/NK1 mRNA is not conserved and primers were limited to only amplify human mRNA; u/d, undetected (n=5). (F) The kidneys of CS/HABMP7- and CS/HAHGF/NK1-injected mice were assessed for BMP7 and HGF expression using antibodies, followed by AlexFluor488-conjugated secondary antibodies (n=3). Original magnification, ×200; scale bar, 100 µm. (G) Kidney-wide expression of BMP7 and HGF expression was assessed and intensity of fluorescence was quantified. Yellow dashed line indicates the cortex-medulla border; asterisks (*) in histogram plots indicate the localization of glomeruli; original magnification, ×100; scale bar, 200 µm. In vivo data are shown as the mean±SD. Statistical significance is shown as: *P≤0.05; **P≤0.01; ***P≤0.001.
Kidney sections were stained with H&E, PAS, Masson’s trichrome, and Sirius Red, and immunostained for collagen IV (Figure 5A), to evaluate tissue architecture and collagen deposition. Numerous tubules displaying atrophy (approximately 60%) were observed in the cortex of UUO and CS/HA groups (Figure 5B), and this correlated with the extent of cast formation in these groups (Figure 5C). Both CS/HABMP7 and CS/HAHGF/NK1 treatments significantly reduced tubular atrophy and cast formation, suggesting renoprotective effects. Masson’s trichrome and Sirius Red staining showed similar trends across stains. Collagen deposition was extensive in the cortex and medulla (Supplemental Figure 11A) of UUO and CS/HA-vehicle groups. CS/HABMP7 and CS/HAHGF/NK1 treatments significantly reduced the amount of visualized collagen in the cortex and medulla, when assessed by Masson’s trichrome (Figure 5D). Sirius Red quantification showed that CS/HAHGF/NK1 delivery attenuated the deposition of collagen fibers to a level comparable to Sham kidneys (Figure 5E). In addition, visualization under polarized light revealed that CS/HABMP7 treatment more effectively reduced collagen fiber thickness than quantity, whereas CS/HAHGF/NK1 delivery more effectively reduced collagen fiber quantity in kidney tissues (Supplemental Figure 11B). Collagen IV deposition (marker of GBM thickening/dysfunction) indicated thickened basement membranes around glomeruli and atrophic tubules. Quantification of collagen IV showed that levels were reduced and were comparable to Sham groups after CS/HAHGF/NK1administration (Figure 5F). CS/HABMP7 NP treatment significantly attenuated collagen IV deposition, but thickened basement membranes were still observable. Gene expression analysis for collagen I, III, and IV reflected the changes seem in tissue sections; these demonstrated that CS/HAHGF/NK1 NP treatments strongly promoted collagen inhibition (Figure 5G). Collectively, these data suggest that CS/HABMP7 and CS/HAHGF/NK1 treatments regenerated kidney architecture and protected against further damage.
Figure 5.
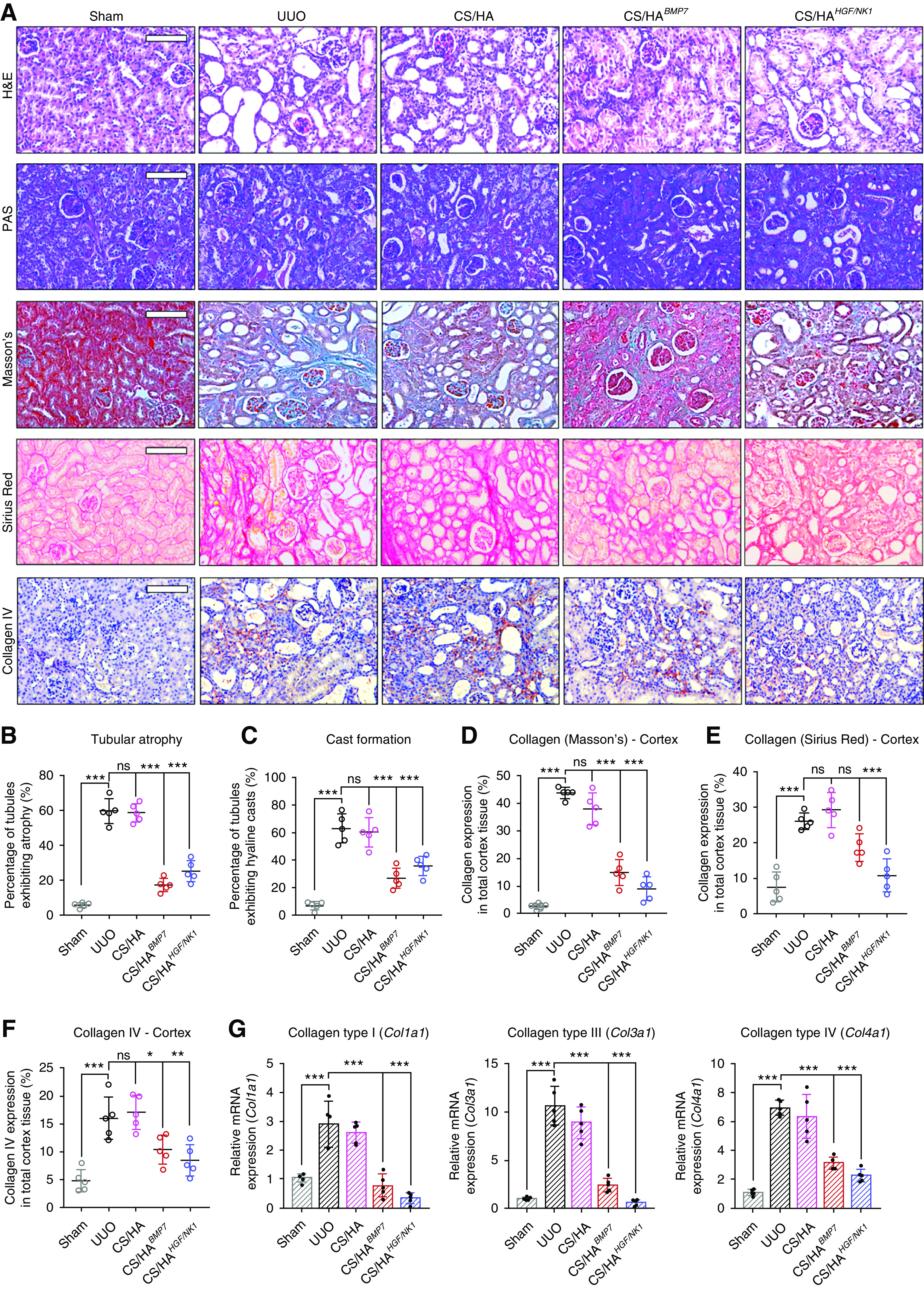
CS/HA-mediated overexpression of BMP7 or HGF/NK1 prevents in vivo development of CKD pathology. (A) Histologic staining of kidney sections 7 days post UUO surgery (n=5). Top row, H&E; second row, PAS; third row, Masson’s trichrome; fourth row, Sirius Red; bottom row, collagen IV immunostaining. Scale bars, 100 µm. (B) Calculated percentage of tubular atrophy and (C) hyaline cast formation, as calculated from averaged three fields of view of cortex images (n=5). (D) Collagen expression in cortex sections as visualized by Masson’s trichrome was quantified by calculating the areas positive for staining (blue) per three separate fields of view (n=5). (E) Collagen expression in cortex sections as visualized by Sirius Red was quantified by calculating the areas positive for staining (red) per three separate fields of view (n=5). (F) Collagen IV expression in cortex sections as visualized by anti-collagen IV immunostaining was quantified by calculating the areas positive for staining (red-brown) per three separate fields of view (n=5). (G) Collagen genes were assessed by qRT-PCR: Col1a1, Col3a1, Col4a1 (n=5). Data are shown as mean±SD, and statistical significance is indicated by: *P≤0.05; **P≤0.01; ***P≤0.001.
