

Abstract
A copper-based complex that contains a sulfonate N-heterocyclic carbene ligand was first reported 15 years ago. Since then, these organometallic entities have proven to be uniquely effective in catalyzing an assortment of enantioselective transformations, including allylic substitutions, conjugate additions, proto-boryl additions to alkenes, boryl and silyl substitutions, hydride–allyl additions to alkenyl boronates, and additions of boron-containing allyl moieties to N-H ketimines. In this review article, we detail the shortcomings in the state-of-the-art that fueled the development of this air stable ligand class, members of which can be prepared on multigram scale. For each reaction class, when relevant, the prior art at the time of the advance involving sulfonate NHC–Cu catalysts and/or subsequent key developments are briefly analyzed, and its relevance to efficient and enantioselective total or formal synthesis of biologically active molecules is underscored. Mechanistic analysis of the structural attributes of sulfonate NHC–Cu catalysts that are responsible for their ability to facilitate transformations with high efficiency as well as regio- and enantioselectivity are detailed. This review contains several formerly undisclosed methodological advances and mechanistic analyses, the latter of which constitute a revision of previously reported proposals.
Keywords: allylic substitutions, allyl additions, boron, catalysis, conjugate additions, copper, enantioselective catalysis, NHC ligands, proto-boryl additions, silicon, sulfonate NHC ligands
Graphical Abstract
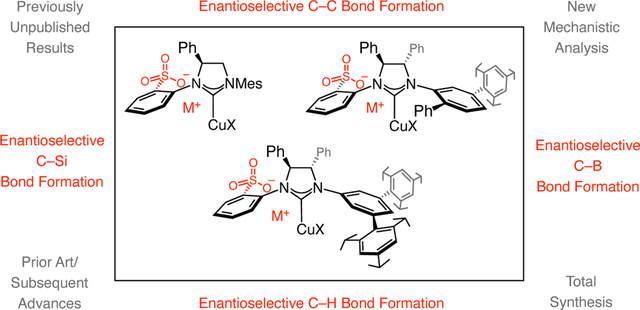
Catalysis by metal bridging: Sulfonate NHC–Cu catalysts promote a variety of transformations with uniquely high efficiency as well as regio-, and enantioselectivity. The origins of the discovery of these chiral complexes, the process that they are adept at catalyzing, comparison with alternative approaches, and different applications to total synthesis are presented. Also provided are mechanistic analyses, which underscore the significance of sulfonate–metal bridges.
“…I believe that every militant chemist can confirm it: that one must distrust the almost-the-same…, the practically identical, the approximate, the or-even, all surrogates and all patchwork. The differences can be small, but they can lead to radically different consequences…; the chemist’s trade consists in good part in being aware of these differences, knowing them close up, and foreseeing their effects.”
Primo Levi in “The Periodic Table”
1. Introduction
Catalysts that can promote the formation of different types of bonds with high efficiency, regio-, diastereo-, and enantioselectivity are central to advances in chemistry, biology, and medicine. The subject of this review is a class of sulfonate NHC–Cu (NHC = N-heterocyclic carbene) complexes that catalyze, often with uniquely high efficiency and selectivity, the generation of C–C, C–B, C–H, and C–Si bonds. For each reaction class, if applicable, the state-of-the-art at the time of application of sulfonate NHC–Cu complexes and/or subsequent related advances are briefly analyzed. Key mechanistic nuances of sulfonate NHC–Cu-catalyzed transformations will also be provided. This review contains previously unpublished methodological progress as well as mechanistic experiments and analyses.
2. State-of-the-Art and Initial Basis for Ligand Design
Two decades ago, we were engaged in the development of NHC–Ru and NHC–Cu catalysts for enantioselective olefin metathesis,[1] allylic substitution (EAS),[2] and conjugate addition (ECA).[3] We designed catalysts with a binaphthol ligand (e.g., NHC(O)–Cu-1a, Scheme 1a), prepared by treatment of the corresponding Ag complex (e.g., NHC(O)–Ag-1a) with a Ru- or Cu-halide species. As a more conformationally flexible system, we prepared and probed the activity of biphenolate complexes (e.g., NHC(O)–Cu-2a, Scheme 1a), which can be generated diastereoselectively upon metal complexation [>98:2 diastereomeric ratio (d.r.)] with the chelating phenoxy group oriented away from the NHC’s proximal phenyl group. The biphenolate catalysts often proved more effective, highlighted by an EAS to form a quaternary carbon stereogenic center (Scheme 1b). Still, a number of notable processes remained low yielding and/or minimally enantioselective (e.g., Scheme 1c).
Scheme 1.
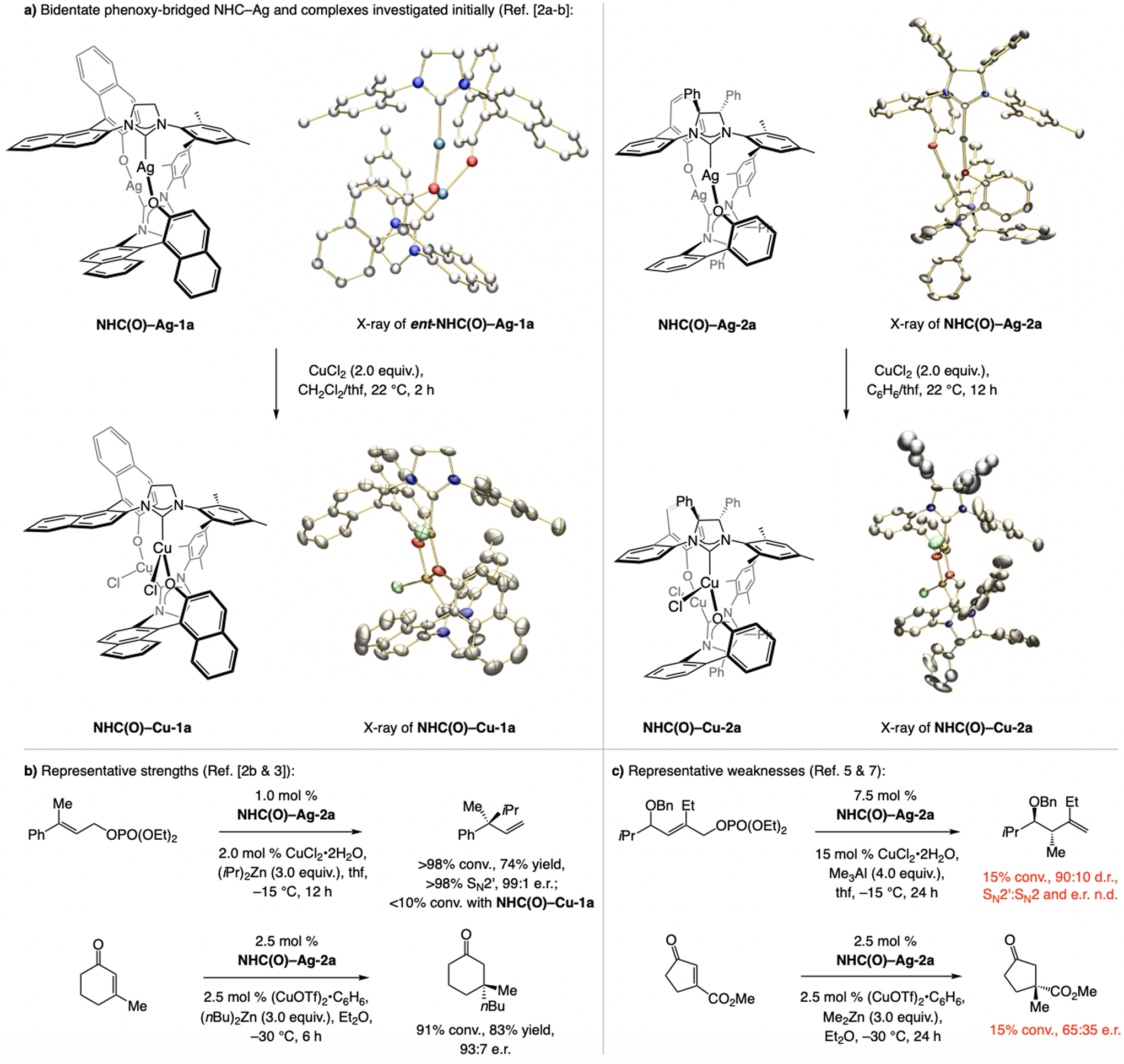
First- and second-generation aryloxy Ag- and Cu-based N-heterocyclic carbene (NHC) complexes used in enantioselective allylic substitutions and conjugate additions. n.d. = not determined.
We reasoned that reactions that proceed via intermediates containing a pendant sulfonate unit might be more efficient due to faster oxidative addition (to generate Cu(III)–alkyl species) and/or reductive elimination (to form a C–C bond; Scheme 2a). These expectations were supported by the studies of Vedernikov, concerning reactions of Pt-alkyl complexes with molecular oxygen, transformations that were shown to be facilitated by an anionic di(2-pyridyl)methanesulfonate ligand (Scheme 2b).[4] Vedernikov proposed that because of the proximal sulfonate group the transition metal’s dz2 electrons should be higher in energy (Scheme 2b), and the electron-withdrawing sulfonate might lower the energy of the vacant dz2 orbital of the Pt(IV) complex, diminishing the barrier to addition of water and subsequent MeOH generation. As we shall see, although our initial suppositions did lead us to pursue and develop an effective new class of NHC–Cu catalysts, the mechanistic basis is probably not as originally envisioned.
Scheme 2.
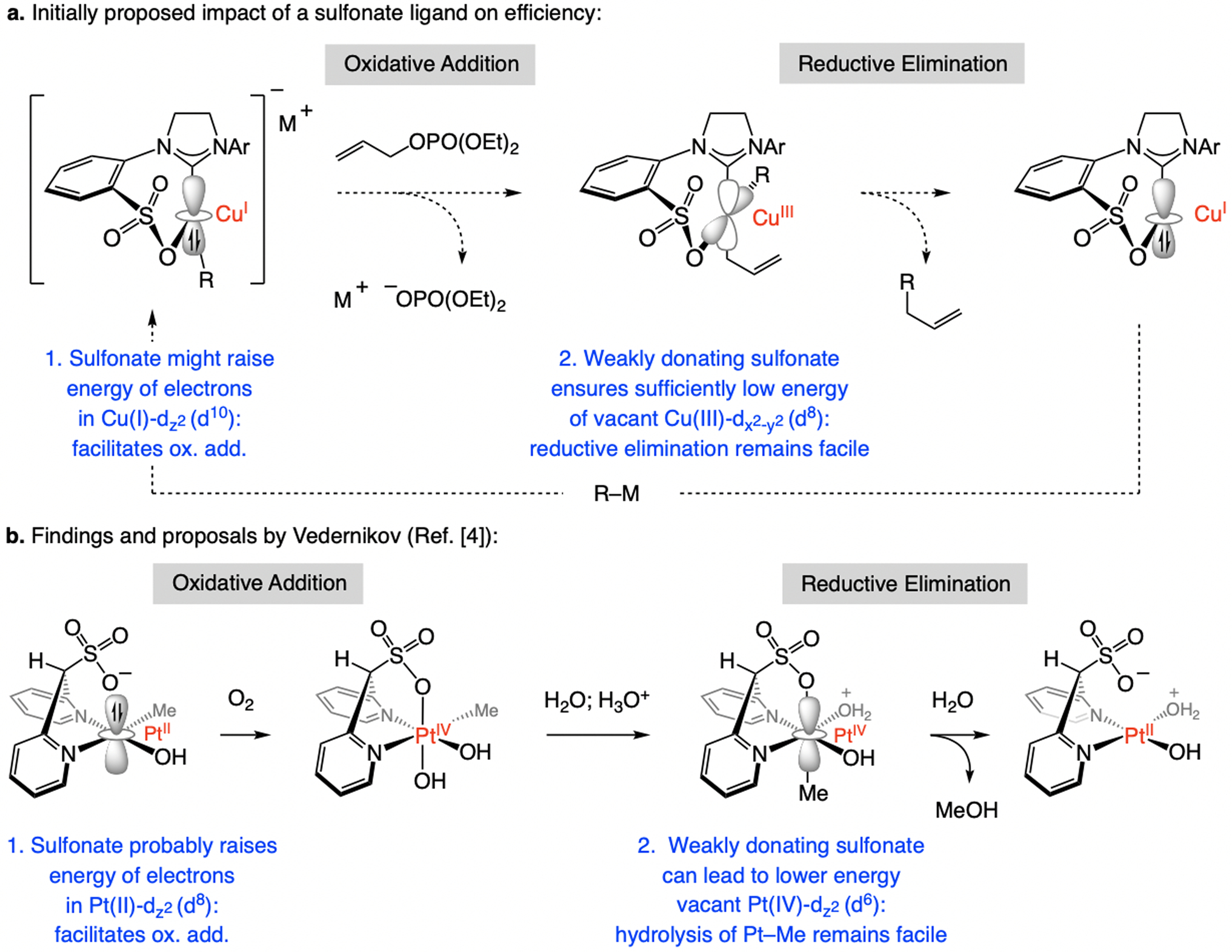
Regarding the influence of a sulfonate NHC ligand on oxidative addition and reductive elimination steps.
3. Sulfonate Imidazolinium Salts and NHC–Ag Complexes
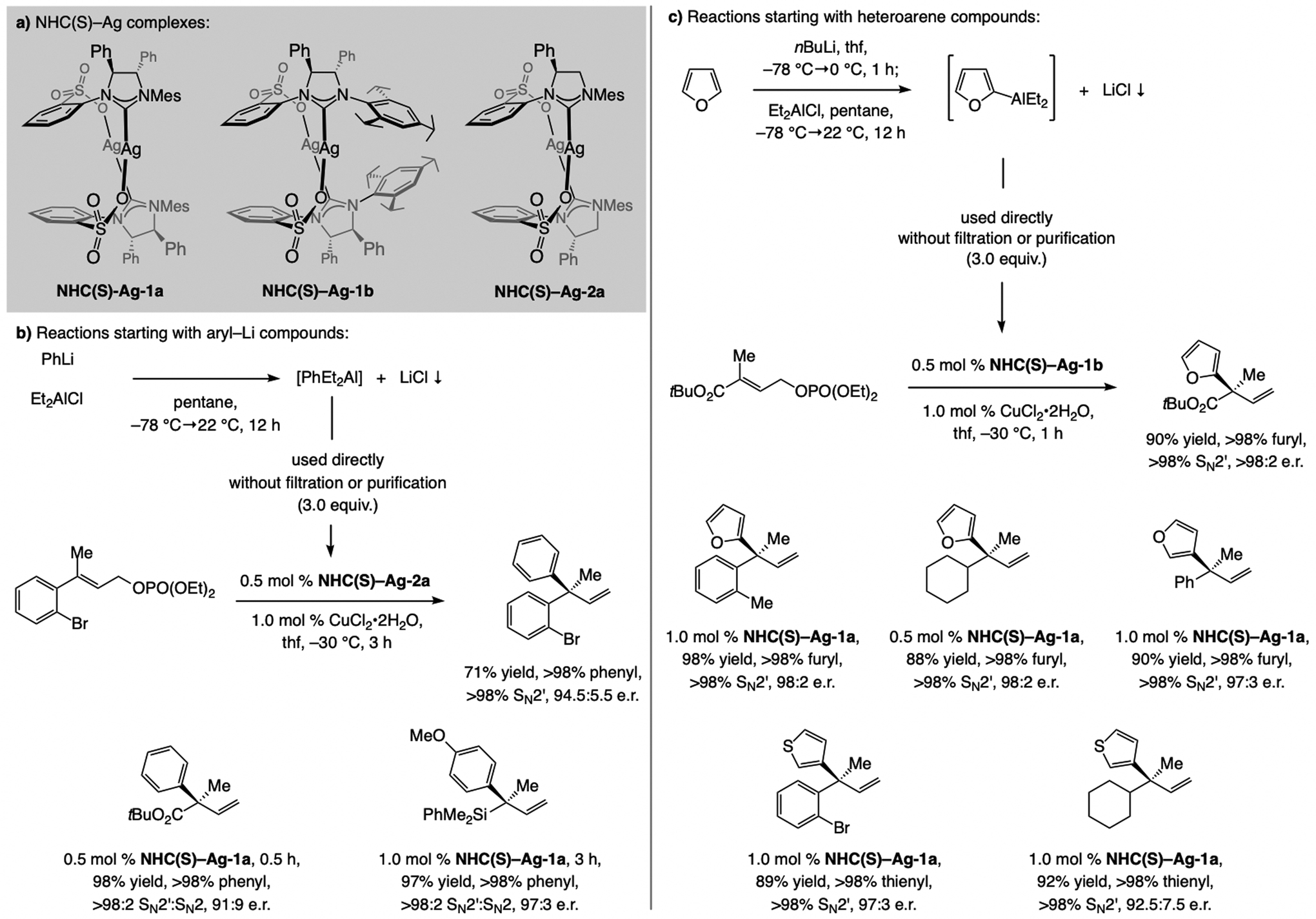
Ligand synthesis. The requisite dianiline sulfonate (Scheme 3) was prepared in multi-gram quantities by sequential catalytic cross-coupling with a commercially available enantiomerically enriched diamine. Subjection of the dianiline to dimethylmethylideneammonium iodide afforded imid(S)-1a (79% yield), likely via the ammonium salt formed by reaction of the amine. Head-to-tail dimer NHC(S)–Ag-1a was then obtained quantitatively and in >98:2 d.r. as an air-stable white solid by subjection of imid(S)-1a to Ag2O and 4Å molecular sieves.[5] Spectroscopic analysis of NHC(S)–Ag-1a indicated altered distribution of electron density compared to the aryloxy complexes (e.g., NHC(O)-Ag-2a).
Scheme 3.
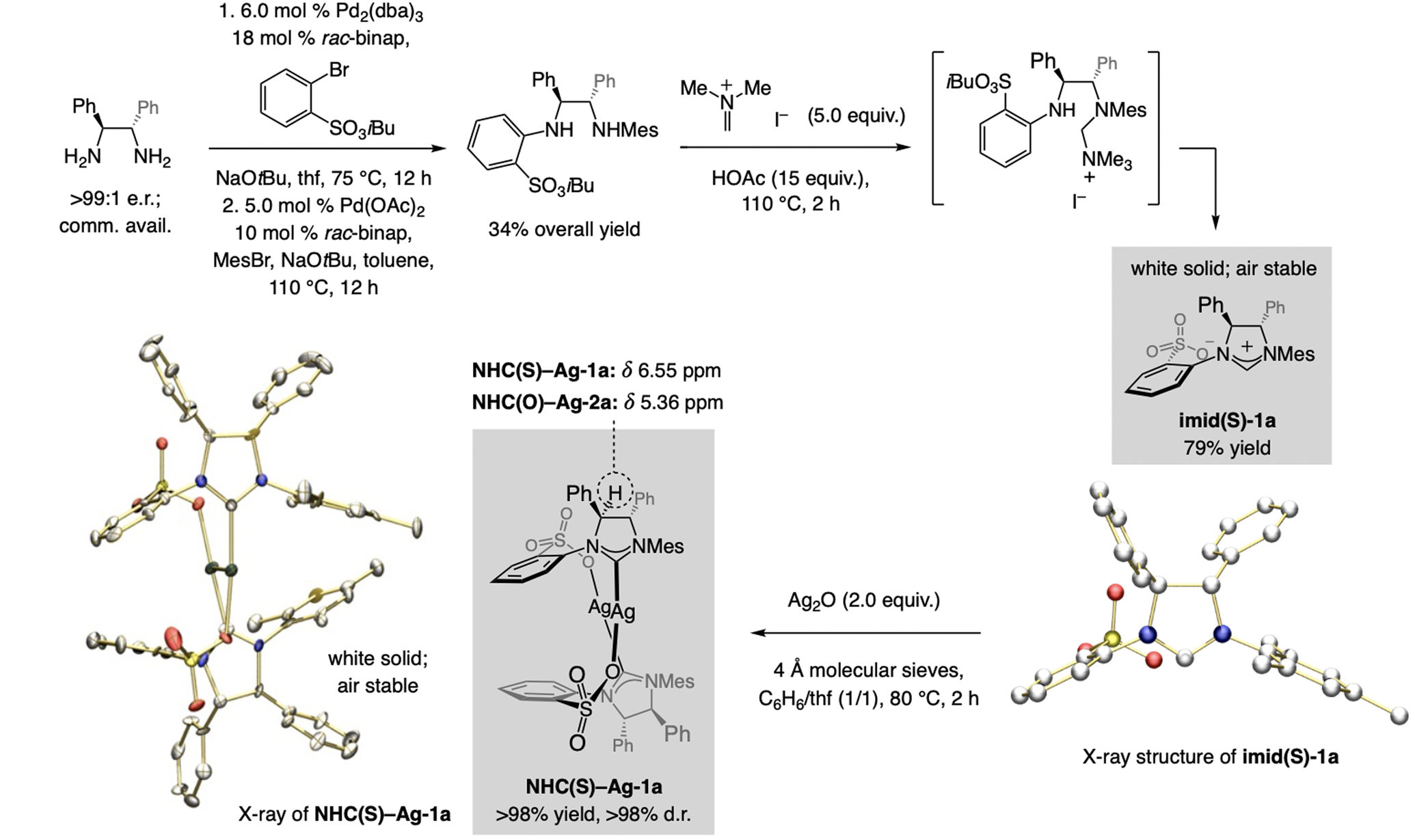
Preparation of a sulfonate imidazolinium salt and the derived NHC–Ag complex.
Ease of preparation and use. Other sulfonate imidazolinium salts are accessible in multi-gram quantities by a similar route (see below). The dimeric NHC–Ag complexes, which should be stored in the dark (e.g., container wrapped with aluminum foil), are indefinitely stable. We have used five-year old samples without encountering a change in efficiency or selectivity. Use of a sulfonate NHC–Cu complex does not require pre-distillation of solvents, or rigorously oxygen-free and anhydrous conditions.
4. Catalytic Enantioselective Allylic Substitution (EAS)
Sulfonate NHC–Cu catalysts are uniquely effective in promoting highly SN2’-selective EAS[6] with organoaluminum, organozinc, or organoboron compounds.
4.1. With Organoaluminum and Organozinc Compounds
4.1.1. With Me3Al.
An early indication suggesting the distinctiveness of sulfonate NHC–Cu catalysts presented itself in a total synthesis of bacinopyrone C (Scheme 4).[7] A key sequence entailed EAS with Me3Al and a bis-phosphate/diastereoselective allylic substitution. Model studies had indicated that under substrate-control the meso isomer is favored (~1.5:1); the stereochemical outcome of the second substitution thus needed to be catalyst-controlled. Reactions with complexes lacking a sulfonate were minimally stereoselective, and it was only through the use of NHC(S)-Ag-1a that we could obtain pure S,S-isomer in 61% yield (pure S,S isomer), 89:11 d.r., and >99:1 enantiomeric ratio (e.r.).
Scheme 4.
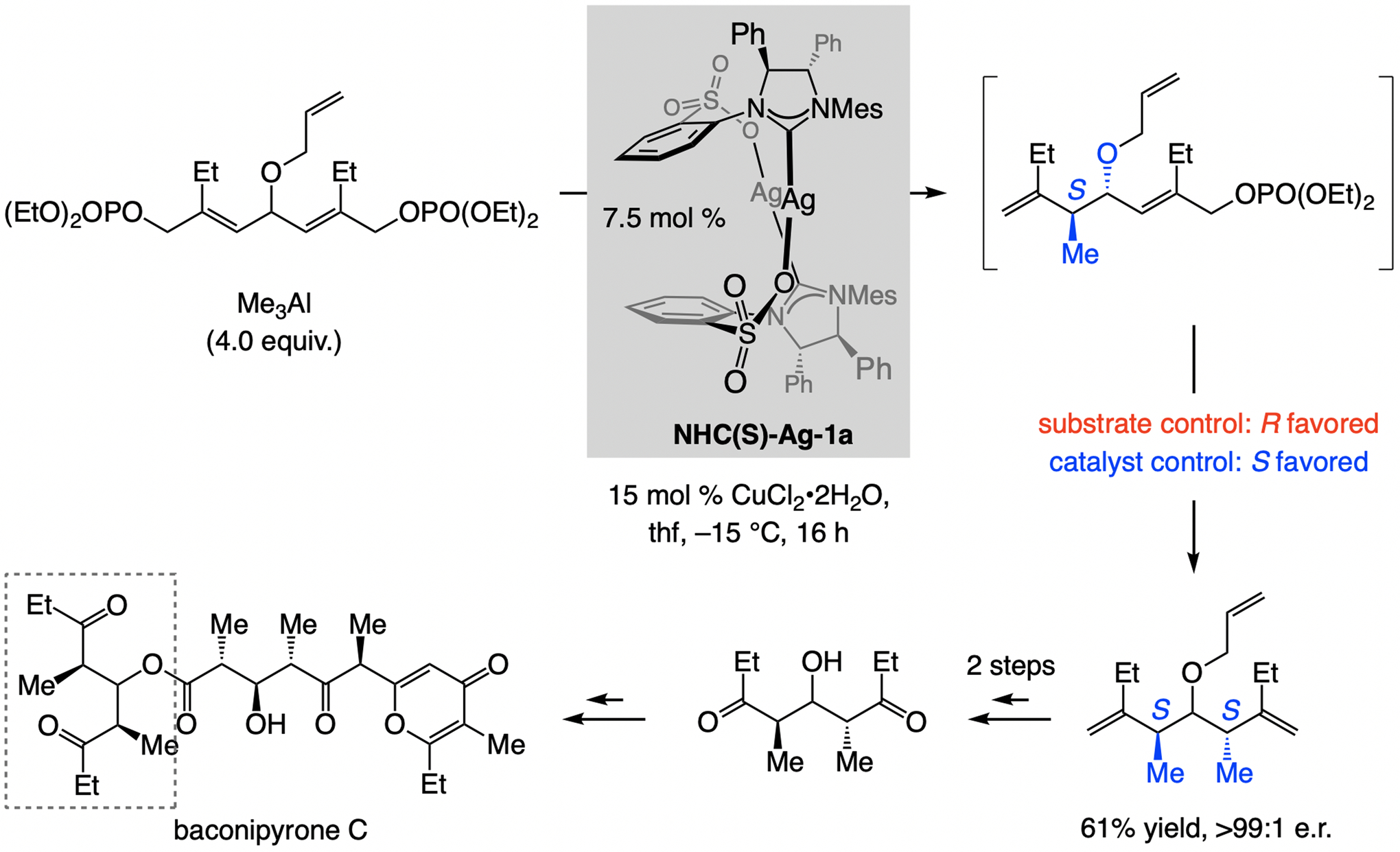
Tandem EAS with Me3Al in a total synthesis of natural product baconipyrone C.
4.1.2. With alkenyl–Al compounds.
The alkenyl–Al compounds (vs. Mg- or Zn-based[8])can be accessed by reaction between a terminal alkyne and diisobutylaluminum hydride (dibal–H; Scheme 5).[9] Although (E)-β-alkenyl–Al compound was easily synthesized by treatment of 1-octyne with dibal–H (Scheme 5),[10] there was no reaction with 1.0 mol % CuCN or CuCl2•2H2O, and only 65% conversion to a mixture of three compounds with one equivalent of CuCN. Nor was any of the desired product detected when Cu complexes derived from an achiral NHC ligand or NHC(O)–Ag-2a were used. Remarkably, however, with just 1.0 mol % of the complex generated from NHC(S)–Ag-1a the desired isomer was isolated in 87% yield, >98% SN2’ selectivity, >98% group selectivity (<2% iBu addn), and >99:1 e.r. In certain cases, the catalyst derived from NHC(S)–Ag-2a, lacking a backbone phenyl group, was somewhat more effective.[11]
Scheme 5.
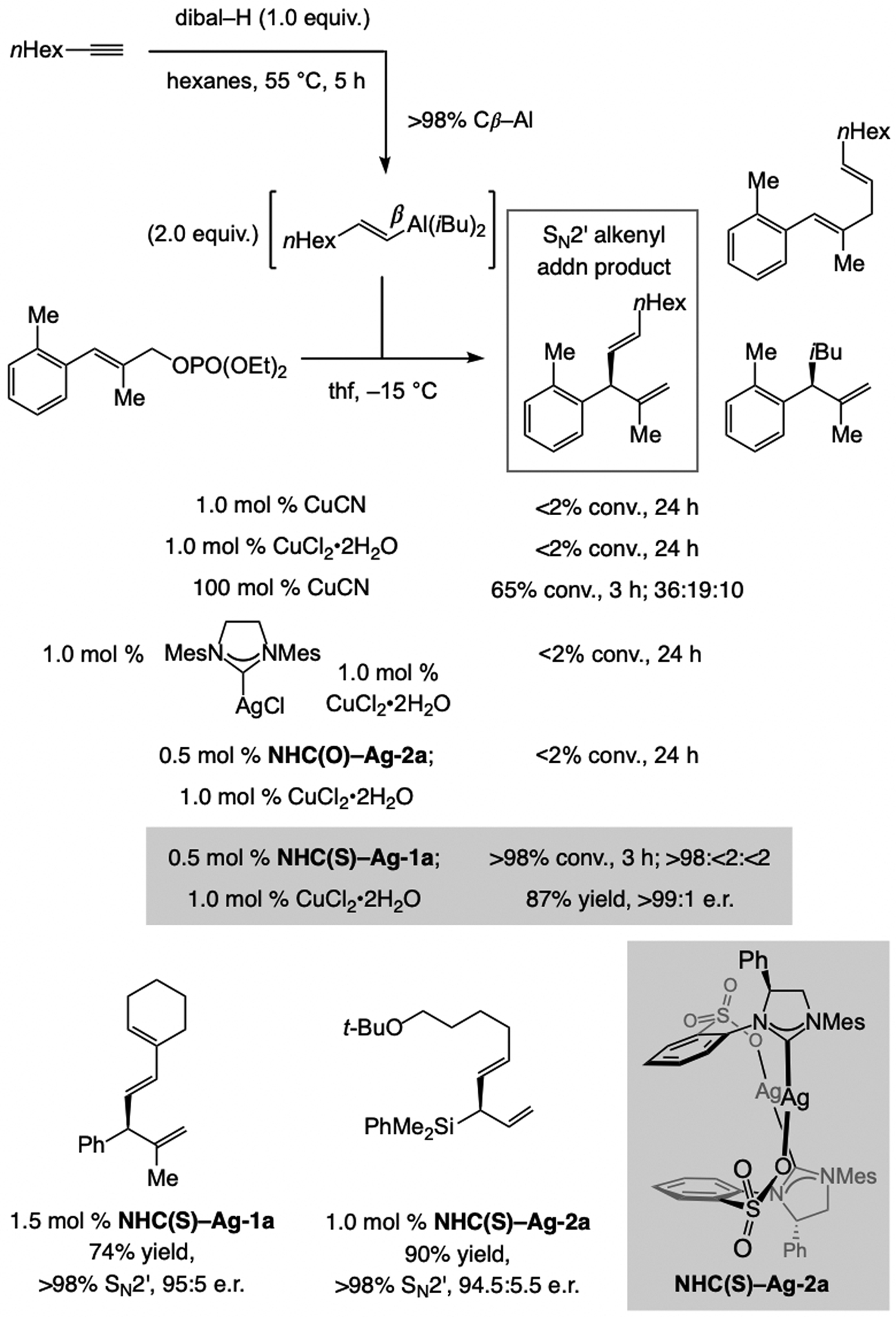
Regio- and stereoselective Al–H addition to an alkyl-substituted terminal alkyne may be followed by a regio- and enantioselective allylic substitution of the in situ generated alkenyl–Al compound (see Scheme 36 for mechanistic analysis).
4.1.3. With Si-substituted alkenyl–Al compounds.
Reaction with dibal–H is inapplicable to aryl- or heteroaryl alkynes, since removal of the more acidic alkynyl proton is competitive with Al–H addition (Scheme 6a). Nor can Z-alkenyl–Al compounds be accessed in this way. These limitations were addressed by the use of silyl-substituted alkynes. The Z-silyl-substituted-alkenyl–Al complex (Scheme 6b) was prepared with complete regio- and stereoselectivity and utilized in EAS [the smaller Me2HSi-substituted alkynes were more efficient (vs. Me3Si)], [12] optimally promoted by the catalyst derived from NHC(S)–Ag-1b. A hexanes/thf mixture is the optimal medium because in pure hexane the E-alkenylsilane isomer is generated preferentially (Scheme 6c). Without a thf molecule bound to the Al center, the initial Z-alkenyl–Al isomer i, in resonance with ii, can isomerize to reduce steric strain. The catalyst derived from NHC(S)–Ag-2c emerged as the most effective for EAS with E-alkenyl–Al compounds (Scheme 6c), allowing for a concise enantioselective synthesis of nyasol (Scheme 6d).
Scheme 6.
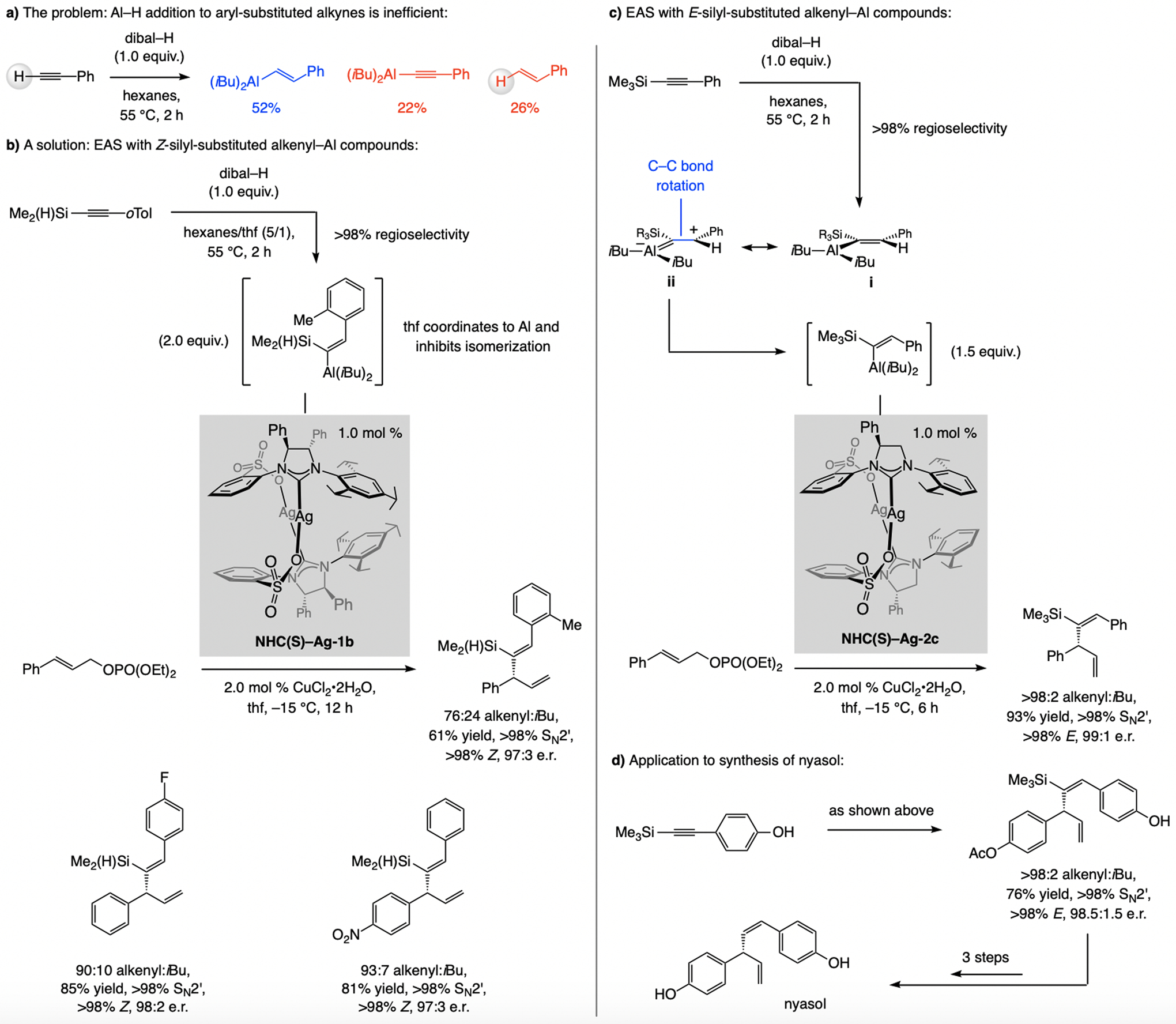
Regio- and E-selective Al–H additions to silyl-substituted aryl alkynes may be used to form (Z)- or (E)-β-alkenyl–Al compounds, applicable to catalytic EAS (see Scheme 37 for mechanistic analysis).
4.1.4. Phosphine–Ni-catalyzed Al–H addition merged with EAS.
Another shortcoming was the low efficiency of EAS with silyl-substituted alkenyl–Al species and trisubstituted olefins to generate a quaternary carbon stereogenic center[13] (Scheme 7a). Similar reactions but with trisubstituted alkenes and non-silyl-containing alkenyl-Al complexes were efficient and regio- and enantioselective,[14] suggesting that the size of the silyl group is problematic. What was needed was a catalytic approach to synthesis of alkenyl–Al compounds directly from an alkyne, one that would be sufficiently fast to render the aforementioned side reactions non-competitive.
Scheme 7.
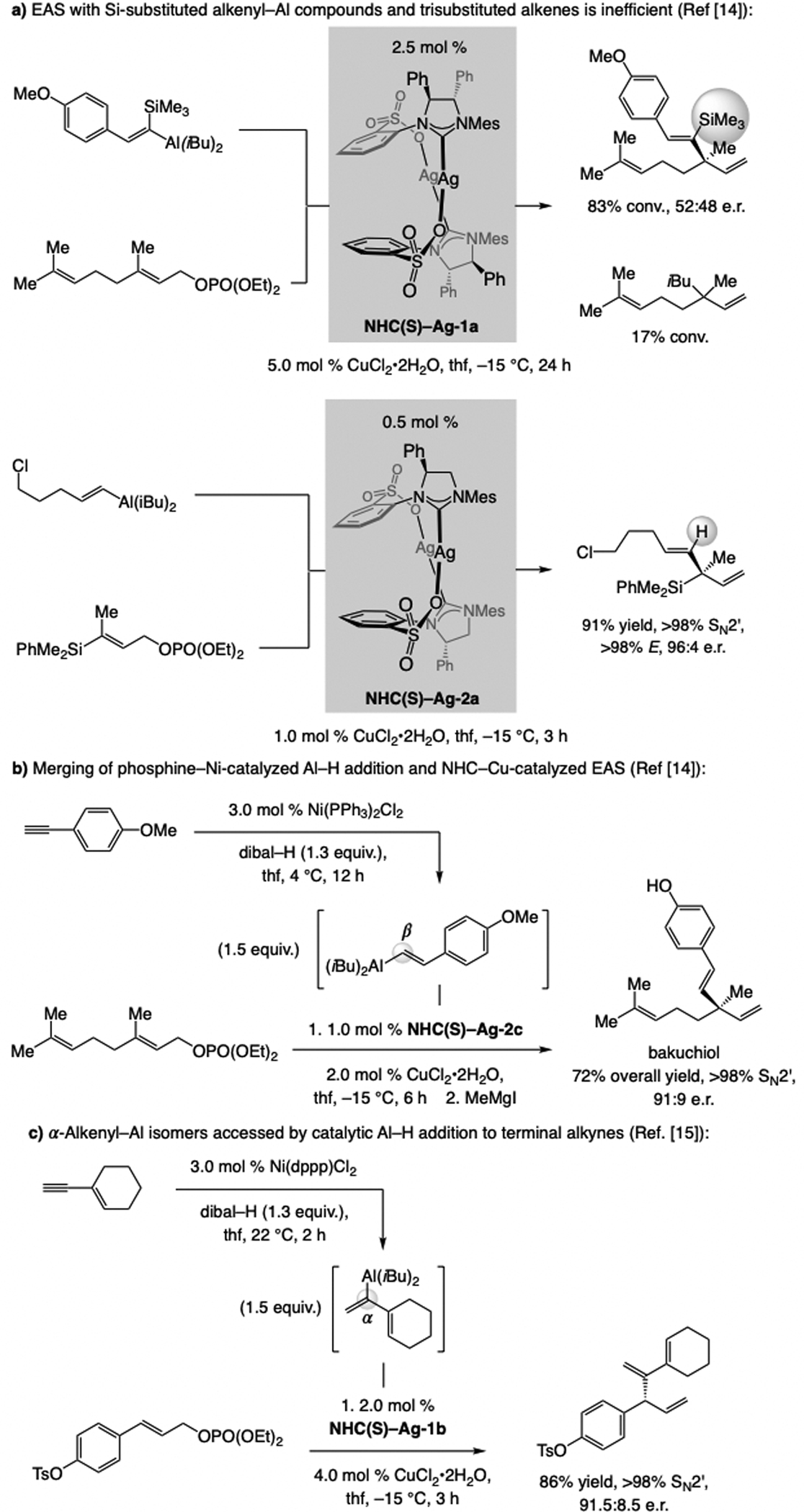
Ni-catalyzed Al–H additions to monosubstituted alkynes to generate Z- or E-alkenyl–Al compounds, and subsequent EAS.
(Ph3P)2Ni-catalyzed Al–H addition[15] to a monosubstituted alkyne turns out to be sufficiently facile to out-compete proton removal (Scheme 7b). A concise synthesis of bakuchiol could thus be achieved involving EAS catalyzed by the NHC–Cu complex derived from NHC(S)–Ag-2c.[16] Equally important, with a bidentate bis-phosphine–Ni catalyst, regioselectivity of the Al–H addition can be reversed and the alternative α-alkenyl–Al isomer can be obtained. The corresponding EAS are similarly efficient, regio- and enantioselective (Scheme 7c).
4.1.5. With alkynyl–Al compounds.
A key corollary to catalytic Al–H addition to alkynes is that by including 5.0 mol % Et3N (Scheme 8a), the alkynyl–Al product can be generated efficiently (<2% alkenyl–Al).[17] The alkynyl–Al compounds may be used in EAS catalyzed by an NHC(S)–Cu complex, affording 1,4-enynes with complete SN2’ selectivity and high e.r.;[18] as far as we know, these are the first reported instances of catalytic EAS with an alkynylmetal compound. Upon treatment of a product with dbu, enantiomerically enriched trisubstituted allenes were formed (Scheme 8a).[19] With the NHC–Cu complex derived from NHC(S)–Ag-2c 1,4-enynes containing a quaternary carbon stereogenic center could be synthesized (Scheme 8b).[20]
Scheme 8.
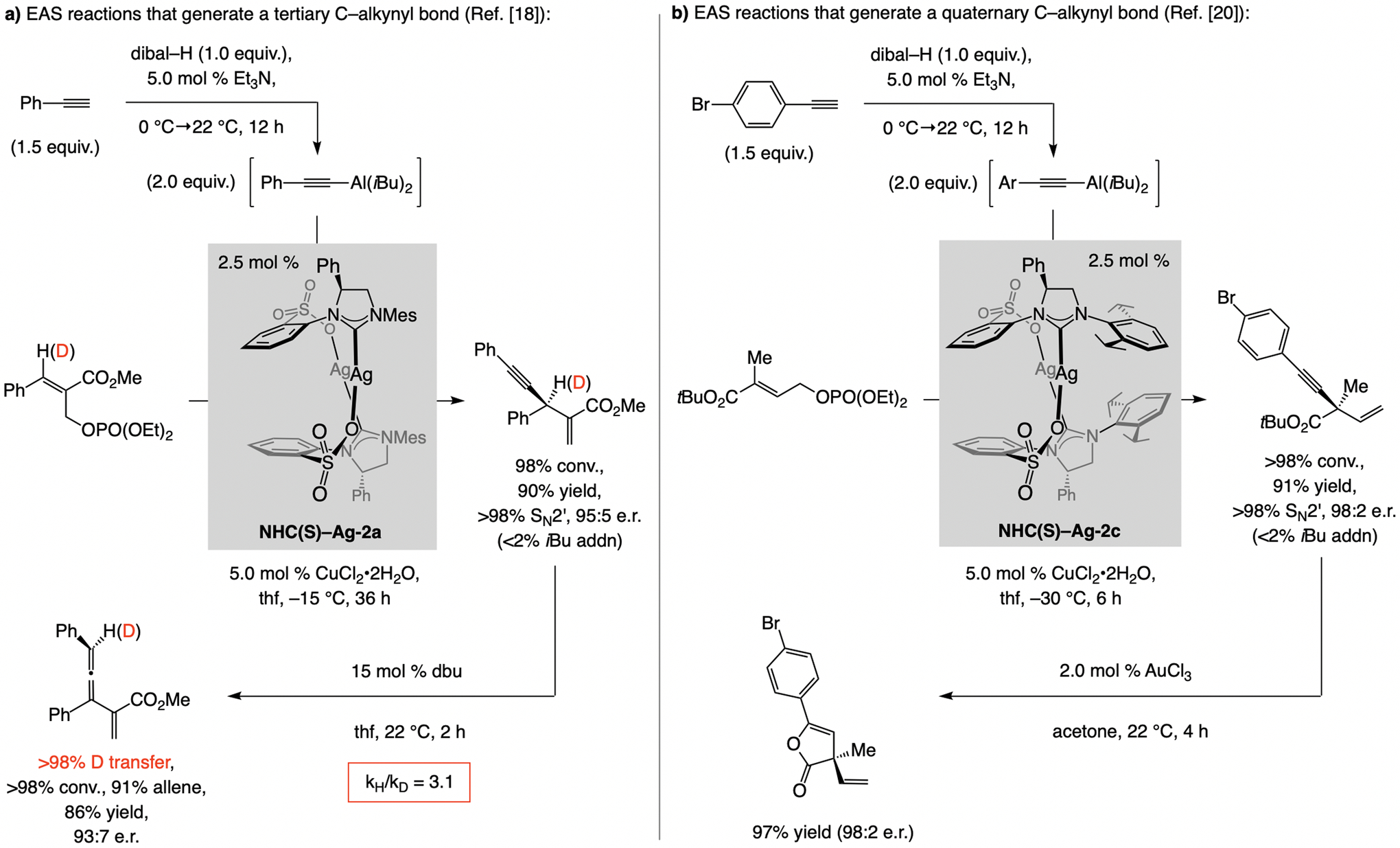
With 5.0 mol % Et3N, reaction of a terminal alkyne with dibal–H affords only the alkynyl–Al compound, which may be used for catalytic EAS.
4.1.6. With aryl–Zn compounds.
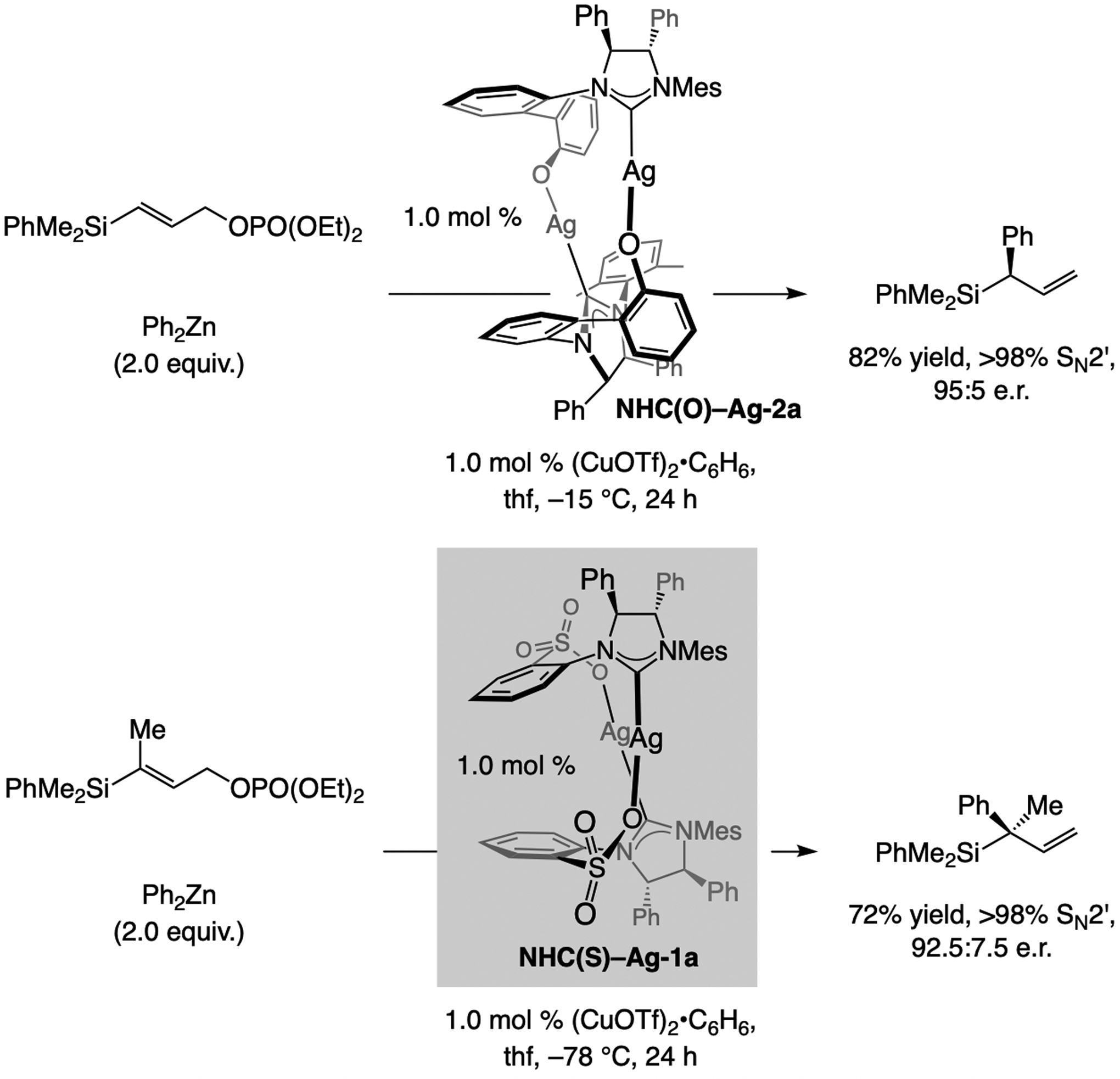
The first examples of EAS, catalyzed by a Cu-based complex, which form a C–aryl bond were reported in 2007. These reactions were promoted by NHC–Cu catalysts and involved silyl-substituted allylic phosphates and (aryl)2Zn reagents (Scheme 9).[2d] The complex derived from NHC(O)–Ag-2a was optimal for generating a tertiary carbon center, but with trisubstituted alkenes NHC(S)–Ag-1a was the most effective.[21,22,23,24]
Scheme 9.
A sulfonate NHC–Cu catalyst is found to be optimal for generating a silyl-substituted quaternary carbon stereogenic center.
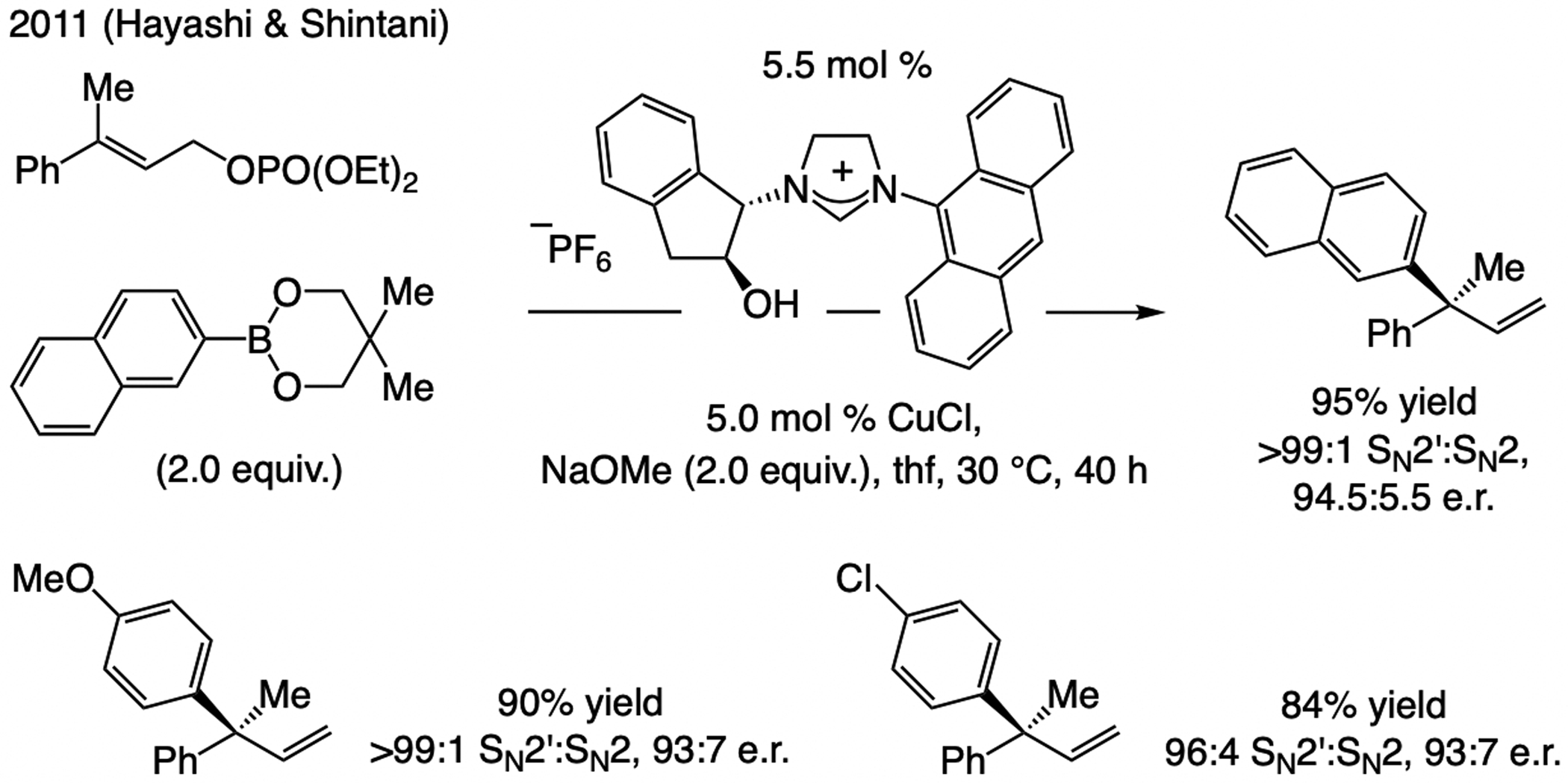
Subsequent advances. In 2011, Hayashi and Shintani[21f] showed that an arylboron compound may be used in NHC–Cu-catalyzed EAS (Scheme 10), affording products with a tertiary or a quaternary carbon center in high e.r. The latter report came on the heels of two 2010 studies involving achiral Cu complexes, one by Ohmiya and Sawamura[25] and another by Lalic,[26] demonstrating the feasibility of using an organoboron compound in allylic substitution. These seminal disclosures provided the necessary blueprint for effecting B/Cu exchange, a crucial step for development of catalytic EAS with organoboron compounds (see below).
Scheme 10.
An early example of EAS with an organoboron compound.
