

Abstract
Perturbations in early life gut microbiota can have long-term impacts on host health. In this study, we investigated antimicrobial-induced temporal changes in diversity, stability, and compositions of gut microbiota in neonatal veal calves, with the objective of identifying microbial markers that predict diarrhea. A total of 220 samples from 63 calves in first 8 weeks of life were used in this study. The results suggest that increase in diversity and stability of gut microbiota over time was a feature of “healthy” (non-diarrheic) calves during early life. Therapeutic antimicrobials delayed the temporal development of diversity and taxa–function robustness (a measure of microbial stability). In addition, predicted genes associated with beta lactam and cationic antimicrobial peptide resistance were more abundant in gut microbiota of calves treated with therapeutic antimicrobials. Random forest machine learning algorithm revealed that Trueperella, Streptococcus, Dorea, uncultured Lachnospiraceae, Ruminococcus 2, and Erysipelatoclostridium may be key microbial markers that can differentiate “healthy” and “unhealthy” (diarrheic) gut microbiota, as they predicted early life diarrhea with an accuracy of 84.3%. Our findings suggest that diarrhea in veal calves may be predicted by the shift in early life gut microbiota, which may provide an opportunity for early intervention (e.g., prebiotics or probiotics) to improve calf health with reduced usage of antimicrobials.
Subject terms: Applied microbiology, Biomarkers
Introduction
Antimicrobials are widely used for both prevention and treatment of common infectious diseases such as diarrhea in food-producing animals during early life [1]. Young animals are particularly susceptible to infections due to their underdeveloped immune system [2] and lack of immunocompetence [3]. Increasing evidence has revealed the importance of early life gut microbiome due to its role in shaping the host immune system [4] and having a long time impact on gut health in adult life [5]. Perturbations in early life microbiome by antimicrobials have been reported to have long-term negative impacts on host immune function, including immunological defects or increased susceptibility to colonization by pathogens, in mice and human infants [6, 7]. Despite these findings in mice and humans, the knowledge of antimicrobial-induced perturbation in gut microbiota of ruminants is scarce, especially for dairy calves. Although reduced fecal microbial diversity has been reported in calves treated with antimicrobials for respiratory disease [8, 9], it is not clear whether the reported reduction in microbial diversity is due to antimicrobial treatment or the disease itself. On the other hand, it is largely unknown how therapeutic antimicrobials affect the temporal development of microbial stability or the ability of given microbial populations to withstand direct perturbations [10] in neonatal ruminants.
The emergence of widespread antimicrobial resistance due to overuse and in some cases misuse of veterinary antimicrobials in food-producing animals has become a global health threat [11]. Some antimicrobial use is necessary to effectively treat diseases in food-producing animals, however, the prevention and control of diseases before an outbreak is more cost-effective [12]. Thus, predicting disease onset based on the gut microbiota composition [13] could provide an opportunity for early intervention with an aim to reduce the use of antimicrobials. Recent findings based on human gut or oral microbiota have enabled the tentative prediction of type-2-diabetes [14], adiposity [15], and tooth decay [16]. Therefore, we speculated that early life gut microbiota in ruminants can predict gut health status as well as onset of early life diarrhea, one of the major health threats to ruminants.
Here, we used the veal calf as an animal model to represent young ruminants. We hypothesized that either diarrhea or the use of therapeutic antimicrobials to treat diarrhea may affect temporal changes in diversity, stability, as well as taxa–function relationship of gut microbiota. In addition, we speculated that the variation in early life microbiota between healthy and diarrheic calves can be exploited to develop predictive markers for diarrhea using a random forest machine learning algorithm.
Materials and methods
Assessment of calf health status and sample collection
Holstein male veal calves sourced from local Ontario (Canada) farms and free from antimicrobials prior to their arrival were enrolled in this study. Upon arrival, all animals were transferred to the experimental farm and individually examined to ensure they were all free of disease, injury, and dehydration. The health condition of each calf was evaluated daily over the 56-day experiment period. The animal experiment was conducted following the instructions of the Canadian Council of Animal Care and the experiment protocol was approved by the Livestock Care Committee of the University of Alberta (AUP00001595). The general appearance, fecal score, rectal temperature, umbilical score, and respiratory score were recorded as reported by Villot et al. [17]. Briefly, a total of 84 calves were enrolled with 42 fed milk replacer (CON) and the other 42 fed milk replacer with Saccharomyces cerevisiae boulardii (SCB). The milk replacer contained 63% of skim milk powder with 260 g/kg crude protein, 160 g/kg crude fat, and 19.2 MJ/kg metabolizable energy on a dry matter basis (Grober Animal Nutrition, Cambridge, ON, Canada). Calves with diarrhea (defined as when the fecal score was ≥2, meaning a fecal sample is either loose but stays on top of mat, or watery and sifts through the mat, for at least 2 days [18]) received 2 l of oral electrolytes (Calf Lyte II, Vetoquinol, Lavaltrie, QC, Canada) once daily over the entire experimental period. If no improvement was observed after electrolyte treatment or if severe diarrhea (dehydration and attitude scores > 0 and fecal score ≥ 2) was detected, calves were treated with trimethoprim sulfa (Trimidox, 0.7 ml/10 kg BW, Vetoquinol) for 3 consecutive days. If a calf was still diarrheic after treatment, it was treated with Excenel (0.2 ml/10 kg BW, Zoetis, Kirkland, QC, Canada) for another 3 consecutive days. Fecal samples (~20 g) were collected directly from the rectum by rectal palpation at 7 (on arrival), then again at 14, 21, 42, and 56 days of age, and were stored at −20 °C immediately after collection.
Classification of health status of calves
Calves that never exhibited diarrhea were classified as “healthy to healthy” (H2H), those that exhibited diarrhea and recovered after treated with electrolyte, indicating that their gut microbiota may be more resistant to diarrhea-induced dysbiosis so that no antimicrobials were needed, were classified as “resistant to healthy” (R2H), and those that exhibited diarrhea and recovered after treated with Trimidox or Excenel, indicating that their gut microbiota may be more susceptible to diarrhea-induced dysbiosis so that antimicrobials were needed, were classified as “susceptible to healthy” (S2H) (Table 1). In other words, calves were classified into three groups depending on the potential perturbations that their fecal microbiota experienced: H2H, no perturbations; R2H, diarrhea as the perturbation; S2H, therapeutic antimicrobials as the perturbation.
Table 1.
