

Abstract
Whether a cell lives or dies is controlled by an array of intercepting and dynamic molecular pathways. Although there is evidence of neuronal loss in Alzheimer’s disease (AD) and multiple programmed cell death (PCD) pathways have been implicated in this process, there has been no comprehensive evaluation of the dominant pathway responsible for cell death in AD. Likewise, the relative dominance of survival and PCD pathways in AD remains unclear. Here, we present the results of hypothesis-driven bioinformatic analysis of PCD and survival pathway activation in paired methylation and expression data from the middle temporal gyrus as well as expression from laser-captured cells from the middle temporal gyrus and hippocampus. The results not only indicate activation of cell death pathways in AD—of which apoptosis is responsible for the largest fraction of upregulated genes—but also of cell survival pathways. These results are indicative of a complex balance between survival and death pathways in AD that future studies should work to delineate at a single cell level.
1. Introduction
Alzheimer’s disease (AD) is the most prevalent form of neurodegeneration (Mayeux and Stern, 2012), conventionally characterized by the accumulation of protein conglomerates intra- and extracellularly (Bierer et al., 1995; Blennow et al., 1996; Serrano-Pozo et al., 2011), impaired synaptic function and loss (Blennow et al., 1996; Forner et al., 2017; Masliah et al., 1994; Scheff et al., 1990; Scheff and Price, 1993; Serrano-Pozo et al., 2011), and neuron death (Andrade-Moraes et al., 2013; Serrano-Pozo et al., 2011; Šimić et al., 1997; Troncoso et al., 1996). Large losses of neurons in the hippocampus and cerebral cortex have been reported in AD (Andrade-Moraes et al., 2013; Šimić et al., 1997) and volumetric changes, presumably caused by neuronal loss, have been observed in the AD brain (Chan et al., 2001; Schuff et al., 2012). Neuronal cultures treated with amyloid beta (Aβ), one of the neurotoxic pathological hallmarks of AD, also show high rates of cell death (Han et al., 2017; Kadowaki et al., 2005; Nieweg et al., 2015). Moreover, the prevalence of cell death markers in AD neurons increase with disease severity, suggesting a direct impact of cell death on cognitive decline and an opportunity for pharmaceutical intervention (Troncoso et al., 1996). That said, the exact cell death pathway responsible for neuron death in AD is an unresolved issue. Apoptosis (Albrecht et al., 2009; Selznick et al., 1999; Silva et al., 2014; Stadelmann et al., 1999; Su et al., 2001; Troncoso et al., 1996), autophagy (Piras et al., 2016; Theofilas et al., 2018), and necroptosis (Caccamo et al., 2017; Ofengeim et al., 2017)—among other pathways—are all potentially responsible for neuron death in AD. However, it remains unclear which pathway is dominant or, potentially, if there is a combinatorial effect between co-activated death pathways.
There are currently twelve recognized programmed cell death (PCD) pathways that can be further divided into three general categories: type I apoptotic, type II autophagic, and type III necrotic PCD (Galluzzi et al., 2018). Of these, apoptosis is perhaps one of the best mechanistically defined PCD pathway and is characterized by caspase activation, chromatin condensation, nuclear fragmentation, and the formation of apoptotic bodies (Elmore, 2007; Galluzzi et al., 2018). Apoptosis can be further separated into two distinct but intersecting pathways: intrinsic apoptosis, which is activated by intracellular stress and results in permeabilization of the mitochondrial membrane through caspase activation (Galluzzi et al., 2018; Green and Llambi, 2015), and extrinsic apoptosis, which is precipitated by extracellular stimuli communicated through transmembrane death receptors and the formation of the death-inducing signaling complex (DISC) (Fulda and Debatin, 2006; Galluzzi et al., 2018). Alternatively, cells can also undergo autophagic or necroptotic PCD. Macroautophagy, hereafter referred to simply as autophagy, is characterized by the formation of double membrane structures, known as autophagosomes, and the digestion of enveloped intracellular material (Galluzzi et al., 2018; Green and Llambi, 2015). Because autophagy is activated under nutrient deficient conditions and can promote cellular survival in some circumstances, it has been contested whether autophagy is truly a PCD pathway (Galluzzi et al., 2018). That said, it has been demonstrated that not only are elements of the autophagic pathway necessary and sufficient to induce cell death but that autophagy can also catalyze the activation of and cooperate with other PCD pathways (Denton et al., 2012; Galluzzi et al., 2018; Green and Llambi, 2015). Necroptosis—a form of necrosis—is distinguished by activation of the RIP kinase cascade (Galluzzi et al., 2018; Li et al., 2012), formation of the necrosome (Galluzzi et al., 2018; Li et al., 2012), and loss of plasma membrane integrity resulting in cell death (Linkermann and Green, 2014).
Evidence suggests that apoptosis is heightened in AD brains (Albrecht et al., 2009; Selznick et al., 1999; Silva et al., 2014; Stadelmann et al., 1999; Su et al., 2001; Troncoso et al., 1996). TUNEL staining, a proxy for apoptosis, is higher in AD neurons (Silva et al., 2014; Troncoso et al., 1996) and levels of caspase 3, an executioner caspase, has also been reported to be elevated in AD neurons from the entorhinal cortex, frontal cortex, and hippocampus (Selznick et al., 1999; Stadelmann et al., 1999; Su et al., 2001). In addition to its function in apoptosis, caspase 3 can also cleave APP, the precursor to the amyloid plaques present in AD (Zhang et al., 2011). Caspases 1, 6, 7, 8, and 9 are also have been suggested to be involved in AD pathogenesis (Albrecht et al., 2009; Burguillos et al., 2011; Matsui et al., 2006; Rohn et al., 2002; Thawkar and Kaur, 2019). Aside from apoptosis, research suggests that autophagy (Piras et al., 2016; Theofilas et al., 2018) and necroptosis (Caccamo et al., 2017; Ofengeim et al., 2017) also occur at a higher rate in AD. LC3, a macroautophagic marker is higher in AD neurons in both familial and sporadic AD cases (Piras et al., 2016; Theofilas et al., 2018) and, in the case of familial AD, these changes correlated with levels of tau hyperphosphorylation (Piras et al., 2016). Transcriptomic and proteomic changes to the RIPK pathway have also been reported in AD neurons, suggesting activation of necroptosis (Caccamo et al., 2017; Ofengeim et al., 2017). This is particularly interesting because, although autophagy can cooperate with other PCD pathways and be activated simultaneously, necroptosis and other PCD pathways are mutually exclusive (Denton et al., 2012; Galluzzi et al., 2018; Green and Llambi, 2015). Additionally, it is also possible neurons are undergoing cell cycle reentry in AD: several cell cycle proteins have been found to be elevated in AD hippocampal neurons while multiple cell cycle inhibitors have been shown to be suppressed (Busser et al., 1988; Yang et al., 2003).
Despite PCD activation and neuron loss in AD, evidence also indicates that neurons can survive for decades with AD pathology (Morsch et al., 1999). It is currently projected that AD development may start significantly earlier—perhaps decades—before clinical presentation (Bloss et al., 2008; Okonkwo et al., 2012; Talboom et al., 2019), suggesting compensatory pathways are activated to counteract the neurotoxic effects of the disease. Multiple intersecting pathways that respond to cytotoxic stress, promote cellular repairs, and inhibit cell death may be candidates for a compensatory mechanism in AD. These include the PI3K/AKT (Brunet et al., 2001; Cardone et al., 1998; Heras-Sandoval et al., 2014; Morita et al., 2015), the MAPK (Kim and Choi, 2015; Wagner and Nebreda, 2009), and NF-κB (Bours et al., 2000; Lawrence, 2009; Nikoletopoulou et al., 2013; Pires et al., 2018) signaling pathways. The PI3K/AKT pathway strongly promotes survival and suppresses apoptosis (Cardone et al., 1998; Heras-Sandoval et al., 2014), regulates autophagy through AKT-mediated phosphorylation of mTOR (Heras-Sandoval et al., 2014; Morita et al., 2015), and suppresses apoptosis directly through phosphorylation of apoptotic proteins (Cardone et al., 1998; Heras-Sandoval et al., 2014) as well as indirectly through regulation of pro-apoptotic gene expression (Brunet et al., 2001). Likewise, both MAPK/ERK and NF-κB pathways can be activated in response to stress and promote cellular survival through indirect and direct mechanisms (Kim and Choi, 2015; Lawrence, 2009; Pires et al., 2018; Wagner and Nebreda, 2009). Although all three signaling pathways discussed above may mitigate the cellular stress present in AD and promote cell survival, they may also lead to neuropathology and, in the case of MAPK and NF-κB, cell death. Hyperactivation of the PI3K/AKT pathway has been reported in AD (Heras-Sandoval et al., 2014) as well as higher levels of phosphorylated proteins downstream of AKT—including tau (Griffin et al., 2005). Moreover, both have been implicated in AD pathogenesis. Expression and phosphorylation of p38, a MAP kinase, increased in neuronal culture exposed to Aβ1–42 and correlated with a decrease in apoptosis (Song et al., 2004) while kinases from the other two branches of the MAPK pathway, JNK and ERK1/2, have also been implicated in AD (Kim and Choi, 2015). Likewise, NF-κB regulates the expression of amyloid processing enzymes β- and γ-secretase (Chami et al., 2012) and is activated in response to Aβ1–42 in neuronal cell culture (Shi et al., 2016; Song et al., 2004), potentially representing a cyclic mechanism of amyloid accumulation in AD.
