

Abstract
The use of biologic agents including anti-tumor necrosis factor monoclonal antibodies followed by anti-integrins and anti-interleukins has drastically changed the treatment paradigm of Crohn’s disease (CD) by improving clinical symptoms and mucosal healing. However, up to 70% of CD patients still eventually undergo surgery mainly due to fibrostenotic strictures. There are no specific anti-fibrotic drugs yet. This review comprehensively addresses the mechanism, prediction, diagnosis and treatment of the fibrostenotic strictures in CD. We also introduce promising anti-fibrotic agents which may be available in the near future and summarize challenges in developing novel therapies to treat fibrostenotic strictures in CD.
Keywords: Crohn disease, Stricture, Fibrostenosis, Intestinal fibrosis, Endoscopic balloon dilatation
INTRODUCTION
At initial diagnosis, most patients with Crohn’s disease (CD) have purely inflammatory phenotype without structural complications (strictures or fistulae); only 10% of CD patients have fibrostenotic strictures [1]. However, approximately 50% of CD patients eventually progress to clinically apparent fibrostenotic strictures during their lifetime [2,3]. The true incidence of fibrostenotic stricture tends to be underestimated owing to the subclinical deposition of extracellular matrix (ECM) over time [1]. It has been suggested that a certain degree of fibrosis exists in nearly all CD patients [2]. Growing evidence indicates that strictures in CD may precede the development of fistula and abscess [4,5].
Surgery will be needed in more than 70% of patients with CD within their lifetime to correct a severe complication such as intestinal obstruction, perforation, fistula and abscess [3,6-9]. Despite of surgery, the rates of postoperative recurrence are high. Endoscopic recurrence by the third postoperative year is reported in 85%–100% of cases [7,10] and fibrostenotic stricture is the most common indication for surgery in CD [3,9]. Surgical resection is related with major morbidity, higher costs, higher risk of unemployment, and poorer quality of life [3,11,12].
Although the introduction of biologics including anti-tumor necrosis factor (TNF), anti-integrin, and anti-interleukin (IL) has modified the disease course of CD in the short term, the long-term outcome of those drugs on the development of fibrostenosis and the need for surgery remains to be elucidated [5]. Fibrostenotic strictures were previously considered an inevitable and irreversible consequence of long-term inflammation in CD patients whose conditions are unresponsive to anti-inflammatory therapies. This paradigm has been changing rapidly due to recent advances in our understanding regarding the process of intestinal fibrosis and the introduction of promising candidates for targeted anti-fibrotic therapy [1].
This review aimed to provide a comprehensive overview of fibrostenotic strictures in CD, including mechanisms and factors that predict its progression, as well as diagnosis and treatment strategies. It also introduces promising anti-fibrotic agents for intestinal fibrosis and discuss the obstacles to be overcome in developing clinically available anti-fibrotic agents for CD stricture.
MECHANISMS OF FIBROSTENOTIC STRICTURE
CD is characterized by chronic and persistent inflammation leading to transmural tissue injury. Intestinal fibrosis results from dysregulated wound healing of the bowel wall [13,14]. In physiologic wound healing, tissue repair mechanisms restore normal structure from damaged tissues by a controlled response [15]. On the other hand, fibrosis results from an aberrant wound healing characterized by excessive deposition of the ECM, which is promoted by an increase in the number of mesenchymal cells, including fibroblasts, myofibroblasts, and smooth muscle cells [1]. When the fibrosis continues and becomes uncontrollable due to repetitive inflammation, the fibroblasts can transdifferentiate into myofibroblasts and in addition a muscular layer thickening caused by smooth muscle hyperplasia and hypertrophy occurs [16,17]. It is assumed that both ECM deposition and muscular thickening has a role in the development of fibrostenotic strictures in CD [18]. The pathophysiological process to fibrotic strictures is summarized in Fig. 1.
Fig. 1.
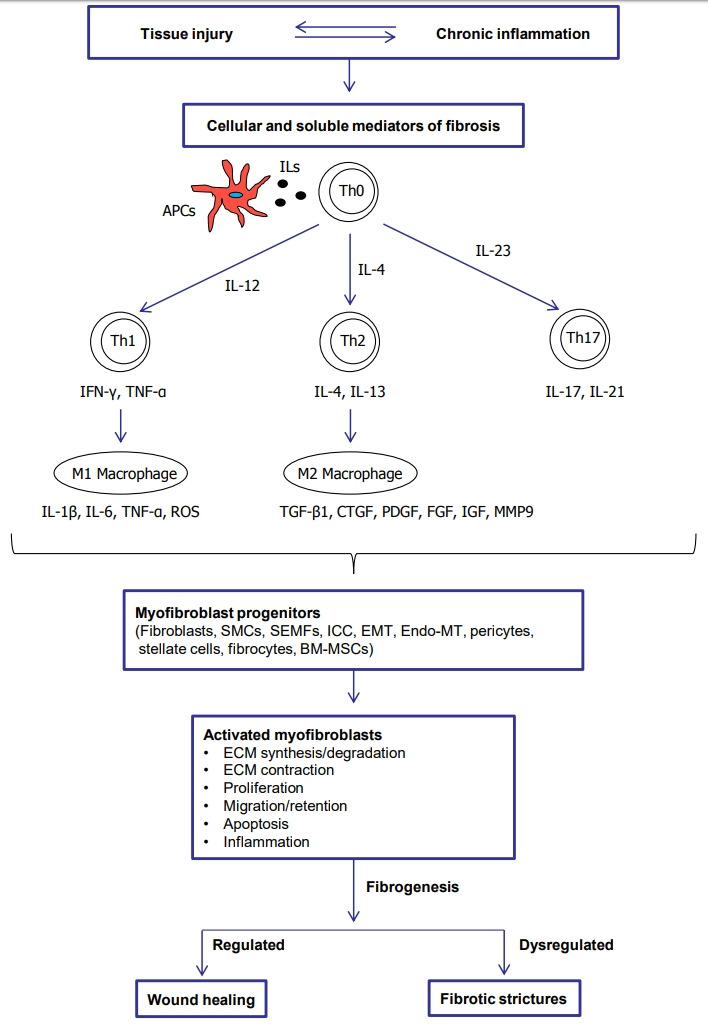
Pathophysiological process to fibrotic strictures. APCs, antigen presenting cells; ILs, interleukins; Th0, naïve T cells; Th, T helper cells; IFN-γ, interferon-γ; TNF-α, tumor necrosis factor α; ROS, reactive oxygen species; TGF-β1, transforming growth factor-β1; CTGF, connective tissue growth factor; PDGF, platelet-derived growth factor; FGF, fibroblast growth factor; IGF, insulin-like growth factor; MMP9, matrix metalloproteinase 9; SMCs, smooth muscle cells; SEMFs, subepithelial myofibroblasts; ICC, interstitial cells of Cajal; EMT, epithelial mesenchymal transition; Endo-MT, endothelial mesenchymal transition; BM-MSCs, bone marrow derived mesenchymal stem cells; ECM, extracellular matrix.
1. Location and Histopathology
The location of fibrostenotic strictures is almost likely to coincide with inflamed lesions in CD [14]. No report to date has described fibrostenotic CD strictures occurring outside areas affected by inflammation [2]. Due to the transmural inflammation in CD, both fibrosis and smooth muscle hyperplasia/hypertrophy can affect all layers of the intestine from mucosa to serosa [13,16,19]. Among these layers, fibrosis appears to be the most prominent in the submucosa, followed by the mucosa, but minimal in the muscularis propria. In contrast, smooth muscle hyperplasia/hypertrophy appears to be predominant in the muscularis mucosa and propria [16]. Histological specimens of CD-associated strictures show thickened muscularis mucosa. The submucosa is thickened with dense collagen accumulation, myofibroblasts, and islands of smooth muscle cells. The muscularis propria is thickened with collagen content interposed between fibers [1,20]. CD can show fibromuscular obliteration of the submucosa, which comprises the fibrosis intermingled with smooth muscle bundles originated from in situ proliferation or migration, accompanied by thickening of the muscularis mucosa and muscularis propria [16,21]. In stricturing CD, the bowel thickening is promoted by several histological alterations, including fibrosis, smooth muscle hyperplasia/hypertrophy, adipose hyperplasia, and edema. As a major contributing factor to intestinal wall thickening in CD, smooth muscle hyperplasia/hypertrophy may be the most critical factor, whereas fibrosis is less significant [16].
2. Cellular and Soluble Mediators of Fibrosis
The 2 key effector cells of intestinal fibrosis are the fibroblasts and myofibroblasts, which produce several ECM proteins [22,23]. In the inflamed intestine, local intestinal mesenchymal cells can transdifferentiate among the 3 phenotypes of fibroblasts (α-smooth muscle actin [α-SMA-]), myofibroblasts (α-SMA+), and smooth muscle cells (α-SMA+ and desmin+) [17]. Myofibroblasts are defined as an activated or differentiated fibroblasts, which have an intermediate phenotype between fibroblasts (ECM production) and smooth muscle cells (contractility) [17,24].
In inflammatory bowel disease (IBD), persistent inflammation results in sustained activation and proliferation of myofibroblasts, leading to the ECM over-accumulation. Human intestinal smooth muscle cells (HISMCs) also have a critical role in intestinal fibrosis, responding to the chronic inflammation by proliferating and depositing collagen in ECM [25]. Activated myofibroblasts are considered the final effector cells of intestinal fibrosis [24]. The activated myofibroblasts express high levels of SMA and therefore represent a markedly increased capability to contract ECM, which in turn contributes to tissue distortion and stricture [26]. There are 2 types of myofibroblasts in the gut: subepithelial myofibroblasts (SEMFs) [27,28] and interstitial cells of Cajal, which are found between the nerve endings and smooth muscle cells in the submucosa and the muscularis propria [29]. SEMFs have both pro-inflammatory and pro-fibrotic roles depending on surrounding conditions. The SEMFs express Toll-like receptors (TLR-4 and TLR-5), which can mediate the pro-inflammatory role of the SEMFs by inducing IL-6 and IL-8 secretion responding to lipopolysaccharide (TLR-4 ligand) or flagellin (TLR-5 ligand) [30,31]. Meanwhile, the soluble factors from intestinal epithelial cells stimulated with pro-inflammatory cytokines induce pro-fibrotic activity of the SEMFs by enhancing ECM production [32]. The intestinal fibroblasts/myofibroblasts can originate from multiple sources, including the migration and proliferation of local mesenchymal cells, epithelial cells via epithelial mesenchymal transition [33,34]. endothelial cells via endothelial–mesenchymal transition [35], stellate cells [17], pericytes [36], circulating precursors (so-called fibrocytes) [37], and the bone marrow [38].
Macrophages and T helper (Th) cells can promote intestinal fibrosis. M2 macrophages are differentiated in response to IL-4 or IL-13 and produce several pro-fibrotic cytokines including transforming growth factor-β1 (TGF-β1), connective tissue growth factor, platelet-derived growth factor (PDGF), fibroblast growth factor (FGF), and insulin-like growth factor (IGF) [36]. These pro-fibrotic cytokines promote myofibroblast proliferation and ECM production [20,22,36,39]. Th cell subsets (Th17 and Th2) are also potently fibrogenic. The Th17 response produces IL-17 and IL-21, which promote ECM production in intestinal myofibroblasts [39,40]. In the Th2 response characterized by IL-4 and IL-13 production [36,41]. IL-13 might directly and indirectly promote collagen deposition by fibroblasts. As IL-13 receptors are expressed in fibroblasts, IL-13 can directly activate collagen production in fibroblasts. In addition, IL-13 stimulates the secretion of latent TGF-β1 and matrix metalloproteinase 9 (MMP9) by macrophages. The MMP9 activates the latent TGF-β1 by cleaving the latency-associated peptide (LAP) [42].
TGF-β1 is considered a major cytokine in intestinal fibrosis [43]. Activated TGF-β1 transdifferentiates human fibroblasts into myofibroblasts [44] by inducing α-SMA, promotes proliferation [44] and the production of ECM and tissue inhibitors of metalloproteinase-1 (TIMP-1) [43,45], migration [46], contraction [47], and resistance to apoptosis in myofibroblasts [44]. The activation of TGF-β type I receptor by TGF-β1 mediates the phosphorylation of Smad2/3, which combines with Smad4. Subsequently, the Smad complex translocates into the nucleus and induces pro-fibrotic gene expression [48]. TGF-β1-induced fibrogenesis is also mediated by Smad-independent pathways, including phosphorylation of extracellular signal regulated kinase, c-Jun N-terminal kinase, p38 mitogen-activated protein kinase (MAPK), and AKT [26,48-50]. Another Smad-independent pathway is Ras homolog family member/Rho kinase/actin/myocardin-related transcription factor/serum response factor (Rho/ROCK/Actin/MRTF/SRF) pathway [51,52]. TGF-β1-induced activation of Rho and its effector ROCK, which promotes actin polymerization from globular actin to filamentous actin. Activated ROCK results in phosphorylated myosin light chain, which contributes to actin polymerization, stress fiber formation, and contraction. Meanwhile, actin polymerization releases the globular actin from MRTF, and subsequently the free MRTF translocates into the nucleus and binds to SRF. Finally, the MRTF/SRF complex induces fibrogenic genes including collagen, fibronectin, α-SMA, and myosin light-chain kinase [51-53]. The overview of Smad-dependent and Smad-independent TGF-β signaling in intestinal fibrosis is summarized in Fig. 2. TGF-β1 activates myofibroblasts, but conversely, the myofibroblasts can activate the latent TGF-β1. The forces generated by myofibroblast contraction pull on the integrin bound to LAP, which subsequently results in a conformational change of the latent TGF-β1 complex that liberates active TGF-β1 [54]. This mechanism may partially explain the autopropagation of intestinal fibrosis in CD.
Fig. 2.
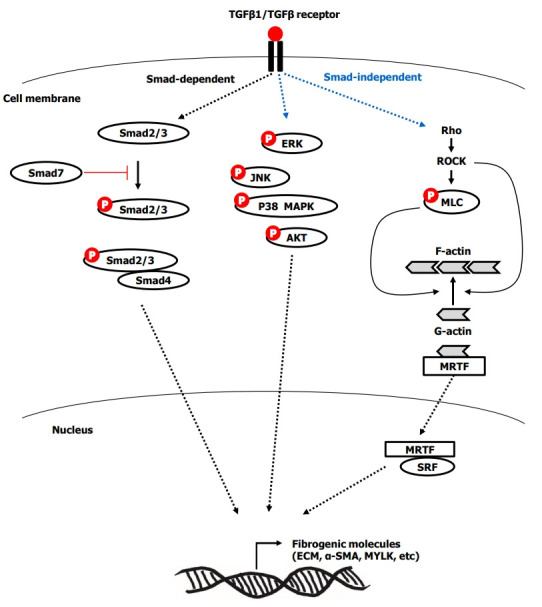
Overview of Smad-dependent and Smad-independent transforming growth factor-β (TGF-β) signaling in intestinal fibrosis. ERK, extracellular signal regulated kinase; JNK, c-Jun N-terminal kinase; p38 MAPK, p38 mitogen-activated protein kinase; Rho, Ras homolog family member; ROCK, Rho kinase; MLC, myosin light chain; F-actin, filamentous actin; G-actin, globular actin; MRTF, myocardin-related transcription factor; SRF, serum response factor; ECM, extracellular matrix; α-SMA, α-smooth muscle actin; MYLK, myosin light-chain kinase.