Prevention of Renal Fibrosis and Rescue of Functional Markers
Immunohistologic staining for PCNA (marker of cell proliferation), α-SMA (marker of myofibroblasts), and fibronectin (marker profibrotic tissue environments) was performed (Figure 6A). PCNA identified marginal upregulation of cell expansion in UUO cortex, compared with Sham groups. Although not statistically significant to UUO or Sham groups, NP treatments displayed a level of PCNA expression more akin to Sham groups (Figure 6B). In the medulla, CS/HABMP7 treatments showed significant increases in PCNA, compared with Sham medulla (Supplemental Figure 12A), reciprocating in vitro results. Elevated α-SMA expression was detected in UUO and CS/HA-vehicle groups, whereas CS/HABMP7 and CS/HAHGF/NK1 treatments significantly reduced α-SMA expression (Figure 6C). This suggested that PCNA+ cells after BMP7 transfection were not myofibroblasts. Fibronectin visualization in UUO and CS/HA-vehicle groups indicated profibrotic ECM. Quantification of fibronectin showed that CS/HABMP7 and CS/HAHGF/NK1 administration inhibited fibronectin, resulting in levels comparable to Sham groups (Figure 6D), allowing us to speculate that the profibrotic tissue microenvironment has been resolved. Evaluation of fibrosis-associated genes (Figure 6E) and proteins (Supplemental Figure 12B) corroborated our findings that CS/HABMP7 and CS/HAHGF/NK1 treatments significantly attenuated UUO-induced and fibrosis-related injury, echoing in vitro results (Figure 6E). Attenuation of α-SMA and fibronectin mRNA, and collagen IV, α-SMA, and fibronectin total protein expression, mirrored our findings of reduced protein expression from tissue section immunostaining and quantification.
Figure 6.
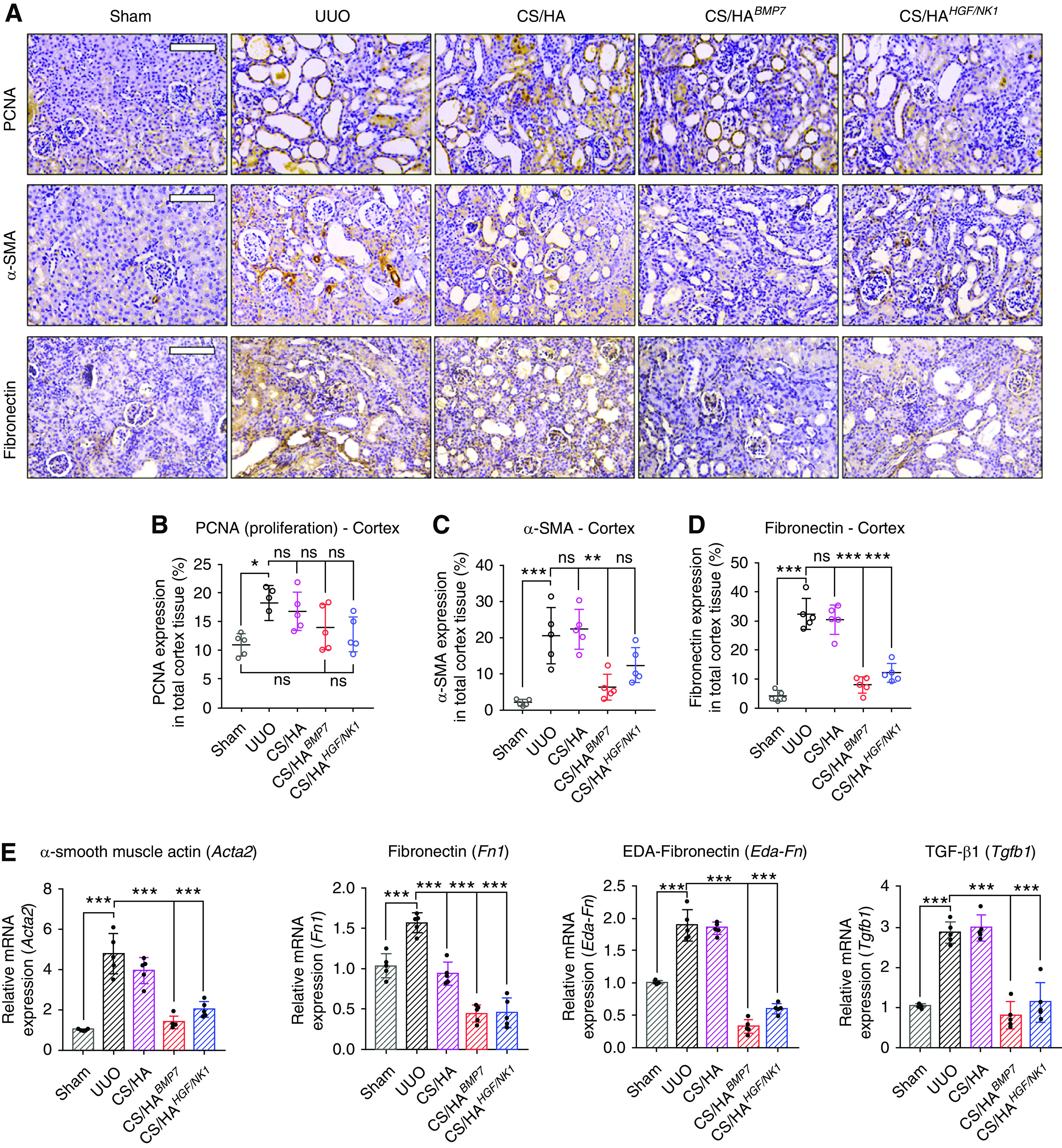
CS/HA NP delivery of BMP7 or HGF/NK1 gene therapy attenuates renal fibrosis. (A) Immunohistology to visualize PCNA (top panel), α-SMA (middle panel), and fibronectin (bottom panel) in kidney cortex sections 7 days post UUO surgery (n=5). Scale bars, 100 µm. The percentage of kidney cortex positive for (B) PCNA, (C) α-SMA, and (D) fibronectin expression was quantified from three separate fields of view (n=5). (E) A panel of fibrosis-associated genes was assessed by qRT-PCR: Acta2, Fn1, Eda-Fn, Tgfb1 (n=5). Data are shown as mean±SD, and statistical significance is indicated by: *P≤0.05; **P≤0.01; ***P≤0.001.