Understanding the combinatorial roles of PCD and stress-activated survival pathways in AD is critical to understanding the origin and progression of the disease. To date, no comprehensive census of PCD or stress-activated survival pathway activation in AD has been conducted. In order to survey pathway activation in AD, we compiled gene lists from nine pathways curated databases (Fabregat et al., 2017, 2014; Kanehisa et al., 2017, 2016; Mi et al., 2010; Thomas et al., 2003) and tested respective pathway activation in AD in multiple -omic datasets, using gene expression and methylation of pathway components as a proxy for regulated pathway activation/suppression. All pathways selected have defined roles in either PCD or acute stress-activated survival—hereafter referred to simply as “death” and “survival” pathways, respectively. Additionally, senescence was included among the survival pathways because evidence suggests that a form of neuronal senescence occurs in AD (Baker and Petersen, 2018; Chinta et al., 2015; Tan et al., 2014) and activation of senescence in AD may be related to cellular stress (Baker and Petersen, 2018; Chinta et al., 2015; Rufini et al., 2013) but senescent pathway activation alone is insufficient to cause cell death (Galluzzi et al., 2018). The data tested included paired DNA methylation and gene expression data from the middle temporal gyrus from age-matched AD cases and non-diseased (ND) controls (Piras et al., 2019), gene expression from laser-captured micro-dissected (LCM) neurons from the middle temporal gyrus and hippocampus from AD and ND brains (Liang et al., 2007), and RNA sequence data from LCM hippocampal neurons and glia (Mastroeni et al., 2017). The integration of these data allows for an investigation of 1) the regulatory and comparative significance of DNA methylation to death and survival pathway activation in AD, 2) neuron-specific changes in death and survival pathways across brain regions, and 3) cell-type specific changes to the selected pathways in AD. Together, this analysis represents a robust endeavor to approximate pathway activation among multiple death and survival pathways, illuminating a complex balance between cell death and survival in AD.
2. Materials and Methods
2.1. Study Approval and Consent
Written informed consent for autopsy was obtained for all cases in compliance with institutional guidelines of Banner Sun Health Research Institute. Banner Sun Health Research Institute review board approved this study including recruitment, enrollment, and autopsy procedures. All donors and their respective next-of-kin consented to brain autopsy for the purpose of research analysis as participants in the Arizona Study of Aging and Neurodegenerative Disorders/Banner Sun Health Research Institute Brain and Body Donation program (Beach et al., 2015).
2.2. Sample Description
Banner Sun Health Research Institute Brain and Body Donation Program provided 404 samples from the middle temporal gyrus (area 21) consisting of 225 cases with Alzheimer’s disease (AD) and 179 non-demented (ND) cases. The tissue was collected within hours of death (average postmortem delay = 2.6 hours) and stored at −80 °C. The diagnosis of AD was based on clinical assessment and subsequent examination postmortem by a board-certified neuropathologist, with AD defined as “intermediate” or “high” classification with National Institute on Aging Reagan Institute criteria (Ball et al., 1997). More AD cases are female than male (55.1% compared to 42.5% of ND cases) and the mean age of the AD cases was 83.28 years +/− 8.00 (compared to 84.18 +/− 7.68 in ND). There was no significant difference in the age distribution (χ2 = 51.57, p-value = 0.148) between AD and ND samples.
2.3. Hypothesis Testing: Pathway and Gene Selection
Rather than conduct an unbiased analysis, only genes from pathways of interest with robust curated pathways were selected for analysis and compared among the datasets, allowing for a hypothesis-testing approach (Figure 1). Five established cell death (intrinsic apoptosis, extrinsic apoptosis, necroptosis, necrosis, and autophagy) (Denton et al., 2012; Elmore, 2007; Galluzzi et al., 2018; Green and Llambi, 2015; Linkermann and Green, 2014) and three survival pathways (PI3K/AKT, NF-κB, and JNK signaling axes) were identified from existing literature for their respective influence on cell death or survival in the event of acute stress (Brunet et al., 2001; Cardone et al., 1998; Heras-Sandoval et al., 2014; Kim and Choi, 2015; Lawrence, 2009; Morita et al., 2015; Pires et al., 2018; Wagner and Nebreda, 2009). Nine-hundred and twenty-eight genes were selected from 15 curated pathways representing eight pathways pooled from three databases (KEGG (Kanehisa et al., 2017, 2016), Reactome (Fabregat et al., 2014), and PANTHER (Mi et al., 2010; Thomas et al., 2003)). Minimal gene overlap is present between death and survival pathways (Supplemental Fig 3B) or amongst individual pathways (Supplemental Fig 3C–D), indicating that the selected pathways each represent a unique set of genes distinguishable from other pathways. An atlas of significant genes from pathways selected from this study, their pathway ID, and the database of origin can be found in Supplemental Table 1. These genes of interest were then analyzed in each of the datasets and only significantly differentially methylated or expressed genes (adjusted p-values < 0.05) were compared among the datasets.
Fig 1.
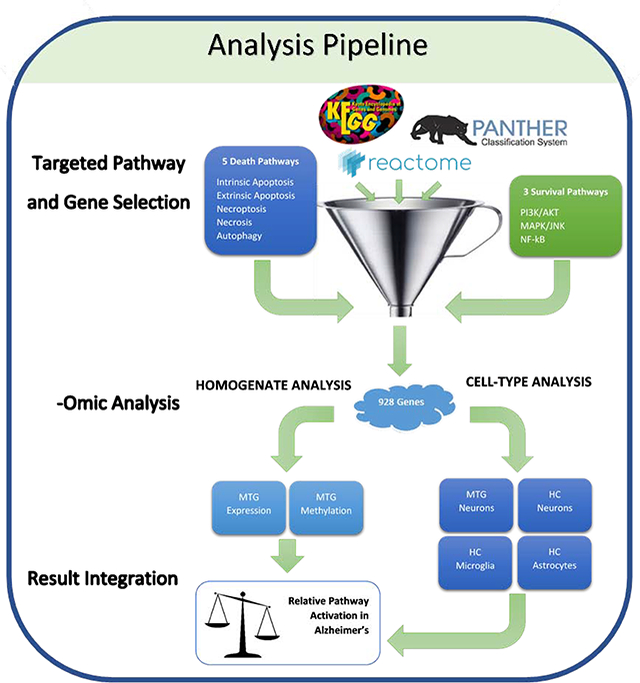
Hypothesis-Testing Analysis Pipeline. Curated genes from three databases (KEGG, Reactome, and PANTHER) were retrieved for eight pathways selected for their role in either promoting or hindering cell death. These unique 928 genes of interest were then analyzed in one of six datasets that can be divided into two wings: 1) analysis of homogenate expression and methylation data from the middle temporal gyrus (MTG) and, 2), cell type specific expression data from LCM cells from the MTG and hippocampus (HC). From these six datasets, the methylation/expression levels from only the significant genes of interest were compared between the datasets. Relative pathway activation in AD was inferred by the methylation/expression status of significant components contributed by each of the six datasets, allowing for a pathway-rather than gene-centric interpretation of the results.