4.1.7. With aryl–Al compounds.
EAS with (aryl)2Zn or (aryl)3Al compounds is not desirable owing to unfavorable atom-economy. A better alternative entails the use of aryl- or heteroaryl-Al(alkyl)2 compounds, generated in situ by treatment of an organolithium or a Grignard reagent with a Cl–Al(alkyl)2 species. Accordingly, reactions with Ph(Et)2Al, generated in situ from a mixture of PhLi and Et2AlCl, and a trisubstituted allylic phosphate in the presence of NHC(S)-Ag-1a-b, or NHC(S)-Ag-2a (Scheme 11a) furnish products containing a quaternary carbon stereogenic center efficiently with >98% aryl-transfer and SN2’ selectivity and typically in >95:5 e.r. (Scheme 11a–b).[27] A heterocyclic moiety can be introduced similarly, and in some cases direct regioselective deprotonation gives the desired heteroaryl–Li species (Scheme 11c).
Scheme 11.
EAS with aryl–Al and heteroaryl–Al compounds, prepared in situ from the corresponding organolithium compounds.
4.2. With Organoboron Compounds
A limitation of reactions with organometallic reagents is that polar functional groups are not tolerated (e.g., aldehydes and ketones). Furthermore, addition of an allenyl or a propargyl group would necessitate difficult-to-access and/or unstable organometallic entities. An effective solution is to use an organoboron reagent.
The issue of Cu/B exchange rate. A challenge associated with designing catalytic EAS with a less reactive organoboron compound is how to form an organocopper intermediate efficiently. As noted above, a convenient solution involves rapid Cu/B exchange with a metal alkoxide (Scheme 12), driven by the strength of the resulting B–O bond. Nonetheless, the inherent basicity of a metal alkoxide can at times cause undesirable side reactions and/or complications in functional group tolerance issues.[28]
Scheme 12.
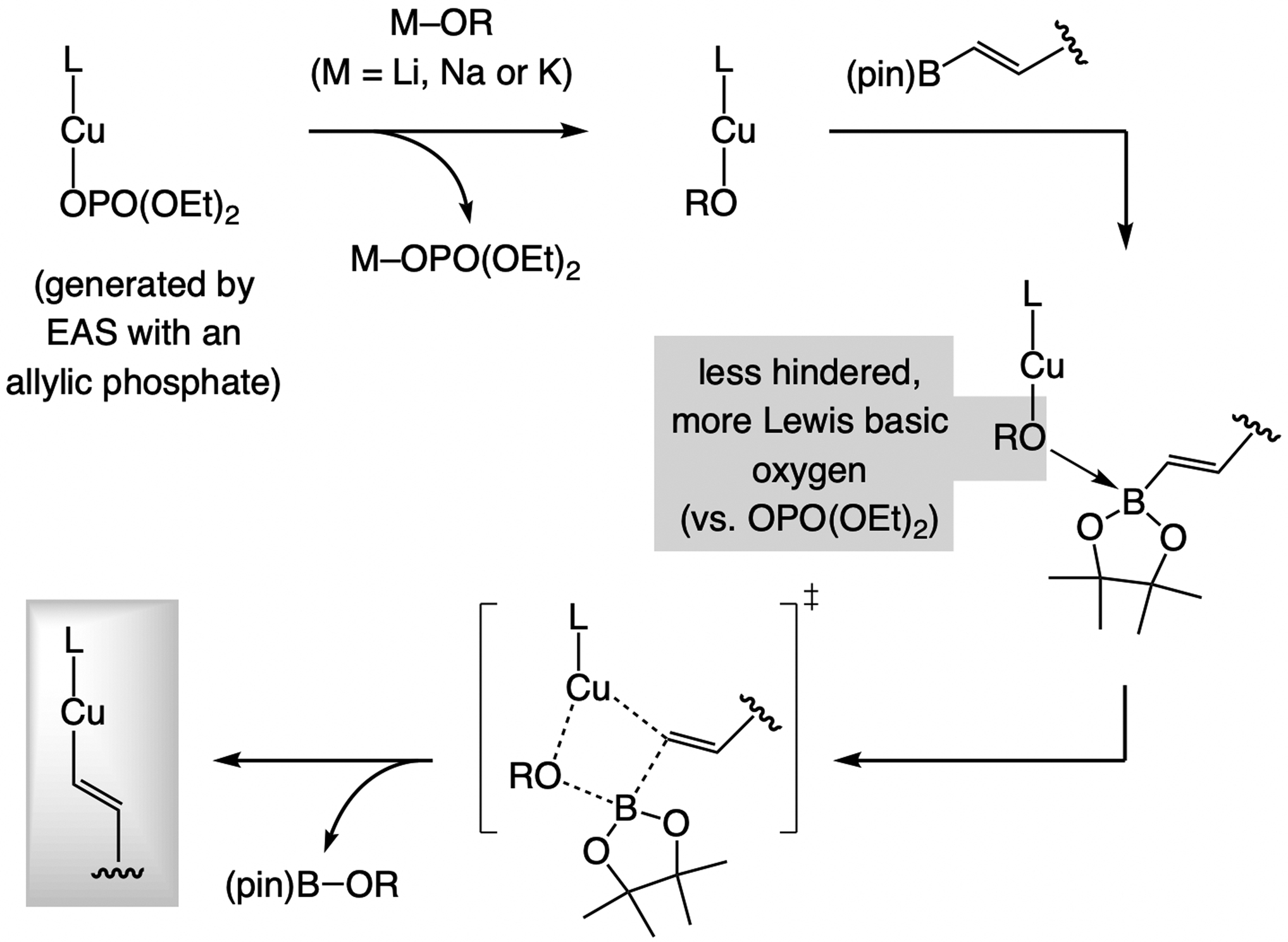
The role of a metal–alkoxide in Cu/B exchange and the formation of an alkenyl–B(pin) compound.
4.2.1. With an allenyl boronate.
Allenes are a versatile set of compounds that are attracting increasing attention in reaction development.[29] Methods that generate such allene-containing products are thus in high demand.
4.2.1.1. SN2’-selective reactions.
In 2012, we reported a sulfonate NHC–Cu-catalyzed EAS, involving commercially available allenyl–B(pin) (Scheme 13).[30] These investigations led to identification of a key subset of sulfonate NHC ligands. Unlike other ligand classes (e.g., imid-1a, imid(O)-1a, or imid(O)-3a) reactions with sulfonate NHC–Cu catalysts (e.g., derived from imid(S)-1a) were highly SN2’-selective, but enantioselectivities were low (66:34 e.r.). Based on mechanistic arguments (later debunked; see Scheme 32), we established that with imid(S)-3a, containing an NHC ligand that bears large C3 and C5 substituents, EAS was not only highly SN2’-selective, the desired product was generated in ≥95:5 e.r. (Scheme 13a). Aryl-, alkyl- (even t-butyl-), and silyl-substituted allylic phosphates proved to be suitable substrates. Reactions with trisubstituted allylic phosphates were similarly high yielding, and SN2’- and enantioselective (Scheme 13b). Two other features distinguish this set of processes: 1) For transformations involving a disubstituted alkene, the catalyst derived from imid(S)-3a is optimal, whereas with a trisubstituted olefin it is best to use imid(S)-1a. 2) Different enantiotopic alkene faces are favored for disubstituted and trisubstituted alkenes (Scheme 13a–b). In the course of these investigations we discovered that, depending on the ligand, NHC–Cu-catalyzed proto-boryl addition to an allene may be used to access either of the alkenyl–B(pin) regioisomers.[30,31]
Scheme 13.
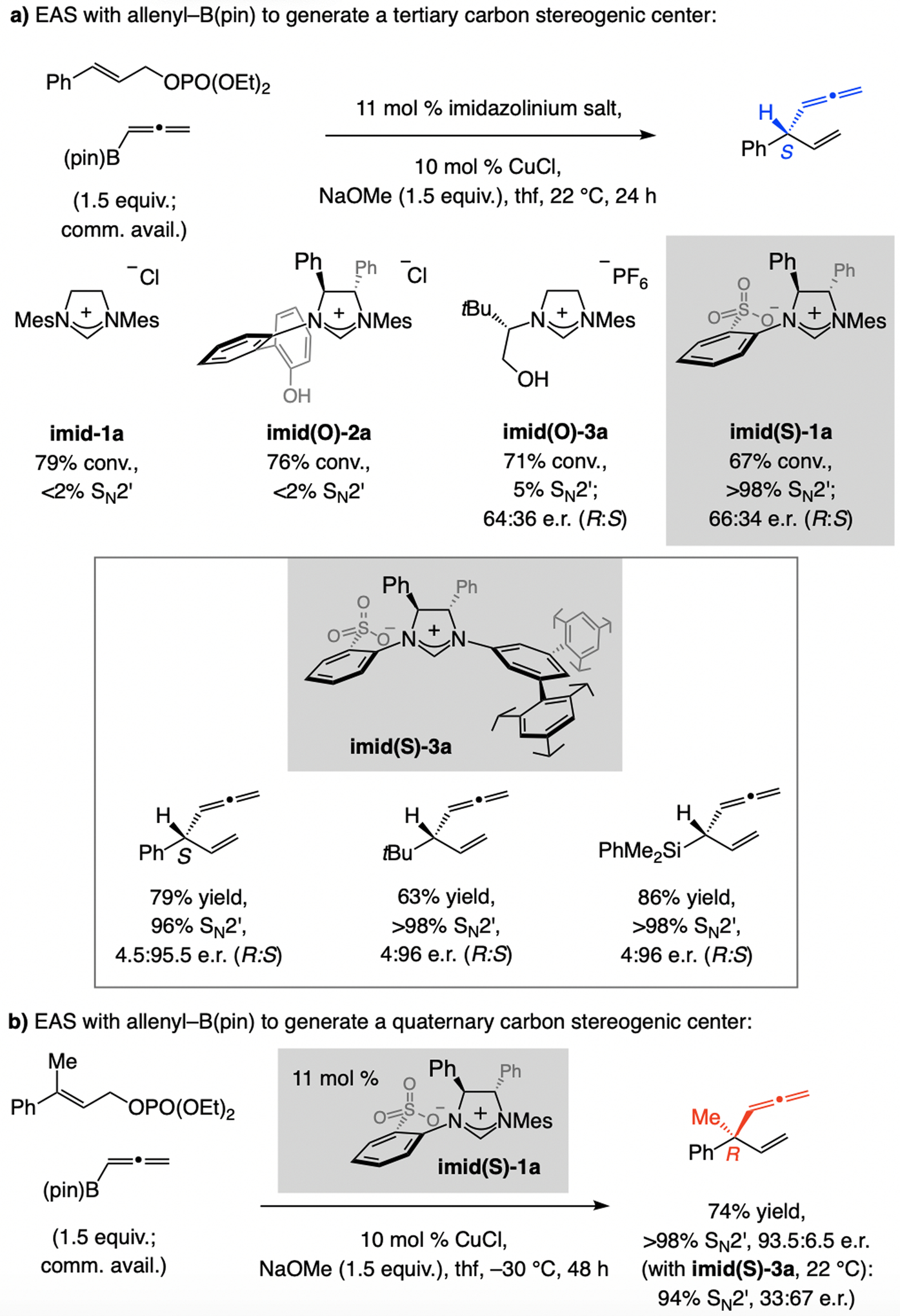
EAS with allenyl–B(pin), affording products that might contain a tertiary or a quaternary carbon stereogenic center (see Schemes 32 for mechanistic analysis).
Scheme 32.
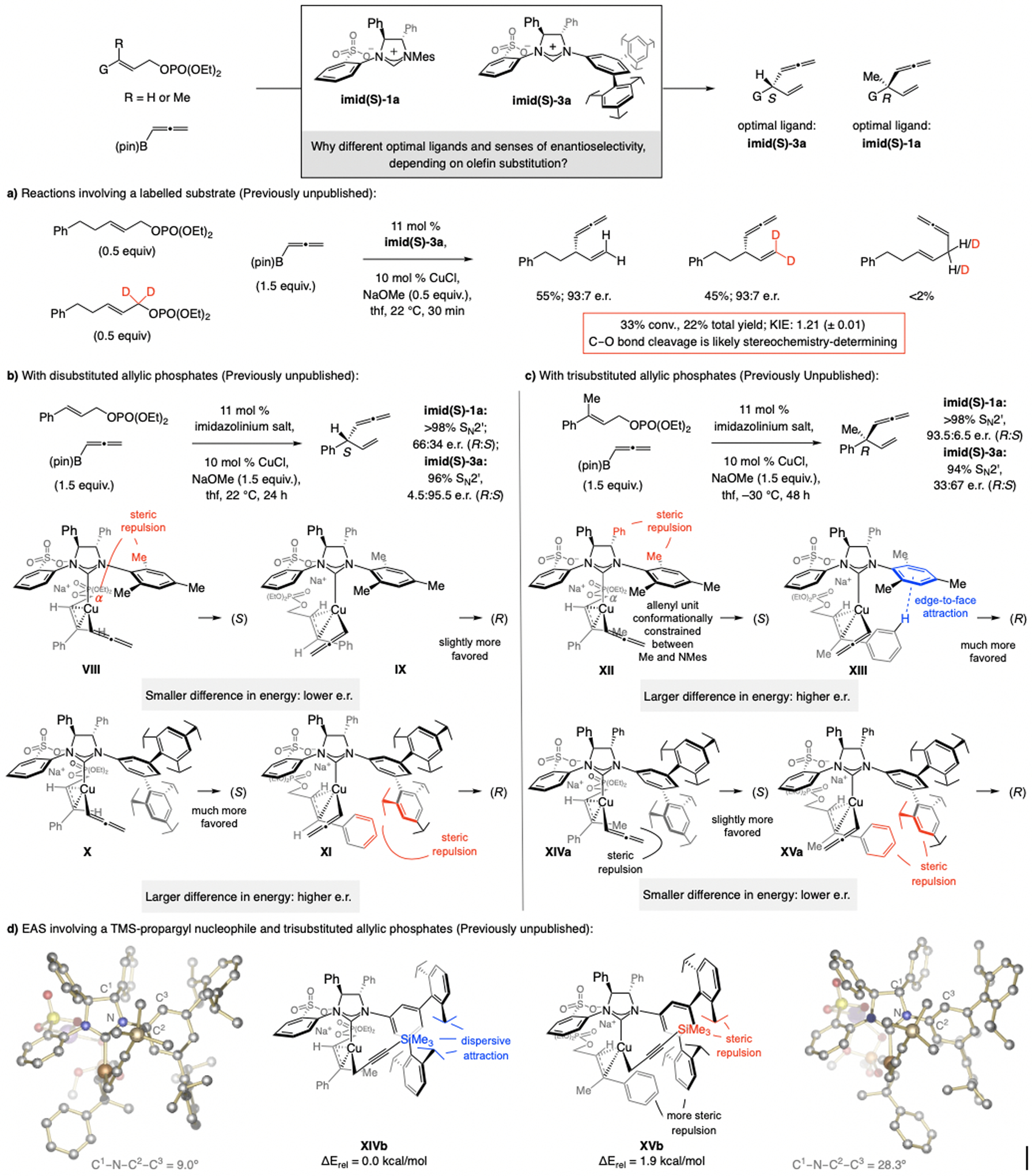
The stereochemistry-determining step and origin of selectivity in EAS with allenyl–B(pin) (see Schemes 13 and 19 for methodology). Reactions performed under N2 atm.; conv. (±2%) determined by analysis of the 1H NMR spectra of the unpurified product mixtures; yields (±5%) are for purified products. Previously unpublished KIE data and DFT analysis; see the Supporting Information for details.
A subsequent advance. Carreira has shown that allenylic substitutions with a racemic homoallenylic carbonates and an alkylzinc halide proceed with high regio- and enantioselectivity [Eq. (1); r.r. = regioisomeric ratio].[32] Reactions with alkyl-substituted substrates were inefficient.
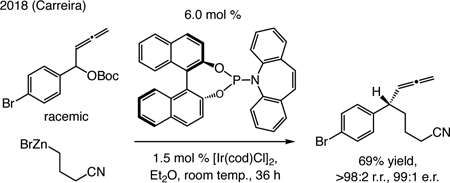 |
(1) |
4.2.1.2. SN2”-selective reactions.
Allenyl–B(pin) can be used in catalytic enantioselective SN2”-selective substitutions, a scarcely investigated class of transformations (Scheme 14). This approach was conceived based on the concept of the initially formed intermediate may undergo 3,3’-reductive elimination.[33] Whereas transformations with catalysts derived from other ligand types afforded the desired SN2” products, those with sulfonate NHC complexes were highly enantioselective, with the catalyst derived from imid(S)-4b being optimal.[34] Enantioselective synthesis of an intermediate formerly used in a racemic synthesis of fasicularin highlights the utility of the approach.
Scheme 14.
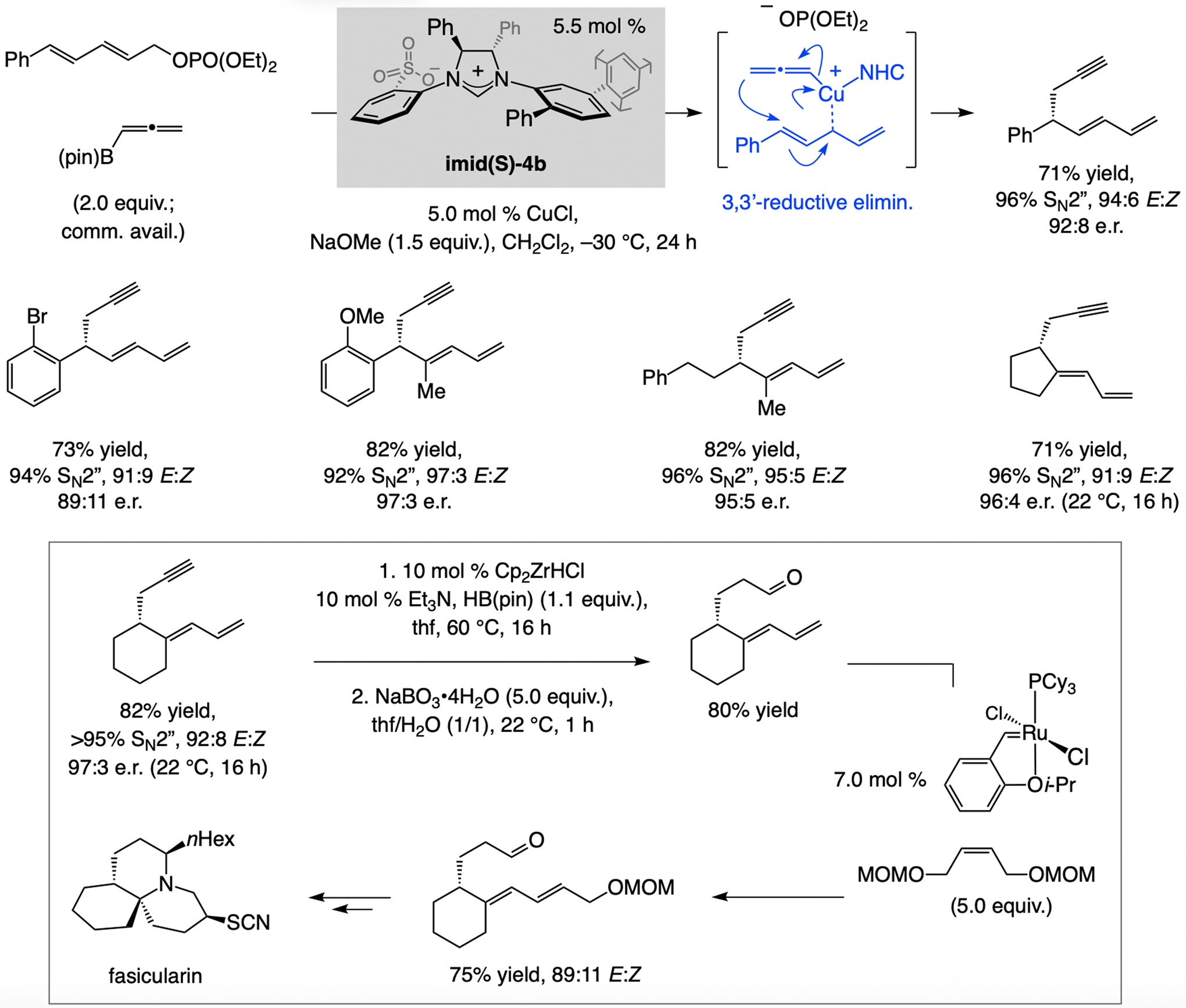
Enantioselective SN2” substitution with allenyl–B(pin) (see Scheme 33 for mechanistic analysis).
4.2.2. With alkenyl boronates.
EAS with alkenyl–B(pin) compounds is a valuable process because Z-alkenyl units cannot otherwise be easily introduced. Several alkenyl–B(pin) compounds are commercially available, and others can be prepared with high E or Z selectivity by reported catalytic protocols.[35,36]
Related advances. The aforementioned Hayashi/Shintani[21f] disclosure included an example of an alkenyl boronate being utilized (Scheme 15a). After the first cases of sulfonate NHC–Cu-catalyzed EAS with alkenyl–B(pin) compounds appeared (see Scheme 17, below), Carreira reported EAS catalyzed by a phosphoramidite–Ir complex involving racemic secondary allylic alcohol substrates and alkenyl(trifluoro)boron potassium salts as latent nucleophiles (Scheme 15b).[37] An activated leaving group was not needed; nonetheless, high catalyst loading aside, the method is confined to aryl-substituted allylic alcohols and formation of tertiary carbon stereogenic centers, two equivalents of HF must be present, and fluoroboryl compounds are typically accessed from alkenyl–B(pin) precursors.
Scheme 15.
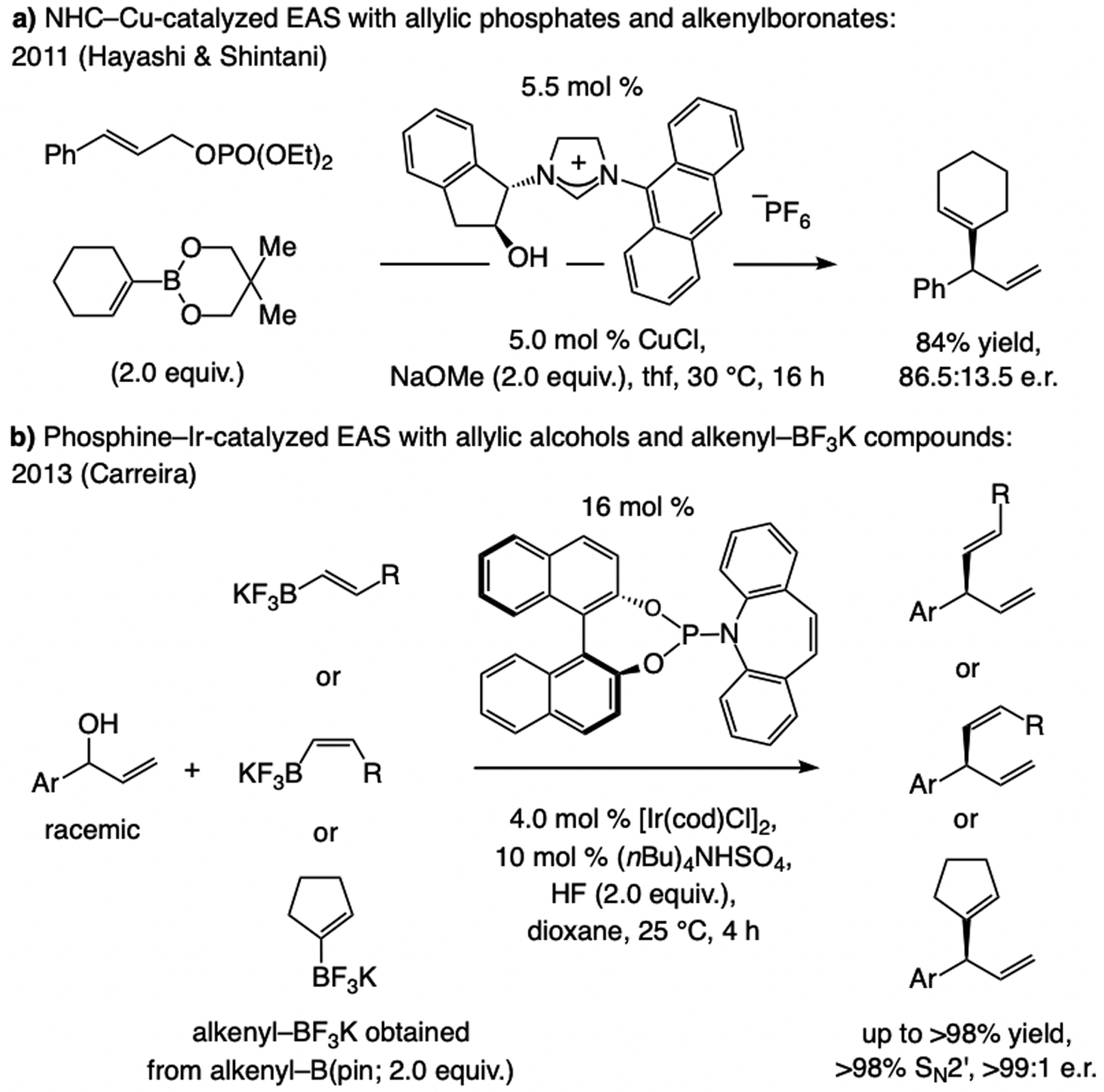
EAS with different acceptor molecules and different alkenyl boronates.
Scheme 17.
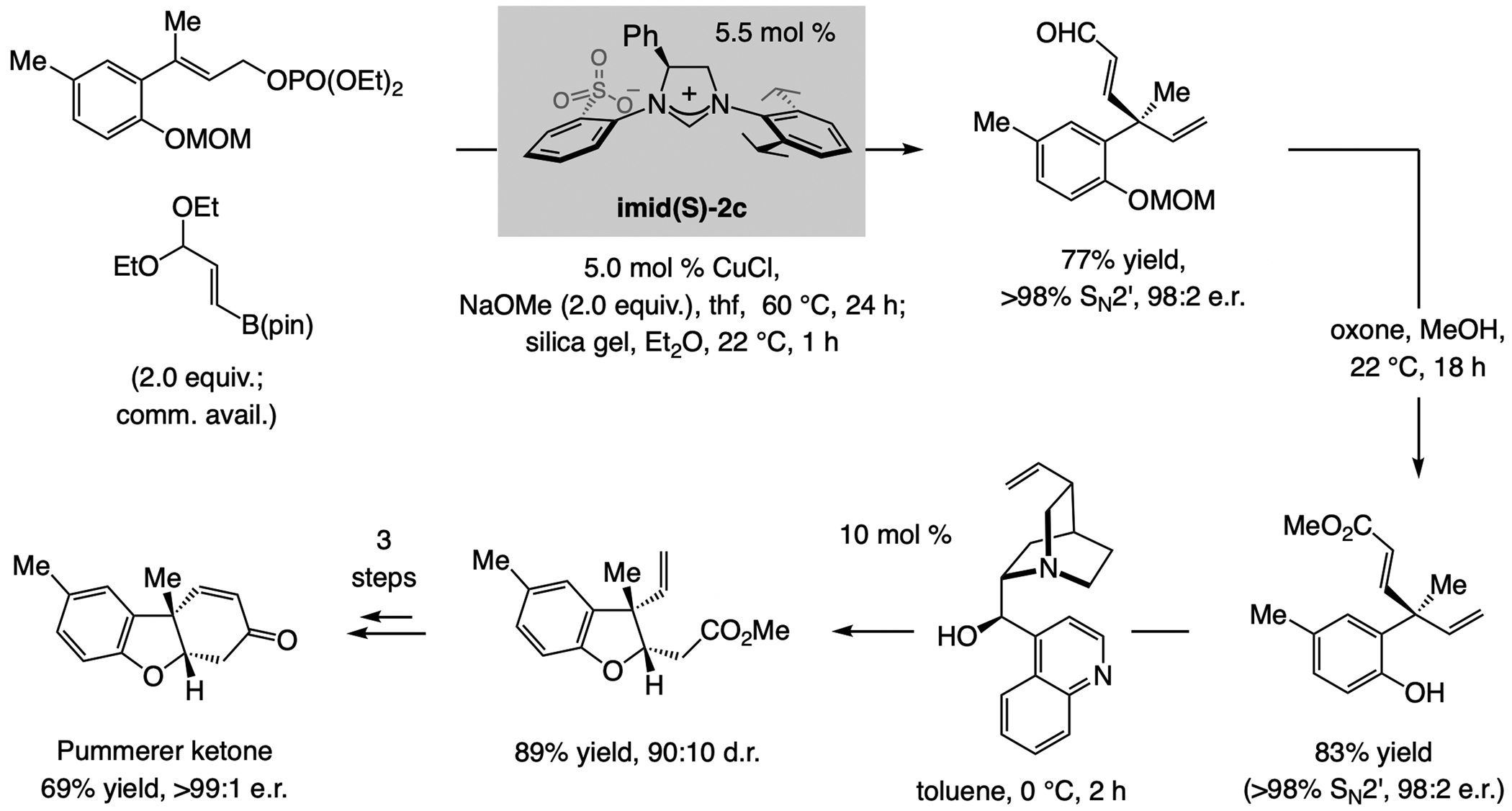
EAS with an alkenyl–B(pin) reagent and generates a quaternary carbon stereogenic center.
Sulfonate NHC–Cu catalysts promote EAS with alkenyl–B(pin) compounds (Scheme 16a), many of which are commercially available or can be synthesized by catalytic procedures (e.g., cross-metathesis or proto-boryl additions to alkynes; see below).[38] Products bearing an E-enal can be generated by mild acidic workup, those that contain an E-enoate may be accessed, and the sensitive benzylic stereogenic center may be retained (>98% enantiospecificity). Cyclic trisubstituted alkenyl–B(pin) compounds can be used, highlighted by a concise synthesis of semburin (Scheme 16b). Z-Alkenyl–B(pin) can be synthesized in a number of different ways,[36] including catalytic stereoselective cross-metathesis,[39] as demonstrated by a four-step synthesis of nyasol (Scheme 16c).
Scheme 16.
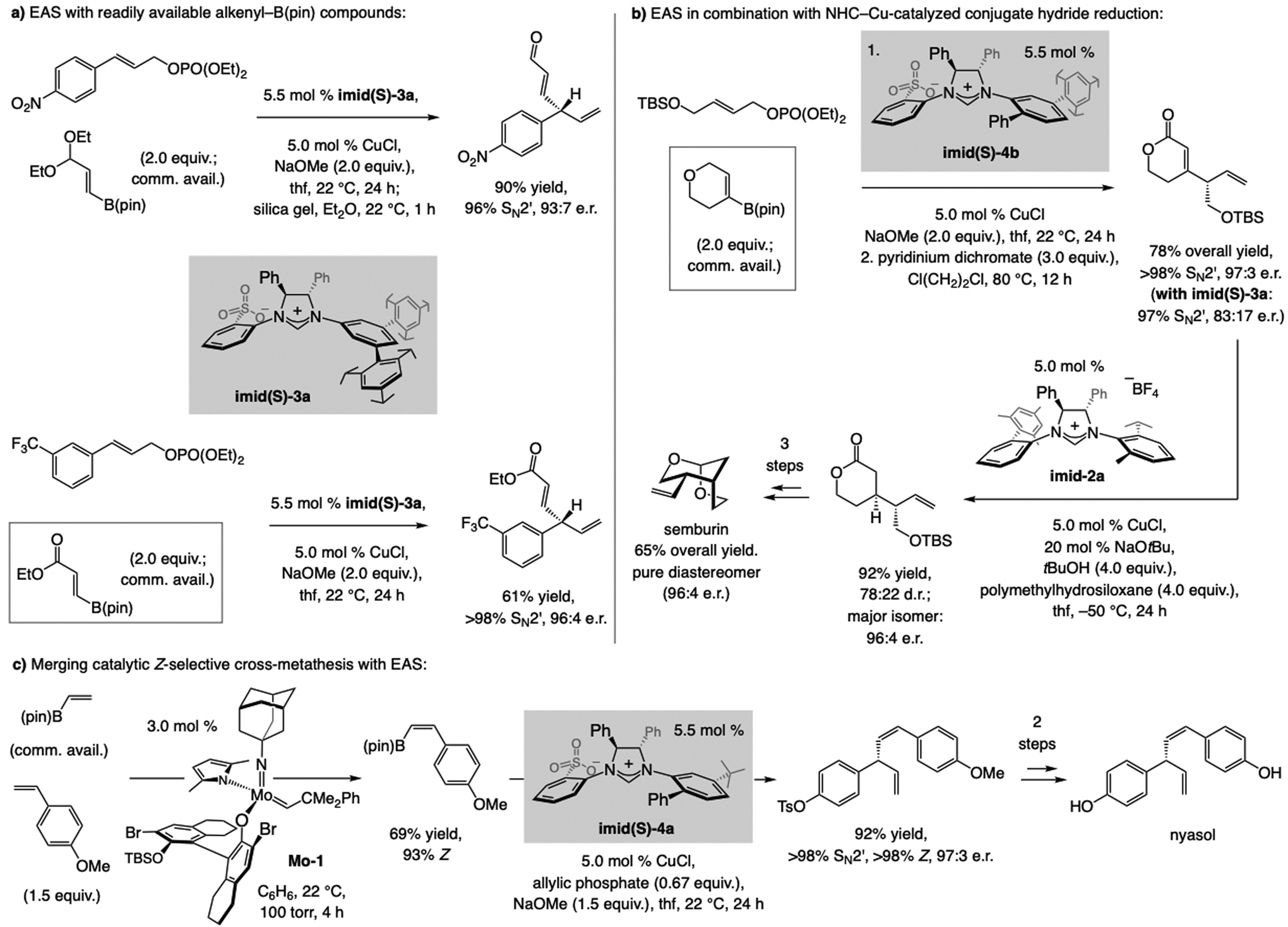
EAS with readily accessible alkenyl–B(pin) compounds. Enantiomerically enriched products that contain an aldehyde, a carboxylic ester, or a Z-alkenyl group can thus be readily synthesized.
EAS with trisubstituted allylic phosphates afford 1,4-dienes with a quaternary carbon stereogenic center. In the application to Pummerer ketone (Scheme 17),[40] the catalyst derived from imid(S)-2c was optimal. The importance of the availability of different members within the sulfonate NHC ligand class is underscored by the fact that in the transformations involving an alkenyl–B(pin) compound, four different sulfonate NHC–Cu complexes were needed for maximum enantioselectivity (e.g., <70:30 e.r. with imid(S)-2c and 1,2-disubstituted allylic phosphates).
A subsequent advance. Fletcher has introduced a set of efficient enantioselective reactions, catalyzed by a binap–Rh complex and involving racemic cyclic allylic chlorides and alkenyl boronic acids (Scheme 18).[41]
Scheme 18.
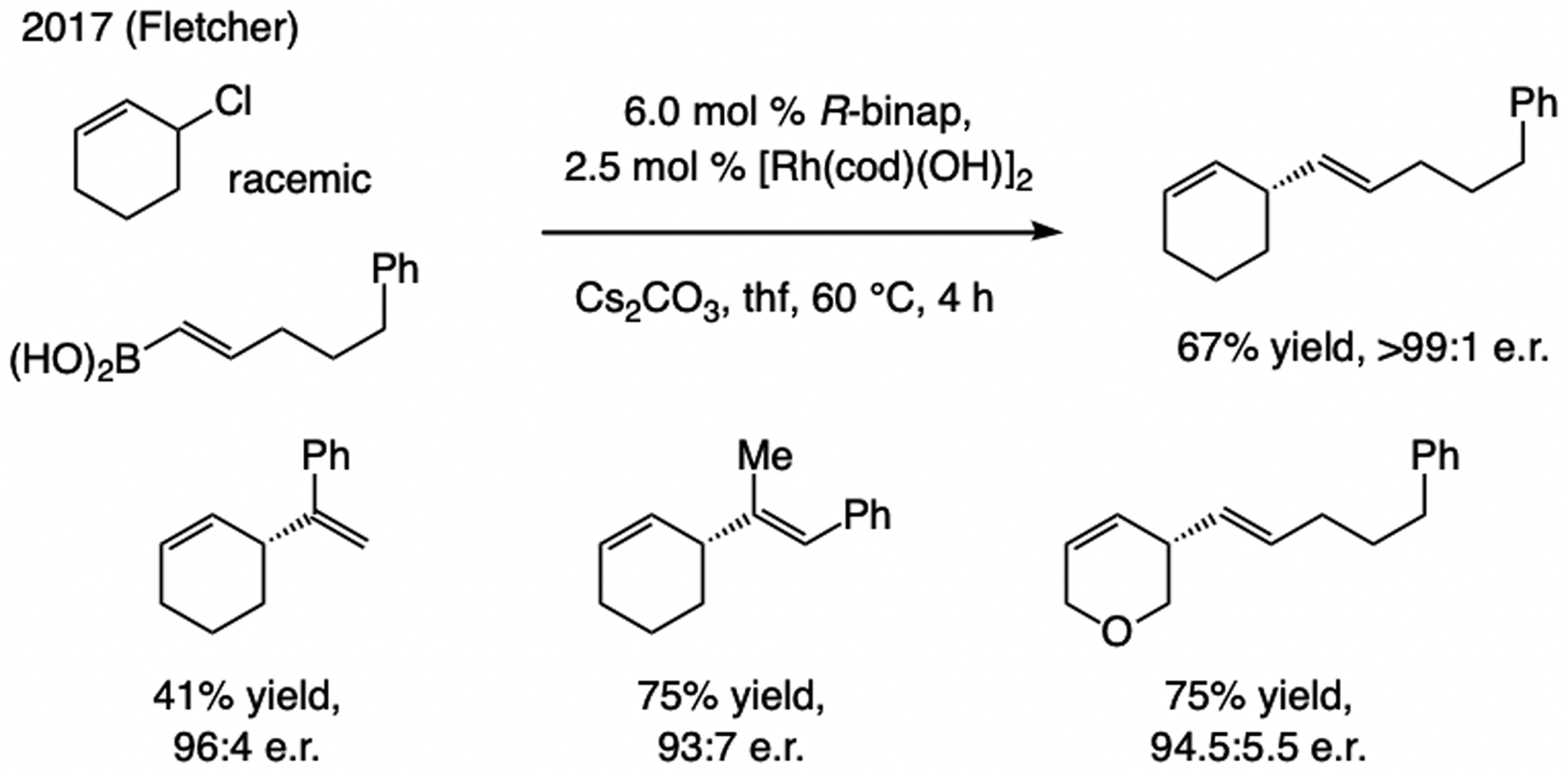
A bis-phosphine–Rh complex promotes EAS with racemic cyclic allylic chlorides and alkenyl–boronic acids.
4.2.3. With a silyl-substituted propargyl boronate.
EAS with a propargyl moiety can be catalyzed by a sulfonate NHC–Cu complex.[42] With disubstituted allylic phosphates, reactions involve an easily accessible silyl-protected propargyl–B(pin) compound (Scheme 19a). The propargylic product isomer is favored. The application to formal synthesis of plakinic acid A demonstrates the feasibility of chemo- and stereoselective alteration of the alkenyl or alkynyl units within a 1,5-enyne product. Chemo- and E-selective alkene metathesis and NHC–Cu-catalyzed proto-boryl addition to the terminal alkyne afforded the substrate for another SN2’- and stereoselective allylic substitution, this time with imid(S)-4b.
Scheme 19.
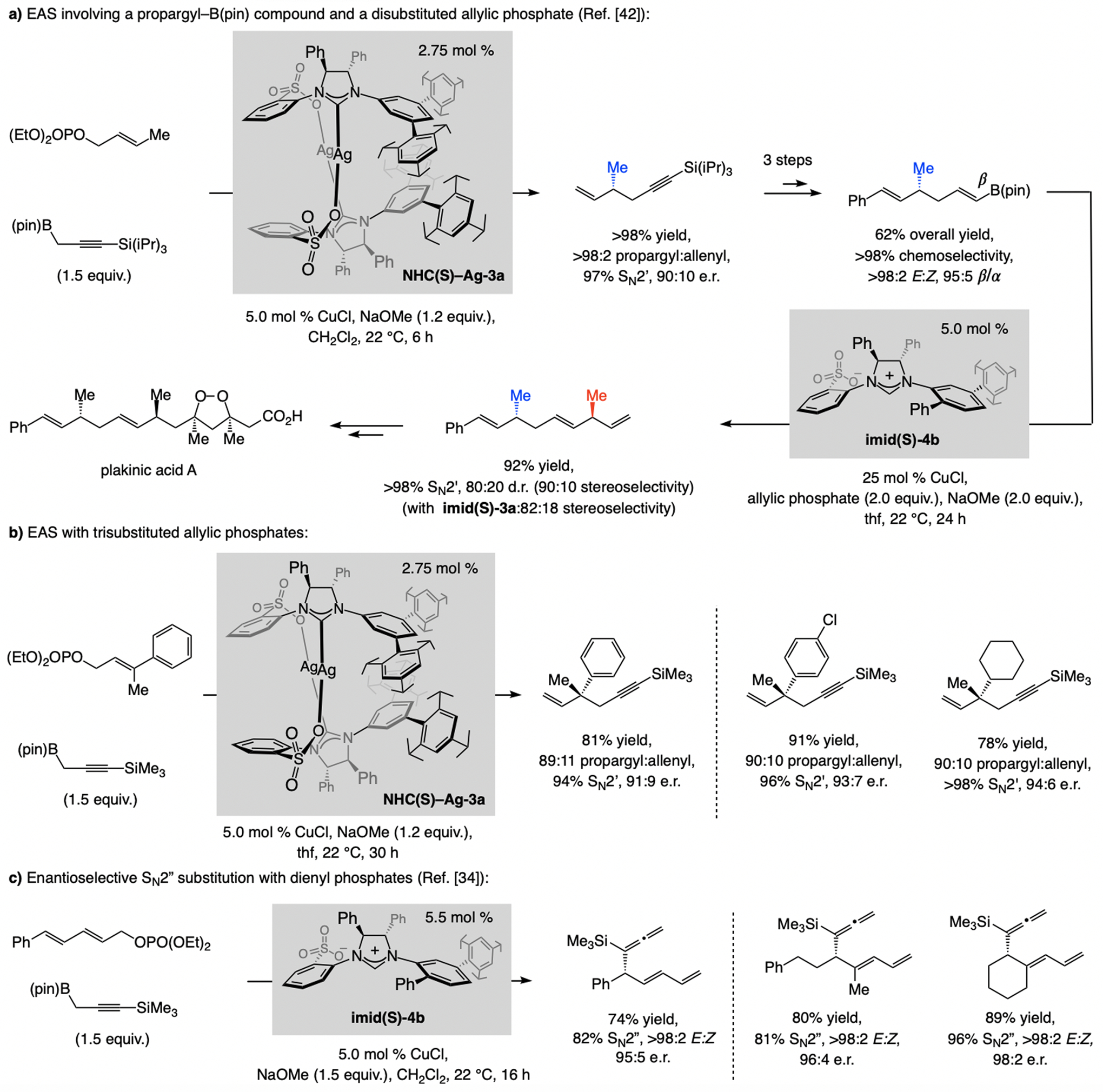
EAS and enantioselective SN2” substitution reactions involving a propargyl–B(pin) compound may be used to generate tertiary or quaternary carbon stereogenic centers.
1,5-Enynes with a quaternary carbon stereogenic center were accessed in the same way (Scheme 19b). Reactions were slower (30 vs. 6 h), and enantioselectivities lower, but aryl- as well as alkyl-substituted allylic phosphates were effective substrates. A single NHC–Cu complex proved to be optimal for EAS for di- and trisubstituted alkene electrophiles, unlike the transformations with allenyl–B(pin) (Scheme 13), or alkenyl–B(pin) compounds (Schemes 16–17). The same catalyst and silyl-protected propargyl-B(pin) were recently utilized in the development of the first method for enantioselective SN2”-selective substitutions (Scheme 19c).[34]
4.2.4. With a bis(boryl)methane.
Unlike alkenyl boronates, EAS with alkyl–B(pin) compounds are inefficient (<10% conv.); reactions are fast only with a more nucleophilic organoboron compound [e.g., an (alkyl)3borane].[43] This is likely because of higher favorability of the transition state for Cu/B exchange (Scheme 20; better stabilization of electron density). A second boronate unit, with a partially vacant p orbital, can therefore facilitate Cu/B exchange, suggesting commercially available bis[(pinacolato)boryl]methane,[44,45] as a suitable reagent.
Scheme 20.
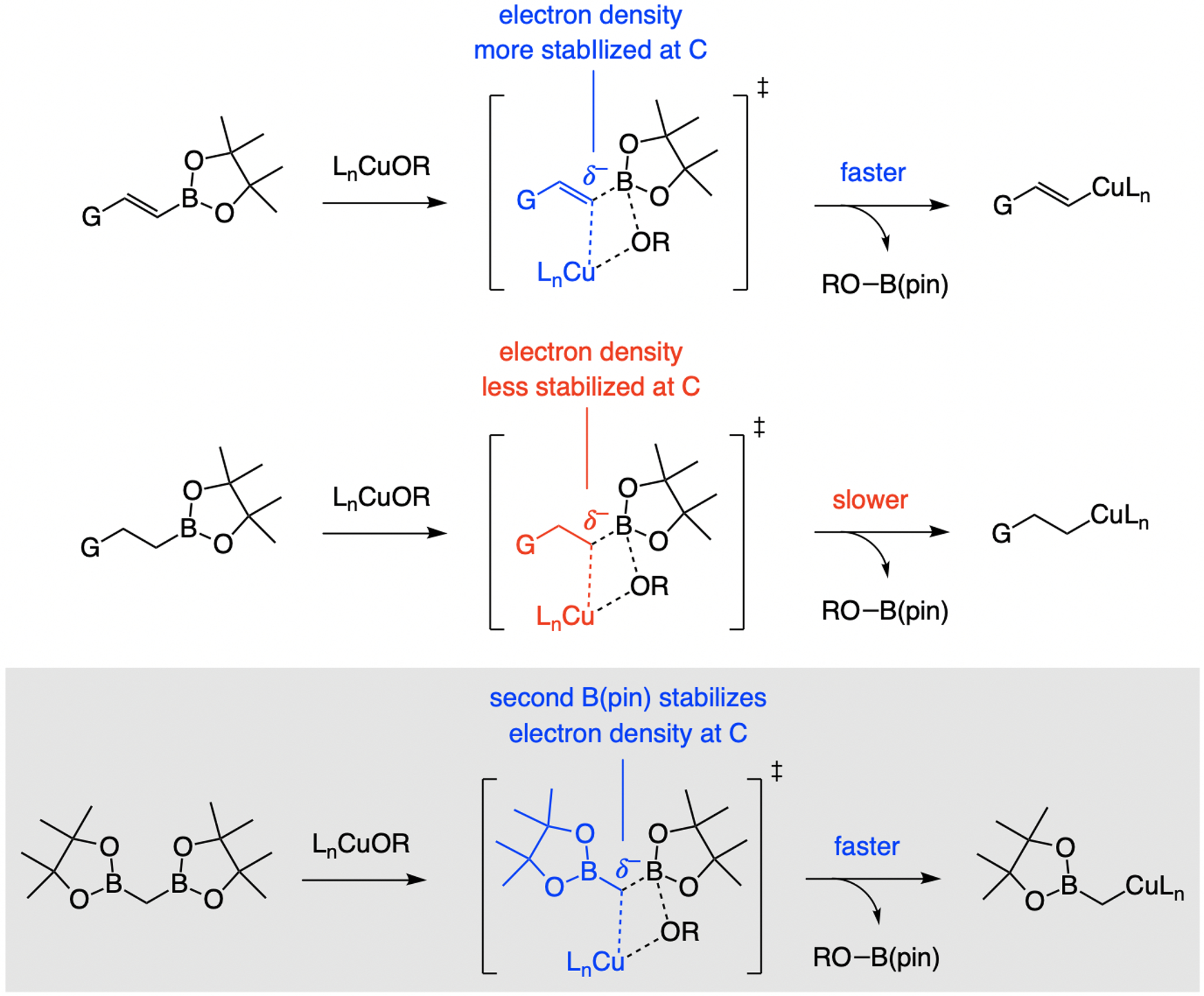
Rate of Cu/B exchange is impacted by how well the developing charge at the carbon center can be stabilized.
4.2.4.1. With disubstituted alkenes.
Only a sulfonate NHC–Cu catalyst delivered high SN2’ selectivity in these cases (Scheme 21);[46] with a mono- or bis-phosphine (e.g., binap or josiphos) or other NHC ligands SN2-addition dominated (80% to >98%). Regioselectivity is higher with the NHC–Ag complex probably because there is no competition from a ligand-free Cu complex. In the application to a synthesis of rhopaloic acid enantioselectivity of the initial step was moderate with NHC(S)-Ag-3a (84:16 e.r.), but the complex derived from NHC(S)–Ag-4b afforded the desired product in 96:4 e.r. Boron–hydride addition to the terminal alkene and chemoselective cross-coupling generated the triene side chain. The efficiency of the ensuing cross-coupling is due to participation of the neighboring hydroxy group.[47]
Scheme 21.
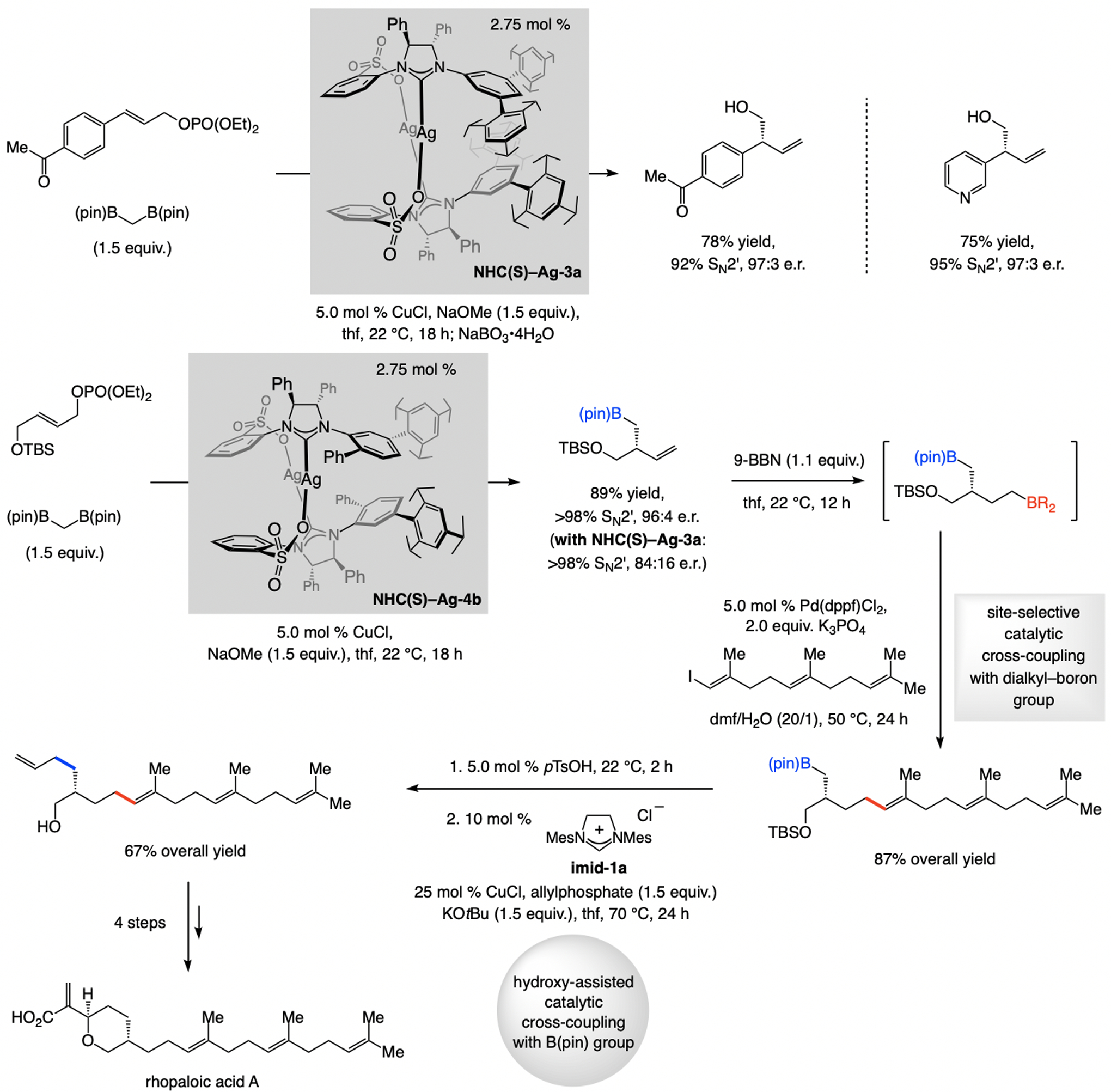
EAS with bis[(pinacolato)boryl]methane furnish enantiomerically enriched alkyl–B(pin)-containing terminal alkenes (see Scheme 34 for mechanistic analysis).
A subsequent advance. In 2016 Chen and Niu disclosed a phosphoramidite–Ir-catalyzed EAS (Eq. [2])[45] for which 1.7 mol % of a Ag salt is needed. This system is analogous to that used by Carreira for EAS with alkylzinc halides and secondary allenyl Boc-esters (see Eq. [1]).
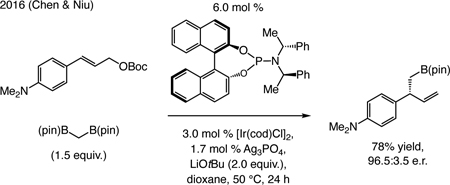 |
(2) |
4.2.4.2. With trisubstituted alkenes.
The catalyst derived from NHC(S)–Ag-5a is optimal for reactions with allylic phosphates with a C2 ester moiety (previously unpublished; Scheme 22a).[48] Only with one particular NAr moiety high SN2’ selectivity was attained. Equally striking, only the achiral (linear) product was generated with other types of NHC or bisphosphine ligands. The method is applicable to aryl- and alkyl-substituted substrates. Investigation of EAS processes that generate products with a quaternary carbon stereogenic center (previously unpublished; Scheme 22b)[49] revealed that reactions with achiral NHC–Cu complexes or those derived from a chiral bisphosphine ligand (e.g., binap or dppp) strongly favor the SN2-addition pathway (>98%). The combination of a Z-allylic phosphate and NHC(S)-Ag-5b is optimal (see Scheme 34 for mechanistic analysis). The approach has appreciable scope (Scheme 22b), albeit with lower e.r. for alkyl-substituted starting materials.
Scheme 22.