Grouping information of veal calves used in the current study.
| Calf ID | Group | Age of mortality (day) | Age of first diarrhea case (day) | Age of sample collection (day) | ||||
|---|---|---|---|---|---|---|---|---|
| 7 | 14 | 21 | 42 | 56 | ||||
| 3 | CON–H2H | – | – | T + RF (H) | T + RF (H) | T + RF (H) | T | T |
| 5 | (n = 13) | – | – | T + RF (H) | T + RF (H) | T + RF (H) | T | T |
| 26 | – | – | T + RF (H) | T + RF (H) | T + RF (H) | T | T | |
| 36 | – | – | T + RF (H) | T + RF (H) | T + RF (H) | T | T | |
| 37 | – | – | T + RF (H) | T + RF (H) | T + RF (H) | T | T | |
| 43 | – | – | T + RF (H) | T + RF (H) | T + RF (H) | T | T | |
| 44 | – | – | T + RF (H) | T + RF (H) | T + RF (H) | T | T | |
| 47 | – | – | T + RF (H) | T + RF (H) | T + RF (H) | T | T | |
| 54 | – | – | T + RF (H) | T + RF (H) | T + RF (H) | T | T | |
| 69 | – | – | T + RF (H) | T + RF (H) | T + RF (H) | T | T | |
| 52 | – | – | T + RF (H) | T + RF (H) | T + RF (H) | T | T | |
| 71 | – | – | T + RF (H) | T + RF (H) | T + RF (H) | T | T | |
| 76 | – | – | T + RF (H) | T + RF (H) | T + RF (H) | T | T | |
| 1 | CON–R2H | – | 14 | T + RF (U) | T | T | T | T |
| 2 | (n = 18) | – | 12 | T + RF (U) | T | T | T | T |
| 10 | – | 8 | T + RF (U) | T | T | T | T | |
| 13 | 24 | 11 | T + RF (U) | T | T | |||
| 18 | – | 10 | T + RF (U) | T | T | T | T | |
| 20 | 24 | 26 | T + RF (U) | T + RF (U) | T + RF (U) | |||
| 21 | – | 8 | T + RF (U) | T | T | T | T | |
| 27 | – | 14 | T + RF (U) | T | T | T | T | |
| 29 | – | 12 | T + RF (U) | T | T | T | T | |
| 34 | – | 12 | T + RF (U) | T | T | T | T | |
| 35 | – | 18 | T + RF (U) | T + RF (U) | T | T | T | |
| 46 | – | 9 | T + RF (U) | T | T | T | T | |
| 55 | – | 11 | T + RF (U) | T | T | T | T | |
| 60 | – | 12 | T + RF (U) | T | T | T | T | |
| 68 | – | 12 | T + RF (U) | T | T | T | T | |
| 70 | – | 13 | T + RF (U) | T | T | T | T | |
| 77 | – | 8 | T + RF (U) | T | T | T | T | |
| 78 | – | 11 | T + RF (U) | T | T | T | T | |
| 4 | CON–S2H | 7 | 10 | RF (U) | ||||
| 11 | (n = 11) | 15 | 12 | RF (U) | ||||
| 12 | – | 8 | T + RF (U) | T | T | T | T | |
| 19 | 12 | 8 | RF (U) | |||||
| 28 | – | 13 | T + RF (U) | T | T | T | T | |
| 45 | – | 11 | T + RF (U) | T | T | T | T | |
| 53 | – | 12 | RF (U) | T | T | T | T | |
| 61 | – | 15 | T + RF (U) | T + RF (U) | T | T | T | |
| 62 | – | 13 | T + RF (U) | T | T | T | T | |
| 63 | – | 11 | T + RF (U) | T | T | T | T | |
| 79 | – | 19 | T + RF (U) | T | T | T | T | |
| 6 | SCB–R2H | – | 12 | RF (U) | ||||
| 15 | (n = 15) | – | 12 | RF (U) | ||||
| 16 | – | 8 | RF (U) | |||||
| 17 | – | 12 | RF (U) | |||||
| 22 | – | 9 | RF (U) | |||||
| 25 | – | 10 | RF (U) | |||||
| 38 | – | 11 | RF (U) | |||||
| 49 | – | 12 | RF (U) | |||||
| 50 | – | 11 | RF (U) | |||||
| 66 | – | 8 | RF (U) | |||||
| 72 | – | 12 | RF (U) | |||||
| 73 | – | 8 | RF (U) | |||||
| 81 | – | 12 | RF (U) | |||||
| 82 | – | 12 | RF (U) | |||||
| 83 | – | 11 | RF (U) | |||||
| 7 | SCB–S2H | – | 32 | RF (U) | RF (U) | RF (U) | ||
| 24 | (n = 6) | 27 | 13 | RF (U) | ||||
| 40 | – | 12 | RF (U) | |||||
| 41 | – | 12 | RF (U) | |||||
| 42 | 22 | 19 | RF (U) | RF (U) | ||||
| 57 | – | 32 | RF (U) | RF (U) | RF (U) | |||
CON: calves fed milk replacer only; SCB: calves fed milk replacer and supplemented with 5 g of live Saccharomyces cerevisiae boulardii per day; T: samples used for analysis of temporal changes in microbial diversity, stability, composition, and taxa–function robustness; RF: samples used for random forest machine learning algorithm. (H) and (U) mean 'healthy' and 'unhealthy' microbiota, respectively; T + RF: samples used for both analysis of temporal changes in microbial diversity, stability, composition, and taxa–function robustness, and random forest machine learning algorithm.
H2H healthy to healthy: calves never exhibited diarrhea, R2H resistant to healthy: calves exhibited diarrhea but not treated with Trimidox or Excenel, S2H susceptible to healthy: calves exhibited diarrhea and treated with Trimidox or Excenel.
In this study, we did not include any individual calf supplemented with SCB in H2H group. This is because at 7 days of age no Saccharomyces boulardii was supplemented to calves in SCB group. Considering that supplementing SCB effectively decreased the incidence of diarrhea (SCB, 50.0%, 21/42 vs. CON, 69.1%, 29/42) [17], we were not sure if healthy calves in SCB group could still be healthy if SCB had not been supplemented. Therefore, only 21 calves exhibiting diarrhea in SCB group were enrolled in the current study, as SCB is a nonpathogenic biotherapeutic agent [19] and it should not contribute to the incidence of diarrhea with the dosage supplemented. Seven calves died during the experiment (Table 1) and the remaining 56 calves were healthy at 56 days of age.
DNA extraction, PCR amplification, and sequencing
Total DNA was extracted from fecal samples (standardized by DM concentration) using the repeated bead beating plus column method [20]. Briefly, 0.2 ± 0.1 g of sample was washed twice with TE buffer following with supplementation with cell lysis buffer containing 20% SDS, and physical disruption at 5000 rpm for 3 min using Biospec Mini Beads Beater 8 (BioSpec, Bartlesville, OK), incubation at 70 °C for 15 min and centrifugation at 16,000 × g for 5 min. The bead-beating, incubation, and centrifugation procedures were repeated, and impurities were removed from the supernatant using 10 M ammonium acetate, followed by DNA precipitation using isopropanol. After precipitation, DNA was purified with QIAmp fast DNA stool mini kit (Qiagen Inc, Germantown, MD). DNA quantity and purity were checked using NanoDrop 1000 spectrophotometer (Nanodrop Technologies, Wilmington, DE).
Amplification of V1–V3 hypervariable regions of the 16S rRNA gene for amplicon sequencing was conducted with 27F and 515R primers (27F-CS1F: ACACTGACGACATGGTTCTACAGAGTTTGATCMTGGCTCAG, 515R-CS2R: TACGGTAGCAGAGACTTGGTCTCCGCGGCKGCTGGCAC) [21]. The amplicon DNA with targeted size (~500 bp) was purified from 1% agarose gel using QIAEX II gel extraction kit (Qiagen Science, MD). The quality and quantity of purified PCR products were checked using a NanoDrop 1000 (NanoDrop Technologies, Wilmington, DE) to ensure that the concentration of DNA of all samples was above 25 ng/μl. In addition, negative controls without sample DNA were also sequenced. The amplicons were sequenced at Genome Quebec at McGill University (Montreal, QC, Canada) using Illumina’s MiSeq platform (2 × 300, pair end).