2.4. Methylation Microarray Preparation
Approximately ~76 mg of MTG from each of 404 cases was treated with lysis buffer to decompose the tissue (100 mM Tris.HCl pH 8.5, 5mM EDTA, 0.2% SDS, 200 mM NaCl, 100 ug/ml Proteinase K (Sigma)) and incubated overnight at 55°C. A handheld pestle mixer (Kontes) was used periodically during incubation to further disassemble the tissue. The samples were treated with 4 uL of RNase A (Qiagen, 19101) at room temperature for 30 minutes. An equal volume of phenol/chloroform/isoamyl alcohol (Sigma, P3803) was added to each sample and samples were vortexed and subsequently rocked for five minutes. The samples were then centrifuged at room temperature for 10 minutes (10,000 RPM). The aqueous phase was collected and treated with ethanol overnight at −20°C. The precipitate was isolated and resuspended in 50 uL of TE buffer (pH 8.0) and stored at −20 °C. One microgram (1 ug) of extracted genomic DNA was treated with bisulfite. Bisulfite-treated DNA was then amplified using whole-genome amplification using a hexamer primer. Samples were randomly assorted among nine Illumina Infinium 450K arrays and annealed. The chips were read using the iScan System.
2.5. Methylation Microarray Normalization and Analysis
The raw light-intensity values were extracted from the .idat files using R (version 3.4.2) and normalized using the minfi (version 1.23.4) (Aryee et al., 2014) and sva (version 3.26.0) (Gagnon-Bartsch et al., 2013; Gagnon-Bartsch and Speed, 2012) packages. Bisulfite conversion rate was assessed per sample via the wateRmelon package (version 1.28.0) and the distribution of the bisulfite control probes were graphically assessed using minfi. A histogram of the sample bisulfite conversion rate is available in supplemental figures (Fig S1). Poorly performing probes (detection p-value > 0.05) and probes located on either sex chromosome were removed from the dataset (remaining samples = 404; remaining probes = 389,849. Methylation data from the nine arrays were combined and functional normalization, which was employed to remove unwanted variation using the Illumina 450K control probes, in concert with background normalization, performed using the NOOB algorithm (minfi) (Aryee et al., 2014). In addition to functional normalization, the ComBat algorithm was used to remove batch effects from the data (sva version: 3.30.1) (Gagnon-Bartsch and Speed, 2012). Differentially methylated positions (DMPs) and regions (DMRs) related to diagnosis (AD or ND) were identified using the minfi dmpfinder and bumphunter algorithms, respectively. Benjamini-Hochberg and q-values were used to filter significantly differentially methylated loci prior to being cross-referenced with genes of interest. All significant sites CpGs were cross-referenced with the potential confounding SNPs provided by Illumina. Rather than removing the CpGs from the analysis, significant probes with a confounding SNP within 10 base pairs of the CpG and a minor allele frequency greater than 0.05 were marked in the supplemental materials (Supplemental Table 2). Raw and normalized DNA methylation data have been deposited in the Gene Expression Omnibus (GEO) database (accession: GSE134379).
2.6. Acquisition and Analysis of Opensource Datasets
In addition to the methylation data, three additional expression datasets were retrieved either through GEO or the supplemental data of the associated publication. The accessory datasets include expression data from the middle temporal gyrus derived from a subset of the samples used in the methylation array (GSE132903) and LCM neurons from the same brain region (GSE5281) as well expression array data from multiple cell types laser-capture micro-dissected from the hippocampus (Mastroeni et al., 2017) (Table 1). No additional normalization or batch correction was performed after acquisition. Linear models were constructed in R with gene expression as a function of patient diagnosis (AD or ND) using the eBayes function from the limma package. In the case of the LCM hippocampus data, significant findings reported by the original authors were simply integrated into the analysis. All p-values were FDR-corrected and reported besides expression changes in supplemental data (Supplemental Tables 3–8).
Table 1. Data Source Summary.
All additional data sources used in the analysis can be accessed via public databases (GSE132903 and GSE5281) or through the original publication (Mastroeni et al., 2017).
| ID | Brain Region | Cell Type | Sample Size (AD/ND) | Platform | Citation |
|---|---|---|---|---|---|
| GSE132903 | Middle temporal gyrus | Homogenate | 97/98 | Illumina HumanHT-12 V4.0 Array | (Piras et al., 2019) |
| GSE5281 | Middle temporal gyrus and Hippocampus | Neurons | 10/13 16/12 | Affymetrix hgU133 Plus 2.0 Array | (Liang et al., 2007) |
| Hippocampus | Neurons, Astrocytes, Microglia | 6/6 | Illumina HiSeq 2000 | (Mastroeni et al., 2017) |
3. Results
3.1. DNA Methylation Changes in Death and Survival Pathways in AD Middle Temporal Gyrus
Of the 1112 unique genes (2190 Illumina 450K probes) associated with a statistically significantly differentially methylated CpG in the AD middle temporal gyrus (adjusted p-value < 0.05), 72 genes (97 probes) fell within at least one of the nine curated cell death and survival pathways selected for the study. Among the 72 significant differentially methylated genes from the curated pathways, there was no significant trend in methylation status (χ2 = 0.06; p-value = 0.8084). Further, there was no significant discrepancy between hypo- and hypermethylation between death and survival pathways among these genes (χ2 = 1.41; p-value = 0.2348). Most of the significantly differentially methylated genes are exclusively included in the selected survival—rather than death—pathways (survival = 70.6%; death = 20.6%; both = 8.8%). Aside from MAPK pathway, in which the majority of genes (60.0%) are hypermethylated, the remaining three survival pathways consist of mostly hypomethylated genes in AD (NF-κB = 75.0%; senescence = 71.4%; PI3K/AKT = 62.5%), suggesting the possibility that these genes have higher expression levels in AD (Fig 2A). Interestingly, the reverse is true for the majority of the programmed cell death genes. Aside from the necrotic and necroptotic pathways, in which all the significant genes are hypomethylated (necroptosis = 4; necrosis = 2), at least fifty percent of genes in the remaining pathways are hypermethylated (autophagic cell death = 80.0%; extrinsic apoptosis = 72.7%; intrinsic apoptosis = 50.0%), indicating potential suppression of these pathways in AD.
Fig 2.

Integration of Middle Temporal Gyrus Methylation and Expression Data. Relative methylation changes per pathway are represented in Figure 2A for all genes associated with a statistically significant differentially methylated CpG (FDR < 0.05). Hypomethylated genes (AD methylation < ND) are counted below the x-axis while hypermethylated genes (AD > ND) are reported above. Figure 2B is a scatterplot of 49 genes that are both significantly differentially methylated and expressed (FDR < 0.05) in the AD middle temporal gyrus. Genes are shaped and colored depending on pathway type (death = red circles; survival = blue squares; both = purple triangles). The relationship between methylation (hyper- or hypomethylated) and expression (increased or decreased) is significantly correlated for this subset of genes (Pearson’s r = −0.32, p-value = 0.0234). A table of congruent significant genes (hypermethylated and decreased expression or hypomethylated and increased expression) is provided in Figure 2C.
3.2. Correlated Gene Methylation and Expression in AD Middle Temporal Gyrus
Expression data were generated from the middle temporal gyrus of half of the same samples used in the methylation study using an Illumina HT12 V4 beadchip (Piras et al., 2019). Five- hundred and forty-five of the statistically significant differentially expressed genes intersected with at least one of the selected pathways used in this study. Of these, 58.0% and 42.0% displayed an increase and decrease, respectively, in expression in AD at statistically significant levels (χ2 = 14.24; p-value = 0.0002). The majority of the differentially expressed genes fell within the MAPK/ERK pathway (218 genes) followed by senescence (85), extrinsic apoptosis (56), and intrinsic apoptosis (55) (Fig 3A). In the majority of the cell death and survival pathways, excluding autophagic death and the MAPK/ERK pathway, the number of genes with elevated expression was higher than those with lower expression per pathway (Fig 3A). Both cell death and survival pathways were statistically significantly more likely to contain genes with heightened expression in AD (cell death pathways: χ2 = 12.97, p-value = 0.0003; survival pathways: χ2 = 6.05, p-value = 0.0139).
Fig. 3.

Gene Expression Changes in Middle Temporal Gyrus (MTG) and Hippocampus (HC) in AD. The bar plots represent number of genes with an increased (above the x-axis) or decreased (below) per pathway in AD. Results derived from the same study are encircled: MTG homogenate expression (GSEl32903) (Piras et al., 2019), LCM neuron expression from the MTG and HC (GSE5281) (Liang et al., 2007), and LCM glia and neuron expression from the HC(Mastroeni et al., 2017). Note that the significance cutoff for Figures 3A-C is FDR < 0.05 but, due to limited significant results in the HC glia and neurons, an unadjusted alpha of 0.05 was applied to genes in Figures 3D-F.