The IL-13/IL-13R/TGF-β/Smad signaling is considered a main pathway of intestinal fibrosis, which has been shown in chronic trinitrobenzene sulfonic acid (TNBS) colitis [55]. The immune response of chronic TNBS colitis can be divided into 3 phases. Intrarectal TNBS delivery induces the first phase, acute colitis with increased production of Th1 cytokines, IL-12p70, and interferon-γ, which is followed by the second phase, chronic colitis with Th17 response (IL-23 and IL-17). The last phase is characterized by Th2 response (IL-4 and IL-13) accompanied by TGF-β1 and collagen production. In the late phase of chronic TNBS colitis, IL-13 induces the production of TGF-β1 in macrophages by inducing and binding to IL-13 receptor (IL-13Rα2); subsequently, TGF-β1 induces collagen production by Smad2/3 phosphorylation [41,56,57].
The putative role of IL-36α/IL-36R signaling in fibrostenotic strictures in CD was recently identified, and targeting IL-36R signaling has emerged as a promising strategy for treatment of intestinal fibrosis in CD patients [58]. The expression levels of IL-36α are increased in the fibrostenotic tissue of CD patients. IL-36α secreted from inflammatory macrophages appears to bind to IL-36R of intestinal myofibroblasts located adjacent to macrophages and subsequently induces collagen VI production and proliferation in myofibroblasts [58-60]. Knockout of the IL-36R gene or neutralizing of the IL-36R reduced intestinal fibrosis in mouse models [58]. TNF like cytokine 1A (TL1A) secreted from immune cells binds to death domain receptor 3 (DR3) expressed in intestinal myofibroblasts, and consequently increases collagen production and proliferation in myofibroblasts, leading to intestinal fibrosis in CD strictures [61,62]. TL1A is highly expressed in the fibrotic tissue of CD patients and a genetic variant of the TL1A gene, which increases TL1A expression, is associated with a higher risk of fibrotic strictures in CD [61,63]. Transgenic mice with constitutive TL1A expression showed increased intestinal fibrosis that worsened under colitogenic conditions [62,63]. More recently, anti-TL1A antibody injection reduced collagen deposition in a mouse intestinal fibrosis model [64].
3. Role of Gut Microbiome
The gut microbiome might be a unique pro-fibrotic factor that may distinguish intestinal from other organ fibrosis [14]. Emerging evidence has underlined the significance of epithelial barrier dysfunction in the pathogenesis of IBD [65]. The altered barrier function can result in bacterial translocation into the lamina propria, and microbial antigens can stimulate immune and non-immune cells to produce pro-fibrotic factors [14]. Essentially, all intestinal immune and non-immune cells can respond to pathogen-associated molecular patterns through pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs) and nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs) [66,67]. Previous studies have shown that gut microbes and their products can induce intestinal fibrosis. Preliminary evidence showed that the TLR5 ligand flagellin, which exists on nearly all flagellated bacteria, promotes ECM production and proliferation of fibroblasts [68,69]. Recently, flagellin from adherent-invasive Escherichia coli (AIEC), which is frequently isolated from the ileal tissue of CD patients and may contribute to the development of CD, was able to lead to intestinal fibrosis by binding to TLR5 expressed in intestinal epithelium, which subsequently induce the expression of IL-33 and its receptor (ST2), leading to the production of IL-13 and TGF-β in intestinal epithelial cells or T cells [70]. The blockade of IL-33/ST2 signaling by anti-ST2 blocking antibody significantly ameliorated intestinal fibrosis induced by co-infection of AIEC and Salmonella, which suggests a novel anti-fibrotic therapy targeting fibrogenic bacteria and its downstream signaling [70]. In addition, serum antibody to flagellin (anti-CBir1 flagellin), which shows an aberrant adaptive immunity to luminal commensal bacteria, is significantly increased in CD patients [71]. Serum anti-CBir1 expression was independently associated with complicated CD including fibrostenotic diseases [71,72]. Besides TLRs, NLR, an intracellular PRR that binds bacterial cell wall component (muramyl dipeptide), is associated with fibrostenotic CD. NOD2 polymorphism may be associated with major genetic susceptibility for CD [73]. Carrying at least one NOD2 variant was associated with increased risk of fibrostenotic CD, which suggests that bacterial components might promote intestinal fibrosis by altered recognition of NOD2 variants [74].
The pro-fibrotic activity of the gut microbiota has been proven in several animal models of intestinal fibrosis. The injection of a bacterial cell wall component (peptidoglycan polysaccharides), feces, or anaerobic bacteria into the bowel wall induces intestinal fibrosis [75,76]. The oral delivery of Salmonella typhimurium induces chronic inflammation with fibrosis in the cecum and colon [77]. These observations indicate that the gut microbiome, at least in part, might initiate fibrogenesis in the gut. However, whether the gut microbiome will contribute substantially and continuously to fibrosis progression remains to be determined.
4. Matrix Stiffness Regulating Fibrosis Autopropagation
The ECM is both a consequence of fibrosis and a critical contributor to intestinal fibrosis by matrix stiffness [14]. ECM stiffness extensively affects cellular behaviors. Cells on a soft matrix are generally rounded, minimally adhesive and proliferative, and prone to apoptosis. In contrast, cells on a stiff matrix are spreading, adhesive, proliferative, and fibrogenic [78]. This phenomenon has also been proven in human intestinal myofibroblasts. Matrix stiffness, gradually appearing in fibrotic tissue of CD, induces a morphological transformation of round to stellate shape, focal adhesion formation, cellular proliferation, and collagen and α-SMA production, even in the absence of inflammatory stimulation [79]. These results may explain the autopropagation of fibrosis without inflammation in CD strictures, which is not diminished by the use of anti-inflammatory agents [11,79,80]. In the T cell transfer model of colitis, anti-TNF monotherapy significantly reduced gut inflammation and the colonic production of pro-inflammatory cytokines but did not significantly reduce collagen deposition or TGF-β1 production [11]. In a previous study of S. typhimurium–infected mice, early levofloxacin eradication after Salmonella infection partially prevented intestinal fibrosis progression. However, delayed eradication failed to prevent intestinal fibrosis progression. This finding indicates that there is a time point at which fibrosis is reversible, at least to a certain degree, if the microbial trigger is suppressed at an early time point, suggesting a role of the early “top-down” therapeutic approach to preventing fibrosis. In addition, this result shows that fibrosis is initiated early after inflammation, persists, and auto-propagative in the absence of inflammation and that the matrix stiffness may contribute to this process [79,80].
5. Mesenteric Fat and Intestinal Fibrosis
Emerging evidence has suggested that mesenteric fat has a crucial role in the pathogenesis of inflammation and fibrostenosis in CD. In CD, longitudinal ulcerations are located mainly alongside the mesenteric border and the increased TNF-α production by adipocytes in mesenteric fat may contribute to transmural inflammation and subsequent ulceration [81]. In addition, “creeping fat” in CD, which refers to hypertrophic mesenteric fat around the inflamed bowel, is considered associated with other soft tissue alterations including fibrosis, smooth muscle hypertrophy, and subsequent stricture formation in CD [82]. A previous study reported that increased levels of adipokines in the creeping fat promote a shift toward a dominance of M2 macrophages within the macrophage compartment [83]. M2 macrophages secrete large amounts of TGF-β1, a cytokine with anti-inflammatory as well as pro-fibrotic functions [84]. Thus, the mesenteric fat in CD might not only be a protective enveloping barrier with the potential to restrict the infiltration of intestinal inflammation, they may also contribute to fibrosis by promoting pro-fibrotic M2 macrophages [83,85]. Our preliminary data show that creeping fat–derived free fatty acids, but not adipokines, increase the proliferation of human intestinal fibroblasts and HISMCs. Compared to UC and normal mesenteric fat, whole creeping fat and its conditioned medium significantly induced the proliferation of HISMCs [85,86]. Taken together, these data suggest that mesenteric fat may contribute to the development of intestinal fibrosis and smooth muscle hyperplasia.
6. Molecular Features of Myofibroblasts from Strictured Gut in CD
The molecular features of myofibroblasts derived from strictured gut in CD (strictured myofibroblasts) are distinct from those in myofibroblasts from non-strictured gut in CD (non-strictured myofibroblasts). Strictured myofibroblasts show higher TGF-β transcripts, higher phosphorylated Smad2/3, lower Smad7, and increased collagen production than non-strictured and normal myofibroblasts [87]. The increased Smad2/3 activity may be correlated with the downregulation of inhibitory Smad, Smad7, which inhibits TGF-β signaling. Smad7 forms a stable complex with the phosphorylated TGF-β type I receptor and therefore inhibits the binding and phosphorylation of Smad2/3 to it [88,89]. Moreover, Smad7 degrades TGF-β type I receptor through ubiquitination and consequently suppress TGF-β signaling [90]. Preliminary data showed that the silenced Smad7 in strictured myofibroblasts and subsequent sustained Smad3 activity occur due to epigenetic regulation of microRNA-21 (miR-21), which degrades Smad7 mRNA [91]. Conversely, the myofibroblasts isolated from the inflamed mucosa in CD show higher Smad7 production and subsequently lower phosphorylated Smad2/3, than those of strictured and normal myofibroblasts [87]. Smad7 is also overexpressed in lamina propria mononuclear cells (LPMCs), referring to dendritic cells, macrophages, lymphocytes, and natural killer cells, isolated from the inflamed mucosa of CD. In normal intestinal LPMCs responding to inflammatory condition, TGF-β signaling down-regulates nuclear factor-κB (NF-κB) activation by inducing NF-κB inhibitor α (IkBα), which binds to NF-κB and inhibits its activity. In contrast, in the inflamed gut in IBD, intestinal LPMCs express high levels of Smad7, which subsequently sustains high NF-κB activity, thereby expanding the local inflammatory response [88,92]. Furthermore, in intestinal myofibroblasts, TGF-β1 increases the transcriptional activity of IkBα, and Smad7 overexpression abolishes this effect [92]. The newly developed oral Smad7 antisense oligonucleotide (mongersen), which showed clinical efficacy for the treatment of active CD, inhibits Smad7 production, and restores anti-inflammatory TGF-β1 signaling by increasing IkBα expression in LPMCs isolated from the inflamed mucosa of CD [92,93]. Inhibiting Smad7 can promote anti-inflammatory signaling in LPMCs but paradoxically might promote pro-fibrotic signaling and fibrosis. Therefore, a further study is required to determine whether the Smad7 inhibition strategy may aggravate intestinal fibrosis in CD [94].
PREDICTION OF FIBROSTENOTIC STRICTURES
Currently, there are no specific predictors of fibrostenotic strictures in CD. To identify the reliable risk factors for fibrostenosing CD, the patient group with fibrostenosing CD should be accurately defined. However, currently available diagnostic modalities cannot determine the presence or degree of intestinal fibrosis. Therefore, previously identified predictors for fibrostenotic strictures including clinical, genetic, and serologic have not been strictly specific for fibrostenosis; rather, they predict a complicated or disabling CD (stricturing/fistulizing disease, need for surgery/early biologic therapy) [1,18,19]. On the other hand, several epigenetic markers including microRNAs have the potential to be used as specific predictors of stricturing CD [95,96]. However, the epigenetic markers are not currently recommended for the prediction of fibrostenotic CD.
1. Clinical, Genetic, and Serologic Factors Can Predict Complicated Disease
Several clinical factors, including the features at CD diagnosis (<40 years of age, perianal disease, need for steroids, weight loss >5 kg), early use of anti-inflammatory agents (azathioprine [AZA], anti-TNF therapy), disease location (ileal disease, extensive small bowel disease, perianal disease), and specific history (smoking, prior appendectomy) have been suggested as predictive of a more aggressive or complicated disease rather than as specific predictive factors of fibrostenotic stricture [1,19,97-101]. In a population-based study, the cumulative risk of developing stricturing and/or penetrating complications was approximately 50% at 20 years after diagnosis. Among the baseline clinical factors studied, disease location, such as upper gastrointestinal or ileal involvement, was strongly associated with progression toward strictures or penetrating disease. The shorter luminal diameter of the small bowel is a possible explanation for this observation [101,102].
Genetic, epigenetic, and serologic biomarkers have been evaluated as predictors of stricturing CD. The presence of several genetic variants, including the NOD2 gene or autophagy-related-16L1 (ATG16L1) gene, are connected with small bowel CD and a fibrostenotic phenotype [19]. Carrying at least one NOD2 variant increased the risk of stricturing CD (odds ratio [OR], 1.94; 95% confidence interval [CI], 1.61–2.34) [74]. In addition, CD patients carrying NOD2 gene variants are more likely to undergo surgery due to stricturing disease [103]. However, both NOD2 and ATG16L1 polymorphisms are known to be associated with dysfunction of the Paneth cells, which are localized to the terminal ileum and protect the epithelial barrier by secreting the anti-microbial peptide defensin [104]. Therefore, these genetic variants are considered risk factors associated with ileal CD or Paneth cell dysfunction rather than specific risk factors for fibrostenotic stricture. Furthermore, the presence of 2 NOD2 mutations predicted a 41% increase in the risk of complicated disease (stricturing or fistulizing) and a 58% increase in the risk of surgery [105]. These data suggest that whether NOD2 mutations actually contribute to the fibrostenosis or merely reflects the increased incidence of ileal disease or complicated disease remains to be elucidated [20,106,107]. CD patients have a dysregulated and stronger immune response to resident luminal microbial components, which induce anti-microbial antibodies in the circulation of patients as serologic markers. In particular, serologic anti-glycan antibodies against various microbial carbohydrate epitopes are useful for distinguishing CD from UC and have been suggested as a promising tool to stratify CD patients at a high risk of rapid progression and need for surgery [108]. These include the first discovered anti-Saccharomyces cerevisiae antibody, which exists in 50%–70% of CD patients, but other antiglycan antibodies, including CBir1 (flagellin CBir1), OmpC (E. coli outer membrane protein C), or I2 (Pseudomonas-associated sequence I2) have been identified [108]. Theses markers can qualitatively and quantitatively predict the disease progression to fibrostenosing/fistulizing phenotypes and increased need for surgery. However, none of them can differentiate fibrostenotic from penetrating behavior or accurately predict fibrostenosing CD [19].