Analysis of taxonomic and functional composition
Sequencing data were processed using QIIME2 (version 2019.7; ref. [22]). Paired sequences were demultiplexed with “demux” plugin and subjected to quality control using “dada2” plugin [23]. To eliminate potential sequence contaminants, three amplicon sequence variants (ASVs) (two were identified as E. coli strains and the third was identified as an uncultured bacterium) identified in negative controls were removed from all samples. Then, 60 ASVs inversely correlated with DNA concentration were further removed using the “decontam” package in R [24]. Finally, taxonomy was assigned to filtered ASVs using a pretrained QIIME2-compatible SILVA 132 database with 99% identity for bacteria and assigned taxonomies to representative sequences. The number of shared bacterial genera was visualized using a Venn diagram [25]. To investigate how shifts in microbial composition impact the functional composition, microbial functionality (protein coding genes) at each day of age was predicted using the “q2-picrust2” plugin in QIIME2 [26].
Assessment of temporal changes in microbial diversity and stability
The temporal changes in microbial diversity and stability were evaluated using fecal microbiota from the three groups of CON calves only. Fecal microbiota SCB group was excluded due to the concern of potential interaction between SCB and diarrhea/therapeutic antimicrobials. Shannon index and Bray–Curtis distance matrix were computed with a sample depth of 2714 using the “diversity” plugin in QIIME2. Bray–Curtis distance between two successive samples was further computed using the “q2-longitudinal” plugin in QIIME2 [27]. The coefficient of variation (CV) was calculated for Shannon indices for each individual over time with lower CV values indicating relatively stable alpha diversity among individuals and vice versa.
Taxa–function robustness was used to assess microbial stability, which defines the degree to which a shift/perturbation in a community’s taxonomic composition will impact its functional capacities [28]. To quantitatively compare taxa–function robustness between communities, varying perturbations of each community’s taxonomic composition was simulated and the average shift in the functional profile of a community as a power function of the taxonomic perturbation magnitude (taxa–function response curve; Fig. S1a) was established as follows:
where t indicates the magnitude of simulated taxonomic perturbation, f indicates the expected shift in functional capacity. Two coefficients in the function, a (attenuation) and b (buffering), were used to directly evaluate taxa–function robustness. Attenuation is defined as inversely proportional to the response curve slope, indicating the expected rate at which increases in the taxonomic perturbation magnitude are expected to increase functional profile shifts [28]. Buffering indicates how large a perturbation must be before a functional profile shift becomes noticeable and approaches the expected shift magnitude defined by attenuation (Fig. S1b) [28]. Therefore, the higher the attenuation and buffering values, the more stable the functional capacity of a community.
Predictive modeling of early life diarrhea and validation of markers
One major aim of the current study was to investigate if early life microbiota could be applied to predict diarrhea in neonatal calves, the incidence of which occurred mostly between 7 and 21 days of age [29]. Therefore, the microbiota collected from H2H calves at 7, 14, and 21 days of age were defined as “healthy” (n = 39 from CON), while those collected from R2H and S2H calves prior to the onset of diarrhea in both CON (n = 33) and SCB groups (n = 26) were defined as “unhealthy” (Table 1).
To establish the model for predicting diarrhea, random forest algorithms (“randomForest” package in R) were used to identify “healthy” and “unhealthy” microbiota based on the relative abundance of selected bacterial genera. The accuracy of selected bacterial genera was then assessed by area under the receiver-operating characteristic (AUC) (“roc.curve” package) in R. To further minimize the potential over-fitting in the model, a threefold cross-validation approach (“trainControl” package in R) was applied [30]. The threefold cross-validation method splits 98 samples into three disjoint (nonoverlapping) subsets with similar number of samples (e.g. 33, 33, and 32). Then, two subsets (containing 66/65 samples) were used to train the model and the other subset (containing 32/33 samples, totally different from two subsets for training) was used to test the results. The threefold cross-validation was completed until accuracy was determined for each permutation and combination of samples in the subsets, and then an overall accuracy was estimated. The AUC of the optimized model was calculated using “roc.curve” package in R.
The difference in relative abundance of certain taxa between two conditions may not necessarily reflect the difference in their true abundance due to the lack of total microbial load [31]. We further performed differential abundance analysis to identify and rank the microbial markers that were truly different between “healthy” and “unhealthy” microbiota using multinomial regression as implemented in TensorFlow [32]. Such analysis ranks the microbes that are changing the most relative to each other between two conditions, and the ranks of relative differentials are identical to the ranks of absolute differentials in abundance [33].
Statistical analyses
Temporal changes in Shannon index, Bray–Curtis distance between 2 successive days of age, attenuation, and buffering values within each group were assessed using the nonparametric Friedman test with calves as repeated measures, and multiple comparison was conducted using Wilcoxon signed-rank test. Differences in CV of Shannon index, the relative abundance of fecal bacterial genera, and predicted genes at each days of age among three groups of calves, were assessed using the nonparametric Kruskal–Wallis test, and multiple comparison was conducted based on Mann–Whitney U test. The P value of multiple comparison of each test was adjusted into false discovery rate using the Benjamini-Hochbert (BH) algorithm [34]. Significant difference was declared at P ≤ 0.05 and tendencies at 0.05 < P ≤ 0.10.
Nucleotide sequence accession numbers
All DNA sequences were deposited in the NCBI sequence read archive and are accessible under the project number PRJNA506828.
Results
Temporal changes in microbial diversity and stability of gut microbiota within each group
For gut microbiota in H2H calves, Shannon index was higher at 21 (P = 0.029), 42 (P = 0.029), and 56 (P = 0.030) than 7 days of age and was higher at 21 (P = 0.029) than 14 days of age (Fig. 1a). For R2H calves, Shannon index was higher at 21 (P = 0.002), 42 (P = 0.005), and 56 (P = 0.002) than at both 7 and 14 days of age (Fig. 1b). For S2H calves, Shannon index was higher at 42 (P = 0.006) than 7 days of age (Fig. 1c). The CV of Shannon index in R2H calves was higher than that in S2H (P = 0.024) calves (Fig. S2).
Fig. 1. Temporal changes of Shannon index in gut microbiota.

a Calves that never exhibited diarrhea (H2H). b Calves that exhibited diarrhea but not treated with Trimidox or Excenel (R2H). c Calves that exhibited diarrhea and treated with Trimidox or Excenel (S2H). Statistical analysis was done by Friedman test and multiple comparisons were performed based on Wilcoxon signed-rank test and subjected to “BH” P value adjustment (*P ≤ 0.05).
The temporal changes in microbial variability were then investigated by assessing beta diversity (Bray–Curtis distance) between two successive days of age from the same calf changed over time among three groups of CON calves. Bray–Curtis distance between 42 vs. 21 days of age was significantly lower than that between 14 vs. 7 days of age in H2H (P = 0.014) (Fig. 2a) and R2H (P < 0.001) (Fig. 2b) calves. On the other hand, the Bray–Curtis distance between 56 vs. 42 days of age was significantly lower than that between 14 vs. 7 age in S2H calves (P = 0.014) (Fig. 2c).
Fig. 2. Temporal changes of Bray–Curtis distance between two successive days of age at sampling in gut microbiota.
