

Abstract
Neurofibrillary tangles (NFTs) composed of hyperphosphorylated and misfolded tau protein are a pathological hallmark of Alzheimer’s disease and other tauopathy conditions. Tau is predominantly an intraneuronal protein but is also secreted in physiological and pathological conditions. The extracellular tau has been implicated in the seeding and propagation of tau pathology and is the prime target of the current tau immunotherapy. However, truncated tau species lacking the microtubule binding repeat (MTBR) domains essential for seeding have been shown to undergo active secretion and the mechanisms and functional consequences of the various extracellular tau are poorly understood. We report here that the transcription factor EB (TFEB), a master regulator of lysosomal biogenesis, plays an essential role in the lysosomal exocytosis of selected tau species. TFEB loss-of-function significantly reduced the levels of interstitial fluid (ISF) tau in PS19 mice expressing P301S mutant tau and in conditioned media of mutant tau expressing primary neurons, while the secretion of endogenous wild-type tau was not affected. Mechanistically we found that TFEB regulates the secretion of truncated mutant tau lacking MTBR and this process is dependent on the lysosomal calcium channel TRPML1. Consistent with the seeding-incompetent nature of the truncated tau and supporting the concept that TFEB-mediated lysosomal exocytosis promotes cellular clearance, we show that reduced ISF tau in the absence of TFEB is associated with enhanced intraneuronal pathology and accelerated spreading. Our results support the idea that TFEB-mediated tau exocytosis serves as a clearance mechanism to reduce intracellular tau under pathological conditions and that effective tau immunotherapy should devoid targeting these extracellular tau species.
Introduction
Tauopathies consist of a group of diseases including frontotemporal dementias and the most common form Alzheimer’s Disease (AD), and are characterized by the accumulation of intracellular neurofibrillary tangles (NFTs) composed of aggregates of hyperphosphorylated and misfolded pathological tau protein and extensive neurodegeneration1–3. To date, the majority of AD clinical trials have focused on Aβ. Unfortunately, such therapies have been unsuccessful so far4–6. One possible shortcoming of Aβ-based therapies is that they may need to target Aβ prior to the accumulation of significant tau pathology in order to have a clear effect. Once neocortical tau pathology ensues, this correlates with cognitive decline more closely than Aβ accumulation7, 8. Thus, there is a growing interest in developing tau-based therapies for AD and other tauopathy diseases9.
Although tau is predominantly an intraneuronal protein, analysis of post-mortem AD brains document that the NFT pathology progresses in a hierarchical, stereotyped pattern beginning in the transentorhinal cortex and eventually spreading to synaptically connected brain regions such as the hippocampus and, later, the neocortex10. Consistent with this observation, studies using cellular and mouse models demonstrate that tau aggregates can cross the cell membrane and seed tau pathology, followed by subsequent spreading to other cells of synaptically connected regions, resulting in the cell-to-cell transfer or prion-like propagation of tau pathology11–16. This pattern of tau spreading implicates a possible role of extracellular tau species in disease progression. Lending support for this notion, tau can be detected in the brain interstitial fluid (ISF) of mutant human tau transgenic mice and its littermate controls by in vivo microdialysis17, and neuronal released tau has been shown to be capable of propagating tau pathology18. Further, passive immunization with antibodies designed to target extracellular tau reduced tau pathology in tauopathy mouse models19–21. Directly relevant to human disease, tau proteins are present in the cerebrospinal fluid (CSF) where the levels of both tau and phospho-tau are elevated in AD patients22. Accordingly, anti-tau antibodies are currently being tested in clinical trials as disease-modifying therapies for tauopathy conditions9.
Effective immunotherapy calls for the need to understand the mechanisms of tau release and to target the seeding and spreading competent tau. Both passive release secondary to neuronal cell death and active tau secretion have been proposed to contribute to the extracellular pool of tau; the latter may be mediated via direct translocation from the cytoplasm across the plasma membrane, endosomal/lysosomal-dependent exocytosis, or exosomal secretion23–25. However, despite the intense interest, the molecular mechanisms mediating tau exocytosis and their functional roles in physiological or pathological conditions have not been well-delineated. Adding to the complexity is the presence of multiple tau species due to alternative splicing, post-translational modifications and proteolytic processing2, 3, 26–28. These distinct tau species may be subject to different intracellular routing and extracellular release and may exhibit differential properties with regards to aggregation and spreading.
While both insoluble tau aggregates and certain soluble monomeric or oligomeric tau species are shown to exhibit seeding and spreading properties18, 29, 30, sequences within the microtubule-binding repeat (MTBR) region of tau are known to be essential for nucleation2. Intriguingly, tau detected from human CSF, iPSC-derived neurons and mouse neuronal cultures are found predominately truncated prior to the MTBR, thus likely seeding and aggregation incompetent31–33. These findings beg the questions as to where these truncated tau are generated, how are they secreted, and what are the functional consequences of these secreted tau species?
Here we present evidence that the Transcription Factor EB (TFEB), a master regulator of lysosomal biogenesis shown to have a potent effect on tau clearance34, 35, plays an essential role in mediating the release of mutant, MTBR truncated, tau in vitro and in PS19 transgenic mice. This activity is dependent on its lysosomal target Mucolipin TRP channel 1 (TRPML1) and is positively correlated with tau clearance. Loss of TFEB leads to reduced ISF tau and increased tau pathology and spreading.
Materials and Methods
Mice
The tau P301S transgenic mouse line (PS19) and Nestin-Cre transgenic mouse line were purchased from the Jackson Laboratory. Tcfeb flox mice were described previously36. Both genders were used in the study and randomly assigned to groups. Investigators were blinded to group identities during data collection and analysis. The sample size was determined according to previous studies35, 37. All procedures were performed based on the protocols approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine.
Reagents
PHF1 and MC1 antibodies were gifts from Peter Davies (Feinstein Institute for Medical Research). Other antibodies and reagents were: AT8 (Invitrogen, MN1020), tau (K9JA) (DAKO, A0024), Tau5 (Thermo Fisher Scientific, AHB0042), biotinylated BT2 (Thermo Fisher Scientific, MN1010), TFEB (Proteintech, 13372–1-AP), γ-tubulin (Sigma, T6557), TRPML1(Sigma, AV35307), FLAG (Sigma, F1804), CTSD (Santa Cruz Biotechnology, sc-6487), CDK5 (Santa Cruz Biotechnology, sc-6247), HDAC9 (Abcam, ab59718), HRP-conjugated secondary antibodies (EMD Millipore, AP100P, AP307P), IRDye secondary antibodies (LI-COR, 926–32211, 926–32212, 926–68070, 926–68073), Alexflour-conjugated secondary antibodies (Thermo Fisher Scientific, A21202, A21422, A21206, A31572), Streptavidin-HRP kit (R&D systems, DY998), lipofectamine 3000 (Thermo Fisher Scientific, L3000015), protease and phosphatase inhibitor cocktails (Roche, 04693116001, 04906845001), ECL (Pierce, 32106, 34096), NP-40 (Thermo Fisher Scientific, 85124), QVD (ApexBio, A1901), Hepes (Thermo Fisher Scientific, 15630080), GlutaMAX Supplement (Thermo Fisher Scientific, 35050061), Pen/Strep (Thermo Fisher Scientific, 15140122), neurobasal medium (Thermo Fisher Scientific, 21103049), B-27 supplement (Thermo Fisher Scientific, 17504044), Pierce TMB Substrate Kit (Thermo Fisher Scientific, 34021), tau protein ladder (Sigma, T7951). The tau P301L expression vector and the associated AAV virus were gifts from Leonard Petrucelli (Mayo Clinic). MCOLN1-HA was a gift from Craig Montell (Addgene plasmid # 18825). CTRL-siRNA and MCOLN1-siRNA (Santa Cruz Biotechnology, sc-37007, sc-44519). ML-SI1 (Sigma, G1421), YM201636 (Sigma, 524611-M). The rTg4510 mice brain lysate was described in the previous publication38.
Cellular assays
The TFEB knockout and TFEB/MITF/TFE3 triple knockout HeLa cell lines were kind gifts from Richard Youle (National Institutes of Health)39. TFEB overexpression HeLa cell line was a gift from Marco Sardiello (Baylor College of Medicine). The transfections were performed according to the product’s manual and apoptosis inhibitor QVD was added into the culture medium to minimize the influence of cell death. Two days after transfection, the medium was collected, followed by 2,000 × g for 10 minutes and subjected to 10,000 × g for 30 minutes to remove floating cells and cellular debris respectively. Cells were lysed in buffer (TBS with 1% NP-40 and protease and phosphatase inhibitor cocktails). For primary neuronal culture, the pups at postnatal day 0 (P0) were used for neuronal culture. The forebrain was dissected in pre-cold dissection medium (HBBS with 10 mM Hepes, 1 × GlutaMA Supplement, and 1% vol/vol Pen/Strep), and was digested in 2.5% trypsin at 37 ℃ for 15 minutes, followed by adding trypsin inhibitor and DNase. The digested tissue was centrifuged at 1,000 g for 10 minutes and resuspended in neuronal culture medium (neurobasal media with B-27 supplement, 1 × GlutaMAX supplement, and 1% vol/vol Pen/Strep). For AAV infection, the virus was added to the neuronal culture at MOI of 1 × 104 immediately after neurons were seeded. The medium on primary neurons was changed after 16 hours, the fresh medium of half the total volume of the well was added. For the seeding assay, 10% brain homogenate from 7-month old rTg4510 mice was added into the medium at day 10 after culturing. At day 18, the cells were fixed for immunostaining.