2. Epigenetic Biomarker May Predict Fibrostenotic Stricture
miRNA are small, noncoding RNAs that repress specific target gene expression post-transcriptionally as an epigenetic mechanism. A previous study found that miRNA-200b, which was shown to suppress in vitro fibrosis by inhibiting epithelial–mesenchymal transition, is overexpressed in the circulation of fibrostenotic CD patients; thus, the authors suggested that serum miRNA-200b could be a candidate biomarker for fibrostenotic CD [109]. In addition, the expression of miRNA-29b, which inhibits TGF-β-induced collagen production in CD myofibroblasts, is down-regulated in the serum and strictured mucosa in fibrostenotic CD patients [95]. Serum miRNA-19 level is decreased in CD patients with a stricturing phenotype than in control CD patients. Of note, the association between miRNA-19 and stricturing phenotype is independent of confounding clinical variables including ileal location and disease duration, which might show the usefulness of miRNAs as specific biomarkers of stricturing CD [110]. Taken together, miRNAs, which can be measured in the serum and mucosal tissue of CD patients, might be attractive candidate future biomarkers of fibrostenosing CD [3]. However, whether the change of expression in specific miRNAs is a cause or result of intestinal fibrosis in CD remains poorly understood and requires further study.
3. Cross-Sectional Imaging, Endoscopy, and Histology are Not Predictive of Fibrostenotic Stricture
No current imaging study accurately predicts the stricturing phenotype of CD. Barium contrast studies and cross-sectional imaging studies (ultrasound [US], computed tomography enterography [CTE], and magnetic resonance enterography [MRE]), are diagnostic but not predictive of strictures [19]. Severe endoscopic lesions in CD patients, defined as extensive and deep ulcerations, appear to be associated with a worse clinical course and higher rates of penetrating complications and surgery [111]. However, whether the severe endoscopic lesions predict the stricturing phenotype has not been proven. Presently, no histological findings can specifically predict fibrostenosing CD.
DIAGNOSIS OF FIBROSTENOTIC STRICTURES
Strictures are usually detected when patients develop clinical symptoms of intestinal obstruction. Patients often then undergo noninvasive diagnostic modalities (barium contrast studies, cross-sectional imaging) followed by invasive modalities (endoscopy, histology) or biomarkers (fecal calprotectin, C-reactive protein [CRP]). Among these, cross-sectional imaging techniques such as US, CTE, and MRE are most preferred due to the high diagnostic accuracy of stricture, but they are not specific for fibrostenotic stricture. Three key features (luminal narrowing, wall thickening, and pre-stenotic dilation) are usually used for stricture detection. However, US, CTE, and MRE use highly heterogeneous definitions for these 3 features [112].
1. Complete Overlap Between Inflammation and Fibrosis in CD Strictures
CD-associated strictures can be classified into inflammatory and fibrotic type to determine whether anti-inflammatory agent is required. However, this differentiation appears unrealistic since most strictures have an almost complete overlap in inflammatory and fibrotic components [2,19]. In histologic evaluation, few strictures were classified as completely inflammatory or completely fibrotic; inflammation and fibrosis overlap each other with different degrees in most strictures [113]. This overlap could make it difficult to define the presence and degree of fibrosis using cross-sectional imaging [1,19]. Surprisingly, CD-associated strictures without the evidence of inflammation on CTE findings could not predict the presence of fibrosis. Conversely, strictures with CTE findings of the most active inflammation also had the greatest extent of fibrosis detected on histology [113]. Barium contrast studies and cross-sectional imaging modalities can be used for the diagnosis of CD-associated stricture. However, no diagnostic modality is currently able to specifically detect or quantify the fibrotic component of the stricture [19].
2. Cross-Sectional Imaging (US, CTE, and MRE)
US (transabdominal), CTE, and MRE are reported to have high sensitivity and specificity for the diagnosis of CD-associated strictures in several studies which used histopathology as a reference standard (US: [112,114,115] 80%–100% sensitivity and 63%–75% specificity; CTE: [112,116] 100% sensitivity and 100% specificity; MRE: [112,117-119] 75%–100% sensitivity and 91%–96% specificity). In contrast, in another study which measured the accuracy of MRE using balloon-assisted enteroscopic finding as a reference standard, MRE was less sensitive for detecting CD-associated stricture (58.8% sensitivity for small bowel stenosis which cannot be passed by the scope). However, this study excluded severe disease, defined as a Crohn’s Disease Activity Index score greater than 450 or serum CRP level greater than 10 mg/dL [120].
US is inexpensive and well-tolerated by patients. However, US diagnostic performance is assumed to be operator-dependent. In addition, the difficulty visualizing the upper segments (duodenum, jejunum) and the lack of a panoramic view compared with CTE and MRE reduced the sensitivity of US for small bowel CD [1,121]. US is inaccurate or insufficiently validated to reliably detect or quantify the fibrotic stricture. US elastography is under exploration to specifically evaluate fibrotic components by measuring bowel stiffness ratios between the mesenteric tissue and the bowel wall. A bowel stiffness ratio ≥2 in patients with CD could predict anti-TNF therapy failure and surgery [112,122].
CTE is superior to MRE with the advantages of higher resolution, rapid diagnosis, lower cost, and wider availability but requires radiation exposure [107]. CTE findings of strictures including luminal narrowing and wall thickening in the absence of intestinal wall inflammation such as mucosal hyperenhancement (including mural stratification), mesenteric hypervascularity (the so-called comb sign), and mesenteric fat stranding are presumed to indicate a fibrotic stricture [113,123]. However, these radiologic findings suspected of fibrosis were not able to predict the histologic fibrosis. Paradoxically, the only feature on CTE to predict fibrosis was mesenteric hypervascularity, which was also correlated with the presence of bowel wall inflammation [113]. Therefore, CTE cannot currently differentiate inflammatory from fibrotic strictures, and it might be attributed to that inflammation and fibrosis in CD strictures do not exist as mutually exclusive histologic types [113].
MRE carries lower resolution with motion artefacts, takes more time, carries higher cost, and is not readily available compared with CTE. However, MRE has superior soft tissue contrast for bowel wall tissue characterization and does not require radiation. In addition, a newer generation of MRE features higher resolution and a faster diagnosis [107]. Therefore, in the context of discriminating fibrotic components from strictures, MRE could be superior to CTE or US. Delayed enhancement MRE (also known as dynamic contrast MR imaging), the currently most advanced imaging technique available for fibrostenosing CD, can differentiate severe fibrosis from mild to moderate fibrosis by delayed gadolinium enhancement within fibrotic stricture [124]. In fibrostenotic stricture, the submucosa and muscularis propria (deep layers) have more dense tissue, less edema, and reduced vascularity compared with the mucosa (superficial layers) [125,126]. Therefore, the intravenous gadolinium diffuses into the extravascular tissue more slowly in the deep layers than in the superficial layers. A previous study using the delayed enhancement MRE defined “the percentage of gadolinium enhancement gain between 70 seconds and 7 minutes,” which is determined by measuring bowel wall signal intensity (WSI) using the following formula: ([WSI 7 minutes–WSI 70 seconds]/[WSI 70 seconds])×100. The study found that the percentage of enhancement gain correlated with degree of fibrosis (P<0.01) and can differentiate mild to moderate from severe fibrosis with a high sensitivity and specificity independent of the degree of coexisting inflammation. However, the low performance in discriminating among none, mild, and moderate fibrosis might limit the use of delayed enhancement MRE [1,124,127].
Novel magnetic resonance (MR) techniques to detect fibrotic stricture include diffusion-weighted imaging (DWI)-MRE and magnetization transfer-MR (MT-MR). DWI can be completed quickly without intravenous contrast and provide a quantitative index, for example, apparent diffusion coefficient (ADC), which reflects the rate of water diffusion through the tissues. Inflamed bowel tissue has a higher cellular density limiting water diffusion and is expressed by a low ADC and a high signal on MR images [13,128,129]. In a study of 20 CD patients undergoing surgery, there was a trend toward a decrease in ADC values being correlated with acute inflammation, but this was not significant. Interestingly, the decrease in ADC values was significantly (P=0.023) associated with histologic fibrosis, which suggests that the increase in fibrosis decreases the extracellular space and, therefore, restricts the diffusion of water molecules in the strictured bowel wall [130]. MT-MR is an advanced technique that can quantify collagen deposition as an increased signal (MT ratio), thereby providing a more direct measurement of fibrosis [131,132]. A recent study compared the ability of delayed enhancement MRE, DWI, and MT-MR to detect and quantify fibrosis in 31 CD patients with small bowel strictures using histologic evaluation as a reference. MT ratio was strongly correlated with fibrosis score but not inflammatory score and could discriminate among non-fibrotic and mild, moderate, and severe fibrotic bowel segments. The MT ratio was assumed the most accurate at distinguishing non-fibrotic from fibrotic lesions among the 3 MR imaging techniques as demonstrated by an area under the receiver operating characteristic curve (0.981 of MT ratio vs. 0.86 of ADC, and 0.646 of enhancement gain %) [127]. Therefore, MT-MR appears to be the most promising modality for evaluating the presence and degree of fibrostenotic stricture, but further validation is required for its routine clinical use. Despite its wide use there is no validated MR or CT score for stricturing disease. A global expert group recently devised definitions for strictures on cross-sectional imaging [4] that are currently being converted into indices through the Stenosis Therapy and Research (STAR) consortium. An additional challenge remains the missing gold standard. Histopathology is usually used to correlate imaging findings with the degree of inflammation or fibrosis. There are more than a dozen different stricture histopathology scores in the literature [133]; however, none have been validated. Ongoing work is building a novel and reliability tested histopathology score for small bowel stricturing CD.
3. Clinical Symptoms, Biomarkers, Barium Contrast Studies, Endoscopy, and Histology
Clinical symptoms of stricturing CD such as abdominal distension, cramping, dietary restrictions, nausea, vomiting, abdominal pain, and post-prandial abdominal pain are not correlated with the presence or degree of strictures on cross-sectional imaging or endoscopy [4]. Fibrotic strictures may develop for long periods with no or only mild symptoms [19]. Approximately, 20% of CD patients with small bowel stricture are asymptomatic [112]. However, no biomarkers (genetic, epigenetic, biochemical, and serologic) are currently recommended to determine the overall fibrotic burden of a CD patient with high accuracy in clinical practice [1]. Fecal calprotectin could be a useful marker for scheduling re-evaluation by endoscopy and/or cross-sectional imaging because it better correlates with endoscopic and histologic inflammation than CRP or symptoms. However, as with CRP, fecal calprotectin levels may be normal in patients with endoscopically active CD, particularly ileal disease [18]. Moreover, fecal calprotectin can be used to predict endoscopic recurrence after surgery for CD stricture, but it is not validated for its diagnostic utility in CD stricture [13]. Conventional barium contrast studies, including small bowel follow-through (duodenum) or enteroclysis (jejunum), can determine stricture extension and location but are being progressively abandoned due to the disadvantages of long duration (>2 hours) and the corresponding long radiation exposure. Endoscopy has a critical role in assessing the achievement of mucosal healing, which is highly correlated with the better long-term outcomes in CD (relapse, recurrence, hospitalization, and surgery) as the severity of endoscopic lesions in luminal inflammation correlates with the severity of transmural changes assessed by cross-sectional imaging [112,134]. However, endoscopy has a limited role in the diagnosis of fibrostenosis in CD as the endoscopic lesions may not be closely correlated with the transmural fibrostenosis. Endoscopic biopsies are superficial and unable to measure the amount of transmural fibrostenosis. The heterogeneity of distribution in inflammation and fibrosis within a stricture can lead to the sampling error of biopsies, which in turn limit the accurate diagnosis of fibrosis [112]. Not all strictures are accessible by endoscopy. The severe luminal narrowing by fibrostenosis makes the endoscopic approach difficult and can lead to incomplete evaluation of the fibrotic lesion, particularly in the small bowel [18]. At the present time, no validated endoscopic and histopathologic scoring system is available to grade intestinal fibrosis severity [19]. The Lémann Index, which is calculated by several diagnostic modalities (cross-sectional imaging, endoscopy, history taking, and physical examination), has been validated to quantify cumulative structural bowel damage in CD [135]. However, this scoring system is a measure of comprehensive bowel damage, including strictures, fistula/abscess, and surgical resections; therefore, it is not specific for strictures.
TREATMENT OF FIBROSTENOTIC STRICTURES
CD patients with obstructive symptoms should be hospitalized followed by initial conservative treatment to decompress the bowel and cross-sectional imaging (CTE, MRE) to assess the nature of strictures. Following treatment options could be medical, endoscopic, and surgical [13,19].
1. Initial Management and Assessment of Inflammatory Component
CD patients with suspected intestinal obstruction should be initially treated with nasogastric decompression, bowel rest, hydration, and electrolyte replacement. Subsequent cross-sectional study is required to assess the presence and feature of stricture such as number, location, length, and contraindications for endoscopic treatment and strictureplasty since they affect the subsequent treatment options [19]. Furthermore, the degree of the inflammatory component should be evaluated using cross-sectional study and several biomarkers (CRP, erythrocyte sedimentation rate, and fecal calprotectin). Anti-inflammatory agents such as intravenous hydrocortisone and subsequent appropriate maintenance therapy should be considered if the stricture has an inflammatory component [13,112]. However, even if the presence of an inflammatory component is not completely identified, anti-inflammatory agents may be attempted in the clinical setting. Several lines of evidence might support this decision. First, a clear distinction between tissue inflammation and fibrosis based on currently available diagnostic modalities is not possible and most strictures have an almost complete overlap between inflammatory and fibrotic components [19]. The inflammatory biomarkers may be normal in the presence of mucosal or transmural inflammation [18]. Second, although the proportion of the inflammatory component is relatively minimal, the clinical significance of the reduced bowel wall edema and obstructive symptoms by anti-inflammatory therapy might be substantial. For example, theoretically, the 17% of decrease in bowel wall thickness could significantly increase the luminal area by 625% (luminal diameter is 2.5 times bigger), which shows that small decrease of bowel wall thickness in the stricture could significantly improve the obstructive symptoms due to significant increase of luminal area. The relationship of percentage decrease in wall thickness and increase in luminal diameter depends on the “starting” wall thickness [107]. The symptom-free interval (>8 months) after the initial obstruction episode significantly reduces the risk of surgery, indicating that the control of obstructive symptoms by proper anti-inflammatory therapy can ultimately prevent surgery [136]. Third, the anti-inflammatory agent itself might be useful as a diagnostic tool to identify the presence of the inflammatory component by the medical response.