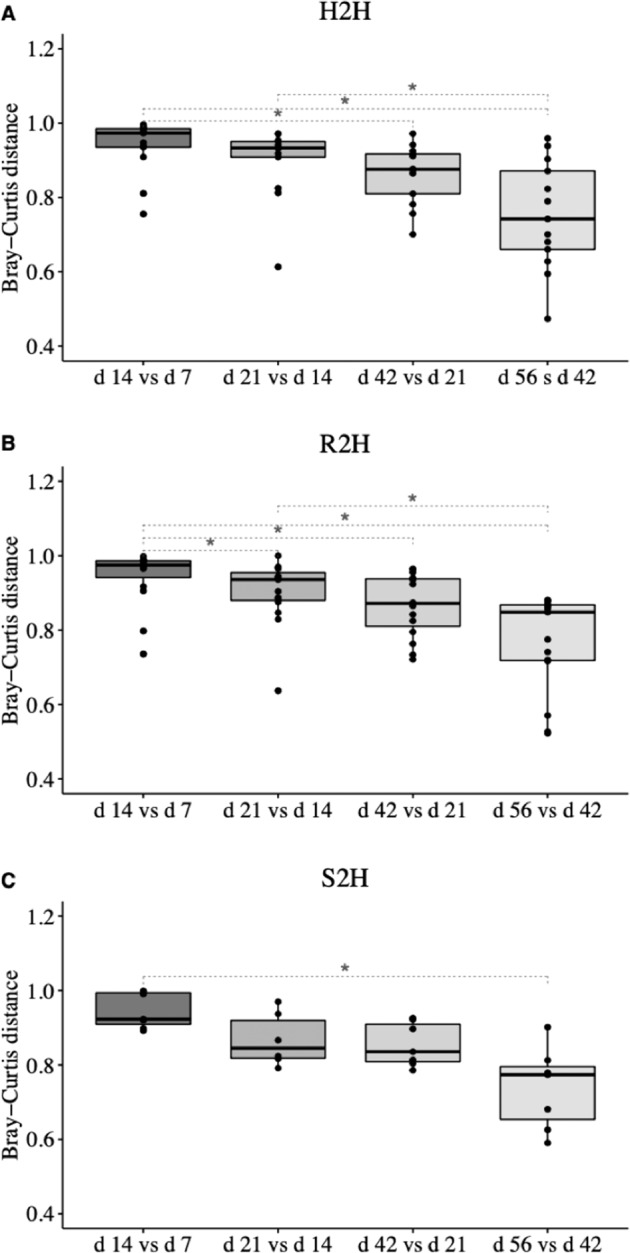
a Calves that never exhibited diarrhea (H2H); b calves that exhibited diarrhea but not treated with Trimidox or Excenel (R2H); c calves that exhibited diarrhea and treated with Trimidox or Excenel (S2H). Statistical analysis was done by Friedman test and multiple comparisons were performed based on Wilcoxon signed-rank test and subjected to “BH” P value adjustment (*P ≤ 0.05).
Furthermore, temporal stability of microbiota was evaluated by attenuation and buffering values. The attenuation value of gut microbiota was higher at 21 (P < 0.001) and 42 (P < 0.001) than 7 days of age in H2H calves, at 42 than 7 days of age (P = 0.025) in R2H calves, and was not different among all age groups in S2H calves (P = 0.179) (Fig. 3a–c). The buffering value was not different among age in either H2H (P = 0.077), R2H (P = 0.371), or S2H (P = 0.195) calves (Fig. 3d–f).
Fig. 3. Temporal changes of attenuation and buffering values in gut microbiota.

a Attenuation value of calves that never exhibited diarrhea (H2H). b Attenuation value of calves that exhibited diarrhea but not treated with Trimidox or Excenel (R2H). c Attenuation value of calves that exhibited diarrhea and treated with Trimidox or Excenel (S2H). d Buffering value of calves that never exhibited diarrhea (H2H). e Buffering value of calves that exhibited diarrhea but not treated with Trimidox or Excenel (R2H). f Buffering value of calves that exhibited diarrhea and treated with Trimidox or Excenel (S2H). Statistical analysis was done by Friedman test and multiple comparisons were performed based on Wilcoxon signed-rank test and subjected to “BH” P value adjustment (*P ≤ 0.05).
Comparison of temporal changes in taxonomic composition and predicted genes of gut microbiota
A total of 11,498,965 high-quality reads were generated from 220 fecal samples for the 63 calves. After quality control and removal of potential contaminants, the remaining 3,529,111 reads were assigned to 8331 ASVs, with an average of 16,568 ± 448 (average ± standard error) reads and 140 ± 4 ASVs per sample based on a 99% nucleotide sequence similarity. At each age, a total of 22, 30, 34, 31, and 31 bacterial genera were identified (defined as the taxon with the relative abundance >0.1% and present in more than half of the samples within each age group) in H2H calves (Fig. S3a), 22, 23, 38, 28, and 42 bacterial genera were identified in R2H calves (Fig. S3b), while 25, 28, 33, 35, and 39 bacterial genera were identified in S2H calves (Fig. S3c), at 7, 14, 21, 42, and 56 days of age, respectively.
A total of 17, 20, 21, 19, and 28 bacterial genera were shared among three groups of calves at 7, 14, 21, 42, and 56 days of age, respectively (Table S1). At 14 days of age, the relative abundance of Blautia (P = 0.058; Fig. S4a) tended to be higher, while that of Escherichia–Shigella (P = 0.067; Fig. S4b) tended to be lower in H2H than R2H calves. At 42 days of age, the relative abundance of unclassified Ruminococcaceae was higher in H2H than that in R2H calves (P = 0.021; Fig. S4c). No difference was observed in the relative abundance of any bacterial genus shared among the three groups of veal calves at 7, 21, or 56 days of age.
Based on the results of PICRUSt2, the relative abundance of three predicted protein coding genes associated with beta-lactam resistance, including “bla regulator protein blaR1” (K02172), “penicillin-binding protein 1” (K12552), and “penicillin-binding protein 3” (K12553), as well as one predicted gene associated with cationic antimicrobial peptide resistance, “aureolysin” (K01401), was significantly different among calves at 56 days of age. Specifically, the relative abundance of K02172 (P = 0.007 and 0.034; Fig. S5a), K12552 (P = 0.012 and 0.036; Fig. S5b), K12553 (P = 0.014 and 0.035; Fig. S5c), and K01401 (P = 0.012 and 0.035; Fig. S5d) was higher in S2H than both H2H and R2H calves, respectively.
Prediction of disease onset based on bacterial markers in early gut microbiota
The random forest supervised machine learning algorithm was applied to construct the model for predicting early life diarrhea in veal calves. Fifty-two bacterial genera were excluded prior to the establishment of model because their relative abundances were significantly affected by sampling days (age) in H2H calves (Table S1). The remaining 21 genera whose relative abundance did not change among sampling days were able to differentiate “healthy” and “unhealthy” microbiota with high accuracy (AUC = 0.713, Fig. S6a). Based on threefold cross-validation (Fig. S6b), it revealed that 6 of those 21 bacterial genera, including Trueperella, Streptococcus, Dorea, uncultured Lachnospiraceae, Ruminococcus 2, and Erysipelatoclostridium, mostly contributed to the discrimination power of health status with improved performance (AUC = 0.800, Fig. S6a). The relative rank of relative abundance of those six microbial markers was plotted against the relative rank of performance represented by AUC (Fig. 4a). Using the profiles of the six bacterial markers as independent variables, the random forest model exhibited an accuracy of 62.0% and 84.3% for predicting “healthy” and “unhealthy” gut microbiota, respectively (Fig. 4b). The relative abundance of uncultured Lachnospiraceae (P < 0.001), Ruminococcus 2 (P < 0.001), and Dorea (P = 0.025) was higher, and Streptococcus (P = 0.010) and Erysipelatoclostridium (P = 0.093) tended to be higher in “healthy” microbiota, while that of Trueperella (P < 0.033) was higher in “unhealthy” microbiota (Fig. S7).