Induced human pluripotent stem cell (iPSC)-derived neurons
The iPSC experiment was performed in the Washington University School of Medicine. The informed consent was approved by the Washington University School of Medicine Institutional Review Board and Ethics Committee (IRB 201104178 and 201306108). The consent allowed for use of tissue by all parties, commercial and academic, for the purposes of research but not for use in human therapy.
The creation and characterization of the iPSC with MAPT p.R406W and its genome-edited isogenic control cells were reported previously40, 41. To generate neurons, iPSCs were harvested for neural aggregate formation and dissociated with Accutase (MP Biomedicals) and pelleted by centrifugation. Cells were resuspended in mTesR1 supplemented with the ROCK inhibitor, Y-27632 (10 μM) and transferred to a v-bottom 96-well plate40. Neurospheres were cultured for 5 days with daily media change in Neural Induction Medium (NIM; Stemcell Technologies). Spheres were then transferred to a pre-coated with poly-Ornithin (PLO)/Laminin (Millpore-Sigma) plate with daily media changes. Neural rosettes were harvested on fay 5 with Neural Rosette Selection reagent (Stemcell Technologies). NPCs were cultured on PLO and Laminin-coated plates and terminal differentiation was initiated with the addition of cortical maturation media (Neurobasal Media (Life Technologies) supplemented with B27 (GIBCO), BDNF (Peprotech), GDNF (Peprotech), cAMP (Sigma) and L-glutamate (Sigma). Neurons were maintained for 6 weeks prior to Torin1 treatment. After 6 weeks in culture, media from iPSC-neurons maintained in a 48-well plate were gently washed with DMEM/F12 and treated with 250 nM Torin1 or DMSO control. After 5 hours of treatment, cell culture media was collected, centrifuged at 1000 rpm for 10 minutes at 4°C to remove cell debris and flash frozen. Cells were collected in PBS and pelleted. Culture media and lysate were IPed by Tau1 (mouse monoclonal, provided by Nicholas Kanaan) and HJ8.5 antibodies followed by tau ELISA.
Lysosomal immunoprecipitation (LysoIP)
The LysoIP experiment was performed as previously described42. Neurons infected with AAV vectors expressing human tau P301L and LAMP1-FLAG were collected with KPBS at 18 days in vitro (DIV)35, 37. The homogenate was spun down at 1000 × g for 2 minutes and the supernatant was incubated with 200 μl of prewashed beads on an end-over-end rotator for 20 minutes. After 3 times wash with KPBS, beads were incubated with 80 μl lysis buffer (KPBS with 1% Triton-X100) for 10 minutes, the supernatant was separated from beads and collected as lysosomal fraction followed by tau ELISA.
In vivo tau microdialysis
In vivo microdialysis experiments to measure brain ISF tau levels from awake and freely moving mice were performed as described43. To avoid tissue damage, the ISF tau collection was initiated 48 hours after probe implantation when ISF tau concentrations are stable. ISF samples were collected in a refrigerated fraction collector and analyzed by a tau sandwich ELISA using Tau5 as a capture antibody and biotinylated BT2 as a detection antibody as detailed in17, 43.
Tau ELISA
The tau levels in microdialysis, culture medium, and cellular lysates were measured in tau sandwich ELISA based on previous publications17, 32, 44. Two μg of Tau5 (aa 210–241) or K9JA (aa 243–441) antibody was coated per well in 96-well plate and incubated at 4 ℃ for 16 hours. The plate was blocked with PBS with 1% BSA for 60 minutes at room temperate. Tau protein ladder was diluted as standards. The samples and standards were loaded and incubated at 4 ℃ for 16 hours. Then, the plate was incubated with biotinylated BT2 (aa 194–198) antibody at the concentration of 0.3 μg/ml for 2 hours at room temperate, followed by incubation of Streptavidin-HRP for 45 minutes at room temperature. The plate was then developed by Pierce TMB Substrate Kit (Thermo Fisher Scientific) and read on SpectraMax i3x Multi-Mode Microplate Reader (Molecular Devices).
Tau spreading assay
The tau spreading assay was performed as described previously37. Briefly, PS19 and PS19, TFEB cKO mice were stereotaxically injected with equal amount (2 μl) of 10% brain homogenate from 7-month old rTg4510 mice at 2–3 months of age into one hippocampal hemisphere (lacunosum moleculare layer of the hippocampus and molecular layer of the dentate gyrus, bregma, −2.5 mm; lateral, −2 mm, and depth, −1.8 mm). The mice were euthanized six weeks later for immunofluorescence staining to evaluate tau spreading.
Immunofluorescence staining
Mice were perfused with 4% paraformaldehyde (PFA) in TBS, followed by overnight fixation. The PFA was replaced with 30% sucrose in TBS. After dehydration, brain tissues were cut into 30 μm sections for staining. For primary cultured neuronal, cells were fixed in 4% PFA for 20 minutes at room temperature. Brain sections or cultured cells were incubated with primary antibodies in TBS with 0.4% Triton X-100 and 2% donkey serum overnight, followed by washing and incubating with AlexFlour-conjugated secondary antibodies. Brain sections or cells were imaged by confocal microscopy (Leica SPE) or EVOS Cell Imaging System (Thermo Fisher Scientific).
Western blotting
The forebrain tissues or cells were lysed in buffer (TBS with 1% NP-40, 1% sodium deoxycholic acid, 0.1% sodium dodecyl sulfate, and protease and phosphatase inhibitor cocktails). Lysates were sonicated and centrifuged at 20,000 × g for 10 minutes. Supernatants were boiled with loading buffer, used for SDS-PAGE and transferred to nitrocellulose membrane. After incubation with primary and secondary antibodies, the membranes were incubated with ECL and developed by Chemidoc image system (Bio-Rad) or Odyssey image system (LI-COR).
Statistics
All data are presented as mean ± SEM. Power analysis was performed using a confidence interval of α=0.05. Two groups comparisons were analyzed using two-tailed Student’s t-test, multiple comparisons were analyzed using one-way or two-way ANOVA followed by post hoc tests as indicated in the figure legends using GraphPad Prism. P-values less than or equal to 0.05 were considered statistically significant. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Results
TFEB loss-of-function reduces ISF tau in PS19 mice
We reported earlier that AAV-mediated neuronal TFEB expression robustly reduced pathological tau proteins and NFT pathology35. Since TFEB has been shown to promote cellular clearance through both autophagy and lysosomal exocytosis45, we wondered whether TFEB may regulate tau exocytosis in addition to the autophagy-lysosomal pathway. We thus created CNS-specific TFEB knockout (cKO) by crossing a Tcfeb floxed allele with Nestin-Cre transgenic mice36, using Cre-negative mice as controls (Ctrl), and bred these with PS19 tau transgenic mice to generate Ctrl, cKO, PS19 and PS19, cKO mice. Quantitative PCR (qPCR) and Western blot analyses showed that TFEB mRNA and protein levels were reduced by about 80% in TFEB cKO mouse brains compared to the controls (Fig. S1a,b and Fig. 1b). The cKO mice are viable and fertile and do not exhibit overt abnormalities (data not shown). Immunostaining of brain sections using anti-NeuN, -p62, and -GFAP antibodies revealed no significant alterations between TFEB cKO and their littermate controls (Fig. S1c). This is consistent with the comparable expression of selected TFEB target genes (Fig. S1d). These may be due to the compensation by TFEB homologs MITF and TFE3.
Fig. 1. TFEB loss-of-function reduces ISF tau in PS19, but not wild-type mice.