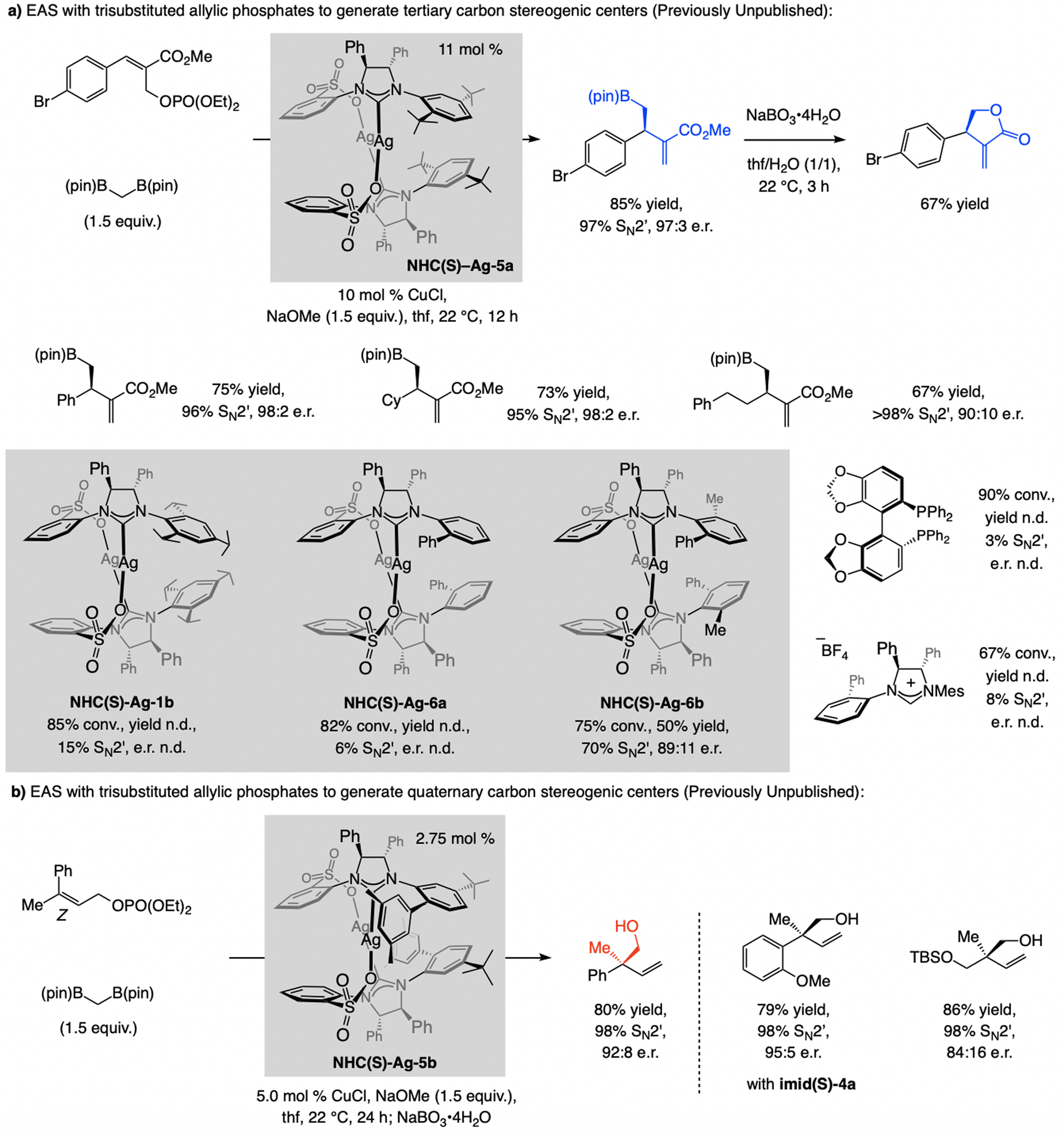
EAS with bis[(pinacolato)boryl]methane and trisubstituted allylic phosphates (see Scheme 34 for mechanistic analysis). Previously unpublished results: reactions performed under N2 atm.; conv. and SN2’ selectivity (±2%) determined by analysis of the 1H NMR spectra of unpurified product mixtures; yields (±5%) correspond to purified products. See the Supporting Information for details. n.d. = not determined.
Scheme 34.
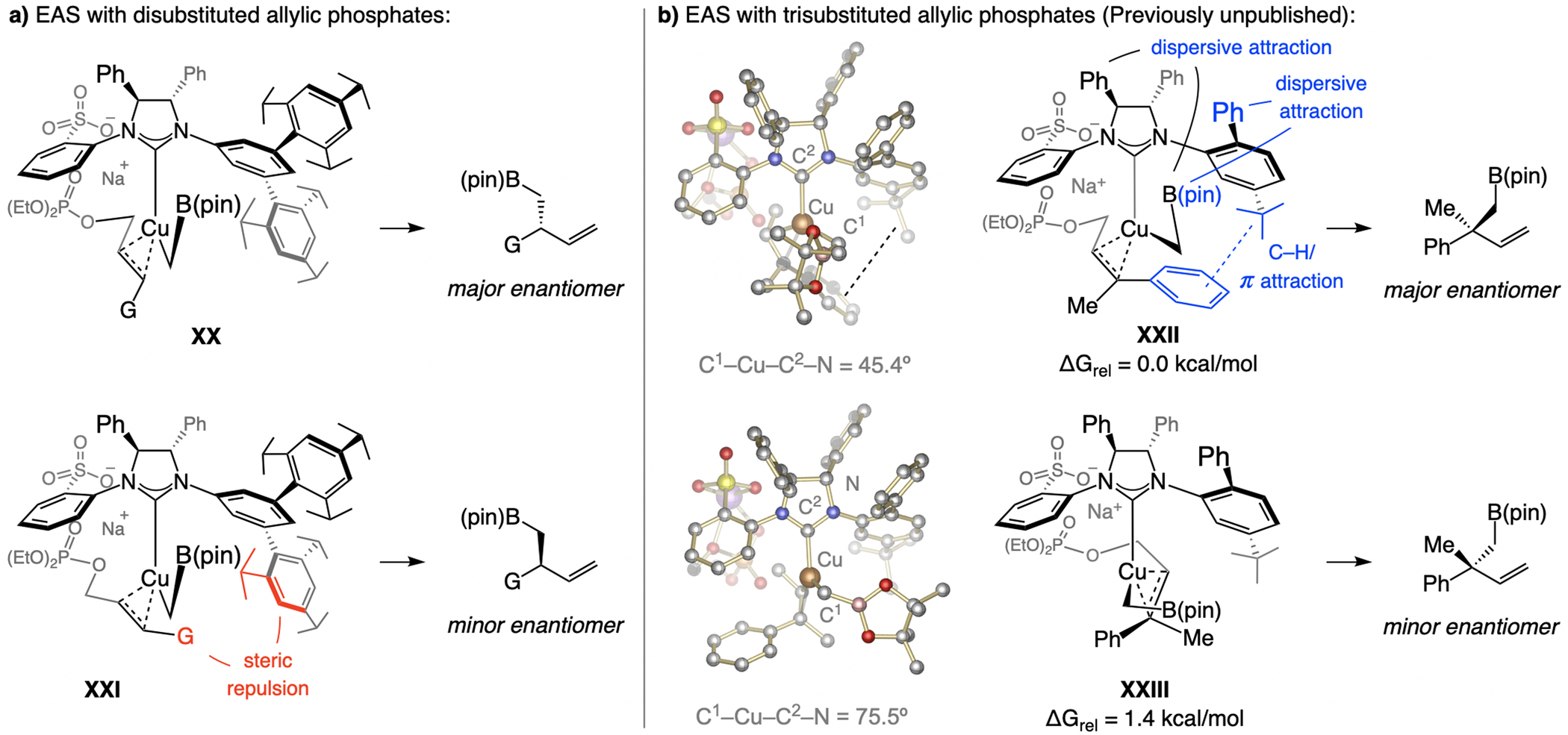
High enantioselectivity in some transformations is due to a combination of sulfonate bridging and dispersive attraction (see Schemes 21–22 for methodology). DFT at the M06L/Def2-TZVPP//M06L/Def2-SVP level. Previously unpublished mechanistic analysis; see the Supporting Information for details of the DFT studies.
4.3. With alkylmagnesium halides and F3C-substituted olefins.
Catalytic EAS with E-trifluoromethyl-substituted olefins was disclosed by Shi in 2018 (Scheme 23).[50] The Cu complex derived from imid(S)-2c was the most effective. Inclusion of PhB(neo) (neo = neopentyl) improved yield and e.r., likely because the borate is the alkylating agent.[51]
Scheme 23.
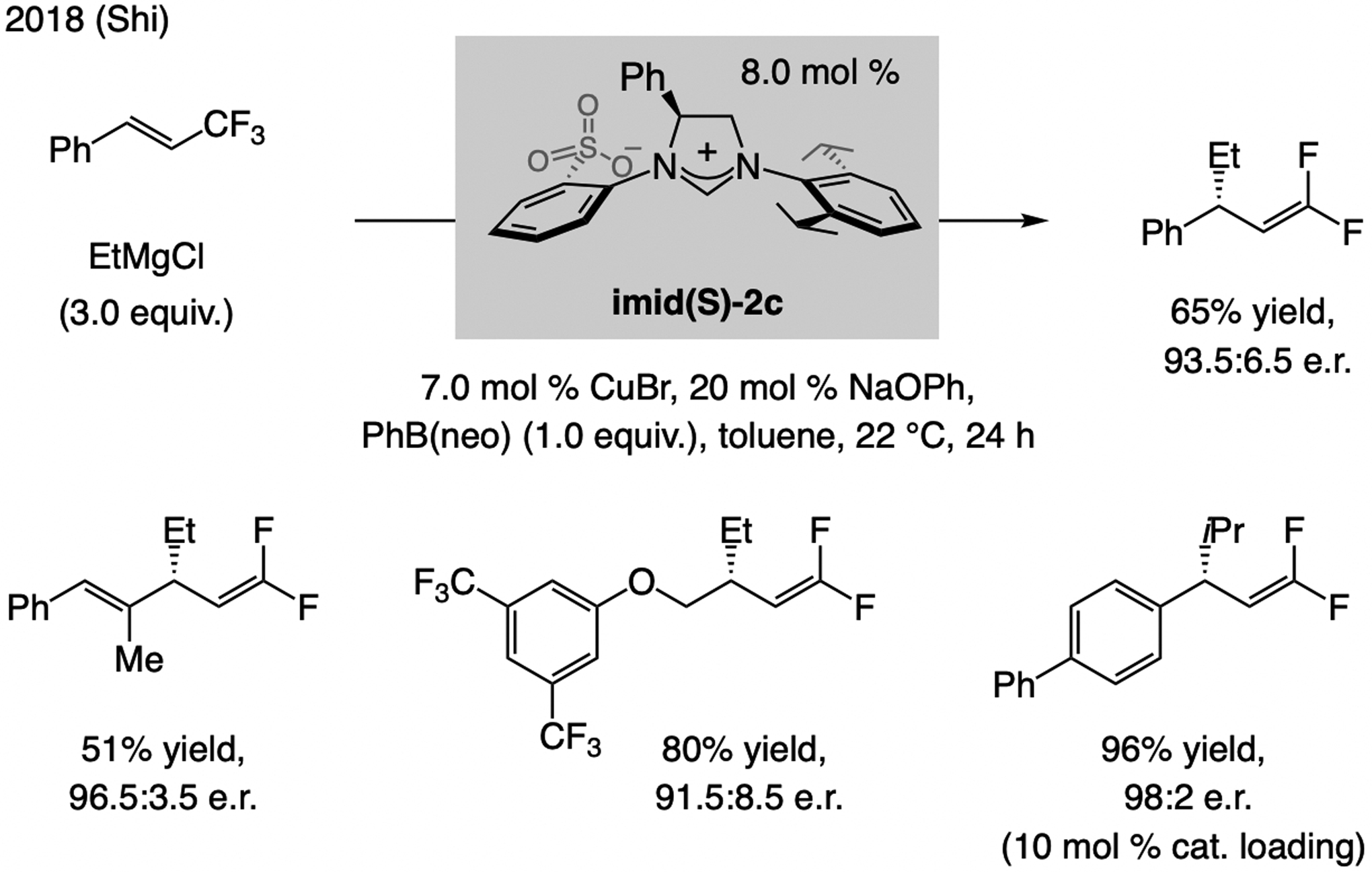
EAS with F3C-substituted alkenes and alkyl–MgCl compounds.
4.4. Multicomponent Processes
4.4.1. With vinyl–B(pin) as latent nucleophile.
Subsequent to the development of catalytic multicomponent processes that begin with Cu–boryl addition to an alkene,[52] related transformation entailing an initial Cu–H addition began to appear.[53]
State-of-the-Art, circa 2016. Yun reported in 2016 a sequential bis-phosphine–Cu–H addition to an alkenyl–B(pin)/allylic substitution (Scheme 24).[54] Reaction with an E-1,2-disubstituted allylic phosphate, however, was less diastereo- and enantioselective.
Scheme 24.
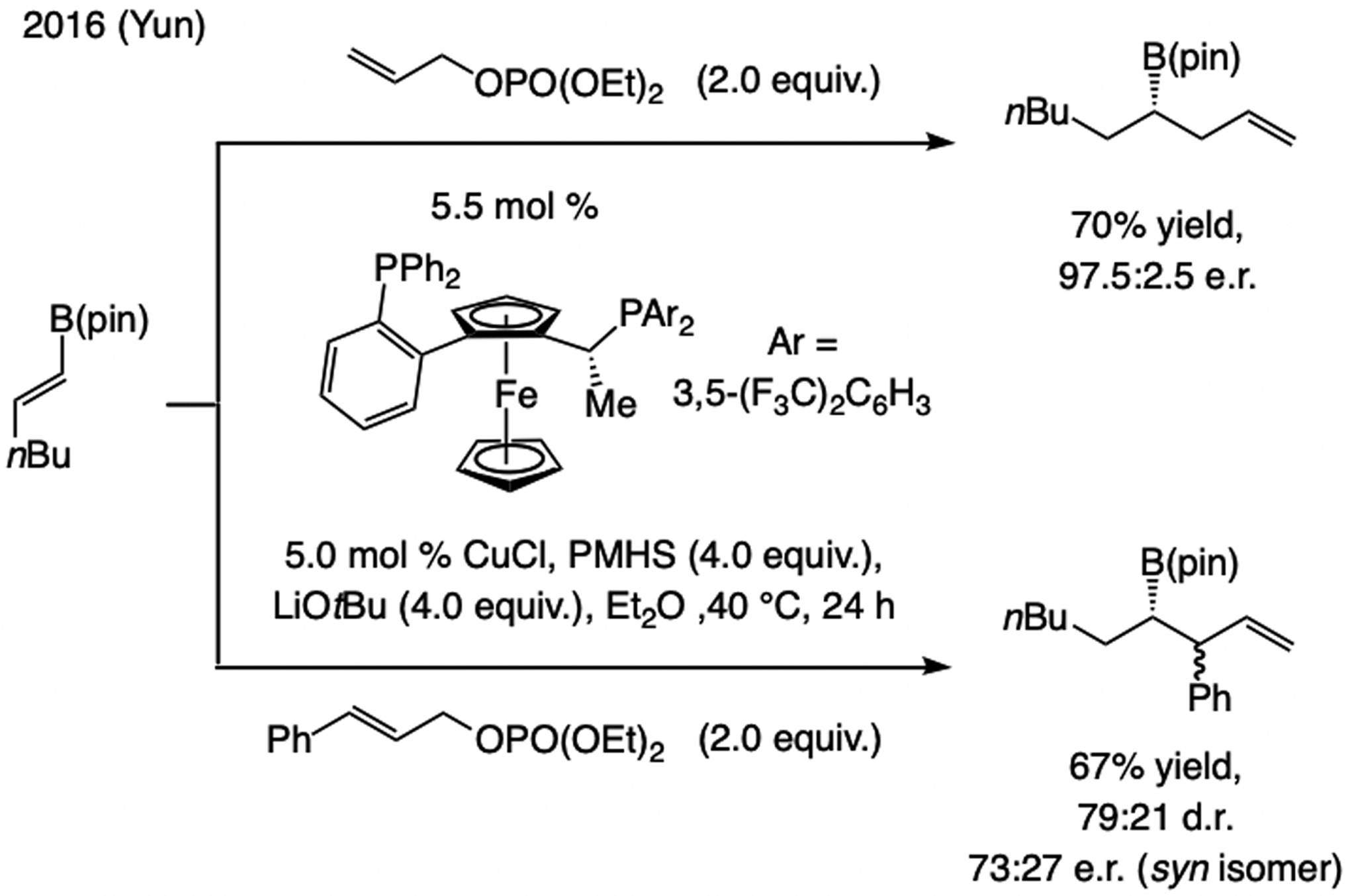
Regio- and enantioselective Cu–H addition to an aryl or boronate-substituted alkene followed by allylic substitution.
The process involving regio- and enantioselective Cu–H addition to vinyl–B(pin) followed by diastereoselective allylic substitution furnishes homoallyl–B(pin) products (Scheme 25a). Bis-phosphine–Cu complexes, optimal for Cu–H addition to aryl olefins and E-1,2-alkyl–B(pin) compounds, were low yielding and/or minimally enantioselective, whereas with complexes derived from imid(S)-1a and imid(S)-3a transformations were highly SN2’-, diastereo-, and enantioselective.[55] Aryl-, heteroaryl-, or alkyl-substituted allylic phosphates were suitable electrophiles (Scheme 25b). These transformations thus constitute an enantioselective formation of a Cu–substituted carbon stereogenic center (see also Scheme 66b).
Scheme 25.
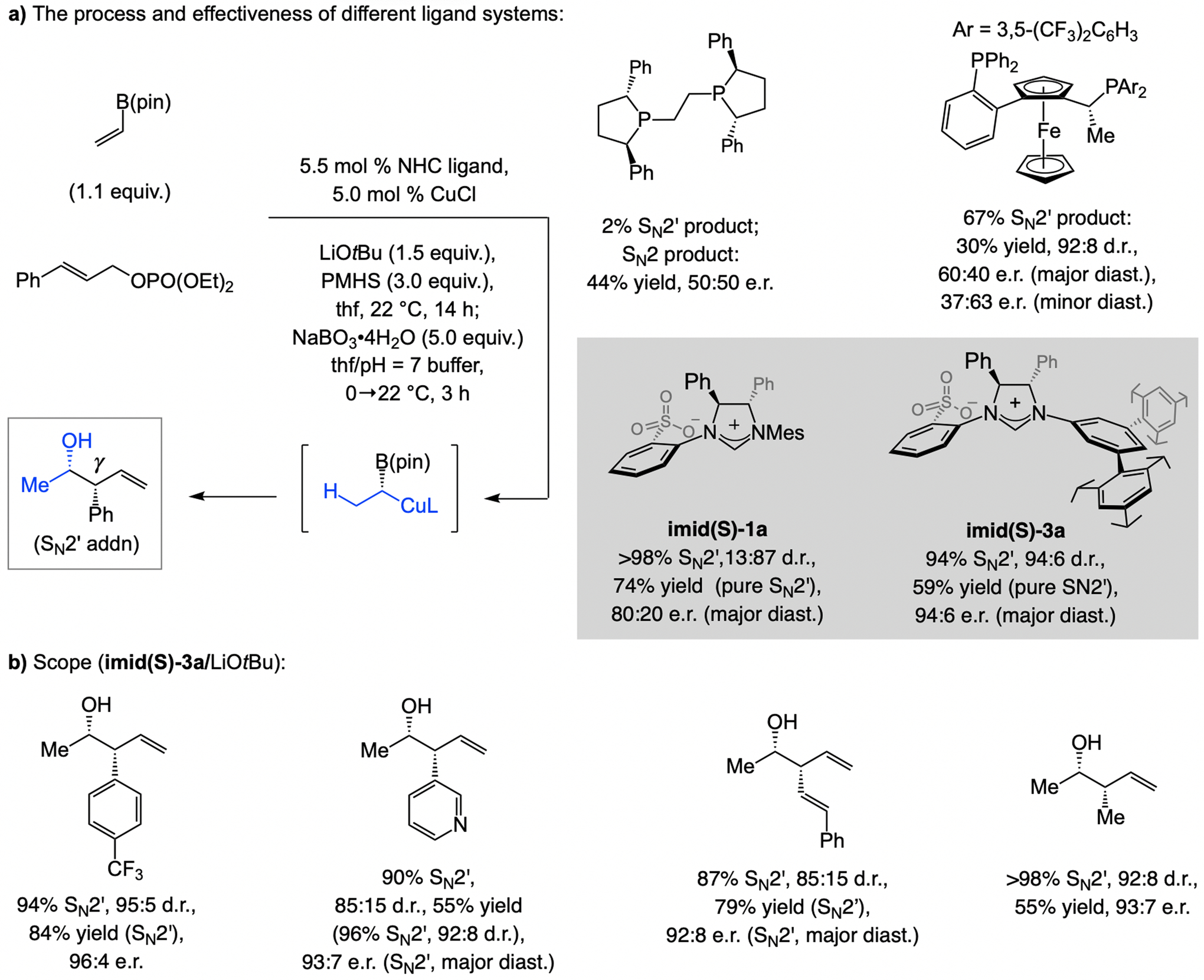
Sulfonate NHC–Cu catalysts promote multicomponent enantioselective Cu–H addition to vinyl–B(pin)/SN2’-selective and diastereoselective allylic substitution. See Scheme 35 for mechanistic analysis.
Scheme 66.
Catalytic enantioselective proto-boryl additions to 1,1-disubstituted alkenes.
4.4.2. With alkynes as latent nucleophiles.
In 2017, Zhang and Xiong reported EAS promoted by the catalyst derived from imid(S)-3a.[56] These processes commence with in situ formation of an alkenyl–Cu intermediate by regioselective Cu–H addition to an alkyne (Scheme 26), and were exceptionally SN2’-selective, affording a single trisubstituted alkene isomer in up to 99:1 e.r. Nevertheless, the alkyne and the alkene substrate needed to be aryl-substituted. No desired product was observed with a 1,2-disubstituted allylic phosphate (i.e., R = H). With a less substituted/more reactive electrophile, competitive reaction with the Cu–H complex, affording the terminal alkene, is likely.
Scheme 26.
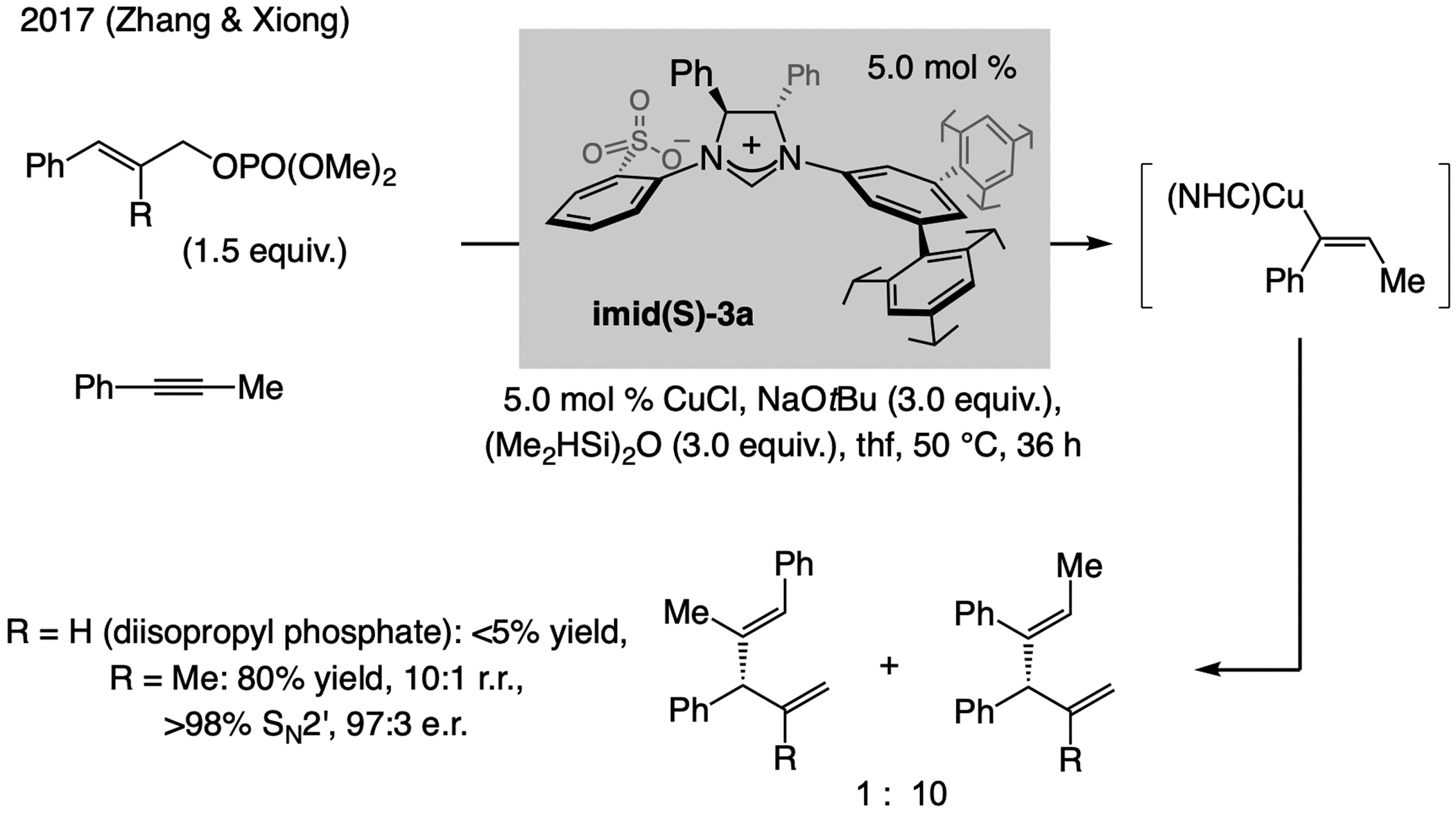
Multicomponent EAS beginning with a regioselective Cu–H addition to an alkyne. r.r. = regioisomeric ratio.
In 2019, Fañanás-Mastral outlined a process involving a terminal alkyne, an allylic bromide, and B2(pin)2, catalyzed by a complex derived from imid(S)-1a (Scheme 27).[57] Reactions with aryl- and alkenyl-substituted alkynes were efficient, and an example of a cyclopropyl alkyne was provided, but a transformation with an n-alkyl-bearing acetylene was minimally regioselective. Synthesis of formyl-substituted vinyl cyclopropane demonstrates versatlity.
Scheme 27.
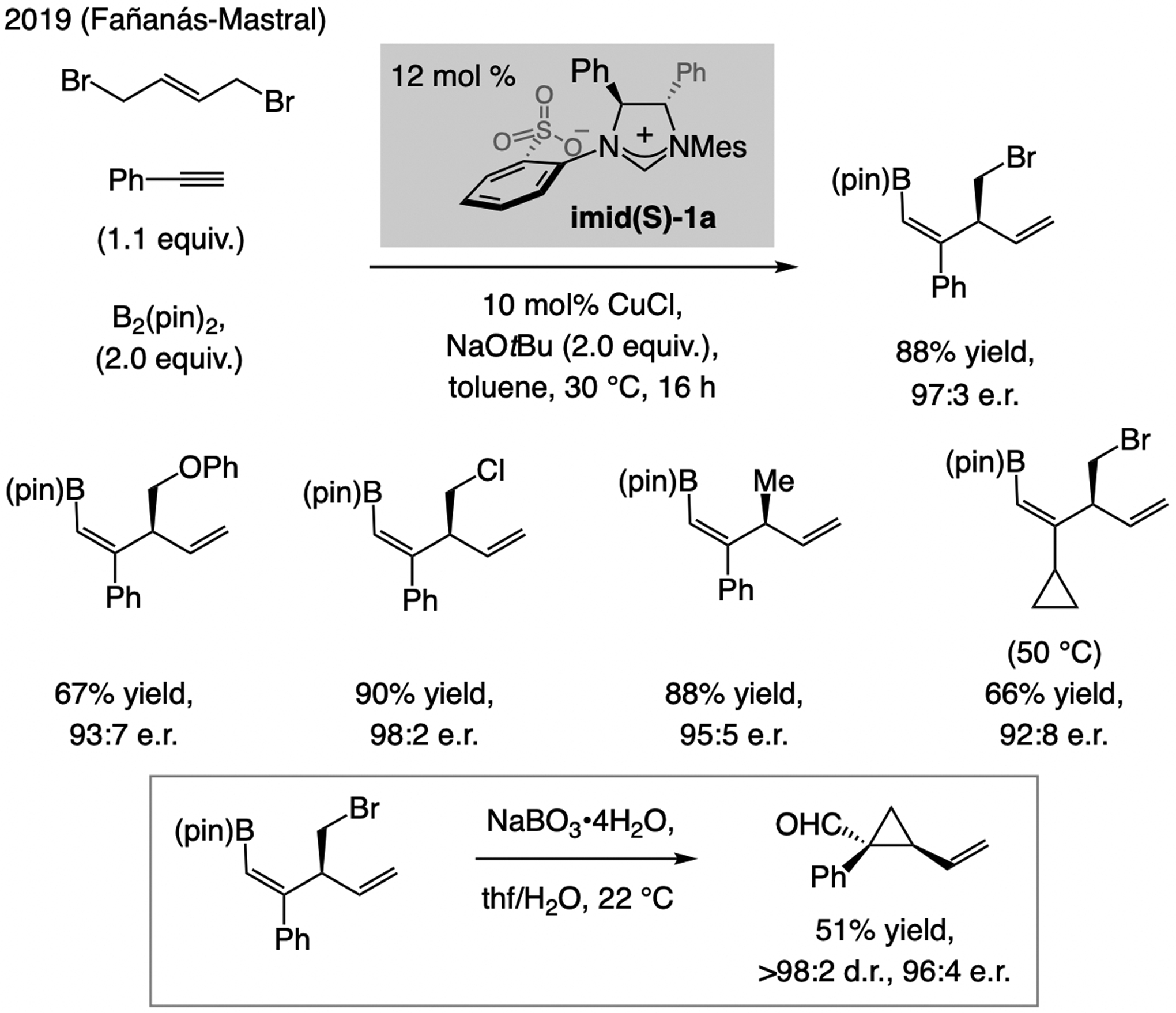
Sulfonate NHC–Cu catalysts promote multicomponent processes involving a Cu-B(pin) addition to alkynes. See Scheme 36 for mechanistic analysis.
4.4.3. With aryl-substituted allenes as latent nucleophiles.
In 2019, Xiong reported a Cu-H-catalyzed process involving aryl-substituted allenes and allylic phosphates (Scheme 28).[58] Regio- and enantioselectivities were high with the imid(S)-3a-derived catalyst. Reactions with 1,1-disubstituted aryl allenes or trisubstituted allylic phosphates gave products bearing a trisubstituted olefin or a quaternary carbon stereogenic center. Nonetheless, the allylic phosphate and the allene were aryl-substituted, limiting applicability. Only a 2:1 Z:E mixture was generated with an alkyl-substituted allene (e.r. not disclosed).
Scheme 28.
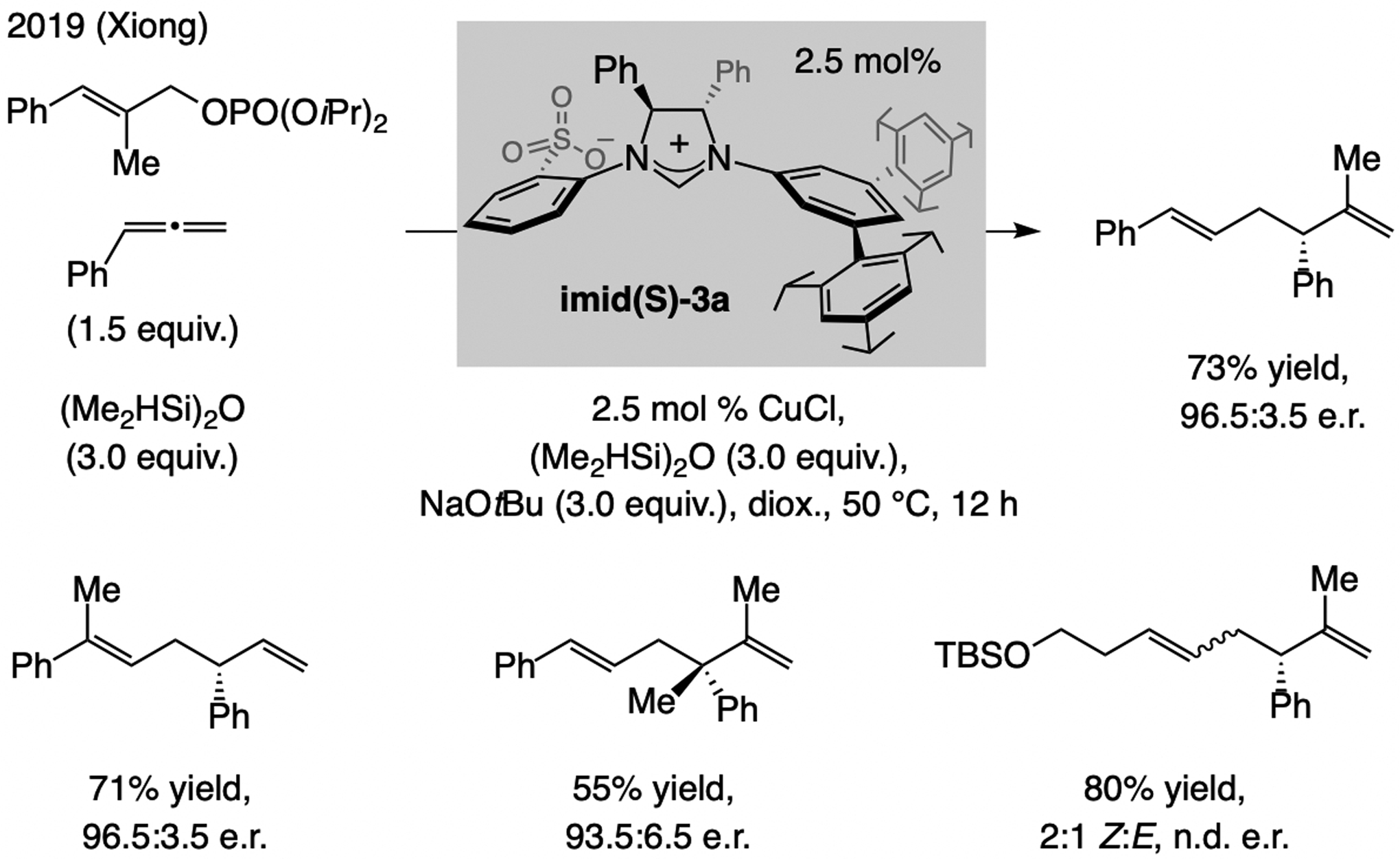
Multicomponent EAS involving Cu-H addition to an allene. n.d. = not determined.
4.4.4. With boron-substituted allenes as latent nucleophiles.
A mechanistically unusual and more versatile multicomponent EAS was disclosed soon after (Scheme 29). The process begins with regioselective NHC–Cu–H addition to allenyl–B(pin) compound, uncommon in that the more common Cu/B exchange is not the major pathway. The resulting Cu–allyl species then reacted with an allylic phosphate.[59] The method’s scope renders it readily applicable to complex molecule synthesis, as highlighted in the synthesis of the acyclic fragment of pumiliotoxin B (Scheme 29a). 1,1-Disubstituted allenyl–B(pin) compound was thus transformed to the desired product with the Cu-based complex derived from imid(S)-3a. More than a gram of the 1,5-diene, bearing a terminal olefin with an allylic stereogenic center as well as a readily modifiable trisubstituted alkenyl–B(pin) moiety, was prepared efficiently and stereoselectively. For total synthesis of netamine C (Scheme 29b) the same sulfonate NHC–Cu complex was used at three separate junctions: for a catalytic chemo-, regio-, and enantioselective multicomponent EAS, a catalyst-controlled diastereoselective EAS with an alkyl–ZnBr compound (reaction with the E allylic phosphate was less stereoselective), and another multicomponent EAS to secure a 1,3-relative stereochemistry. By using an E-allylic phosphate and (R)-imid(S)-3a, an authentic sample of the initially proposed netamine isomer was prepared, allowing for confirmation of the stereochemical assignment.
Scheme 29.
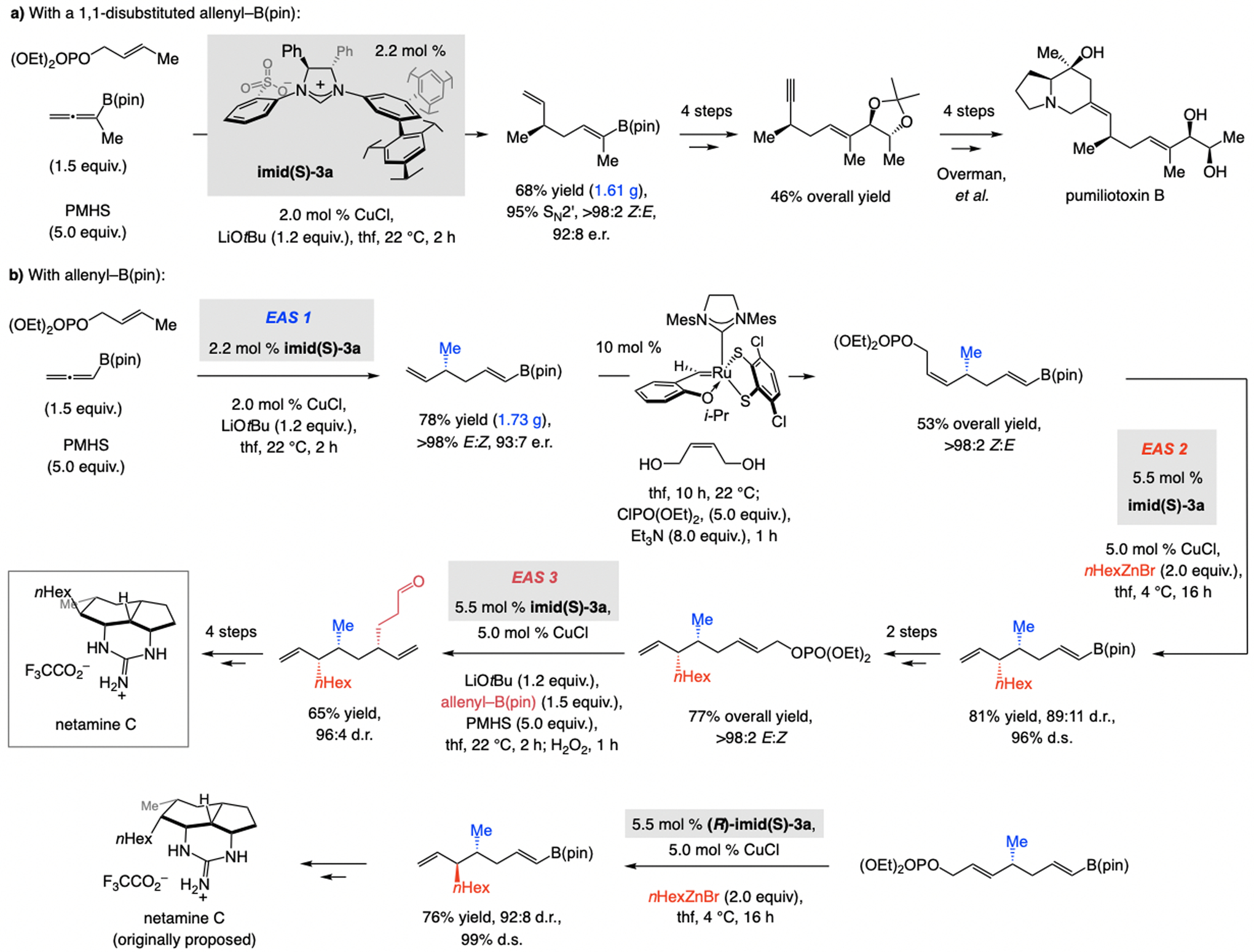
Multicomponent EAS commencing with Cu–H addition to boryl-substituted allenes (vs. the more common Cu/B exchange) and applications to complex molecule synthesis. d.s. = diastereospecificity.
4.5. Mechanistic Analysis
4.5.1. Are sulfonate NHC–Cu complexes bidentate or monodentate?
Although an X-ray structure for a sulfonate NHC–Cu complex has remained elusive, we were able to secure an X-ray structure for a Zn(II)- and an Al(III)-based species (NHC(S)-Zn and NHC(S)-Al, Scheme 30a).[60] These indicate that, to minimize steric repulsion, the sulfonate moiety is oriented syn to the nearby phenyl group (within the NHC backbone); nOe studies indicate that the same holds true in solution. Complexes NHC(S)-Zn and NHC(S)-Al catalyze EAS (no Cu salt; Scheme 30b). Control experiments indicated that the NHC–Zn complex probably serves as a Lewis acid activator, suggesting complex I as a plausible stereochemical model. We hypothesized that because the sulfonate group is oriented towards the Ph group, a binding pocket is made available for substrate coordination, and that the pseudo-equatorial S=O bond might be a sufficiently Lewis basic site to bind and deliver the (alkyl)2Zn compound.
Scheme 30.
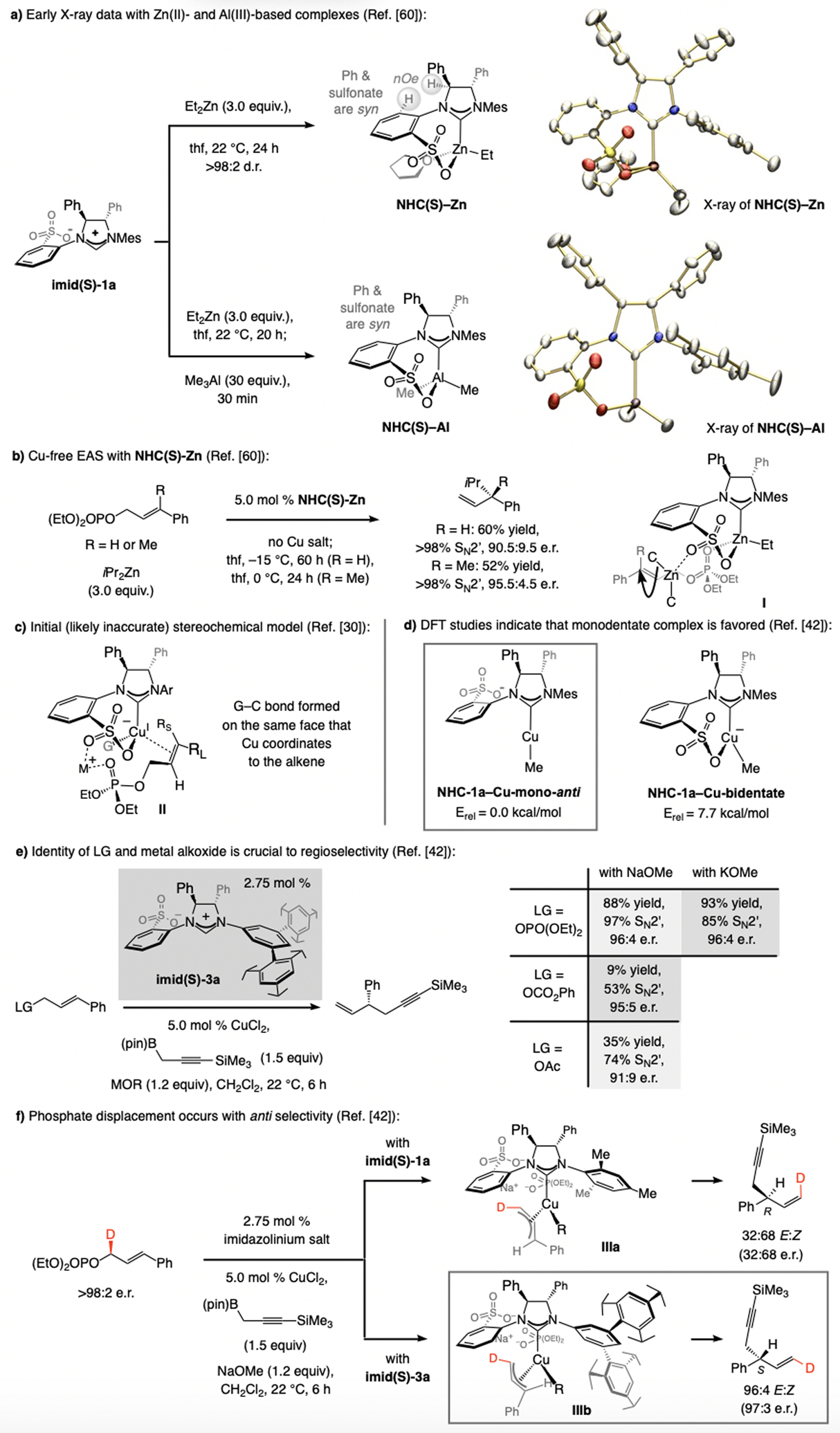
X-ray structures of Zn(II)- and Al(III)-based complexes, the initial stereochemical model, results of DFT studies at the ω-B97XD/Def2-TZVPPTHF(SMD)//ω-B97XD/Def2-SVP level), and key experimental data. LG = leaving group.
We extended the above model to Cu-based systems (II, Scheme 30c), proposing that the less encumbered quadrant might accommodate the alkyl moiety to allow for a metal bridge to form involving the departing phosphate group. (For steric maps, see the Supporting Information.) The high SN2’ selectivity,[61] e.r., and related trends could thus be accounted for. Nevertheless, a consistent model is not necessarily the most plausible one; as we shall see, an unwise assumption made was that the Cu(I) center is similarly Lewis acidic as a Zn(II) or an Al(III) site.
DFT studies showed that an anionic monodentate sulfonate NHC–Cu complex, electronically more closely related to Ag(I) (see Scheme 3; vs. Zn and Al), is energetically favored (Scheme 30d).[62] Experimental data showed the Lewis basicity of a leaving group and the Lewis acidity of the cation in a metal alkoxide impact SN2’ selectivity (Scheme 30e), supporting the involvement of metal bridging, which, while in line with the intermediacy of a complex such as II, does not rule out a monodentate sulfonate NHC–Cu species (IIIa-b, Scheme 30f). One aspect that distinguishes reactions of a bi- vs. a monodentate complex is that in II the C–C bond is formed on the same face as that from which the phosphate group is displaced (syn addition), whereas in IIIa-b addition occurs at the opposite face (anti addition). Studies with enantiomerically enriched D-substituted allylic phosphates, where the stereochemistry of the labeled alkene and the Z:E ratios are reflected in the e.r. values, indicate that, congruent with the DFT calculations (Scheme 30d), reactions likely proceed via a monodentate complex (IIIa-b, Scheme 30f). [42,63]
4.5.2. The stereochemistry-determining step.
Allylic substitution can occur in two ways. 1) After formation of the Cu–alkene complex (IV → V, Scheme 31a), Cu–alkyl intermediate VI may be formed by migratory insertion. 2) The transformation might proceed via π-allyl complex VII (oxidative addition; see Scheme 30f). DFT studies (model sulfonate NHC), carried out with the C-O distance in tsCu-Xadd constrained to 1.50 Å (to avoid C–O bond cleavage; Scheme 31b), indicate that the π-allyl route is generally more favored, particularly for methyl (28.5 vs. 6.7 kcal/mol), phenyl (25.4 vs. 8.6 kcal/mol), silyl (19.2 vs. 5.3 kcal/mol), and hydride (13.7 vs. 7.2 kcal/mol) additions. (The same preference applies to boryl substitution, but with a narrower energy gap (8.5 vs. 4.9 kcal/mol for migratory insertion vs. π-allyl generation; see Section 7)).
Scheme 31.
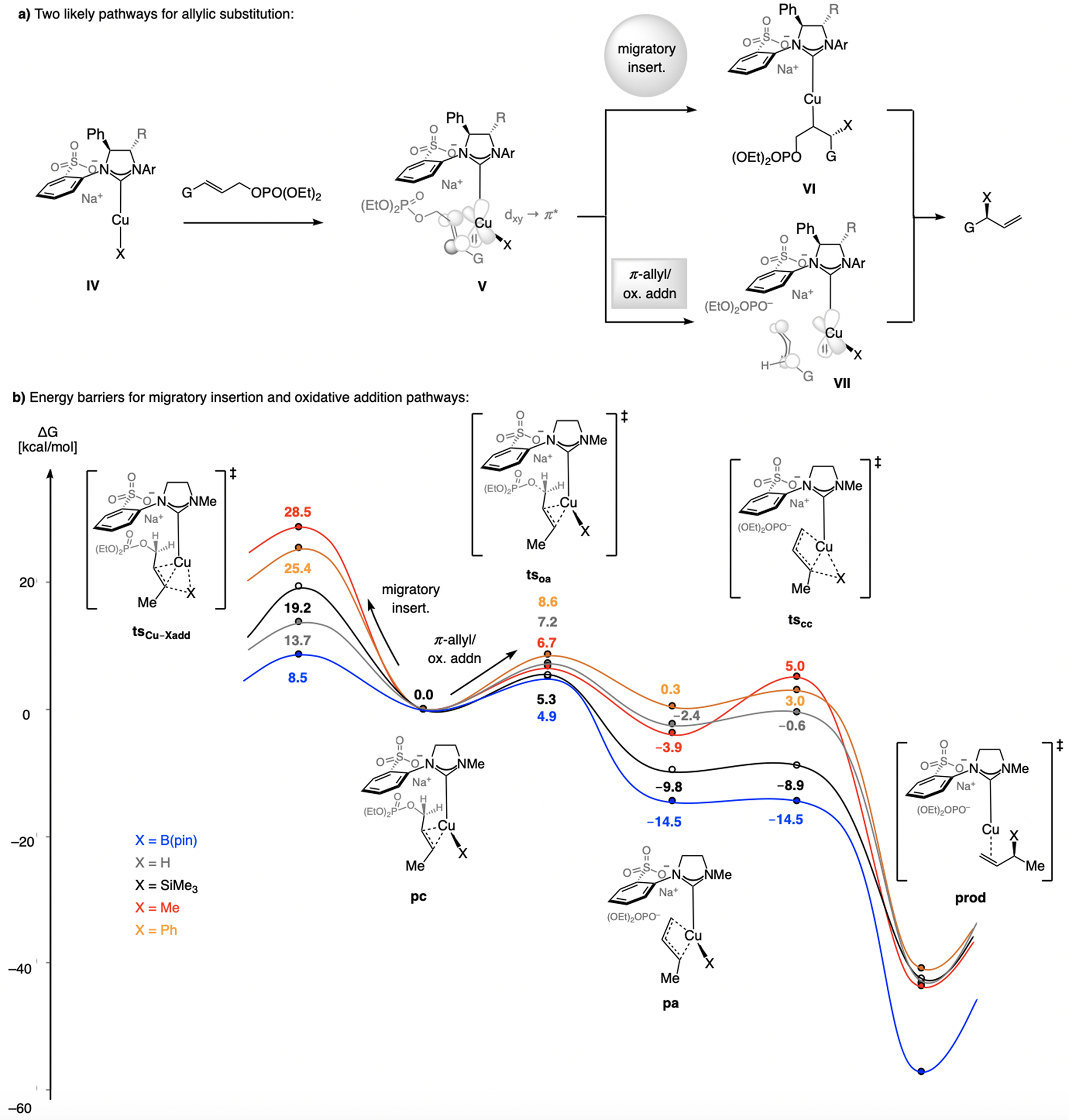
Evaluation of two EAS pathways [M06/def2-TZVPP//M06L/def2-SVP level of theory in CH2Cl2 (SMD solvation model)]; C-O bond constrained in tsCu-Xadd to 1.50 Å to circumvent C–O bond rupture. Previously unpublished analysis; see the Supporting Information for details. ts = transition state; pc = p complex; oa = oxidative addition; pa = π-allyl complex; cc = Cu complex; prod = final product.
4.5.3. Allenyl substitutions.
The intermediacy of a π-allyl complex (see Scheme 31b) is supported by the secondary kinetic isotope effect (KIE) data (1.21 (±0.01); Scheme 32a),[64] indicating that C–O bond cleavage, or generation of a cationic Cu π-allyl intermediate, is stereochemistry-determining. The suggested models account for the observed selectivity profiles and trends (see below).
Why is imid(S)-3a optimal for EAS of allenyl–B(pin) with disubstituted alkenes, whereas imid(S)-1a is the superior choice with trisubstituted allylic phosphates? And why do these processes proceed with the opposite sense of enantioselectivity (Scheme 32)? With NMes-substituted imid(S)-1a, reactions can proceed via energetically similar VIII and IX (Scheme 32b), engendering low e.r. On the other hand, with imid(S)-3a, bearing an NAr moiety with sizeable C3 and C5 substituents, in which rotation around the C–N bond is less hindered, reaction via X should be preferred (vs. XI), generating the (S) enantiomer.
With a trisubstituted alkene (Scheme 32c), EAS with imid(S)-1a can occur via XIII, favoring the formation of an (R) isomer. Reaction via XII likely suffers from steric repulsion caused by the allenyl moiety being lodged between the NMes moiety and the substrate Me group (unlike VIII, where there is a greater conformational mobility). There is a smaller energy gap between XIVa and XVa, corresponding to intermediates with the catalyst derived from imid(S)-3a. With a Cu–propargyl complex, e.r. is high regardless of the substitution at the olefin (Scheme 32d). This may be attributed to restricted rotation around the ligand’s NAr bond, partly owing to dispersive attraction between the silyl moiety and the NHC’s iPr substituents (C1–N–C2–C3 dihedral angle = 9.0° in XIVb). The alkene’s Me unit thus interacts with the π-cloud of the meta-aryl substituent, allowing it to avoid interaction with the iPr groups. In XVb, steric repulsion between the alkene’s Ph ring and the NAr moiety causes the C1–N–C2–C3 dihedral angle to expand (28.3°), converting an otherwise dispersive attraction between the silyl and the NHC’s iPr substituents (see XIVb) into a repulsive interaction involving the silyl unit and one of the iPr groups (see XVb). As we shall see, the trend where additions to di- and trisubstituted alkenes afford the opposite product enantiomers in reactions with sulfonate NHC–Cu complexes is not limited to this set of reactions (see also Scheme 34). The above mechanistic analysis constitutes a revision of the originally published mechanistic scenario.[30]
4.5.4. SN2”-selective substitutions.
The idea that C–O bond rupture is stereochemistry-determining in SN2”-selective reactions is supported by KIE data (1.11 (±0.01), Scheme 33a). Furthermore, KIE is negligible for EAS with a dienyl phosphate labeled at the site of C–C bond formation, indicating this step is not likely to impact the stereochemical outcome (Scheme 33a).[34]
Scheme 33.