Assessment of a panel of kidney injury marker genes corroborated our findings that CS/HABMP7 and CS/HAHGF/NK1 treatments significantly attenuated UUO-induced and fibrosis-related injury, echoing in vitro results. Both BMP7 and HGF/NK1 therapies significantly attenuated kidney damage markers (Kim1, Ngal, Mmp2, Mmp9, Il6, Tnfa) (Figure 7A), while preserving kidney function markers (Nphs1, Nphs2, Cdh1) (Figure 7B). BMP7 therapy was shown to be more effective at inhibiting Kim1 and Mmp9 expression. The presence of inflammatory cells preludes and drives progressive fibrosis. Staining for the presence of CD68+ proinflammatory macrophages indicated that there was an influx of invading macrophages in UUO injury. However, after CS/HABMP7 and CS/HAHGF/NK1 administration, CD68+ macrophage numbers were approximately halved, compared with untreated and CS/HA NP vehicle-treated UUO kidneys (Supplemental Figure 13). Changes in macrophage numbers after CS/HApDNA NP treatments were also reflected by the inhibition of UUO-induced Il6 and Tnfa mRNA expression. Interestingly, HGF/NK1 delivery induced elevated expression of nephrin, podocin, and E-cadherin, highly suggestive of undescribed roles in maintaining specialized cell phenotype and the renal filtration barrier. Surprisingly, our routine measurements of BUN (Supplemental Figure 14A) and serum creatinine (Supplemental Figure 14B) showed that both were reduced to levels comparable to Sham animals in CS/HABMP7 and CS/HAHGF/NK1 groups. Although presently we cannot confirm whether these readings are associated with rescued kidney function in UUO models, we speculate that the elevated BMP7 and HGF/NK1 protein expression identified in unligated control kidneys may have contributed to improved measurements. UUO injuries induced expression of liver enzyme functional markers, commonly associated with liver dysfunction (Supplemental Figure 14C). CS/HABMP7 and CS/HAHGF/NK1 treatment groups effectively attenuated the expression of these markers. No tissue changes or pathology was observable in major organs of mice receiving NP treatments (Supplemental Figure 15). Therefore, whether liver enzyme expression data are resultant of resolved renal fibrosis or a synergistic effect of NP delivery to liver tissue remains unclear. Overall, these data supported our histopathologic observations that biosafe CS/HABMP7 and CS/HAHGF/NK1 iv treatments alleviated and prevented UUO-induced renal fibrosis through attenuation of pathology-associated gene upregulation and the safeguarding of functional specialist genes.
Figure 7.
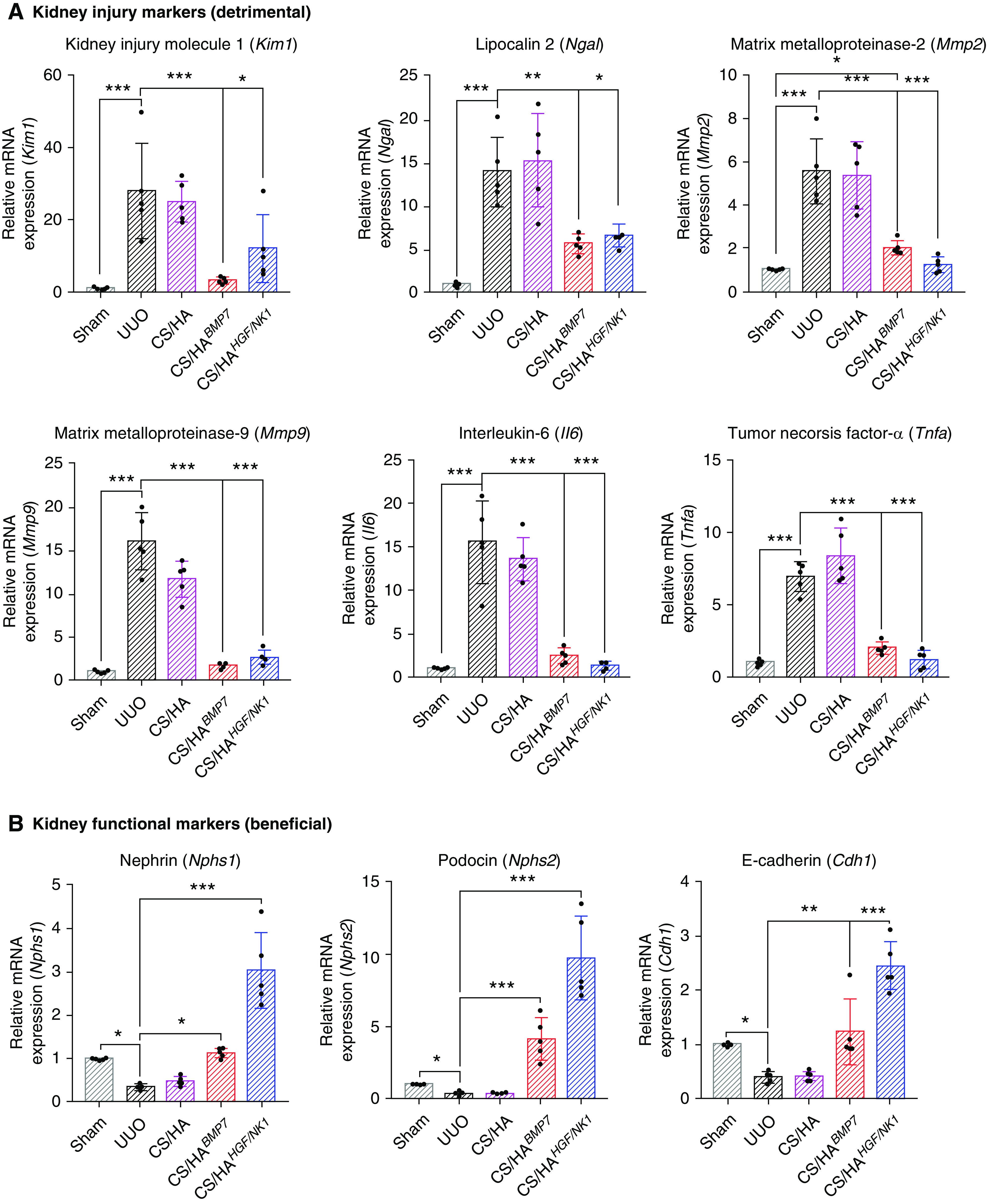
CS/HA NP delivery of BMP7 or HGF/NK1 gene therapy attenuates kidney injury–associated gene expression and protects healthy kidney gene expression. (A) A panel of kidney injury–associated genes (detrimental) was assessed by qRT-PCR: Kim1, Ngal, Mmp2, Mmp9, Il6, Tnfa (n=5). (B) A panel of genes associated with healthy kidney function (beneficial) was assessed by qRT-PCR: Nphs1, Nphs2, Cdh1 (n=5). Data are shown as mean±SD, and statistical significance is indicated by: *P≤0.05; **P≤0.01; ***P≤0.001.