2. No Specific Medical Therapy for Fibrostenotic Strictures Exist
Currently, no specific medicine can prevent or reverse fibrostenotic stricture in CD [19]. although 5-aminosalicylic acid (5-ASA) may be an option to maintain post-surgical remission but not medically induced remission in CD [137-140]. However, classical 5-ASA does not appear to have anti-fibrotic activities in IBD, although a novel 5-ASA analog (GED), with strong affinity for peroxisome proliferator-activated receptor gamma (PPARγ) and potent anti-inflammatory properties greater than 5-ASA, has shown anti-fibrotic activity via PPARγ activation [141]. Corticosteroids are potent inhibitors of collagen synthesis in a variety of tissues, and its anti-fibrotic effects may inhibit wound healing [25,142]. However, methylprednisolone did not significantly reduce TGF-β1 levels or collagen content in TNBS-induced chronic colitis in rats [143]. Additionally, in HISMCs, collagen expression is not inhibited by corticosteroids; rather, it is increased at certain concentrations. In contrast, in fibroblasts, collagen expression is inhibited in a dose-dependent manner by corticosteroids. This complicated observation indicates that the anti-fibrotic effects of corticosteroids might be tissue- or cell-specific [25] and partially account for the contradictory results in 2 studies that evaluated the efficacy of the endoscopic injection of triamcinolone into strictures after endoscopic balloon dilation (EBD) [13]. Taken together, the anti-fibrotic effects of corticosteroids in stricturing CD is questionable; therefore, corticosteroid injection after EBD is not currently recommended [13,19].
Purine analogs (AZA, 6-mercaptopurine [6-MP]) are effective for the maintenance of surgically induced remission in CD by preventing postoperative recurrence [144]. Purine analogs also effectively maintain medically induced remission in CD by reducing the relapse following the discontinuation of anti-TNF [145]. Stricturing behavior and no use of purine analogs were independent risk factors of relapse following anti-TNF discontinuation in CD patients, suggesting the role of purine analogs that might be able to suppress stricture progression [145]. Although these studies were not designed to evaluate the direct effect of purine analogs on intestinal fibrosis, these findings support the hypothesis that purine analogs might prevent or delay the progression of fibrosis in stricturing CD [48,142]. There is currently no evidence that AZA/6-MP may be effective for symptomatic stricturing CD [18]. A total of 72 patients with sub-occlusive ileocecal CD, which was responsive to intravenous hydrocortisone in the initial admission, were randomized to AZA or mesalazine treatment over 36 months. In the AZA group, 38.9% were re-admitted for intestinal obstruction and 22.2% for bowel resection (vs. 75% and 52%, respectively, in the mesalazine group) [138]. These data suggest that long-term use of AZA in ileocecal CD patients with intestinal sub-occlusion can reduce the proportion of re-hospitalizations for intestinal obstruction or bowel resection. However, further studies are required to demonstrate the direct effect of purine analogs alone in fibrostenotic CD.
The theoretical concern that anti-TNF infliximab could paradoxically increase the risk of fibrostenotic stricture by rapid mucosal healing was overcome by the ACCENT I infliximab maintenance trial which did not show an increased risk of the clinical occurrence of strictures [146]. A recent prospective study reported that the use of adalimumab was effective in two-thirds of cases of symptomatic stricturing CD. More than half of the patients were free of surgery 4 years after treatment initiation [147]. However, the surgery-free interval is still short and data on the anti-fibrotic effect of anti-TNF are lacking. A plausible explanation for this finding is that the impairment of wound healing in the mucosa or submucosa of stricturing CD is likely to be improved by anti-TNF, whereas deeper, transmural fibrosis cannot be resolved by these drugs [23]. In 2017, the American Gastroenterological Association recommended anti-TNF and/or thiopurines as pharmacological prophylaxis within 8 weeks of surgery for disease recurrence based on the previous results that the administration of anti-TNF for several weeks postoperative effectively reduces recurrence with an effect that is greater than those of purine analogs [148,149]. In addition, combination therapy of anti-TNF and an immunomodulator following EBD significantly reduced the need for repeated dilatation [150]. Taken together, despite the absence of anti-fibrotic effects, anti-TNF appears to be effective for symptomatic stricturing CD as well as a preventive option for recurrence after surgery or EBD.
In a multicenter study of a gut-selective integrin inhibitor vedolizumab for anti-TNF-naïve CD patients (n=50), 24% of whom had a stricturing phenotype. A high proportion (84%) of subjects demonstrated a clinical response at week 14. In particular, the CD behavior (non-stricturing non-penetrating, stricturing, and penetrating) was not significantly associated with the likelihood of clinical response at week 14 (P=0.47), suggesting that vedolizumab may be effective for stricturing CD [151,152]. However, long-term outcomes specific to stricturing CD are lacking. Furthermore, the anti-p40 subunit of IL-12 and IL-23 (ustekinumab) has been proven effective for inducing and maintaining the clinical response and remission in patients with CD failing anti-TNF therapy (n=167), of whom 33.5% had the stricturing phenotype. In the analyses of clinical factors associated with ustekinumab response, patients with the stricturing phenotype showed lower rates of clinical response (OR, 0.29; 95% CI, 0.12–0.72) [153]. However, current data are insufficient to determine the efficacy of ustekinumab or vedolizumab on stricturing CD.
Collectively, some of the current medical options (aminosalicylates, steroids, immunosuppressive agents, and biologic agents) could be effective for treating symptomatic stricturing CD or preventing postoperative recurrence in CD, but none proved a direct anti-fibrotic effect in CD strictures [19].
3. Endoscopic Management
Patients with stricturing CD who do not respond well to medical therapy require endoscopic or surgical intervention. The cumulative evidence supports the application of EBD as the first-line therapy for the treatment of selected CD strictures since it is less invasive and has a high technical success rate (approximately 90%; defined as the passage of the scope without resistance) and a favorable short- and long-term clinical outcome with an acceptable rate of major complication (3%–5% per procedure) [13,19,154,155]. The short-term clinical outcome (i.e., clinical success [156]) is generally defined as the improvement in clinical obstructive symptoms and the long-term clinical outcome comprises the surgery-free survival (interval from the first EBD to stricture-related surgery) or intervention-free survival (interval from the first EBD to the first event of either additional EBD or surgery) [4,154,157]. The major complication comprises major bleeding or perforation requiring admission, transfusion, and surgical intervention [157]. EBD was able to increase the surgery-free survival in two-thirds of patients during the follow-up period of 24 months [13]. In a retrospective study of CD patients with ileocolonic anastomosis stricture treated with EBD or surgery, EBD could delay the need for surgery by 6.45 years and had a lower complication rate, suggesting the benefit of EBD as a bridge between medical treatment and surgery. However, the long-term outcome is likely to be more favorable in the surgery group than in the EBD group since EBD requires frequent procedures and has a shorter surgery-free survival [156,158]. EBD is indicated when strictures are ≤5 cm long, non-angulated, and without complications such as fistula, abscess, phlegmon, high-grade dysplasia, or malignancy. A stricture length of ≤5 cm was related to a longer surgery-free survival, and every-1 cm increase in stricture length increased the risk of surgery by 8% [155].
There is currently no standardized EBD techniques regarding maximal balloon diameter, duration, pressure of balloon inflation, number of dilations per patient or session, and graded/non-graded dilation. These various areas of operator-dependent heterogeneity in the technical aspects of EBD make inter-study comparisons difficult in systemic reviews and might lead to the results that any technical features of EBD do not affect short- or long-term outcomes or complication rates [13,18,155,159]. In a recent expert consensus of CD strictures, the following items were considered appropriate for EBD: 18 mm as the maximal balloon diameter; a balloon inflation time of at least 1 minute; and 5 cm as the maximum stricture length that should be dilated; thus, graded-through-the-scope balloons should be preferred [4]. EBD is not contraindicated for strictures due to local inflammation or ulceration at the stricture site since it did not affect the short- or long-term outcomes or complication rates [19,160].
Self-expandable metal stents have been proposed in the treatment of CD in cases of strictures longer than 5 cm or EBD failure, considering their slow radial expansion force compared to EBD, which may be more advantageous in long strictures. However, the rate of procedure-related complications, particularly stent migration, is too high to make this option practically recommendable to treat CD strictures at this time [5,19,161,162].
4. Surgical Management (Bowel Resection and Strictureplasty)
Despite the appropriate therapy, approximately, two-thirds of CD patients with strictures do not respond to medical therapy within 5 years [163], and 75% of patients who undergo EBD due to symptomatic CD strictures ultimately require surgery within 5 years [164]. When medical or endoscopic therapy fails or is contraindicated, surgery (limited bowel resection and strictureplasty) should be considered [18,19]. In particular, early bowel resection should be the preferred option in high-risk patients with isolated ileocecal CD stricture since it prevents subsequent complications (fistula and obstruction) more effectively than prolonged medical treatment. Previous studies reported that early surgical resection in isolated ileocecal CD was related with a lower risk of repeated surgery, longer surgery-free survival, or less use of steroids than the control group, suggesting that the surgical timing for localized ileocecal stricturing CD is relevant to the long-term surgical outcome [19,165-167]. To preserve bowel length and reduce the anastomotic leakage, strictureplasty can be considered in cases of multiple strictures over a long length of the bowel, previous extensive bowel resection, and short bowel syndrome [1,18]. Strictureplasty is generally performed with a longitudinal incision along the anti-mesenteric border and specific suture method on the strictured area [168]. Depending on the length of the stricture segment, Heineke-Mikulicz (most common; 5–10 cm), Finney (“U” shape fold; 10–25 cm), and Michelassi (side-to-side longitudinal enteroenterostomy; >10 cm) strictureplasties are indicated [13]. The strictureplasty is contraindicated in cases of concomitant features such as an abscess, fistula, dysplasia, or malignancy [2]. Four percent of patients who underwent strictureplasties had septic complications including anastomotic leaks, fistula, and abscess. The strictureplasty site was involved in the septic complications in about 78% of patients with sepsis [169]. Strictureplasty is preferred for small bowel strictures and generally not recommended for colonic stricture in CD due to the potential presence of a malignancy [19]. Strictureplasty has been shown to be a safe and effective technique comparable to bowel resection and utilized during the past 30 years in cases of stricturing CD [169]. Based on this evidence, we suggest this treatment algorithm for patients with stricturing CD (Fig. 3).
Fig. 3.
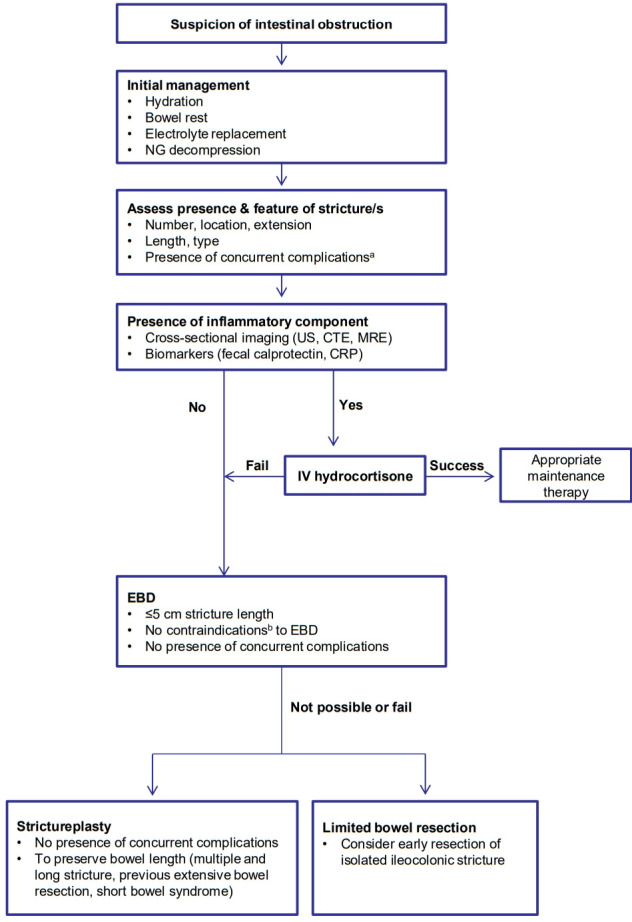
Suggested treatment algorithm for stricturing Crohn’s disease. a Concurrent complications include: fistula, abscess, phlegmon, dysplasia, or malignancy. b Contraindications include: outside reach of endoscopy, stricture type (angulated, spindle-shaped, or asymmetric), or stricture location at proximity of the penetrating complication. NG, nasogastric; US, ultrasound; CTE, computed tomography; enterography; MRE, magnetic resonance enterography; IV, intravenous; CRP, C-reactive protein; EBD, endoscopic balloon dilatation.
FUTURE PERSPECTIVES
Recent advances in our understanding of the cellular and molecular mechanisms of fibrosis are gradually changing the traditional concept that intestinal fibrosis is unavoidable and irreversible [22,170]. CD patients who undergo strictureplasty have shown the reversibility of stricturing CD. Serial US evaluations of the previous strictureplasty site in CD patients showed a decreased or normalized thickening of the diseased bowel wall with lower recurrence rates [171]. Furthermore, a study including a total of 1,112 patients with CD who underwent strictureplasty reported that the recurrence at the previous strictureplasty site was noted in only 3% of total patients with recurrence, while most recurrences occurred at non-strictureplasty sites [170,172]. We hypothesized that a reduced mechanical and antigenic (microbial and food antigen) pressure after successful increase of luminal diameter, leading to a reduced inflammatory stimulation, may be associated with subsequent myofibroblast inactivation and in turn prevent fibrosis progression [23,170]. In addition, a growing body of evidence regarding the reversibility of fibrosis from other organs including the liver, skin, kidney, lung, and heart, provides the concept that intestinal fibrosis is also reversible [170]. The fibrotic tissue is not a fixed, hypo-metabolic tissue; rather, it is a dynamic, hyper-metabolic tissue in which a continuous matrix turnover occurs with the synthesis and degradation of various ECM components by several enzymes such as MMPs and TIMPs. Thus, even the severe established fibrotic strictures in CD might be reversed by modification of the matrix turnover [87,170]. Based on the premise of reversibility of intestinal fibrosis, multiple potential mechanisms and anti-fibrotic agents have been identified, but no specific anti-fibrotic therapy is currently available for fibrostenotic strictures in CD.