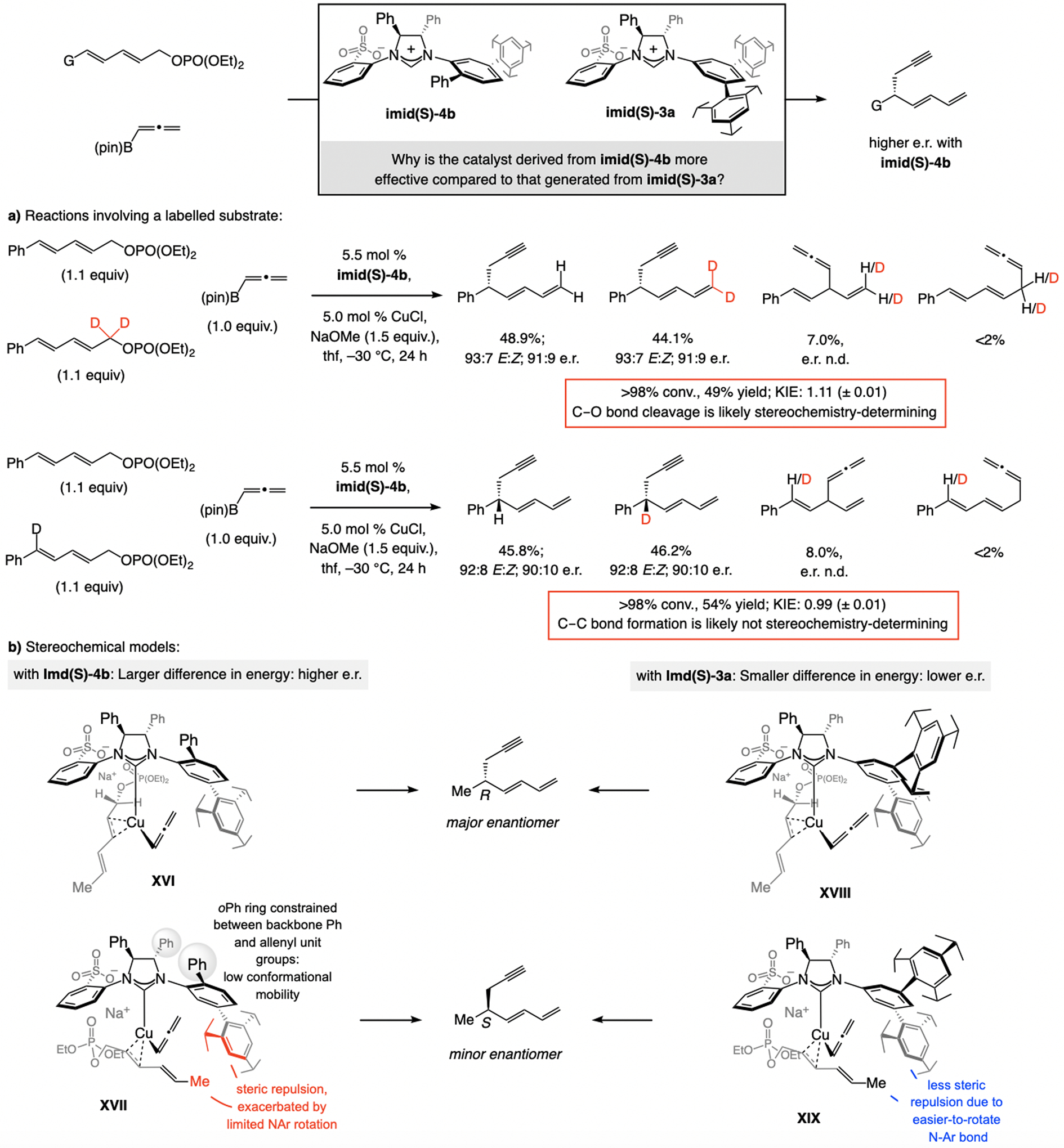
Additional support for C–O bond cleavage being stereochemistry-determining and related stereochemical models (see Scheme 14 for methodology). DFT at the MN15/Def2-TZVPP//M06L/Def2-SVP level. n.d. = not determined.
The higher e.r. with imid(S)-4b (vs. imid(S)-3a) might be due to restricted rotation around the N-Ar bond in XVI and XVII (from imid(S)-4b), arising from repulsive interaction between the ortho-phenyl group with the allenyl ligand, on the one hand, and the NHC backbone, on the other. This enhances the steric pressure caused by the proximity of the substrate’s alkenyl substituent (HC=CMe in XVII) and the (iPr)3-aryl ring. Because the NAr rings in complexes XVIII and XIX (from imid(S)-3a) lack an ortho aryl unit, rotation around the C–N bond is less costly, and steric strain between the substrate’s alkenyl substituent and those of the NAr ring can be avoided. The energy difference between XVIII and XIX is thus less and the e.r. lower (vs. via XVI and XVII).
4.5.5. With bis[(pinacolato)boryl]methane.
While steric factors offer a rationale for why e.r. is higher in EAS with bis[(pinacolato)boryl]methane and disubstituted alkenes (XX-XXI, Scheme 34a), edge-to-face/π-π association might also impact transformations with Z-trisubstituted allylic phosphates (Scheme 34b). Dispersive forces,[65] namely, the association between an aromatic p cloud and the nearby C–H bonds of a tBu group, or between the ortho-phenyl substituent and the B(pin) moiety in XXII, could be involved. The difference in the dihedral angle values (Scheme 34b) support the presence of such interactions, which in XXIII, seem to play a lesser role.[66]
4.5.6. Sulfonate NHC–Cu catalyst can control enantio- and diastereoselectivity.
A model may be proposed based on DFT studies (Scheme 35; for DFT studies regarding the identity of the stereochemistry-determining step, see Scheme 31b). Cu–H addition with imid(S)-3a might involve association of a pinacolato oxygen with Li+ (XXIV). With the complex derived from imid(S)-1a, there is likely to be steric repulsion between the B(pin) moiety and an o-methyl substituent (XXV), diminishing enantiofacial selectivity. The preferential allylic substitution pathway likely occurs via a complex with a Li+ bridging chelate, wherein steric repulsion between the substrates aryl substituent and the NHC’s large NAr group is minimal (XXVI). The sulfonate-Li-phosphate bridge is probably also responsible for the high SN2’-selectivity. The sulfonate NHC–Cu catalyst thus controls the enantioselectivity as well as the diastereochemical outcome.
Scheme 35.
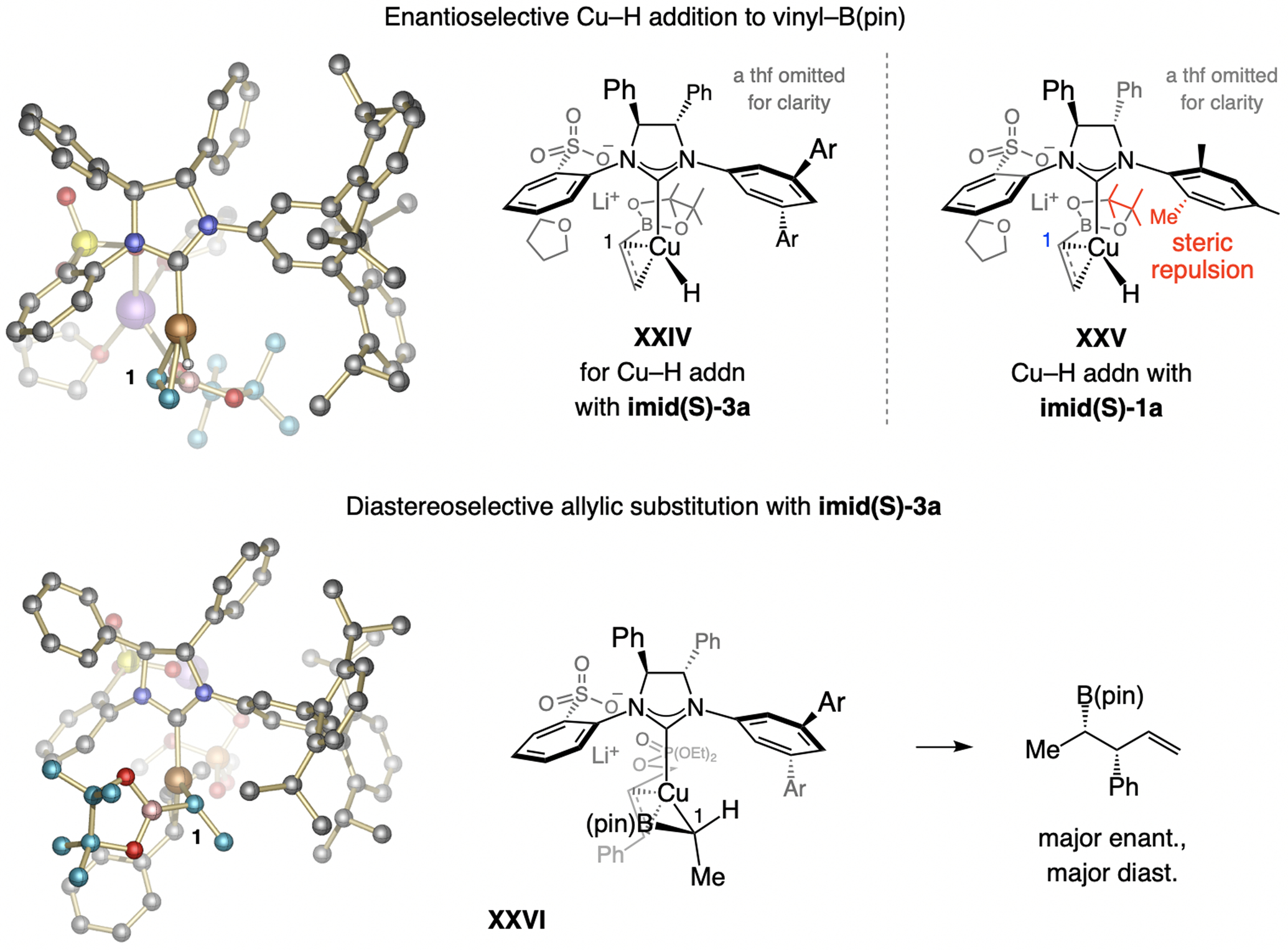
The sulfonate moiety is likely responsible for the high d.r. and e.r. in Cu–H-catalyzed multicomponent EAS reactions (see Scheme 25 for methodology). DFT at the M06L/Def2-TZVPP//M06L/Def2-SVP level.
4.5.7. With organoaluminum compounds.
Metal bridging likely applies to Al(III)-based systems as well (Scheme 36a–b).[67] Whether it is with a di- or a trisubstituted alkene substrate (XXVII-XXVIII, Scheme 36a, and XXIX-XXX, Scheme 36b), steric factors can impact e.r. With a smaller β-alkenyl unit, steric repulsion between the catalyst and the substrate becomes a stereocontrolling element: the larger substituent (Ph) occupies the less hindered quadrant below the NAr group, while Cα is positioned to avoid steric repulsion with the o-Me unit. With a trisubstituted alkene, steric pressure owing to propinquity of Cα and the NHC ligand (XXX) is likely more destabilizing than with the substrate’s Me unit. That is, as a result of interaction with Cu during π-allyl formation, Cα is closer to the ligand than the Me group (Cα–Cu = 2.63 Å vs. CMe–Cu = 3.06 Å). DFT studies by Fañanás-Mastral[57] (XXXI- XXXII, Scheme 36c) indicate that a S=O…Na…Br bridge might well be responsible for the high e.r. in the corresponding multicomponent reactions (see Scheme 27).
Scheme 36.
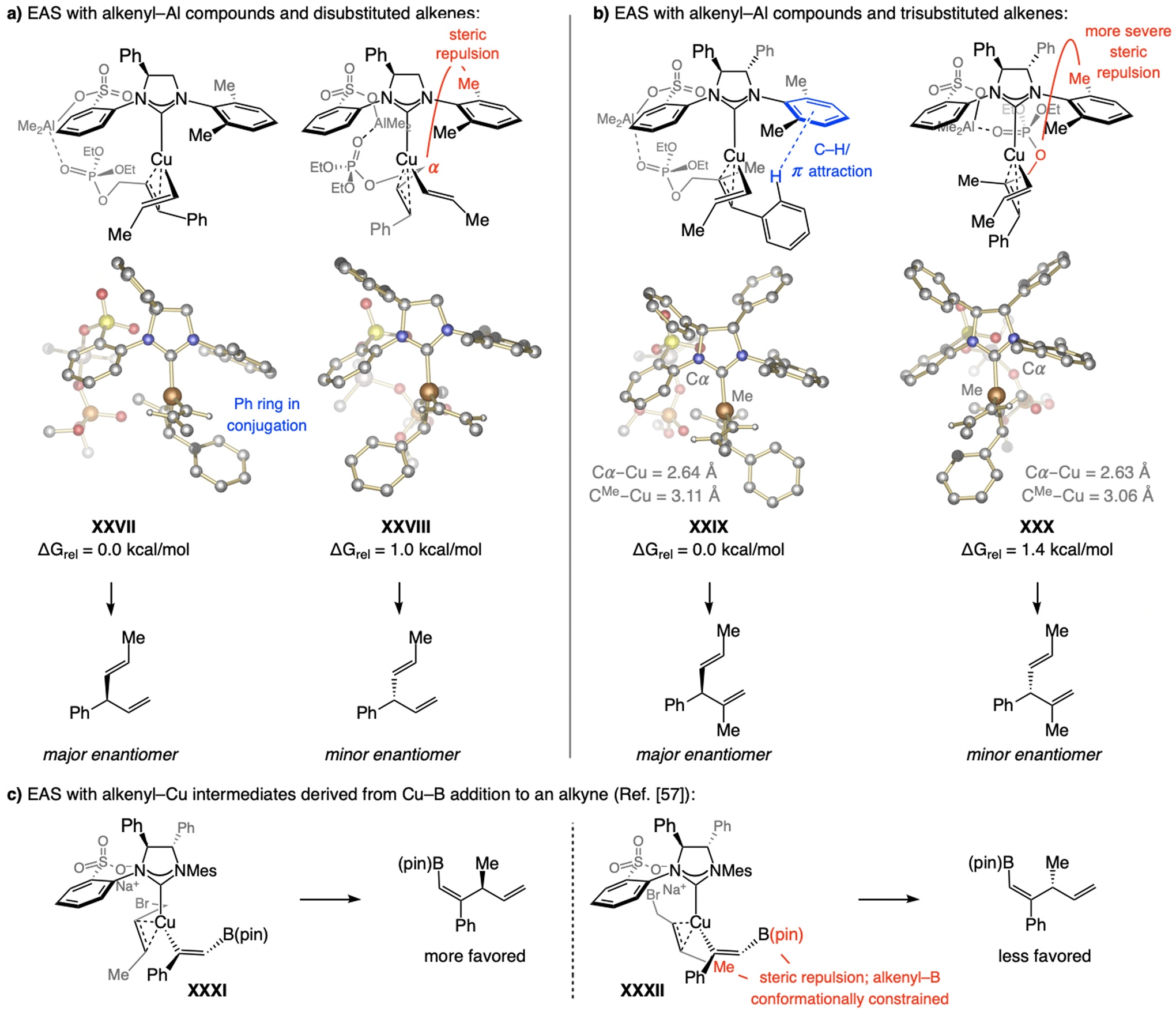
Rationale for EAS of alkenyl moieties derived from Cu/Al exchange or formed by Cu–B addition to an alkyne (see Schemes 5 and 27 for methodology). DFT at the M06L/Def2-TZVPP//M06L/Def2-SVP level. Previously unpublished analyses; see the Supporting Information for details.
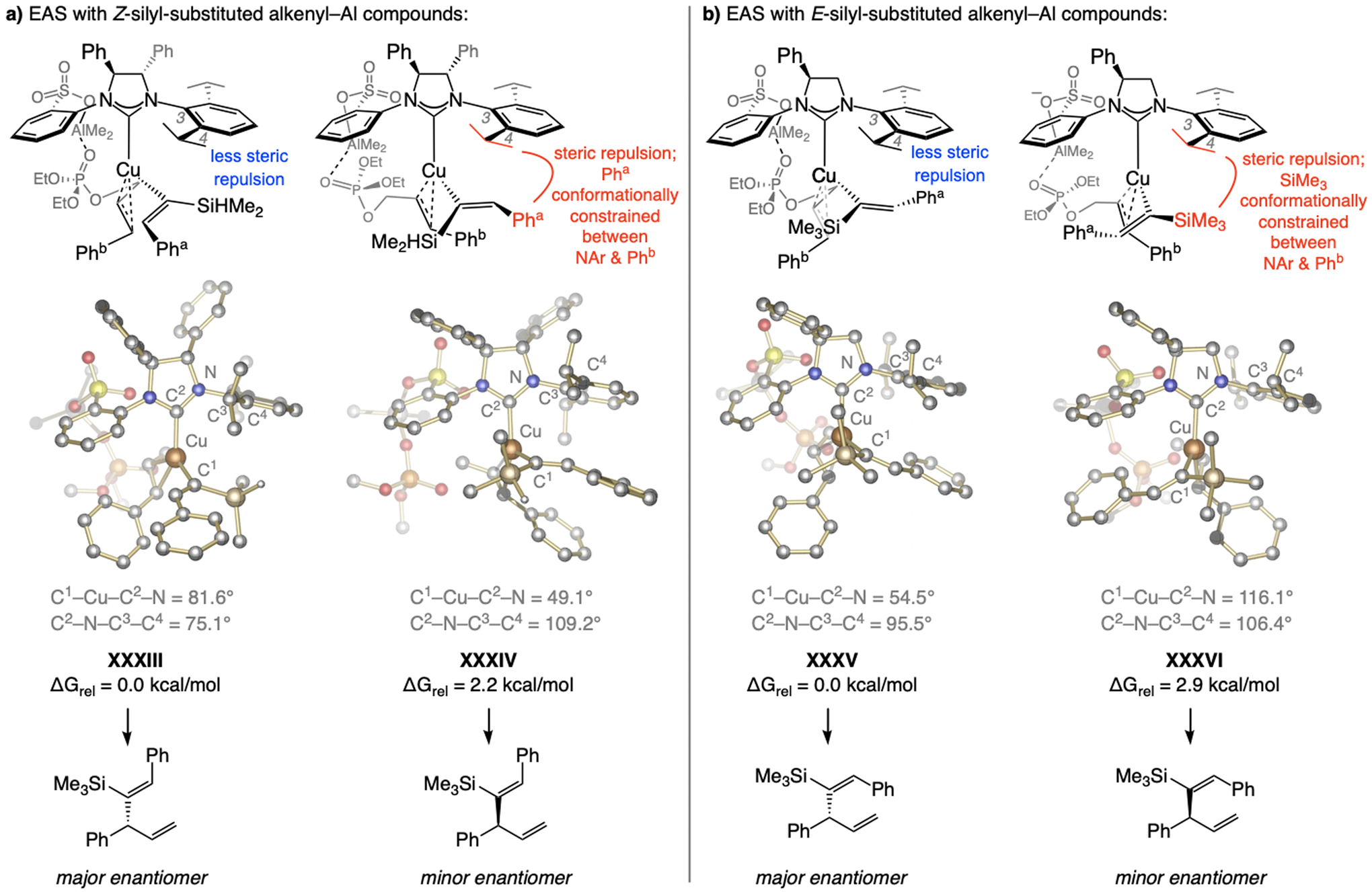
Not surprisingly, for silyl-substituted Z-alkenyl–Al compounds steric factors become dominant (Scheme 37). With Z-alkenylmetal isomers, the allylic phosphate substituent (Phb, XXXIII, Scheme 37a) and the NHC’s iPr-substituted aryl unit point in opposite directions, placing the silyl group below the NAr moiety. Although this engenders steric pressure, it is less than in the alternative XXXIV. The phenyl ring of the alkenyl moiety (Pha) in the latter is constrained between Phb on the electrophile and the NAr group on the NHC, as reflected by the expanded C2–N–C3–C4 dihedral angle (109.2° vs. 75.1° in XXXIII).
Scheme 37.
Rationale for EAS with silyl-substituted alkenyl–Al compounds (see Scheme 6 for methodology). DFT at the M06L/Def2-TZVPP//M06L/Def2-SVP level. Previously unpublished analyses; see the Supporting Information for details of the DFT studies.
With a larger E-alkenyl–Al compound, the more diminutive sulfonate NHC ligands that bear a single phenyl moiety are more effective (Scheme 37b). The C1–Cu–C2–N dihedral angle in XXXV contracts further (54.5° vs. 116.1° in XXXVI) to minimize repulsion between the silyl moiety and the sulfonate-bearing NAr ring. In the higher energy XXXVI, the silyl unit is forced to sit between Phb and the NAr group, giving rise to a wider C2–N–C3–C4 dihedral angle (106.4° in XXXVI vs. 95.5° in XXXV, respectively).
5. Catalytic Enantioselective Conjugate Addition (ECA) [68]
5.1. Additions of Alkyl Moieties to Trisubstituted Enones
State-of-the-art, circa 2007. Alexakis reported[69] in 2005 that phosphoramidite–Cu complexes promote ECA of Me3Al or Et3Al to β-substituted cyclohexenones (Scheme 38). We later showed that, with an amino acid derived Cu complex, ECA of (alkyl)2Zn compounds to trisubstituted nitroalkenes[70] (a rare case involving acyclic substrates) and tetrasubstituted cyclic keto-esters[71] can be carried out. In a related investigation, Fillion utilized a phosphoramidite ligand for ECA of (alkyl)2Zn compounds to arylalkylidene derivatives of Meldrum’s acid.[72] In 2006, we showed that the combination of NHC(O)–Ag-2a and (CuOTf)2•C6H6 may be used to catalyze ECA of (alkyl)2Zn and (aryl)2Zn compounds to β-substituted cyclic enones.[3] Mauduit and Alexakis illustrated that β,β’-disubstituted cyclohexenones may be prepared by ECA of an alkyl Grignard reagent with an NHC–Cu catalyst.[73]
Scheme 38.
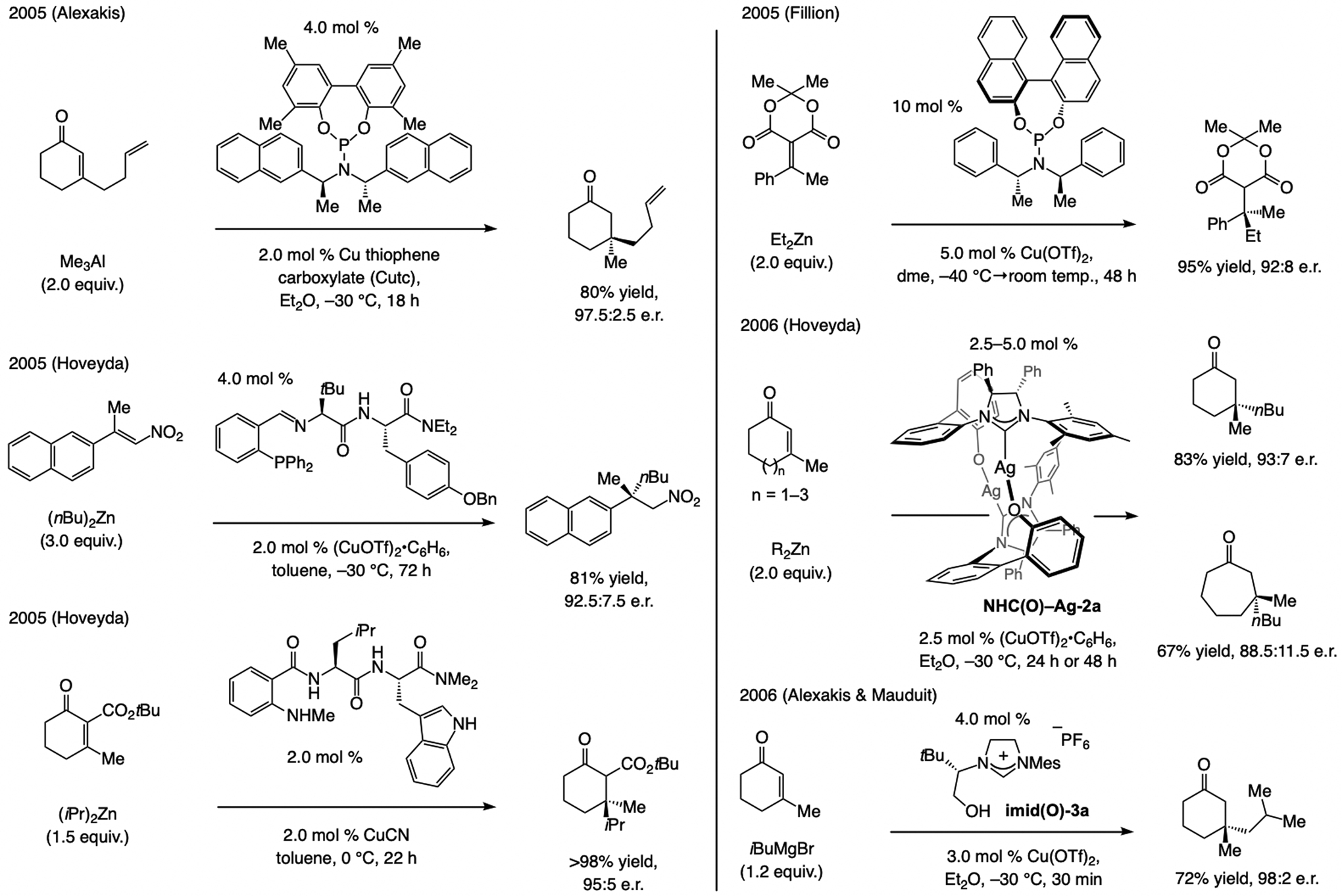
The state-of-the-art in catalytic ECA reactions that generate a quaternary carbon stereogenic center, circa 2006. tc = thiophene-2-carboxylate.
Nonetheless, several major shortcomings remained. High catalyst loadings (e.g., 10 mol %) and/or extended reaction times (e.g., 48–72 h) aside, the scope remained limited. Often, activated electrophiles were required (e.g., nitroalkenes, keto-esters, Medrum’s acid derivative), and, in most cases, results were limited to additions to cyclohexenones. With the catalyst derived from NHC(O)–Ag-2a (Scheme 38), ECA of (alkyl)2Zn compounds, but not Me2Zn, to six-, seven- and eight-membered ring enones were efficient. Reactions of cyclopentenones were lower yielding and e.r. values were not as high in the case of medium ring enones.
5.1.1. With organozinc compounds.
β-Ester-substituted cyclic enones are challenging substrates because the electron-withdrawing carboxylic ester group enhances electron density at the site of C–C bond formation (Scheme 39). The complex derived from NHC(O)-Ag-2a, which promotes ECA to β-alkyl-substituted enones, is ineffective in these cases. In contrast, ECA of organozinc compounds, including Me2Zn and Ph2Zn, to β-ester-substituted cyclic enones can be catalyzed efficiently and enantioselectively by the complex derived from NHC(S)-Ag-1a and (CuOTf)2•C6H6.[5] Gin has used this approach in a total synthesis of nominine,[74] where the initial product was converted to the corresponding enol triflate. The carboxylic ester was then reduced to the derived aldehyde, and an ensuing catalytic cross-coupling afforded an alkenyl nitrile.
Scheme 39.
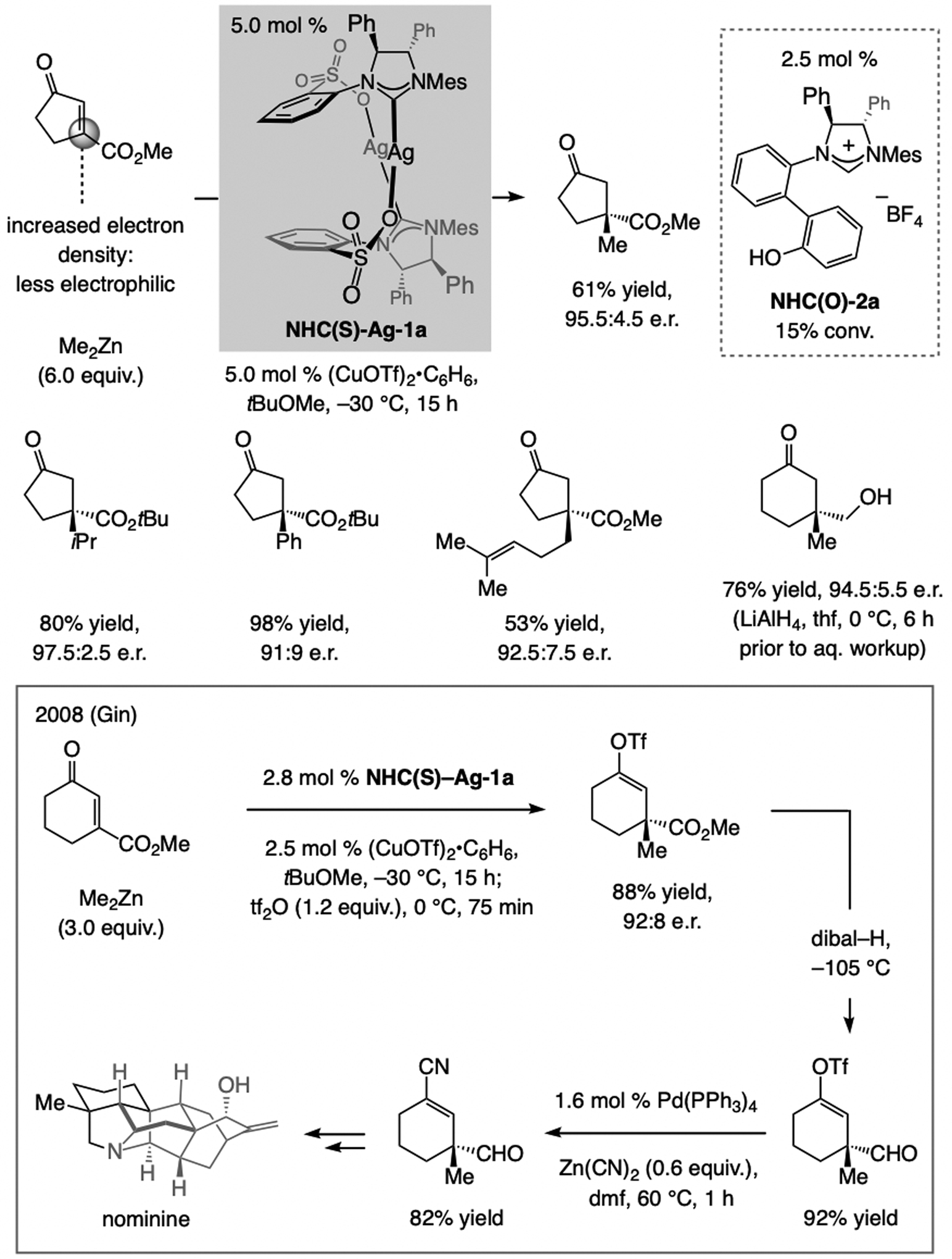
ECA of (alkyl)2Zn reagents to cyclic β-carboxylic enones and application to total synthesis of nominine by Gin et al.
5.1.2. With alkylaluminum compounds.
Cyclopentenones are a difficult substrate class in catalytic ECA.[75] The combination of the NHC–Cu catalyst derived from NHC(S)–Ag-2d and the more Lewis acidic/nucleophilic (alkyl)3Al compounds [vs. (alkyl)2Zn reagents] provides an attractive solution to this problem (Scheme 40a).[76] β-Substituted cyclohexenones and larger rings may be used. The application to total synthesis of clavirolide C,[77] via the enol silane derived from trapping of ECA of Me3Al to a β-substituted cyclopentenone, highlights the utility of this approach (Scheme 40b). Two relevant recent applications, disclosed by Snyder, relate to enantioselective total synthesis of complex polycyclic natural products, conidiogenones and waihoensene.[78] In both instances, ECA was performed on multi-gram scale. In one case, the imidazolinium salt was recovered (>98 % yield) by filtration of the reaction mixture and treatment of the resulting cake with a 9/1 CH2Cl2/MeOH mixture; this material was re-used.
Scheme 40.
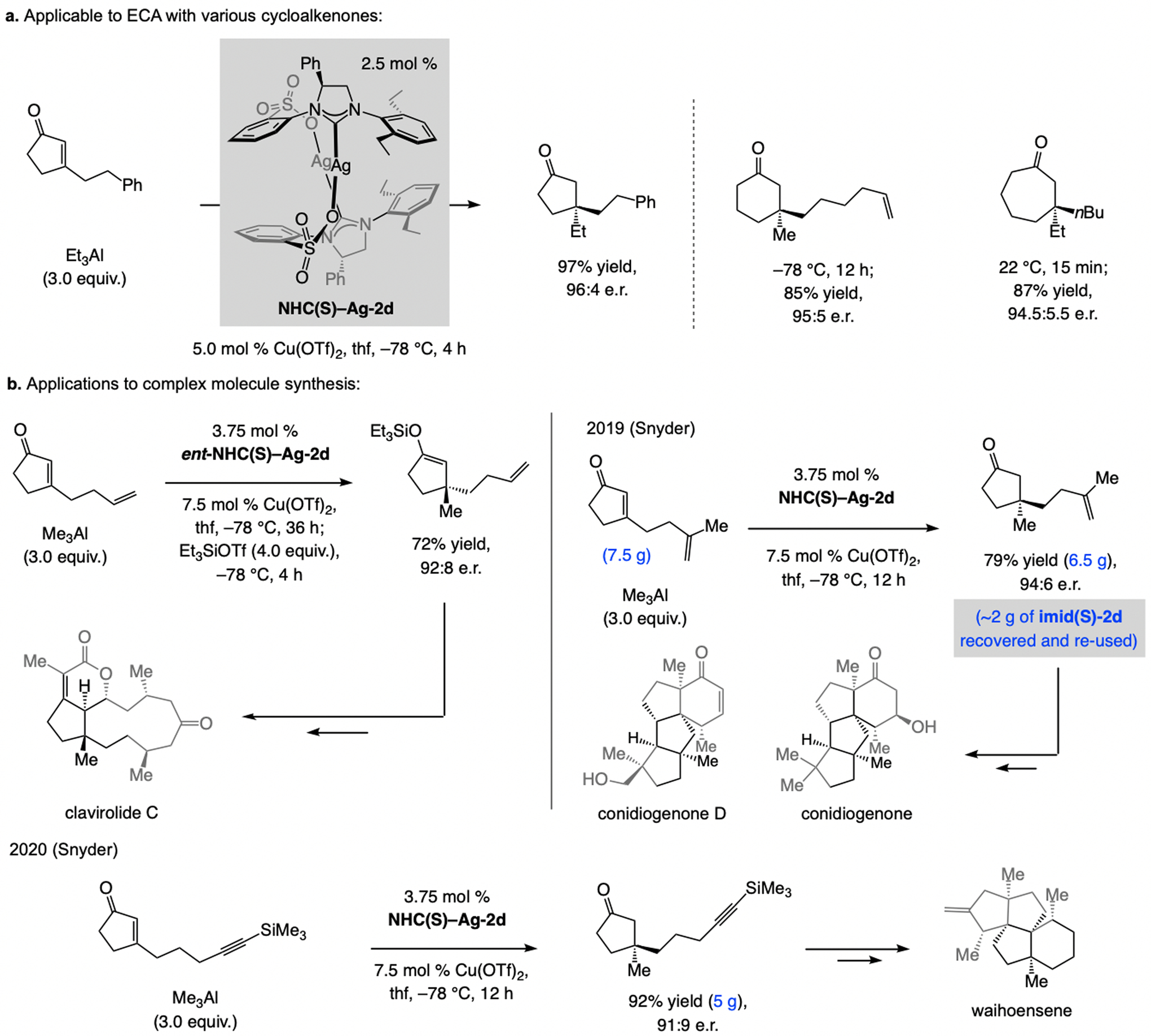
ECA with β-substituted cyclic enones and (alkyl)3Al reagents and applications to total synthesis of structurally complex bioactive natural products.
Catalytic ECA to acyclic trisubstituted alkenes are uncommon (see Scheme 38). The lack of ring strain leads to diminished reactivity and, in the case of disubstituted enones, the possibility of reaction through the s-cis or the s-trans conformer can lead to low e.r. In such instances, having access to a collection of catalyst candidates becomes important, as underscored by the finding that for ECA of (alkyl)3Al compounds to acyclic trisubstituted enones (Scheme 41), the catalyst derived from the previously unknown NHC(S)-Ag-1e delivers the highest e.r. (0.5 mol %).[79] Oxidation by commercial bleach[80] afforded the carboxylic acid derivatives.
Scheme 41.
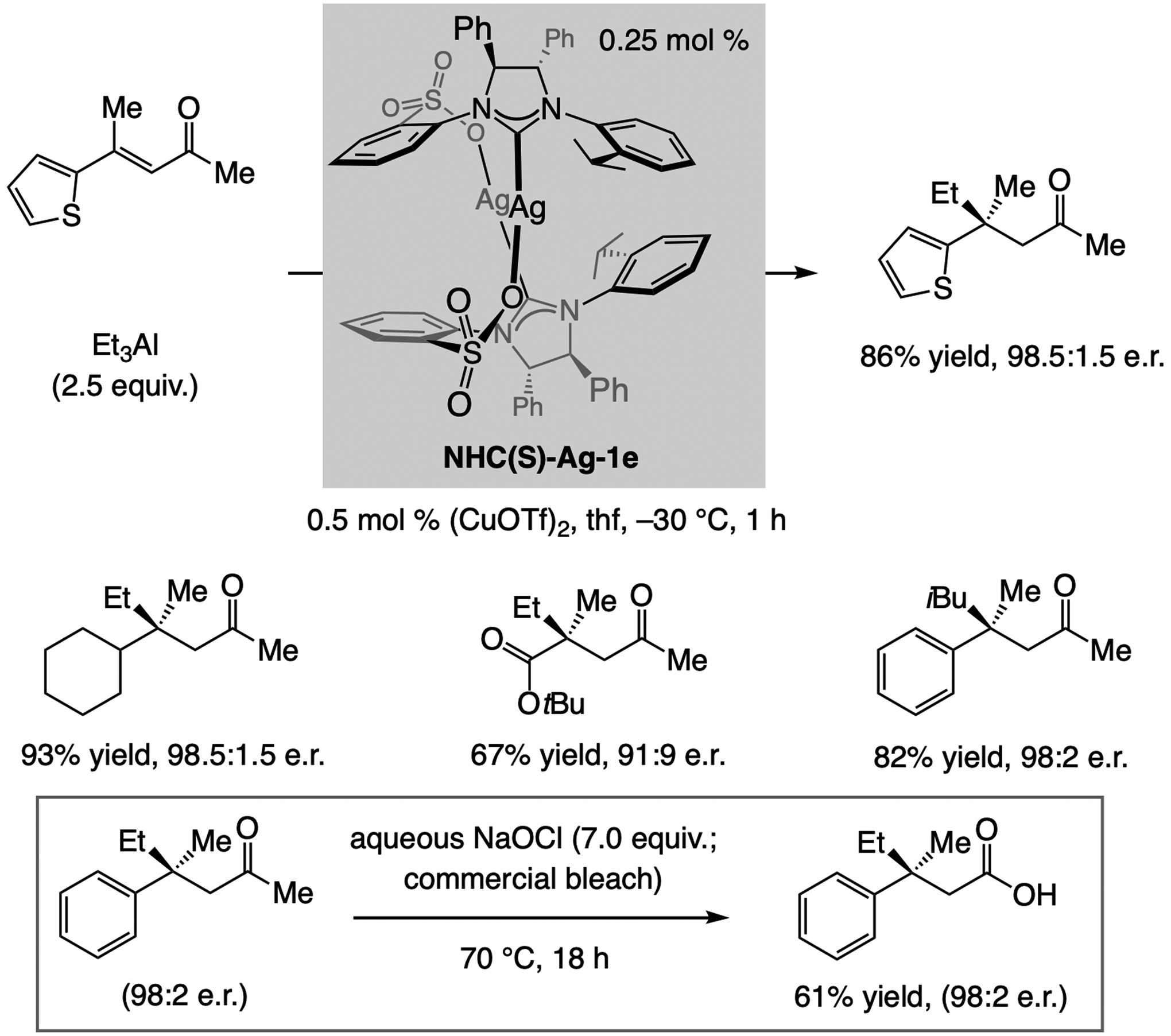
ECA of (alkyl)3Al compounds to acyclic trisubstituted enones.
Related advances. Also in 2013, Endo and Shibata reported that the complex formed by mixing 5.0 mol % CuCl2•2H2O and 10 mol % 2-diphenylphosphine-substituted binaphthol catalyzes ECA of Me3Al to linear enones (Scheme 42).[81] Yields and enantioselectivities were high, but the majority of substrates contained an aryl-substituted alkene and carbonyl. In 2017, Fletcher outlined a protocol entailing in situ generation of an alkyl–zirconocene compound. β-Substituted enones were effective substrates for ECA to β-substituted cyclopentenones and cyclohexenones.[82,83]
Scheme 42.
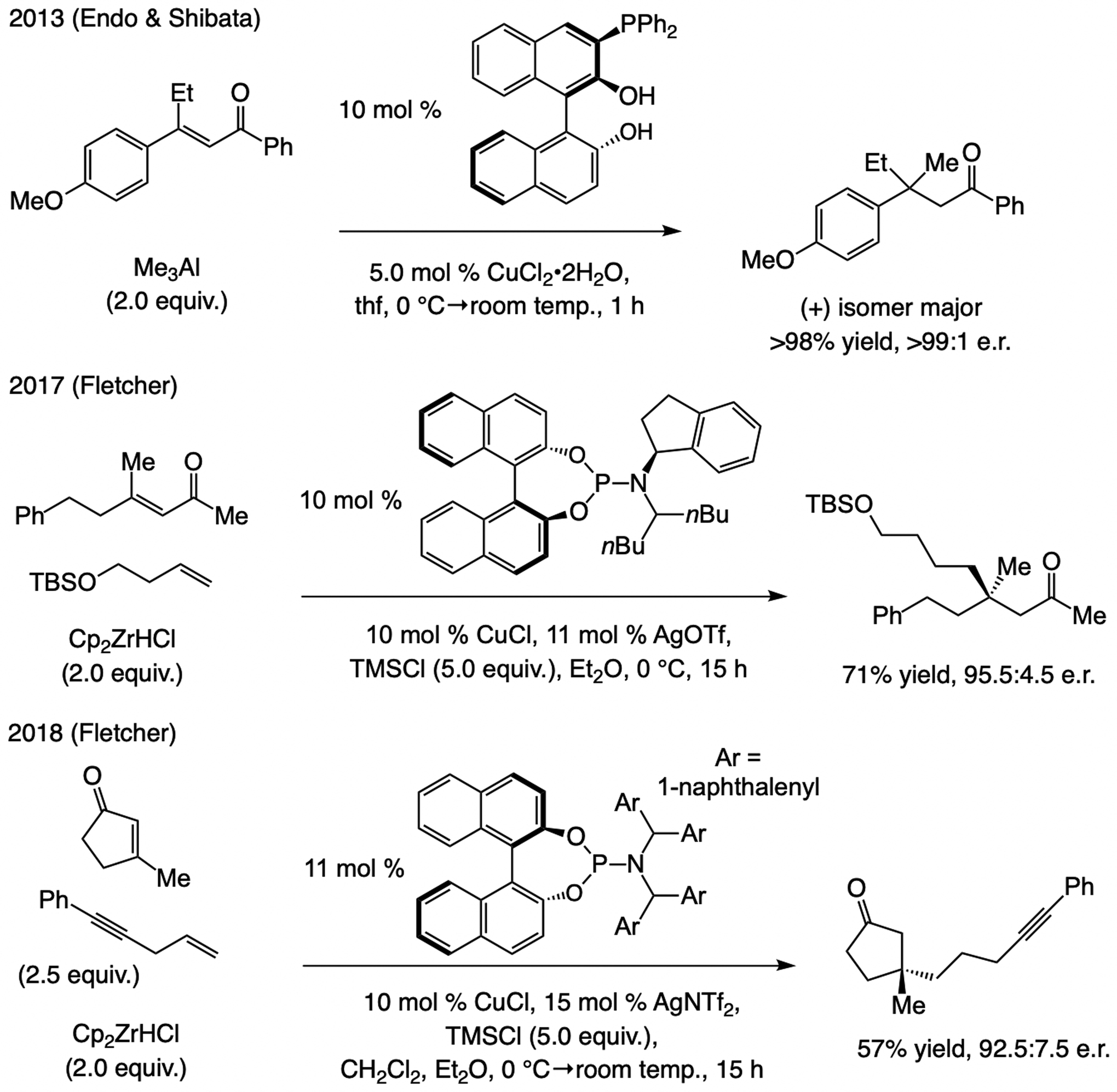
ECA of (alkyl)3Al and alkyl–zirconocene compounds to trisubstituted enones.
5.2. Additions of Alkenyl Groups to Disubstituted Enones
State-of-the-art, circa 2019. Rh-based catalysts. The first examples of ECA of an alkenyl group to an acyclic α,β-unsaturated carbonyl compound, catalyzed by a bisphosphine–Rh complex, were disclosed by Hayashi in 1998 (Scheme 43a).[84,85] In 2009, Hayashi and Shintani demonstrated that Rh-based catalysts bearing a chiral bicyclic diene ligand can facilitate ECA of alkenyl boronic acids.[86] In 2004, Inoue and Oi disclosed that ECA of alkenylzirconocene compounds (formed by Zr–H addition to an alkyne), can be catalyzed by a bisphosphine–Rh complex (Scheme 43b).[87] Also in 2004, Hayashi reported a multicomponent process, promoted by a binap–Rh complex, resulting in regio- and stereoselective Si–H addition to a terminal alkyne followed by ECA with the derived alkenylsilane compound (Scheme 43c).[88] In a 2007 report, Hayashi, Hiyama, and Shintani showed that alkenylsilane compounds may be used in ECA reactions catalyzed by a diene–Rh complex.[89]
Scheme 43.
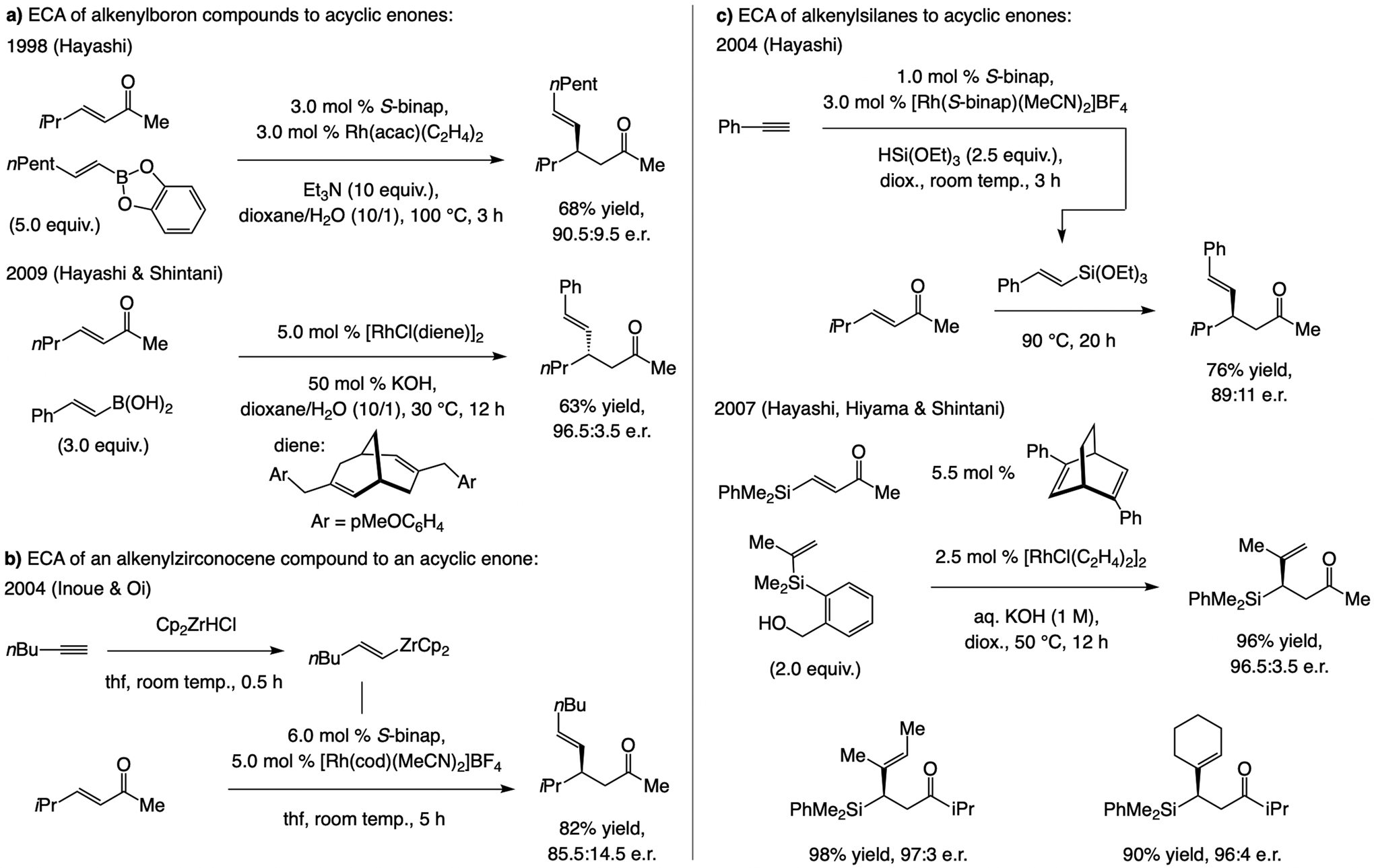
ECA with alkenylboronate, alkenylzirconocene, and alkenylsilane compounds and Rh-based complexes.
Non-organometallic catalysts.[90] In 2007 Chong illustrated that % 3,3’-diiodo-binaphthol can catalyze ECA of an alkenyl–B(OMe)2 compound to an acyclic dienone (Scheme 44).[91] An iminium-based catalyst has been used by MacMillan to promote the 1,4-addition of alkenyl trifluoroborate salts to enals, although an equivalent of HF (severely corrosive) was needed.[92] A protocol by Takemoto[93] and another by Sugiura[94] may be used to access related ECA products in high e.r., but only if an allylic hydroxy directing group and an aryl-substituted alkenyl boronic acid, respectively, is used. May has shown that with 3,3’-bis-penta(fluorophenyl)binaphthol an alkenyl boronic acid may be used towards the same end,[95] but under harsher conditions (110 °C). Kudo has shown that a peptidic catalyst may be used to facilitate similar processes (Scheme 44).[96]
Scheme 44.
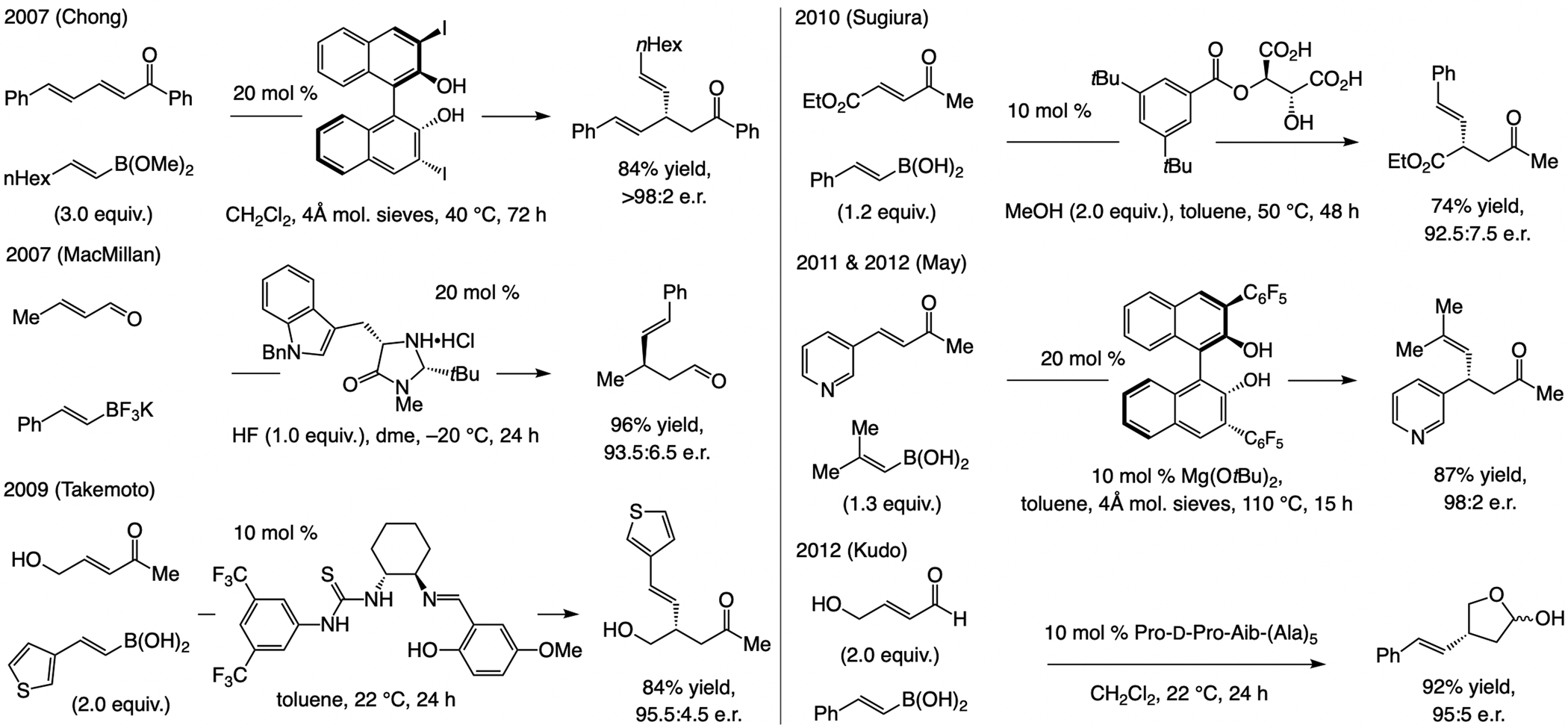
Organic molecules have been used to catalyze ECA of alkenylboron compounds to acyclic enones.
Cu-based catalysts. In a 2005 disclosure, concerned largely with phosphoramidite–Cu-catalyzed additions of Me3Al and Et3Al to cyclohexenone, Alexakis and Woodward included one example of an ECA with an alkenyl–Al compound (Scheme 45).[97] Schmalz has utilized taddolate-based phosphine–phosphonite ligands for ECA of Grignard reagents to cyclic enones.[98] In 2009, we identified a C1-symmetric NHC–Cu catalyst for ECA of alkenylsilanes to cyclic enones,[99] and subsequently, Alexakis and Woodward reported on additions of alkenyl–Al species to N-Cbz-protected dihydropiperidones, [100] α,β-unsaturated lactams, [101] and cyclohexenones (Scheme 45).[102]
Scheme 45.
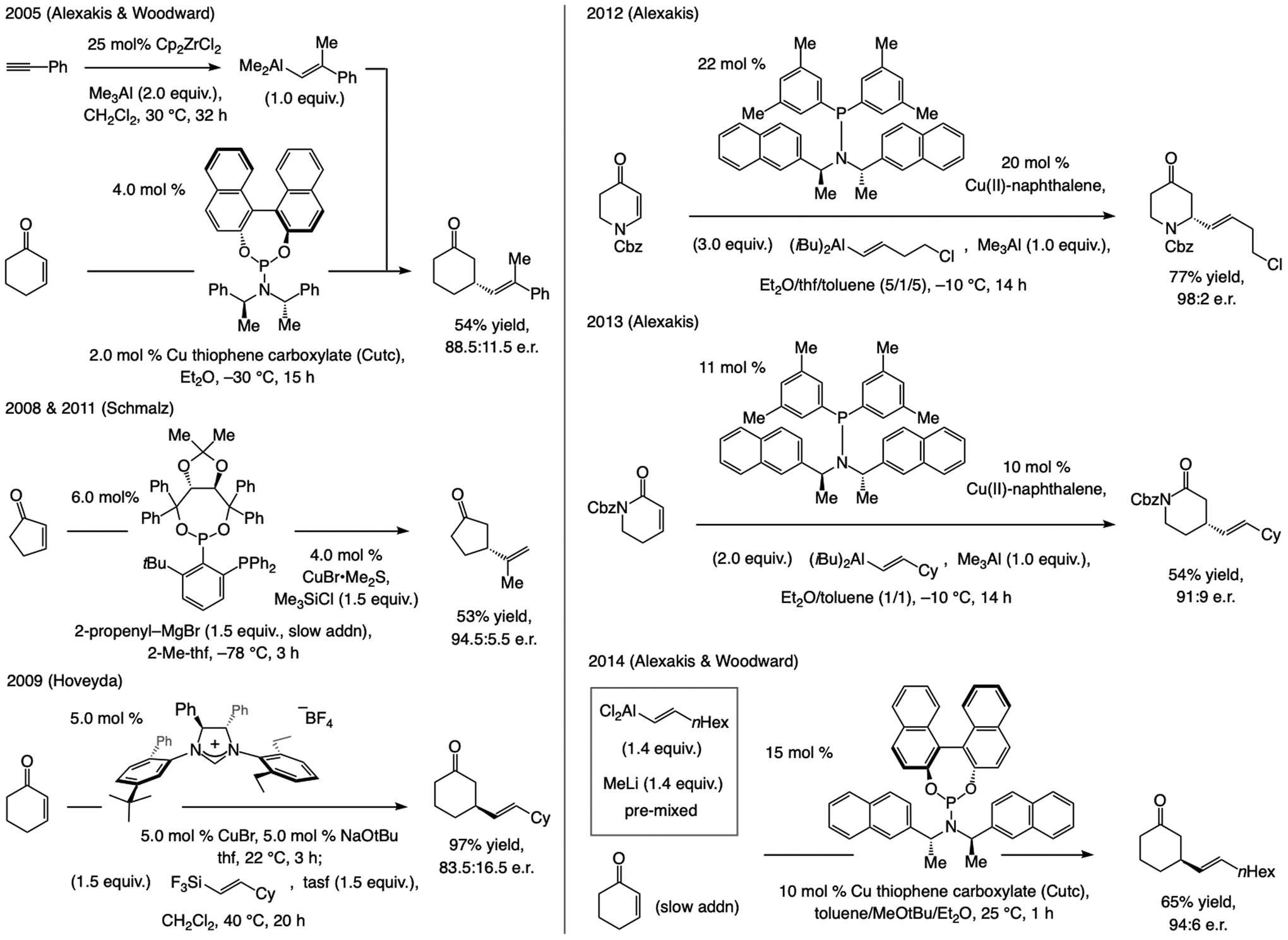
ECA of an alkenyl group with Cu-based catalysts have largely involved cyclic enones. Abbreviation: tasf = tris(dimethylamino)sulfonium difluorotrimethylsilicate.