Discussion
This investigation utilized biocompatible M-HA–coated L-CS/pDNA NPs to enhance delivery of antifibrotic genes for CKD therapy. CS/HABMP7 NPs promoted proliferation, CS/HAHGF/NK1 NPs promoted migration, and both prevented TGF-β1–driven myofibroblast differentiation in vitro. When administered by iv tail injection, UUO-induced renal fibrosis was inhibited; collagen accumulation was eliminated; α-SMA+ cells were reduced; GBM structure was protected; and renal function was rescued (Figure 8).
Figure 8.

Schematic diagram of CS/HA-mediated delivery of BMP7 or HGF/NK1 and the renoprotective outcomes. When administered by iv injection, NPs accumulate within kidney glomeruli and successfully traverse the GBM to transfect E-cadherin+ tubule epithelial cells and PDGFRα+ mesangial and fibroblast cells within proximity to glomeruli. The paracrine production of BMP7 or HGF/NK1 results in an antifibrotic and regenerative tissue microenvironment, attenuating UUO-induced myofibroblast formation and the deposition of disorganized collagen-rich ECM, while protecting epithelial tubular cell phenotype and function. Consequently, CS/HA-mediated BMP7 and HGF/NK1 gene therapy rescues the structure and markers of functional kidney tissue.
iv injection is preferable to avoid invasive complications associated with typically favored intraperitoneal injection. iv-administered L-CS and 50% N-acetylated–CS selectively accumulated within kidneys.14–16 Here, 15% N-acetylated 50-kDa L-CS NPs had favorable DNA encapsulation, cytocompatibility, charge, and size. Given stark contrasts in wet (approximately 240 nm) and dry (approximately 50 nm) diameters, it can be speculated that size varies depending on surrounding tonicity. It is possible that NPs with reduced water content could traverse 50–100-nm fenestrae of glomerular capillary endothelium,44,45 more so in nephropathic lesions in renal disease.45,46 Recently, another CS/HA NP system was reported to mitigate AKI after iv injection into mice.47 These negatively charged NPs were synthesized by combining CS with an HA and antioxidant peptide mixture. Although kidney retention was elevated in AKI, the spleen, liver, lung, and heart tissues also had NP accumulation. In our investigation, we sought to minimize off-target uptake by employing a differential layer-by-layer synthesis method and taking additional steps to tune NP properties. L-CS was mixed with pDNA to take advantage of electrostatic interactions, to create tightly complexed L-CS NP cores with strong DNA-protectant properties and capacity for low-pH–induced destabilization for endosomal escape. M-HA was used to optimize coating of L-CSpDNA NPs, providing camouflage from circulating macrophage recognition of CS and reducing NP surface charge, but maintaining an overall positive net charge to promote accumulation in glomeruli due to electrostatic attraction to the GBM.17–19 M-HA of 80–100 kDa was chosen to benefit higher-affinity binding to CD44 receptor clusters. Our results showed that there was 43%, 43%, and 14% of total tissue transfection in the kidneys, liver, and spleen, respectively. Liu et al.47 did not investigate cell-specific uptake of their NPs. In this study, we demonstrated that NPs were predominantly internalized by E-cadherin+ tubule epithelial cells and PDGFRα+ cell clusters in and surrounding the glomeruli, indicating that our CS/HA NPs could traverse the GBM and enter the cortical interstitium. A possible route for future investigations would be the improvement of cell-specific transfection in injured kidneys.
In renal fibrosis, infiltrated resident fibroblasts/myofibroblasts,4 activated mesangial cells, or dysfunctional cells that underwent epithelial/endothelial-mesenchymal transition (EMT)48 may have elevated probability to be the recipient cells. Indeed, we showed prominent inhibition of α-SMA and collagen production around glomeruli and tubules. Our experimental design relied on the paracrine production of antifibrotic cytokines to influence the tissue microenvironment, suppress myofibroblast activity, and promote regenerative mechanisms; in vitro data suggested that paracrine activity would persist for at least 8–10 days, thereby lengthening time between dosing in a theoretic clinical setting. The UUO mouse models used in this study displayed aggressive interstitial fibrosis within 3–7 days. Beyond 7 days, kidney tissue deteriorated rapidly, thereby preventing longer-term evaluation. As such, upon further optimization to improve kidney-specific uptake, our NP system could be utilized in longer-term, larger-animal UUO models or in other models of kidney disease, such as ischemic reperfusion injuries or diabetic nephropathies, to improve clinical relevance.
We hypothesized that M-HA–coated L-CS NPs would increase internalization and transfection into CD44+ cells. CD44 is the principal receptor for HA with CS-binding capacity,49 and Cd44 knockdown suggested that CS/HA NP internalization was CD44-dependent. HA binding affinity depends on CD44 clustering and HA mol wt.23,36 Endogenously expressed high-mol wt–HA cell glycocalyces induce CD44 clustering, increasing overall affinity for HA.23 The HA glycocalyx is involved in myofibroblast and profibrotic cell phenotype stabilization but can be disrupted by exogenous addition of lower-mol wt HA.50,51 It was reported that CD44 receptors have optimal binding affinity to approximately 80-kDa M-HA.36 Therefore, we speculate that our CS/HA NPs could have a similar effect to exogenous addition of HA, providing therapeutic delivery while interfering with HA glycocalyces, concomitantly destabilizing myofibroblast or profibrotic phenotypes. In addition, CS/HA NPs may have positively regulated epithelial cell activity,52 indicated by pDNA-independent increases in BMP7 and CD44 expression. Overall, CS/HA NPs could be effective in targeting therapeutic delivery to CD44+-expressing profibrotic cells, due to abundant cell surface and clustered CD44, and the receptor’s role in endocytosis. Despite failure of vehicle treatments in preventing fibrosis, physiologic activity of CS/HA NPs warrants address in future studies, particularly elucidation of effects on cells involved in renal injury recovery, such as tubule epithelial cells and leukocytes.