Because the expression data from the middle temporal gyrus were derived from a randomly selected subset of the samples used to generate the methylation data, it is particularly interesting to compare the two for the purpose of identifying genes that are both differentially methylated and expressed in AD (adjusted p-value < 0.05). Forty-nine genes (68.1% of the significant methylation and 8.4% of the significant expression results) from the selected pathways were both significantly differentially methylated and expressed in AD. Within these genes, there is a significant negative correlation between methylation and expression (Pearson r = −0.32, p-value = 0.0234). Approximately three quarters of the shared forty-nine genes had changes in expression that were consistent with the methylation profiles of the genes (hypomethylation and increased expression vs. hypermethylation in decreased expression) (Fig 2B). The majority of the congruent genes (hypomethylation and increased expression or hypermethylation and decreased expression) fall within one of the survival pathways (65.3%), particularly MAPK/ERK signaling (Fig 2C).
Because DNA methylation can influence gene regulation and expression, the consistency between methylation and expression in this subset of genes may be indicative of a consistent regulatory change in AD rather than stochastic dysregulation. Moreover, the preponderance of congruent survival genes (65.3%)—genes that are both significantly hypomethylated and have increased expression or hypermethylated and have decreased expression—between the middle temporal gyrus methylation and expression results may be suggestive of systematic regulatory changes of these pathways at the epigenomic level. Such genes include regulators of the NF-κB pathway activation, IKBKE and IFIH1, which are both hypomethylated/increased in AD as well as multiple receptors that promote the activation of MAPK/ERK and PI3K/AKT signaling (CACNA1G, CACNA2D1, RAC1, RASGRP1, SYNGAP1), all of which are hypermethylated/decreased in AD. Although a smaller proportion of the congruent genes are involved in death pathways, those that are (hypo/inc: STAT3; hyper/dec.: MADD, PRKCB, PRKCD) are potent regulators of cell death. Together, these results are indicative of a systematic and regulated changes in activity of death and survival pathways in AD.
3.3. Expression Profiles from LCM Neurons Mirror Homogenate Data
To further isolate the neuron-specific changes in AD, gene expression data from LCM neurons from the middle temporal gyrus and hippocampus were acquired from GEO (accession = GSE5281) (Liang et al., 2007) and analyzed for AD-related expression changes. In total, 284 (increased expression in MTG = 32.7%) and 150 (increased expression in HC = 62.7%) genes in LCM neurons from middle temporal gyrus and hippocampus, respectively, were significantly differentially expressed (FDR < 0.05) in AD and fell within one of the selected pathways. Except for the necrosis pathway, most of the genes in selected pathways were downregulated in AD middle temporal gyrus neurons (Fig 3B), mirroring the expression changes in survival pathway genes observed in middle temporal gyrus homogenate data (Fig 3A). In contrast, all the pathways in the LCM neuron hippocampus data have more upregulated genes than not (Fig 3C). Ninety-three genes were significantly differentially expressed in both the hippocampus and middle temporal gyrus neurons and, of these, the expression of 75% were significantly positively correlated among brain regions (Pearson r = 0.59, p-value = 3.818E-10). Senescence (15 genes) and intrinsic apoptosis (7 genes) were enriched among the shared upregulated genes while MAPK genes (25 genes) were enriched in the shared downregulated genes.
One hundred and eighty-seven significantly differentially expressed genes (p-adjusted value < 0.05) were shared between the middle temporal gyrus homogenate and LCM middle temporal gyrus neuron data. The log2-transformed fold change among the shared genes was significantly positively correlated (Pearson’s r = 0.6030; p-value < 2.2E-16) and over three quarters of the genes had consistent directional change in expression between the two datasets (increased expression: 53; decreased: 91) (Fig 4A). Among the shared genes, expression changes were significantly related to pathway type (survival, death, or both) (χ2 = 13.00; p-value =0.0015). Forty-nine percent of shared upregulated genes are involved in survival pathways (χ2 = programmed cell death pathways (χ2 = 40.07; p-value = 1.995E-09), illustrating possible survival pathway activation and death pathway transcriptomic suppression middle temporal gyrus neurons in AD. Notably, several regulators of cell cycle and senescence are included among the upregulated genes (ANAPC16, ETS1, CCND1, CMGC, CDKN2C, FZR1) in addition to CFLAR, whose gene product has been implicated in directly preventing caspase-mediated apoptosis (Fig 4B). Likewise, several cell death genes—particularly those involved in the regulation and initiation of intrinsic apoptosis—are decreased in AD neurons (BCL2L2, BCL2L10, CDKN2A, PRKCB, PRKCE, YWHAZ, and YWHAH) (Fig 4B). These data are indicative that the activation of survival and inhibition of death pathways in the middle temporal gyrus homogenate results have, to some extent, a neuronal source.
Fig 4.

Integration of Expression Data from the Hippocampus and Middle Temporal Gyrus. The log2 transformed fold-change (log2FC) of shared significant genes (adjusted p-value < 0.05) between MTG homogenate (GSE132903) and LCM MTG neurons (GSE5281) are significantly positively correlated (Pearson’s r = 0.6030; p-value < 2.2E-16) (Fig 4A). Genes with positively correlated expression (Q1 = increased and Q3 = decreased) are represented as word clouds in Figure 4B. Word color corresponds to gene pathway type (survival = blue, death = read, both = purple) and size is the product of the log2 fold change in between the datasets. Data are reported similarly for LCM MTG and HC neurons (GSE5281) (Fig 4C-D).
3.4. Death and Survival Pathways in LCM Neurons and Glia
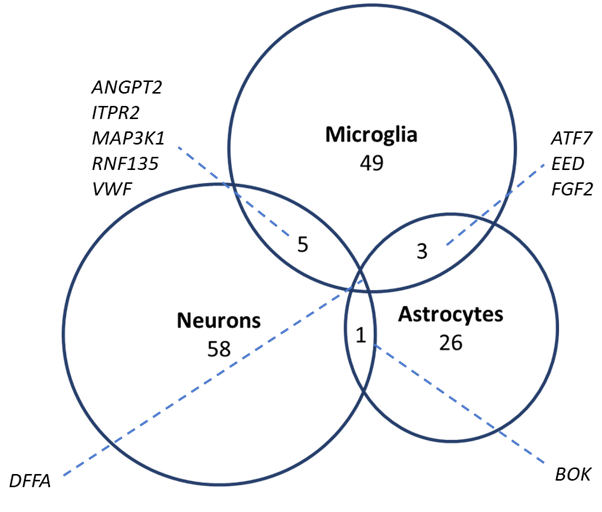
The middle temporal gyrus homogenate data represents an amalgam of expression profiles from multiple cell types besides neurons. In order to approximate the contribution of nonneuronal cells to the effect on cell death and survival pathway expression in the middle temporal gyrus homogenate data, genes from the selected pathways were cross-referenced with the significant gene lists from hippocampal neurons, astrocytes, and microglia RNA sequencing data (Mastroeni et al., 2017). Because few significant genes were identified (microglia: SRC; neurons: MCM5 and IFIH1), the enrichment analysis was expanded to include nominal genes (unadjusted p-value < 0.05). Across all three cell types, the MAPK pathway had the highest level of nominally differentially number of genes (neurons: 26, astrocytes: 10, microglia: 26) (Fig 3D–F). Parallel to the middle temporal gyrus dataset, the death pathway genes were mostly downregulated in neurons and astrocytes (Fig 3E–F). Likewise, the trend of increased expression in survival pathway gene expression in all three cell types mirrored the middle temporal gyrus homogenate data. Similarly, there was minimal overlap among the nominally differentially expressed AD genes in the hippocampal neuron and glial data (Fig S5). Only one gene, DFFA, was differentially expressed in all three cell types (HC neurons logFC: −2.16; HC microglia log2FC: −2.63; HC astrocytes log2FC: 4.33). Despite, the lack of overlap among the hippocampal datasets, a large proportion of differentially expressed genes in the hippocampus overlap with the significant results from the middle temporal gyrus homogenate data. Overall, 48.3%, 41.5%, and 35.5% of the nominally differentially expressed genes from the hippocampal microglia, neurons, and astrocytes, respectively, were significantly differentially expressed in the middle temporal gyrus homogenate expression data. In all three cell types, most genes that intersect the middle temporal gyrus homogenate data are within the MAPK pathway (microglia: 10 genes; neurons: 8; astrocytes: 5). However, in both microglia and neurons, the senescence pathway was also enriched (13.3% and 21.4% of nominal genes overlapping with the middle temporal gyrus in microglia and neurons, respectively).