ECA of alkenyl groups with Cu-based catalysts has therefore largely involved cyclic enones. There are only three reports, one by Hayashi and Shintani (2011),[103] and two more recently by Meng (2017–18),[104] where acyclic electrophiles were examined (Scheme 46). A distinguishing aspect of these transformations is that relatively mild alkenylboronates can be used, albeit along with highly activated diesters.
Scheme 46.
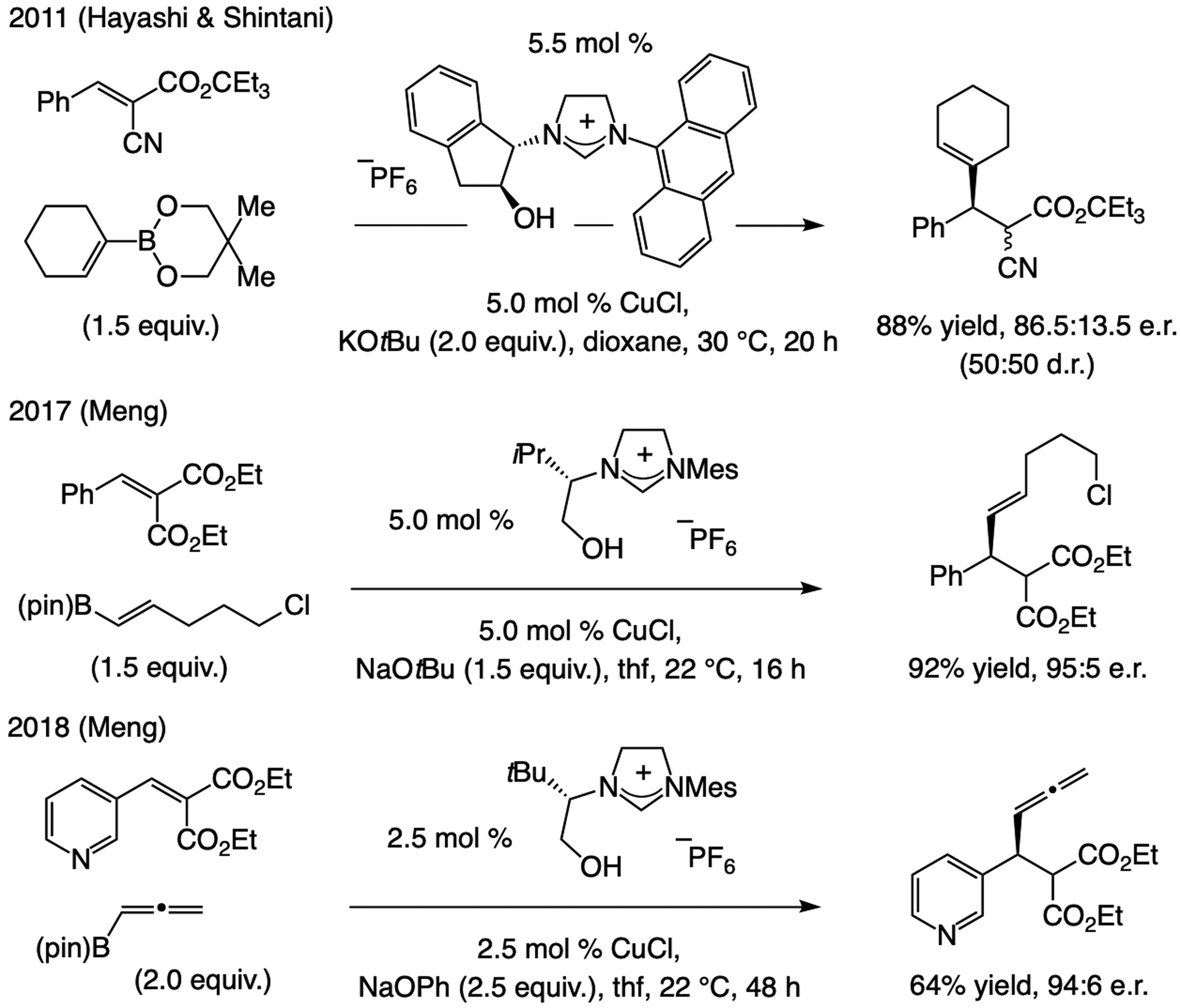
ECA of an alkenyl unit to an acyclic α,β-unsaturated carbonyl.
5.2.1. With alkenylaluminum compounds.
A sulfonate NHC–Cu complex may be used for ECA of an alkenyl–Al species to an acyclic enone (Schemes 47 and 53).[105] One entails reactions with silyl-substituted alkenyl–Al compounds (Scheme 47a), formed by regioselective addition of dibal–H to an alkyne. Transformations with Me2HSi-substituted variants are generally higher yielding (vs. Me3Si-substituted, which afforded up to 30% iBu addition byproducts). Efficiency hinges on electronic attributes of the electrophilic partner, which also influences the identity of the most effective catalyst. ECA with electron-neutral enones is more effective with imid(S)-3a. The electronic features of the aryl group in an alkenyl–Al reagent have less of an impact on e.r., although addition of a less nucleophilic p-trifluoromethylphenyl-substituted alkenyl–Al species was lower yielding. With p-methoxyphenyl-substituted alkenyl–Al compounds, yield and e.r. improved when imid(S)-6a was used. The catalyst derived from imid(S)-2c is optimal for ECA of E-silyl-substituted alkenyl–Al compounds (Scheme 47a). The utility of the approach is highlighted by a facile and exceptionally diastereoselective intramolecular silyl-hydride addition to a ketone (Scheme 47b). Catalytic ECA can be performed with β-or α-alkenyl–Al compounds, generated in situ by phosphine–Ni-catalyzed Al–H addition to an alkyne (Scheme 47c).[15] ECA with alkyl-substituted enones is inefficient (~20% conv.), probably due to diminished electrophilicity.
Scheme 47.
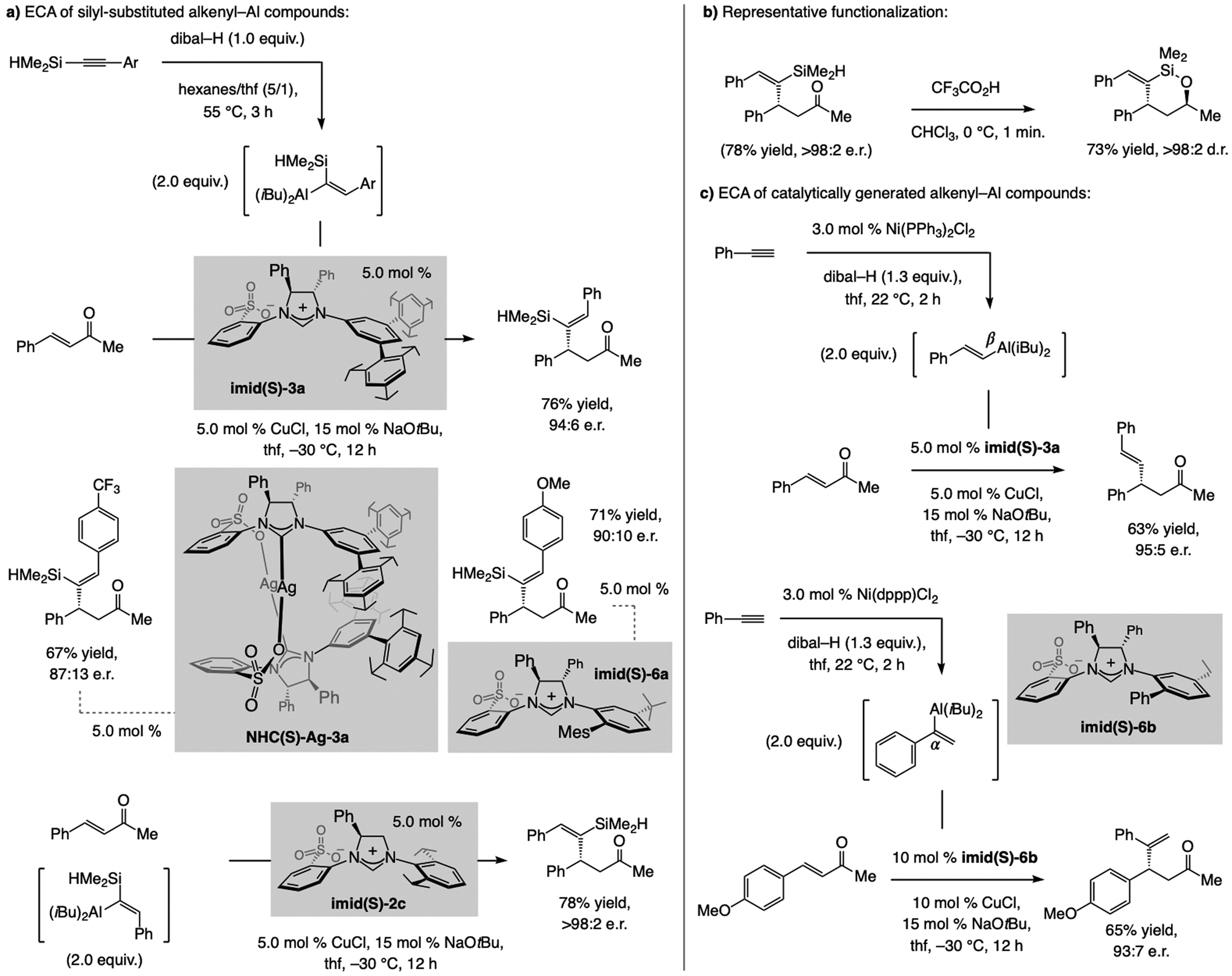
ECA of alkenyl–Al compounds to acyclic enones. Depending on the substrate, a different sulfonate-containing NHC–Cu catalyst might prove to be optimal (see Scheme 61 for mechanistic analysis).
Scheme 53.
ECA of alkenyl–Al compounds to β-substituted cyclohexenones with aminophosphinite–Cu complexes.
5.3. Additions of Alkenyl Moieties to Trisubstituted Enones
State-of-the-art, circa 2010. ECA of an alkenyl moiety to generate a quaternary carbon stereogenic center was first reported by Carretero [Eq. (3)],[106] involving alkenylboronic acids and a bis-phosphine–Rh catalyst. While only α,β-unsaturated pyridylsulfones bearing an aryl moiety served as electrophiles, aryl-substituted alkenyl boronic acids were also used.
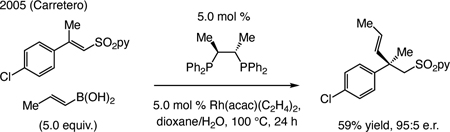 |
(3) |
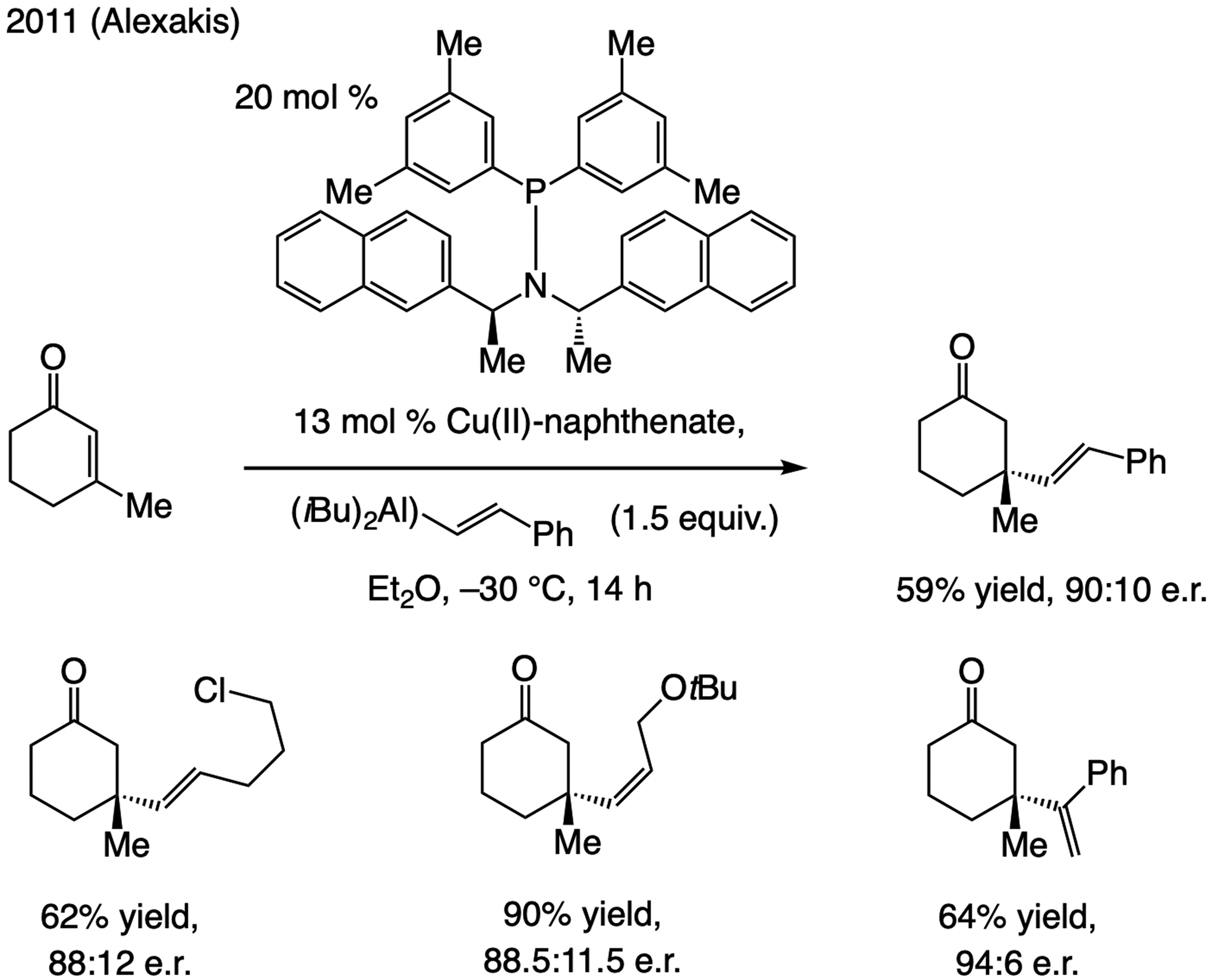
At the time we initiated our studies (2010), there were two related reports by Alexakis (Scheme 48). The first dealt largely with ECA of Me3Al and Et3Al, including one example with an alkenyl–Al compound and β-methylcyclohexenone.[107] A more detailed study appeared later,[108] demonstrating that with a modified ligand structure, transformations were more efficient (10 mol % vs. 30 mol % loading). Enantioselectivities were higher with sterically demanding 1,2- and 1,1-disubstituted alkenyl–Al compounds, but, as usual, ECA to cyclopentenones was problematic. As will be discussed below, conjugate additions of arylboronic acids to β-substituted cyclic enones, catalyzed by enantiomerically pure Rh-based complexes are known,[109] but, to the best of our knowledge, none leads to incorporation of an alkenyl group.
Scheme 48.
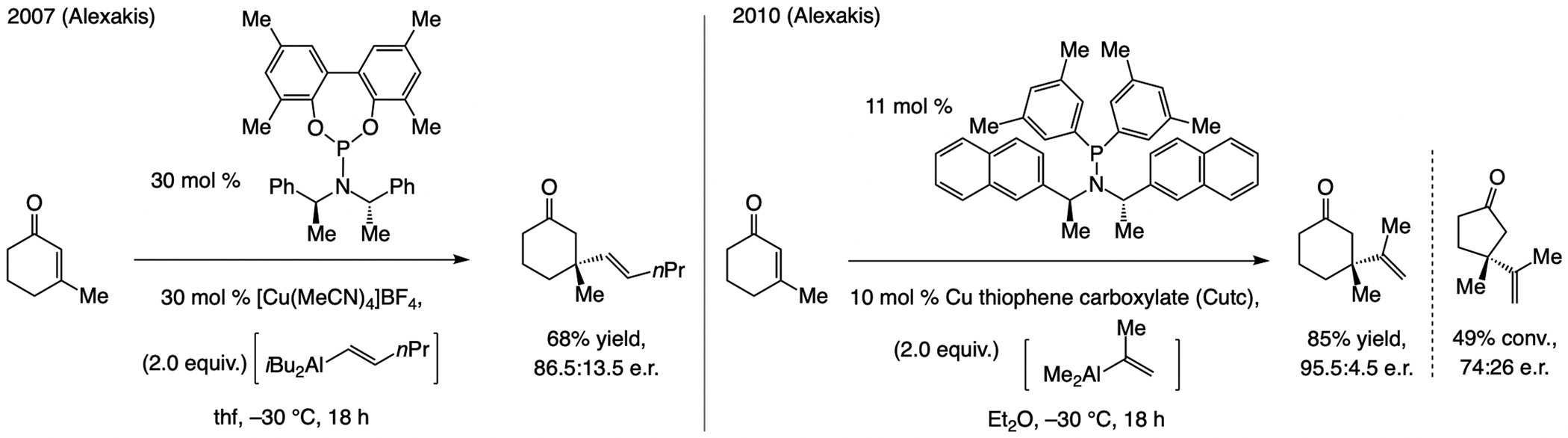
The state-of-the-art in ECA of alkenyl units to enones, circa 2010.
5.3.2. With alkenyl–Al compounds.
Sulfonate NHC–Cu catalysts promote ECA of an E-1,2-disubstituted alkenyl–Al compound to β-substituted cyclopentenyl or cyclohexenyl enones efficiently but in moderate e.r. (Scheme 49a). In contrast, with 1,1-disubstituted alkenyl–Al reagents, the catalyst derived from NHC(S)–Ag-2d promotes ECA to cyclopentenones in higher yield and e.r. than reported previously. [110] Overman has applied this strategy in a total synthesis of trans-clerodane (Scheme 49b).[111]
Scheme 49.
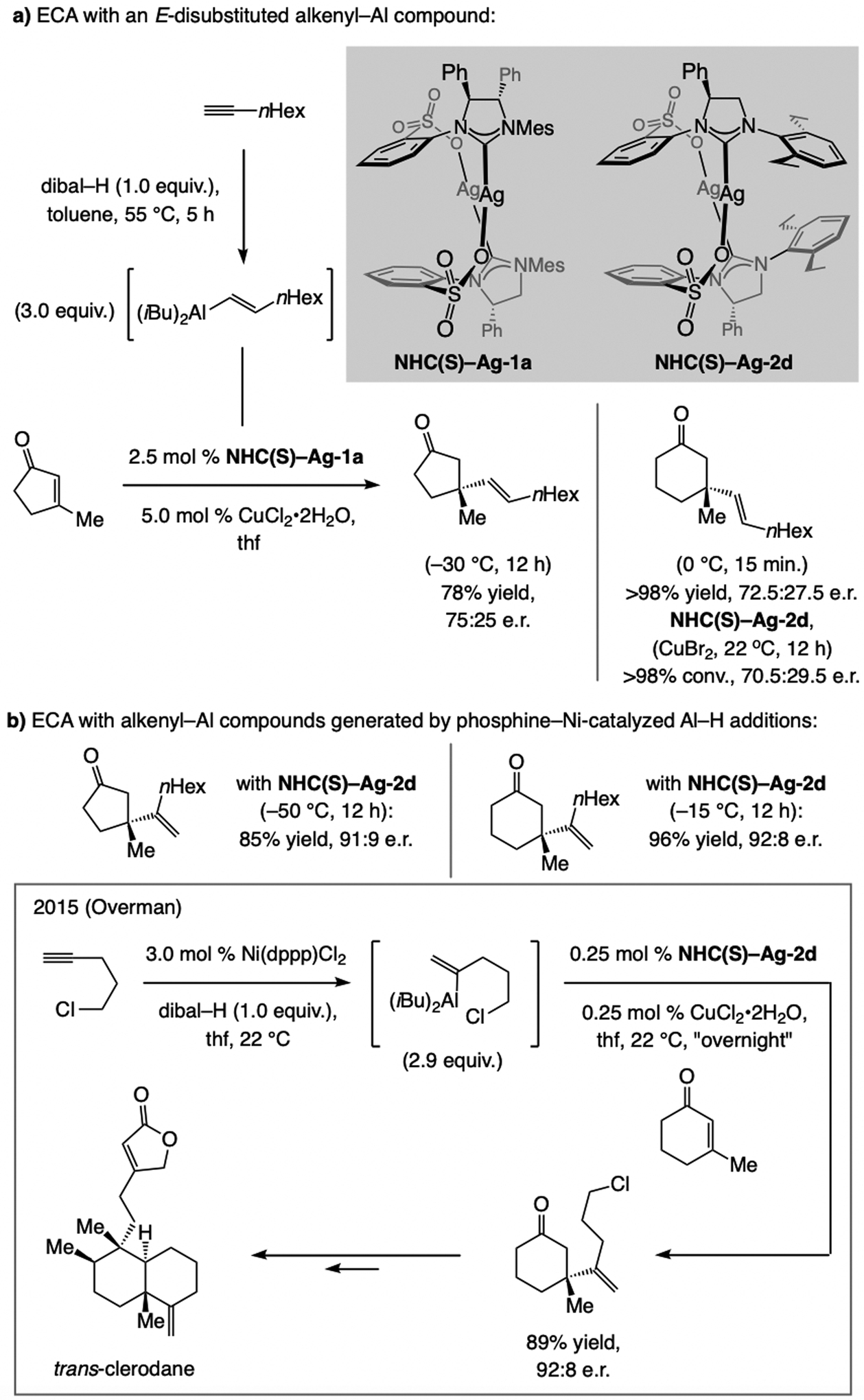
ECA of alkenyl–Al compounds with sulfonate NHC–Cu catalysts and application to total synthesis of trans-clerodane by Overman et al.
When 1-phenyl-substituted alkenyl–Al was used, reactions with sulfonate-containing complexes were moderately enantioselective (Scheme 50).[112] Unlike the aforementioned cases, however, with the complex derived from NHC(O)–Ag-2a, reactions were faster and more enantioselective. What is more, despite containing the same diamine backbone enantiomer, the opposite product enantiomers were generated. Such dichotomies, revealed by catalyst screening, point to subtle mechanistic nuances that are hard to predict.
Scheme 50.
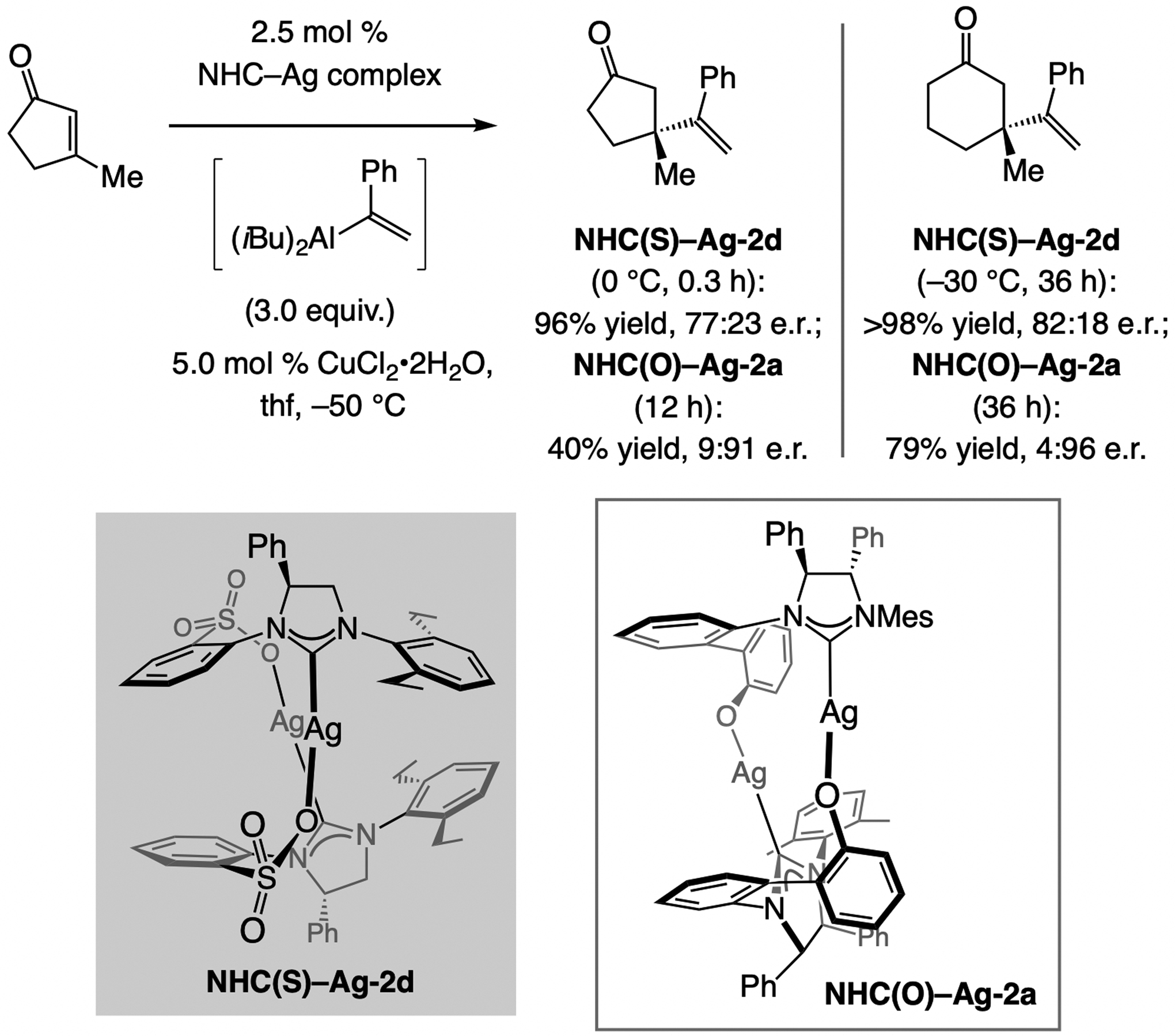
ECA of a 1-phenyl-substituted alkenyl–Al compound. Reactions performed under N2 atm.; conv. (±2%) determined by analysis of the 1H NMR spectra of the unpurified product mixtures; yields (±2%) correspond to purified products, and enantioselectivity determined by GC analysis. Previously unpublished results; see the Supporting Information for details.
Sulfonate NHC–Cu complexes are effective (e.g., vs. phenoxy analogues; Scheme 50) in promoting ECA of silyl-substituted alkenyl–Al compounds to trisubstituted alkenes.[113] Steric pressure associated with the formation of the silylalkenyl–C bond formation can lead to the transfer of an i-butyl group from the (iBu)2alkenyl–Al compound (Scheme 51). Reaction with a phenoxy NHC ligand favors a different enantiomer compared to the sulfonate variants (see Scheme 59 for further analysis). Comparison of the results with NHC(S)-Ag-1a, NHC(S)-Ag-2a, and NHC(S)-Ag-2d again highlights the importance of subtle structural variations. The silyl-substituted alkenyl moiety offer several advantages. Treatment of a product with m-CPBA and then formic acid afforded the β-acyl compound, or the silyl moiety can be swapped with an iodide with complete retention of alkene stereochemistry. The silyl group may be replaced by a C–H bond, generating products that would be expected from ECA of a 1,2-disubstituted alkenyl–Al compound, but in much higher e.r. The application to synthesis of an alkene isomer of cytotoxic agent riccardiphenol B (Scheme 51) highlights utility.[114]
Scheme 51.
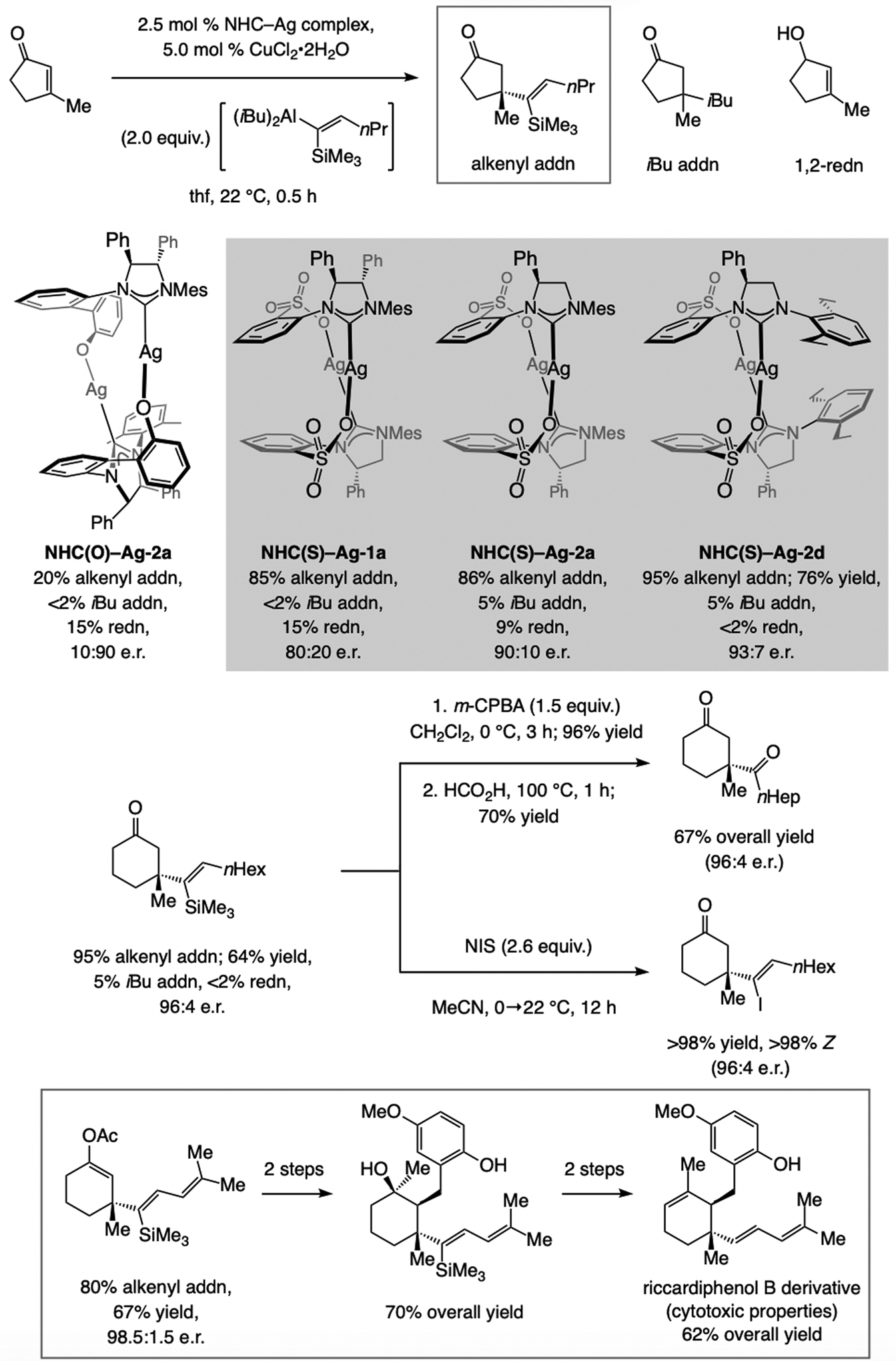
ECA of silyl-substituted alkenyl–Al compounds to β-substituted cyclic enones (for mechanistic analysis, see Scheme 59). NIS = N-iodosuccinimide.
Scheme 59.
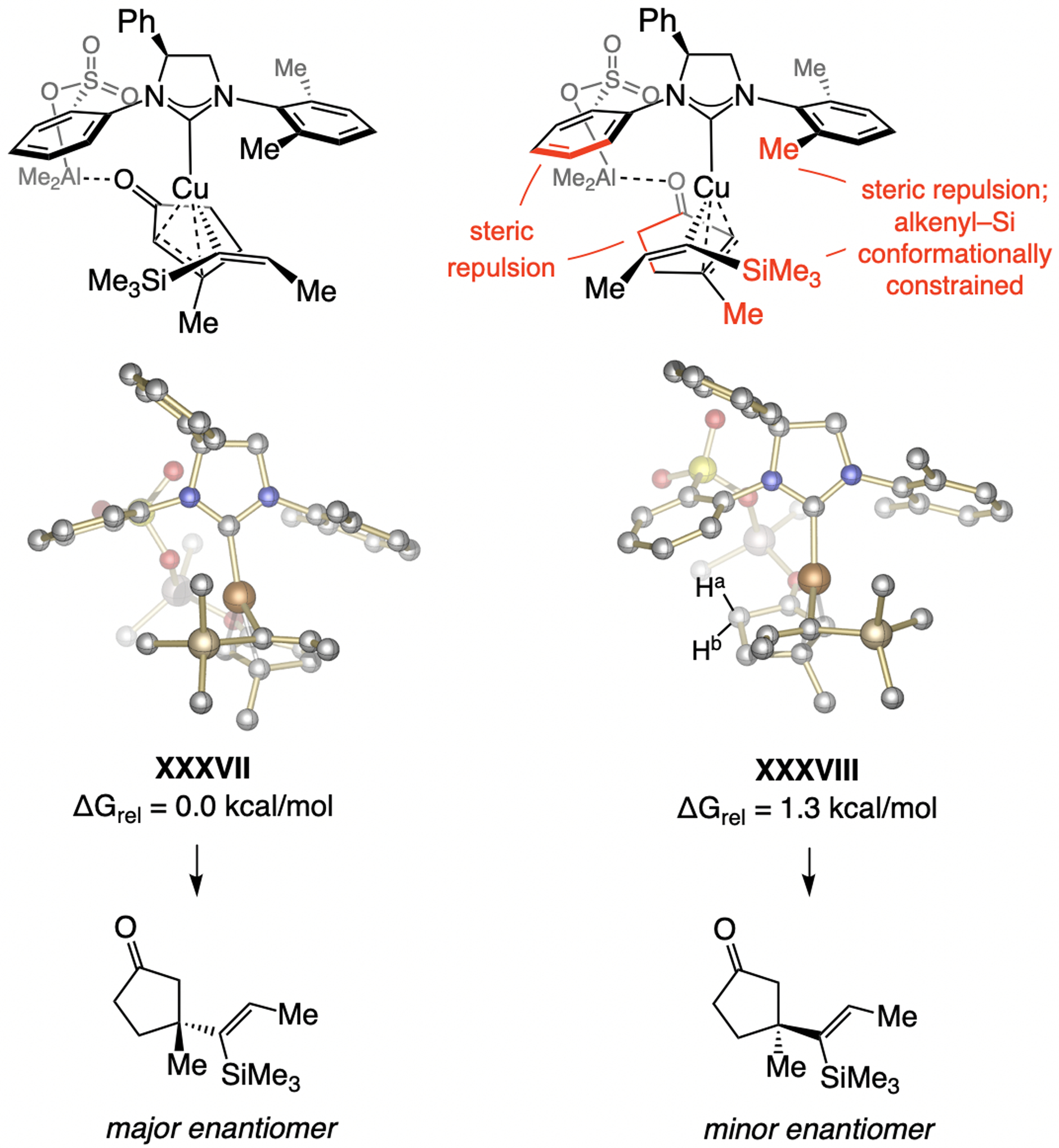
Rationale for high e.r. in ECA with silyl-substituted alkenyl–Al compounds (see Scheme 51 for methodology). DFT at the M06L/Def2-TZVPP//M06L/Def2-SVP level. Previously unpublished analysis; see the Supporting Information for details of the DFT studies.
Sulfonate NHC–Cu complexes catalyze ECA of 1,2-disubstituted alkenyl–Al compounds to acyclic enones, affording products containing a quaternary carbon stereogenic center (Scheme 52).[115] The organometallic reagent and the trisubstituted linear enone were accessed catalytically, efficiently and with high stereoselectivity. Reactions performed with the NHC–Cu catalyst derived from ent-NHC(S)–Ag-2b or ent-NHC(S)–Ag-2a (for 1,2- and 1,1-disubstituted alkenyl–Al compounds, respectively) afforded products in high e.r. Enantioselective synthesis of an intermediate to enokipodin B demonstrated applicability.[116] The sense of enantioselectivity in the above transformations is at times opposite to that observed for ECA of alkenyl–Al compounds to disubstituted acyclic enones (see Scheme 47; see Scheme 61 for analysis).
Scheme 52.
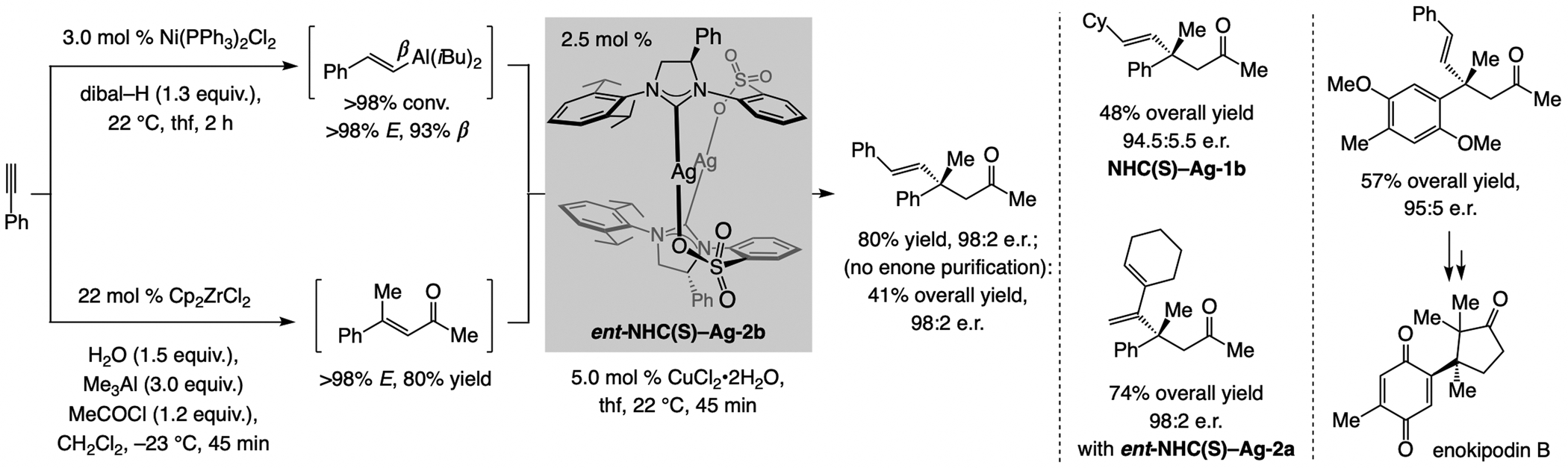
ECA of alkenyl–Al compounds to acyclic trisubstituted enones proceeds with the opposite enantioselectivity compared to disubstituted substrates (Scheme 47). See Scheme 61 for mechanistic analysis.
Scheme 61.
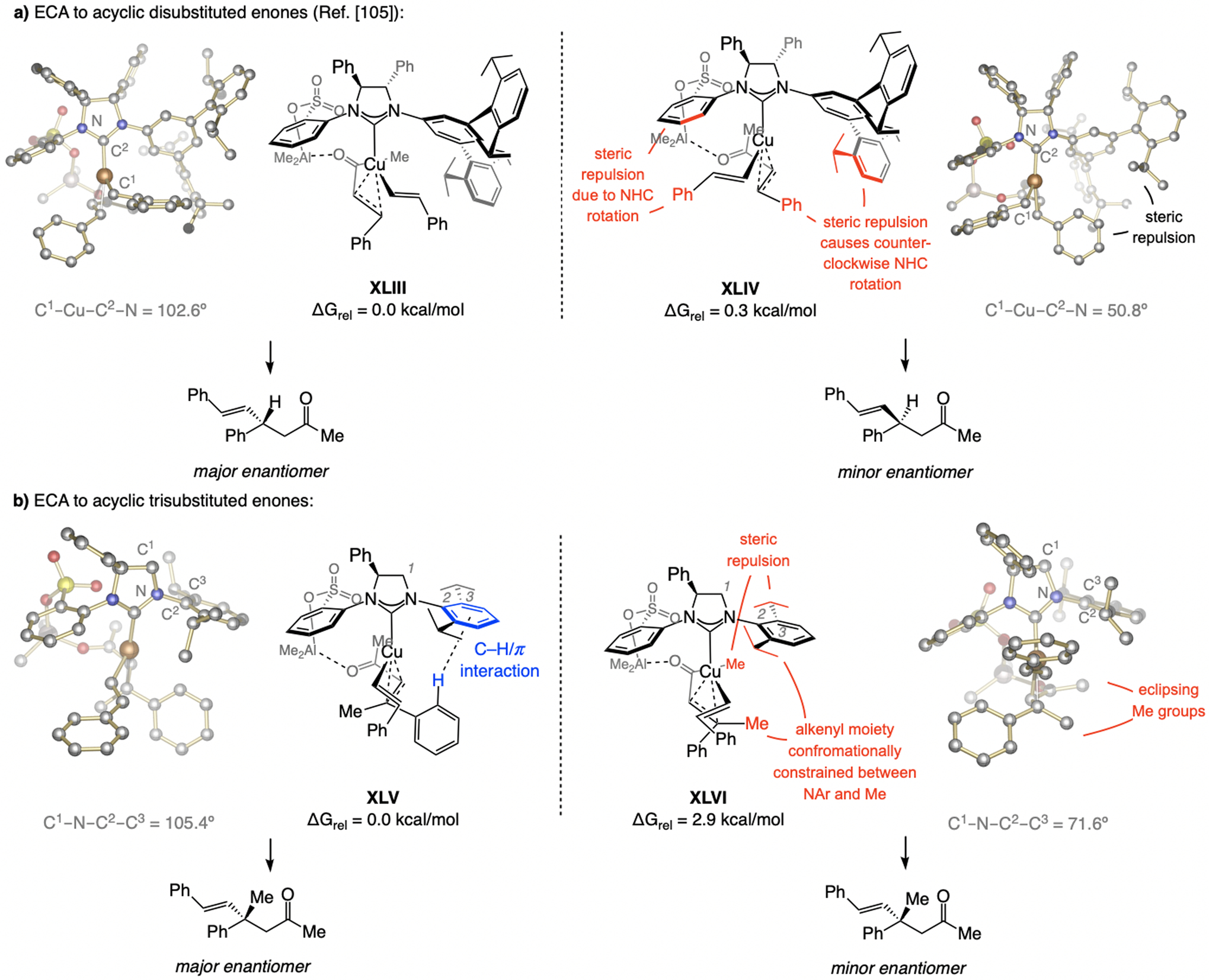
Rationale for high e.r. in ECA of β-alkenyl–Al compounds (see Schemes 47 and 53 for methodology). DFT at the M06L/Def2-TZVPP//M06L/Def2-SVP level. See the Supporting Information for details.
Subsequent advances. Aminophosphinite–Cu complexes have been used to promote ECA of alkenyl–Al compounds to cyclohexenones (Scheme 53).[117] Enantioselectivities were higher with 1,1-disubstituted alkenyl–Al species, similar to those involving a 1,1-disubstituted organoaluminum compound compared to when sulfonate NHC–Cu catalysts were used (see Schemes 51 and 53, respectively). Nonetheless, catalyst loading was higher (20 vs. 5.0 mol % NHC), and additions to other ring sizes, such as cyclopentenones, were significantly less enantioselective. Data regarding reactions with acyclic enones were not provided.
5.4. Additions of Aryl and Heteroaryl Moieties to Trisubstituted Enones
State-of-the-art, circa 2008. The first instances of ECA of an aryl moiety was disclosed in 2006 by Hayashi and involved a bisphosphine–Rh-catalyzed transformation between 3-substituted maleimides and arylboronic acids (Scheme 54); [118] a key aspect of this study was the dependence of the regioselectivity on the chiral ligand used (preferential formation of the more congested quaternary carbon). Later the same year, we showed that an NHC–Cu complex derived from NHC(O)-Ag-2a may be used to promote ECA of (aryl)2Zn compounds to cyclic enones; [119] while reactions with six- and seven-membered ring enones were efficient and afforded products in useful levels of enantioselectivity, the same did not apply to five- and eight-membered substrates. Despite these advances, notable shortcomings remained; a major problem was the absence of effective methods that are applicable to cyclopentenones, an important set of transformations for applications to synthesis of bioactive molecules.
Scheme 54.
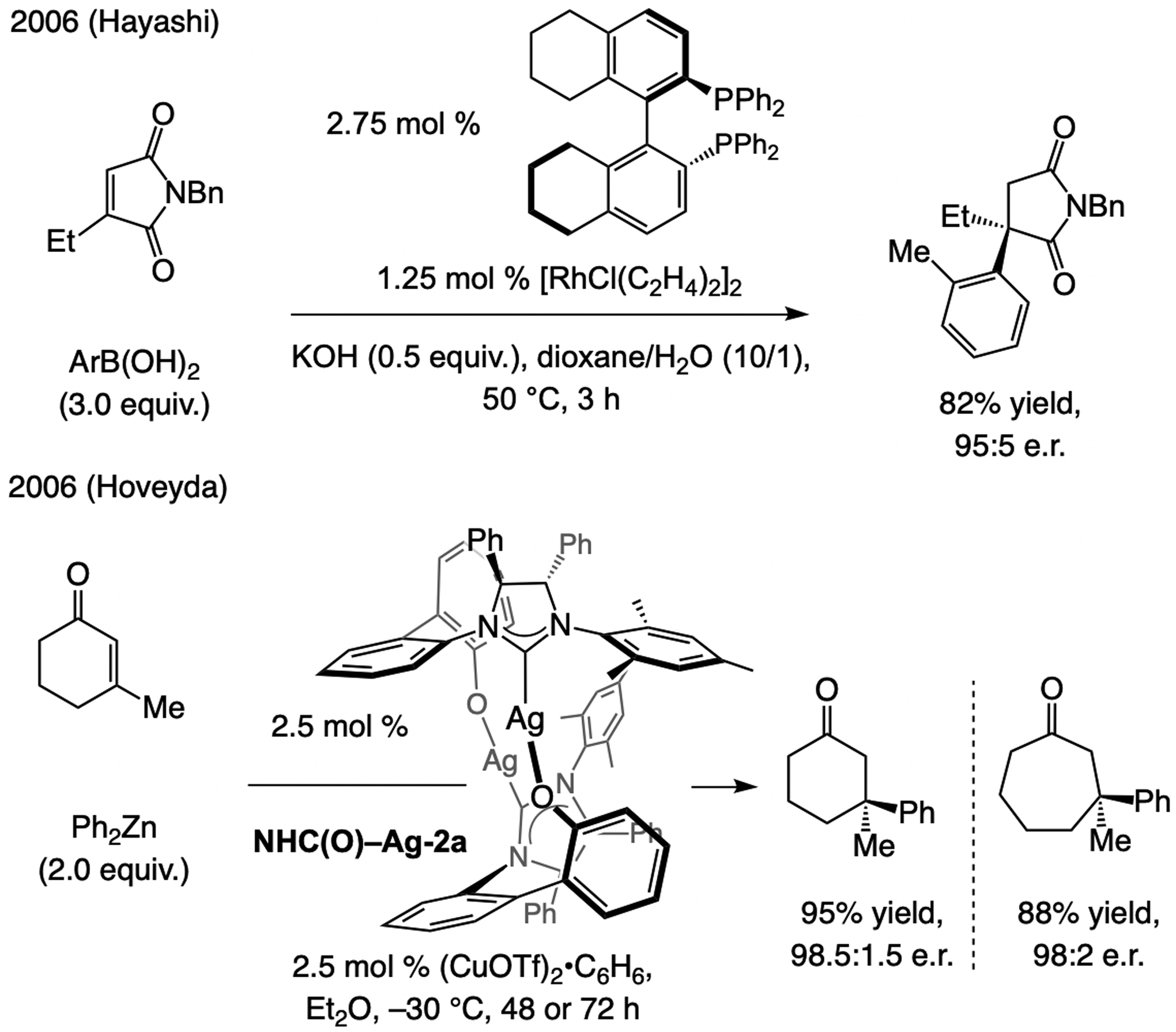
The first examples of ECA of an aryl moiety to cyclic β-substituted enones.
5.4.1. Effective catalysts for ECA to cyclopentenones.
In 2008, we established that sulfonate NHC–Cu catalysts promote ECA of in situ generated (Me)2aryl–Al compounds to β-substituted enones efficiently but with low e.r. (Scheme 55). These studies also led us to discover that when the catalyst derived from NHC(O)–Ag-2a is used, the same transformations are efficient and highly enantioselective, including when cyclopentenones were used.[76] Similar to ECA of silyl-substituted alkenyl–Al compounds (see Scheme 51), the phenoxy and sulfonate NHC–Cu complexes generate products with the opposite sense of enantioselectivity (see Scheme 60 for analysis). Later in the same year, Alexakis demonstrated that phosphoramidite–Cu and aminophosphinite–Cu complexes promote ECA of (alkyl)2aryl–Al compounds to β-substituted cyclic enones, including the more challenging cyclopentenones (Scheme 56).[120]
Scheme 55.
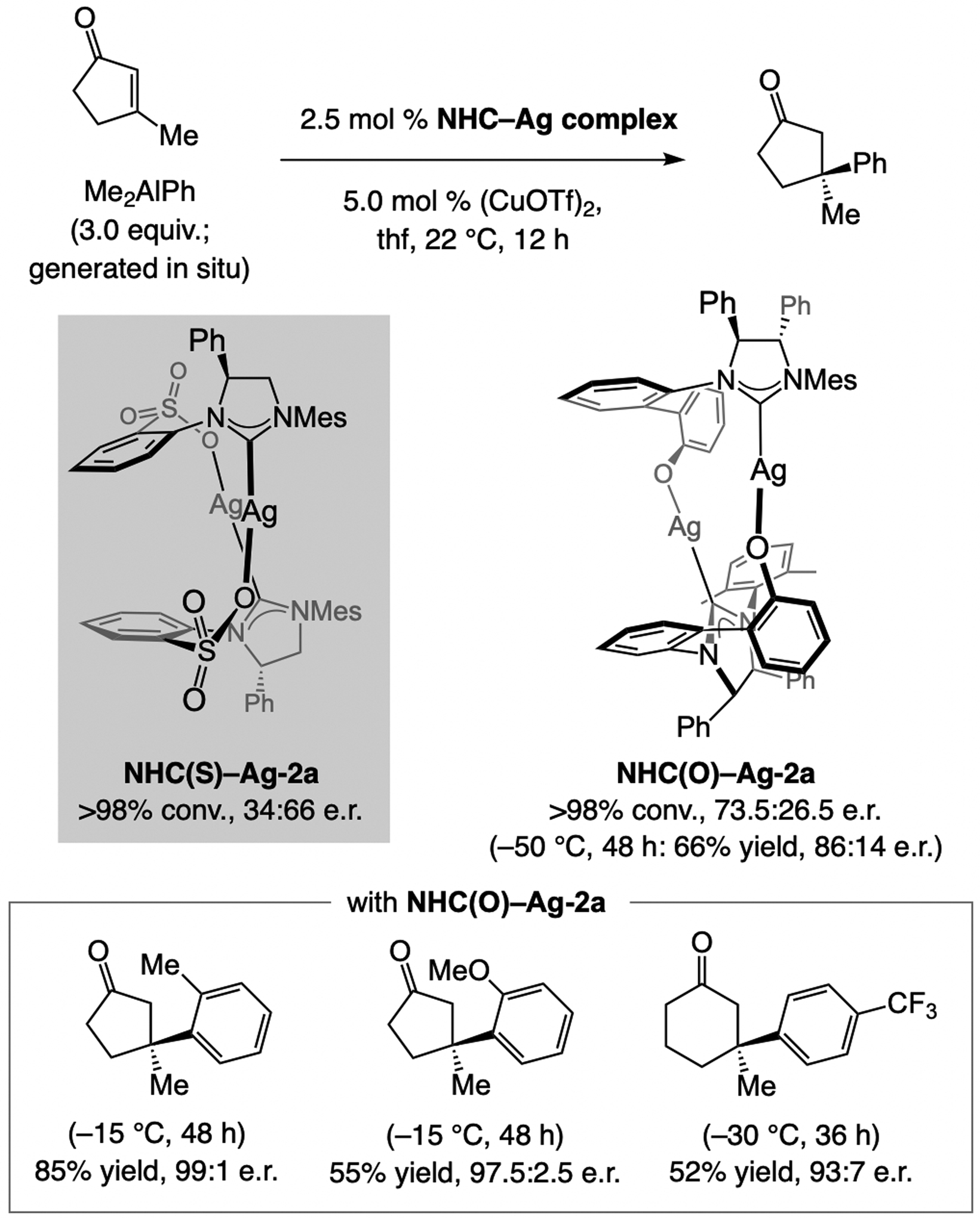
ECA of di(alkyl)aryl–Al compounds to β-substituted cyclic enones is more enantioselective with a phenoxy NHC ligand (for mechanistic analysis, see Scheme 60).
Scheme 60.
