

Abstract
Cutaneous mast cell tumours (MCTs) are common skin neoplasms in dogs. MicroRNAs (miRNAs) are post-transcriptional regulators involved in several cellular processes, and they can function as tumour promoters or suppressors. However, the role of miRNAs in canine MCTs has not yet been elucidated. Thus, the current study aimed to characterize miRNA profiles and to assess their value as biomarkers for MCTs. miRNA expression profiles were assessed in formalin-fixed, paraffin-embedded samples by next-generation sequencing. Ten samples were MCT tissues, and 7 were healthy adjacent tissues. Nine dysregulated miRNAs (DE-miRNAs) were then validated using RT-qPCR in a larger group of MCT samples, allowing the calculation of ROC curves and performance of multiple factor analysis (MFA). Pathway enrichment analysis was performed to investigate miRNA biological functions. The results showed that the expression of 63 miRNAs (18 up- and 45 downregulated) was significantly affected in MCTs. Five DE-miRNAs, namely, miR-21-5p, miR-92a-3p, miR-338, miR-379 and miR-885, were validated by RT-qPCR. The diagnostic accuracy of a panel of 3 DE-miRNAs—miR-21, miR-379 and miR-885—exhibited increased efficiency in discriminating animals with MCTs (AUC = 0.9854) and animals with lymph node metastasis (AUC = 0.8923). Multiple factor analysis revealed clusters based on nodal metastasis. Gene Ontology and KEGG analyses confirmed that the DE-miRNAs were involved in cell proliferation, survival and metastasis pathways. In conclusion, the present study demonstrated that the miRNA expression profile is changed in the MCT microenvironment, suggesting the involvement of the altered miRNAs in the epigenetic regulation of MCTs and identifying miR-21, miR-379 and miR-885 as promising biomarkers.
Subject terms: Skin cancer, miRNAs
Introduction
Cutaneous mast cell tumours (MCTs) are a common skin neoplasm in dogs, accounting for up to 21% of all skin tumours1. Mast cell tumours originate from uncontrolled proliferation of neoplastic mast cells in cutaneous and subcutaneous tissues and usually occur as single tumours but sometimes as multiple tumours2. The clinical phenotype of MCTs ranges from relatively benign to highly malignant tumours that can spread to the local lymph nodes, liver and spleen3,4. Their diagnosis and prognosis are usually based on tumour grading as determined by the Patnaik5 and Kiupel4,6 systems, and tumour staging, including evaluation of draining lymph nodes7. In addition. the proliferation rate (Ki-67), c-kit proto-oncogene mutations and KIT expression are significant prognostic factors7,8. The pathogenesis and aetiology of MCTs are poorly understood, and the main causes that lead to MCT carcinogenesis remain elusive9.
Canine MCTs have been recently investigated at the molecular level using next-generation RNA sequencing10, and two distinct tumour subtypes have been identified with differential expression of 71 genes involved in cell proliferation processes. However, no information about the epigenetic regulation of MCT development, such as regulation mediated by microRNAs, is available.
MicroRNAs (miRNAs) are small non-coding RNAs that are involved in post-transcriptional regulation and thus affect virtually every cellular process. MiRNAs regulate mRNA translation, leading to modulation of protein expression. MiRNA dysregulation occurs during the pathogenesis of several diseases, including cancer, and leads to disruption of pathways that play a role in cancer initiation and progression11. MiRNAs can act as oncomiRs, targeting tumour suppressor genes and promoting tumour progression, or as tumour suppressors, although overall miRNA dysregulation is a hallmark of cancer12–14. The role of miRNAs in canine MCT is still unknown. The present study aims to close this gap by assessing the miRNA expression profile of canine MCTs using next-generation sequencing, investigating whether miRNAs are dysregulated in the MCT microenvironment, and identifying links between miRNAs and their target genes and relevant biological processes.
Results
Determination of miRNA profiles and identification of DE-miRNAs in healthy margins versus MCTs
After RNA extraction, small RNA libraries were generated, pooled, concentrated and size selected on a non-denaturing acrylamide gel (≈ 141 bp bands). After elution from the gel, the size-selected libraries were quantified and sequenced on a NextSeq 500 sequencer (Illumina). The resulting number of reads per sample varied from 53,000 to 29,000,000. Eight MCT samples with insufficiently high mapping rates were excluded from further analysis. For two dogs (numbers 8 and 15 in Table 1), the results for the tumours and matched healthy adjacent margins were reported.
Table 1.