4. Discussion
Many death pathways have been suggested to be responsible for neuron death in AD, including apoptosis, autophagy, and necroptosis (Albrecht et al., 2009; Caccamo et al., 2017; Ofengeim et al., 2017; Piras et al., 2016; Selznick et al., 1999; Silva et al., 2014; Stadelmann et al., 1999; Su et al., 2001; Theofilas et al., 2018; Troncoso et al., 1996). Of the curated cell death pathways selected for this study, intrinsic and extrinsic apoptosis combined had the highest number of significant genes across all the datasets (Figs 2 and 3). These genes include essential pro-apoptotic genes such as CASP8 and BID, which are both hypomethylated and have higher expression in AD cases in the middle temporal gyrus homogenate data (Supplemental Table 3). Multiple caspases (CASP2, CASP3, CASP6, and CASP7), members of the Bcl2 gene family (BBC3 and BAK1), and p53 associated genes (TP53AP1 and TPF3BP2) were expressed at significantly higher levels in the AD middle temporal gyrus. Additionally, other pro-apoptotic genes (APAF1, APIP, ATF4, DDIT3, DFFB, GZMB, HRK) were expressed at significantly elevated in AD middle temporal gyrus while multiple anti-apoptotic genes have significantly reduced expression in the AD (BAG1, BCL2L10, BCL2L2, PRKCE, YWAHB, YWHAE, YWHAG, YWHAH). The single neuron expression profiles from both the hippocampus and the middle temporal gyrus are reflective of similar changes in expression among apoptotic genes (Supplemental Tables 4–5). Together, these results are suggestive of an upregulation of apoptotic pathways in AD neurons. In contrast, although some autophagic (ATG3, ATG101, ATG12, ATG16L2, MLST8, RHEB, STK11, SQSTM1) and necroptotic (RIPK1 and MLKL) genes were also upregulated in AD, far fewer were significant than in the apoptotic pathways (Figs 2 and 3), suggesting that apoptosis may be more dominant in AD neurons than the other forms of cell death examined in this study. This does not exclude an assistive role for autophagy which has been previously demonstrated to activate and cooperate with apoptosis (Denton et al., 2012; Galluzzi et al., 2017; Green and Llambi, 2015)—although this is not necessarily the case for necroptosis. The inhibitory, mutually exclusive relationship between apoptosis and necroptosis (Galluzzi et al., 2018) and the relatively small number of significant pro-necroptotic genes found in the analysis suggests that in AD necroptosis accounts for a smaller percentage of cell death than apoptosis.
Survival pathways also display patterns of DNA methylation and gene expression that suggest activation in AD. In particular, the MAPK pathway had the largest number of significant genes across all the datasets (Figs 2 and 3). In the middle temporal gyrus homogenate data, multiple MAP kinases (MAPK1, MAPK3, MAPK7, MAPK10, MAPK14) and MAPK-activated kinases (MAPKAP2 and MAP3K11) are present at significantly higher levels in AD in addition to a number of genes that promote MAPK pathway activity, including ARRB1, ARRB2, BRAF, ERRB2–4, KSR2, RAF1, RASA1–3, and WDR83 (Supplemental Table 3). Additionally, essential genes in the PI3K/AKT and NF-κB are also upregulated, suggesting possible activation of these pathways in AD. Multiple subunits of the PI3K/AKT complex (PI3KAP1, PIK3CA, PIK3CD, PIK3CG, PIK3R1, PIK3R2) are expressed at significantly higher levels in the middle temporal gyrus homogenate data in addition to the NF-κB activator IFIH1, which is also hypomethylated, and two subunits of the NF-κB complex (NFKB1 and NFKBIA). Increased expression of activators of the MAPK, PI3K/AKT, and NF-κB pathways are reflected in the LCM neurons from the middle temporal gyrus and, to a lesser extent, hippocampus (Supplemental Tables 4–5). Senescence, which was included among the survival pathways because it is activated in response to stress but does not necessarily result in cell death, had one of the highest proportions of genes with increased expression across the examined datasets (Fig 3). Multiple genes that regulate the cell cycle or promote senescence were hypomethylated in AD in the middle temporal gyrus (CDK2, CDKN1B, SERPINE1) or demonstrated higher expression in AD across multiple datasets (middle temporal gyrus homogenate: ANAPC4, ANAPC11, ANAPC16, CCND1, CCND3, CCNE, CDC16, CDC25A, CDC27, CDK2, CDK4, CDK6, CDKN1, CDKN2B, CDKN2C, CHEK2, FZR1, RAD9A, SERPINE1; middle temporal gyrus neurons: ANAPC5, ANAPC16, CCNA1, CCNB2, CDK1, CDK6, CDKN2A; hippocampal neurons: ANAPC5, CDC25A, CDK6), supporting the hypothesis of possible cell cycle reentry and a senescent-like state in AD neurons (Baker and Petersen, 2018; Busser et al., 1988; Chinta et al., 2015; Tan et al., 2014; Yang et al., 2003). That said, most of the datasets examined also had a sizeable number of significant genes with lower expression from all four pathways (Fig 3), indicating the activation of these pathways is not ubiquitous or absolute. Instead, combined with the upregulated cell death genes explored in the previous section, these findings suggest a dynamic interplay between cell survival and death pathway activation within the AD brain.
The high-throughput integrative nature of this study that lent power to simultaneously evaluate nearly a dozen pathways across multiple datasets has natural limitations. All of the data employed were derived from either homogenates (Piras et al., 2019) or LCM cells that were pooled per sample prior to array preparation (Liang et al., 2007) or sequencing (Mastroeni et al., 2017). Thus, it is unclear whether the tension between cell death and survival pathways indicated by the data is the result of a balance occurring within individual cells or among a population of cells each in a different stage of disease progression. It is indisputably the case that different cell types have distinct roles in AD progression and, to some extent, this is reflected in the differential expression patterns of nominal genes in the LCM neurons and glia (Fig 3 D–F). However, the minimal number of significant genes, which was likely a result of limited sample size (Mastroeni et al., 2017), made it difficult to assess this effect. Likewise, the lack of significant genes in the LCM hippocampal neuron sequencing data as well as platform differences are likely responsible for the lack of overlap between the hippocampal neuron datasets (Supplemental Figure 4). Finally, all the data are reflective of a static snapshots of gene methylation and expression. Thus, it is ill-equipped to probe the dynamism of and interactions among these pathways or investigate trends in pathway activity over time. For instance, when activated under acute stress, both NF-κB and MAPK have a positive effect on cell survival while chronic activation over an extended period of time of these pathways can result in cell death (Bours et al., 2000; Lawrence, 2009; Wagner and Nebreda, 2009)—a transition that this study is ill-equipped to delineate. A complete understanding of the interaction and contribution to AD progression of the pathways examined in this study can only result from rigorous experimentation.
This study presents a unique hypothesis-testing approach of a selected set of curated pathways—rather than a global inventory of significant genes—performed on multiple homogenate and cell type specific datasets. The results, although not conclusive, suggest a complex balance between survival and death pathway activity among populations of cells and, perhaps, within a single cell in AD (Fig 5). Further laboratory work is necessary to delineate the extent of pathway activation in AD, define potential transitional points in pathway activity during disease progression, and determine whether survival and death pathways are coactivated in individual cells in AD. We believe our findings provide a useful framework for future work that, instead of looking at singularly at one pathway, addresses global interactions among multiple pathways on a cellular and tissue level, leading to a comprehensive molecular understanding of the balance between cell death and survival in AD.
Fig 5.

Balance Between Cell Death and Survival Pathway Activation in AD Neurons. Arrows represent the relative contribution of pathway upregulation (represented by green arrows above the x-axis) and downregulation (red arrows below the x-axis) in AD neurons. The size of arrows is proportional to the average pathway contribution to total significant genes from the middle temporal gyrus homogenate methylation and expression results (Sections 3.1 and 3.2) in conjunction with those from LCM neurons (Section 3.3). On a cellular level, we hypothesize a transition from elevated survival to death pathway activity in AD neurons as the disease progresses—especially as the MAPK and NF-κβ pathways that, under acute stress conditions promote cell survival, are activated chronically and begin to favor neuron death.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Five PCD pathways contain upregulated genes in Alzheimer’s disease (AD) across multiple datasets
Of these pathways, apoptosis has the largest proportion of upregulated genes
Pathways attributed to cell survival also contain genes that are upregulated in AD
These results suggest a balance between cell death and survival pathway activity in AD
Acknowledgements
The work reported here was supported by National Institute on Aging grant AG R01 036400 to PDC and Medical Research Council NIRG-15-321390 to DM. Additionally, we are grateful to the Banner Sun Health Research Institute Brain and Body Donation Program of Sun City, Arizona for the provision of human brain tissue. The Brain and Body Donation Program has been supported by the National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinson’s Disease and Related Disorders), the National Institute on Aging (P30 AG19610 Arizona Alzheimer’s Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer’s Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05-901 and 1001 to the Arizona Parkinson’s Disease Consortium) and the Michael J. Fox Foundation for Parkinson’s Research.