Fig. 4. Prediction of health status based on fecal microbial markers.

a The relationship between the contributions of the six most discriminant bacterial genera to classification of microbiota as well as their relative abundance. b Accuracy of the random forest model established based on the six most discriminant bacterial genera for discriminating “healthy” (n = 39) and “unhealthy” (n = 59) microbiota. High indicates a probability above 0.50, and low indicates a probability below 0.50.
To further investigate if the difference in the relative abundance of those six microbial markers between “healthy” and “unhealthy” microbiota also existed in their absolute abundance, log-ratio and differential ranking techniques were applied. The log ratio between “unhealthy” and “healthy” microbiota of 178 bacterial genera was ranked by performing multinomial regression (Table S2). The results showed that Trueperella had low ranks (log ratio between “unhealthy” and “healthy” was 1.74), while Erysipelatoclostridium, Ruminococcus 2, Streptococcus, Dorea, and uncultured Lachnospiraceae had high ranks (log ratio between “unhealthy” and “healthy” was –0.07, –0.24, –0.36, –0.43, and –1.43, respectively) (Fig. 5). The difference in ranks between those taxa revealed that Trueperella was more prevalent in “unhealthy” microbiota, while the other five bacterial genera were more prevalent in “healthy” microbiota relative to other taxa.
Fig. 5. The ranks of bacterial genera between “healthy” and “unhealthy” gut microbiota estimated using multinomial regression.
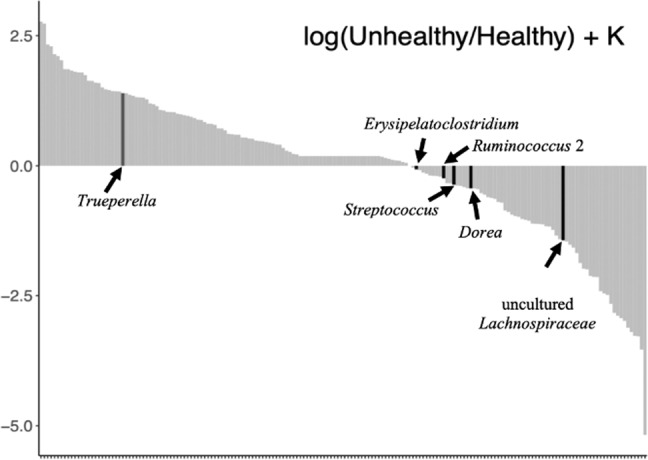
The y-axis represents the log-fold change that is known up to some bias constant K, and the x-axis numerically orders the ranks of each genus in the analysis. Truperella is postively associated with 'unhealthy' microbiota while Erysipelatoclostridium, Ruminococcus 2, Streptococcus, Dorea, and uncultured Lachnospiraceae are positively associated with 'healthy' microbiota.
Discussion
Increase in diversity and stability at an early age has been characterized as a feature of establishment of healthy gut microbiota in humans [35, 36]. In the current study, we observed an increase in Shannon index of gut microbiota from 7 to 56 days of age in both H2H and R2H calves. Such findings are similar to the changes in fecal microbiota in healthy dairy calves during first 8 weeks of age [37, 38]. However, the detected changes in gut microbial diversity in healthy calves of previous studies were largely confounded with diets, as those calves were fed different diets during their experimental trials [37, 38]. To overcome such limitations, the calves in the current study were fed only milk replacer (same amount under the same age), and therefore we speculate that host (age and growth) was the main factor driving the development of microbial diversity in early life of these veal calves. Although the temporal changes in microbial diversity of gut microbiota in R2H calves was similar to those in H2H calves, higher CV of Shannon index in R2H than H2H calves suggests that perturbations due to diarrhea in R2H calves may cause fluctuation to the development in microbial diversity. The use of therapeutic antimicrobials, on the other hand, “delayed” the temporal development of microbial diversity of gut microbiota from 7 to 21 days of age in S2H calves, which may be explained by the loss of microorganisms sensitive to antimicrobials as previously reported in human gut microbiota [39]. In addition, the decrease in Bray–Curtis distance between two successive samples reached significant at 42 and 21 days of age, respectively, in H2H and R2H groups, but at 56 days of age in S2H group, suggesting that it may take much longer time for microbiota to become less variable due to the use of antimicrobials. Those findings suggest that therapeutic antimicrobial treatment had more severe negative impact on the temporal development of microbial diversity and stability than diarrhea during the first 21 days of life in veal calves. The insignificant Shannon indices among R2H, S2H, and H2H calves at 56 days of age may be explained by the relatively long interval after disease/the use of therapeutic antimicrobials, during which the microbial diversity recovered as observed in humans [40, 41]. However, it should be noted that perturbation such as disease and antimicrobial treatment may also lead to an alternative stable but “dysbiosis” microbial community [42]. How those perturbations and altered microbial community affect the health and productivity of calves in their later life deserves further investigation.
To understand how those perturbations affect fecal microbiome (taxonomic and functional composition), we first compared the bacterial composition among H2H, R2H, and S2H calves. As most of the diarrhea incidence occurred in veal calves between 7 and 21 days of age, we speculated that the lower relative abundance of Blautia and higher relative abundance of Escherichia–Shigella at 14 days of age in R2H than H2H calves may be associated with diarrhea. High prevalence of Blautia has been reported in colon and fecal microbiota of healthy neonatal swine [43] and human infants [44, 45]. On the contrary, the reduction in the abundance of this genus was reported in fecal samples of dogs with diarrhea [46] and enteropathy [47]. Blautia utilizes polysaccharides that other gut microorganisms cannot degrade [48] and produces butyrate, one of the major SCFA that maintains gut health via regulation of the immune system [49] and/or maintenance of the intestinal epithelial barrier [50]. On the other hand, many species of Escherichia–Shigella, e.g. E. coli [51] and S. flexneri [52], are widely accepted as the major pathogens to cause bovine diarrhea. Although there was no difference in the relative abundance of any bacterial genus at any age between H2H and S2H calves, the detected higher relative abundance of predicted genes involved in antimicrobial resistance in S2H compared with H2H calves suggests that the changes of microbiota in response to the antimicrobials may occur not necessarily at compositional level but at functional level [53]. It has been reported that short-term antimicrobial treatment drove the gut microbiota to a more resilient state to future antimicrobial treatment in adult human due to the persistence of these increased antimicrobial-resistant genes for years [40]. In this regard, higher relative abundance of predicted microbial genes associated with antimicrobial resistance (bla regulator protein blaR1, penicillin-binding protein 1 and 3, aureolysin) at 56 days of age in H2S calves may potentially increase the risk of failure of future antimicrobial treatment.