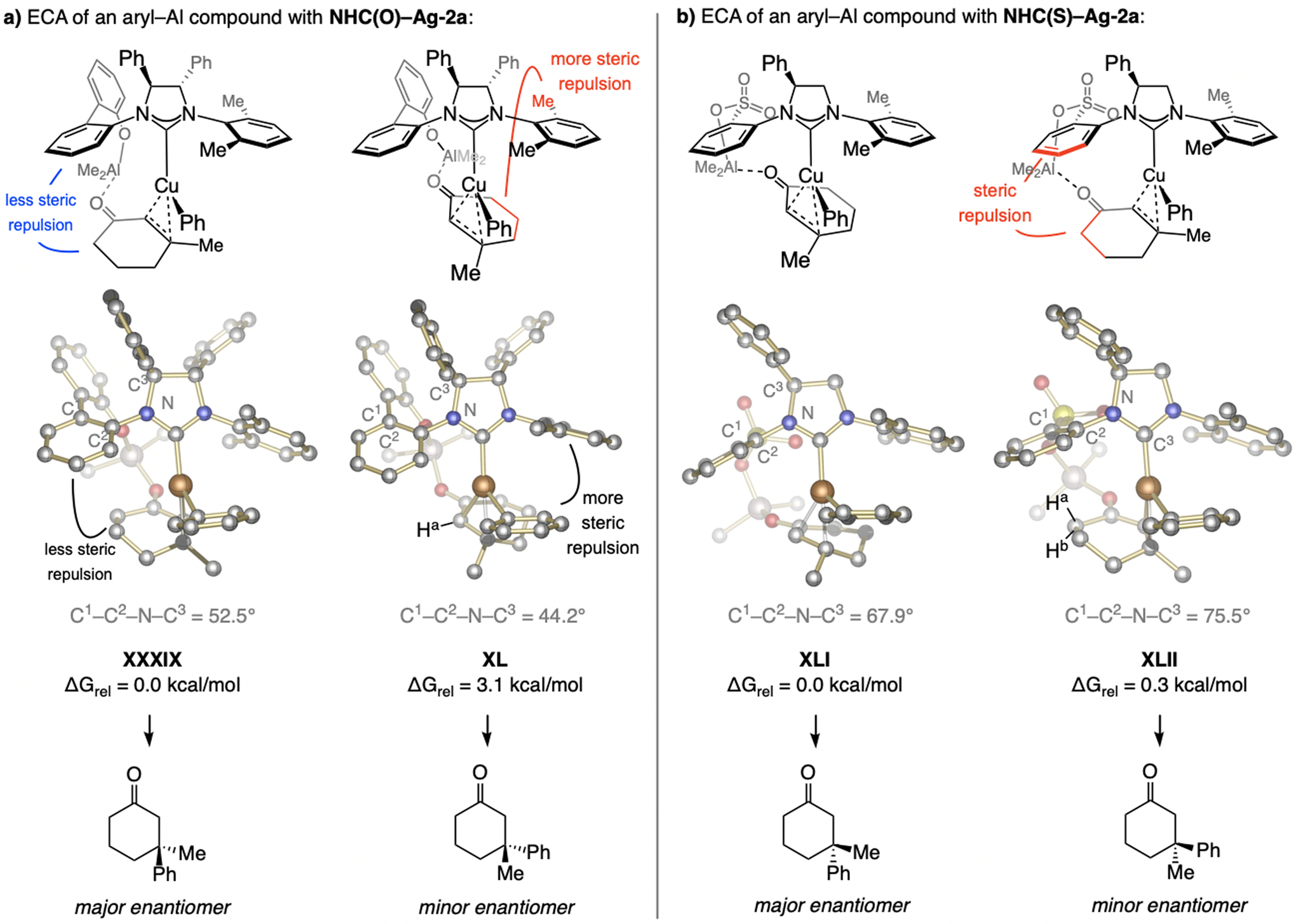
Rationale for high enantioselectivity in ECA with aryl–Al compounds (see Scheme 55 for methodology). DFT at the M06L/Def2-TZVPP//M06L/Def2-SVP level. Previously unpublished analysis; see the Supporting Information for details of the DFT studies.
Scheme 56.
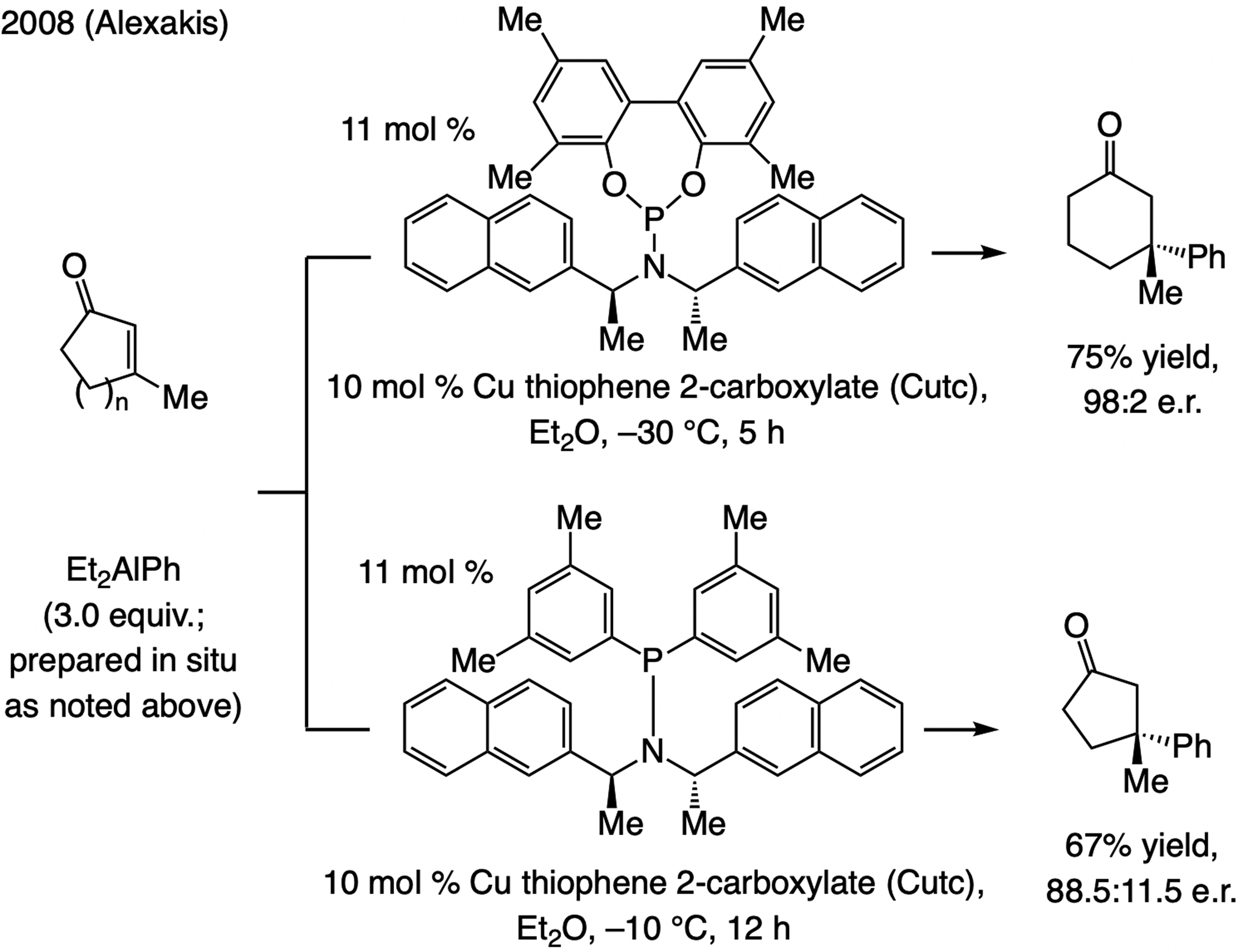
Phosphoramidite–Cu-catalyzed ECA of aryl moieties to β-substituted cyclic enones, including cyclopentenones.
Subsequent advances and prior art regarding acyclic enones. Rh- and Pd-based catalysts have been developed for ECA of arylboroxins [(ArBO)3] and arylboronic acids, respectively, to β-substituted cyclic enones, including cyclopentenones and lactones (Scheme 57).[121] An advantage of these methods is the compatibility of organoboron compounds to some of the commonly occurring polar functional groups.[122] Equally important, the challenge of ECA with acyclic trisubstituted enones was addressed for the first time. Nonetheless, the need for precious metal salts aside, there were no extant cases involving a heteroaryl moiety.
Scheme 57.

ECA of organoboron compounds to β-substituted enones catalyzed by Rh- or Pd-based catalysts.
5.4.2. With aryl- and heteroaryl-Al compounds.
Sulfonate NHC–Cu catalysts promote ECA of (Me)2heteroaryl- and (Me)2aryl–Al compounds to acyclic trisubstituted enones[79] (Scheme 58). The necessary reagent may be formed in situ by subjection of an aryl–Li compound to Me2AlCl. In some instances, minor amounts of methyl-addition products can form, likely due to steric pressure associated with transferring a sizeable aryl or heteroaryl moiety. As far as we know, these represent the first examples of ECA of a heteroaryl moiety to trisubstituted enones. Conversion to the derived carboxylic acids, obtained in high yield and applied to synthesis of a serotonin receptor inhibitor, illustrates applicability of the approach.
Scheme 58.
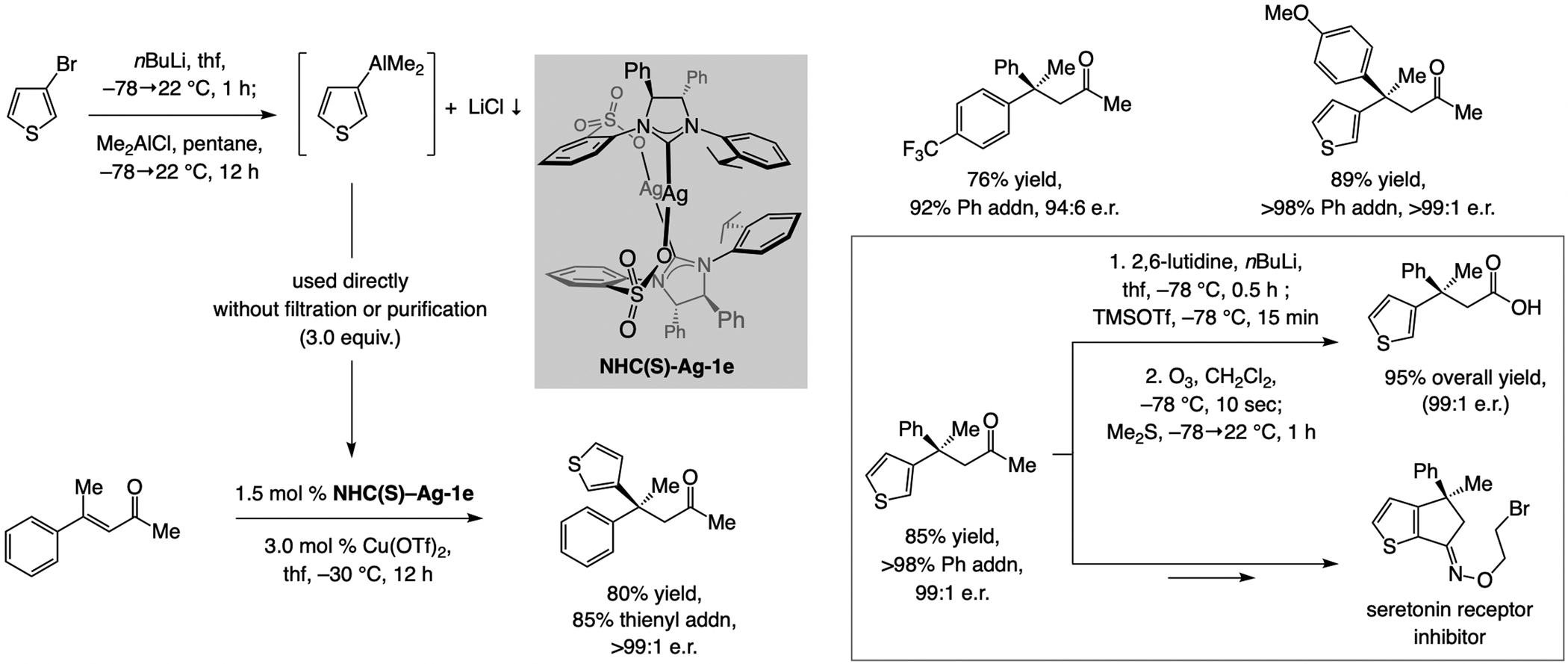
ECA of aryl- and heteroaryl–Al compounds to acyclic trisubstituted enones.
5.5. Mechanistic Analysis
DFT studies were performed to assemble a plausible set of stereochemical models. A salt bridge involving the sulfonate moiety again appears to be central to enantioselectivity for ECA of Al-based compounds. The results of these previously unpublished investigations are presented below (see the Supporting Information for details).
5.5.1. Cyclic enones
5.5.1.1. Origin of enantioselectivity.
The energetically preferred mode of ECA is probably a cyclic substrate/catalyst complex that includes ArSO3…Al…O=C complexation (Scheme 59). DFT investigations suggest that transformation via an ensemble such as XXXVIII, where the ring is oriented towards the sulfonate-bearing aryl moiety is higher in energy. This is probably largely because the smaller Al bridge, in turn enhancing steric strain in XXXVIII (see Scheme 60 and the Supporting Information for details). The higher e.r. with NHC ligands with a single phenyl substituent might be because the adjacent NAr group can more readily rotate, allowing for further minimization of unfavorable interactions with the substrate (see XXXVII).
5.5.1.2. Why is the sense of enantioselectivity opposite to phenoxy NHC–Cu catalysts?
Reactions with phenoxy NHC–Cu catalysts likely involve intermediates with a bridging fragment that is part of a larger ring compared to that generated within a sulfonate. The steric pressure caused by the enone ring orienting towards the phenoxy-bearing NAr group would hence be lower (compare XXXIX vs. XL, Scheme 60a). Reaction via XL, in which the ring occupies the space below the symmetrical NAr moiety, thus becomes sterically more demanding (repulsion with the rear o-Me group). This is particularly the case when there is a nearby phenyl group, which limits the ability of the symmetrical NAr group to alter its conformation. The situation is different with a sulfonate complex (Scheme 60b) because orienting the substrate structure towards the sulfonate-bearing NAr ring (see XLII) leads to higher steric pressure, as revealed by the larger C1–C2–N–C3 dihedral angle in XLII (75.5° and 67.9° in XLI compared to a smaller angle in XXXIX and XL). The opposite enantiomer is thus favored with a sulfonate catalyst. ECA of (alkyl)2Zn compounds proceed with a preference for addition to the opposite enantiotopic face of a cyclic enone with NHC(S)-Ag-1a (see ester-containing cyclic enones in Schemes 39). This might be attributed to a different coordination geometry of the Lewis acidic metal (Me2Al+ vs. MeZn+).[123] (See the Supporting Information for further discussion).
5.5.2. Acyclic enones
Similar to EAS (e.g., Scheme 32), ECA reactions involving acyclic di- and trisubstituted enones at times proceed with the opposite predominant sense of enantioselectivity. A telling instance corresponds to the transformations with β- or α-alkenyl–Al compounds (see Schemes 47 and 53). DFT studies indicate that in the case of disubstituted enones and β-alkenyl–Al compounds catalyzed by the complex derived from imid(S)-3a, reaction via XLIII (Scheme 61a) is preferred over XLIV, wherein there is steric repulsion between the alkenyl substituent (Ph) and a triisopropylphenyl moiety of the NHC. Such strain can lead to counter-clockwise rotation of the NHC, in turn inducing steric pressure between the alkenyl phenyl and the sulfonate-bearing aryl group. This explains the change in the C1–Cu–C2–N dihedral angle (102.6° in XLIII vs. 50.8° in XLIV). In contrast, with a trisubstituted enone (Scheme 61b), the pathway via XLV, wherein there is an attractive C–H/aryl-p interaction,[65] is favored; in the alternative XLVI, there is steric repulsion between the methyl ketone and one of the ortho isopropyl of the NAr moiety of the NHC, causing the C1–N–C2–C3 dihedral angle to contract (105.4° in XLV vs. 71.6° in XLVI); this, together with the substrate Me group, limits the space available for the alkenyl moiety. These models explain why, with disubstituted enones, the catalyst derived from imid(S)-3a, with a C3- and C5-substituted NAr moiety, is optimal, whereas with trisubstituted enones, the imid(S)-2b is the most effective ligand. The above stereochemical model represents a revision of those formerly reported.[115] Similar arguments may account for the outcome of the reactions with α-alkenyl–Al compounds (see the Supporting Information for further detail).
6. Enantioselective C–B and C–H Bond Forming Reactions
Different Cu-based catalysts have been developed for net enantioselective addition of a B–H unit across an alkene (Scheme 62b–c).[124] These transformations complement or offer an alternative to the earlier processes, promoted by a Rh- or Ir-based complex (Scheme 62a).
Scheme 62.
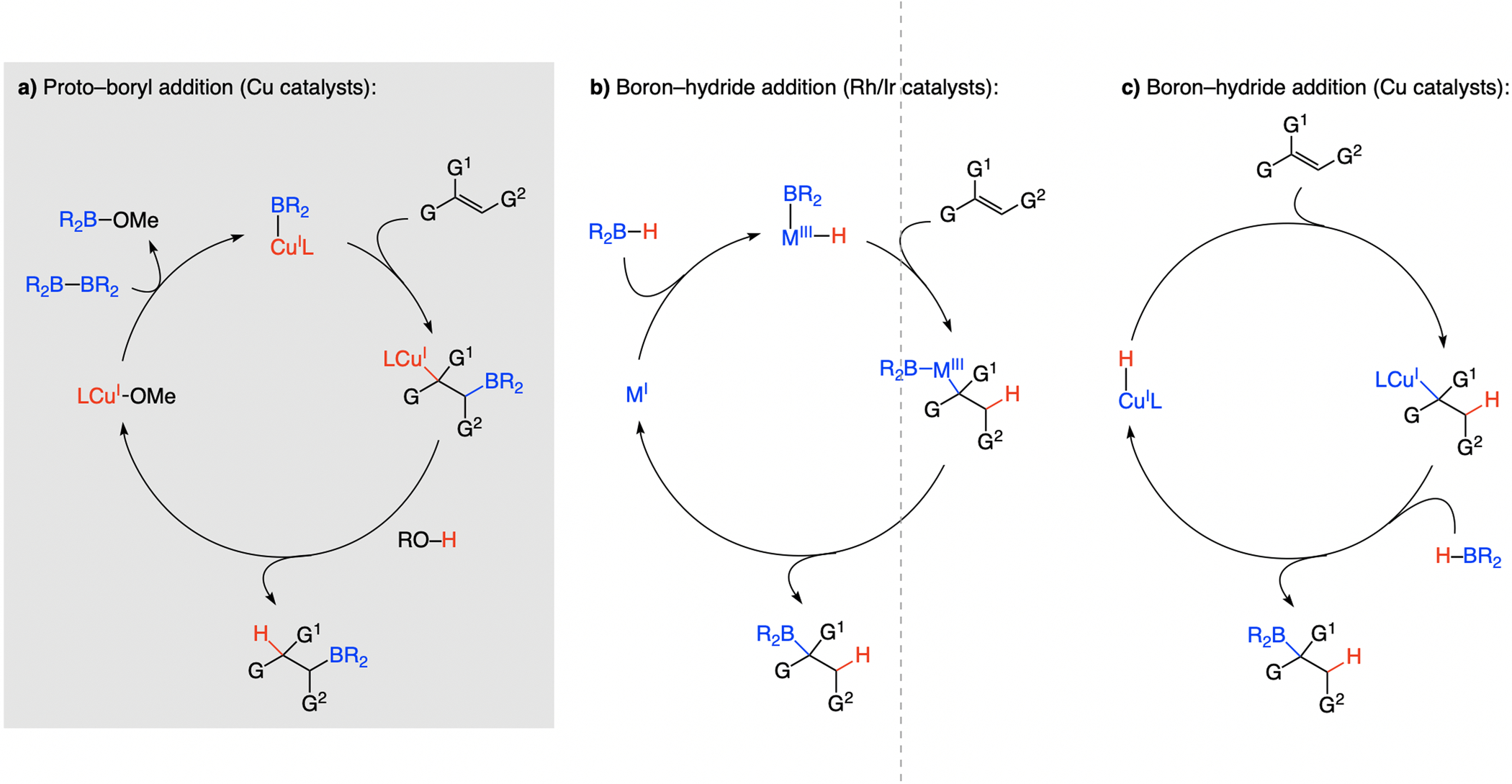
Different pathways by which an H and a B atom can be added to an alkene.
6.1. Catalytic Copper-Boryl Addition to Alkenes.
Sulfonate NHC–Cu complexes played an early role in the development of enantioselective Cu–B(pin) additions to alkenes.
6.1.1. Three pathways for B–H addition to alkenes.
Cu–boryl addition to an alkene may be followed by protonolysis of the resulting Cu–C bond by an alcohol, furnishing an organoboron product (Scheme 62a). This process was conceived[125] partly based on the work by Sadighi [126] regarding regioselective addition of NHC–Cu–B(pin) complexes to aryl alkenes, illustrating a strong preference for benzylic Cu–C formation. Another relevant finding was that of Yun,[127] who showed that re-generation of a Cu–alkoxide can be pomoted by an alcohol. Proto-boryl additions afford products that are complementary to catalytic boron–hydride additions to olefins.[128] Depending on the olefin substitution pattern, the process might either generate a stereogenic boryl-substituted carbon or a constitute an enantioselective C–H bond formation (see below).
In phosphine–Rh- or phosphine–Ir-catalyzed processes (Scheme 62b), addition of the complex derived from oxidative insertion into a B–H bond forms a benzylic C–metal and a homobenzylic C–H bond (vs. initial C–B(pin) bond formation in proto-boryl addition, Scheme 62a), after which the C–B bond is generated by reductive elimination (vs. protonolysis of the Cu–C bond). A third mechanism, introduced by Yun[129] (Scheme 62c), entails Cu–H addition to an alkene, affording a benzylic Cu–C bond. Thus, catalytic proto-boryl addition (Scheme 62a) may be used to generate organoboron regioisomers that are inaccessible by boron–hydride additions (Scheme 62b–c).
6.1.2. State-of-the-art in catalytic enantioselective boron–hydride addition to acyclic 1,2-disubstituted alkenes, circa 2009.
At the time catalytic enantioselective proto-boryl addition (Scheme 62a) was first reported,[125] a limited number of catalytic boron–hydride addition protocols were known (Scheme 63), the earlier methods being developed by Brown (1995)[130] and Guiry (2000).[131] As was noted (Scheme 62b), the products bear a benzylic C–B bond, and thus, a stereoisomerically pure alkene substrate is not required. Takacs has shown that phosphine–Rh-catalyzed boron–hydride additions can be directed by a secondary amide, affording β-boryl carbonyl products in high e.r.[132]
Scheme 63.
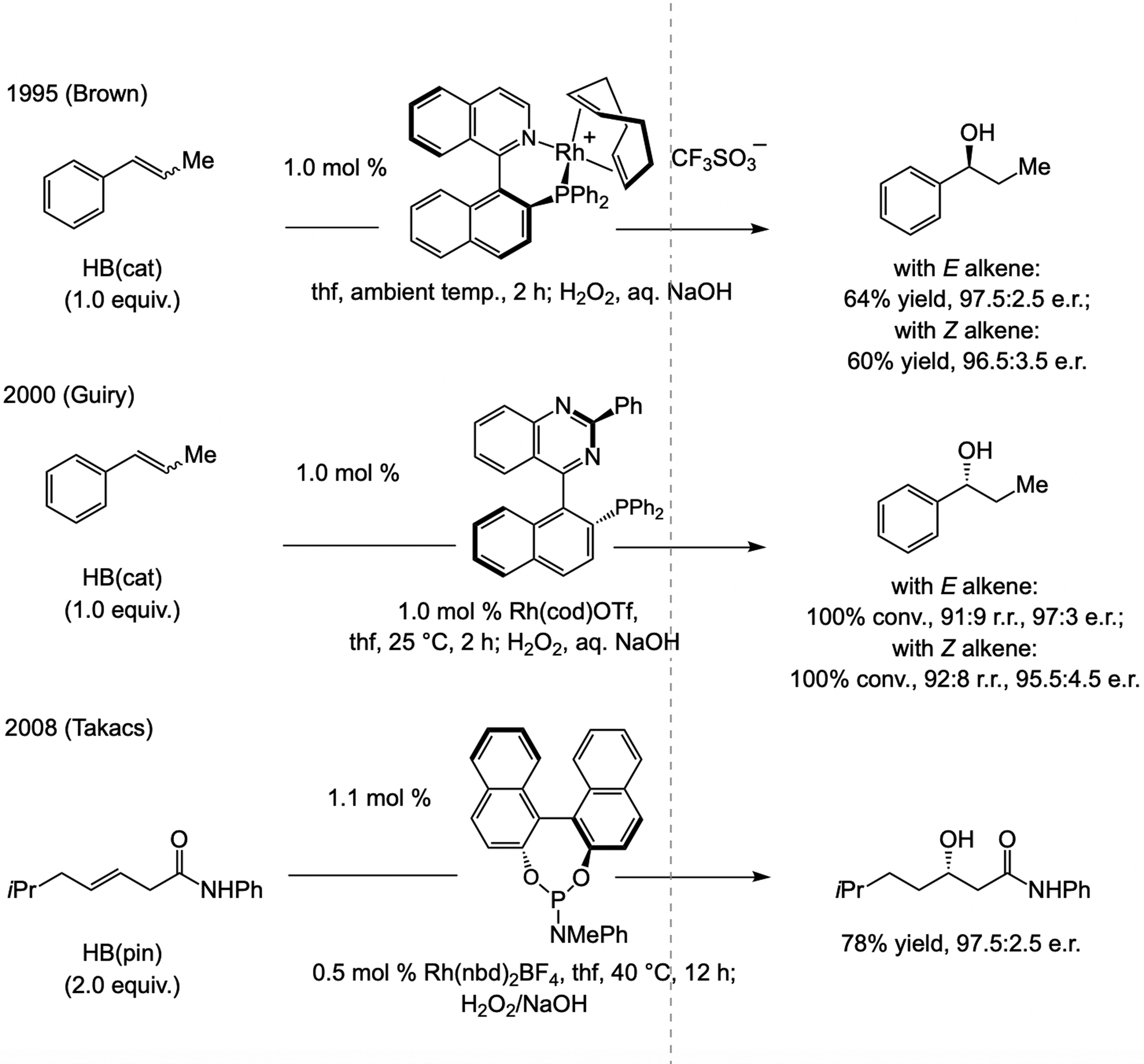
Catalytic enantioselective boron–hydride additions to 1,2-disubsdtituted linear alkenes. cat = catecholate.
6.1.3. Proto–boryl additions with sulfonate NHC–Cu catalysts
6.1.3.1. With 1,2-disubstituted aryl olefins (enantioselective C–B bond formation).
The first instances of enantioselective proto–boryl addition to an alkene were catalyzed by a sulfonate NHC–Cu complex (Scheme 64a).[125] The Cu–B moiety adds with syn stereochemistry and the C–B bond is formed exclusively at the homobenzylic site. With cyclic aryl alkenes (Scheme 64b), C1-symmetric NHC–Cu complex proved to be more effective. As stated in the original report in 2009,[125] an important attribute of the transformation – one that distinguishes it from a boron–hydride addition (e.g., Scheme 63) – is that intermediate Cu–alkyl compounds (or those obtained by a related route) may be trapped by a C- or an N-based electrophile (vs. a proton). Advances during the last 10 years have validated this prediction.[133]
Scheme 64.
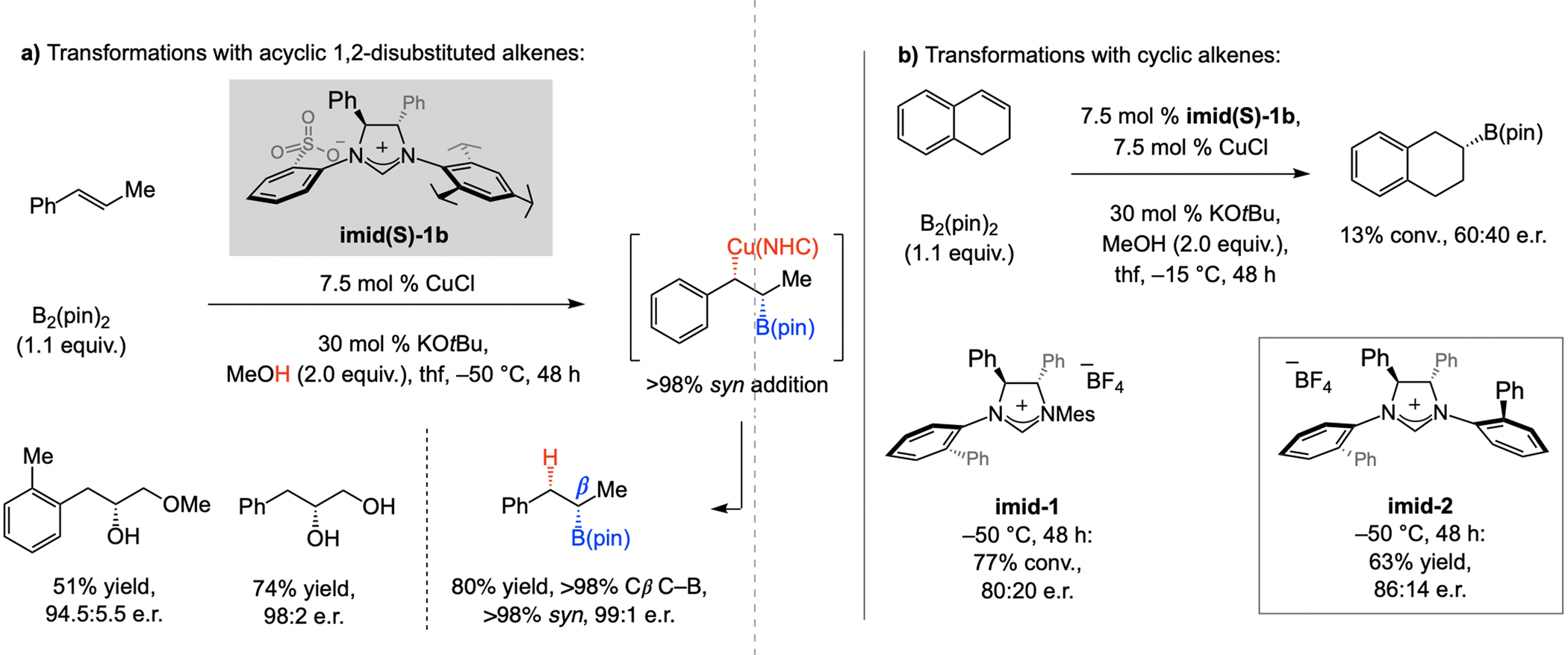
The first examples of enantioselective proto-boryl additions to alkenes.
Subsequent advances. Catalytic proto-boryl additions have been performed with strained cycloalkenes.[134] There have also been several disclosures regarding catalytic enantioselective boron–hydride additions to 1,2-disubstituted alkenes (Scheme 65). Among them is an advance by Yun.[129] In 2016, Hartwig demonstrated that an E-1,2-disubstituted alkene that is homoallylic to a benzoate, aryl ether, tertiary tosyl amide, or a silyl ether undergoes highly regio- and enantioselective boron–hydride addition in the presence of dtbm-segphos–Cu complex and HB(pin).[135] The Cu–C bond was formed preferentially at the site proximal to the electron-withdrawing group. Despite the label “directed”, however, the influence of the heteroatomic substituents seems to be mostly inductive (i.e., no temporal catalyst or reagent/substrate association[136]). Catalytic enantioselective boron–hydride additions to E-1,2-disubstituted enynes, affording trisubstituted allenyl–B(pin) compounds, were introduced later (Scheme 62c).[137]
Scheme 65.
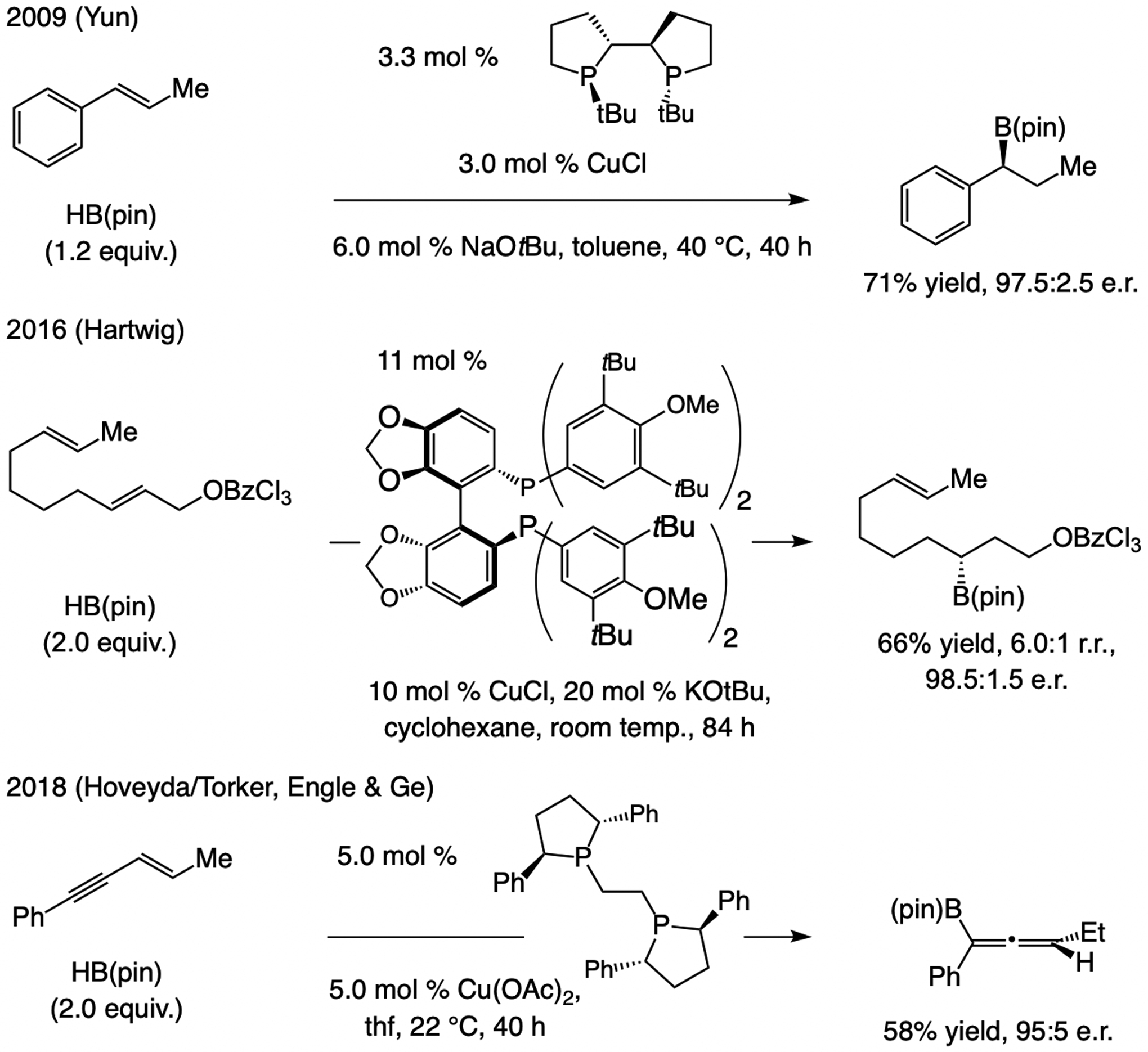
Catalytic enantioselective boron–hydride additions to E-1,2-disubstituted olefins. r.r. = regioisomeric ratio.
6.1.3.2. With 1,2-disubstituted aryl olefins (enantioselective C–H bond formation)
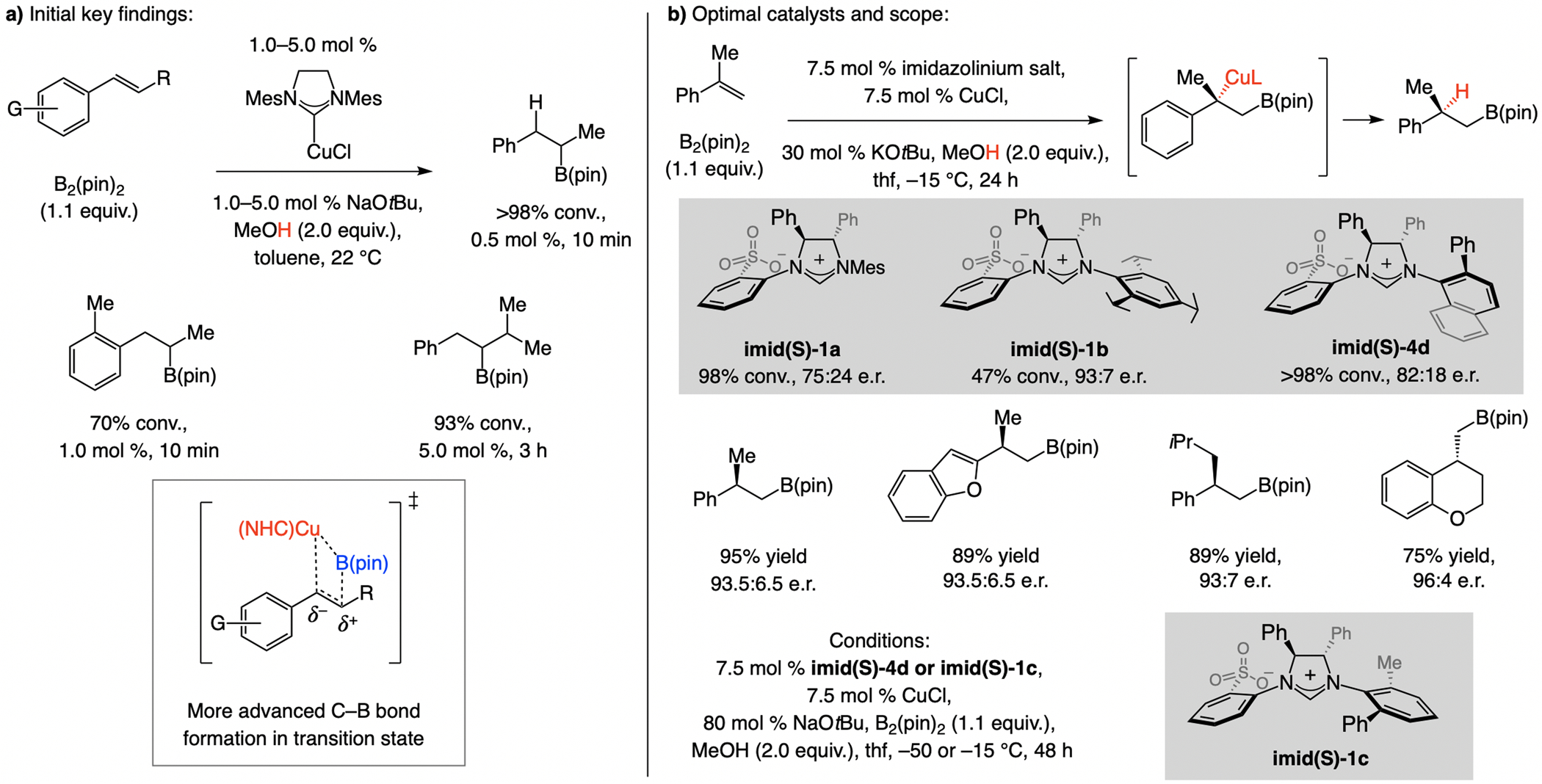
Earlier advances. 1,1-Disubstituted alkenes are challenging substrates in enantioselective synthesis,[138] and related boron–hydride additions are uncommon. An early advance was disclosed in 2010 by Mazet [Eq. (4)].[139]
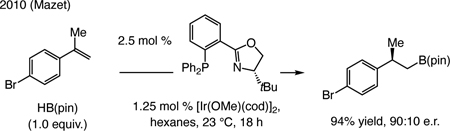 |
(4) |
With sulfonate NHC–Cu catalysts. Initial investigations showed that the rate of Cu–B addition to an alkene is slower with a larger homobenzylic substituent (Scheme 66a),[125] suggesting a dissymmetric transition structure with C–B bond formation is more advanced (vs. Cu–C bond). This implied that addition to 1,1-disubstituted alkenes, via a Cu-substituted quaternary carbon center, should be possible. Enantioselective Cu–B(pin) additions to 1,1-disubstituted aryl alkenes are indeed efficient (Scheme 66b).[140] Although the NHC–Cu complex derived from imid(S)-1b emerged as highly enantioselective with α-methylstyrene (93:7 e.r.), the reaction proceeded faster with the less sizeable imid(S)-1a (98% vs. 47% conv.). The best yield/e.r. combination was thus obtained with catalysts derived from imid(S)-4d and imid(S)-1c.
Subsequent advances. There has been much progress regarding catalytic enantioselective boron–hydride addition to 1,1-disubstituted alkenes (Scheme 67). Takacs has introduced amide-directed additions with phosphine–Rh-based catalysts.[141] Effective chiral catalysts containing earth abundant metals have been identified, including the Co- and Fe-based systems introduced by Huang[142] and Lu,[143] respectively. Yun developed the first non-directed approach to catalytic enantioselective boron–hydride additions to alkyl-substituted olefins.[144] Hong and Shi have provided an alternative regio- and enantioselective method for catalytic proto-boryl additions to aliphatic terminal olefins [Eq. (5)].[145] The latter discoveries are noteworthy because electron-rich alkenes are inherently less reactive for Cu–H or Cu–B addition.
 |
(5) |
Scheme 67.
The latest advances in catalytic enantioselective boron–hydride additions to 1,1-disubstituted alkenes.
Mechanistic analysis. While the available data indicate that the sulfonate moiety is crucial for high enantioselectivity, it probably has less of an impact on reactivity. The results of DFT studies indicate that there is likely no significant sulfonate/substrate interaction (thus the anionic model, Scheme 68). The sulfonate NHC–Cu complex can accommodate the B(pin) moiety at the same quadrant as the sulfonate, perhaps facilitated by electrostatic attraction between an aryl C–H, causing the SO3 group to move into closer proximity of the ortho C–H bonds of the NHC’s phenyl moiety (see the view from the back of the complex, Scheme 68a). The front/left pocket (as drawn) can accommodate a substrate molecule. Accordingly, the major isomer would be formed via XLVII or XLIX, where the phenyl group is positioned within the less encumbered front pocket. In XLVIII and L the position of the phenyl unit engenders considerable steric repulsion with the NAr unit.
Scheme 68.
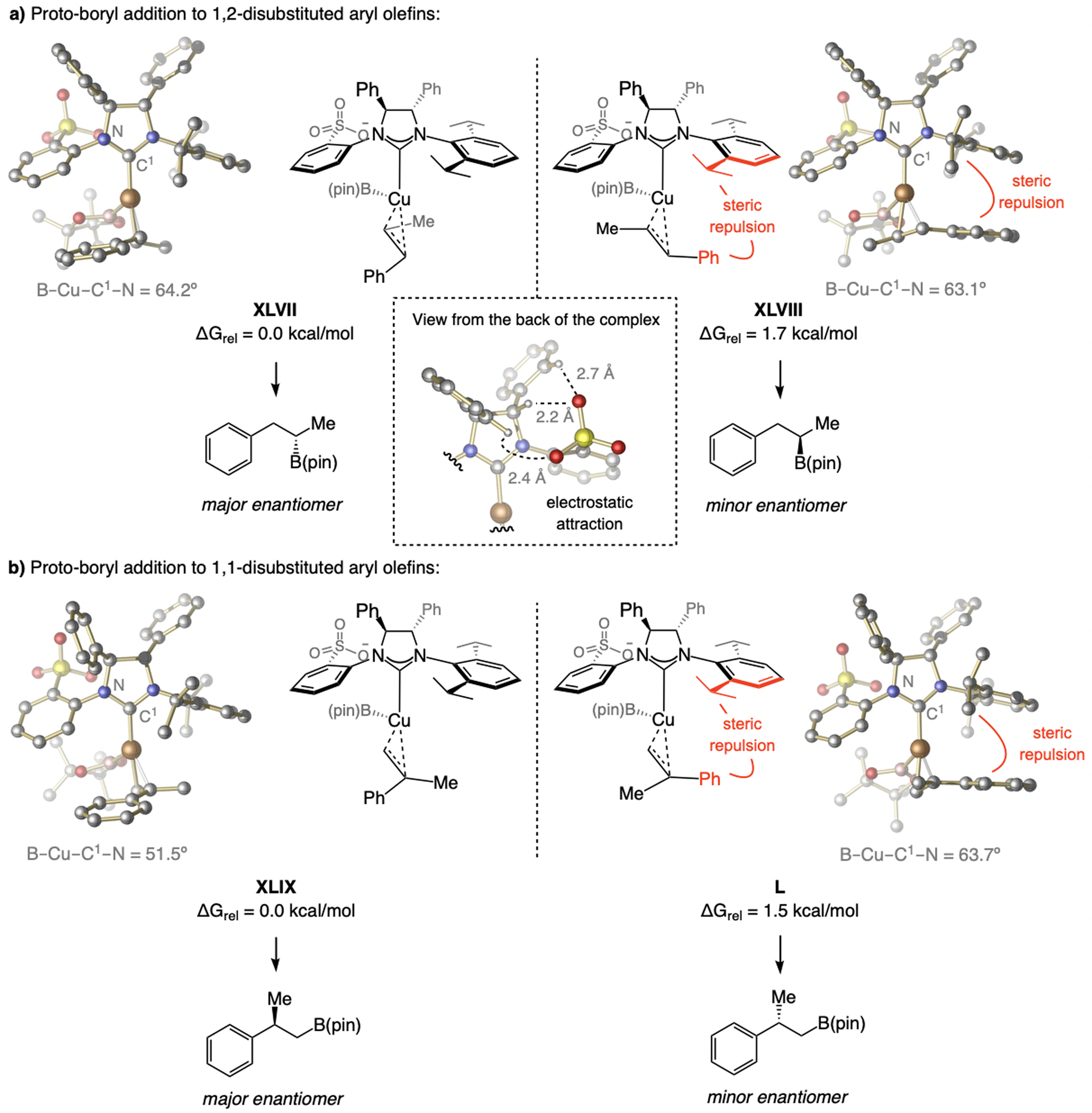
Stereochemical models accounting for the high e.r. in proto-boryl additions to aryl alkenes. DFT with the anionic model at the M06L/Def2-TZVPP//M06L/Def2-SVP level. Previously unpublished analysis; see the Supporting Information for details of the DFT studies.
6.1.3.3. With alkenyl boronates (enantioselective C–B bond formation).
Because of the stronger stabilization of electron density at the carbon center of the incipient Cu–C bond (see Scheme 66a), Cu–B addition to an electron-deficient alkene is relatively facile. Cu–B(pin) addition to an alkenyl–B(pin) is efficient (Scheme 69) [146] and regioselective with the Cu complex derived from imid(S)-1b(<2% geminal isomer). A terminal alkyne may undergo sequential proto-boryl additions, first generating an E-β-alkenyl–B(pin) compound, the substrate for an ensuing regio- and enantioselective process. Control experiments indicate that Cu–B addition to an α-alkenyl–B(pin) compound, to afford a geminal diboronate, is much less efficient. Therefore, by strongly favoring an initial β-selective addition, the sulfonate NHC ligand is key to reactions with enynes. We have applied this method to a synthesis of antioxidant plasmalogen C18 (plasm)-16:0 (PC).[147] It is worth noting that catalytic diboryl addition to an alkene [148] would be inefficient, due to competitive π-allyl formation.
Scheme 69.
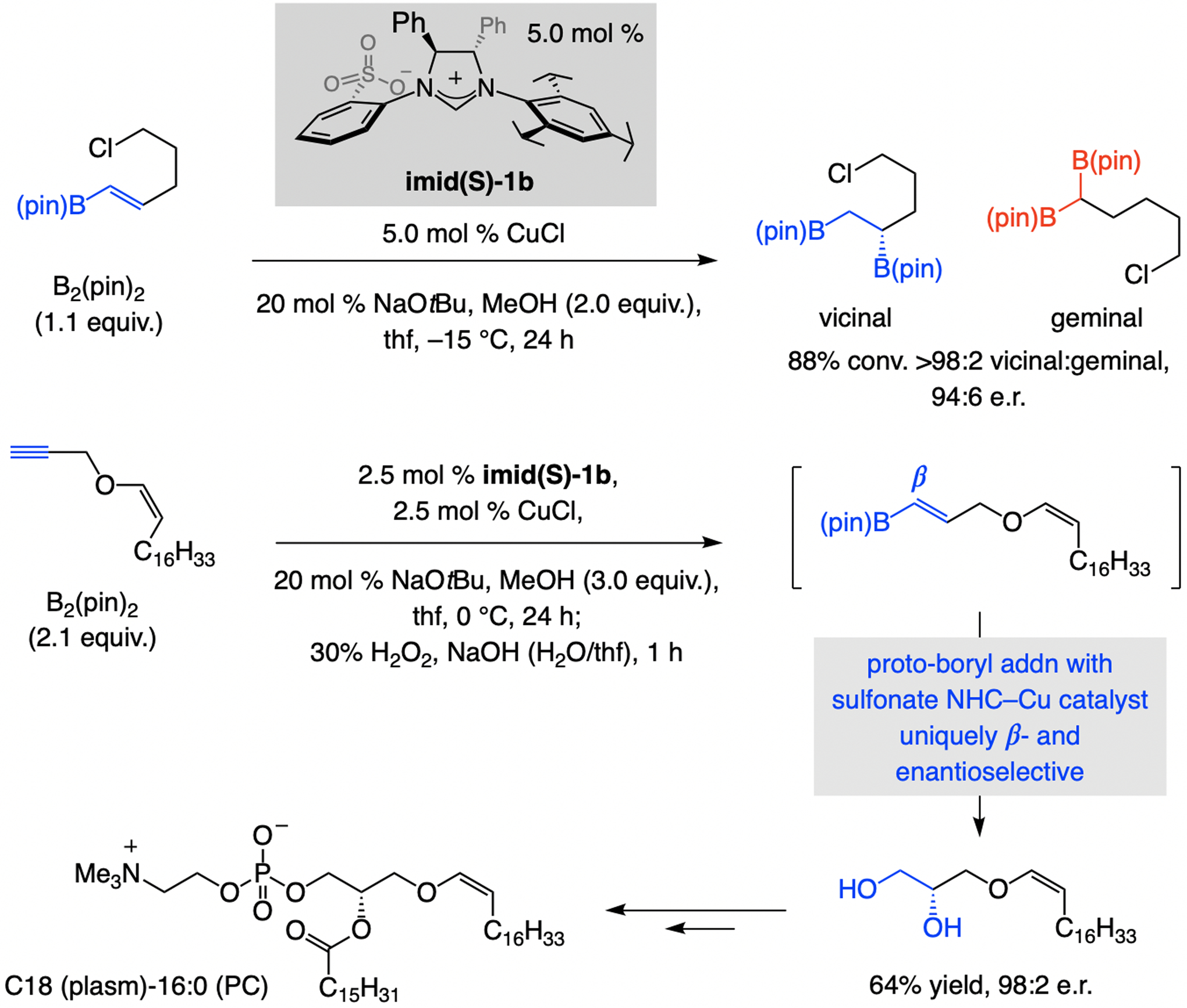
Chemo-, regio- and enantioselective sequential catalytic proto-boryl additions to alkynes.
Most recently, a sulfonate NHC–Cu complex was shown by Yun to be effective in promoting regio- and enantioselective Cu–B addition to an alkenyl–B(dan) moiety, followed by Cu/Pd ligand exchange and cross-coupling with aryl bromide (Eq. 6)]. [149] Considering that the same Cu-based catalyst was used a decade earlier for Cu–B additions to β-alkyl styrenes,[,146] the mechanistic principles outlined above (see Scheme 66) are likely applicable here as well.
 |
(6) |
6.1.3.4. With alkenyl silanes (enantioselective C–B bond formation).
NHC–Cu-catalyzed E- and β-selective proto-silyl addition to a terminal alkyne may be followed by regio- and enantioselective proto-boryl addition to the resulting alkenyl silane (Scheme 70).[150] The regiochemical outcome hinges on whether the alkenyl silane substrate is alkyl- or aryl-substituted. In the aliphatic case, where a non-sulfonate NHC–Cu catalyst is optimal, accumulation of electron density at the carbon of the incipient Cu–C bond is better accommodated by a silyl group, resulting in preferential formation of products with vicinal C–Si and Cu–B bonds. With aryl-substituted alkenylsilanes, as in the synthesis of antibacterial agent brugueirol A, the aromatic ring can better stabilize the developing negative charge, and the geminal isomer is formed preferentially (see Scheme 68).
Scheme 70.
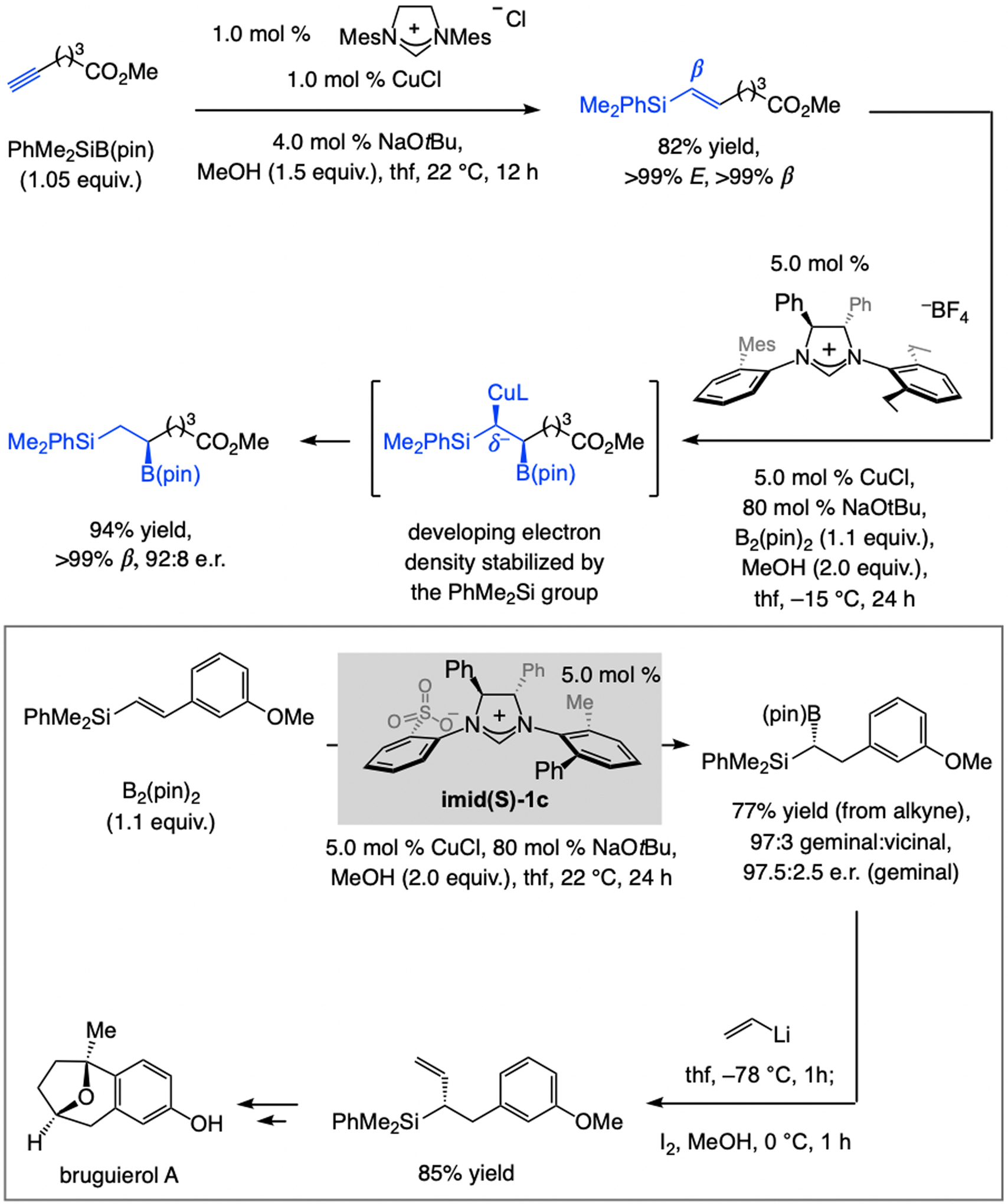
Catalytic regio- and enantioselective proto-boryl addition to an alkenylsilanes.