There are numerous pDNA gene therapy studies in animal models,53,54 and clinical trials of DNA vaccines and gene therapy are underway.55 Here, we used nonviral and noninvasive pDNA delivery of genes selected for their demonstrated antifibrotic activity. BMP7 has essential roles in kidney morphogenesis56 and in supporting adult tubular epithelium in kidneys.57,58 BMP7 expression is lost in renal disease,59,60 whereas exogenous BMP7 stimulated renal repair, including in UUO61,62 among other models.60,63,64 Mechanisms of BMP7-mediated protection have not been fully elucidated. Inhibition of TGF-β1/Smad signaling is commonly proposed,22,63,65 and recently BMP7 was shown to induce CD44 variant 7/8 expression, which facilitated internalization of the HA glycocalyx and reversed myofibroblast differentiation.22,28 In this study, we confirmed BMP7 prevention of myofibroblasts in vitro and reduction of α-SMA+ cells and fibrotic ECM in vivo. Myofibroblast reversal benefits from the maintenance of local cell populations to remodel the otherwise difficult-to-infiltrate fibrotic ECM. Our approach led to paracrine production of an antifibrotic microenvironment, resulting in a regenerative tissue environment benefiting heterotypic and cooperating cell populations required for kidney functionality.
HGF has roles in kidney development and regeneration,66–68 as evidenced by the reparative effect of HGF transgenic mesothelial cell sheets transplanted into rat UUO models.69 Short t1/2 and difficulty in purification are obstacles in recombinant HGF therapeutic development, and difficulties in upscaled manufacture of large, full-length HGF-encoding plasmids have restricted HGF gene therapy. Here, we used CS/HA NPs to deliver shorter and more readily cloned HGF/NK1 plasmids to kidneys. Few studies have established definitive roles for HGF/NK variants. HGF/NK1 knockdown was shown to exacerbate myofibroblast formation.29 We showed that HGF/NK1 resulted in myofibroblast attenuation and drastically reduced collagen synthesis and deposition. BMP7 therapy reduced α-SMA+ cells and induced PCNA+ renal cells, resulting in the regeneration of tubules and in-line with previously reported capabilities of BMP7 to reverse myofibroblast differentiation22,28,63 and promote epithelial tubule integrity.58 HGF/NK1 resulted in significant upregulation of kidney epithelial cell markers, supporting reports of HGF roles in recovery of nephrin70 and tubular cell survival.71 Indeed, we may have identified a potent renoprotective factor, and this finding will inform detailed investigations into HGF/NK variant stimulation on primary and highly specialized renal cells, such as tubular epithelial cells and podocytes.
Successful clinical application of NP-based regenerative therapies will benefit from adaptions enabling the noninvasive diagnosis, treatment, and prognostic monitoring of CKD and tracking of NP fate. Our results revealed that the majority of positive transfection occurred in kidneys and liver. Here, we used simplistic crosslinking and layer-by-layer assembly techniques to facilitate NP fabrication without large deviations in size. Further modifications to improve kidney retention of NPs will undoubtedly affect size/charge and could inevitably be detrimental. Therefore, nontoxic base materials, compatible with sophisticated assembly techniques for kidney-targeting NPs, should be considered. Alternatively, incorporation of NPs into transplanted biomaterials for controlled release, such as those we previously explored,72 may avoid off-target organ transfection, albeit eliminating noninvasive aspects. When considering realistic progression to clinical studies, improved kidney- and cell-specific targeting continue to be imperative requirements.
In summary, we developed natural, biopolymer-based NP antifibrotic gene carriers for noninvasive CKD therapy. Using L-CS enabled facile NP synthesis, and NP uptake by kidneys. M-HA coatings significantly improved CD44-dependent internalization, transfection efficiency, and cytocompatibility. BMP7 delivery prevented the upregulation of profibrotic markers and regenerated tubules. HGF/NK1 delivery resulted in effective attenuation and removal of collagen, and renoprotective effects on epithelial cells. Both treatments prevented the development of UUO-induced fibrosis and ameliorated renal markers of dysfunction. The use of biomimetic and biocompatible polymers as noninvasive, nonviral gene carriers offers an accessible and modifiable approach for safe and effective antifibrotic gene therapy. Importantly, our results determined the plausibility of using multifunctional biopolymer NPs for future developments in gene-mediated therapeutics.
Disclosures
All authors have nothing to disclose.
Funding
National Natural Science Foundation of China (NSFC) International Young Scholar Research Fellowship (81850410552), NSFC Key National Project Fund (81830060), NSFC Project Fund (81701840), Open Funding of the State Key Laboratory of Kidney Diseases (KF-2018-10), and National Key Research and Development Program of China (2016YFA0101002).
Supplementary Material
Acknowledgments
Dr. Adam C. Midgley designed the research project, performed the majority of the experiments, interpreted and analyzed data, and wrote the manuscript. Dr. Yongzhen Wei assisted and performed in vitro and in vivo immunostaining and histologic staining experiments. Dr. Dashuai Zhu performed the majority of animal surgical procedures and tissue sectioning. Dr. Fangli Gao and Dr. Chuangnian Zhang provided advice and assisted in the optimization of nanoparticle synthesis. Dr. Anila Khalique, Dr. Hongyu Yan, and Dr. Wenya Luo performed select experiments and histologic analysis. Dr. Huan Jiang assisted in nanoparticle characterization and sample imaging. Dr. Xiangsheng Liu and Dr. Chao Yang upscaled and authenticated the plasmids used in the study. Dr. Jiasen Guo, Dr. Wen Ning, and Dr. Kai Wang gave advice and assisted in the optimization of the animal models. Dr. Guowei Feng, Dr. Xueyuan Bai, Dr. Qiang Zhao, and Dr. Deling Kong gave expert advice and discussed the results and research direction throughout the project term.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2019111160/-/DCSupplemental.
Supplemental Table 1. Custom primers and siRNA.
Supplemental Table 2. Physiochemical properties of finalized NPs.
Supplemental Table 3. Statistical analyses for CS and CS/HA NP effects on cell proliferation and migration.
Supplemental Table 4. Statistical analyses for CS/HA and CS/HApDNA NP effects on cell proliferation and migration.