Changes in the abundance of members could result in a minor or major alteration to a community’s functional profile, depending on the stability of microbiota [54]. Stability relates to the concept that stable equilibrium status exists for a microbial community, in which disturbances and temporal dynamics of individual members cause changes, but the community as a whole has the ability to restore such changes to the original status [55]. A community with higher stability is more resilient to external perturbations so its functional profile is less likely to change. Based on the attenuation and buffering values of three group of calves, higher attenuation implies that smaller functional shifts are expected in a community given similar levels of perturbation, while higher buffering indicates that larger perturbations are needed before a significant functional shift could be observed [28]. No temporal change in buffering was observed in either group, indicating that similar level of perturbations is required to cause a substantial functional shift regardless of sampling time, disease, or antimicrobial treatment. However, our results suggest that in H2H calves, the temporal development of stability was featured by a significant increase in attenuation value from 7 to 21 days of age, a pattern that was not observed in R2H or S2H calves. Higher attenuation in H2H calves may ensure the maintenance of functional capacities of the fecal microbial community in the face of ecological dysbiosis during early life. In comparison, the increase in attenuation value in R2H calves may be “delayed,” as it was not different between 21 and 7 days of age. The potential delay in the increase of attenuation over time in R2H calves may reflect gradual recovery of temporal stability after disease (diarrhea). Although we expected similar delay of the increase in attenuation in S2H as in R2H calves, the increase in attenuation was not observed in S2H calves. As it was reported that antimicrobial treatment may disrupt microbial community function [25], our results suggest that antimicrobial treatment may have a more negative impact on the temporal development of stability of gut microbiota compared with disease (such as diarrhea). Future studies using shotgun metagenomics may be able to determine the factors driving microbial resilience during early life, which is crucial for designing strategies to minimize the negative impact of antimicrobial treatment on microbial stability of veal calves. Despite those findings, it should be noted that further increase in sample size may be needed to validate those differences, as early life gut microbiota is featured by high between-individual variabilities indicated by the high SEM of relative abundance of bacterial genera (Table S1).
Considering the negative effect of perturbation due to diarrhea/antimicrobial treatment on temporal development of microbial diversity and stability, early prediction of diarrhea based on microbial markers may be a promising method to avoid such perturbations. Our results revealed higher prevalence of butyrate-producing bacteria belonging to families Lachnospiraceae (uncultured Lachnospiraceae and Dorea) and Ruminococcaceae (Ruminococcus 2) in healthy calves. Those bacterial genera were also reported in “healthy” microbiota in human beings or animals. Specifically, the relative abundance of R. bromii was higher [56], while that of Dorea was lower [57] in gut microbiota of healthy human subjects compared with Crohn’s disease patients. In addition, Erysipelatoclostridium was reported to be highly abundant in the hindgut digesta of healthy calves at 7 days of age [58]. Streptococcus, which was also enriched in “healthy” microbiota, contains strains such as S. infantarius and S. equines in bovine fecal samples, exhibited anti-Salmonella activities [59]. Our results highlight the potential of using these butyrate-producing bacteria as microbial markers for identifying healthy (high prevalence) or unhealthy (low prevalence) in young ruminants. On the contrary, Trueperella was reported as an opportunistic pathogen that may cause diverse pyogenic infections such as liver abscessation [60] and gastroenteritis [61] in cattle. Although the function of Trueperella has not been well studied in the gastrointestinal tract of calves, it can be speculated that this bacterial genus may potentially cause infections to in the gastrointestinal tract of calves and therefore be associated with the prevalence of diarrhea. Future studies are needed to investigate the mechanism of action of those microbial markers on the modulation of gut health in young ruminants.
The development of sequencing techniques and modeling algorithms facilitates the application of microbiota to predict the risk of disease outbreaks, which is one of the goals of microbiome research [12]. However, studies are still limited, and to our knowledge no study has been conducted in the prediction of disease onset based on gut microbiota in young ruminants. Teng et al. [16] exploited microbial indicators of dental caries (Prevotella spp.) after excluding the age-discriminatory taxa, yielding a predicting accuracy of 81%. More recently, it is reported that disease initiation can be diagnosed using gut microbial indicators with 95% accuracy in shrimp [62]. In this study, we initially tried to establish the model based on 72 samples from CON calves (39 healthy and 33 unhealthy) only. However, the prediction accuracy for “unhealthy” microbiota was <70% (data not shown). After including the 26 samples from SCB calves in the model, the prediction accuracy of “unhealthy” microbiota reached 84.3%, suggesting that early life microbiota has the potential to be employed for disease (such as diarrhea) prediction in young ruminants. It should be noted that in spite of the prediction model established by us and others, it is still challenging to predict human and animal diseases using microbial markers in practice. One of the reasons may be due to the fact that the “relative abundance” of microbial markers was used in those model establishing studies, which is difficult to compare among studies and make reproducible analyses due to the constraints of compositional data [63]. Recently, a novel method, differential abundance analysis, may address the issue of misinterpretation of compositional data and provide insight into the absolute change in microbiota between two conditions, as the ranks of relative differentials are identical to the ranks of absolute differentials [33]. By applying this method, it is suggested that Trueperella was positively associated with “unhealthy” microbiota, while the other five bacterial genera were positively associated with “healthy” microbiota. This further highlighted that the difference in the relative abundance of those six markers correctly reflects their absolute difference between “healthy” and “unhealthy” microbiota. One of the limitations of our study is that for most of diarrheic calves, only one sampling time (7 days of age) was available prior to incidence of diarrhea, which may not be sufficient to reflect the changes in gut microbiota during disease processes. Therefore, studies are needed to verify if and how those microbial markers play a role in the process of diarrhea in newborn calves with more sampling time points and different populations to determine their prediction specificity of diarrhea in young ruminants. It is also important to be noted that it may not be easy to choose a “optimal” or even “suitable” external cohort for validation of the random forest model for the current study. Due to the heterogeneity of DNA extraction, library preparation, sequencing, and bioinformatic techniques, meta-analyses among studies [64], the standardized microbial analysis pipeline should be developed in order to test the robustness of microbial classifiers developed based on machine learning algorithm such as the random forest model among studies.
Conclusion
Increased microbial diversity and stability are important features of temporal development of gut microbiota in healthy calves. However, the use of therapeutic antimicrobials for treating diarrhea resulted in delayed temporal development of diversity and stability of early life gut microbiota especially during 7 to 21 days of age, as well as increased relative abundance of predicted genes related to antimicrobial resistance at 56 days of age, suggesting that therapeutic antimicrobials may have negative impact on the long-term health of calves. Future studies of the relationship between changes in microbial composition/function in response to antimicrobials and gut health and productivity of calves is warranted using functional analysis such as metagenomics and/or culturomics to elucidate the mode of action of bacterial markers on the modulation of gut health of young ruminants. Using the random forest model, this study showed a promise to predict diarrhea in calves using gut microbiota. However, samples with unknown health status should be used to assess the accuracy of the current model as well as validation in different populations and animal herds in the future. Regardless, our study highlights the possibility of employing early life microbiota for predicting other types of disease or infections in neonatal food-producing animals.
Supplementary information
Acknowledgements
This work was funded by the Natural Sciences and Engineering Research Council of Canada (NSERC), Lallemand Animal Nutrition, Grober Animal Nutrition, Westgen, BC Dairy Association, Alberta Milk, Sask-Milk, and Dairy Farmers of Manitoba and by the Agricultural Science and Technology Innovation Program of the Chinese Academy of Agricultural Sciences and Chinese Scholarship Council Scholarship. We appreciate technical support and guidance from Drs A. Eng and E. Borenstein (University of Washington), F. Chaucheyras-Durand, M. Castex, and A. Aguilar (Lallemand Animal Nutrition), and A. Kerr and H. Copland (Grober Animal Nutrition).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41396-020-0678-3) contains supplementary material, which is available to authorized users.