Subsequent advances. Morken has demonstrated[151] that vicinal diboron compounds can be prepared by catalytic B–B addition to alkenes (Scheme 71),[152] and Yun has shown[153] that differentiable geminal diboronates may be accessed regio- and enantioselectively by bisphosphine–Cu-catalyzed boron–hydride addition to alkenyl–B(dan) compounds (dan = naphthalene-1,8-diaminato). Meek has merged regio- and enantioselective Cu–B(pin) addition to alkenyl–B(pin) moieties with trapping of the Cu–alkyl intermediate with aldehydes.[154] Morken has further illustrated that[155] catalytic Si–H addition to an E-alkenyl–B(pin) compound can lead to enantiomerically enriched geminal borosilanes.
Scheme 71.
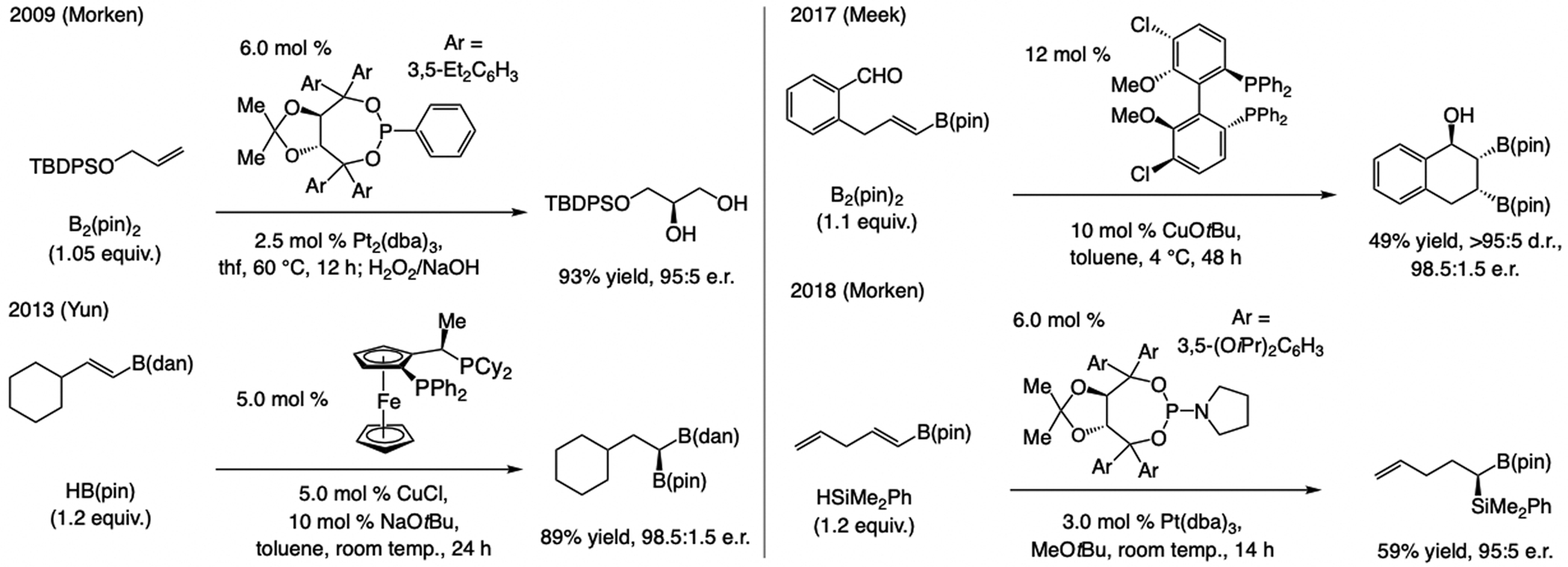
Catalytic enantioselective diboryl additions to alkenes, and boron–hydride and silyl–hydride additions to alkenyl boronates. dan = naphthalene-1,8-diaminato.
Vicinal diboryl products may be synthesized by regio- and enantioselective Cu–B(pin) addition to vinyl–B(dan) or Cu–B(dan) addition to vinyl–B(pin)/allylic substitution (Scheme 72).[156] Bis-phosphine–Cu-based catalysts are most effective in these cases. Because a C–B(pin) is considerably more reactive than a C–B(dan) bond, products can be chemoselectively modified.
Scheme 72.
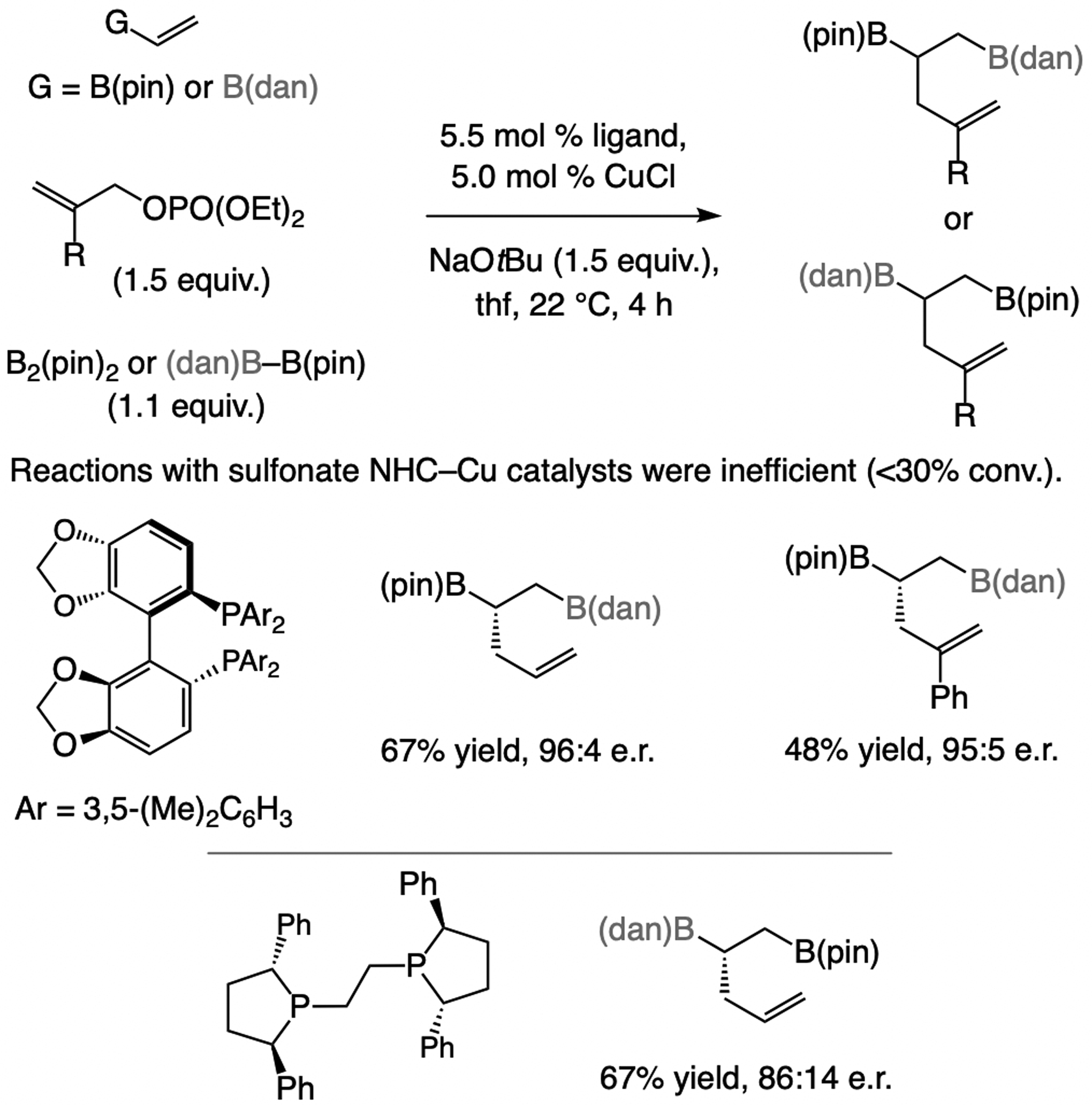
Cu–boryl addition to alkenyl boronates followed by allylic substitution. dan = naphthalene-1,8-diaminato.
6.1.3.5. With allenes (enantioselective C–H bond formation).
This set of enantioselective reactions is promoted by catalysts derived from imid(S)-1b and imid(S)-1d (Scheme 73),[157] and probably proceed via a six-membered ring transition state, represented by LI, leading to γ-selective C–H bond formation. Although the highest e.r. by a P-based ligand was by the use of segphos, transformations with sulfonate-bearing NHC–Cu catalysts were more efficient. These processes belong to a small class of catalytic reactions that include enantioselective protonation of an allylmetal intermediate,[158] furnishing masked ketones with an α-stereogenic center. The alkenyl–B(pin) moiety can be oxidized to a carboxylic acid (see naproxen synthesis, Scheme 73).
Scheme 73.
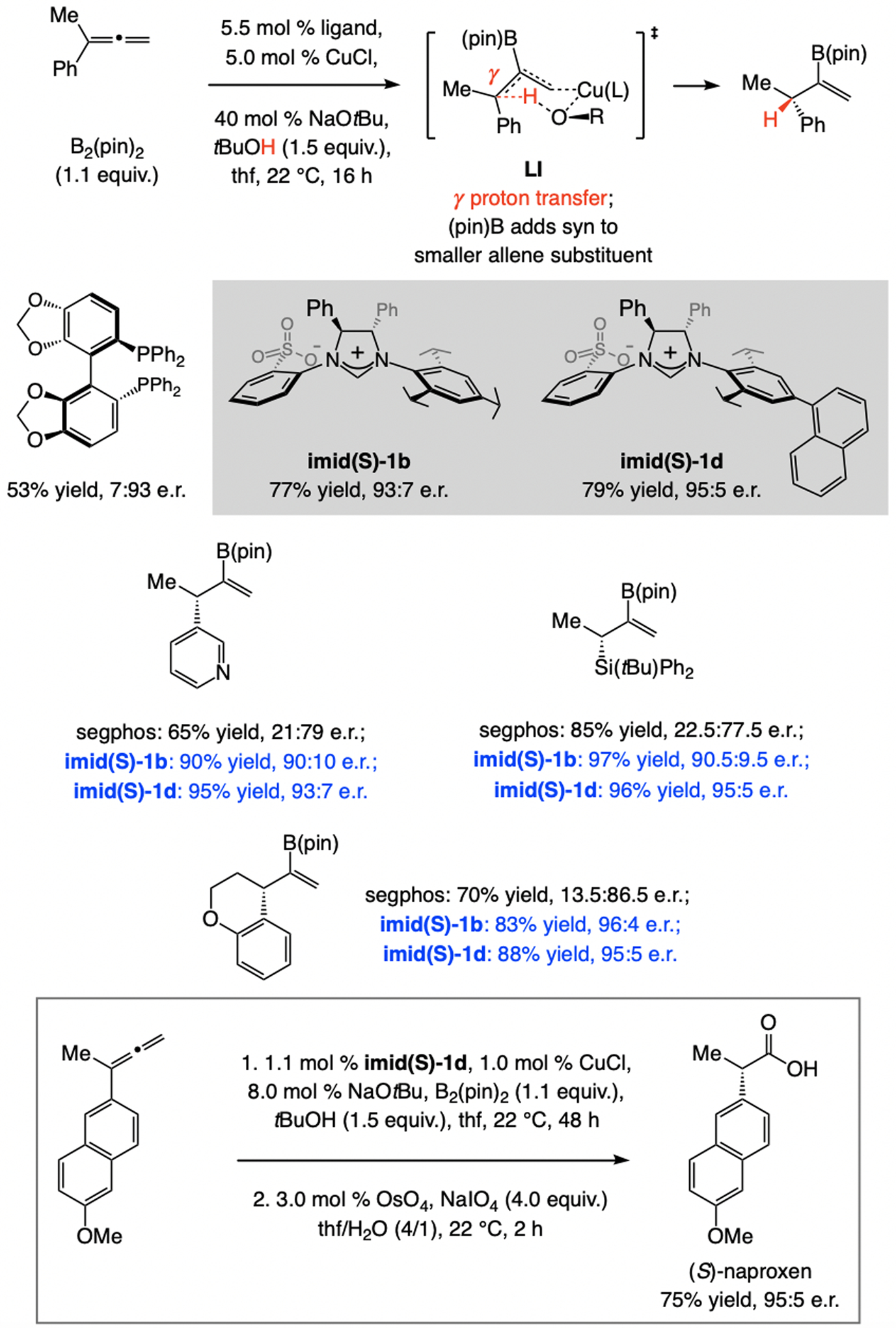
Regio- and enantioselective proto-boryl additions to 1,1-disubstituted allenes.
7. Catalytic Enantioselective Boryl Substitution
7.1. State-of-the-Art, circa 2010.
The first catalytic enantioselective boryl substitution leading to allylboronate products was disclosed in 2007 by Ito and Sawamura (Scheme 74).[159] Reactions were between a Z-allylic carbonate and B2(pin)2, catalyzed by a bis-phosphine–Cu complex (low e.r. with E isomers). Conversion was minimal with substrates bearing a larger moiety (e.g., iPr-substituted). A year later, the same team showed that Cu–B(pin) addition to an alkenyl silane preferentially affords products with geminal Cu–C and C–Si bonds; carbonate displacement afforded the corresponding cyclopropane.[160]
Scheme 74.
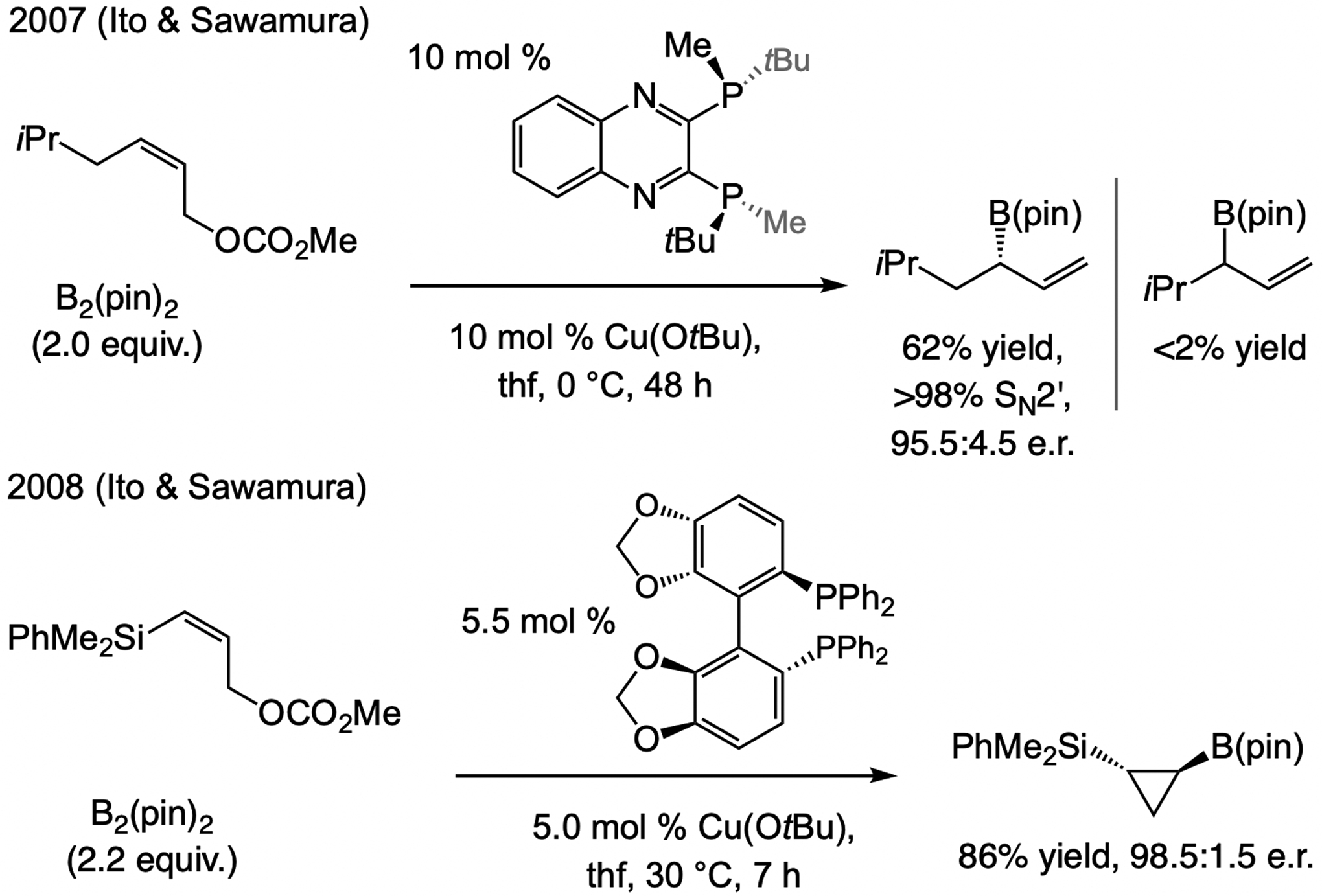
The first examples of catalytic enantioselective boryl substitution, and an approach leading to cyclopropyl products.
7.2. With Sulfonate NHC–Cu Catalysts.
The complex derived from imid(S)-2c promotes regio- and enantioselective boryl substitution with alkyl-substituted allylic carbonates (Scheme 75a).[161] Unlike the earlier protocols (see Scheme 74), an E- or a Z-alkene can be used to access either product enantiomer, consistent with a step other than C–B bond formation being stereochemistry-determining. Product stereochemistry is thus probably controlled by the position of the carbonate group, taking place at a relatively extended C…B distance. (The enantiomers derived from E- and Z-alkene isomers in Scheme 75a constitute a revision of the absolute stereochemical identity of the products.) Allylic carbonates bearing a hindered substituent can be used (e.g., Cy-substituted). A marked attribute that distinguishes sulfonate NHC–Cu catalysts in this arena is that reactions with trisubstituted allylic carbonates can be effected efficiently and with high enantioselectivity, furnishing allyl–B(pin) products with a B-substituted quaternary stereogenic carbon center (Scheme 75b).[161] Sterically demanding alkyl- and aryl-substituted allylic carbonates emerged as suitable starting materials. An application entails α-selective addition of an enantiomerically enriched allyl boronate to a phosphinoylimine, a process that takes place with inversion of stereochemistry. [162]
Scheme 75.
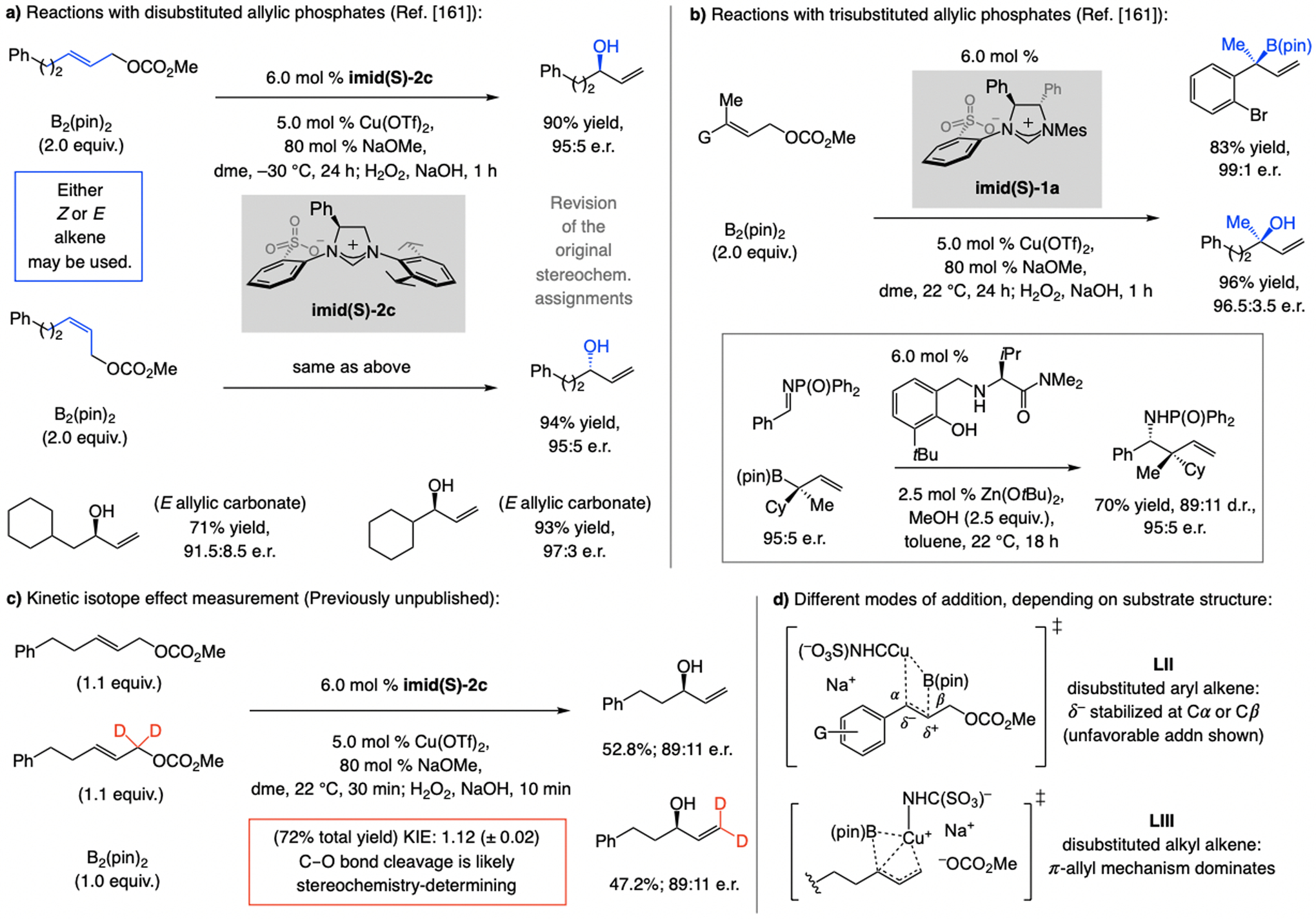
Sulfonate NHC–Cu-catalyzed boryl substitution with a di- or a trisubstituted alkene to generate allylic boronates in high e.r., and related KIE data regarding substitution (previously unpublished). Reactions performed under N2 atm.; conv. (±2%) determined by analysis of the 1H NMR spectra of the unpurified mixtures; yields (±5%) of purified products. See the Supporting Information for details.
Mechanistic Analysis. KIE measurements (Scheme 75c; previously unpublished) indicate that C–O bond cleavage is probably stereochemistry-determining (see the Supporting Information for a stereochemical model). However, as was noted earlier (see Scheme 31b), DFT studies indicate the energy gap between a migratory insertion and a π-allyl generation is the narrowest for C–B bond formation (8.5 vs. 4.9 kcal/mol). Either mechanism might therefore be operative, perhaps depending on the substrate type. The regiochemical differences can be explained if a Cu–B addition pathway is invoked for reactions of disubstituted aryl olefins, where a benzylic Cu–Cα bond (LII, Scheme 75d) would likely be formed, affording an intermediate that cannot be converted to an allylboronate product. With alkyl-1,2-substituted carbonates (LIII), the electronically favored a π-allyl mechanism steers the B(pin) group into the allylic position (SN2’). For trisubstituted olefins, regardless of whether the substrate is aryl- or alkyl-substituted, transformation via the initial formation of a π-allyl complex/C–O cleavage is probably more favored (less sterically demanding).
Subsequent advances. Cu-based complexes containing a six-membered NHC ligand have been shown by McQuade (Scheme 76)[163] to be effective boryl substitution catalysts. This approach is stereoconvergent: E- and the Z-allylic ether substrates afford the same product enantiomer, a finding that is consistent with C–B bond formation being stereochemistry-determining. Reactions in which a fluorine atom is the leaving group has received attention recently, allowing for recent development of catalytic methods for synthesis of γ,γ-gem-difluoroallylboronates[164] or allylic boronates that contain a trisubstituted alkenyl fluoride (Scheme 76).[165] These latter studies showed that sulfonate NHC–Cu catalysts are effective with substrates bearing a primary alcohol. [166]
Scheme 76.
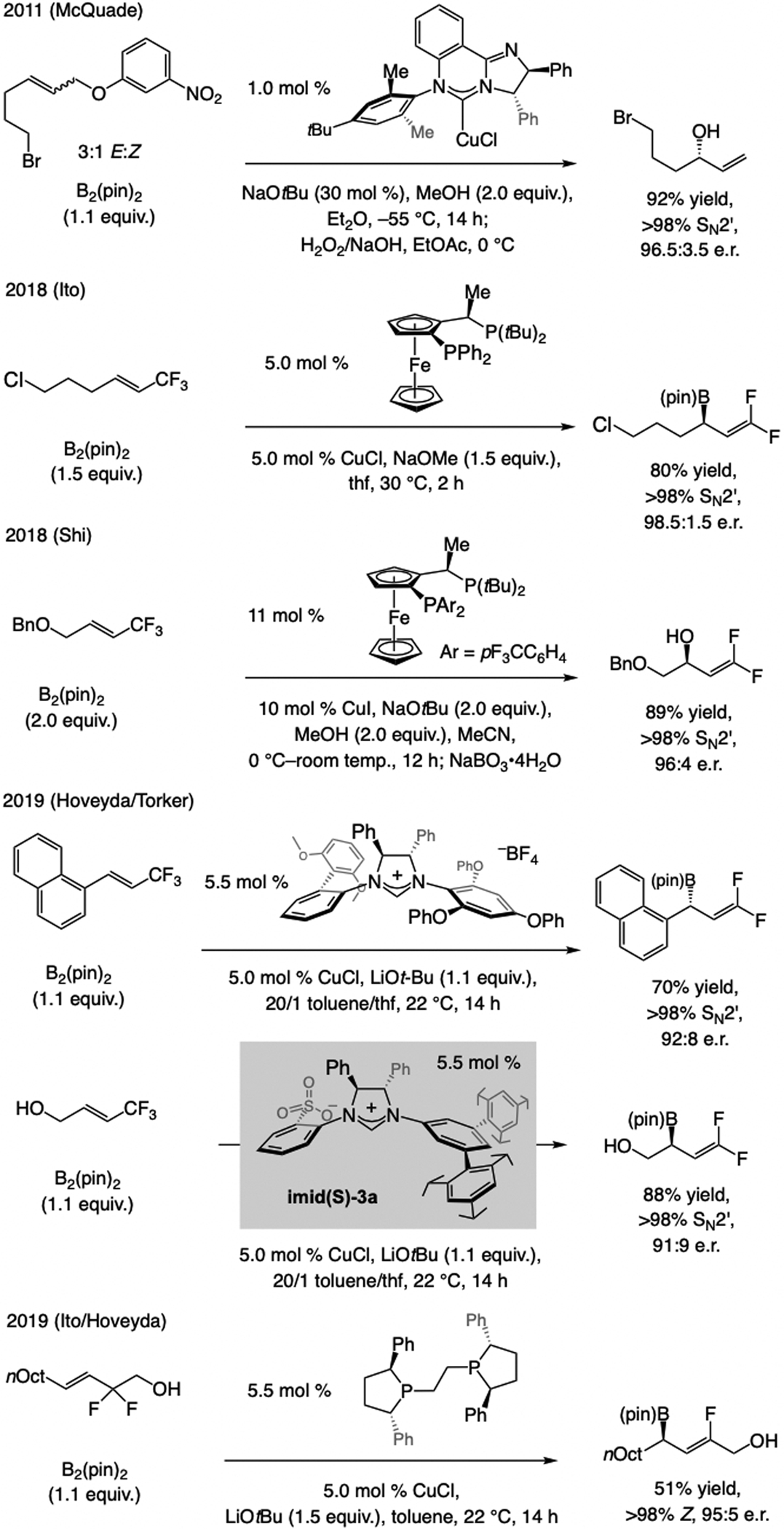
Advances in catalytic enantioselective boryl substitution include those that generate F-substituted alkenes.
8. Catalytic Enantioselective Silyl Substitution
Prior Art. In 1994, Hayashi and Yanagi reported that reaction of an allyl chloride with PhCl2Si–SiMe3 can be facilitated by a bisphosphine–Pd complex, leading to transfer of the PhCl2Si moiety (Scheme 77);[167] workup with basic EtOH afforded the allylic diethoxysilane products. Enantioselectivity was high in some instances, but not regioselectivity. As noted earlier (see Schemes 7 and 9), catalytic EAS with alkenylsilane may be used to access enantiomerically enriched allylsilanes. In 2013, Oestreich presented a more general strategy,[168] involving the use of a six-membered NHC–Cu catalyst, allylic phosphates, and Me2PhSi–B(pin). Soon afterwards, Hayashi and Shintani demonstrated that, with the same set of substrates and borosilane compounds, a different NHC–Cu complex can be used to generate allylsilanes enantioselectively.[169]
Scheme 77.
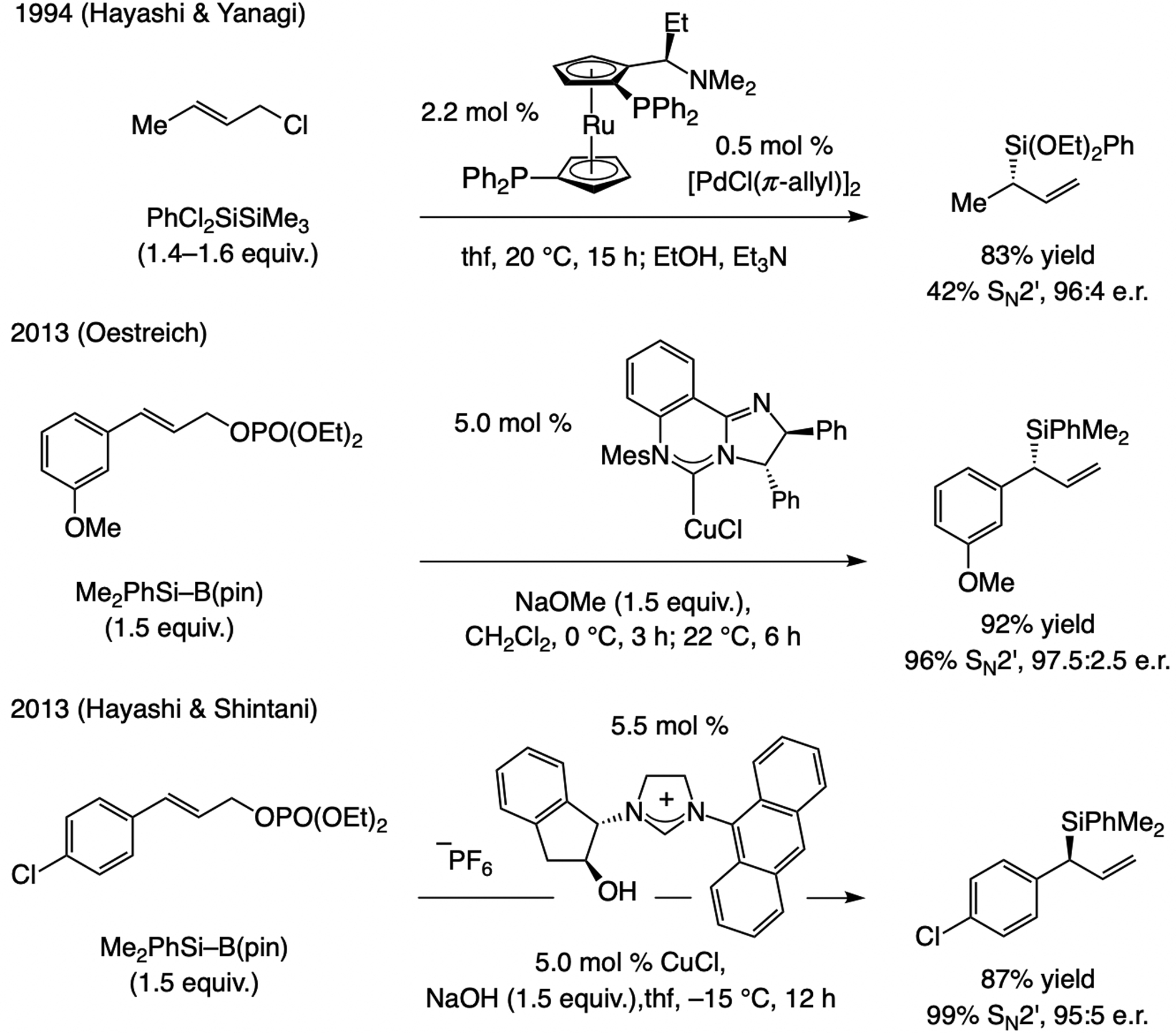
Enantioselective silyl substitution with Pd- and Cu-based catalysts.
With a Sulfonate NHC–Cu Catalyst. With 1.0 mol % of the NHC–Cu complex derived from imid(S)-1a reaction can be promoted between 1,2-disubstituted allylic phosphates and PhMe2Si–B(pin) (Scheme 78; previously unpublished).[170] Aryl- as well as alkyl-substituted substrates react with high SN2’ and enantioselectivity.
Scheme 78.
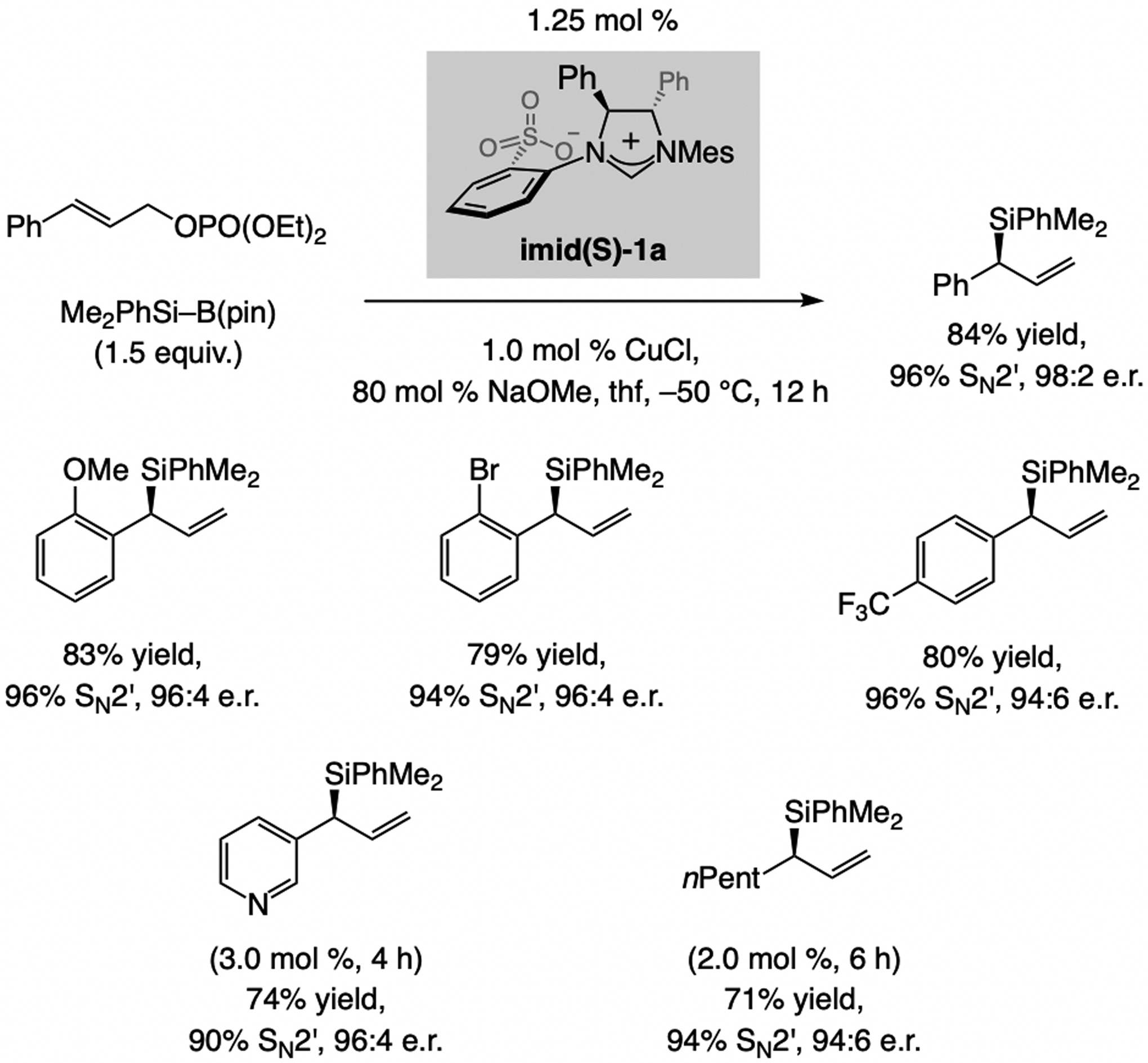
Enantioselective silyl substitution with a sulfonate NHC–Cu catalyst. Reactions performed under N2 atm.; conv. and SN2’ selectivity (±2%) determined by analysis of the 1H NMR spectra of the unpurified product mixtures; yields (±5%) of the purified products; enantioselectivity determined by HPLC analysis. Previously unpublished results; see the Supporting Information for details.
Catalytic silyl substitutions that afford γ,γ-gem-difluoroallylsilanes were recently introduced (Scheme 79a).[51] In line with the C–B bond forming reactions, processes involving sulfonate NHC–Cu catalysts, while efficient, were less enantioselective. Nonetheless, reaction with a PMB-protected alcohol starting material was most efficient with the catalyst derived from imid(S)-3a (80% conv., 60% yield, 93:7 e.r. with the C2-symmetric NHC–Cu complex; in contrast, with the alcohol substrate and catalyst derived from imid(S)-3a: 85% conv., 66% yield, 88:12 e.r.).
Scheme 79.
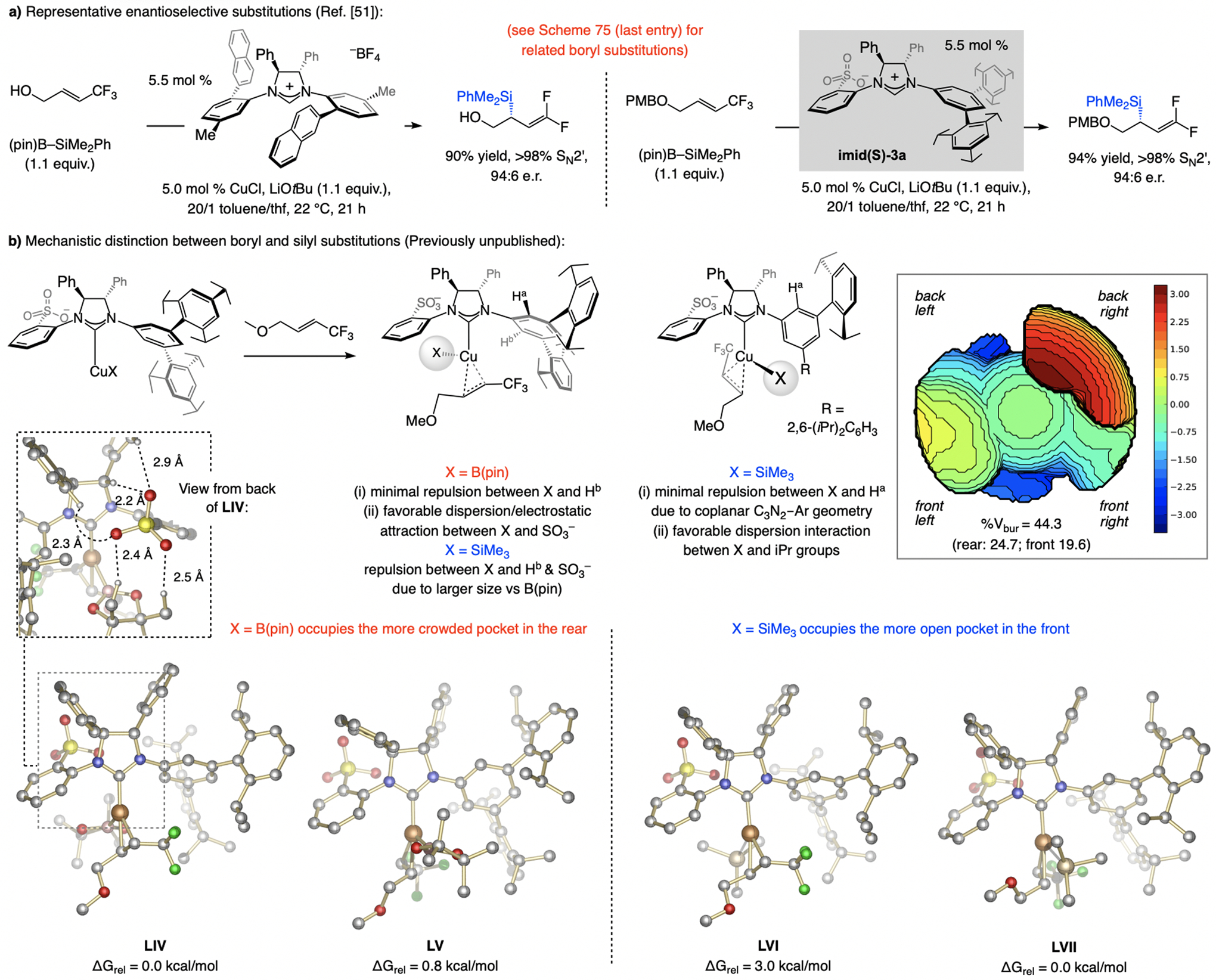
A sulfonate NHC–Cu complex can promote highly regio- and enantioselective silyl substitution to generates γ,γ-gem-difluoroallylsilanes, underscoring mechanistic differences with the related boryl substitutions. Previously unpublished analysis; see the Supporting Information for details of the DFT studies.
Mechanistic analysis. Notably, boryl (Scheme 76) and silyl substitutions (Scheme 79a) afford products with the opposite sense of enantioselectivity. The map of steric effects[171] for imid(S)-3a reveals a more accessible front pocket (% Vbur = 19.6 vs. 24.7 in the rear). For C–B addition, there is minimal repulsion between an ortho hydrogen atom (Hb) of the NHC’s NAr moiety and the B(pin) moiety in the rear, while favorable dispersive associations or electrostatic attraction with the sulfonate anion might also be a factor, as suggested by the close SO3…..H–C contacts of 2.2–2.9 Å (see view from back of LIV, Scheme 79b). The only space available for the larger silyl group is at the front of the complex (LVII); as such, the steric repulsion with the sulfonate and Hb, which exists in LVI, can be avoided. In LVII, repulsion with Ha can be further minimized owing to the near-coplanar orientation of the sizeable 3,5-bis-(2,4,6-triisopropylphenyl)phenyl fragment (with respect to the NHC). Thus, dispersive affinity between the silyl and the NHC’s isopropyl groups can dominate repulsive interactions. Similar features (dispersive attraction and conformational mobility around the NAr bond) have been put forward on the basis of DFT studies regarding Cu–H addition to allenyl–B(pin) compounds (see Scheme 29a).[59]
9. Enantioselective Allyl Additions to NH-Ketimines
State-of-the-Art, circa 2017. Unlike aldimines,[172] reactions with the significantly less reactive ketimines are uncommon.[173] The small number of the existing methods available in 2017 require the use of N-protected/activated imines (Scheme 80). In 2006, Shibasaki and Kanai reported on enantioselective addition of allyl–B(pin) to N-benzyl ketimines, catalyzed by 20 mol % of a bis-phosphine (cyclopentyl-duphos) and 10 mol % CuF2•2H2O. Enantioselectivities were high, alkyl-substituted substrates were suitable, and the unprotected amine was obtained after two steps (2-iodoxybenzoic acid and hydrolysis with aq. HCl). [174] Shibasaki and Kumagai later outlined a protocol for enantioselective addition of an allylic cyanide to N-phosphinoyl ketimines, catalyzed by 10 mol % of a bis-phosphine ligand and 10 mol % of a Cu salt. Unmasking of the N-H amine was accomplished by treatment with 12N aqueous solution of HCl. [175] Finally, through a set of transformations catalyzed by phosphoramidite–Pd complex, Trost was able to promote diastereo- and enantioselective cycloaddition of trimethylenemethane to N-tosyl-ketimines; the approach is applicable to aryl- and alkyl-substituted ketimines, but removal of the tosyl group would require harsh reductive conditions (issue not addressed).[176] Later in 2012, Lam showed that a dienyl–Rh complex can facilitate enantioselective allyl addition to cyclic aldimines and ketimines; unprotected amines were then generated by a two-step sequence.[177] In 2016, Zhang disclosed an enantioselective Morita-Baylis-Hillman type process catalyzed by a phosphine and involving N-benzyl ketimines; enantioselectivities were high and α-tertiary NH2-amines were obtained after acidic workup (Scheme 80).[178]
Scheme 80.
Catalytic enantioselective additions of allylic moieties to N-protected ketimines.
With Allenes, B2(pin)2, and N-H Ketimines. The ability of different Cu-based catalysts to promote diastereo- and enantioselective addition of 2-B(pin)-substituted allyl groups to N-H ketimines varies widely (Scheme 81a). Unlike when a bis-phosphine ligand is used, with imid(S)-3a, e.r. is high. [179] Whereas the desired N-H ketimine was formed in 95:5 e.r. with 3,5-diaryl-substituted imid(S)-3a as the catalyst precursor, with N-mesityl-containing imid(S)-1a, the product was isolated in only 55:45 e.r. With the sulfonate NHC–Cu catalyst derived from imid(S)-3a, on the other hand, heteroaryl- and alkyl-substituted N-H ketimines were converted to α-tertiary amines efficiently and selectively (Scheme 81b); application to synthesis of the core structure of a class of compounds with anti-Alzheimer activity highlights the utility of the protocol.
Scheme 81.
Catalytic diastereo- and enantioselective multicomponent additions of boryl-substituted allyl moieties to N-H ketimines.
Mechanistic analysis. DFT studies point to the interaction of a cationic Na bridge between the sulfonate and the nitrogen atom of the imine (LVIII-LIX). The B(pin) moiety is probably oriented in the left/front quadrant and away from the sizeable 3,5-disubstituted aryl group, in LVIII. This is in contrast to LIX, where there is steric strain caused by the proximity of the B(pin) and the chiral ligand’s NAr groups; such interactions are diminished with mesityl-substituted NHC–Cu catalyst derived from imid(S)-1a, which is congruent with the observed low e.r. (55:45).
Subsequent advances. In 2019, the research groups of Wang and Zhang/Niu reported catalytic protocols for enantioselective synthesis of E-trifluoromethyl-substituted α-tertiary homoallylic NH2-amines, catalyzed by a phosphine–Ir complex (Scheme 82).[180] Most recently, we demonstrated that reactions of trifluoromethyl-substituted silyl ketimines with Z-allyl boronate compounds may be promoted by an aminophenol-boryl catalyst, directly affording unprotected α-tertiary amine in high enantiomeric purity. [181] It should be noted that trifluoromethyl-substituted NH-ketimines are unstable and therefore not suitable for sulfonate NHC–Cu-catalyzed processes (see Scheme 81; see also Zhang, Scheme 80).
Scheme 82.
Catalytic diastereo- and enantioselective strategies for synthesis of trifluoromethyl-substituted α-tertiary NH2-amines.
11. Conclusion and Outlook
We provide significant evidence supporting the contention that sulfonate NHC–Cu complexes are uniquely effective catalysts for a range of enantioselective processes, ranging from different types of C–C bond forming transformations to those that generate an assortment of C–B, C–H, and C–Si linkages. In many instances, sulfonate NHC–Cu catalysts offer degrees of reactivity and/or selectivity (regio- and/or enantioselectivity) that are not within the reach of catalysts derived from other ligand types, NHC or otherwise.
Experimental and computational data are provided that offer a mechanistic basis for the observed reactivity and selectivity trends. All underscore the central role of the sulfonate moiety. The experimental and computational results indicate that, in most cases, the sulfonate unit is to provide a temporal metal bridge that connects the catalyst structure with a Lewis basic site within the substrate molecule; this additional interaction belies the unusually high efficiency, regio- and enantioselectivities.
There is probably more to the story, however. For instance, the corresponding carboxylate NHC–Cu complex[182] is far less effective (Scheme 83), suggesting that a chelation site alone does not suffice. One advantage of a sulfonate might be in its being C3-symmetric: unlike a carboxylate group where rotation around the C–C bond disrupts/alters resonance between the aryl moiety and the carbonyl group, rotation around the C–S bond is inconsequential. These considerations beg the question of how would a phosphate NHC–Cu complex perform?
Scheme 83.
A carboxylate NHC–Cu complex is less effective (vs. sulfonate variants). See the Supporting Information for details.
Looking back, one cannot help but be astonished what substantial an effect a subtle structural modification can have on a catalyst’s performance. This, combined with the fact that less than 3 kcal/mol separates a highly enantioselective reaction from one generating racemic mixtures (i.e., about the same barrier as C–C bond rotation in ethane), underscores the importance of being able to modify a catalyst easily. This review provides further testament to the importance of mechanistic understanding. Otherwise, each discovery, by chance or screening, remains just that, a stroke of luck or a feat of engineering, confined only to the present with little investment toward mechanistically guided a future advance. We are then left with only the hope that we might, somehow, find a way out of future puzzles. Only true science can free us from such a precarious predicament.
Supplementary Material
Acknowledgments.
We thank the NIH (GM-47480 and GM-130395) for financial support. Additional funding was provided by Pfizer, AstraZeneca, Bristol-Myers Squibb, Schering-Plough, Novartis, the Fullbright Foundation, the Spanish Ministry of Science, and the Vanderslice, and LaMattina Endowment funds. We are deeply indebted to Dr. Carl Baxter, Prof. Dennis G. Gillingham, Prof. Yunmi Lee, Dr. Tricia May, Dr. Fang Gao, Prof. Byunghyuck Jung, Dr. Katsuhiro Akiyama, Dr. Hwanjong Jang, Prof. Jennifer A. Dabrowski, Prof. Fanke Meng, Prof. Aikomari Guzman-Martinez, Dr. Adil R. Zhugralin, Dr. Rosa Corberán, Dr. Nicholas W. Mszar, Prof. Simon J. Meek, Prof. Kang-sang Lee, Dr. Monica A. Fitzgerald, Dr. Juan del Pozo, Dr. Filippo Romiti, Dr. James L. Carr, Dr. Fredrik Haeffner, Dr. Matthew T. Villaume, Dr. Kyoko Mandai, Dr. Kevin P. McGrath, Dr. Aran K. Hubbell, Ms. Mikiko Akiyama, Dr. Stephanie A. Kazane, and Dr. Damian P. Santos for their intellectual and experimental contributions. We are grateful to Dr. Bo Li, Dr. Kyoko Mandai, and Dr. Malte S. Mikus for their assistance in obtaining X-ray structures, and thank Dr. J. del Pozo, Dr. Y. Huang, and Dr. Y. Sun for helpful suggestions regarding the manuscript.
Contributor Information
Amir H. Hoveyda, Department of Chemistry, Merkert Chemistry Center, Boston College, Chestnut Hill, MA 02467 (USA) Supramolecular Science and Engineering Institute, University of Strasbourg, CNRS, 67000 Strasbourg (France).
Sebastian Torker, Supramolecular Science and Engineering Institute, University of Strasbourg, CNRS, 67000 Strasbourg (France).
References and Footnotes:
- [1].a) van Veldhuizen JJ, Garber SB, Kingsbury JS, Hoveyda AH, J. Am. Chem. Soc 2002, 124, 4954–4955; [DOI] [PubMed] [Google Scholar]; b) van Veldhuizen JJ, Gillingham DG, Garber SB, Kataoka O, Hoveyda AH, J. Am. Chem. Soc 2003, 125, 12502–12508; [DOI] [PubMed] [Google Scholar]; c) Gillingham DG, Kataoka O, Garber SB, Hoveyda AH, J. Am. Chem. Soc 2004, 126, 12288–12290. [DOI] [PubMed] [Google Scholar]
- [2].a) Larsen AO, Leu W, Oberhuber C. Nieto, Campbell JE, Hoveyda AH, J. Am. Chem. Soc 2004, 126, 11130–11131; [DOI] [PubMed] [Google Scholar]; b) Van Veldhuizen JJ, Campbell JE, Guidici RE, Hoveyda AH, J. Am. Chem. Soc 2005, 127, 6877–6882; [DOI] [PubMed] [Google Scholar]; c) Lee Y, Hoveyda AH, J. Am. Chem. Soc 2006, 128, 15604–15605; [DOI] [PubMed] [Google Scholar]; d) Kacprzynski MA, May TL, Kazane SA, Hoveyda AH, Angew. Chem. Int. Ed 2007, 46, 4554–4558. [DOI] [PubMed] [Google Scholar]
- [3].Lee K.-s., Brown MK, Hird AW, Hoveyda AH, J. Am. Chem. Soc 2006, 128, 7182–7184. [DOI] [PubMed] [Google Scholar]
- [4].a) Vedernikov AN, Fettinger JC, Mohr F, J. Am. Chem. Soc 2004, 126, 11160–11161. [DOI] [PubMed] [Google Scholar]; For related review articles, see:; b) Vedernikov AN, Chem. Commun 2009, 4781–4790; [DOI] [PubMed] [Google Scholar]; c) Vedernikov AN, Acc. Chem. Res 2012, 45, 803–813. [DOI] [PubMed] [Google Scholar]
- [5].Brown KM, May TL, Baxter CA, Hoveyda AH, Angew. Chem. Int. Ed 2007, 46, 1097–1100. [DOI] [PubMed] [Google Scholar]
- [6].For reviews on catalytic EAS reactions with “hard” alkyl- or arylmetal-based reagents, see:; (a) Hoveyda AH, Hird AW, Kacprzynski MA, Chem. Commun 2004, 1779–1785; [DOI] [PubMed] [Google Scholar]; b) Yorimitsu H, Oshima K, Angew. Chem. Int. Ed 2005, 44, 4435–4439; [DOI] [PubMed] [Google Scholar]; c) Alexakis A, Bäckvall JE, Krause N, Pàmies O, Diéguez M, Chem. Rev 2008, 108, 2796–2823; [DOI] [PubMed] [Google Scholar]; d) Helmchen G, Kazmaier U, Förster S in Catalytic Asymmetric Synthesis (Ojima I, Ed.); Wiley, 2010, 497–641; [Google Scholar]; e) Baslé O, Denicourt-Nowicki A, Crévisy C, Mauduit M in Copper-Catalyzed Asymmetric Synthesis (Alexakis A, Krause N, Woodward S, Eds); VCH–Wiley, 2014, pp. 85–125. [Google Scholar]; For a review regarding applications of catalytic EAS to natural product synthesis, see:; f) Calvo BC, Buter J Minnaard AJ in Copper-Catalyzed Asymmetric Synthesis (Alexakis A, Krause N, Woodward S, Eds); VCH–Wiley, 2014, pp. 373–447. [Google Scholar]
- [7].Gillingham DG, Hoveyda AH, Angew. Chem. Int. Ed 2007, 46, 3860–3864. [DOI] [PubMed] [Google Scholar]
- [8]. For examples of EAS reactions involving dialkyl- and diarylzinc reagents where sulfonate-containing NHC–Cu complexes were used to generate allylsilane products, see: ref. [2d].