Summary of clinical and histopathological data.
| Breed | Sex | Age (years) | Tumor location | Size (cm) | Grade | Lymph node statuse | ||
|---|---|---|---|---|---|---|---|---|
| Patnaik | Kiupel | |||||||
| 1 | American Staffordshire Terriera,d | Male | 6 | Trunk | 1 | II | Low | HN1 |
| 2 | Bernese | Female | 4 | Limb | 2.5 | II | Low | HN2 |
| 3 | Boxer | Male | 8 | Limb | 2.2 | II | Low | HN2 |
| 4 | Dachshund | Female | 9 | Trunk | 0.8 | II | Low | HN0 |
| 5 | Dogoa,b | Male | 6 | Limb | 2 | II | Low | HN2 |
| 6 | English settera,b | Female | 6 | Trunk | 3 | II | Low | HN2 |
| 7 | English settera,c,d | Female | 11 | Trunk | 5 | II | Low | HN3 |
| 8 | Italian pointera,b | Male | 5.5 | Trunk | 4 | II | Low | HN1 |
| 9 | Jack Russella,b | Male | 13 | Head | 5 | II | High | HN2 |
| 10 | Labrador | Male | 1 | Head | 1 | I | Low | HN0 |
| 11 | Labrador d | Male | 10 | Scrotum | 2 | II | Low | HN0 |
| 12 | Labrador | Male | 9 | Trunk | 0.6 | II | Low | HN2 |
| 13 | Labrador | Female | 6 | Trunk | 3.5 | II | Low | HN2 |
| 14 | Mixed breeda,b,c | Female | 11 | Trunk | 3 | II | Low | HN0 |
| 15 | Mixed breeda,b | Female | 6 | Trunk | 4 | II | Low | HN2 |
| 16 | Mixed breeda,b | Male | 11 | Limb | 3 | III | High | HN2 |
| 17 | Mixed breeda,c,d | Female | 8 | Limb | 3 | III | High | – |
| 18 | Mixed breedc | Male | 12 | Neck | 7 | II | Low | HN3 |
| 19 | Pug | Male | 3.5 | Head | 1 | II | Low | HN2 |
| 20 | Tosa inu | Male | 4 | Trunk | 3 | II | Low | HN2 |
| 21 | Weimaraner | Male | 7 | trunk | 2 | II | Low | HN2 |
aMCT samples sequenced using NGS.
bHealthy margins sequenced using NGS.
c$amples in which miRNAs selected for the RT-qPCR validation step were not detected.
dSamples for which healthy margins were not collected.
eClassification system proposed by Weishaar et al.7. HN histological node, NGS next-generation sequencing.
A count table was used to identify differentially expressed miRNAs via DESeq2 analysis15. The DESeq threshold was set by discarding low-expression miRNAs having an average count of 2 or less. This analysis revealed the expression of 246 and 116 Canis familiaris (cfa) miRNAs in healthy margins and MCTs, respectively.
The expression profiles of sequenced samples were used to carry out cluster analysis. Samples were grouped into two clusters: healthy tissues and MCTs (Fig. 1a).
Figure 1.
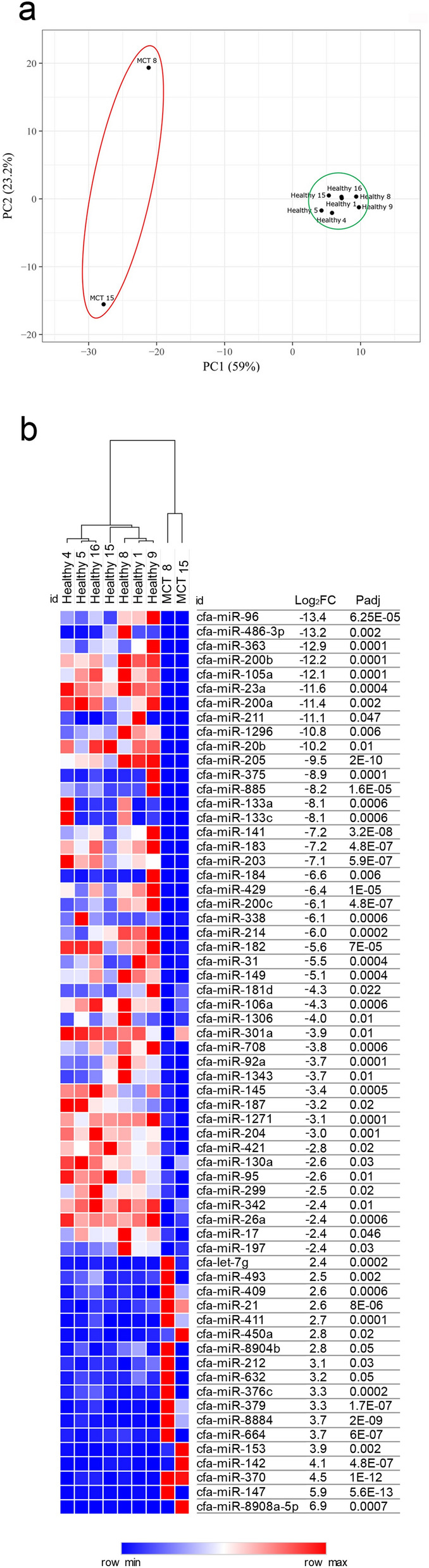
NGS results. (a) Principal component analysis (PCA) of sequenced samples. Two-dimensional PCA was used to determine whether MCTs (red circle) could be differentiated from healthy (green circle) samples. (b) Identification of DE-miRNAs between MCTs and healthy samples. Heat map and table displaying the fold change and Padj of DE-miRNAs.