1. Promising Anti-fibrotic Agents for Intestinal Fibrosis
The small molecules pirfenidone and nintedanib were recently approved by the U.S. Food and Drug Administration for the treatment of idiopathic pulmonary fibrosis [1]. Pirfenidone showed a combined anti-inflammatory, anti-oxidant, and anti-fibrotic effect by inhibiting TNF-α, reactive oxygen species, and TGF-β signaling in pulmonary fibrosis [173,174]. More recently, several results have suggested that oral pirfenidone prevents intestinal fibrosis by inhibiting fibroblast proliferation and differentiation by suppressing TGF-β1-induced fibrosis signaling observed in both in in vitro and in vivo intestinal fibrosis models [175-177]. In a study using gut-derived fibroblasts from CD patients, pirfenidone inhibited cellular proliferation and MMP-3 production in a dose-dependent manner, which supports the anti-fibrotic role of pirfenidone in stricturing CD [178]. Nintedanib is a multi-target small molecule inhibitor against 3 receptor tyrosine kinases (PDGF, FGF, and vascular endothelial growth factor receptors). Nintedanib appears to have anti-fibrotic effects by inhibiting TGF-β1-induced proliferation, differentiation, migration, and contraction in fibroblasts [47,179]. In clinical trials, pirfenidone and nintedanib have prevented the decrease in pulmonary function (forced vital capacity) and the risk of acute respiratory deteriorations, causing high morbidity and mortality in patients with idiopathic pulmonary fibrosis [180]. This finding suggests that pirfenidone and nintedanib should be evaluated as new treatment option to prevent intestinal fibrosis in CD strictures.
The role of Rho/ROCK/Actin/MRTF/SRF signaling, which is induced by TGF-β1 or matrix stiffness, has been revealed in intestinal fibrosis [11,51,53,181]. ROCK is activated in inflamed and fibrotic tissue in CD. The rectal delivery of a novel small molecule ROCK inhibitor (AMA0825) reversed the established intestinal fibrosis in 2 different murine models of fibrosis by diminishing the TGF-β1-induced MRTF and p38 MAPK activation and increasing autophagy in fibroblasts. Interestingly, AMA0825 alone did not improve histologic inflammation and production of the pro-inflammatory cytokines, suggesting that the anti-fibrotic effect of AMA0825 is mediated by a fibrosis-specific mechanism but not an anti-inflammatory mechanism. In addition, combining AMA0825 with anti-TNF not only prevented fibrosis but also ameliorated inflammation, emphasizing the importance of combination therapy [11]. These findings indicate that combining anti-TNF with local AMA0825 therapy, should be evaluated further as a therapeutic strategy for treatment of stricturing CD [181]. Moreover, as mentioned in the previous section, neutralizing antibodies designed to block IL-36α/IL-36R signaling or TL1A/DR3 signaling, which are activated in patients with CD strictures, are now available for clinical studies and emerge as promising therapeutic strategies for intestinal fibrosis in CD. Anti-IL36R antibodies and anti-TL1A antibodies are currently tested in a clinical phase 2 trial in UC patients (NCT03482635 and NCT04090411, respectively).
2. Challenges in Developing Clinically Available Anti-fibrotic Agents for CD Stricture
To make use of the above described anti-fibrotic mechanisms and novel anti-fibrotic agents as clinically available option for CD stricture, multiple obstacles must be overcome.
First, the ideal anti-fibrotic agents for CD stricture should be developed by targeting specific molecules or signaling that is unique in fibrotic tissue in CD and thus should not cause any side effects in other organs. However, to date, no specific target for intestinal fibrosis has been identified [182]. Second, currently, no specific predictors currently allow for an early identification of CD patients with a high risk of stricture development. Some clinical factors and genetic/serologic biomarkers can be useful for predicting complicated CD with a disabling disease course. However, none are able to discriminate stricturing from penetrating behavior to stratify and enroll CD patients with a high risk of strictures into clinical trials [14,101]. Third, no clinically useful biomarkers or imaging techniques can accurately determine and quantify the overall fibrotic burden of a stricturing CD. The absence of reliable modalities to define fibrosis has led to substantial heterogeneity in definitions of stricturing CD [4,112]. Furthermore, this makes it difficult to assess the response to anti-fibrotic therapy in clinical trials. Cross-sectional imaging techniques including US, CTE, and MRE can detect intestinal strictures or assess the degree of inflammation, but cannot determine fibrosis grade. To meet the urgent need of end points that can be used to assess the efficacy of anti-fibrotic agents under investigation, several groups of IBD study have reached expert consensus [4,183]. Recently, the need for intervention (EBD or surgery) within 24–48 weeks has been proposed as an appropriate end point to assess anti-fibrotic agents in pharmacological trials of patients with stricturing CD in an expert consensus [4]. The development of appropriate trial endpoints, including a patient-reported outcome tool, radiology indices, and histopathology index are now underway in a global effort through the STAR consortium to pave the way for clinical trials in this arena and tackle one of the largest remaining unmet needs.
CONCLUSIONS
Although advances in the treatment of CD have provided the improvement of clinical symptoms and the resolution of inflammation, the most patients still develop structural bowel damage including strictures, fistulae, and abscess requiring surgical resection, which can cause disability and impact social or professional life [184]. Parallel to this, the treatment paradigm in CD is moving from the control of clinical symptoms and inflammation toward modifying the natural history by reducing structural damage and disability. In this regard, intestinal fibrosis is one of the fastest-growing fields within IBD research. Over the past few decades, remarkable progress has been made in the field of fibrosis mechanisms in IBD, including the feature of fibrosis progressing independently of inflammation in response to matrix stiffness, the role of microbiome-and mesenteric fat–influencing fibrosis, and reversibility of fibrosis. Based on these novel mechanisms, several molecular candidates are under evaluation for the diagnosis and treatment of intestinal fibrosis. Advances have been hampered by the lack of validated clinical trial endpoints. This is currently being addressed by the STAR consortium to allow for testing of novel anti-fibrotic drugs in the near future.
Footnotes
FINANCIAL SUPPORT
The National Research Foundation of Korea (NRF) grant funded by the South Korea government (Ministry of Science and ICT, No. 2019R1H1A1035601); the Basic Science Research Program through the NRF funded by the Ministry of Science, ICT & Future Planning (No. 2015R1C1A1A02037048).
CONFLICT OF INTEREST
No potential conflicts of interest relevant to this article was reported.
AUTHOR CONTRIBUTION
Conceptualization: Yoo JH. Funding acquisition: Yoo JH. Project administration: Yoo JH. Writing - original draft: Yoo JH. Writing - review and editing: Yoo JH, Rieder F, Holubar S. Approval of final manuscript: all authors.
REFERENCES
- 1.Rieder F, Fiocchi C, Rogler G. Mechanisms, management, and treatment of fibrosis in patients with inflammatory bowel diseases. Gastroenterology. 2017;152:340–350. doi: 10.1053/j.gastro.2016.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rieder F. Managing intestinal fibrosis in patients with inflammatory bowel disease. Gastroenterol Hepatol (N Y) 2018;14:120–122. [PMC free article] [PubMed] [Google Scholar]
- 3.Lewis A, Nijhuis A, Mehta S, et al. Intestinal fibrosis in Crohn’s disease: role of microRNAs as fibrogenic modulators, serum biomarkers, and therapeutic targets. Inflamm Bowel Dis. 2015;21:1141–1150. doi: 10.1097/MIB.0000000000000298. [DOI] [PubMed] [Google Scholar]
- 4.Rieder F, Bettenworth D, Ma C, et al. An expert consensus to standardise definitions, diagnosis and treatment targets for anti-fibrotic stricture therapies in Crohn’s disease. Aliment Pharmacol Ther. 2018;48:347–357. doi: 10.1111/apt.14853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shen B. Interventional IBD: the role of endoscopist in the multidisciplinary team management of IBD. Inflamm Bowel Dis. 2018;24:298–309. doi: 10.1093/ibd/izx058. [DOI] [PubMed] [Google Scholar]
- 6.Munkholm P, Langholz E, Davidsen M, Binder V. Disease activity courses in a regional cohort of Crohn’s disease patients. Scand J Gastroenterol. 1995;30:699–706. doi: 10.3109/00365529509096316. [DOI] [PubMed] [Google Scholar]
- 7.Gklavas A, Dellaportas D, Papaconstantinou I. Risk factors for postoperative recurrence of Crohn’s disease with emphasis on surgical predictors. Ann Gastroenterol. 2017;30:598–612. doi: 10.20524/aog.2017.0195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peyrin-Biroulet L, Harmsen WS, Tremaine WJ, Zinsmeister AR, Sandborn WJ, Loftus EV., Jr Surgery in a population-based cohort of Crohn’s disease from Olmsted County, Minnesota (1970-2004) Am J Gastroenterol. 2012;107:1693–1701. doi: 10.1038/ajg.2012.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Toh JW, Stewart P, Rickard MJ, Leong R, Wang N, Young CJ. Indications and surgical options for small bowel, large bowel and perianal Crohn’s disease. World J Gastroenterol. 2016;22:8892–8904. doi: 10.3748/wjg.v22.i40.8892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buisson A, Chevaux JB, Allen PB, Bommelaer G, Peyrin-Biroulet L. Review article: the natural history of postoperative Crohn’s disease recurrence. Aliment Pharmacol Ther. 2012;35:625–633. doi: 10.1111/j.1365-2036.2012.05002.x. [DOI] [PubMed] [Google Scholar]
- 11.Holvoet T, Devriese S, Castermans K, et al. Treatment of intestinal fibrosis in experimental inflammatory bowel disease by the pleiotropic actions of a local Rho kinase inhibitor. Gastroenterology. 2017;153:1054–1067. doi: 10.1053/j.gastro.2017.06.013. [DOI] [PubMed] [Google Scholar]
- 12.De Buck van Overstraeten A, Wolthuis A, D’Hoore A. Surgery for Crohn’s disease in the era of biologicals: a reduced need or delayed verdict? World J Gastroenterol. 2012;18:3828–3832. doi: 10.3748/wjg.v18.i29.3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan WP, Mourad F, Leong RW. Crohn’s disease associated strictures. J Gastroenterol Hepatol. 2018;33:998–1008. doi: 10.1111/jgh.14119. [DOI] [PubMed] [Google Scholar]
- 14.Bettenworth D, Rieder F. Pathogenesis of intestinal fibrosis in inflammatory bowel disease and perspectives for therapeutic implication. Dig Dis. 2017;35:25–31. doi: 10.1159/000449079. [DOI] [PubMed] [Google Scholar]
- 15.Latella G, Di Gregorio J, Flati V, Rieder F, Lawrance IC. Mechanisms of initiation and progression of intestinal fibrosis in IBD. Scand J Gastroenterol. 2015;50:53–65. doi: 10.3109/00365521.2014.968863. [DOI] [PubMed] [Google Scholar]
- 16.Chen W, Lu C, Hirota C, Iacucci M, Ghosh S, Gui X. Smooth muscle hyperplasia/hypertrophy is the most prominent histological change in Crohn’s fibrostenosing bowel strictures: a semiquantitative analysis by using a novel histological grading scheme. J Crohns Colitis. 2017;11:92–104. doi: 10.1093/ecco-jcc/jjw126. [DOI] [PubMed] [Google Scholar]
- 17.Rieder F, Fiocchi C. Intestinal fibrosis in inflammatory bowel disease: current knowledge and future perspectives. J Crohns Colitis. 2008;2:279–290. doi: 10.1016/j.crohns.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 18.Malgras B, Pautrat K, Dray X, et al. Multidisciplinary management of gastrointestinal fibrotic stenosis in Crohn’s disease. Dig Dis Sci. 2015;60:1152–1168. doi: 10.1007/s10620-014-3421-y. [DOI] [PubMed] [Google Scholar]
- 19.Rieder F, Latella G, Magro F, et al. European Crohn’s and Colitis Organisation topical review on prediction, diagnosis and management of fibrostenosing Crohn’s disease. J Crohns Colitis. 2016;10:873–885. doi: 10.1093/ecco-jcc/jjw055. [DOI] [PubMed] [Google Scholar]
- 20.Burke JP, Mulsow JJ, O’Keane C, Docherty NG, Watson RW, O’Connell PR. Fibrogenesis in Crohn’s disease. Am J Gastroenterol. 2007;102:439–448. doi: 10.1111/j.1572-0241.2006.01010.x. [DOI] [PubMed] [Google Scholar]
- 21.Shepherd NA, Jass JR, Duval I, Moskowitz RL, Nicholls RJ, Morson BC. Restorative proctocolectomy with ileal reservoir: pathological and histochemical study of mucosal biopsy specimens. J Clin Pathol. 1987;40:601–607. doi: 10.1136/jcp.40.6.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rieder F, Fiocchi C. Intestinal fibrosis in IBD: a dynamic, multifactorial process. Nat Rev Gastroenterol Hepatol. 2009;6:228–235. doi: 10.1038/nrgastro.2009.31. [DOI] [PubMed] [Google Scholar]
- 23.Lenti MV, Di Sabatino A. Intestinal fibrosis. Mol Aspects Med. 2019;65:100–109. doi: 10.1016/j.mam.2018.10.003. [DOI] [PubMed] [Google Scholar]
- 24.Valatas V, Filidou E, Drygiannakis I, Kolios G. Stromal and immune cells in gut fibrosis: the myofibroblast and the scarface. Ann Gastroenterol. 2017;30:393–404. doi: 10.20524/aog.2017.0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Graham MF, Willey A, Adams J, Diegelmann RF. Corticosteroids increase procollagen gene expression, synthesis, and secretion by human intestinal smooth muscle cells. Gastroenterology. 1995;109:1454–1461. doi: 10.1016/0016-5085(95)90630-4. [DOI] [PubMed] [Google Scholar]