References
- 1.Mathew AG, Cissell R, Liamthong S. Antibiotic resistance in bacteria associated with food animals: a United States perspective of livestock production. Foodborne Pathog Dis. 2007;4:115–33. doi: 10.1089/fpd.2006.0066. [DOI] [PubMed] [Google Scholar]
- 2.Bauer E, Williams BA, Smidt H, Verstegen MW, Mosenthin R. Influence of the gastrointestinal microbiota on development of the immune system in young animals. Curr Issues Intest Microbiol. 2006;7:35–52. [PubMed] [Google Scholar]
- 3.Hulbert LE, Moisá SJ. Stress, immunity, and the management of calves. J Dairy Sci. 2016;99:3199–216. doi: 10.3168/jds.2015-10198. [DOI] [PubMed] [Google Scholar]
- 4.Gensollen T, Iyer SS, Kasper DL, Blumberg RS. How colonization by microbiota in early life shapes the immune system. Science. 2016;352:539–44. doi: 10.1126/science.aad9378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kerr CA, Grice DM, Tran CD, Bauer DC, Li D, Hendry P, et al. Early life events influence whole-of-life metabolic health via gut microflora and gut permeability. Crit Rev Microbiol. 2015;41:326–40.. doi: 10.3109/1040841X.2013.837863. [DOI] [PubMed] [Google Scholar]
- 6.Zeissig S, Blumberg RS. Life at the beginning: perturbation of the microbiota by antibiotics in early life and its role in health and disease. Nat Immunol. 2014;5:307–10. doi: 10.1038/ni.2847. [DOI] [PubMed] [Google Scholar]
- 7.Cox LM, Blaser MJ. Antibiotics in early life and obesity. Nat Rev Endocrinol. 2015;11:182–90.. doi: 10.1038/nrendo.2014.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oikonomou G, Teixeira AG, Foditsch C, Bicalho ML, Machado VS, Bicalho RC. Fecal microbial diversity in pre-weaned dairy calves as described by pyrosequencing of metagenomic 16S rDNA. Associations of Faecalibacterium species with health and growth. PloS ONE. 2013;8:e63157. doi: 10.1371/journal.pone.0063157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oultram J, Phipps E, Teixeira AG, Foditsch C, Bicalho ML, Machado VS, et al. Effects of antibiotics (oxytetracycline, florfenicol or tulathromycin) on neonatal calves’ faecal microbial diversity. Vet Rec. 2015;177:598. doi: 10.1136/vr.103320. [DOI] [PubMed] [Google Scholar]
- 10.Beisner BE, Haydon DT, Cuddington K. Alternative stable states in ecology. Front Ecol Environ. 2003;1:376–82. [Google Scholar]
- 11.McEachran AD, Blackwell BR, Hanson JD, Wooten KJ, Mayer GD, Cox SB, et al. Antibiotics, bacteria, and antibiotic resistance genes: aerial transport from cattle feed yards via particulate matter. Environ Health Persp. 2015;123:337–43.. doi: 10.1289/ehp.1408555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cho Y, Yoon K-J. An overview of calf diarrhea-infectious etiology, diagnosis, and intervention. J Vet Sci. 2014;15:1–17. doi: 10.4142/jvs.2014.15.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knights D, Parfrey LW, Zaneveld J, Lozupone C, Knight R. Human-associated microbial signatures: examining their predictive value. Cell Host Microbe. 2011;10:292–6. doi: 10.1016/j.chom.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- 15.Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500:541–6. doi: 10.1038/nature12506. [DOI] [PubMed] [Google Scholar]
- 16.Teng F, Yang F, Huang S, Bo C, Xu ZZ, Amir A, et al. Prediction of early childhood caries via spatial-temporal variations of oral microbiota. Cell Host Microbe. 2015;18:296–306. doi: 10.1016/j.chom.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 17.Villot C, Ma T, Renaud D, Hosseini-Ghaffari M, Gibson DJ, Skidmore A, et al. Saccharomyces cerevisiae boulardii CNCM I-1079 affects health, growth and fecal microbiota in milk-fed veal calves. J Dairy Sci. 2019;102:7011–25.. doi: 10.3168/jds.2018-16149. [DOI] [PubMed] [Google Scholar]
- 18.Lesmeister K, Heinrichs A. Effects of corn processing on growth characteristics, rumen development, and rumen parameters in neonatal dairy calves. J Dairy Sci. 2004;87:3439–50.. doi: 10.3168/jds.S0022-0302(04)73479-7. [DOI] [PubMed] [Google Scholar]
- 19.Buts JP, De Keyser N. Effects of Saccharomyces boulardii on intestinal mucosa. Dig Dis Sci. 2006;51:1485–92.. doi: 10.1007/s10620-005-9016-x. [DOI] [PubMed] [Google Scholar]
- 20.Yu Z, Morrison M. Comparisons of different hypervariable regions of rrs genes for use in fingerprinting of microbial communities by PCR-denaturing gradient gel electrophoresis. Appl Environ Micro. 2004;70:4800–6. doi: 10.1128/AEM.70.8.4800-4806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kroes I, Lepp PW, Relman DA. Bacterial diversity within the human subgingival crevice. Proc Natl Acad Sci USA. 1999;96:14547–52. doi: 10.1073/pnas.96.25.14547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME2. Nat Biotechnol. 2019;37:852–7. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davis NM, Proctor DM, Holmes SP, Relman DA, Callahan BJ. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome. 2018;6:226. doi: 10.1186/s40168-018-0605-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heberle H, Meirelles GV, da Silva FR, Telles GP, Minghim R. InteractiVenn: a web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015;16:169. doi: 10.1186/s12859-015-0611-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Douglas GM, Maffei VJ, Zaneveld J, Yurgel SN, Brown JR, Taylor CM, et al. PICRUSt2: an improved and extensible approach for metagenome inference. BioRxiv. 2019:672295.
- 27.Bokulich NA, Dillon MR, Zhang Y, Rideout JR, Bolyen E, Li H, et al. q2-longitudinal: longitudinal and paired-sample analyses of microbiome data. mSystems. 2018;3:e00219–18. doi: 10.1128/mSystems.00219-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eng A, Borenstein E. Taxa-function robustness in microbial communities. Microbiome. 2018;6:45. doi: 10.1186/s40168-018-0425-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Urie NJ, Lombard JE, Shivley CB, Kopral CA, Adams AE, Earleywine TJ, et al. Preweaned heifer management on US dairy operations: part V. Factors associated with morbidity and mortality in preweaned dairy heifer calves. J Dairy Sci. 2018;101:9229–44.. doi: 10.3168/jds.2017-14019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cawley GC, Talbot NL. On over-fitting in model selection and subsequent selection bias in performance evaluation. J Mach Learn Res. 2010;11:2079–107. [Google Scholar]
- 31.Vandeputte D, Kathagen G, D’hoe K, Vieira-Silva S, Valles-Colomer M, Sabino J, et al. Quantitative microbiome profiling links gut community variation to microbial load. Nature. 2017;551:507–11.. doi: 10.1038/nature24460. [DOI] [PubMed] [Google Scholar]
- 32.Abadi M, Barham P, Chen J, Chen Z, Davis A, Dean J, et al. Tensorflow: a system for large-scale machine learning. In: 12th USENIX symposium on operating systems design and implementation (OSDI 16); USENIX Association, Savannah, GA, 2016. p. 265–83.