To rank the most differentially expressed miRNAs (DE-miRNAs) between healthy and MCT samples, the results of differential expression analysis performed with DESeq2 were further filtered a more stringent cut-off criteria of an adjusted P value of 0.01 and an absolute log2FC of 2.4. This filtering allowed the identification of sixty-three miRNAs whose abundance differed significantly between MCT and healthy samples, demonstrating that 45 miRNAs were downregulated with a log2FC of between − 2.4 and − 13.4 and 18 miRNAs were upregulated with a log2FC of between 2.4 and 6.9 (Fig. 1b).
Quantification of DE-miRNAs in healthy versus MCT samples by RT-qPCR
To validate the NGS results and measure the abundances of DE-miRNAs in MCTs, RT-qPCR was performed on both the sequenced samples and a separate group of 11 MCTs and related healthy adjacent (normal) tissue samples. To validate the sequencing results, 9 DE-miRNAs—miR-370, miR-379, miR-92a, miR-21, miR-26a, miR-342, miR-885, miR-375 and miR-338—were selected based on the fold change and read count values. MiR-122, miR-128 and miR-101 were quantified as controls for normalization. The artificial spike-in cel-miR-39 was used as the internal control. MiRNAs selected for the validation step were detected in almost all samples, except for sample numbers 7, 14, 17 and 18 (Table 1). The results are presented in Fig. 2. The RT-qPCR results confirmed that five miRNAs were differentially regulated in healthy adjacent margin tissues versus MCTs. In detail, miR-21 (P = 0.004, log2FCMCT/Healthy = 2.84) and miR-379 (P = 0.0005, log2FCMCT/Healthy = 2.61) were upregulated, while miR-885 (P = 0.008, log2FCMCT/Healthy = − 2.53), miR-338 (P = 0.025, log2FCMCT/Healthy = − 0.86) and miR-92a (P = 0.021, log2FCMCT/Healthy = − 0.78) were downregulated in MCT samples compared to healthy margin samples. Conversely, miR-26a, miR-342, miR-370 and miR-375 did not exhibit statistically significant differences between the groups.
Figure 2.

Box plots of DE-miRNAs in MCTs compared with healthy margins. Significance was accepted at P < 0.05 (*), P < 0.01 (**) and P < 0.001 (***). The black lines inside the boxes denote the medians. The whiskers indicate variability outside the upper and lower quartiles.
Diagnostic value of DE-miRNAs in dogs with MCT
Receiver operating characteristic (ROC) analysis was used to assess the diagnostic value of DE-miRNAs as biomarkers to further discriminate between MCT and healthy adjacent tissue. To confirm the diagnostic efficacy of each miRNA, the associated area under the curve (AUC) was calculated. Table 2 shows a summary of the diagnostic performance of each DE-miRNA and shows combinations of three DE-miRNAs. The AUC was fair for miR-92a (AUC = 0.7427) and miR-338 (AUC = 0.7339) and excellent for miR-21 (AUC = 0.9825), miR-379 (AUC = 0.9211) and miR-885 (AUC = 0.9181) (Fig. 3).
Table 2.