Supplemental Figure 1. NP core and coat optimization is dependent on the physiochemical properties of CS used.
Supplemental Figure 2. Inclusion of 2% w/w pDNA within CS cores results in more compact CS/HApDNA NPs with reduced surface charge.
Supplemental Figure 3. The stability and DNA release of CS/HA NPs is dependent on storage conditions and polymer-degrading enzymatic activity.
Supplemental Figure 4. Cellular viability and transfection efficiency are optimal at 10 µg/ml CS/HA NPs loaded with 2% w/w pDNA.
Supplemental Figure 5. CS/HA transfection is predominantly CD44-mediated.
Supplemental Figure 6. CS/HApDNA NP treatments have additional beneficial effects on fibroblast activity and phenotype.
Supplemental Figure 7. CS/HA transfection of BMP7 or HGF/NK1 has antifibrotic activity in mouse kidney fibroblasts (MKFs).
Supplemental Figure 8. CS/HApDNA NP treatments induce gene expression predominantly in cortical tissue tubules and interstitial space surrounding glomeruli.
Supplemental Figure 9. Extensive fibrosis develops in UUO mouse models after 7 days.
Supplemental Figure 10. CS/HApDNA NP treatments induce gene expression predominantly in cortical tissue tubules and interstitial space surrounding glomeruli.
Supplemental Figure 11. Increases in pathologic markers in UUO kidney medulla sections are inhibited by CS/HApDNA NP treatments.
Supplemental Figure 12. Increases in fibrosis markers in UUO kidney medulla sections are inhibited by CS/HApDNA NP treatments.
Supplemental Figure 13. Macrophage infiltration resulting from UUO injury is attenuated by CS/HApDNA NP treatment.
Supplemental Figure 14. The UUO induction of elevated kidney and liver tissue functional markers is attenuated by CS/HApDNA NP treatment.
Supplemental Figure 15. CS/HApDNA NP treatment biodistribution has no adverse or observable effects on major organ appearance or architecture.
References
- 1.Jha V, Garcia-Garcia G, Iseki K, Li Z, Naicker S, Plattner B, et al. : Chronic kidney disease: Global dimension and perspectives [published correction appears in Lancet 382: 208, 2013]. Lancet 382: 260–272, 2013. [DOI] [PubMed] [Google Scholar]
- 2.Hallan SI: Cardiovascular disease prevention in CKD. Am J Kidney Dis 64: 326–328, 2014. [DOI] [PubMed] [Google Scholar]
- 3.Collins AJ, Foley RN, Herzog C, Chavers B, Gilbertson D, Herzog C, et al. : US renal data system 2012 annual data report. Am J Kidney Dis 61[Suppl 1]: A7, e1-A7, e476, 2013. [DOI] [PubMed] [Google Scholar]
- 4.Sun YB, Qu X, Caruana G, Li J: The origin of renal fibroblasts/myofibroblasts and the signals that trigger fibrosis. Differentiation 92: 102–107, 2016. [DOI] [PubMed] [Google Scholar]
- 5.Ruiz-Ortega M, Rayego-Mateos S, Lamas S, Ortiz A, Rodrigues-Diez RR: Targeting the progression of chronic kidney disease. Nat Rev Nephrol 16: 269–288, 2020. [DOI] [PubMed] [Google Scholar]
- 6.Chang CF, Lin SZ, Chiang YH, Morales M, Chou J, Lein P, et al. : Intravenous administration of bone morphogenetic protein-7 after ischemia improves motor function in stroke rats. Stroke 34: 558–564, 2003. [DOI] [PubMed] [Google Scholar]
- 7.Urbina P, Singla DK: BMP-7 attenuates adverse cardiac remodeling mediated through M2 macrophages in prediabetic cardiomyopathy. Am J Physiol Heart Circ Physiol 307: H762–H772, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson RC, Leopold JA, Loscalzo J: Vascular calcification: Pathobiological mechanisms and clinical implications. Circ Res 99: 1044–1059, 2006. [DOI] [PubMed] [Google Scholar]
- 9.Thomas CE, Ehrhardt A, Kay MA: Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet 4: 346–358, 2003. [DOI] [PubMed] [Google Scholar]
- 10.Dreaden EC, Alkilany AM, Huang X, Murphy CJ, El-Sayed MA: The golden age: Gold nanoparticles for biomedicine. Chem Soc Rev 41: 2740–2779, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loureiro A, Azoia NG, Gomes AC, Cavaco-Paulo A: Albumin-based nanodevices as drug carriers. Curr Pharm Des 22: 1371–1390, 2016. [DOI] [PubMed] [Google Scholar]
- 12.Yao CG, Martins PN: Nanotechnology applications in transplantation medicine. Transplantation 104: 682–693, 2019. [DOI] [PubMed] [Google Scholar]
- 13.Shariatinia Z, Jalali AM: Chitosan-based hydrogels: Preparation, properties and applications. Int J Biol Macromol 115: 194–220, 2018. [DOI] [PubMed] [Google Scholar]
- 14.Yuan ZX, Sun X, Gong T, Ding H, Fu Y, Zhang ZR: Randomly 50% N-acetylated low molecular weight chitosan as a novel renal targeting carrier. J Drug Target 15: 269–278, 2007. [DOI] [PubMed] [Google Scholar]
- 15.Chou CK, Chen SM, Li YC, Huang TC, Lee JA: Low-molecular-weight chitosan scavenges methylglyoxal and N (ε)-(carboxyethyl)lysine, the major factors contributing to the pathogenesis of nephropathy. Springerplus 4: 312, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geng X, Zhang M, Lai X, Tan L, Liu J, Yu M, et al. : Small-sized cationic miRi-PCNPs selectively target the kidneys for high-efficiency antifibrosis treatment. Adv Healthc Mater 7: e1800558, 2018. [DOI] [PubMed] [Google Scholar]
- 17.Zuckerman JE, Choi CH, Han H, Davis ME: Polycation-siRNA nanoparticles can disassemble at the kidney glomerular basement membrane. Proc Natl Acad Sci U S A 109: 3137–3142, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bennett KM, Beeman SC, Baldelomar EJ, Zhang M, Wu T, Hann BD, et al. : Use of cationized ferritin nanoparticles to measure renal glomerular microstructure with MRI. Methods Mol Biol 1397: 67–79, 2016. [DOI] [PubMed] [Google Scholar]
- 19.Zuckerman JE, Davis ME: Targeting therapeutics to the glomerulus with nanoparticles. Adv Chronic Kidney Dis 20: 500–507, 2013. [DOI] [PubMed] [Google Scholar]
- 20.Rayahin JE, Buhrman JS, Zhang Y, Koh TJ, Gemeinhart RA: High and low molecular weight hyaluronic acid differentially influence macrophage activation. ACS Biomater Sci Eng 1: 481–493, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lemarchand C, Gref R, Couvreur P: Polysaccharide-decorated nanoparticles. Eur J Pharm Biopharm 58: 327–341, 2004. [DOI] [PubMed] [Google Scholar]