Footnotes
Disclosure Statement
The authors declare that they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albrecht S, Bogdanovic N, Ghetti B, Winblad B, LeBlanc AC, 2009. Caspase-6 activation in familial alzheimer disease brains carrying amyloid precursor protein or presenilin i or presenilin II mutations. J. Neuropathol. Exp. Neurol 68, 1282–1293. 10.1097/NEN.0b013e3181c1da10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade-Moraes CH, Oliveira-Pinto AV, Castro-Fonseca E, da Silva CG, Guimaraes DM, Szczupak D, Parente-Bruno DR, Carvalho LRB, Polichiso L, Gomes BV, Oliveira LM, Rodriguez RD, Leite REP, Ferretti-Rebustini REL, Jacob-Filho W, Pasqualucci CA, Grinberg LT, Lent R, 2013. Cell number changes in Alzheimer’s disease relate to dementia, not to plaques and tangles. Brain 136, 3738–3752. 10.1093/brain/awt273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA, 2014. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369. 10.1093/bioinformatics/btu049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Petersen RC, 2018. Cellular senescence in brain aging and neurodegenerative diseases: evidence and perspectives. J. Clin. Invest 128, 1208–1216. 10.1172/JCI95145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball M, Braak H, Goethe JW, Coleman P, Dickson D, Gambetti P, Hansen L, Hyman B, Jellinger K, Perl D, Powers J, Price J, Trojanowski JQ, Wisniewski H, Phelps C, Khachaturian Z, 1997. Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Neurobiol. Aging 18, S1–2. [PubMed] [Google Scholar]
- Beach TG, Adler CH, Sue LI, Serrano G, Shill HA, Walker DG, Lue L, Roher AE, Dugger BN, Maarouf C, Birdsill AC, Intorcia A, Saxon-Labelle M, Pullen J, Scroggins A, Filon J, Scott S, Hoffman B, Garcia A, Caviness JN, Hentz JG, Driver-Dunckley E, Jacobson SA, Davis KJ, Belden CM, Long KE, Malek-Ahmadi M, Powell JJ, Gale LD, Nicholson LR, Caselli RJ, Woodruff BK, Rapscak SZ, Ahern GL, Shi J, Burke AD, Reiman EM, Sabbagh MN, 2015. Arizona Study of Aging and Neurodegenerative Disorders and Brain and Body Donation Program. Neuropathology 35, 354–389. 10.1111/neup.12189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierer LM, Hof PR, Purohit DP, Carlin L, Schmeidler J, Davis KL, Perl DP, 1995. Neocortical Neurofibrillary Tangles Correlate With Dementia Severity in Alzheimer’s Disease. Arch. Neurol 52, 81–88. 10.1001/archneur.1995.00540250089017 [DOI] [PubMed] [Google Scholar]
- Blennow K, Bogdanovic N, Alafuzoff I, Ekman R, Davidsson P, 1996. Synaptic pathology in Alzheimer’s disease: relation to severity of dementia, but not to senile plaques, neurofibrillary tangles, or the ApoE4 allele. J. Neural Transm 103, 603–618. 10.1007/BF01273157 [DOI] [PubMed] [Google Scholar]
- Bloss CS, Delis DC, Salmon DP, Bondi MW, 2008. Decreased cognition in children with risk factors for Alzheimer’s disease. Biol. Psychiatry 64, 904–906. 10.1016/j.biopsych.2008.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bours V, Bentires-Alj M, Hellin A-C, Viatour P, Robe P, Delhalle S, Benoit V, Merville M-P, 2000. Nuclear factor-KB, cancer, and apoptosis. Biochem. Pharmacol 60, 1085–1089. [DOI] [PubMed] [Google Scholar]
- Brunet A, Datta SR, Greenberg ME, 2001. Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr. Opin. Neurobiol 11, 297–305. [DOI] [PubMed] [Google Scholar]
- Burguillos MA, Deierborg T, Kavanagh E, Persson A, Hajji N, Garcia-Quintanilla A, Cano J, Brundin P, Englund E, Venero JL, Joseph B, 2011. Caspase signalling controls microglia activation and neurotoxicity. Nature 472, 319–324. 10.1038/nature09788 [DOI] [PubMed] [Google Scholar]
- Busser J, Geldmacher DS, Herrup K, 1988. Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer’s disease brain. J. Neurosci 18, 2801–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccamo A, Branca C, Piras IS, Ferreira E, Huentelman MJ, Liang WS, Readhead B, Dudley JT, Spangenberg EE, Green KN, Belfiore R, Winslow W, Oddo S, 2017. Necroptosis activation in Alzheimer’s disease. Nat. Neurosci 20, 1236–1246. 10.1038/nn.4608 [DOI] [PubMed] [Google Scholar]
- Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S, Reed JC, 1998. Regulation of cell death protease caspase-9 by phosphorylation. Science 282, 1318–1321. 10.1126/science.282.5392.1318 [DOI] [PubMed] [Google Scholar]
- Chami L, Buggia-Prevot V, Duplan E, Delprete D, Chami M, Peyron J-F, Checler F, 2012. Nuclear Factor-κβ Regulates βAPP and β- and γ-Secretases Differently at Physiological and Supraphysiological Aβ Concentrations. J. Biol. Chem 287, 24573–24584. 10.1074/jbc.M111.333054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan D, Fox NC, Jenkins R, Scahill RI, Crum WR, Rossor MN, 2001. Rates of global and regional cerebral atrophy in AD and frontotemporal dementia. Neurology 57, 1756–1763. 10.1212/wnl.57.10.1756 [DOI] [PubMed] [Google Scholar]
- Chinta SJ, Woods G, Rane A, Demaria M, Campisi J, Andersen JK, 2015. Cellular senescence and the aging brain. Exp. Gerontol 68, 3–7. https://doi.Org/10.1016/j.exger.2014.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton D, Nicolson S, Kumar S, 2012. Cell death by autophagy: facts and apparent artefacts. Cell Death Differ. 19, 87–95. 10.1038/cdd.2011.146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore S, 2007. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol 35, 495–516. 10.1080/01926230701320337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabregat A, Sidiropoulos K, Garapati P, Gillespie M, Hausmann K, Haw R, Jassal B, Jupe S, Korninger F, McKay S, Matthews L, May B, Milacic M, Rothfels K, Shamovsky V, Webber M, Weiser J, Williams M, Wu G, Stein L, Hermjakob H, D’Eustachio P, 2017. The reactome pathway knowledgebase. Nucleic Acids Res. 46 10.1093/nar/gkv1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabregat A, Sidiropoulos K, Garapati P, Gillespie M, Hausmann K, Haw R, Jassal B, Jupe S, Korninger F, McKay S, Matthews L, May B, Milacic M, Rothfels K, Shamovsky V, Webber M, Weiser J, Williams M, Wu G, Stein L, Hermjakob H, D’Eustachio P, 2014. The reactome pathway knowledgebase. Nucleic Acids Res. 42, D472–D477. 10.1093/nar/gkv1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forner S, Baglietto-Vargas D, Martini AC, Trujillo-Estrada L, LaFerla FM, 2017. Synaptic Impairment in Alzheimer’s Disease: A Dysregulated Symphony. Trends Neurosci. 40, 347–357. 10.1016/j.tins.2017.04.002 [DOI] [PubMed] [Google Scholar]
- Fulda S, Debatin KM, 2006. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 25, 4798–4811. [DOI] [PubMed] [Google Scholar]
- Gagnon-Bartsch JA, Jacob L, Speed TP, 2013. Removing Unwanted Variation from High Dimensional Data with Negative Controls. Tech. Reports from Dep. Stat. Univ. California, Berkeley: 1–112. [Google Scholar]