The area under the curve (AUC), sensitivity and specificity values of DE-miRNAs.
| miRNA | AUC | 95% CI | P-value | Cut-off | Sensitivity | 1-Specificity | |
|---|---|---|---|---|---|---|---|
| Downregulated | miR-885 | 0.9181 | 0.8276–1.000 | < 0.0001 | 0.0357 | 0.8889 | 0.9474 |
| miR-92a | 0.7427 | 0.5925–0.8929 | = 0.0015 | 0.814 | 0.7222 | 0.6842 | |
| miR-338 | 0.7339 | 0.5827–0.8851 | = 0.0024 | 1.7878 | 0.6111 | 0.7895 | |
| Upregulated | miR-21 | 0.9825 | 0.9825–0.9825 | < 0.0001 | 1.6250 | 0.9444 | 0.9474 |
| miR-379 | 0.9211 | 0.8328–1.000 | < 0.0001 | 11.5688 | 1.000 | 0.7895 | |
| W-AV* | miR-379 + miR-21 + miR-885 | 0.9854 | 0.9854–0.9854 | < 0.0001 | 0.1654 | 1.000 | 0.9444 |
| W-AV-HN** | miR-379 + miR-21 + miR-885 | 0.8923 | 0.759–1.000 | < 0.0001 | 0.5528 | 0.9231 | 0.8000 |
*W-AV = weighted average relative quantification of miR-379 + miR-21 + miR-885 in healthy versus MCT samples.
**W-AV-HN = weighted average relative quantification of miR-379 + miR-21 + miR-885 in HN0/1 versus HN2 samples.
Figure 3.

Receiver operating characteristic (ROC) curve analysis of DE-miRNAs. (a) AUC of miR-885; (b) AUC of miR-92a; (c) AUC of miR-338; (d) AUC of miR-21; (e) AUC of miR-379. AUC area under the curve, CI confidence interval.
Discriminant analysis was used to further investigate the potential for improving diagnostic performance by analysing multiple DE-miRNAs. Statistical analysis was performed to examine the weighted average relative quantification (RQ) values of the miRNAs with an AUC of > 0.9 (miR-21, miR-379 and miR-885) (Fig. 4). The median expression levels were 0.0301 (range 0.0069–0.9334) and 0.99998 (range 0.3485–1) in healthy margin and MCT samples, respectively (Fig. 4a). The predicted probability of being able to discriminate a sample as positive based on the logit model [logit = 1/(1 + exp (− (− 4.92611–1.31822 × expression level of miR-885 + 0.40746 × expression level of miR-379 + 0.86787 expression level of miR-21)))] was used to construct the ROC curve (Fig. 4b). The AUC for the panel of these three DE-miRNAs was 0.9854 (95% CI 0.9854–0.9854) with a cut-off value of 0.1654, and a sensitivity of 100% and a specificity of 94.4%.
Figure 4.

The average expression of the DE-miRNAs with AUC > 0.9, including miR-379, miR-21 and miR-885. (a) The weighted average relative quantification (RQ) values of DE-miRNAs in healthy versus MCT samples (a) and ROC curve analysis performed using the logit model, for healthy versus MCT samples (b). AUC, area under the curve; CI, confidence interval. The black lines denote the medians. **P < 0.001; ***P < 0.0001.
Potential of the miRNA panel for the detection of nodal metastases
Excised lymph nodes were categorized in accordance with the Weishaar classification system for nodal metastases (2014)7, and the potential of the three-miRNA panel to discriminate patients with and without lymph node involvement was evaluated. Two groups, namely, HN0/1, including non-metastatic and pre-metastatic samples, and HN2, including early metastatic samples, were included for further analysis (Table 1). The weighted average relative quantification (RQ) values of the miRNA panel (miR-21, miR-379 and miR-885) were calculated (Fig. 5). The median expression levels were 0.3179 (range 0.0071–0.8858) and 0.9424 (range 0.3741–1) in the HN0/1 and HN2 groups, respectively (Fig. 5a). The predicted probability of being able to discriminate a sample as metastatic based on the logit model [logit = 1/(1 + exp (−(− 1.58980–7.91569 × expression level of miR-885 + 0.13130 × expression level of miR-379 + 0.05084 expression level of miR-21)))] was used to construct the ROC curve (Fig. 5b). The AUC for the panel of these three DE-miRNAs was 0.8923 (95% CI 0.759–1.000) with a cut-off value of 0.5528, a sensitivity of 92.3% and a specificity of 80%. MFA identifies individuals with similar profiles who are close to each other on the factor map. Collectively, the components F1 and F2 explained 68.53% of the total variance in the data (Fig. 5c). The first component (F1) explained 42.76% of the variance and separated the HN0/1 group from the HN2 group according to lymph node involvement. The second component (F2) explained 25.77%, discriminating non-metastatic HN0 samples (samples 4 and 10) in the upper right panel from high-grade early metastatic HN2 samples (samples 16 and 9) in the lower-left panel (Supplementary Table S1).
Figure 5.