- 22.Midgley AC, Duggal L, Jenkins R, Hascall V, Steadman R, Phillips AO, et al. : Hyaluronan regulates bone morphogenetic protein-7-dependent prevention and reversal of myofibroblast phenotype. J Biol Chem 290: 11218–11234, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Midgley AC, Rogers M, Hallett MB, Clayton A, Bowen T, Phillips AO, et al. : Transforming growth factor-β1 (TGF-β1)-stimulated fibroblast to myofibroblast differentiation is mediated by hyaluronan (HA)-facilitated epidermal growth factor receptor (EGFR) and CD44 co-localization in lipid rafts. J Biol Chem 288: 14824–14838, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kastner S, Thomas GJ, Jenkins RH, Davies M, Steadman R: Hyaluronan induces the selective accumulation of matrix- and cell-associated proteoglycans by mesangial cells. Am J Pathol 171: 1811–1821, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones SG, Ito T, Phillips AO: Regulation of proximal tubular epithelial cell CD44-mediated binding and internalisation of hyaluronan. Int J Biochem Cell Biol 35: 1361–1377, 2003. [DOI] [PubMed] [Google Scholar]
- 26.Greco RM, Iocono JA, Ehrlich HP: Hyaluronic acid stimulates human fibroblast proliferation within a collagen matrix. J Cell Physiol 177: 465–473, 1998. [DOI] [PubMed] [Google Scholar]
- 27.Ooki T, Murata-Kamiya N, Takahashi-Kanemitsu A, Wu W, Hatakeyama M: High-molecular-weight hyaluronan is a Hippo pathway ligand directing cell density-dependent growth inhibition via PAR1b. Dev Cell 49: 590–604.e9, 2019 [DOI] [PubMed] [Google Scholar]
- 28.Midgley AC, Oltean S, Hascall V, Woods EL, Steadman R, Phillips AO, et al. : Nuclear hyaluronidase 2 drives alternative splicing of CD44 pre-mRNA to determine profibrotic or antifibrotic cell phenotype. Sci Signal 10: eaao1822, 2017. [DOI] [PubMed] [Google Scholar]
- 29.Dally J, Khan JS, Voisey A, Charalambous C, John HL, Woods EL, et al. : Hepatocyte growth factor mediates enhanced wound healing responses and resistance to transforming growth factor-β1-driven myofibroblast differentiation in oral mucosal fibroblasts. Int J Mol Sci 18: 1843, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sinuani I, Averbukh Z, Gitelman I, Rapoport MJ, Sandbank J, Albeck M, et al. : Mesangial cells initiate compensatory renal tubular hypertrophy via IL-10-induced TGF-beta secretion: Effect of the immunomodulator AS101 on this process. Am J Physiol Renal Physiol 291: F384–F394, 2006. [DOI] [PubMed] [Google Scholar]
- 31.Park MJ, Bae CS, Lim SK, Kim DI, Lim JC, Kim JC, et al. : Effect of protopanaxadiol derivatives in high glucose-induced fibronectin expression in primary cultured rat mesangial cells: Role of mitogen-activated protein kinases and Akt. Arch Pharm Res 33: 151–157, 2010. [DOI] [PubMed] [Google Scholar]
- 32.Wang Y, Jia L, Hu Z, Entman ML, Mitch WE, Wang Y: AMP-activated protein kinase/myocardin-related transcription factor-A signaling regulates fibroblast activation and renal fibrosis. Kidney Int 93: 81–94, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abul Kalam M, Khan AA, Khan S, Almalik A, Alshamsan A: Optimizing indomethacin-loaded chitosan nanoparticle size, encapsulation, and release using Box-Behnken experimental design. Int J Biol Macromol 87: 329–340, 2016. [DOI] [PubMed] [Google Scholar]
- 34.Almalik A, Alradwan I, Majrashi MA, Alsaffar BA, Algarni AT, Alsuabeyl MS, et al. : Cellular responses of hyaluronic acid-coated chitosan nanoparticles. Toxicol Res (Camb) 7: 942–950, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Almalik A, Day PJ, Tirelli N: HA-coated chitosan nanoparticles for CD44-mediated nucleic acid delivery. Macromol Biosci 13: 1671–1680, 2013. [DOI] [PubMed] [Google Scholar]
- 36.Wolny PM, Banerji S, Gounou C, Brisson AR, Day AJ, Jackson DG, et al. : Analysis of CD44-hyaluronan interactions in an artificial membrane system: Insights into the distinct binding properties of high and low molecular weight hyaluronan. J Biol Chem 285: 30170–30180, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tammi R, Rilla K, Pienimaki JP, MacCallum DK, Hogg M, Luukkonen M, et al. : Hyaluronan enters keratinocytes by a novel endocytic route for catabolism. J Biol Chem 276: 35111–35122, 2001. [DOI] [PubMed] [Google Scholar]
- 38.Wynn TA, Ramalingam TR: Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat Med 18: 1028–1040, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eddy AA: Molecular basis of renal fibrosis. Pediatr Nephrol 15: 290–301, 2000. [DOI] [PubMed] [Google Scholar]
- 40.Gabbiani G: The myofibroblast in wound healing and fibrocontractive diseases. J Pathol 200: 500–503, 2003. [DOI] [PubMed] [Google Scholar]
- 41.Darby IA, Laverdet B, Bonté F, Desmoulière A: Fibroblasts and myofibroblasts in wound healing. Clin Cosmet Investig Dermatol 7: 301–311, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA: Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 3: 349–363, 2002. [DOI] [PubMed] [Google Scholar]
- 43.Stephenson LA, Haney LB, Hussaini IM, Karns LR, Glass WF 2nd: Regulation of smooth muscle alpha-actin expression and hypertrophy in cultured mesangial cells. Kidney Int 54: 1175–1187, 1998. [DOI] [PubMed] [Google Scholar]
- 44.Ballermann BJ: Glomerular endothelial cell differentiation. Kidney Int 67: 1668–1671, 2005. [DOI] [PubMed] [Google Scholar]
- 45.Ichimura K, Stan RV, Kurihara H, Sakai T: Glomerular endothelial cells form diaphragms during development and pathologic conditions. J Am Soc Nephrol 19: 1463–1471, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Toyoda M, Najafian B, Kim Y, Caramori ML, Mauer M: Podocyte detachment and reduced glomerular capillary endothelial fenestration in human type 1 diabetic nephropathy. Diabetes 56: 2155–2160, 2007. [DOI] [PubMed] [Google Scholar]