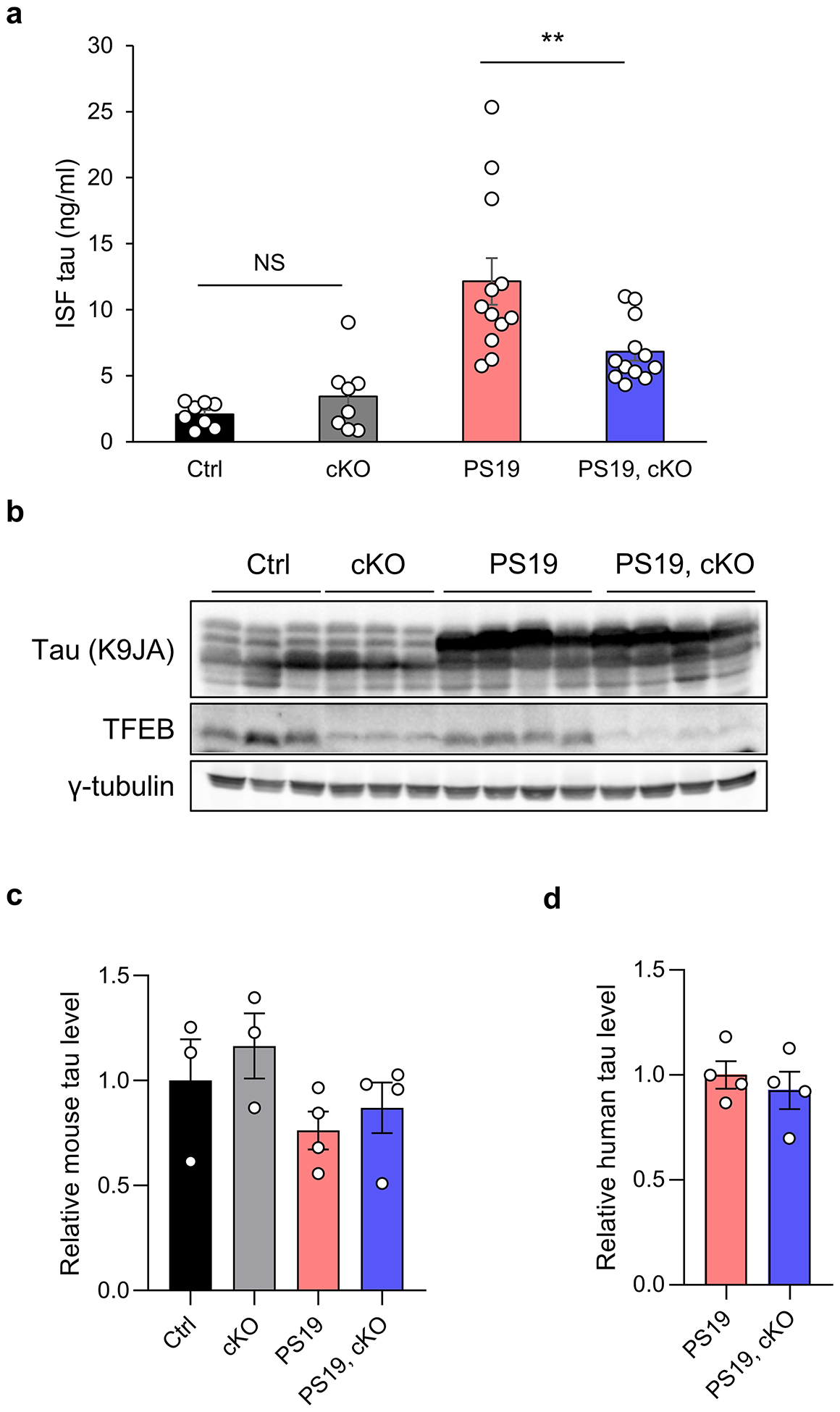
(a) ISF tau levels in brains of WT, TFEB cKO, PS19, and PS19, TFEB cKO mice at 4- to 5-month of age measured by ELISA. Two ways ANOVA followed by Sidak’s multiple comparisons test. (n=8 for WT and TFEB cKO groups; n=12 for PS19 and PS19, TFEB cKO groups). (b) Representative Western blot image of tau (K9JA) and TFEB protein levels in brains of WT, TFEB cKO, PS19, and PS19, TFEB cKO mice at 4- to 5-month of age. (c) Quantification of mouse tau levels in (b). Two ways ANOVA followed by Sidak’s multiple comparisons test. (d) Quantification of human tau levels in (b). Two tailed Student’s t-test. Data are presented as mean ± SEM (n=3 for WT and TFEB cKO groups; n=4 for PS19 and PS19, TFEB cKO groups). Data are presented as mean ± SEM. NS, not significant; **P ≤ 0.01.
We measured the tau levels in the interstitial fluid (ISF) of awake, free moving mice of the four genotypes using in vivo microdialysis17. To avoid the confounding effect of intracellular tau aggregation on ISF tau, we performed the analysis at 4 to 5-month of age prior to the development of tau pathology. To minimize the tau released by tissue damage and cell death, the ISF tau collection was initiated 48 hours after probe implantation when ISF tau levels were stabilized. We found that TFEB cKO had no effect on endogenous mouse ISF tau (Fig. 1a, WT vs cKO), but significantly reduced the ISF tau in PS19 mice (Fig. 1a, PS19 vs PS19, cKO). Western blot analysis of brain lysates using tau (K9JA) (Fig. 1b–d) and PHF tau (Fig. S1e,f) antibodies showed comparable levels of mouse and human tau in the presence or absence of TFEB at this age, supporting the notion that changes of ISF tau represent a primary TFEB-mediated effect rather than consequences of intracellular tau alterations. These results demonstrate that TFEB plays a physiological role in mediating mutant tau release in vivo.
TFEB mediates the release of truncated mutant tau in vitro
The tau microtubule binding repeat (MTBR) sequences are known to facilitate its self-aggregation. The ISF tau is detected using an enzyme-linked immunosorbent assay (ELISA) that recognizes the mid-domain region of tau (Tau5, aa 210–241 and BT2, aa 194–198, Fig. 2a), therefore, does not distinguish between MTBR truncated or MTBR containing forms. We sought to evaluate the tau species secreted by TFEB further in primary cultured neurons using both the mid-domain ELISA and an antibody combination that recognizes the MTBR-containing tau (K9JA, aa 243–441, and BT2, Fig. 2a and Fig. S2a). We prepared primary neuronal cultures derived from TFEB cKO mice and their littermate controls. Similar to the in vivo results, measurement of secreted and intracellular mouse endogenous tau in the two cultures using the mid-domain ELISA did not observe any appreciable differences (Fig. 2b–d). We thus infected the cultures with AAV-human tau containing the P301L mutation, which led to robust expression of mutant human tau seven days after infection (Fig. S2b), and measured the tau levels in both the conditioned media and in cell lysates using both ELISA systems. Consistent with the recent reports that actively released tau is MTBR truncated32, 33, we found that the extracellular tau detected by the K9JA/BT2 ELISA was drastically lower compared to that detected by the mid-domain ELISA (Tau5/BT2) (Fig. 2e), although the intracellular tau levels detected by the two ELISAs were comparable (Fig. 2f). Further, only the mid-domain-positive extracellular tau was reduced in TFEB cKO neurons (Fig. 2e). This is associated with increased intracellular tau (Fig. 2f), and a reduced ratio of extra/intracellular tau (Fig. 2g). The results combined demonstrate that TFEB mediates the release of MTBR truncated tau in cultured primary neurons and possibly in PS19 mouse brains.
Fig. 2. TFEB mediates the release of truncated mutant tau in primary neurons.
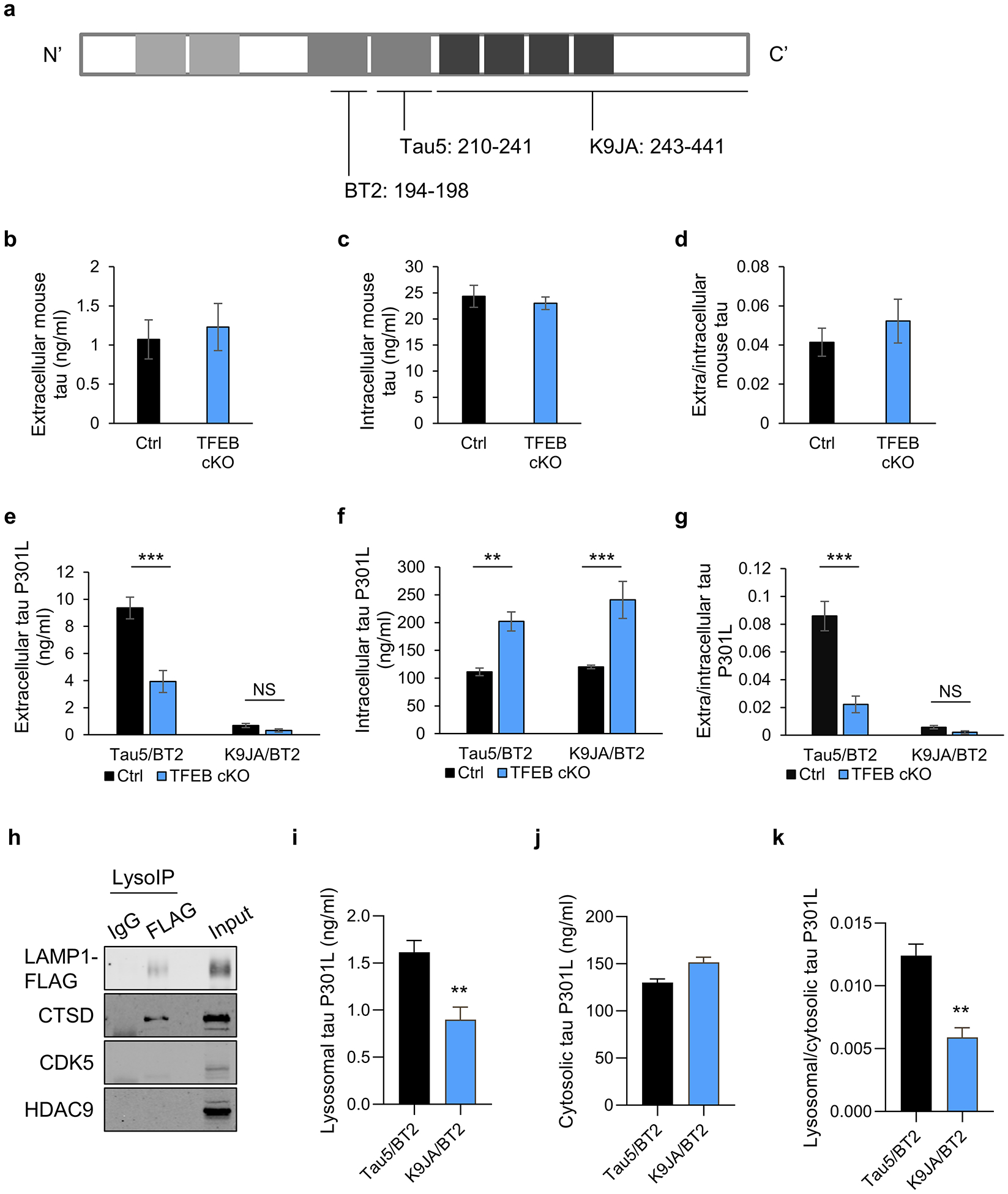
(a) Schematic of tau protein and epitopes of tau antibodies used for ELISA: Tau5 (aa 210–241), K9JA (aa 243–441), and BT2 (aa 194–198). (b-d) Extracellular (b) and intracellular (c) tau levels from cultured media or cell lysates, respectively, of WT and TFEB cKO neurons at 18 days in vitro (DIV) and quantified by ELISA (Tau5/BT2 combination). (d) Extra/intracellular tau ratio. Two tailed Student’s t-test. (n=6 of 2 experiments). (e-g) Extracellular (e) and intracellular (f) tau levels from cultured media or cell lysates of AAV-tau-P301L infected WT and TFEB cKO neurons at 18 days in vitro (DIV) and quantified by ELISA (Tau5/BT2 or K9JA/BT2 combination). (g) Extra/intracellular tau ratio. Two ways ANOVA followed by Sidak’s multiple comparisons test. (n=6 of 2 experiments). (h) Biochemical characterization of lysosomes isolated by anti-FLAG immunoprecipitation (LysoIP) using antibodies against: lysosomal membrane LAMP1, lysosomal lumen CTSD and non-lysosomal cytosolic protein CDK5 and nuclear protein HDAC9. (i-k) Lysosomal (i) and cytosolic (j) tau levels from AAV-tau-P301L infected WT and TFEB cKO neurons at 18 days in vitro (DIV) and quantified by ELISA (Tau5/BT2 or K9JA/BT2 combination). (k) Lysosomal/cytosolic tau ratio. Two tailed Student’s t-test. (n=4 of 2 experiments). Data are presented as mean ± SEM. NS, not significant; **P ≤ 0.01; ***P ≤ 0.001.