- 26.Koo JB, Nam MO, Jung Y, et al. Anti-fibrogenic effect of PPAR-γ agonists in human intestinal myofibroblasts. BMC Gastroenterol. 2017;17:73. doi: 10.1186/s12876-017-0627-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Powell DW, Mifflin RC, Valentich JD, Crowe SE, Saada JI, West AB. Myofibroblasts: II. intestinal subepithelial myofibroblasts. Am J Physiol. 1999:277–C183-C201. doi: 10.1152/ajpcell.1999.277.2.C183. [DOI] [PubMed] [Google Scholar]
- 28.Mifflin RC, Pinchuk IV, Saada JI, Powell DW. Intestinal myofibroblasts: targets for stem cell therapy. Am J Physiol Gastrointest Liver Physiol. 2011:300–G684-G696. doi: 10.1152/ajpgi.00474.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mostafa RM, Moustafa YM, Hamdy H. Interstitial cells of Cajal, the Maestro in health and disease. World J Gastroenterol. 2010;16:3239–3248. doi: 10.3748/wjg.v16.i26.3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Otte JM, Rosenberg IM, Podolsky DK. Intestinal myofibroblasts in innate immune responses of the intestine. Gastroenterology. 2003;124:1866–1878. doi: 10.1016/s0016-5085(03)00403-7. [DOI] [PubMed] [Google Scholar]
- 31.Zawahir S, Li G, Banerjee A, Shiu J, Blanchard TG, Okogbule-Wonodi AC. Inflammatory and immune activation in intestinal myofibroblasts is developmentally regulated. J Interferon Cytokine Res. 2015;35:634–640. doi: 10.1089/jir.2014.0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drygiannakis I, Valatas V, Sfakianaki O, et al. Proinflammatory cytokines induce crosstalk between colonic epithelial cells and subepithelial myofibroblasts: implication in intestinal fibrosis. J Crohns Colitis. 2013;7:286–300. doi: 10.1016/j.crohns.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 33.Scharl M, Huber N, Lang S, Fürst A, Jehle E, Rogler G. Hallmarks of epithelial to mesenchymal transition are detectable in Crohn’s disease associated intestinal fibrosis. Clin Transl Med. 2015;4:1. doi: 10.1186/s40169-015-0046-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Micallef L, Vedrenne N, Billet F, Coulomb B, Darby IA, Desmoulière A. The myofibroblast, multiple origins for major roles in normal and pathological tissue repair. Fibrogenesis Tissue Repair. 2012;5(Suppl 1):S5. doi: 10.1186/1755-1536-5-S1-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rieder F, Kessler SP, West GA, et al. Inflammation-induced endothelial-to-mesenchymal transition: a novel mechanism of intestinal fibrosis. Am J Pathol. 2011;179:2660–2673. doi: 10.1016/j.ajpath.2011.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lawrance IC, Rogler G, Bamias G, et al. Cellular and molecular mediators of intestinal fibrosis. J Crohns Colitis. 2017;11:1491–1503. doi: 10.1016/j.crohns.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uehara H, Nakagawa T, Katsuno T, et al. Emergence of fibrocytes showing morphological changes in the inflamed colonic mucosa. Dig Dis Sci. 2010;55:253–260. doi: 10.1007/s10620-009-0730-7. [DOI] [PubMed] [Google Scholar]
- 38.Brittan M, Chance V, Elia G, et al. A regenerative role for bone marrow following experimental colitis: contribution to neovasculogenesis and myofibroblasts. Gastroenterology. 2005;128:1984–1995. doi: 10.1053/j.gastro.2005.03.028. [DOI] [PubMed] [Google Scholar]
- 39.Fiocchi C, Lund PK. Themes in fibrosis and gastrointestinal inflammation. Am J Physiol Gastrointest Liver Physiol. 2011:300–G677-G683. doi: 10.1152/ajpgi.00104.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Biancheri P, Pender SL, Ammoscato F, et al. The role of interleukin 17 in Crohn’s disease-associated intestinal fibrosis. Fibrogenesis Tissue Repair. 2013;6:13. doi: 10.1186/1755-1536-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fichtner-Feigl S, Strober W, Kawakami K, Puri RK, Kitani A. IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 production and fibrosis. Nat Med. 2006;12:99–106. doi: 10.1038/nm1332. [DOI] [PubMed] [Google Scholar]
- 42.Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol. 2004;4:583–594. doi: 10.1038/nri1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Medina C, Santos-Martinez MJ, Santana A, et al. Transforming growth factor-beta type 1 receptor (ALK5) and Smad proteins mediate TIMP-1 and collagen synthesis in experimental intestinal fibrosis. J Pathol. 2011;224:461–472. doi: 10.1002/path.2870. [DOI] [PubMed] [Google Scholar]
- 44.Sanders YY, Cui Z, Le Saux CJ, et al. SMAD-independent down-regulation of caveolin-1 by TGF-beta: effects on proliferation and survival of myofibroblasts. PLoS One. 2015;10:e0116995. doi: 10.1371/journal.pone.0116995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lawrance IC, Maxwell L, Doe W. Altered response of intestinal mucosal fibroblasts to profibrogenic cytokines in inflammatory bowel disease. Inflamm Bowel Dis. 2001;7:226–236. doi: 10.1097/00054725-200108000-00008. [DOI] [PubMed] [Google Scholar]
- 46.Leeb SN, Vogl D, Falk W, Schölmerich J, Rogler G, Gelbmann CM. Regulation of migration of human colonic myofibroblasts. Growth Factors. 2002;20:81–91. doi: 10.1080/08977190290031941. [DOI] [PubMed] [Google Scholar]
- 47.Lin X, Wen J, Liu R, Gao W, Qu B, Yu M. Nintedanib inhibits TGF-beta-induced myofibroblast transdifferentiation in human Tenon’s fibroblasts. Mol Vis. 2018;24:789–800. [PMC free article] [PubMed] [Google Scholar]
- 48.Latella G, Sferra R, Speca S, Vetuschi A, Gaudio E. Can we prevent, reduce or reverse intestinal fibrosis in IBD? Eur Rev Med Pharmacol Sci. 2013;17:1283–1304. [PubMed] [Google Scholar]
- 49.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 50.Mu Y, Gudey SK, Landström M. Non-Smad signaling pathways. Cell Tissue Res. 2012;347:11–20. doi: 10.1007/s00441-011-1201-y. [DOI] [PubMed] [Google Scholar]
- 51.Johnson LA, Rodansky ES, Haak AJ, Larsen SD, Neubig RR, Higgins PD. Novel Rho/MRTF/SRF inhibitors block matrix-stiffness and TGF-beta-induced fibrogenesis in human colonic myofibroblasts. Inflamm Bowel Dis. 2014;20:154–165. doi: 10.1097/01.MIB.0000437615.98881.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Small EM. The actin-MRTF-SRF gene regulatory axis and myofibroblast differentiation. J Cardiovasc Transl Res. 2012;5:794–804. doi: 10.1007/s12265-012-9397-0. [DOI] [PubMed] [Google Scholar]
- 53.Choi YJ, Koo JB, Kim HY, et al. Umbilical cord/placenta-derived mesenchymal stem cells inhibit fibrogenic activation in human intestinal myofibroblasts via inhibition of myocardin-related transcription factor A. Stem Cell Res Ther. 2019;10:291. doi: 10.1186/s13287-019-1385-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hinz B. Tissue stiffness, latent TGF-beta1 activation, and mechanical signal transduction: implications for the pathogenesis and treatment of fibrosis. Curr Rheumatol Rep. 2009;11:120–126. doi: 10.1007/s11926-009-0017-1. [DOI] [PubMed] [Google Scholar]
- 55.Latella G, Rogler G, Bamias G, et al. Results of the 4th Scientific Workshop of the ECCO (I): pathophysiology of intestinal fibrosis in IBD. J Crohns Colitis. 2014;8:1147–1165. doi: 10.1016/j.crohns.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 56.Fichtner-Feigl S, Fuss IJ, Young CA, et al. Induction of IL-13 triggers TGF-beta1-dependent tissue fibrosis in chronic 2,4,6- trinitrobenzene sulfonic acid colitis. J Immunol. 2007;178:5859–5870. doi: 10.4049/jimmunol.178.9.5859. [DOI] [PubMed] [Google Scholar]
- 57.Fichtner-Feigl S, Young CA, Kitani A, Geissler EK, Schlitt HJ, Strober W. IL-13 signaling via IL-13R alpha2 induces major downstream fibrogenic factors mediating fibrosis in chronic TNBS colitis. Gastroenterology. 2008;135:2003–2013. doi: 10.1053/j.gastro.2008.08.055. [DOI] [PubMed] [Google Scholar]
- 58.Scheibe K, Kersten C, Schmied A, et al. Inhibiting interleukin 36 receptor signaling reduces fibrosis in mice with chronic intestinal inflammation. Gastroenterology. 2019;156:1082–1097. doi: 10.1053/j.gastro.2018.11.029. [DOI] [PubMed] [Google Scholar]
- 59.Mao R, Rieder F. Cooling down the hot potato: anti-interleukin 36 therapy prevents and treats experimental intestinal fibrosis. Gastroenterology. 2019;156:871–873. doi: 10.1053/j.gastro.2019.02.007. [DOI] [PubMed] [Google Scholar]
- 60.Scheibe K, Backert I, Wirtz S, et al. IL-36R signalling activates intestinal epithelial cells and fibroblasts and promotes mucosal healing in vivo. Gut. 2017;66:823–838. doi: 10.1136/gutjnl-2015-310374. [DOI] [PubMed] [Google Scholar]
- 61.Shih DQ, Zheng L, Zhang X, et al. Inhibition of a novel fibrogenic factor Tl1a reverses established colonic fibrosis. Mucosal Immunol. 2014;7:1492–1503. doi: 10.1038/mi.2014.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shih DQ, Barrett R, Zhang X, et al. Constitutive TL1A (TNFSF15) expression on lymphoid or myeloid cells leads to mild intestinal inflammation and fibrosis. PLoS One. 2011;6:e16090. doi: 10.1371/journal.pone.0016090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Barrett R, Zhang X, Koon HW, et al. Constitutive TL1A expression under colitogenic conditions modulates the severity and location of gut mucosal inflammation and induces fibrostenosis. Am J Pathol. 2012;180:636–649. doi: 10.1016/j.ajpath.2011.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li H, Song J, Niu G, et al. TL1A blocking ameliorates intestinal fibrosis in the T cell transfer model of chronic colitis in mice. Pathol Res Pract. 2018;214:217–227. doi: 10.1016/j.prp.2017.11.017. [DOI] [PubMed] [Google Scholar]
- 65.Okamoto R, Watanabe M. Role of epithelial cells in the pathogenesis and treatment of inflammatory bowel disease. J Gastroenterol. 2016;51:11–21. doi: 10.1007/s00535-015-1098-4. [DOI] [PubMed] [Google Scholar]
- 66.Rieder F. The gut microbiome in intestinal fibrosis: environmental protector or provocateur? Sci Transl Med. 2013;5:190–ps10. doi: 10.1126/scitranslmed.3004731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 68.Rieder F, Bhilocha S, Schirbel A, et al. Activation of Toll-like receptor (TLR) 5 induces a pro-fibrogenic phenotype on human intestinal myofibroblasts (HIF): a novel pathway mediated by caspase 1. Gastroenterology. 2011;140:S–114. [Google Scholar]
- 69.Hasan UA, Trinchieri G, Vlach J. Toll-like receptor signaling stimulates cell cycle entry and progression in fibroblasts. J Biol Chem. 2005;280:20620–20627. doi: 10.1074/jbc.M500877200. [DOI] [PubMed] [Google Scholar]
- 70.Imai J, Kitamoto S, Sugihara K, et al. Flagellin-mediated activation of IL-33-ST2 signaling by a pathobiont promotes intestinal fibrosis. Mucosal Immunol. 2019;12:632–643. doi: 10.1038/s41385-019-0138-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lodes MJ, Cong Y, Elson CO, et al. Bacterial flagellin is a dominant antigen in Crohn disease. J Clin Invest. 2004;113:1296–1306. doi: 10.1172/JCI20295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Targan SR, Landers CJ, Yang H, et al. Antibodies to CBir1 flagellin define a unique response that is associated independently with complicated Crohn’s disease. Gastroenterology. 2005;128:2020–2028. doi: 10.1053/j.gastro.2005.03.046. [DOI] [PubMed] [Google Scholar]
- 73.Hugot JP, Chamaillard M, Zouali H, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 74.Economou M, Trikalinos TA, Loizou KT, Tsianos EV, Ioannidis JP. Differential effects of NOD2 variants on Crohn’s disease risk and phenotype in diverse populations: a metaanalysis. Am J Gastroenterol. 2004;99:2393–2404. doi: 10.1111/j.1572-0241.2004.40304.x. [DOI] [PubMed] [Google Scholar]
- 75.Mourelle M, Salas A, Guarner F, Crespo E, García-Lafuente A, Malagelada JR. Stimulation of transforming growth factor beta1 by enteric bacteria in the pathogenesis of rat intestinal fibrosis. Gastroenterology. 1998;114:519–526. doi: 10.1016/s0016-5085(98)70535-9. [DOI] [PubMed] [Google Scholar]
- 76.Pucilowska JB, Williams KL, Lund PK. Fibrogenesis. IV. Fibrosis and inflammatory bowel disease: cellular mediators and animal models. Am J Physiol Gastrointest Liver Physiol. 2000:279–G653-G659. doi: 10.1152/ajpgi.2000.279.4.G653. [DOI] [PubMed] [Google Scholar]
- 77.Rieder F, Kessler S, Sans M, Fiocchi C. Animal models of intestinal fibrosis: new tools for the understanding of pathogenesis and therapy of human disease. Am J Physiol Gastrointest Liver Physiol. 2012:303–G786-G801. doi: 10.1152/ajpgi.00059.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wells RG. The role of matrix stiffness in regulating cell behavior. Hepatology. 2008;47:1394–1400. doi: 10.1002/hep.22193. [DOI] [PubMed] [Google Scholar]
- 79.Johnson LA, Rodansky ES, Sauder KL, et al. Matrix stiffness corresponding to strictured bowel induces a fibrogenic response in human colonic fibroblasts. Inflamm Bowel Dis. 2013;19:891–903. doi: 10.1097/MIB.0b013e3182813297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Johnson LA, Luke A, Sauder K, Moons DS, Horowitz JC, Higgins PD. Intestinal fibrosis is reduced by early elimination of inflammation in a mouse model of IBD: impact of a “top-down” approach to intestinal fibrosis in mice. Inflamm Bowel Dis. 2012;18:460–471. doi: 10.1002/ibd.21812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Peyrin-Biroulet L, Chamaillard M, Gonzalez F, et al. Mesenteric fat in Crohn’s disease: a pathogenetic hallmark or an innocent bystander? Gut. 2007;56:577–583. doi: 10.1136/gut.2005.082925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sheehan AL, Warren BF, Gear MW, Shepherd NA. Fat-wrapping in Crohn’s disease: pathological basis and relevance to surgical practice. Br J Surg. 1992;79:955–958. doi: 10.1002/bjs.1800790934. [DOI] [PubMed] [Google Scholar]