- Gagnon-Bartsch JA, Speed TP, 2012. Using control genes to correct for unwanted variation in microarray data. Biostatistics 13, 539–552. 10.1093/biostatistics/kxr034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, Choi AM, Chu CT, Codogno P, Colombo MI, Cuervo AM, Debnath J, Deretic V, Dikic I, Eskelinen E-L, Fimia GM, Fulda S, Gewirtz DA, Green DR, Hansen M, Harper JW, Jaattela M, Johansen T, Juhasz G, Kimmelman AC, Kraft C, Ktistakis NT, Kumar S, Levine B, Lopez-Otin C, Madeo F, Martens S, Martinez J, Melendez A, Mizushima N, Munz C, Murphy LO, Penninger JM, Piacentini M, Reggiori F, Rubinsztein DC, Ryan KM, Santambrogio L, Scorrano L, Simon AK, Simon H-U, Simonsen A, Tavernarakis N, Tooze SA, Yoshimori T, Yuan J, Yue Z, Zhong Q, Kroemer G, 2017. Molecular definitions of autophagy and related processes. EMBO J. 36, 1811–1836. 10.15252/embj.201796697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, Annicchiarico-Petruzzelli M, Antonov AV, Arama E, Baehrecke EH, Barlev NA, Bazan NG, Bernassola F, Bertrand MJM, Bianchi K, Blagosklonny MV, Blomgren K, Borner C, Boya P, Brenner C, Campanella M, Candi E, Carmona-Gutierrez D, Cecconi F, Chan FK-M, Chandel NS, Cheng EH, Chipuk JE, Cidlowski JA, Ciechanover A, Cohen GM, Conrad M, Cubillos-Ruiz JR, Czabotar PE, D’Angiolella V, Dawson TM, Dawson VL, De Laurenzi V, De Maria R, Debatin K-M, DeBerardinis RJ, Deshmukh M, Di Daniele N, Di Virgilio F, Dixit VM, Dixon SJ, Duckett CS, Dynlacht BD, El-Deiry WS, Elrod JW, Fimia GM, Fulda S, García-Sáez AJ, Garg AD, Garrido C, Gavathiotis E, Golstein P, Gottlieb E, Green DR, Greene LA, Gronemeyer H, Gross A, Hajnoczky G, Hardwick JM, Harris IS, Hengartner MO, Hetz C, Ichijo H, Jäättelä M, Joseph B, Jost PJ, Juin PP, Kaiser WJ, Karin M, Kaufmann T, Kepp O, Kimchi A, Kitsis RN, Klionsky DJ, Knight RA, Kumar S, Lee SW, Lemasters JJ, Levine B, Linkermann A, Lipton SA, Lockshin RA, López-Otín C, Lowe SW, Luedde T, Lugli E, MacFarlane M, Madeo F, Malewicz M, Malorni W, Manic G, Marine J-C, Martin SJ, Martinou J-C, Medema JP, Mehlen P, Meier P, Melino S, Miao EA, Molkentin JD, Moll UM, Muñoz-Pinedo C, Nagata S, Nuñez G, Oberst A, Oren M, Overholtzer M, Pagano M, Panaretakis T, Pasparakis M, Penninger JM, Pereira DM, Pervaiz S, Peter ME, Piacentini M, Pinton P, Prehn JHM, Puthalakath H, Rabinovich GA, Rehm M, Rizzuto R, Rodrigues CMP, Rubinsztein DC, Rudel T, Ryan KM, Sayan E, Scorrano L, Shao F, Shi Y, Silke J, Simon H-U, Sistigu A, Stockwell BR, Strasser A, Szabadkai G, Tait SWG, Tang D, Tavernarakis N, Thorburn A, Tsujimoto Y, Turk B, Vanden Berghe T, Vandenabeele P, Vander Heiden MG, Villunger A, Virgin HW, Vousden KH, Vucic D, Wagner EF, Walczak H, Wallach D, Wang Y, Wells JA, Wood W, Yuan J, Zakeri Z, Zhivotovsky B, Zitvogel L, Melino G, Kroemer G, 2018. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 25, 486–541. 10.1038/s41418-017-0012-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Llambi F, 2015. Cell death signaling. Cold Spring Harb. Perspect. Biol 7 10.1101/cshperspect.a006080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin RJ, Moloney A, Kelliher M, Johnston JA, Ravid R, Dockery P, O’Connor R, O’Neill C, 2005. Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer’s disease pathology. J. Neurochem 93, 105–117. 10.1111/j.1471-4159.2004.02949.x [DOI] [PubMed] [Google Scholar]
- Han X-J, Hu Y-Y, Yang Z-J, Jiang L-P, Shi S-L, Li Y-R, Guo M-Y, Wu H-L, Wan Y-Y, 2017. Amyloid β−42 induces neuronal apoptosis by targeting mitochondria. Mol. Med. Rep 16, 4521–4528. 10.3892/mmr.2017.7203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heras-Sandoval D, Pérez-Rojas JM, Hernández-Damián J, Pedraza-Chaverri J, 2014. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell. Signal 26, 2694–2701. [DOI] [PubMed] [Google Scholar]
- Kadowaki H, Nishitoh H, Urano F, Sadamitsu C, Matsuzawa A, Takeda K, Masutani H, Yodoi J, Urano Y, Nagano T, Ichijo H, 2005. Amyloid beta induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ. 12, 19–24. 10.1038/sj.cdd.4401528 [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K, 2017. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, SAto Y, Kawashima M, Furumichi M, Tanabe M, 2016. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EK, Choi E-J, 2015. Compromised MAPK signaling in human diseases: an update. Arch. Toxicol 89, 867–882. 10.1007/s00204-015-1472-2 [DOI] [PubMed] [Google Scholar]
- Lawrence T, 2009. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol 1, 1–11. 10.1101/cshperspect.a001651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao Y-S, Damko E, Moquin D, Walz T, McDermott A, Chan FK-M, Wu H, 2012. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150, 339–350. 10.1016/j.cell.2012.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang WS, Dunckley T, Beach TG, Grover A, Mastroeni D, Walker DG, Caselli RJ, Kukull WA, McKeel D, Morris JC, Hulette C, Schmechel D, Alexander GE, Reiman EM, Rogers J, Stephan DA, 2007. Gene expression profiles in anatomically and functionally distinct regions of the normal aged human brain. Physiol. Genomics 28, 311–322. 10.1152/physiolgenomics.00208.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkermann A, Green DR, 2014. Necroptosis. N. Engl. J. Med 370, 455–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Hansen L, DeTeresa R, Alford M, Terry R, 1994. Synaptic and neuritic alterations during the progression of Alzheimer’s disease. Neurosci. Lett 174, 67–72. 10.1016/0304-3940(94)90121-x [DOI] [PubMed] [Google Scholar]
- Mastroeni D, Sekar S, Nolz J, Delvaux E, Lunnon K, Mill J, Liang WS, Coleman PD, 2017. ANK1 is upregulated in laser captured microglia in Alzheimer’s brain; the importance of addressing cellular heterogeneity. PLoS One 12, 1–11. 10.1371/journal.pone.0177814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Ramasamy K, Ingelsson M, Fukumoto H, Conrad C, Frosch MP, Irizarry MC, Yuan J, Hyman BT, 2006. Coordinated expression of caspase 8, 3 and 7 mRNA in temporal cortex of Alzheimer disease: relationship to formic acid extractable abeta42 levels. J. Neuropathol. Exp. Neurol 65, 508–515. 10.1097/01.jnen.0000229238.05748.12 [DOI] [PubMed] [Google Scholar]
- Mayeux R, Stern Y, 2012. Epidemiology of Alzheimer Disease. Cold Spring Harb. Perspect. Med 2, a006239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Dong Q, Muruganujan A, Gaudet P, Lewis S, Thomas PD, 2010. PANTHER version 7: improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Res. 38, D204–D210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita M, Gravel S-P, Hulea L, Larsson O, Pollak M, St-Pierre J, Topisirovic I, 2015. mTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle 14, 473–480. 10.4161/15384101.2014.991572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morsch R, Simon W, Coleman PD, 1999. Neurons may live for decades with neurofibrillary tangles. J. Neuropathoogy Exp. Neurol 58, 188–197. [DOI] [PubMed] [Google Scholar]
- Nieweg K, Andreyeva A, van Stegen B, Tanriöver G, Gottmann K, 2015. Alzheimer’s disease-related amyloid-β induces synaptotoxicity in human iPS cell-derived neurons. Cell Death Dis. 6, e1709–e1709. 10.1038/cddis.2015.72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N, 2013. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 1833, 3448–3459. https://doi.Org/10.1016/j.bbamcr.2013.06.001 [DOI] [PubMed] [Google Scholar]