- 33.Morton JT, Marotz C, Washburne A, JSilverman J, Zaramela LS, Edlund A, et al. Establishing microbial composition measurement standards with reference frames. Nat Commun. 2019;10:2719. doi: 10.1038/s41467-019-10656-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- 35.Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–7. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO. Development of the human infant intestinal microbiota. PLoS Biol. 2007;5:e177. doi: 10.1371/journal.pbio.0050177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klein-Jöbstl D, Schornsteiner E, Mann E, Wagner M, Drillich M, Schmitz-Esser S. Pyrosequencing reveals diverse fecal microbiota in Simmental calves during early development. Front Microbiol. 2014;5:622. doi: 10.3389/fmicb.2014.00622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dill-McFarland KA, Breaker JD, Suen G. Microbial succession in the gastrointestinal tract of dairy cows from 2 weeks to first lactation. Sci Rep. 2017;7:40864. doi: 10.1038/srep40864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Panda S, El khader I, Casellas F, Vivancos JL, Cors MG, Santiago A, et al. Short-term effect of antibiotics on human gut microbiota. PLoS ONE. 2014;9:e95476. doi: 10.1371/journal.pone.0095476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jakobsson HE, Jernberg C, Andersson AF, Sjölund-Karlsson M, Jansson JK, Engstrand L. Short-term antibiotic treatment has differing long-term impacts on the human throat and gut microbiome. PloS ONE. 2010;5:e9836. doi: 10.1371/journal.pone.0009836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pérez-Cobas AE, Gosalbes MJ, Friedrichs A, Knecht H, Artacho A, Eismann K, et al. Gut microbiota disturbance during antibiotic therapy: a multi-omic approach. Gut. 2013;62:1591–601. doi: 10.1136/gutjnl-2012-303184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sommer F, Anderson JM, Bharti R, Raes J, Rosenstiel P. The resilience of the intestinal microbiota influences health and disease. Nat Rev Microbiol. 2017;15:630–8. doi: 10.1038/nrmicro.2017.58. [DOI] [PubMed] [Google Scholar]
- 43.Saraf MK, Piccolo BD, Bowlin AK, Mercer KE, LeRoith T, Chintapalli SV, et al. Formula diet driven microbiota shifts tryptophan metabolism from serotonin to tryptamine in neonatal porcine colon. Microbiome. 2017;5:77. doi: 10.1186/s40168-017-0297-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jost T, Lacroix C, Braegger CP, Rochat F, Chassard C. Vertical mother–neonate transfer of maternal gut bacteria via breastfeeding. Environ Microbiol. 2013;16:2891–904. doi: 10.1111/1462-2920.12238. [DOI] [PubMed] [Google Scholar]
- 45.Sagheddu V, Patrone V, Miragoli F, Morelli L. Abundance and diversity of hydrogenotrophic microorganisms in the infant gut before the weaning period assessed by denaturing gradient gel electrophoresis and quantitative PCR. Front Nutr. 2017;4:29. doi: 10.3389/fnut.2017.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Minamoto Y, Dhanani N, Markel ME, Steiner JM, Suchodolski JS. Prevalence of Clostridium perfringens, Clostridium perfringens enterotoxin and dysbiosis in fecal samples of dogs with diarrhea. Vet Microbiol. 2014;174:463–73. doi: 10.1016/j.vetmic.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 47.AlShawaqfeh MK, Wajid B, Minamoto Y, Markel M, Lidbury JA, Steiner JM, et al. A dysbiosis index to assess microbial changes in fecal samples of dogs with chronic inflammatory enteropathy. FEMS Microbiol Ecol. 2017;93:fix136. doi: 10.1093/femsec/fix136. [DOI] [PubMed] [Google Scholar]
- 48.Flint HJ, Bayer EA, Rincon MT, Lamed R, White BA. Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis. Nat Rev Microbiol. 2008;6:121–31. doi: 10.1038/nrmicro1817. [DOI] [PubMed] [Google Scholar]
- 49.Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446–50. doi: 10.1038/nature12721. [DOI] [PubMed] [Google Scholar]
- 50.Ruemmele FM, Schwartz S, Seidman EG, Dionne S, Levy E, Lentze MJ. Butyrate induced Caco-2 cell apoptosis is mediated via the mitochondrial pathway. Gut. 2003;52:94–100. doi: 10.1136/gut.52.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bartels CJM, Holzhauer M, Jorritsma R, Swart WAJM, Lam TJGM. Prevalence, prediction and risk factors of enteropathogens in normal and non-normal faeces of young Dutch dairy calves. Prev Vet Med. 2010;93:162–9. doi: 10.1016/j.prevetmed.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu Z, Cao M, Zhou X, Li B, Zhang J. Epidemic characterization and molecular genotyping of Shigella flexneri isolated from calves with diarrhea in Northwest China. Antimicrob Resist Infect Control. 2017;6:92. [DOI] [PMC free article] [PubMed]
- 53.Ma X, Zhang Q, Zheng M, Gao Y, Yuan T, Hale L, et al. Microbial functional traits are sensitive indicators of mild disturbance by lamb grazing. ISME J. 2019;13:1370–3. doi: 10.1038/s41396-019-0354-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moya A, Ferrer M. Functional redundancy-induced stability of gut microbiota subjected to disturbance. Trends Microbiol. 2016;24:402–13. doi: 10.1016/j.tim.2016.02.002. [DOI] [PubMed] [Google Scholar]
- 55.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;13:220–30. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mondot S, Kang S, Furet JP, de Cárcer AD, McSweeney C, Morrison M, et al. Highlighting new phylogenetic specificities of Crohn’s disease microbiota. Inflamm Bowel Dis. 2011;17:185–92. doi: 10.1002/ibd.21436. [DOI] [PubMed] [Google Scholar]
- 57.Sokol H, Jegou S, McQuitty C, Straub M, Leducq V, Landman C, et al. Specificities of the intestinal microbiota in patients with inflammatory bowel disease and Clostridium difficile infection. Gut Microbes. 2018;9:55–60. doi: 10.1080/19490976.2017.1361092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Song Y, Malmuthuge N, Steele MA, Guan LL. Shift of hindgut microbiota and microbial short chain fatty acids profiles in dairy calves from birth to pre-weaning. FEMS Microbiol Ecol. 2018;93:fix179. doi: 10.1093/femsec/fix179. [DOI] [PubMed] [Google Scholar]
- 59.Adetoye A, Pinloche E, Adeniyi BA, Ayeni FA. Characterization and anti-salmonella activities of lactic acid bacteria isolated from cattle faeces. BMC Microbiol. 2018;18:96. doi: 10.1186/s12866-018-1248-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rzewuska M, Kwiecień E, Chrobak-Chmiel D, Kizerwetter-Świda M, Stefańska I, Gieryńska M. Pathogenicity and virulence of Trueperella pyogenes: a review. Int J Mol Sci. 2019;20:2737. doi: 10.3390/ijms20112737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hurst CJ. Opportunistic bacteria associated with mammalian livestock disease. In: The connections between ecology and infectious disease. Cham: Springer; 2018. p. 185–238.
- 62.Xiong J, Zhu J, Dai W, Dong C, Qiu Q, Li C. Integrating gut microbiota immaturity and disease‐discriminatory taxa to diagnose the initiation and severity of shrimp disease. Environ Microbiol. 2017;19:1490–501.. doi: 10.1111/1462-2920.13701. [DOI] [PubMed] [Google Scholar]
- 63.Gloor GB, Wu JR, Pawlowsky-Glahn V, Egozcue JJ. It’s all relative: analyzing microbiome data as compositions. Ann Epidemiol. 2016;26:322–9. doi: 10.1016/j.annepidem.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 64.Langille MG. Exploring linkages between taxonomic and functional profiles of the human microbiome. MSystems. 2018;3:e00163–17. doi: 10.1128/mSystems.00163-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.