- 83.Kredel LI, Batra A, Stroh T, et al. Adipokines from local fat cells shape the macrophage compartment of the creeping fat in Crohn’s disease. Gut. 2013;62:852–862. doi: 10.1136/gutjnl-2011-301424. [DOI] [PubMed] [Google Scholar]
- 84.Lech M, Anders HJ. Macrophages and fibrosis: how resident and infiltrating mononuclear phagocytes orchestrate all phases of tissue injury and repair. Biochim Biophys Acta. 2013;1832:989–997. doi: 10.1016/j.bbadis.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 85.Mao R, Kurada S, Gordon IO, et al. The mesenteric fat and intestinal muscle interface: creeping fat influencing stricture formation in Crohn’s disease. Inflamm Bowel Dis. 2019;25:421–426. doi: 10.1093/ibd/izy331. [DOI] [PubMed] [Google Scholar]
- 86.Rieder F, Doyon G, Ouyang Z, West G, Fiocchi C. 573 Adipocyte and preadipocyte derived-mediators induce a PRO-fibrogenic phenotype in human intestinal mesenchymal cells: a novel link between fat and intestinal fibrosis. Gastroenterology. 2014;146:S–106. [Google Scholar]
- 87.Di Sabatino A, Jackson CL, Pickard KM, et al. Transforming growth factor beta signalling and matrix metalloproteinases in the mucosa overlying Crohn’s disease strictures. Gut. 2009;58:777–789. doi: 10.1136/gut.2008.149096. [DOI] [PubMed] [Google Scholar]
- 88.Monteleone G, Pallone F, MacDonald TT. Smad7 in TGF-beta-mediated negative regulation of gut inflammation. Trends Immunol. 2004;25:513–517. doi: 10.1016/j.it.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 89.Monteleone G, Kumberova A, Croft NM, McKenzie C, Steer HW, MacDonald TT. Blocking Smad7 restores TGF-beta1 signaling in chronic inflammatory bowel disease. J Clin Invest. 2001;108:601–609. doi: 10.1172/JCI12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Biancheri P, Giuffrida P, Docena GH, MacDonald TT, Corazza GR, Di Sabatino A. The role of transforming growth factor (TGF)-beta in modulating the immune response and fibrogenesis in the gut. Cytokine Growth Factor Rev. 2014;25:45–55. doi: 10.1016/j.cytogfr.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 91.Li C, Kuemmerle JF. Tu1772: epigenetic silencing of Smad7 contributes to fibrosis in stricturing Crohn’s disease. Gastroenterology. 2018;154:S–1015. [Google Scholar]
- 92.Monteleone G, Mann J, Monteleone I, et al. A failure of transforming growth factor-beta1 negative regulation maintains sustained NF-kappaB activation in gut inflammation. J Biol Chem. 2004;279:3925–3932. doi: 10.1074/jbc.M303654200. [DOI] [PubMed] [Google Scholar]
- 93.Kennedy BW. Mongersen: an oral SMAD7 antisense oligonucleotide, and Crohn’s disease. N Engl J Med. 2015;372:2461. doi: 10.1056/NEJMc1504845. [DOI] [PubMed] [Google Scholar]
- 94.Zorzi F, Calabrese E, Monteleone I, et al. A phase 1 open-label trial shows that smad7 antisense oligonucleotide (GED0301) does not increase the risk of small bowel strictures in Crohn’s disease. Aliment Pharmacol Ther. 2012;36:850–857. doi: 10.1111/apt.12051. [DOI] [PubMed] [Google Scholar]
- 95.Nijhuis A, Biancheri P, Lewis A, et al. In Crohn’s disease fibrosis-reduced expression of the miR-29 family enhances collagen expression in intestinal fibroblasts. Clin Sci (Lond) 2014;127:341–350. doi: 10.1042/CS20140048. [DOI] [PubMed] [Google Scholar]
- 96.Lewis A, Mehta S, Hanna LN, et al. Low serum levels of microRNA-19 are associated with a stricturing Crohn’s disease phenotype. Inflamm Bowel Dis. 2015;21:1926–1934. doi: 10.1097/MIB.0000000000000443. [DOI] [PubMed] [Google Scholar]
- 97.Tarrant KM, Barclay ML, Frampton CM, Gearry RB. Perianal disease predict changes in Crohn’s disease phenotype: results of a population-based study of inflammatory bowel disease phenotype. Am J Gastroenterol. 2008;103:3082–3093. doi: 10.1111/j.1572-0241.2008.02212.x. [DOI] [PubMed] [Google Scholar]
- 98.Romberg-Camps MJ, Dagnelie PC, Kester AD, et al. Influence of phenotype at diagnosis and of other potential prognostic factors on the course of inflammatory bowel disease. Am J Gastroenterol. 2009;104:371–383. doi: 10.1038/ajg.2008.38. [DOI] [PubMed] [Google Scholar]
- 99.Lakatos PL, Czegledi Z, Szamosi T, et al. Perianal disease, small bowel disease, smoking, prior steroid or early azathioprine/biological therapy are predictors of disease behavior change in patients with Crohn’s disease. World J Gastroenterol. 2009;15:3504–3510. doi: 10.3748/wjg.15.3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rieder F, Lawrance IC, Leite A, Sans M. Predictors of fibrostenotic Crohn’s disease. Inflamm Bowel Dis. 2011;17:2000–2007. doi: 10.1002/ibd.21627. [DOI] [PubMed] [Google Scholar]
- 101.Thia KT, Sandborn WJ, Harmsen WS, Zinsmeister AR, Loftus EV., Jr Risk factors associated with progression to intestinal complications of Crohn’s disease in a population-based cohort. Gastroenterology. 2010;139:1147–1155. doi: 10.1053/j.gastro.2010.06.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Louis E, Michel V, Hugot JP, et al. Early development of stricturing or penetrating pattern in Crohn’s disease is influenced by disease location, number of flares, and smoking but not by NOD2/CARD15 genotype. Gut. 2003;52:552–557. doi: 10.1136/gut.52.4.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Alvarez-Lobos M, Arostegui JI, Sans M, et al. Crohn’s disease patients carrying Nod2/CARD15 gene variants have an increased and early need for first surgery due to stricturing disease and higher rate of surgical recurrence. Ann Surg. 2005;242:693–700. doi: 10.1097/01.sla.0000186173.14696.ea. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bevins CL, Salzman NH. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol. 2011;9:356–368. doi: 10.1038/nrmicro2546. [DOI] [PubMed] [Google Scholar]
- 105.Adler J, Rangwalla SC, Dwamena BA, Higgins PD. The prognostic power of the NOD2 genotype for complicated Crohn’s disease: a meta-analysis. Am J Gastroenterol. 2011;106:699–712. doi: 10.1038/ajg.2011.19. [DOI] [PubMed] [Google Scholar]
- 106.Brant SR, Picco MF, Achkar JP, et al. Defining complex contributions of NOD2/CARD15 gene mutations, age at onset, and tobacco use on Crohn’s disease phenotypes. Inflamm Bowel Dis. 2003;9:281–289. doi: 10.1097/00054725-200309000-00001. [DOI] [PubMed] [Google Scholar]
- 107.Rieder F, Zimmermann EM, Remzi FH, Sandborn WJ. Crohn’s disease complicated by strictures: a systematic review. Gut. 2013;62:1072–1084. doi: 10.1136/gutjnl-2012-304353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lakatos PL, Papp M, Rieder F. Serologic antiglycan antibodies in inflammatory bowel disease. Am J Gastroenterol. 2011;106:406–412. doi: 10.1038/ajg.2010.505. [DOI] [PubMed] [Google Scholar]
- 109.Chen Y, Ge W, Xu L, et al. miR-200b is involved in intestinal fibrosis of Crohn’s disease. Int J Mol Med. 2012;29:601–606. doi: 10.3892/ijmm.2012.894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Giuffrida P, Pinzani M, Corazza GR, Di Sabatino A. Biomarkers of intestinal fibrosis: one step towards clinical trials for stricturing inflammatory bowel disease. United European Gastroenterol J. 2016;4:523–530. doi: 10.1177/2050640616640160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Allez M, Lemann M, Bonnet J, Cattan P, Jian R, Modigliani R. Long term outcome of patients with active Crohn’s disease exhibiting extensive and deep ulcerations at colonoscopy. Am J Gastroenterol. 2002;97:947–953. doi: 10.1111/j.1572-0241.2002.05614.x. [DOI] [PubMed] [Google Scholar]
- 112.Bettenworth D, Bokemeyer A, Baker M, et al. Assessment of Crohn’s disease-associated small bowel strictures and fibrosis on cross-sectional imaging: a systematic review. Gut. 2019;68:1115–1126. doi: 10.1136/gutjnl-2018-318081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Adler J, Punglia DR, Dillman JR, et al. Computed tomography enterography findings correlate with tissue inflammation, not fibrosis in resected small bowel Crohn’s disease. Inflamm Bowel Dis. 2012;18:849–856. doi: 10.1002/ibd.21801. [DOI] [PubMed] [Google Scholar]
- 114.Maconi G, Carsana L, Fociani P, et al. Small bowel stenosis in Crohn’s disease: clinical, biochemical and ultrasonographic evaluation of histological features. Aliment Pharmacol Ther. 2003;18:749–756. doi: 10.1046/j.1365-2036.2003.01673.x. [DOI] [PubMed] [Google Scholar]
- 115.Pallotta N, Vincoli G, Montesani C, et al. Small intestine contrast ultrasonography (SICUS) for the detection of small bowel complications in Crohn’s disease: a prospective comparative study versus intraoperative findings. Inflamm Bowel Dis. 2012;18:74–84. doi: 10.1002/ibd.21678. [DOI] [PubMed] [Google Scholar]
- 116.Vogel J, da Luz Moreira A, Baker M, et al. CT enterography for Crohn’s disease: accurate preoperative diagnostic imaging. Dis Colon Rectum. 2007;50:1761–1769. doi: 10.1007/s10350-007-9005-6. [DOI] [PubMed] [Google Scholar]
- 117.Kumar S, Hakim A, Alexakis C, et al. Small intestinal contrast ultrasonography for the detection of small bowel complications in Crohn’s disease: correlation with intraoperative findings and magnetic resonance enterography. J Gastroenterol Hepatol. 2015;30:86–91. doi: 10.1111/jgh.12724. [DOI] [PubMed] [Google Scholar]
- 118.Pous-Serrano S, Frasson M, Palasí Giménez R, et al. Accuracy of magnetic resonance enterography in the preoperative assessment of patients with Crohn’s disease of the small bowel. Colorectal Dis. 2017:19–O126-O133. doi: 10.1111/codi.13613. [DOI] [PubMed] [Google Scholar]
- 119.Sinha R, Murphy P, Sanders S, et al. Diagnostic accuracy of high-resolution MR enterography in Crohn’s disease: comparison with surgical and pathological specimen. Clin Radiol. 2013;68:917–927. doi: 10.1016/j.crad.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 120.Takenaka K, Ohtsuka K, Kitazume Y, et al. Comparison of magnetic resonance and balloon enteroscopic examination of the small intestine in patients with Crohn’s disease. Gastroenterology. 2014;147:334–342. doi: 10.1053/j.gastro.2014.04.008. [DOI] [PubMed] [Google Scholar]
- 121.Castiglione F, Mainenti PP, De Palma GD, et al. Noninvasive diagnosis of small bowel Crohn’s disease: direct comparison of bowel sonography and magnetic resonance enterography. Inflamm Bowel Dis. 2013;19:991–998. doi: 10.1097/MIB.0b013e3182802b87. [DOI] [PubMed] [Google Scholar]
- 122.Orlando S, Fraquelli M, Coletta M, et al. Ultrasound elasticity imaging predicts therapeutic outcomes of patients with Crohn’s disease treated with anti-tumour necrosis factor antibodies. J Crohns Colitis. 2018;12:63–70. doi: 10.1093/ecco-jcc/jjx116. [DOI] [PubMed] [Google Scholar]
- 123.Liu YB, Liang CH, Zhang ZL, et al. Crohn disease of small bowel: multidetector row CT with CT enteroclysis, dynamic contrast enhancement, CT angiography, and 3D imaging. Abdom Imaging. 2006;31:668–674. doi: 10.1007/s00261-006-9092-1. [DOI] [PubMed] [Google Scholar]
- 124.Rimola J, Planell N, Rodríguez S, et al. Characterization of inflammation and fibrosis in Crohn’s disease lesions by magnetic resonance imaging. Am J Gastroenterol. 2015;110:432–440. doi: 10.1038/ajg.2014.424. [DOI] [PubMed] [Google Scholar]
- 125.Gramlich T, Petras RE. Pathology of inflammatory bowel disease. Semin Pediatr Surg. 2007;16:154–163. doi: 10.1053/j.sempedsurg.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 126.Brahme F, Lindström C. A comparative radiographic and pathological study of intestinal vaso-architecture in Crohn’s disease and in ulcerative colitis. Gut. 1970;11:928–940. doi: 10.1136/gut.11.11.928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Li XH, Mao R, Huang SY, et al. Characterization of degree of intestinal fibrosis in patients with Crohn disease by using magnetization transfer MR imaging. Radiology. 2018;287:494–503. doi: 10.1148/radiol.2017171221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Allocca M, Fiorino G, Bonifacio C, Peyrin-Biroulet L, Danese S. Noninvasive multimodal methods to differentiate inflamed vs fibrotic strictures in patients with Crohn’s disease. Clin Gastroenterol Hepatol. 2019;17:2397–2415. doi: 10.1016/j.cgh.2019.04.025. [DOI] [PubMed] [Google Scholar]
- 129.Choi SH, Kim KW, Lee JY, Kim KJ, Park SH. Diffusion-weighted magnetic resonance enterography for evaluating bowel inflammation in Crohn’s disease: a systematic review and meta-analysis. Inflamm Bowel Dis. 2016;22:669–679. doi: 10.1097/MIB.0000000000000607. [DOI] [PubMed] [Google Scholar]
- 130.Tielbeek JA, Ziech ML, Li Z, et al. Evaluation of conventional, dynamic contrast enhanced and diffusion weighted MRI for quantitative Crohn’s disease assessment with histopathology of surgical specimens. Eur Radiol. 2014;24:619–629. doi: 10.1007/s00330-013-3015-7. [DOI] [PubMed] [Google Scholar]
- 131.Adler J, Swanson SD, Schmiedlin-Ren P, et al. Magnetization transfer helps detect intestinal fibrosis in an animal model of Crohn disease. Radiology. 2011;259:127–135. doi: 10.1148/radiol.10091648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Moy MP, Sauk J, Gee MS. The role of MR enterography in assessing Crohn’s disease activity and treatment response. Gastroenterol Res Pract. 2016;2016:8168695. doi: 10.1155/2016/8168695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gordon IO, Bettenworth D, Bokemeyer A, et al. Histopathology scoring systems of stenosis associated with small bowel Crohn’s disease: a systematic review. Gastroenterology. 2020;158:137–150. doi: 10.1053/j.gastro.2019.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Benitez JM, Meuwis MA, Reenaers C, van Kemseke C, Meunier P, Louis E. Role of endoscopy, cross-sectional imaging and biomarkers in Crohn’s disease monitoring. Gut. 2013;62:1806–1816. doi: 10.1136/gutjnl-2012-303957. [DOI] [PubMed] [Google Scholar]