- Ofengeim D, Mazzitelli S, Ito Y, DeWitt JP, Mifflin L, Zou C, Das S, Adiconis X, Chen H, Zhu H, Kelliher MA, Levin JZ, Yuan J, 2017. RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc. Natl. Acad. Sci 114, E8788–E8797. 10.1073/PNAS.1714175114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okonkwo OC, Xu G, Dowling NM, Bendlin BB, Larue A, Hermann BP, Koscik R, Jonaitis E, Rowley HA, Carlsson CM, Asthana S, Sager MA, Johnson SC, 2012. Family history of Alzheimer disease predicts hippocampal atrophy in healthy middle-aged adults. Neurology 78, 1769–1776. 10.1212/WNL.0b013e3182583047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piras A, Collin L, Grüninger F, Graff C, Rönnbäck A, 2016. Autophagic and lysosomal defects in human tauopathies: analysis of post-mortem brain from patients with familial Alzheimer disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol. Commun 4 10.1186/s40478-016-0292-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piras IS, Krate J, Delvaux E, Nolz J, Mastroeni DF, Persico AM, Jepsen WM, Beach TG, Huentelman MJ, Coleman PD, 2019. Transcriptome Changes in the Alzheimer’s Disease Middle Temporal Gyrus: Importance of RNA Metabolism and Mitochondria-Associated Membrane Genes. J. Alzheimers. Dis 70, 691–713. 10.3233/JAD-181113 [DOI] [PubMed] [Google Scholar]
- Pires BRB, Silva RCMC, Ferreira GM, Abdelhay E, 2018. NF-kappaB: Two Sides of the Same Coin. Genes (Basel). 9 10.3390/genes9010024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohn TT, Rissman RA, Davis MC, Kim YE, Cotman CW, Head E, 2002. Caspase-9 activation and caspase cleavage of tau in the Alzheimer’s disease brain. Neurobiol. Dis 11, 341–354. 10.1006/nbdi.2002.0549 [DOI] [PubMed] [Google Scholar]
- Rufini A, Tucci P, Celardo I, Melino G, 2013. Senescence and aging: the critical roles of p53. Oncogene 32, 5129–5143. 10.1038/onc.2012.640 [DOI] [PubMed] [Google Scholar]
- Scheff SW, DeKosky ST, Price DA, 1990. Quantitative assessment of cortical synaptic density in Alzheimer’s disease. Neurobiol. Aging 11, 29–37. 10.1016/0197-4580(90)90059-9 [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA, 1993. Synapse loss in the temporal lobe in Alzheimer’s disease. Ann. Neurol 33, 190–199. 10.1002/ana.410330209 [DOI] [PubMed] [Google Scholar]
- Schuff N, Tosun D, Insel PS, Chiang GC, Truran D, Aisen PS, Jack CR Jr, Weiner MW, Initiative ADN, 2012. Nonlinear time course of brain volume loss in cognitively normal and impaired elders. Neurobiol. Aging 33, 845–855. 10.1016/j.neurobiolaging.2010.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selznick LA, Holtzman DM, Han BH, Gokden M, Srinivasan AN, Johnson EMJ, Roth KA, 1999. In situ immunodetection of neuronal caspase-3 activation in Alzheimer disease. J. Neuropathol. Exp. Neurol 58, 1020–1026. 10.1097/00005072-199909000-00012 [DOI] [PubMed] [Google Scholar]
- Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT, 2011. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med 1, a006189 10.1101/cshperspect.a006189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z-M, Han Y-W, Han X-H, Zhang K, Chang Y-N, Hu Z-M, Qi H-X, Ting C, Zhen Z, Hong W, 2016. Upstream regulators and downstream effectors of NF-κB in Alzheimer’s disease. J. Neurol. Sci 366, 127–134. [DOI] [PubMed] [Google Scholar]
- Silva ART, Santos ACF, Farfel JM, Grinberg LT, Ferretti REL, Campos AHJFM, Cunha IW, Begnami MD, Rocha RM, Carraro DM, de Braganca Pereira CA, Jacob-Filho W, Brentani H, 2014. Repair of oxidative DNA damage, cell-cycle regulation and neuronal death may influence the clinical manifestation of Alzheimer’s disease. PLoS One 9, e99897 10.1371/journal.pone.0099897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šimić G, Kostović I, Winblad B, Bogdanović N, 1997. Volume and number of neurons of the human hippocampal formation in normal aging and Alzheimer’s disease. J. Comp. Neurol 379, 482–494. [DOI] [PubMed] [Google Scholar]
- Song YS, Park HJ, Kim SY, Lee SH, Yoo HS, Lee HS, Lee MK, Oh KW, Kang S-K, Lee SE, Hong JT, 2004. Protective role of Bcl-2 on β-amyloid-induced cell death of differentiated PC12 cells: reduction of NF-κB and p38 MAP kinase activation. Neurosci. Res 49, 69–80. [DOI] [PubMed] [Google Scholar]
- Stadelmann C, Deckwerth TL, Srinivasan A, Bancher C, Bruck W, Jellinger K, Lassmann H, 1999. Activation of Caspase-3 in Single Neurons and Autophagic Granules of Granulovacuolar Degeneration in Alzheimer’s Disease: Evidence for Apoptotic Cell Death. Am. J. Pathol 155, 1459–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su JH, Zhao M, Anderson AJ, Srinivasan A, Cotman CW, 2001. Activated caspase-3 expression in Alzheimer’s and aged control brain: correlation with Alzheimer pathology. Brain Res. 898, 350–357. 10.1016/s0006-8993(01)02018-2 [DOI] [PubMed] [Google Scholar]
- Talboom JS, Haberg A, De Both MD, Naymik MA, Schrauwen I, Lewis CR, Bertinelli SF, Hammersland C, Fritz MA, Myers AJ, Hay M, Barnes CA, Glisky E, Ryan L, Huentelman MJ, 2019. Family history of Alzheimer’s disease alters cognition and is modified by medical and genetic factors. Elife 8 10.7554/eLife.46179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan FCC, Hutchison ER, Eitan E, Mattson MP, 2014. Are there roles for brain cell senescence in aging and neurodegenerative disorders? Biogerontology 15, 643–660. 10.1007/s10522-014-9532-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thawkar BS, Kaur G, 2019. Inhibitors of NF-kappaB and P2X7/NLRP3/Caspase 1 pathway in microglia: Novel therapeutic opportunities in neuroinflammation induced early-stage Alzheimer’s disease. J. Neuroimmunol 326, 62–74. 10.1016/j.jneuroim.2018.11.010 [DOI] [PubMed] [Google Scholar]
- Theofilas P, Ehrenberg AJ, Nguy A, Thackrey JM, Dunlop S, Mejia MB, Alho AT, Paraizo Leite RE, Rodriguez RD, Suemoto CK, Nascimento CF, Chin M, Medina-Cleghorn D, Cuervo AM, Arkin M, Seeley WW, Miller BL, Nitrini R, Pasqualucci CA, Filho WJ, Rueb U, Neuhaus J, Heinsen H, Grinberg LT, 2018. Probing the correlation of neuronal loss, neurofibrillary tangles, and cell death markers across the Alzheimer’s disease Braak stages: a quantitative study in humans. Neurobiol. Aging 61, 1–12. 10.1016/j.neurobiolaging.2017.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, 2003. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 13, 2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troncoso JC, Sukhov RR, Kawas CH, Koliatsos VE, 1996. In situ labeling of dying cortical neurons in normal aging and in Alzheimer’s disease: correlations with senile plaques and disease progression. J. Neuropathol. Exp. Neurol 55, 1134–1142. 10.1097/00005072-199611000-00004 [DOI] [PubMed] [Google Scholar]
- Wagner EF, Nebreda ÁR, 2009. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 9, 537–549. 10.1038/nrc2694 [DOI] [PubMed] [Google Scholar]
- Yang Y, Mufson Elliott, J., Herrup K, 2003. Neuronal Cell Death Is Preceded by Cell Cycle Events at All Stages of Alzheimer’s Disease. J. Neurosci 23, 2557–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Thompson R, Zhang H, Xu H, 2011. APP processing in Alzheimer’s disease. Mol. Brain 4, 3 10.1186/1756-6606-4-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.