- 47.Liu D, Jin F, Shu G, Xu X, Qi J, Kang X, et al. : Enhanced efficiency of mitochondria-targeted peptide SS-31 for acute kidney injury by pH-responsive and AKI-kidney targeted nanopolyplexes. Biomaterials 211: 57–67, 2019. [DOI] [PubMed] [Google Scholar]
- 48.Cruz-Solbes AS, Youker K: Epithelial to mesenchymal transition (EMT) and endothelial to mesenchymal transition (EndMT): Role and implications in kidney fibrosis. Results Probl Cell Differ 60: 345–372, 2017. [DOI] [PubMed] [Google Scholar]
- 49.Chang PH, Sekine K, Chao HM, Hsu SH, Chern E: Chitosan promotes cancer progression and stem cell properties in association with Wnt signaling in colon and hepatocellular carcinoma cells. Sci Rep 8: 45751, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Webber J, Meran S, Steadman R, Phillips A: Hyaluronan orchestrates transforming growth factor-beta1-dependent maintenance of myofibroblast phenotype. J Biol Chem 284: 9083–9092, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Webber J, Jenkins RH, Meran S, Phillips A, Steadman R: Modulation of TGFbeta1-dependent myofibroblast differentiation by hyaluronan. Am J Pathol 175: 148–160, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ito T, Williams JD, Fraser D, Phillips AO: Hyaluronan attenuates transforming growth factor-beta1-mediated signaling in renal proximal tubular epithelial cells. Am J Pathol 164: 1979–1988, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Song Y, Morales L, Malik AS, Mead AF, Greer CD, Mitchell MA, et al. : Non-immunogenic utrophin gene therapy for the treatment of muscular dystrophy animal models. Nat Med 25: 1505–1511, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Murakami T, Sunada Y: Plasmid DNA gene therapy by electroporation: Principles and recent advances. Curr Gene Ther 11: 447–456, 2011. [DOI] [PubMed] [Google Scholar]
- 55.Ginn SL, Amaya AK, Alexander IE, Edelstein M, Abedi MR: Gene therapy clinical trials worldwide to 2017: An update [published correction appears in J Gene Med 21: e3124, 2019]. J Gene Med 20: e3015, 2018. [DOI] [PubMed] [Google Scholar]
- 56.Tsujimura T, Idei M, Yoshikawa M, Takase O, Hishikawa K: Roles and regulation of bone morphogenetic protein-7 in kidney development and diseases. World J Stem Cells 8: 288–296, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gould SE, Day M, Jones SS, Dorai H: BMP-7 regulates chemokine, cytokine, and hemodynamic gene expression in proximal tubule cells. Kidney Int 61: 51–60, 2002. [DOI] [PubMed] [Google Scholar]
- 58.Kopp JB: BMP-7 and the proximal tubule. Kidney Int 61: 351–352, 2002. [DOI] [PubMed] [Google Scholar]
- 59.Manson SR, Song JB, Guo Q, Liapis H, Austin PF: Cell type specific changes in BMP-7 expression contribute to the progression of kidney disease in patients with obstructive uropathy. J Urol 193[Suppl]: 1860–1869, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang S, Chen Q, Simon TC, Strebeck F, Chaudhary L, Morrissey J, et al. : Bone morphogenic protein-7 (BMP-7), a novel therapy for diabetic nephropathy. Kidney Int 63: 2037–2049, 2003. [DOI] [PubMed] [Google Scholar]
- 61.Manson SR, Niederhoff RA, Hruska KA, Austin PF: Endogenous BMP-7 is a critical molecular determinant of the reversibility of obstruction-induced renal injuries. Am J Physiol Renal Physiol 301: F1293–F1302, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Higgins DF, Ewart LM, Masterson E, Tennant S, Grebnev G, Prunotto M, et al. : BMP7-induced-Pten inhibits Akt and prevents renal fibrosis. Biochim Biophys Acta Mol Basis Dis 1863: 3095–3104, 2017. [DOI] [PubMed] [Google Scholar]
- 63.Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, et al. : BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med 9: 964–968, 2003. [DOI] [PubMed] [Google Scholar]
- 64.Zeisberg M, Bottiglio C, Kumar N, Maeshima Y, Strutz F, Müller GA, et al. : Bone morphogenic protein-7 inhibits progression of chronic renal fibrosis associated with two genetic mouse models. Am J Physiol Renal Physiol 285: F1060–F1067, 2003. [DOI] [PubMed] [Google Scholar]
- 65.Yu Z, Zai-Chun X, Wun-Lun H, Yun-Yun Z: BMP-7 attenuates TGF-β1-Induced fibronectin secretion and apoptosis of NRK-52E cells by the suppression of miRNA-21. Oncol Res 23: 147–154, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dai C, Saleem MA, Holzman LB, Mathieson P, Liu Y: Hepatocyte growth factor signaling ameliorates podocyte injury and proteinuria. Kidney Int 77: 962–973, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mizuno S, Matsumoto K, Nakamura T: HGF as a renotrophic and anti-fibrotic regulator in chronic renal disease. Front Biosci 13: 7072–7086, 2008. [DOI] [PubMed] [Google Scholar]
- 68.Cheng G, Tang X, Zhang J: Hepatocyte growth factor exerts beneficial effects on mice with type II diabetes-induced chronic renal failure via the NF-κB pathway. Mol Med Rep 18: 3389–3396, 2018. [DOI] [PubMed] [Google Scholar]
- 69.Oka M, Sekiya S, Sakiyama R, Shimizu T, Nitta K: Hepatocyte growth factor-secreting mesothelial cell sheets suppress progressive fibrosis in a rat model of CKD. J Am Soc Nephrol 30: 261–276, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kato T, Mizuno S, Nakamura T: Preservations of nephrin and synaptopodin by recombinant hepatocyte growth factor in podocytes for the attenuations of foot process injury and albuminuria in nephritic mice. Nephrology (Carlton) 16: 310–318, 2011. [DOI] [PubMed] [Google Scholar]
- 71.Gui Y, Lu Q, Gu M, Wang M, Liang Y, Zhu X, et al. : Fibroblast mTOR/PPARγ/HGF axis protects against tubular cell death and acute kidney injury. Cell Death Differ 26: 2774–2789, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Feng G, Zhang J, Li Y, Nie Y, Zhu D, Wang R, et al. : IGF-1 C domain-modified hydrogel enhances cell therapy for AKI. J Am Soc Nephrol 27: 2357–2369, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.