- 135.Pariente B, Cosnes J, Danese S, et al. Development of the Crohn’s disease digestive damage score, the Lémann score. Inflamm Bowel Dis. 2011;17:1415–1422. doi: 10.1002/ibd.21506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yaffe BH, Korelitz BI. Prognosis for nonoperative management of small-bowel obstruction in Crohn’s disease. J Clin Gastroenterol. 1983;5:211–215. doi: 10.1097/00004836-198306000-00003. [DOI] [PubMed] [Google Scholar]
- 137.Gordon M. 5-Aminosalicylates to maintain remission in Crohn’s disease: interpreting conflicting systematic review evidence. World J Gastrointest Pharmacol Ther. 2017;8:99–102. doi: 10.4292/wjgpt.v8.i2.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.de Souza GS, Vidigal FM, Chebli LA, et al. Effect of azathioprine or mesalazine therapy on incidence of re-hospitalization in sub-occlusive ileocecal Crohn’s disease patients. Med Sci Monit. 2013;19:716–722. doi: 10.12659/MSM.889196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Reinisch W, Angelberger S, Petritsch W, et al. Azathioprine versus mesalazine for prevention of postoperative clinical recurrence in patients with Crohn’s disease with endoscopic recurrence: efficacy and safety results of a randomised, double-blind, double-dummy, multicentre trial. Gut. 2010;59:752–759. doi: 10.1136/gut.2009.194159. [DOI] [PubMed] [Google Scholar]
- 140.Ardizzone S, Maconi G, Sampietro GM, et al. Azathioprine and mesalamine for prevention of relapse after conservative surgery for Crohn’s disease. Gastroenterology. 2004;127:730–740. doi: 10.1053/j.gastro.2004.06.051. [DOI] [PubMed] [Google Scholar]
- 141.Speca S, Rousseaux C, Dubuquoy C, et al. Novel PPARγ modulator GED-0507-34 Levo ameliorates inflammation-driven intestinal fibrosis. Inflamm Bowel Dis. 2016;22:279–292. doi: 10.1097/MIB.0000000000000618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Szabò H, Fiorino G, Spinelli A, et al. Review article: anti-fibrotic agents for the treatment of Crohn’s disease: lessons learnt from other diseases. Aliment Pharmacol Ther. 2010;31:189–201. doi: 10.1111/j.1365-2036.2009.04171.x. [DOI] [PubMed] [Google Scholar]
- 143.Videla S, Vilaseca J, Medina C, et al. Selective inhibition of phosphodiesterase-4 ameliorates chronic colitis and prevents intestinal fibrosis. J Pharmacol Exp Ther. 2006;316:940–945. doi: 10.1124/jpet.105.090837. [DOI] [PubMed] [Google Scholar]
- 144.Peyrin-Biroulet L, Deltenre P, Ardizzone S, et al. Azathioprine and 6-mercaptopurine for the prevention of postoperative recurrence in Crohn’s disease: a meta-analysis. Am J Gastroenterol. 2009;104:2089–2096. doi: 10.1038/ajg.2009.301. [DOI] [PubMed] [Google Scholar]
- 145.Casanova MJ, Chaparro M, García-Sánchez V, et al. Evolution after anti-TNF discontinuation in patients with inflammatory bowel disease: a multicenter long-term follow-up study. Am J Gastroenterol. 2017;112:120–131. doi: 10.1038/ajg.2016.569. [DOI] [PubMed] [Google Scholar]
- 146.Lichtenstein GR, Olson A, Travers S, et al. Factors associated with the development of intestinal strictures or obstructions147. Bouhnik Y, Carbonnel F, Laharie D, et al. Efficacy of adalimumab in patients with Crohn’s disease and symptomatic small bowel stricture: a multicentre, prospective, observational cohort (CREOLE) study. Gut. 2018;67:53–60. doi: 10.1136/gutjnl-2016-312581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Bouhnik Y, Carbonnel F, Laharie D, et al. Efficacy of adalimumab in patients with Crohn’s disease and symptomatic small bowel stricture: a multicentre, prospective, observational cohort (CREOLE) study. Gut. 2018;67:53–60. doi: 10.1136/gutjnl-2016-312581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Fields AC, Melnitchouk N. Medical prophylaxis of post-surgical Crohn’s disease recurrence: towards timely anti-TNF therapy. Dig Dis Sci. 2019;64:7–8. doi: 10.1007/s10620-018-5236-8. [DOI] [PubMed] [Google Scholar]
- 149.Nguyen GC, Loftus EV, Jr, Hirano I, et al. American Gastroenterological Association Institute guideline on the management of Crohn’s disease after surgical resection. Gastroenterology. 2017;152:271–275. doi: 10.1053/j.gastro.2016.10.038. [DOI] [PubMed] [Google Scholar]
- 150.Ding NS, Yip WM, Choi CH, et al. Endoscopic dilatation of Crohn’s anastomotic strictures is effective in the long term, and escalation of medical therapy improves outcomes in the biologic era. J Crohns Colitis. 2016;10:1172–1178. doi: 10.1093/ecco-jcc/jjw072. [DOI] [PubMed] [Google Scholar]
- 151.Kopylov U, Verstockt B, Biedermann L, et al. Effectiveness and safety of vedolizumab in anti-TNF-naïve patients with inflammatory bowel disease-a multicenter retrospective European study. Inflamm Bowel Dis. 2018;24:2442–2451. doi: 10.1093/ibd/izy155. [DOI] [PubMed] [Google Scholar]
- 152.Dulai PS, Singh S, Jiang X, et al. The real-world effectiveness and safety of vedolizumab for moderate-severe Crohn’s disease: results from the US VICTORY consortium. Am J Gastroenterol. 2016;111:1147–1155. doi: 10.1038/ajg.2016.236. [DOI] [PubMed] [Google Scholar]
- 153.Ma C, Fedorak RN, Kaplan GG, et al. Clinical, endoscopic and radiographic outcomes with ustekinumab in medically-refractory Crohn’s disease: real world experience from a multicenter cohort. Aliment Pharmacol Ther. 2017;45:1232–1243. doi: 10.1111/apt.14016. [DOI] [PubMed] [Google Scholar]
- 154.Hirai F, Andoh A, Ueno F, et al. Efficacy of endoscopic balloon dilation for small bowel strictures in patients with Crohn’s disease: a nationwide, multi-centre, open-label, prospective cohort study. J Crohns Colitis. 2018;12:394–401. doi: 10.1093/ecco-jcc/jjx159. [DOI] [PubMed] [Google Scholar]
- 155.Bettenworth D, Gustavsson A, Atreja A, et al. A pooled analysis of efficacy, safety, and long-term outcome of endoscopic balloon dilation therapy for patients with stricturing Crohn’s disease. Inflamm Bowel Dis. 2017;23:133–142. doi: 10.1097/MIB.0000000000000988. [DOI] [PubMed] [Google Scholar]
- 156.Shen B, Kochhar G, Navaneethan U, et al. Role of interventional inflammatory bowel disease in the era of biologic therapy: a position statement from the Global Interventional IBD Group. Gastrointest Endosc. 2019;89:215–237. doi: 10.1016/j.gie.2018.09.045. [DOI] [PubMed] [Google Scholar]
- 157.Chen M, Shen B. Comparable short- and long-term outcomes of colonoscopic balloon dilation of Crohn’s disease and benign non-Crohn’s disease strictures. Inflamm Bowel Dis. 2014;20:1739–1746. doi: 10.1097/MIB.0000000000000145. [DOI] [PubMed] [Google Scholar]
- 158.Lian L, Stocchi L, Remzi FH, Shen B. Comparison of endoscopic dilation vs surgery for anastomotic stricture in patients with Crohn’s disease following ileocolonic resection. Clin Gastroenterol Hepatol. 2017;15:1226–1231. doi: 10.1016/j.cgh.2016.10.030. [DOI] [PubMed] [Google Scholar]
- 159.Bettenworth D, Lopez R, Hindryckx P, Levesque BG, Rieder F. Heterogeneity in endoscopic treatment of Crohn’s disease-associated strictures: an international inflammatory bowel disease specialist survey. J Gastroenterol. 2016;51:939–948. doi: 10.1007/s00535-016-1172-6. [DOI] [PubMed] [Google Scholar]
- 160.Chen M, Shen B. Endoscopic therapy in Crohn’s disease: principle, preparation, and technique. Inflamm Bowel Dis. 2015;21:2222–2240. doi: 10.1097/MIB.0000000000000433. [DOI] [PubMed] [Google Scholar]
- 161.Loras Alastruey C, Andújar Murcia X, Esteve Comas M. The role of stents in the treatment of Crohn’s disease strictures. Endosc Int Open. 2016;4:E301–E308. doi: 10.1055/s-0042-101786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Levine RA, Wasvary H, Kadro O. Endoprosthetic management of refractory ileocolonic anastomotic strictures after resection for Crohn’s disease: report of nine-year follow-up and review of the literature. Inflamm Bowel Dis. 2012;18:506–512. doi: 10.1002/ibd.21739. [DOI] [PubMed] [Google Scholar]
- 163.Campos C, Perrey A, Lambert C, et al. Medical therapies for stricturing Crohn’s disease: efficacy and cross-sectional imaging predictors of therapeutic failure. Dig Dis Sci. 2017;62:1628–1636. doi: 10.1007/s10620-017-4572-4. [DOI] [PubMed] [Google Scholar]
- 164.Morar PS, Faiz O, Warusavitarne J, et al. Systematic review with meta-analysis: endoscopic balloon dilatation for Crohn’s disease strictures. Aliment Pharmacol Ther. 2015;42:1137–1148. doi: 10.1111/apt.13388. [DOI] [PubMed] [Google Scholar]
- 165.Golovics PA, Lakatos L, Nagy A, et al. Is early limited surgery associated with a more benign disease course in Crohn’s disease? World J Gastroenterol. 2013;19:7701–7710. doi: 10.3748/wjg.v19.i43.7701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kulungowski AM, Acker SN, Hoffenberg EJ, Neigut D, Partrick DA. Initial operative treatment of isolated ileal Crohn’s disease in adolescents. Am J Surg. 2015;210:141–145. doi: 10.1016/j.amjsurg.2014.07.009. [DOI] [PubMed] [Google Scholar]
- 167.Latella G, Cocco A, Angelucci E, et al. Clinical course of Crohn’s disease first diagnosed at surgery for acute abdomen. Dig Liver Dis. 2009;41:269–276. doi: 10.1016/j.dld.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 168.Lee CH, Rieder F, Holubar SD. Duodenojejunal bypass and strictureplasty for diffuse small bowel Crohn’s disease with a step-by-step visual guide. Crohns Colitis 360. 2019:1–otz002. [Google Scholar]
- 169.Ambe R, Campbell L, Cagir B. A comprehensive review of strictureplasty techniques in Crohn’s disease: types, indications, comparisons, and safety. J Gastrointest Surg. 2012;16:209–217. doi: 10.1007/s11605-011-1651-2. [DOI] [PubMed] [Google Scholar]
- 170.Bettenworth D, Rieder F. Reversibility of stricturing Crohn’s disease: fact or fiction? Inflamm Bowel Dis. 2016;22:241–247. doi: 10.1097/MIB.0000000000000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Maconi G, Sampietro GM, Cristaldi M, et al. Preoperative characteristics and postoperative behavior of bowel wall on risk of recurrence after conservative surgery in Crohn’s disease: a prospective study. Ann Surg. 2001;233:345–352. doi: 10.1097/00000658-200103000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Yamamoto T, Fazio VW, Tekkis PP. Safety and efficacy of strictureplasty for Crohn’s disease: a systematic review and meta-analysis. Dis Colon Rectum. 2007;50:1968–1986. doi: 10.1007/s10350-007-0279-5. [DOI] [PubMed] [Google Scholar]
- 173.Oku H, Shimizu T, Kawabata T, et al. Antifibrotic action of pirfenidone and prednisolone: different effects on pulmonary cytokines and growth factors in bleomycin-induced murine pulmonary fibrosis. Eur J Pharmacol. 2008;590:400–408. doi: 10.1016/j.ejphar.2008.06.046. [DOI] [PubMed] [Google Scholar]
- 174.Noble PW, Albera C, Bradford WZ, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377:1760–1769. doi: 10.1016/S0140-6736(11)60405-4. [DOI] [PubMed] [Google Scholar]
- 175.Li G, Ren J, Hu Q, et al. Oral pirfenidone protects against fibrosis by inhibiting fibroblast proliferation and TGF-beta signaling in a murine colitis model. Biochem Pharmacol. 2016;117:57–67. doi: 10.1016/j.bcp.2016.08.002. [DOI] [PubMed] [Google Scholar]
- 176.Sun YW, Zhang YY, Ke XJ, Wu XJ, Chen ZF, Chi P. Pirfenidone prevents radiation-induced intestinal fibrosis in rats by inhibiting fibroblast proliferation and differentiation and suppressing the TGF-beta1/Smad/CTGF signaling pathway. Eur J Pharmacol. 2018;822:199–206. doi: 10.1016/j.ejphar.2018.01.027. [DOI] [PubMed] [Google Scholar]
- 177.Meier R, Lutz C, Cosín-Roger J, et al. Decreased fibrogenesis after treatment with pirfenidone in a newly developed mouse model of intestinal fibrosis. Inflamm Bowel Dis. 2016;22:569–582. doi: 10.1097/MIB.0000000000000716. [DOI] [PubMed] [Google Scholar]
- 178.Kadir SI, Wenzel Kragstrup T, Dige A, Kok Jensen S, Dahlerup JF, Kelsen J. Pirfenidone inhibits the proliferation of fibroblasts from patients with active Crohn’s disease. Scand J Gastroenterol. 2016;51:1321–1325. doi: 10.1080/00365521.2016.1185146. [DOI] [PubMed] [Google Scholar]
- 179.Huang J, Beyer C, Palumbo-Zerr K, et al. Nintedanib inhibits fibroblast activation and ameliorates fibrosis in preclinical models of systemic sclerosis. Ann Rheum Dis. 2016;75:883–890. doi: 10.1136/annrheumdis-2014-207109. [DOI] [PubMed] [Google Scholar]
- 180.Maher TM, Strek ME. Antifibrotic therapy for idiopathic pulmonary fibrosis: time to treat. Respir Res. 2019;20:205. doi: 10.1186/s12931-019-1161-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Rieder F. ROCKing the field of intestinal fibrosis or between a ROCK and a hard place? Gastroenterology. 2017;153:895–897. doi: 10.1053/j.gastro.2017.08.056. [DOI] [PubMed] [Google Scholar]
- 182.Bettenworth D, Rieder F. Medical therapy of stricturing Crohn’s disease: what the gut can learn from other organs: a systematic review. Fibrogenesis Tissue Repair. 2014;7:5. doi: 10.1186/1755-1536-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Danese S, Bonovas S, Lopez A, et al. Identification of endpoints for development of antifibrosis drugs for treatment of Crohn’s disease. Gastroenterology. 2018;155:76–87. doi: 10.1053/j.gastro.2018.03.032. [DOI] [PubMed] [Google Scholar]
- 184.Pariente B, Hu S, Bettenworth D, et al. Treatments for Crohn’s disease-associated bowel damage: a systematic review. Clin Gastroenterol Hepatol. 2019;17:847–856. doi: 10.1016/j.cgh.2018.06.043. [DOI] [PubMed] [Google Scholar]