To assess whether the tau truncation occurs within the lysosome, we performed lysosome immunoprecipitation (LysoIP) of primary cultured neurons infected with AAV-tauP301L and LAMP1-FLAG using an anti-FLAG antibody. Western blot analysis showed that the anti-FLAG precipitates, but not the IgG control, contained the lysosomal enzyme Cathepsin D (CTSD). The cytosolic protein CDK5 or the nuclear protein HDAC9 were absent although both can be detected in the input, demonstrating the purity of the isolated lysosomes (Fig. 2h). Using ELISAs that detect mid-domain (Tau5/BT2) and MTBR region (K9JA/BT2), we found that, in the lysosomal preparation, the Tau5/BT2 signal is significantly higher than that of the K9JA/BT2 (Fig. 2i), but the levels are comparable in the cytosolic fraction (Fig. 2j). These results suggest that while the cytosolic tau consists of mostly MTBR-containing form detected by both set of ELISAs, approximately half of the lysosomal tau is MTBR truncated that can only be detected by mid-domain ELISA. These results support the idea that tau truncation occurs in the lysosome.
We next probed the mechanism of TFEB-dependent tau release using HeLa cells deficient in either TFEB (TFEB KO) or all three TFEB family members, namely TFEB, MITF and TFE3 (TKO) (Fig. S3a)39. qPCR analysis showed similar levels of transfected MAPT mRNA in these cells compared to wild-type controls (Fig. S3b). We quantified the extracellular and intracellular tau levels using the mid-domain tau ELISA only, given the positive regulation of these tau species by TFEB. The qPCR analysis revealed that the mRNA levels of TFEB lysosome genes were reduced by about 50% compared to wild-type HeLa cells upon Torin1 treatment which promotes TFEB nuclear translocation and activation (Fig. 3a). Transfection of P301L mutant human tau to these cells and measurement of tau levels in conditioned medium showed that secreted tau was reduced by 53% in TFEB KO and 76% in TKO cells compared to WT HeLa controls (Fig. 3b), while levels of intracellular tau were not significantly affected at the protein level (Fig. 3c), resulting in significant reductions in the extra/intracellular tau ratio in TFEB KO and TKO conditions (Fig. 3d).
Fig. 3. TFEB mediates mutant tau release in HeLa cells and iPSC-derived neurons.
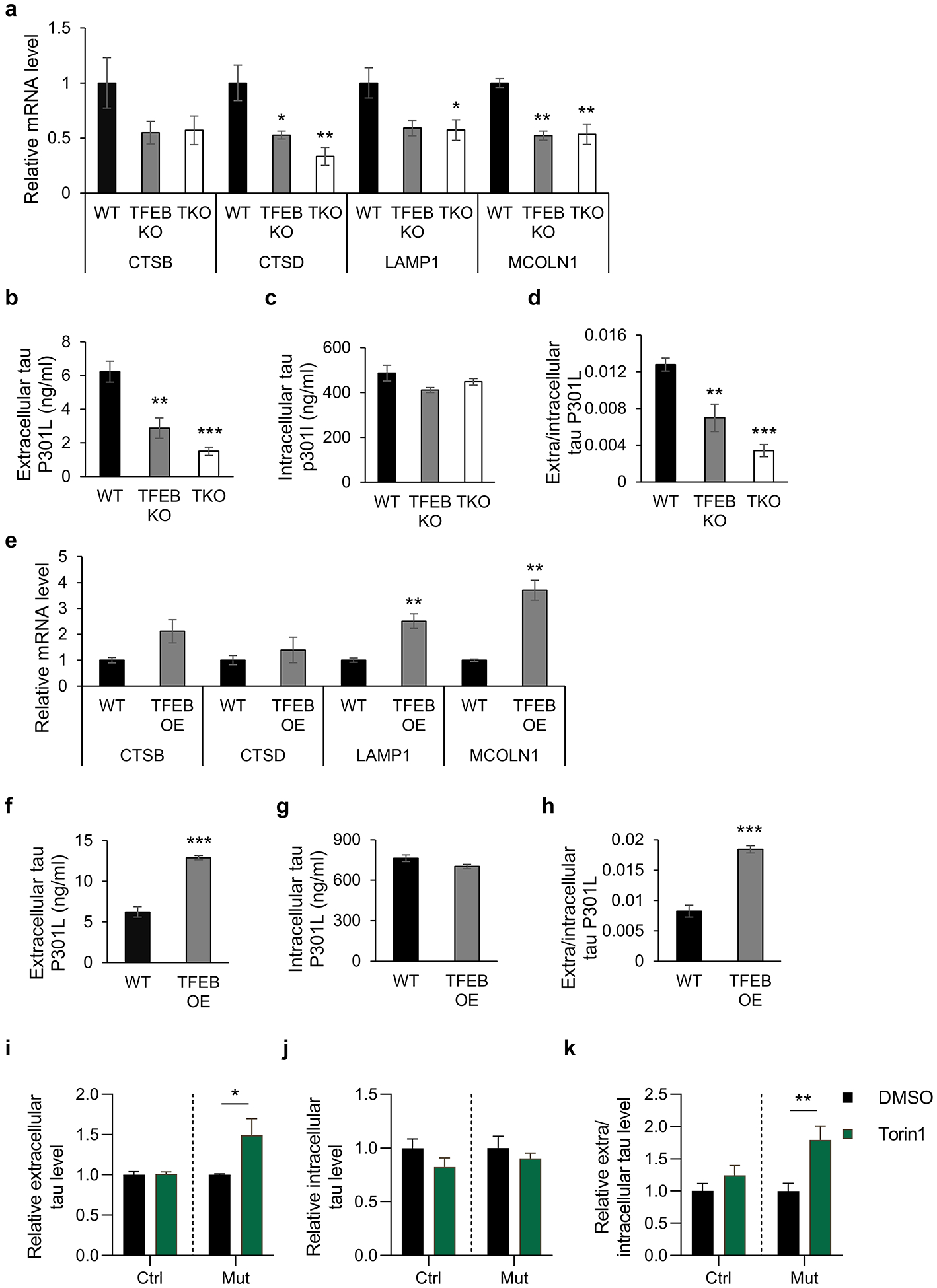
(a) qPCR measurement of mRNA levels of TFEB lysosomal target genes (CTSB, Cathepsin B; CTSD, Cathepsin D; LAMP1, lysosomal-associated membrane protein 1; MCOLN1, Mucolipin 1) in WT, TFEB KO, and TKO HeLa cells treated with 300 nM Torin1 for 6 h. One-way ANOVA followed by Dunnett’s multiple comparisons test. (n=3 of 2 experiments). (b-d) WT, TFEB KO, and TKO HeLa cells were transfected with tau-P301L plasmid. Two days after transfection, culture medium and cell lysates were collected. Extracellular (b) and intracellular (c) tau in culture media and cell lysates were quantified by ELISA respectively. The extra/intracellular tau ratio (d) was calculated. One-way ANOVA followed by Dunnett’s multiple comparisons test. (n=5 of 2 experiments). (e) qPCR measurement of mRNA levels of TFEB lysosomal target genes in WT and TFEB OE HeLa cells. Two tailed Student’s t-test. (n=3 of 2 experiments). (f-h) Extracellular (f) and intracellular (g) tau in culture media and cell lysates were quantified by ELISA respectively. The extra/intracellular tau ratio (h) was calculated. Two tailed Student’s t-test. (n=5 of 2 experiments). (i-k) Relative levels of extracellular (i) or intracellular (j) tau in cultured media or cell lysates of human iPSCs-derived neurons harboring tau R406W mutation (Mut) or its isogenic controls (WT) treated with DMSO or Torin1 (250 nM), immunoprecipitated by Tau1 and HJ8.5 antibodies and quantified by ELISA. The relative extra/intracellular tau ratio (k) was calculated. Two ways ANOVA followed by Sidak’s multiple comparisons test. (n=3–4). Data are presented as mean ± SEM. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
In contrast to TFEB loss-of-function, transfection of the human P301L mutant tau to HeLa cells overexpressing TFEB (Fig. S3c) followed by measurement of tau levels in the conditioned media and cell lysates showed that TFEB overexpression resulted in increased expression of its lysosomal target genes (Fig. 3e). This is associated with significantly higher extracellular tau levels (Fig. 3f). Although there is a trend of reduction in intracellular tau (Fig. 3g), the difference did not reach statistical significance (p=0.067). Nevertheless, both combined led to a highly significant increase in the ratio of extra/intracellular tau (Fig. 3h).