- [9].For a review on hydroaluminations of alkynes and alkenes, see:; Eisch JJ in Comprehensive Organic Synthesis, Vol. 8 (Trost BM, Fleming I, Schreiber SL, Eds), Pergamon, Oxford, 1991, pp. 733–761. [Google Scholar]
- [10].Lee Y, Akiyama K, Gillingham DG, Brown MK, Hoveyda AH, J. Am. Chem. Soc 2008, 130, 446–447. [DOI] [PubMed] [Google Scholar]
- [11].These findings underscore a key principle: a catalyst, which might happen to be chiral, can at times deliver higher efficiency than its achiral analogues. Just because initial examination of a transformation in the absence of a chiral ligand or with an achiral ligand is inefficient, it should not be assumed that a chiral complex is also incapable of delivering high efficiency. For an analysis of the utility of chiral catalysts beyond the goal of achieving high enantioselectivity, see:; Hoveyda AH, Malcolmson SJ, Meek SJ, Zhugralin AR, Angew. Chem. Int. Ed 2010, 49, 34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Akiyama K, Gao F, Hoveyda AH, Angew. Chem. Int. Ed 2010, 49, 419–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].For recent reviews on enantioselective generation of all-carbon quaternary stereogenic centers through catalytic processes, see:; a) Das JP, Marek I, Chem. Commun 2011, 47, 4593–4623; [DOI] [PubMed] [Google Scholar]; b) Feng J, Holmes M, Krische MJ, Chem. Rev 2017, 117, 12564–12580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gao F, McGrath KP, Lee Y, Hoveyda AH, J. Am. Chem. Soc 2010, 132, 14315–14320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gao F, Hoveyda AH, J. Am. Chem. Soc 2010, 132, 10961–10963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].For previous enantioselective syntheses of bakuchiol, see:; a) Takano S, Shimazaki Y, Ogasawara K, Tetrahedron Lett 1990, 31, 3325–3326; [Google Scholar]; b) Du X-L, Chen H-L, Feng H-J, Li Y-C, Helv. Chim. Acta 2008, 91, 371–378; [Google Scholar]; c) Esumi T, Shimizu H, Kashiyama A, Sasaki C, Toyota M, Fukuyama Y, Tetrahedron Lett 2008, 49, 6846–6849; [Google Scholar]; d) Bequette JP, Jungong CS, Novikov AV, Tetrahedron Lett 2009, 50, 6963–6964; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Xu QQ, Zhao Q, Shan GS, Yang XC, Shi QY, Lei X, Tetrahedron 2013, 69, 10739–10746; [Google Scholar]; f) Takao K, Sakamoto S, Touati MA, Kusakawa Y, Tadano K, Molecules 2012, 17, 13330–13344; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Esumi T, Yamamoto C, Fukuyama Y Synlett 2013, 24, 1845–1847; [Google Scholar]; h) Xiong Y, Zhang G, Org. Lett 2016, 18, 5094–5097. [DOI] [PubMed] [Google Scholar]
- [17].a) Binger P, Angew. Chem. Int. Ed 1963, 2, 686; [Google Scholar]; b) Feuvrie C, Blanchet J, Bonin M, Micouin L, Org. Lett 2004, 6, 2333–2336. [DOI] [PubMed] [Google Scholar]
- [18].Dabrowski J, Haeffner F, Hoveyda AH, Angew. Chem. Int. Ed 2013, 52, 7694–7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].For reviews regarding the utility of allenes in chemical synthesis, See:; a) Modern Allene Chemistry (Eds: Krause N, Hashmi ASK), Wiley–VCH, Weinheim, Germany: 2004; [Google Scholar]; b) Brummond KM, DeForrest JE, Synthesis 2007, 6, 795–818; [Google Scholar]; c) López F, Mascareñas JL, Chem. Eur. J 2011, 17, 418–428; [DOI] [PubMed] [Google Scholar]; d) Yu S, Ma S, Angew. Chem. Int. Ed 2012, 51, 3074–3112. [DOI] [PubMed] [Google Scholar]
- [20].Dabrowski J, Gao F, Hoveyda AH, J. Am. Chem. Soc 2011, 133, 4778–4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].For subsequent reports regarding EAS of an aryl moiety, catalyzed by a Cu-based complex, see:; a) Selim KB, Yamada K-i., Tomioka K, Chem. Commun 2008, 5140–5142; [DOI] [PubMed] [Google Scholar]; b) Falciola CA, Alexakis A, Chem. Eur. J 2008, 14, 10615–10627; [DOI] [PubMed] [Google Scholar]; c) Selim KB, Matsumoto Y, Yamada K-I, Tomioka K, Angew. Chem. Int. Ed 2009, 48, 8733–8735; [DOI] [PubMed] [Google Scholar]; d) Polet D, Rathgeb X, Falciola CA, Langlois JB, Hajjiji SE, Alexakis A, Chem. Eur. J 2009, 15, 1205–1206; [DOI] [PubMed] [Google Scholar]; e) Salim KB, Nakanishi H, Matsumoto Y, Yamamoto Y, Yamada K.-i., Tomioka K, J. Org. Chem 2011, 76, 1398–1408; [DOI] [PubMed] [Google Scholar]; f) Shintani R, Takatsu K, Takeda M, Hayashi T, Angew. Chem. Int. Ed 2011, 50, 8656–8659; [DOI] [PubMed] [Google Scholar]; g) Guduguntla S, Hornillos V, Tessier R, Fañanás-Mastral M, Feringa BL, Org. Lett 2016, 18, 252–255; [DOI] [PubMed] [Google Scholar]; h) Goh SS, Guduguntla S, Kikuchi T, Lutz M, Otten E, Fujita M, Feringa BL, J. Am. Chem. Soc 2018, 140, 7052–7055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].For related processes to generate a tertiary carbon stereogenic center, catalyzed by Pd-based complexes, see:; a) Bandini M, Melloni A, Piccinelli F, Sinisi R, Tommasi S, Umanchi-Ronchi A, J. Am. Chem. Soc 2006, 128, 1424–1425; [DOI] [PubMed] [Google Scholar]; b) Cheung HY, Yu W-Y, Lam FL, Au-Yeung T-L, Zhou Z, Chan TH, Chan ASC, Org. Lett 2007, 9, 4295–4298; [DOI] [PubMed] [Google Scholar]; c) Cao Z, Liu Y, Liu Z, Feng X, Zuang M, Du H, Org. Lett 2011, 13, 2164–2167; [DOI] [PubMed] [Google Scholar]; d) Hoshi T, Sasaki K, Sato S, Ishii Y, Suzuki T, Hagiwara H, Org. Lett 2011, 13, 932–935; [DOI] [PubMed] [Google Scholar]; e) Suzuki Y, Nemoto T, Kakugawa K, Hamajima A, Hamada Y, Org. Lett 2012, 14, 2350–2353; [DOI] [PubMed] [Google Scholar]; f) Zhao Z-L, Xu Q-L, Gu Q, Wu X-Y, You S-L, Org. Biomol. Chem 2015, 13, 3086–3092. [DOI] [PubMed] [Google Scholar]
- [23].For reports of EAS of an aryl moiety to generate a tertiary carbon stereogenic center, catalyzed by a Ir-based complex, see:; a) Liu W-B, He H, Dai L-X, You S-L, Org. Lett 2008, 10, 1815–1818; [DOI] [PubMed] [Google Scholar]; b) Huang L, Dai L-X, You S-L, J. Am. Chem. Soc 2016, 138, 5793–5796; [DOI] [PubMed] [Google Scholar]; c) Schafroth MA, Rummelt SM, Sarlah D, Carreira EM, Org. Lett 2017, 19, 3235–3238; [DOI] [PubMed] [Google Scholar]; d) Tan H, Zhang P, Peng F, Yang H, Fu H, Org. Lett 2017, 19, 3775–3778. [DOI] [PubMed] [Google Scholar]
- [24].For related processes that afford a tertiary stereogenic carbon center and are catalyzed by Rh-based complexes, see:; a) Dong L, Xu Y-J, Yuan W-C, Cui X, Cun L-F, Gong L-Z, Eur. J. Org. Chem 2006, 4093–4105; [Google Scholar]; b) Menard F, Chapman TM, Dockendorff C, Lautens M, Org. Lett 2006, 8, 4569–4572; [DOI] [PubMed] [Google Scholar]; c) Kiuchi H, Takahashi D, Funaki K, Sato T, Oi S, Org. Lett 2012, 14, 4502–4505; [DOI] [PubMed] [Google Scholar]; d) Sidera M, Fletcher SP, Nat. Chem 2015, 7, 935–939. [DOI] [PubMed] [Google Scholar]; For related processes to generate a tertiary carbon stereogenic center, catalyzed by Ni-based complexes, see:; e) Gomez-Bengoa E, Heron NM, Didiuk MT, Luchaco CA, Hoveyda AH, J. Am. Chem. Soc 1998, 120, 7649–7650; [Google Scholar]; f) Chung KG, Miyake Y, Uemura S, J. Chem. Soc. Perkin Trans 1 2000, 2725–2729. [Google Scholar]
- [25].Ohmiya H, Yokokawa N, Sawamura M, Org. Lett 2010, 12, 2438–2440. [DOI] [PubMed] [Google Scholar]
- [26].Whittaker AM, Rucker RP, Lalic G, Org. Lett 2010, 12, 3216–3218. [DOI] [PubMed] [Google Scholar]
- [27].Gao F, Lee Y, Mandai K, Hoveyda AH, Angew. Chem. Int. Ed 2010, 49, 8370–8374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].For example, see:; Zhang S, del Pozo J, Romiti F, Mu Y, Torker S, Hoveyda AH, Science 2019, 364, 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].For selected reviews, see:; a) ref. [24].; For catalytic enantioselective cycloadditions involving allenes, see:; b) Wurz RP, Fu GC, J. Am. Chem. Soc 2005, 127, 12234–12235; [DOI] [PubMed] [Google Scholar]; c) Luzungm MR, Mauleón P, Toste FD, J. Am. Chem. Soc 2007, 129, 12402–12403; [DOI] [PubMed] [Google Scholar]; d) Han X, Yao W, Wang T, Tan YR, Yan Z, Kwiatkowski J, Lu Y, Angew. Chem. Int. Ed 2014, 53, 5643–5647. [DOI] [PubMed] [Google Scholar]; For reactions with allenoates, see:; e) Elsner P, Bernardi L, Salla GD, Overgaard J, Jørgensen KA, J. Am. Chem. Soc 2008, 130, 4897–4905. [DOI] [PubMed] [Google Scholar]; For catalytic enantioselective cyclopropane synthesis methods, see:; f) Adams CS, Weatherly CD, Burke EG, Schomaker JM, Chem. Soc. Rev 2014, 43, 3136–3163. [DOI] [PubMed] [Google Scholar]; For catalytic enantioselective modification of allenes, see:; g) Pelz NF, Woodward AR, Burks HE, Sieber JD, Morken JP, J. Am. Chem. Soc 2004, 126, 16328–16329. [DOI] [PubMed] [Google Scholar]; For catalytic enantioselective allylic substitutions, see:; h) Petrone DA, Isomura M, Franzoni I, Rössler SL, Carreira EM, J. Am. Chem. Soc 2018, 140, 4697–4704. [DOI] [PubMed] [Google Scholar]; For dynamic kinetic enantioselective synthesis with allenes, see:; i) Trost BM, Fandrick DR, Dinh DC, J. Am. Chem. Soc 2005, 127, 14186–14187. [DOI] [PubMed] [Google Scholar]; For catalytic enantioselective allenyl additions, see:; j) Wu H, Haeffner F, Hoveyda AH, J. Am. Chem. Soc 2014, 136, 3780–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]; For catalytic enantioselective reaction with allenesand involving a metal-hydride catalyst, see:; k) Osborne JD, Randell-Sly HE, Currie GS, Cowley AR, Willis MC, J. Am. Chem. Soc 2008, 130, 17232–17233; [DOI] [PubMed] [Google Scholar]; l) Han SB, Kim IS, Han H, Krische MJ, J. Am. Chem. Soc 2009, 131, 6916–6917; [DOI] [PMC free article] [PubMed] [Google Scholar]; o) Koschker P, Breit B, Acc. Chem. Res 2016, 49, 1524–1536; [DOI] [PubMed] [Google Scholar]; p) Tsai EY, Liu RY, Yang Y, Buchwald SL, J. Am. Chem. Soc 2018, 140, 2007–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]; For catalytic enantioselective reactions of allenes that proceed via a Cu–boryl species, see:; q) Huang Y, Torker S, Li X, del Pozo J, Hoveyda AH, Angew. Chem. Int. Ed 2019, 58, 2685–2691; [DOI] [PMC free article] [PubMed] [Google Scholar]; r) Zhang S, del Pozo J, Romiti F, Mu Y, Torker S, Hoveyda AH, Science 2019, 364, 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jung B, Hoveyda AH, J. Am. Chem. Soc 2012, 134, 1490–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].(a) Yuan W, Ma S, Adv. Synth. Catal 2012, 354, 1867–1872; [Google Scholar]; b) Meng F; Jung B; Haeffner F; Hoveyda AH Org. Lett 2013, 15, 1414–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Petrone DA, Isomura M, Franzoni I, Rössler SL, Carreira EM, J. Am. Chem. Soc 2018, 140, 4697–4704 [DOI] [PubMed] [Google Scholar]
- [33].Meng F, Li X, Torker S, Shi Y, Shen X, Hoveyda AH, Nature 2016, 537, 387–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhou Y, Shi Y, Torker S, Hoveyda AH, J. Am. Chem. Soc 2018, 140, 16842–16854. [DOI] [PubMed] [Google Scholar]
- [35].For representative catalytic methods for preparation of E-1,2-disubstituted-alkenyl–B(pin) compounds, see:; a) Tucker CE, Davidson J, Knochel P, J. Org. Chem 1992, 57, 3482–3485; [Google Scholar]; b) Takagi J, Takahashi K, Ishiyama T, Miyaura N, J. Am. Chem. Soc 2002, 124, 8001–8006; [DOI] [PubMed] [Google Scholar]; c) Morrill C, Grubbs RH, J. Org. Chem 2003, 68, 6031–6034; [DOI] [PubMed] [Google Scholar]; d) Jang H, Zhugralin AR, Lee Y, Hoveyda AH, J. Am. Chem. Soc 2011, 133, 7859–7871; [DOI] [PubMed] [Google Scholar]; e) Coombs JR, Zhang L, Morken JP, Org. Lett 2015, 17, 1708–1711; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Reid WB, Spillane JJ, Krause SB, Watson DA, J. Am. Chem. Soc 2016, 138, 5539–5542; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Murray SA, Luc ECM, Meek SJ, Org. Lett 2018, 20, 469–472; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Green JC, Zanghi JM, Meek SJ, J. Am. Chem. Soc 2020, 142, 1704–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a review, see:; i) Carreras J, Caballero A, Pérez PJ, Chem. Asian J 2019, 14, 329–343. [DOI] [PubMed] [Google Scholar]
- [36].For representative methods regarding preparation of Z-1,2-disubstituted-alkenyl–B(pin) compounds, see:; a) Roush WR, Ando K, Powers DB, Palkowitz AD, Halterman RL, J. Am. Chem. Soc 1990, 112, 6339–6348; [Google Scholar]; b) Ramachandran PV, Pratihar D, Biswas D, Chem. Commun 2005, 1988–1989; [DOI] [PubMed] [Google Scholar]; c) Molander GA; Ellis NM J. Org. Chem 2008, 73, 6841–6844. [DOI] [PMC free article] [PubMed] [Google Scholar]; For corresponding catalytic strategies, see:; d) Ohmura T, Yamamoto Y, Miyaura N, J. Am. Chem. Soc 2000, 122, 4990–4991; [Google Scholar]; e) Gunanathan C, Hölscher M, Pan F, Leitner W, W. J. Am. Chem. Soc 2012, 134, 14349–14352; [DOI] [PubMed] [Google Scholar]; f) Obligacion JV, Neely JM, Yazdani AN, Pappas I, Chirik PJ, J. Am. Chem. Soc 2015, 137, 5855–5858; [DOI] [PubMed] [Google Scholar]; g) Gorgas N, Alves LG, Stöger B, Martins AM, Veiros LF, Kirchner K, J. Am. Chem. Soc 2017, 139, 8130–8133. [DOI] [PubMed] [Google Scholar]
- [37].Hamilton JY, Sarlah D, Carreira EM, J. Am. Chem. Soc 2013, 135, 994–997. [DOI] [PubMed] [Google Scholar]
- [38].Gao F, Carr JL, Hoveyda AH, J. Am. Chem. Soc 2014, 136, 2149–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].a) Kieswetter ET, O’Brien RV, Yu EC, Meek SJ, Schrock RR, Hoveyda AH, J. Am. Chem. Soc 2013, 135, 6026–6029; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mu Y, Nguyen TT, van der Mei FW, Schrock RR, Hoveyda AH, Angew. Chem. Int. Ed 2019, 58, 5365–5370. [DOI] [PMC free article] [PubMed] [Google Scholar]; For an overview regarding merging of olefin metathesis with other catalytic processes, see:; c) Hoveyda AH, J. Org. Chem 2014, 79, 4763–4792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gao F, Carr JL, Hoveyda AH, Angew. Chem. Int. Ed 2012, 51, 6613–6617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].a) Schäfer P, Palacin T, Sidera M, Fletcher SP, Nat. Commun 2017, 8, 15762. [DOI] [PMC free article] [PubMed] [Google Scholar]; For related earlier work, see:; b) Sidera M, Fletcher SP, Nat. Chem 2015, 7, 935–939. [DOI] [PubMed] [Google Scholar]; For a related transformation, see:; Yap C, Lenagh-Snow GMJ, Karad SN, Lewis W, Diorazio LJ, Lam HW, Angew. Chem. Int. Ed 2017, 56, 8216–8220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Shi Y, Jung B, Torker S, Hoveyda AH, J. Am. Chem. Soc 2015, 137, 8948–8964. [DOI] [PubMed] [Google Scholar]
- [43].A trialkylboron compound, generated in situ by reaction of a monosubstituted olefin and 9–BBN, can be used in bisphosphine–Cu-catalyzed EAS with Z-allylic chlorides. See:; Shido Y, Yoshida M, Tanabe M, Ohmiya H, Sawamura M, J. Am. Chem. Soc 2012, 134, 18573–18576. [DOI] [PubMed] [Google Scholar]
- [44].Matteson DS, Moody RJ, Organometallics 1982, 1, 20–28. [Google Scholar]
- [45].a) Endo K, Ohkubo T, Hirokami M, Shibata T, J. Am. Chem. Soc 2010, 132, 11033–11035; [DOI] [PubMed] [Google Scholar]; b) Endo K, Ohkubo T, Shibata T, Org. Lett 2011, 13, 3368–3371; [DOI] [PubMed] [Google Scholar]; c) Endo K, Ohkubo T, Ishioka T, Shibata T, J. Org. Chem 2012, 77, 4826–4831; [DOI] [PubMed] [Google Scholar]; d) Endo K, Ishioka T, Ohkubo T, Shibata T, J. Org. Chem 2012, 77, 7223–7231; [DOI] [PubMed] [Google Scholar]; e) Zhang Z-Q, Yang C-T, Liang L-J, Xiao B, Lu X, Liu J-H, San Y-Y, Marder TB, Fu Y, Org. Lett 2014, 16, 6342–6345; [DOI] [PubMed] [Google Scholar]; f) Endo K, Ishioka T, Shibata T, Synlett 2014, 25, 2184–2188; [Google Scholar]; g) Sun C, Potter B, Morken JP, J. Am. Chem. Soc 2014, 136, 6534–6537; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Potter B, Szymaniak AA, Edelstein EK, Morken JP, J. Am. Chem. Soc 2014, 136, 17918–17921; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Joannou MV, Moyer BS, Meek SJ, J. Am. Chem. Soc 2015, 137, 6176–6179; [DOI] [PubMed] [Google Scholar]; j) Joannou MV, Moyer BS, Goldfogel MJ, Meek SJ, Angew. Chem. Int. Ed 2015, 54, 14141–14145; [DOI] [PubMed] [Google Scholar]; k) Kim J, Park S, Park J, Cho SH, Angew. Chem. Int. Ed 2016, 55, 1498–1501; [DOI] [PubMed] [Google Scholar]; l) Zhan M, Li RZ, Mou ZD, Cao CG, Liu J, Chen YW, Niu D, ACS Catal 2016, 6, 3381–3386; [Google Scholar]; m) Murray SA, Green JC, Tailor SB, Meek SJ, Angew. Chem. Int. Ed 2016, 55, 9065–9069; [DOI] [PMC free article] [PubMed] [Google Scholar]; n) Kim J, Ko K, Cho SH, Angew. Chem. Int. Ed 2017, 56, 11584–11588; [DOI] [PubMed] [Google Scholar]; o) Murray S, Liang MZ, Meek SJ, J. Am. Chem. Soc 2017, 139, 14061–14064. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a recent summary overview, see:; p) Nallagonda R, Padala K, Masarwa A, Org. Biomol. Chem, 2018, 16, 1050–1064. [DOI] [PubMed] [Google Scholar]
- [46].Shi Y, Hoveyda AH, Angew. Chem. Int. Ed 2016, 55, 3455–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].a) Meng F, Haeffner F, Hoveyda AH, J. Am. Chem. Soc 2014, 136, 11304–11307; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Blaisdell TP, Morken JP J. Am. Chem. Soc 2015, 137, 8712–8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].McGrath KP, Hoveyda AH; , previously unpublished results. See the Supporting Information for details.
- [49].Zhou Y, Huang Z, Shi Y, Hoveyda AH; , previously unpublished results. See the Supporting Information for details.
- [50].Wang M, Pu X, Zhao Y, Wang P, Li Z, Zhu C, Shi Z, J. Am. Chem. Soc 2018, 140, 9061–9065. [DOI] [PubMed] [Google Scholar]
- [51].For a relevant stereochemical model, see:; Paioti PHS, del Pozo J, Mikus MS, Lee J, Koh MJ, Romiti F, Torker S, Hoveyda AH, J. Am. Chem. Soc 2019, 141, 19917–19934. [DOI] [PubMed] [Google Scholar]
- [52].a) Meng F, Jang H, Jung B, Hoveyda AH, Angew. Chem. Int. Ed 2013, 52, 5046–5051; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Matsuda N, Hirano K, Satoh T, Miura M, J. Am. Chem. Soc 2013, 135, 4934–4937; [DOI] [PubMed] [Google Scholar]; c) Ref. [130b].
- [53].For example, see:; a) Ref. [130b];; b) Miki Y, Hirano K, Satoh T, Miura M, Angew. Chem. Int. Ed 2013, 52, 10830–10834; [DOI] [PubMed] [Google Scholar]; c) Shi S-L, Buchwald SL, Nat. Chem 2015, 7, 38–44; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Sakae R, Hirano K, Satoh T, Miura M, Angew. Chem., Int. Ed 2015, 54, 613–617; [DOI] [PubMed] [Google Scholar]; e) Pirnot MT, Wang Y-M, Buchwald SL, Angew. Chem., Int. Ed 2016, 55, 48–57; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Wang H, Yang JC, Buchwald SL, J. Am. Chem. Soc 2017, 139, 8428–8431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Han JT, Jang WJ, Kim N, Yun J, J. Am. Chem. Soc 2016, 138, 15146–15149. [DOI] [PubMed] [Google Scholar]
- [55].Lee J, Torker S, Hoveyda AH, Angew. Chem. Int. Ed 2017, 56, 821–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Xu G, Zhao H, Fu B, Cang A, Zhang G, Zhang Q, Xiong T, Zhang Q, Angew. Chem. Int. Ed 2017, 56, 13130–13134. [DOI] [PubMed] [Google Scholar]
- [57].Rivera-Chao E, Mitxelena M, Varela JA, Fañanás-Mastral M, Angew. Chem. Int. Ed 2019, 58, 18230–18234. [DOI] [PubMed] [Google Scholar]
- [58].Xu G, Fu B, Zhao H, Li Y, Zhang G, Wang Y, Xiong T, Zhang Q, Chem. Sci, 2019, 10, 1802–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Sun Y, Zhou Y, Shi Y, del Pozo J, Torker S, Hoveyda AH, J. Am. Chem. Soc 2019, 141, 12087–12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lee Y, Li B, Hoveyda AH, J. Am. Chem. Soc 2009, 131, 11625–11633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Previously proposed mechanistic models involve two distinct scenarios: 1) Leaving group association with Cu (mainly in cases where soft N, S and P donors are present); see:; a) Breit B, Schmidt Y, Chem. Rev 2008, 108, 2928–2951; [DOI] [PubMed] [Google Scholar]; b) Nagao K, Yokobori U, Makida Y, Ohmiya H, Sawamura M, J. Am. Chem. Soc 2012, 134, 8982–8987. [DOI] [PubMed] [Google Scholar]; 2) Control through electronic factors; see:; c) Yoshikai N, Zhang S-L, Nakamura E, J. Am. Chem. Soc 2008, 130, 12862–12863. [DOI] [PubMed] [Google Scholar]
- [62].Mori S, Hirai A, Nakamura M, Nakamura E, Tetrahedron 2000, 56, 2805–2809. [Google Scholar]
- [63].A related stereochemical model, entailing assisted leaving group displacement by a bifunctional phosphine/amino alcohol ligand has been proposed by Nakamura. See:; Yoshikai N, Miura K, Nakamura E, Adv. Synth. Catal 2009, 351, 1014–1018. [Google Scholar]
- [64].a) Wiberg KB, Chem. Rev 1955, 55, 713–743; [Google Scholar]; b) Westheimer FH, Chem. Rev 1961, 61, 265–273; [Google Scholar]; c) Simmons EM, Hartwig JF, Angew. Chem. Int. Ed 2012, 51, 3066–3072. [DOI] [PubMed] [Google Scholar]
- [65].Wagner JP, Schreiner PR, Angew. Chem. Int. Ed 2015, 54, 12274–12296. [DOI] [PubMed] [Google Scholar]
- [66]. For a stereochemical model regarding the related reaction involving a Na+-carboxylic ester interaction (cf. Scheme 22a), see the Supporting Information.
- [67]. For stereochemical models in connection with EAS involving alkynyl-Al compounds (Scheme 8), see the Supporting Information.
- [68].For recent reviews on catalytic ECA with “hard” alkyl- or arylmetal-based reagents, see:; a) Alexakis A, Krause N, Woodward S in Copper-Catalyzed Asymmetric Synthesis (Alexakis A, Krause N, Woodward S, Eds); VCH-Wiley, 2014, pp. 33–68; [Google Scholar]; b) López F, Minnaard AJ, Feringa BL, Acc. Chem. Res 2007, 40, 179–188; [DOI] [PubMed] [Google Scholar]; c) Ref. [6c];; d) Harutyunyan SR, den Hartog T, Geurts K, Minnaard AJ, Feringa BL, Chem. Rev 2008, 108, 2824–2852; [DOI] [PubMed] [Google Scholar]; e) Jerphagnon T, Pizzuti MG, Minnaard AJ, Feringa BL, Chem. Soc. Rev 2009, 38, 1039–1075; [DOI] [PubMed] [Google Scholar]; f) Hawner C, Alexakis A, Chem. Commun 2010, 46, 7295–7306; [DOI] [PubMed] [Google Scholar]; g) Hargrave JD, Allen JC, Frost CG, Chem. Asian J 2010, 5, 386–396. [DOI] [PubMed] [Google Scholar]
- [69].a) d’Augustin M, Palais L, Alexakis A, Angew. Chem. Int. Ed 2005, 44, 1376–1378. [DOI] [PubMed] [Google Scholar]; For an application of this catalytic approach, see:; b) Mendoza A, Ishihara Y, Baran PS, Nat. Chem 2012, 4, 21–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].a) Wu J, Mampreian DM, Hoveyda AH, J. Am. Chem. Soc 2005, 127, 4584–4585. [DOI] [PubMed] [Google Scholar]; For an application of this method, see:; b) Zeng X, Gao JJ, Song JJ, Ma S, Desrosiers J-N, Mulder JA, Rodriguez S, Herbage MA, Haddad N, Qu B, Fandrick KR, Grinberg N, Lee H, Wei X, Yee NK, Senanayake CH, Angew. Chem. Int. Ed 2014, 53, 12153–12175. [DOI] [PubMed] [Google Scholar]
- [71].Hird AW, Hoveyda AH, J. Am. Chem. Soc 2005, 127, 14988–14989. [DOI] [PubMed] [Google Scholar]
- [72].Fillion E, Wilsily A, J. Am. Chem. Soc 2006, 128, 2774–2775. [DOI] [PubMed] [Google Scholar]
- [73].Martin D, Kehrli S, d’Augustin M, Clavier H, Mauduit M, Alexakis A, J. Am. Chem. Soc 2006, 128, 8416–8417. [DOI] [PubMed] [Google Scholar]
- [74].Peese KM, Gin DY, Chem. Eur. J 2008, 14, 1654–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Degrado SJ, Mizutani H, Hoveyda AH, J. Am. Chem. Soc 2001, 123, 755–756 and references therein. [DOI] [PubMed] [Google Scholar]
- [76].May TL, Brown MK, Hoveyda AH, Angew. Chem. Int. Ed 2008, 47, 7358–7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Brown MK, Hoveyda AH, J. Am. Chem. Soc 2008, 130, 12904–12906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].a) Hu P, Chi HM, DeBacker KC, Gong X, Keim JH, Hsu IT, Snyder SA, Nature 2019, 569, 703–707; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) peng C, Arya P, Zhou Z, Snyder SA, Angew. Chem. Int. Ed DOI: 10.1002/anie.202004177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Dabrowski JA, Villaume MT, Hoveyda AH, Angew. Chem. Int. Ed 2013, 52, 8156–8159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Liskin DV, Valente EJ, Mol J. Structure 2008, 878, 149–154. [Google Scholar]
- [81].Endo K, Hamada D, Yakeishi S, Shibata T, Angew. Chem. Int. Ed 2013, 52, 606–610. [DOI] [PubMed] [Google Scholar]
- [82].Gao Z, Fletcher SP, Chem. Sci, 2017, 8, 641–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].a) Ardkhean R, Mortimore M, Paton RS, Fletcher SP, Chem. Sci 2018, 9, 2628–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a related example, see:; b) Sidera M, Roth PMC, Maksymowicz RM, Fletcher SP, Angew. Chem. Int. Ed 2013, 52, 7995–7999. [DOI] [PubMed] [Google Scholar]
- [84].Takaya Y, Ogasawara M, Hayashi T, Tetrahedron Lett 1998, 39, 8479–8482. [Google Scholar]
- [85].For representative studies of ECA of aryl- and alkenylboron compounds to cyclic enones, promoted by Rh-based catalysts, see:; a) Nakao Y, Chen J, Imanaka H, Hiyama T, Ichikawa Y, Duan W-L, Shintani R, Hayashi T, J. Am. Chem. Soc 2007, 129, 9137–9143; [DOI] [PubMed] [Google Scholar]; b) Lalic G, Corey EJ, Tetrahedron Lett 2008, 49, 4894–4896; [Google Scholar]; c) Shintani R, Ichikawa Y, Takatsu K, Chen F-X, Hayashi T, J. Org. Chem 2009, 74, 869–873; [DOI] [PubMed] [Google Scholar]; d) Westmeier J, Pfaff C, Siewert J, von Zezschwitz P, Adv. Synth. Catal 2013, 355, 2651–2658. [Google Scholar]; For related review articles, see:; e) Berthon G, Hayashi T in Catalytic Asymmetric Synthesis (Ed.: Córdova A), Wiley–VCH, Weinheim, 2010, pp. 1–70; [Google Scholar]; f) Edwards HJ, Hargrave JD, Penrose SD, Frost CG, Chem. Soc. Rev 2010, 39, 2093–2105; [DOI] [PubMed] [Google Scholar]; g) Tian P, Dong H-Q, Lin G-Q, ACS Catal 2012, 2, 95–119; [Google Scholar]; h) Lam H in Organic Reactions, vol. 93 (Ed.: Denmark SE), John Wiley & Sons, New York, 2017, pp. 1–686. [Google Scholar]
- [86].Shintani R, Ichikawa Y, Takutsu K, Chen F-X, Hayashi T, J. Org. Chem 2009, 74, 869–873. [DOI] [PubMed] [Google Scholar]
- [87].Oi S, Sato T, Inoue Y, Tetrahedron Lett 2004, 45, 5051–5055. [Google Scholar]
- [88].Otomaru Y, Hayashi T, Tetrahedron: Asymm 2004, 15, 2647–2651. [Google Scholar]
- [89].a) Shintani R, Ichikawa Y, Hayashi T, Chen J, Nakao Y, Hiyama T, Org. Lett 2007, 9, 4643–4645. [DOI] [PubMed] [Google Scholar]; For related studies, see:; b) Oi S, Taira A, Inoue Y, Org. Lett 2003, 5, 97–99; [DOI] [PubMed] [Google Scholar]; c) Thaler T, Guo L-N, Steib AK, Raducan M, Karaghiosoff K, Mayer P, Knochel P, Org. Lett 2011, 13, 3182–3185. [DOI] [PubMed] [Google Scholar]
- [90].For a relevant review article, see:; Nguyen TN, May JA, Tetrahedron Lett 2017, 58, 1535–1544. [Google Scholar]
- [91].Wu TR, Chong JM, J. Am. Chem. Soc 2007, 129, 4908–4909. [DOI] [PubMed] [Google Scholar]
- [92].Lee S, MacMillan DWC, J. Am. Chem. Soc 2007, 129, 15438–15439. [DOI] [PubMed] [Google Scholar]
- [93].Inokuma T, takasu K, Sakaeda T, Takemoto Y, Org. Lett 2009, 129, 2425–2428. [DOI] [PubMed] [Google Scholar]
- [94].Sugiura M, Tokudomi M, Nakajima M, Chem. Commun 2010, 46, 7799–7800. [DOI] [PubMed] [Google Scholar]
- [95].a) Lundy BJ, Jansone-Popova S, May JA, Org. Lett 2011, 13, 4958–4961; [DOI] [PubMed] [Google Scholar]; b) Le PQ, Nguyen TS, May JA, Org. Lett 2012, 14, 6104–6107. [DOI] [PubMed] [Google Scholar]
- [96].Akagawa K, Sugiyama M, Kudo K, Org. Biomol. Chem, 2012, 10, 4839–4843. [DOI] [PubMed] [Google Scholar]
- [97].Alexakis A, Albrow V, Biswas K, d’Augustin M, Prieto O, Woodward S, Chem. Commun 2005, 2843–2845. [DOI] [PubMed] [Google Scholar]
- [98].a) Robert T, Velder J, Schmalz H-G, Angew. Chem. Int. Ed 2008, 47, 7718–7721; [DOI] [PubMed] [Google Scholar]; b) Naeemi Q, Robert T, Krantz DP, Velder J, Schmalz H-G, Tetrahedron: Asymmetry 2011, 22, 887–892. [Google Scholar]
- [99].Lee K.-s., Hoveyda AH, J. Org. Chem 2009, 74, 4455–4462. [DOI] [PubMed] [Google Scholar]
- [100].Müller D, Alexakis A, Org. Lett 2012, 14, 1842–1845. [DOI] [PubMed] [Google Scholar]
- [101].Cottet P, Müller D, Alexakis A, Org. Lett 2013, 15, 828–831. [DOI] [PubMed] [Google Scholar]
- [102].Wilcox D, Woodward S, Alexakis A, Chem. Commun 2014, 50, 1655–1657. [DOI] [PubMed] [Google Scholar]
- [103].Takatsu K, Shintani R, Hayashi T, Angew. Chem. Int. Ed 2011, 50, 5548–5552. [DOI] [PubMed] [Google Scholar]
- [104].a) Chong Q, Yue Z, Zhang S, Ji C, Cheng F, Zhang H, Hong X, Meng F, ACS Catal 2017, 7, 5693–5698; [Google Scholar]; b) Chong Q, Zhang S, Cheng F, Wang J, Hong X, Meng F, Org. Lett 2018, 20, 6896–6900. [DOI] [PubMed] [Google Scholar]
- [105].McGrath KP, Hubbell AK, Zhou Y, Santos DP, Torker S, Romiti F, Hoveyda AH, Adv. Synth. Catal 2020, 362, 370–375. [Google Scholar]
- [106].Mauleón P, Carretero JC, Chem. Commun 2005, 4961–4963. [DOI] [PubMed] [Google Scholar]
- [107].Vuagnoux-d’Augustin M, Alexakis A, Chem. Eur. J 2007, 13, 9647–9662. [DOI] [PubMed] [Google Scholar]
- [108].Müller D, Hawner C, Tissot M, Palais L, Alexakis A, Synlett 2010, 1694–1698. [Google Scholar]
- [109].Ji J-X, Chan ASC in Catalytic Asymmetric Synthesis (Ojima I, Ed.); Wiley, 2010, 439–495. [Google Scholar]
- [110].May TL, McGrath KP, Hoveyda AH; , previously unpublished results. See the Supporting Information for experimental and analytic details.
- [111].a) Müller DS, Untiedt NL, Dieskau AP, Lackner GL, Overman LE, J. Am. Chem. Soc 2015, 137, 660–663; [DOI] [PubMed] [Google Scholar]; b) Slutskyy Y, Jamison CR, Lackner GL, Müller DS, Dieskau AP, Untiedt NL, Overman LE, J. Org. Chem 2016, 81, 7029–7035. [DOI] [PubMed] [Google Scholar]
- [112].May TL, Hoveyda AH; , previously unpublished results. See the Supporting Information for experimental and analytic details.
- [113].a) May TL, Dabrowski JA, Hoveyda AH, J. Am. Chem. Soc 2011, 133, 736–739; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) May TL, Dabrowski JA, Hoveyda AH, J. Am. Chem. Soc 2014, 136, 10544. [Google Scholar]
- [114].Kumar SK, Amador M, Hidalgo M, Bhat SV, Khan SR, Bioorg. Med. Chem 2005, 13, 2873–2880. [DOI] [PubMed] [Google Scholar]
- [115].McGrath KP, Hoveyda AH, Angew. Chem. Int. Ed 2014, 53, 1910–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Yoshida M, Shoji Y, Shishido K, Org. Lett 2009, 11, 1441–1443. [DOI] [PubMed] [Google Scholar]
- [117].a) Müller D, Tissot M, Alexakis A, Org. Lett 2011, 13, 3040–3043. [DOI] [PubMed] [Google Scholar]; For a similar study, see:; b) Wilcox D, Woodward S, Alexakis A, Chem. Commun 2014, 50, 1655–1657. [DOI] [PubMed] [Google Scholar]
- [118].Shintani R, Duan W-L, Hayashi T, J. Am. Chem. Soc 2006, 128, 5628–5629. [DOI] [PubMed] [Google Scholar]
- [119].Lee K.-s., Brown MK, Hird AW, Hoveyda AH, J. Am. Chem. Soc 2006, 128, 7182–7184. [DOI] [PubMed] [Google Scholar]
- [120].Hawner C, Li K, Cirriez V, Alexakis A, Angew. Chem. Int. Ed 2008, 47, 8211–8214. [DOI] [PubMed] [Google Scholar]
- [121].a) Shintani R, Tsutsumi Y, Nagaosa M, Nishimura T, Hayashi T, J. Am. Chem. Soc 2009, 131, 13588–13589; [DOI] [PubMed] [Google Scholar]; b) Shintani R, Takeda M, Nishimura T, Hayashi T, Angew. Chem. Int. Ed 2010, 49, 3969–3971; [DOI] [PubMed] [Google Scholar]; c) Hawner C, Müller D, Gremaud L, Felouat A, Woodward S, Alexakis A, Angew. Chem. Int. Ed 2010, 49, 7769–7772; [DOI] [PubMed] [Google Scholar]; d) Kikushima K, Holder JC, Gatti M, Stoltz BM, J. Am. Chem. Soc 2011, 133, 6902–6905; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Holder JC, Zhou L, Marziale AN, Liu P, Lan Y, Gatti M, Kikushima K, Houk KN, Stoltz BM, J. Am. Chem. Soc 2013, 135, 14906–15007; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Gottumukkala AL, Matcha K, Lutz M, de Vries JG, Minnaard AJ, Chem. Eur. J 2012, 18, 6907–6914. [DOI] [PubMed] [Google Scholar]; For applications to enantioselective synthesis of natural products, see:; g) Buter J, Moezelaar R, Minnaard AJ, Org. Biomol. Chem 2014, 12, 5883–5890. [DOI] [PubMed] [Google Scholar]
- [122].a) Shintani R, Hayashi T, Org. Lett 2011, 13, 350–352; [DOI] [PubMed] [Google Scholar]; b) Shockley SE, Holder JC, Stoltz BM, Org. Process Res. Dev 2015, 19, 974–981; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Holder JC, Goodman ED, Kikushima K, Gatti M, Marziale AN, Stoltz BM, Tetrahedron 2015, 71, 5781–5792. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a related recent (2017) advance, see:; d) Kadam AA, Ellern A, Stanley LM, Org. Lett 2017, 19, 4062–4065. [DOI] [PubMed] [Google Scholar]
- [123].del Pozo J, Perez-Iglesias M, Alvarez R, Lledos A, Casares JA, Espinet P, ACS Catal 2017, 7, 3575–3583. [Google Scholar]
- [124].Hoveyda AH, Koh MJ, Lee K, Lee J in Organic Reactions (Denmark SE); VCH–Wiley, 2020, pp. 959–1055. [Google Scholar]
- [125].Lee Y, Hoveyda AH, J. Am. Chem. Soc 2009, 131, 3160–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].a) Laitar DS, Müller P, Sadighi JP, J. Am. Chem. Soc 2005, 127, 17196–17197; [DOI] [PubMed] [Google Scholar]; b) Laitar DS, Tsui EY, Sadighi JP, Organometallics 2006, 25, 2405–2408. [Google Scholar]
- [127].Mun S, Yun J-E, Org. Lett 2006, 8, 4887–4889. [DOI] [PubMed] [Google Scholar]
- [128].a) Mannig D, Nöth H, Angew. Chem. Int. Ed. Engl 1985, 24, 878–879. [Google Scholar]; For related reviews, see:; b) Crudden CM, Edwards D, Eur. J. Org. Chem 2003, 4695–4712; [Google Scholar]; c) Carroll A-M, O’Sullivan TP, Guiry PJ, Adv. Synth. Catal 2005, 347, 609–631; [Google Scholar]; d) Chen J, Lu Z, Org. Chem. Front 2018, 5, 260–272; [Google Scholar]; e) Collins BSL, Wilson CM, Myers EL, Aggarwal VK, Angew. Chem. Int. Ed 2017, 56, 11700–11733. [DOI] [PubMed] [Google Scholar]
- [129].a) Noh D, Chea H, Ju J, Yun J, Angew. Chem. Int. Ed 2009, 48, 6062–6064; [DOI] [PubMed] [Google Scholar]; b) Noh D, Yoon SK, Won J, Lee JY, Yun J, Chemistry, Asian J 2011, 6, 1967–1969. [DOI] [PubMed] [Google Scholar]
- [130].Valk JM, Whitlock GA, Layzell TP, Brown JM, Tetrahedron: Asymmetry 1995, 6, 2593–2596; [Google Scholar]; b) Doucet H, Fernandez E, Layzell TP, Brown JM, Chem. Eur. J 1999, 5, 1320–1330. [Google Scholar]
- [131].McCarthy M, Hooper MW, Guiry PJ, Chem. Commun 2000, 1333–1334. [Google Scholar]
- [132].a) Smith SM, Thacker NC, Takacs JM, J. Am. Chem. Soc 2008, 130, 3734–3735; [DOI] [PubMed] [Google Scholar]; b) Yang Z-D, Pal R, Hoang GL, Zeng XC, Takacs JM, ACS Catal 2014, 4, 763–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].For example, see:; a) Matsuda N, Hirano K, Satoh T, Miura M, J. Am. Chem. Soc 2013, 135, 4934–4937; [DOI] [PubMed] [Google Scholar]; b) Zhu S, Niljianskul N, Buchwald SL, J. Am. Chem. Soc 2013, 135, 15746–15749; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Jia T, Cao P, Wang B, Lou Y, Yin X, Wang M, Liao J, J. Am. Chem. Soc 2015, 137, 13760–13763; [DOI] [PubMed] [Google Scholar]; d) Logan KM, Brown MK, Angew. Chem. Int. Ed 2017, 56, 851–855; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Lee J, Radomkit S, Torker S, del Pozo J, Hoveyda AH, Nat. Chem 2018, 10, 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].a) Tian B, Liu Q, Tong X, Tian P, Lin G-Q, Org. Chem. Front 2014, 1, 1116–1122; [Google Scholar]; b) Parra A, Amenós L, Guisán-Ceinos M, López A, García Ruano JL, Tortosa M, J. Am. Chem. Soc 2014, 136, 15833–15836; [DOI] [PubMed] [Google Scholar]; c) Guisán-Ceinos M, Parra A, Martín-Heras V, Tortosa M, Angew. Chem. Int. Ed 2016, 55, 6969–6972. [DOI] [PubMed] [Google Scholar]
- [135].Xi Y, Hartwig JF, J. Am. Chem. Soc 2016, 138, 6703–6706. [DOI] [PubMed] [Google Scholar]
- [136].Hoveyda AH, Evans DA, Fu GC, Chem. Rev 1993, 93, 1307–1370. [Google Scholar]
- [137].a) Huang Y, del Pozo J, Torker S, Hoveyda AH, J. Am. Chem. Soc 2018, 140, 2643–2655; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gao D-W, Xiao Y, Liu Z, Karunananda MK, Chen JS, Engle KM, ACS Catal 2018, 8, 3650–3654; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sang HL, Yu S, Ge S, Org. Chem. Front 2018, 5, 1284–1287. [Google Scholar]
- [138].Thomas SP, Aggarwal VK, Angew. Chem. Int. Ed 2009, 48, 1896–1898. [DOI] [PubMed] [Google Scholar]
- [139].a) Mazet C, Gérard D, Chem. Commun 2011, 47, 298–300. [DOI] [PubMed] [Google Scholar]; For a related study, see:; b) Magre M, Biosca M, Pámies O, Diéguez M, ChemCatChem 2015, 7, 114–120. [Google Scholar]
- [140].Corberán R, Mszar NW, Hoveyda AH, Angew. Chem. Int. Ed 2011, 50, 7079–7082. [DOI] [PubMed] [Google Scholar]
- [141].a) Smith S, Hoang GL, Pal R, Bani Khaled MO, Pelter LSW, Zeng XC, Takacs JM, Chem. Commun 2012, 48, 12180–12182; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Shoba VM, Thacker NC, Bochat AJ, Takacs JM, Angew. Chem. Int. Ed 2016, 55, 1465–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Zhang L, Zuo Z, Wan X, Huang Z, J. Am. Chem. Soc 2014, 136, 15501–15504. [DOI] [PubMed] [Google Scholar]
- [143].a) Zhang H, Lu Z, ACS Catal 2016, 6, 6596–6600; [Google Scholar]; b) Chen J, Xi T, Ren X, Cheng B, Guo J, Lu Z, Org. Chem. Front 2014, 1, 1306–1309. [Google Scholar]
- [144].Jang WJ, Song SM, Moon JH, Lee JY, Yun J, J. Am. Chem. Soc 2017, 139, 13660–13663. [DOI] [PubMed] [Google Scholar]
- [145].a) Cai Y, Yang X-T, Zhang S-Q, Li F, Li Y-Q, Ruan L-X, Hong X, Shi S-L, Angew. Chem. Int. Ed 2018, 57, 1376–1380. [DOI] [PubMed] [Google Scholar]; For a related study, see:; b) Iwamoto H, Imamoto T, Ito H, Nat. Commun 2018, 9, 2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Lee Y, Jang H, Hoveyda AH, J. Am. Chem. Soc 2009, 131, 18234–18235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Meek SJ, O’Brien RV, Llaveria J, Schrock RR, Hoveyda AH, Nature 2011, 471, 461–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].a) Trudeau S, Morgan JB, Shrestha M, Morken JP, J. Org. Chem 2005, 70, 9538–9544. [DOI] [PubMed] [Google Scholar]; For a review article, see:; b) Burks HE, Morken JP, Chem. Commun 2007, 4717–4725. [DOI] [PubMed] [Google Scholar]
- [149].Lee H, Lee S, Yun J, ACS Catal 2020, 10, 2069–2073. [Google Scholar]
- [150].Meng F, Jang H, Hoveyda AH, Chem. Eur. J 2013, 19, 3204–3214. [DOI] [PubMed] [Google Scholar]
- [151].For the latest developments on catalytic enantioselective diboron additions to alkenes, see:; a) Neeve EC, Geier SJ, Mkhalid IA, Westcott SA, Marder TB, Chem. Rev 2016, 116, 9091–9161; [DOI] [PubMed] [Google Scholar]; b) Collins BSL, Wilson CM, Myers EL, Aggarwal VK, Angew. Chem. Int. Ed 2017, 56, 11700–11733. [DOI] [PubMed] [Google Scholar]
- [152].Kliman LT, Mlynarski SN, Morken JP, J. Am. Chem. Soc 2009, 131, 13210–13211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Feng X, Jeon H, Yun J, Angew. Chem. Int. Ed 2013, 52, 3989–3992. [DOI] [PubMed] [Google Scholar]
- [154].Green JC, Joannou MV, Murray SA, Zanghi JM, Meek SJ, ACS Catal 2017, 7, 4441–4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Szymaniak A, Zhang C, Coombs JR, Morken JP, ACS Catal 2018, 8, 2897–2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Radomkit S, Liu Z, Closs A, Mikus MS, Hoveyda AH Tetrahedron 2017, 73, 5011–5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Jang H, Jung B, Hoveyda AH, Org. Lett 2014, 16, 4658–4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].For examples of reactions involving enantioselective protonolysis of an allylmetal compound, see:; a) Nishimura T, Hirabayashi S, Yasuhara Y, Hayashi T, J. Am. Chem. Soc 2006, 128, 2556–2557; [DOI] [PubMed] [Google Scholar]; b) Sawano T, Ou K, Nishimura T, Hayashi T, J. Org. Chem 2013, 78, 8986–8993. [DOI] [PubMed] [Google Scholar]
- [159].a) Ito H, Ito S, Sasaki Y, Matsuura K, Sawamura M, J. Am. Chem. Soc 2007, 129, 14856–14857. [DOI] [PubMed] [Google Scholar]; For related reports, see:; b) Ito H, Kunii S, Sawamura M, Nat. Chem 2010, 2, 972–976; [DOI] [PubMed] [Google Scholar]; c) Yamamoto, Takenouchi Y, Ozaki T, Miya T, Ito H, J. Am. Chem. Soc 2014, 136, 16515–16521. [DOI] [PubMed] [Google Scholar]
- [160].Ito H, Kosaka Y, Nonoyama K, Sasaki Y, Sawamura M, Angew. Chem. Int. Ed 2008, 47, 7424–7427. [DOI] [PubMed] [Google Scholar]
- [161].Guzman-Martinez A, Hoveyda AH, J. Am. Chem. Soc 2010, 132, 10634–10637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Silverio DL, Torker S, Pilyugina T, Vieira EM, Snapper ML, Haeffner F, Hoveyda AH, Nature 2013, 494, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].a) Park JK, Lackey HH, Ondrusek BA, McQuade DT, J. Am. Chem. Soc 2011, 133, 2410–2413. [DOI] [PubMed] [Google Scholar]; For a relevant review article, see:; b) Semba K, Fujihara T, Terao J, Tsuji Y, Tetrahedron, 2015, 71, 2183–2197. [Google Scholar]
- [164].a) Kojima R, Akiyama S, Ito H, Angew. Chem. Int. Ed 2018, 57, 7196–7199; [DOI] [PubMed] [Google Scholar]; b) Gao P, Yuan C, Zhao Y, Shi Z, Chem 2018, 4, 2201–2211; [Google Scholar]; c) Ref. [52].
- [165].Akiyama S, Kubota K, Mikus MS, Paioti PHS, Romiti F, Liu Q, Zhou Y, Hoveyda AH, Ito H, Angew. Chem. Int. Ed 2019, 58, 11998–12003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166]. For stereochemical models regarding Cu–B addition to F3C-substituted alkenes, see ref. [51].
- [167].Hayashi T, Ohno A, Lu S, Matsumoto Y, Fukuyo E, Yanagi K, J. Am. Chem. Soc 1994, 116, 4221–4226. [Google Scholar]
- [168].a) Delvos LB, Vyas DJ, Oestreich M, Angew. Chem. Int. Ed 2013, 52, 4650–4653; [DOI] [PubMed] [Google Scholar]; b) Delvos LB, Hensel A, Oestreich M, Synthesis 2014, 46, 2957–2964. [Google Scholar]; For related processes catalyzed by the same type of NHC–Cu catalysts and with (PhMe2Si)2Zn, see:; c) Hensel A, Oestreich M, Chem. Eur. J 2015, 21, 9062–9065. [DOI] [PubMed] [Google Scholar]
- [169].Takeda M, Shintani R, Hayashi J Org. Chem 2013, 78, 5007–5017. [DOI] [PubMed] [Google Scholar]
- [170].Wu H, Hoveyda AH; , previously unpublished results. See the Supporting Information for experimental and analytical details.
- [171].Falivene L, Credendino R, Poater A, Petta A, Serra L, Oliva R, Scarano V, Cavallo L, Organometallics 2016, 35, 2286–2293. [Google Scholar]
- [172].Clayden J, Donnard M, Lefranc J, Tetlow DJ, Chem DJ. Commun 2011, 47, 4624–4639. [DOI] [PubMed] [Google Scholar]
- [173].Yus M, González-Gómez JC, Foubelo JCF, Chem. Rev 2011, 111, 7774–7854. [DOI] [PubMed] [Google Scholar]
- [174].Wada R, Shibuguschi T, Makino S, Oisaki K, Kanai M, Shibasaki M, J. Am. Chem. Soc 2006, 128, 7687–7691. [DOI] [PubMed] [Google Scholar]
- [175].Yazaki R, Nitabaru T, Kumagai N, Shibasaki M, J. Am. Chem. Soc 2008, 130, 14477–14479. [DOI] [PubMed] [Google Scholar]
- [176].Trost BM, Silverman SM, J. Am. Chem. Soc 2010, 132, 8238–8240. [DOI] [PubMed] [Google Scholar]
- [177].a) Luo Y, Hepburn HB, Chotsaeng N, Lam HW, Angew. Chem. Int. Ed 2012, 51, 8309–8313; [DOI] [PubMed] [Google Scholar]; b) Hepburn HB, Chotsaeng N, Luo Y, Lam HW, Synthesis 2013, 45, 2649–2661. [Google Scholar]
- [178].a) Chen P, Yue Z, Zhang J, Lv X, Wang L, Zhang J, Angew. Chem. Int. Ed 2016, 55, 13316–13320. [DOI] [PubMed] [Google Scholar]; For synthesis of products that contain an E-enoate, see:; b) Chen P, Zhang J, Org. Lett 2017, 19, 6550–6553. [DOI] [PubMed] [Google Scholar]
- [179].Jang H, Romiti F, Torker S, Hoveyda AH, Nat. Chem 2017, 9, 1269–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].a) Shen C, Wang R-Q, Wei L, Wang Z-F, Tao H-Y, Wang C-J, Org. Lett 2019, 21, 6940–6945; [DOI] [PubMed] [Google Scholar]; b) Wang Y, Deng L-F, Zhang X, Niu D, Org. Lett 2019, 21, 6951–6956. [DOI] [PubMed] [Google Scholar]
- [181].Fager DC, Morrison RJ, Hoveyda AH, Angew. Chem. Int. Ed DOI: 10/1002/anie.202001184. [Google Scholar]
- [182].Lee K.-s., Zhou Y, Hoveyda AH; , previously unpublished data. See the Supporting Information for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.