Although the above studies employed the use of multiple systems, including living mouse models, primary cultured neurons and HeLa cell lines, one common caveat is that the mutant tau is overexpressed. To address whether the TFEB-mediated tau exocytosis is relevant under physiological conditions, we chose to examine the tau dynamics in human iPSCs-derived neurons from a frontotemporal dementia patient expressing the R406W mutant tau and its genome-edited isogenic controls40, 41. Cortical neurons derived from the iPSCs were maintained for 6 weeks and then treated with DMSO or 250 nM Torin1 for 5 hours to induce TFEB activation. Quantification of tau in conditioned media and cell lysates by mid-domain ELISA showed that, similar to the overexpression system, Torin1 treatment induced the release of the mutant tau but not wild-type tau (Fig. 3i). The minor but insignificant reduction of the intracellular tau (Fig. 3j) contributed to the highly significant increases of extra/intracellular ratio of mutant tau (Fig. 3k). Thus, the results combined support a specific regulation of TFEB in mediating the release of mutant but not wild-type tau under physiological or overexpression conditions.
TFEB-mediated tau release is dependent on TRPML1
The lysosomal Ca2+ channel Mucolipin TRP channel 1 (TRPML1, encoded by MCOLN1) is a TFEB target that has been reported to mediate lysosomal exocytosis downstream of TFEB45, 46. We therefore tested whether TFEB-mediated tau release is dependent on TRPML1. Upon co-transfection of empty vector (CMV) or MCOLN1 expression vector with the P301L tau construct to WT and TKO HeLa cells, we showed that overexpression of MCOLN1 had no overt effect on extracellular tau in WT cells (Fig. 4a, WT). Levels of secreted tau were reduced in vector-transfected TKO cells compared to that of WT cells as expected (Fig. 4a, WT CMV vs TKO CMV). MCOLN1 expression in TKO cells significantly upregulated extracellular tau levels and the extra/intracellular tau ratio (Fig. 4a, c, CMV vs MCOLN1, TKO), without exerting an appreciable effect on intracellular tau (Fig. 4b). Thus, TRPML1 mediates tau exocytosis downstream of TFEB. Conversely, while siRNA knockdown of MCOLN1 (Fig. S4a,b) had no appreciable effect in WT HeLa cells (Fig. 4d, WT), it partially normalized elevated extracellular tau (Fig. 4d) and the ratio of extra/intracellular tau (Fig. 4f) under TFEB overexpression (TFEB OE) conditions. Intracellular tau remained constant across groups (Fig. 4e). Since Ca2+ is required for TRPML1-mediated lysosomal fusion with the plasma membrane during lysosomal exocytosis46, we next tested whether intracellular Ca2+ is involved in tau exocytosis by treating the cells with ionomycin which is known to increase intracellular Ca2+ levels. The result showed that ionomycin significantly increased extracellular tau and the ratio of extra/intracellular tau without affecting intracellular tau (Fig. S4c).
Fig. 4. TFEB-mediated mutant tau release requires TRPML1.
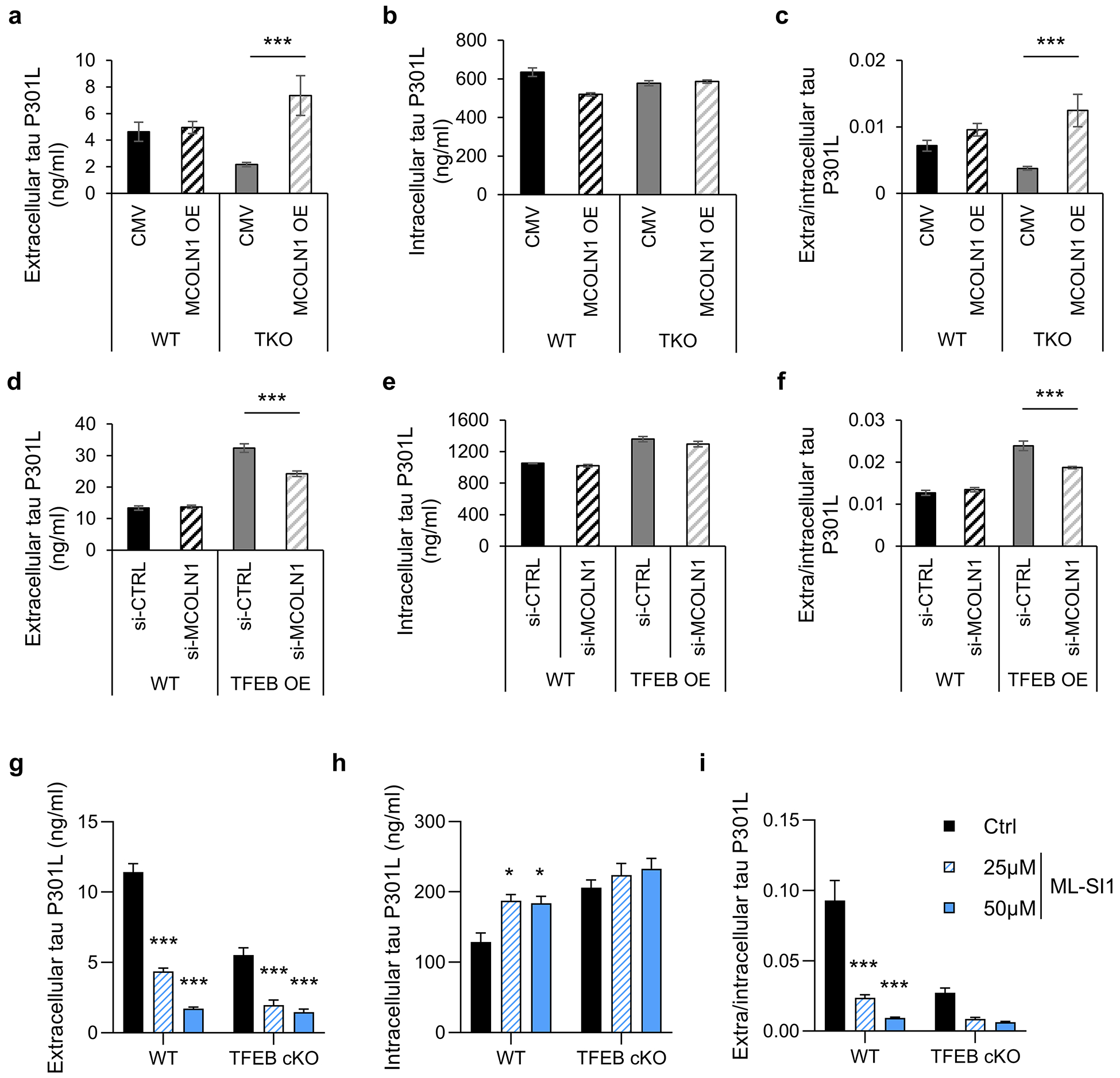
(a-c) WT and TKO HeLa cells were co-transfected with tau-P301L and empty vector (CMV) or MCOLN1. Two days after transfection, culture medium and cell lysate were collected. Extracellular (a) and intracellular (b) tau in culture medium and cell lysates were quantified by ELISA respectively. The extra/intracellular tau ratio (c) was calculated. Two ways ANOVA followed by Sidak’s multiple comparisons test. (n=5 of 2 experiments). (d-f) WT and TFEB OE HeLa cells were co-transfected with tau-P301L and control (CTRL) siRNA or MCOLN1 siRNA. Two days after transfection, culture media and cell lysate were collected. Extracellular (d) and intracellular (e) tau in culture media and cell lysates were quantified by ELISA. The extra/intracellular tau ratio (f) was calculated. Two ways ANOVA followed by Sidak’s multiple comparisons test. (n=5 of 2 experiments). (g-i) WT and TFEB cKO primary cultured neurons infected with AAV-tau-P301L were treated with ML-SI1 for 2 hours. Extracellular (g) and intracellular (h) tau in culture medium and cell lysates were quantified by ELISA respectively. The extra/intracellular tau ratio (i) was calculated. Two ways ANOVA followed by Sidak’s multiple comparisons test. (n=4 of 2 experiments). Data are presented as mean ± SEM. *P ≤ 0.05; ***P ≤ 0.001.
To validate that TRPML1 also mediates tau release in neurons, we treated the AAV-tau-P301L infected primary cultured neurons with TRPML1 specific inhibitors ML-SI1 or YM201636. The results showed the both ML-SI1 (Fig. 4g–i) and YM201636 (Fig. S4d) treatment inhibited tau release. Interestingly, while we did not detect appreciable effect of MCOLN1 overexpression or knockdown in P301L tau transfected wild-type HeLa cells, significant reductions of tau release can be observed in wild-type neurons expressing human P301L tau, which was further reduced in TFEB cKO cells (Fig. 4g). This could be caused by the low efficiency of tau secretion in HeLa cells or the incomplete inactivation in the case of MCOLN1 siRNA.
To provide further evidence that TRPML1 mediates the release of lysosomal tau, we performed LysoIP to isolate lysosomal tau after ML-SI1 treatment and measured tau levels using the mid-domain (Tau5/BT2, Fig. S4e) or MTBR-containing (K9JA/BT2, Fig. S4f) ELISAs. The results showed that ML-SI1 treatment specifically increased the mid-domain tau enriched in the lysosome, suggesting that TRPML1 inactivation leads to the accumulation of lysosomal tau.
Reduced tau release by TFEB loss-of-function is associated with augmented intracellular pathology and spreading
To examine the functional consequence of TFEB KO in pathological tau accumulation, we first seeded the AAV-tau-P301L infected neurons with lysates collected from rTg4510 mice to induce the formation of hyperphosphorylated and misfolded tau37, 38. Compared to the littermate controls, TFEB cKO robustly increased MC1-positive insoluble misfolded tau (MC1, Fig. 5a,b) and AT8-positive insoluble phospho-tau (AT8, Fig. S5a,b). These findings indicate that TFEB-mediated mutant tau exocytosis is inversely correlated with its intraneuronal accumulation in a seeding paradigm.
Fig. 5. TFEB cKO augments tau pathology in vitro and in vivo.

(a) Representative images of immunostaining showing that TFEB cKO increases insoluble misfolded tau (MC1) in seeded primary mouse neurons. WT and TFEB cKO neurons were infected with AAV-tau-P301L at 0 DIV and brain lysate from 7-month rTg4510 mice was added into culture medium at 10 DIV. At 18 DIV, neurons were treated with 1% Triton-X100 to remove soluble proteins during fixation. Blue, DAPI; green, MC1. Scale bar: 200 μm. (b) Quantitative analysis of (a). Two-tailed Student’s t-test. (n=4 of 2 experiments). (c) Representative Western blot image of phospho-tau (AT8) and tau (K9JA) protein levels in brains of PS19, and PS19, TFEB cKO mice at 12-month of age. (d) Quantification of (c). Two tailed Student’s t-test. (n=6/group). (e) Representative Western blot image of phospho-tau (AT8) and tau (K9JA) protein levels in cytosolic pools in brains of PS19, and PS19, TFEB cKO mice at 10-month of age. (f) Quantification of (e). Two tailed Student’s t-test. (n=5/group). (g) Representative Western blot image of phospho-tau (AT8) and tau (K9JA) protein levels in synaptosomal pools in brains of PS19, and PS19, TFEB cKO mice at 10-month of age. (h) Quantification of (g). Two tailed Student’s t-test. (n=5/group). (i) Representative immunofluorescence images of the hippocampus using the AT8 antibody. Scale bar: 20 μm. (j) Quantification of AT8-positive area in (i). Two tailed Student’s t-test. (n=7/group). Data are presented as mean ± SEM. *P ≤ 0.05; **P ≤ 0.01.
To understand the functional role of TFEB-mediated tau exocytosis in tau pathology in vivo, we performed biochemical and immunofluorescence staining of PS19 and PS19, TFEB cKO mice at 12 months of age. While tau (K9JA) levels remained constant, there were modest but significant increases of AT8-positive phospho-tau in TFEB-deficient samples (Fig. 5c,d). Increases in AT8-positive tau can be detected in both cytosolic (Fig. 5e,f) and synaptosomal preparations (Fig. 5g,h and Fig. S5c). Consistent with the immunoblotting, immunostaining also revealed significantly higher AT8 immunofluorescence intensity in PS19, TFEB cKO samples compared to the PS19 mice (Fig. 5i,j).
The tau pathology is thought to spread through a trans-synaptic mechanism. Since synaptosomal AT8-positivivity is increased in the absence of TFEB, we next evaluated TFEB’s role in a tau seeding and spreading model by injecting the brain lysate of rTg4510 tau transgenic mice into the lacunosum-moleculare of one hippocampal hemisphere of 2- to 3-month-old PS19 or PS19, TFEB cKO mice (Fig. S6a)15, 37, which has been documented to induce tau pathology through synaptically-connected regions from the ipsilateral CA1 to lateral CA3, followed by the caudal entorhinal cortex, then the contralateral CA337, 47. To minimize the possible confounding effect of seeding, we chose ipsilateral and contralateral CA3 distant from the injection site to assess tau pathology through spreading. Immunofluorescence analysis of 6 weeks post-injection showed that MC1-positive tau could be detected in both ipsilateral and contralateral hippocampus in PS19 mice, whereas MC1 immunoreactivity was significantly increased in PS19, TFEB cKO mice (Fig. 6a,b). No MC1 signal was detectable in wild-type mice injected with rTg4510 lysate or PS19 mice injected with wild-type lysate (Fig. S6b). Thus, reduced ISF tau in PS19, TFEB cKO mice is correlated with elevated phospho-tau pathology and spreading.
Fig. 6. TFEB cKO increases pathological tau spreading in vivo.
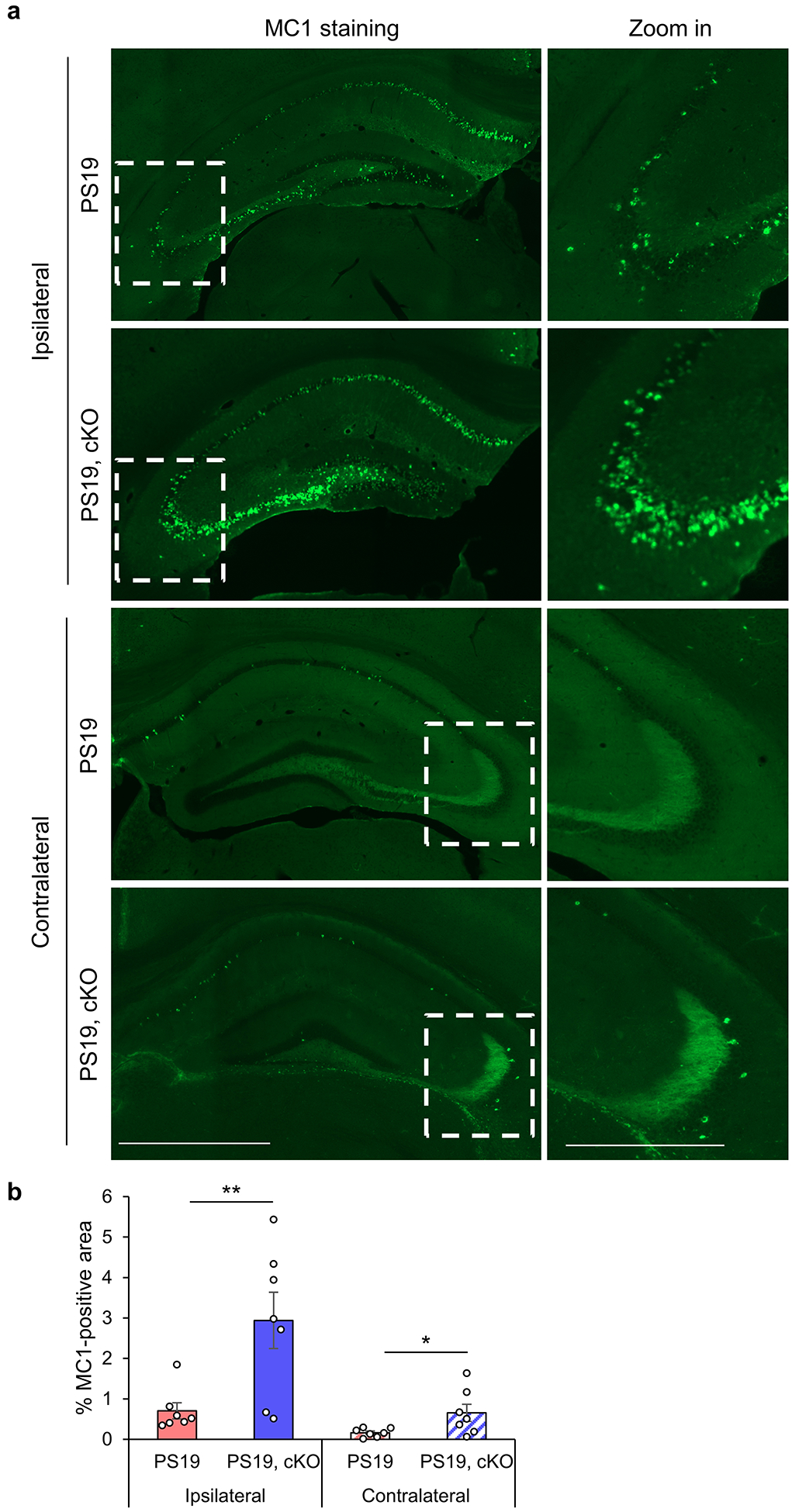
(a) Representative immunofluorescence images of the ipsilateral and contralateral hippocampus using the MC1 antibody. PS19 and PS19, TFEB cKO mice were injected with 2 μl of 7-month-old rTg4510 brain lysate at 2- to 3-month of age. Pathological tau spreading was analyzed 6-week after injection. Scale bar: 200 μm; 100 μm in zoom in. (b) Quantitative analysis of MC1-positive areas in the ipsilateral and contralateral hippocampus of PS19 and PS19, TFEB cKO mice. Two tailed Student’s t-test. (n=7/group). Data are presented as mean ± SEM. *P ≤ 0.05.
Discussion
Tau is known to be released from neurons and can be detected in the ISF of mouse models and human CSF. The extracellular tau has been implicated in the prion-like spreading of tau pathology and is the prime target for tau immunotherapy. As such, understanding the mechanisms of neuronal tau release and the functional consequences of extracellular tau species are crucially important from both biological and therapeutic perspective. We report here that TFEB plays an active role in the secretion of predominantly MTBR truncated human mutant tau, but not endogenous wild-type tau, in cultured neurons and in mouse brains, and this process is dependent on lysosomal Ca2+ channel TRPML1. Combined with a general role of TFEB-TRPML1 mediated lysosomal exocytosis in promoting cellular clearance45, we propose a model whereby TFEB-TRPML1 signaling serves as an innate clearance mechanism to remove aberrant intracellular tau through lysosomal exocytosis of seeding incompetent tau. Thus, loss of TFEB leads to reduced release of these mutant tau species and heightened intracellular tau. This in turn promotes the development of neuronal pathology and cell-to-cell propagation. The fact that loss of TFEB is associated with reduced ISF tau in PS19 mice and increased tau pathology and spreading is in keeping with this model.
Our analysis revealed two biochemical characteristics of TFEB-regulated extracellular tau: First, this pool of tau is recognizable by the mid-domain, but not MTBR-containing tau ELISA, suggesting that these tau species are MTBR truncated and seeding incompetent. Our results are consistent with recent publications using ELISAs of various antibody combinations31, 32 and by stable isotope labeling and mass spectrometry33, which collectively demonstrate that extracellular tau released from neuronal cell lines, primary neuronal cultures, and human iPSC-derived neurons, and human CSF samples are predominantly C-terminal truncated and MTBR absent. Our LysoIP experiment suggests that the truncation occurs in the lysosome. In this regard, Wang et al. showed that N2a cells expressing a tau repeat domain construct can be processed by a cytosolic protease and lysosomal cathepsin L28. However, whether these enzymes are responsible for processing full-length tau is not clear and future investigation is required to identify the lysosomal enzymes responsible for tau proteolysis. It is also worth noting that significant MTBR-containing tau can still be detected in the lysosome although the extracellular tau is to the most part truncated. Since lysosomes are heterogeneous and only those close to the plasma membrane undergo fusion and exocytosis, it is tempting to speculate that the fusion-competent lysosomal pool may contain higher percentage of processed tau. In addition, since LAMP1 is also expressed in the late endosome, it is possible that this organelle contains higher level of MTBR-containing tau. Further dissection of these mechanisms requires more advanced technology.
Second, we document here that TFEB deficiency has no effect on endogenous mouse tau secreted from primary neurons or mouse brains. Instead, significant reduction is observed only when TFEB is inactivated in mutant tau expressing neurons or on PS19 tau transgenic background. This finding is further corroborated using iPSC-derived neurons expressing mutant tau and their wild-type isogenic controls. Given that TFEB can be activated by starvation or stress and that tau aggregates are able to induce TFEB nuclear translocation and signaling47, 48, it is possible that the human mutant tau, but not normal wild-type tau, acts as a stress signal to activate TFEB and associated lysosomal exocytosis. Alternatively, the selectivity may be conferred at substrate recognition as both selective autophagy and chaperone-mediated autophagy have been implicated in the interaction with pathological forms of tau through specific receptors or chaperones and deliver them to the lysosome28, 37. It is also possible that the lack of effect in wild-type mice or neurons is due to compensatory activities by TFEB family members. Although the precise mechanism remains to be elucidated, the specific modulation of human mutant tau by TFEB is in agreement with our earlier report that AAV-mediated TFEB overexpression selectively clears pathological tau and attenuates the NFT pathology35.
Besides lysosomal exocytosis, overwhelming evidence demonstrates that TFEB mediates intracellular protein clearance through the autophagy-lysosomal pathway (ALP). Thus, TFEB manipulation is expected to simultaneously affect both pathways and tau release may be downstream of the ALP. Our results that changes of extracellular tau can be attained by manipulating Ca2+ and TRPML1 independent of autophagy supports a primary and active role of tau exocytosis by TFEB rather than secondary to autophagy. Consistent with our finding, Tsunemi et al. recently showed that activation of TRPML1 enhances α-synuclein exocytosis and prevents its intracellular accumulation in iPSC-derived neurons49. Nevertheless, TFEB likely modulates intracellular tau dynamics through a combined effect of autophagy-lysosomal degradation and lysosomal exocytosis. The precise relationship of the two pathways and the contribution of each pathway in tau clearance remains to be deciphered. In addition, impaired lysosomal clearance and pathogenic tau accumulation in the absence of TFEB may influence other cellular pathways, which may in turn modulate tau secretion and spreading.
Our in vitro studies provide mechanistic support for the in vivo observation that PS19 mice lacking TFEB is associated with reduced ISF tau and increased intracellular tau pathology and spreading. However, it is important to note the significant differences between the two systems. Among others, the extracellular tau measured in the conditioned media of cultured neurons is the direct result of neuronal release whereas the ISF tau from the mouse brain represents a combined effect of secretion and uptake from multiple cell types, particularly microglia. In this regard, while some studies reported that microglia mediate extracellular tau uptake and degradation50, 51, others have shown that microglia facilitate tau spreading via exosome secretion of tau52, 53. Since the microdialysis probe used here captures mainly the monomeric and low N tau, but not oligomeric or exosomal tau23, 52, 54, the involvement of microglia in extracellular tau dynamics and pathological spreading is a possibility but not addressed here. Nevertheless, since microglial TFEB is not disturbed in the cKO mice, any contribution by microglia should be consistent between PS19 and PS19, cKO mice.
Our result that the ISF tau and intracellular pathology is inversely correlated may at first glance appear to contradict with Holth et al., which demonstrates that chronic sleep deprivation elevates ISF tau and increases its pathological spreading55. Since the ISF tau is detected by the mid-domain ELISA that does not distinguish between MTBR truncated and full-length tau, the different outcomes could be caused by the different tau species secreted by TFEB and sleep deprivation, but both are captured by the detection method. Indeed, the increased ISF tau by sleep deprivation is likely dictated by neuronal activity43, and activity-dependent neuronal release of full-length tau has been shown to promote the propagation of tau pathology18. Our results that reduced ISF tau in PS19, TFEB cKO mice is associated with increased synaptosomal AT8 positive phospho-tau is in keeping with this idea and indicate that this pool of tau may contribute to augmented spreading. However, additional methods are needed to identify the different ISF tau species and decipher their functional properties.
Given the correlation between NFTs and AD clinical progression, strategies aimed at targeting toxic tau proteins and NFTs are increasingly recognized as promising therapeutic approaches. Our findings have important implications with regard to both extracellular and intracellular tau targeting. In terms of extracellular tau, anti-tau antibodies have been shown to reduce tau pathology in mouse models and are currently being tested in clinical trials as disease-modifying therapies for AD and other tauopathy conditions9, 19–21. Our results, together with published reports32, 33, indicate that a significant amount of extracellular tau are truncated and seeding incompetent. These tau species may bind to antibodies that recognizes the mid-domain region of tau and reduce their therapeutic efficacies. Thus, effective tau immunotherapy should be designed to target the seeding competent tau. With respect to the broader tau therapy, our study uncovers a physiological role of TFEB in mediating tau exocytosis and we present evidence that this pathway acts an active mechanism to promote tau clearance and, therefore, is therapeutically advantageous. This, combined with our previous studies showing beneficial effects of exogenous TFEB in addressing the tau pathology35, 47, support the concept that TFEB has the ability to selectively target the pathological tau species for clearance through potent activation of multiple cellular degradative pathways. Although potential side-effects should be taken into careful consideration, this specificity and efficacy make TFEB an attractive therapeutic target for AD and other tauopathy diseases.
Supplementary Material
Acknowledgements
We are grateful to R. Youle (NIH) for the gift of TFEB KO and TFEB, MITF and TFE3 TKO Hela cells, M. Sardiello for the TFEB overexpressing TFEB Hela line, C. Cook and L. Petrucelli for the tau P301L expression vector and virus, and P. Davies (Feinstein Institute for Medical Research) for PHF1 and MC1 antibodies. We thank F. Stewart, B. Contreras and N. Aithmitti for expert technical assistance and members of the Zheng laboratory for insightful discussions. This project was supported by the Gene Vector Core of Baylor College of Medicine and by grants from the NIH (R01 NS093652, R01 AG020670, and RF1 AG054111 and R01 AG057509 to HZ; R01 AG062734, R56 NS110980 to CMK), the Tau Consortium and the Farrell Family Alzheimer’s Disease Research Fund (to CMK).
Footnotes
Declaration of Interests
The authors declare no competing financial interests.
References
- 1.Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci 2001; 24: 1121–1159. [DOI] [PubMed] [Google Scholar]
- 2.Mandelkow EM, Mandelkow E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harbor perspectives in medicine 2012; 2(7): a006247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gendron TF, Petrucelli L. The role of tau in neurodegeneration. Mol Neurodegener 2009; 4: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wisniewski T, Goni F. Immunotherapeutic approaches for Alzheimer’s disease. Neuron 2015; 85(6): 1162–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Panza F, Lozupone M, Logroscino G, Imbimbo BP. A critical appraisal of amyloid-beta-targeting therapies for Alzheimer disease. Nat Rev Neurol 2019; 15(2): 73–88. [DOI] [PubMed] [Google Scholar]
- 6.Karran E, De Strooper B. The amyloid cascade hypothesis: are we poised for success or failure? J Neurochem 2016; 139 Suppl 2: 237–252. [DOI] [PubMed] [Google Scholar]
- 7.Giannakopoulos P, Herrmann FR, Bussiere T, Bouras C, Kovari E, Perl DP et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 2003; 60(9): 1495–1500. [DOI] [PubMed] [Google Scholar]
- 8.Hanseeuw BJ, Betensky RA, Jacobs HIL, Schultz AP, Sepulcre J, Becker JA et al. Association of Amyloid and Tau With Cognition in Preclinical Alzheimer Disease: A Longitudinal Study. JAMA Neurol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khanna MR, Kovalevich J, Lee VM, Trojanowski JQ, Brunden KR. Therapeutic strategies for the treatment of tauopathies: Hopes and challenges. Alzheimers Dement 2016; 12(10): 1051–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991; 82(4): 239–259. [DOI] [PubMed] [Google Scholar]
- 11.Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem 2009; 284(19): 12845–12852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo JL, Lee VM. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem 2011; 286(17): 15317–15331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol 2009; 11(7): 909–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Calignon A, Polydoro M, Suarez-Calvet M, William C, Adamowicz DH, Kopeikina KJ et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 2012; 73(4): 685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, Lee VM. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J Neurosci 2013; 33(3): 1024–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 2014; 82(6): 1271–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamada K, Cirrito JR, Stewart FR, Jiang H, Finn MB, Holmes BB et al. In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J Neurosci 2011; 31(37): 13110–13117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu JW, Hussaini SA, Bastille IM, Rodriguez GA, Mrejeru A, Rilett K et al. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci 2016; 19(8): 1085–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chai X, Wu S, Murray TK, Kinley R, Cella CV, Sims H et al. Passive immunization with anti-Tau antibodies in two transgenic models: reduction of Tau pathology and delay of disease progression. J Biol Chem 2011; 286(39): 34457–34467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yanamandra K, Jiang H, Mahan TE, Maloney SE, Wozniak DF, Diamond MI et al. Anti-tau antibody reduces insoluble tau and decreases brain atrophy. Ann Clin Transl Neurol 2015; 2(3): 278–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yanamandra K, Kfoury N, Jiang H, Mahan TE, Ma S, Maloney SE et al. Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron 2013; 80(2): 402–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Toledo JB, Xie SX, Trojanowski JQ, Shaw LM. Longitudinal change in CSF Tau and Abeta biomarkers for up to 48 months in ADNI. Acta Neuropathol 2013; 126(5): 659–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saman S, Kim W, Raya M, Visnick Y, Miro S, Saman S et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem 2012; 287(6): 3842–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karch CM, Jeng AT, Goate AM. Extracellular Tau levels are influenced by variability in Tau that is associated with tauopathies. J Biol Chem 2012; 287(51): 42751–42762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chai X, Dage JL, Citron M. Constitutive secretion of tau protein by an unconventional mechanism. Neurobiol Dis 2012; 48(3): 356–366. [DOI] [PubMed] [Google Scholar]
- 26.Morris M, Knudsen GM, Maeda S, Trinidad JC, Ioanoviciu A, Burlingame AL et al. Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat Neurosci 2015; 18(8): 1183–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song L, Lu SX, Ouyang X, Melchor J, Lee J, Terracina G et al. Analysis of tau post-translational modifications in rTg4510 mice, a model of tau pathology. Mol Neurodegener 2015; 10: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y, Martinez-Vicente M, Kruger U, Kaushik S, Wong E, Mandelkow EM et al. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet 2009; 18(21): 4153–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Michel CH, Kumar S, Pinotsi D, Tunnacliffe A, St George-Hyslop P, Mandelkow E et al. Extracellular monomeric tau protein is sufficient to initiate the spread of tau protein pathology. J Biol Chem 2014; 289(2): 956–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mirbaha H, Chen D, Morazova OA, Ruff KM, Sharma AM, Liu X et al. Inert and seed-competent tau monomers suggest structural origins of aggregation. Elife 2018; 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meredith JE Jr., Sankaranarayanan S, Guss V, Lanzetti AJ, Berisha F, Neely RJ et al. Characterization of novel CSF Tau and ptau biomarkers for Alzheimer’s disease. PLoS One 2013; 8(10): e76523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kanmert D, Cantlon A, Muratore CR, Jin M, O’Malley TT, Lee G et al. C-Terminally Truncated Forms of Tau, But Not Full-Length Tau or Its C-Terminal Fragments, Are Released from Neurons Independently of Cell Death. J Neurosci 2015; 35(30): 10851–10865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sato C, Barthelemy NR, Mawuenyega KG, Patterson BW, Gordon BA, Jockel-Balsarotti J et al. Tau Kinetics in Neurons and the Human Central Nervous System. Neuron 2018; 97(6): 1284–1298 e1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA et al. A gene network regulating lysosomal biogenesis and function. Science 2009; 325(5939): 473–477. [DOI] [PubMed] [Google Scholar]
- 35.Polito VA, Li H, Martini-Stoica H, Wang B, Yang L, Xu Y et al. Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Mol Med 2014; 6(9): 1142–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol 2013; 15(6): 647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu Y, Zhang S, Zheng H. The cargo receptor SQSTM1 ameliorates neurofibrillary tangle pathology and spreading through selective targeting of pathological MAPT (microtubule associated protein tau). Autophagy 2019; 15(4): 583–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu Y, Martini-Stoica H, Zheng H. A seeding based cellular assay of tauopathy. Mol Neurodegener 2016; 11: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nezich CL, Wang C, Fogel AI, Youle RJ. MiT/TFE transcription factors are activated during mitophagy downstream of Parkin and Atg5. J Cell Biol 2015; 210(3): 435–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karch CM, Kao AW, Karydas A, Onanuga K, Martinez R, Argouarch A et al. A Comprehensive Resource for Induced Pluripotent Stem Cells from Patients with Primary Tauopathies. Stem Cell Reports 2019; 13(5): 939–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang S, Wen N, Li Z, Dube U, Del Aguila J, Budde J et al. Integrative system biology analyses of CRISPR-edited iPSC-derived neurons and human brains reveal deficiencies of presynaptic signaling in FTLD and PSP. Transl Psychiatry 2018; 8(1): 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abu-Remaileh M, Wyant GA, Kim C, Laqtom NN, Abbasi M, Chan SH et al. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 2017; 358(6364): 807–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamada K, Holth JK, Liao F, Stewart FR, Mahan TE, Jiang H et al. Neuronal activity regulates extracellular tau in vivo. J Exp Med 2014; 211(3): 387–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Croft CL, Wade MA, Kurbatskaya K, Mastrandreas P, Hughes MM, Phillips EC et al. Membrane association and release of wild-type and pathological tau from organotypic brain slice cultures. Cell Death Dis 2017; 8(3): e2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Medina DL, Fraldi A, Bouche V, Annunziata F, Mansueto G, Spampanato C et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell 2011; 21(3): 421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.LaPlante JM, Sun M, Falardeau J, Dai D, Brown EM, Slaugenhaupt SA et al. Lysosomal exocytosis is impaired in mucolipidosis type IV. Mol Genet Metab 2006; 89(4): 339–348. [DOI] [PubMed] [Google Scholar]
- 47.Martini-Stoica H, Cole AL, Swartzlander DB, Chen F, Wan YW, Bajaj L et al. TFEB enhances astroglial uptake of extracellular tau species and reduces tau spreading. J Exp Med 2018; 215(9): 2355–2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martini-Stoica H, Xu Y, Ballabio A, Zheng H. The Autophagy-Lysosomal Pathway in Neurodegeneration: A TFEB Perspective. Trends Neurosci 2016; 39(4): 221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsunemi T, Perez-Rosello T, Ishiguro Y, Yoroisaka A, Jeon S, Hamada K et al. Increased Lysosomal Exocytosis Induced by Lysosomal Ca(2+) Channel Agonists Protects Human Dopaminergic Neurons from alpha-Synuclein Toxicity. J Neurosci 2019; 39(29): 5760–5772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luo W, Liu W, Hu X, Hanna M, Caravaca A, Paul SM. Microglial internalization and degradation of pathological tau is enhanced by an anti-tau monoclonal antibody. Sci Rep 2015; 5: 11161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Audrain M, Haure-Mirande JV, Wang M, Kim SH, Fanutza T, Chakrabarty P et al. Integrative approach to sporadic Alzheimer’s disease: deficiency of TYROBP in a tauopathy mouse model reduces C1q and normalizes clinical phenotype while increasing spread and state of phosphorylation of tau. Mol Psychiatry 2019; 24(9): 1383–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 2015; 18(11): 1584–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hopp SC, Lin Y, Oakley D, Roe AD, DeVos SL, Hanlon D et al. The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. J Neuroinflammation 2018; 15(1): 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Y, Balaji V, Kaniyappan S, Kruger L, Irsen S, Tepper K et al. The release and trans-synaptic transmission of Tau via exosomes. Mol Neurodegener 2017; 12(1): 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Holth JK, Fritschi SK, Wang C, Pedersen NP, Cirrito JR, Mahan TE et al. The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science 2019; 363(6429): 880–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.