The average expression of the DE-miRNAs with AUC > 0.9, including miR-379, miR-21 and miR-885. (a) The weighted average relative quantification (RQ) values of DE-miRNAs in HN0/1 versus HN2 samples; (b) ROC curve analysis performed using the logit model for HN0/1 versus HN2 samples; (c) individual map for Multiple Factor Analysis (MFA): each sample name represents the barycentre of the two positions according to the dataset coloured according to lymph node involvement: HN0/1 (blue) and HN2 (green). AUC area under the curve, CI confidence interval. The black lines denote the medians. **P < 0.001; ***P < 0.0001.
Gene Ontology and pathway enrichment analysis of miRNAs
The MiRWalk 3.0 and DAVID databases were searched to retrieve the candidate target genes of DE-miRNAs and to perform mRNA enrichment analysis, respectively. Of the predicted mRNA targets of downregulated miRNAs, 196 were in the 3′UTR, 45 were in the 5′UTR and 171 were in the CDS. Of the predicted mRNA targets of upregulated miRNAs, 16 were in the 3′UTR, 3 were in the 5′UTR and 11 were in the CDS. The list of candidate target genes is provided in Table 3.
Table 3.
Candidate target genes retrieved from the miRWalk 3.0 database.
| Target genes of downregulated miRNAs | |
| 3′UTR | FOS, POF1B, TP63, PTGES2, CCDC113, LPIN1, IL6ST, GTF2A1, HDAC2, MED19, TET2, PBLD, ZBTB7B, KIF1B, PPP1R3D, CNNM4, MED29, PAFAH1B1, RASAL2, ABL2, PAWR, TMCC3, SMARCA5, KLHL3, CDK5R1, TIA1, PDIK1L, FKBP1A, MACROD2, RAB14, ATG9A, SCN1A, FAM168A, NOL4, HEXIM1, C16orf87, SH3PXD2A, TSFM, ARPC1B, MSL2, RIC1, NFIA, GNG12, ZZZ3, DUSP16, SERINC5, LAMC1, IPO9, HNF4G, YRDC, RPH3A, PPM1B, AAK1, LSM14A, CNOT6, SOX6, ZADH2, NOL4L, NFIB, MAP2K4, NEDD4L, TMEM50B, SLC30A7, LIN54, GNAQ, PALM2, SYNDIG1, KIF3B, JPH2, LPP, PCGF3, RGS3, DAB2IP, TSC1, COL5A1, ATXN1, MARCH4, PAX3, TRIP12, EPC2, MAP3K20, SESTD1, ITGAV, TPBGL, LDLRAD4, TECPR2, TRAF3, ACTC1, UBE2Z, OTUD3, LUZP1, MTMR9, RNF157, SLC9A7, TAF1, ATRX, USP28, GRAMD1B, SRPRA, C21orf91, AFF3, KCNC4, ANP32E, SEC31B, CCDC186, PLEKHA1, PITPNM2, LRCH1, AXL, PPP1R37, CDC42BPA, ZBTB18, GRHL1, PHLPP2, ZFHX3, RASSF3, DYRK2, STRN3, STYX, PPP1R9A, SMURF1, PIK3CB, TFDP2, DCX, TMEM255A, MBNL3, PCDH11Y, DESI1, SHOX, PTPRD, DENND4B, KCNN3, NF2, PLXDC2, GPR158, PIK3R3, EVI5, ERGIC2, RAB3C, AGGF1, BTG2, ELK4, ARID1B, FAM20C, SNX13, ATG14, KCNK10, FOXN3, ATXN3, PCMTD1, SGK3, UBE2W, MMP16, CCNE2, MTDH, NIPAL1, CXCL5, G3BP2, WDFY3, TCF21, LIN28A, GAS2L3, EFR3B, KCNK3, YIPF4, SOCS5, SERTAD2, CEP41, CREB3L2, ELOVL6, SETD7, DCLK2, SH3D19, PPARGC1B, CAMK2A, CPEB4, RNF141, EIF4G2, CHST1, ARRDC3, MAN2A1, FNIP1, SLX4, GLYR1, SNN, ATXN7, ZBTB20, SPOCK2, DNAJB12, CCSER2, GID4, PIP5K1C, SCUBE3, CD2AP, TRAM2, PHF3 |
| 5′UTR | MYLIP, PCMTD1, RRBP1, ELOVL6, E2F3, GAA, AURKA, ANP32E, CCDC186, PAX9, DYNLT3, DENND4B, WASL, SETD5, FAM135A, XKR7, GAN, CNOT2, RABGAP1L, ZNF287, IDH1, NR4A3, TBC1D19, PLEKHB2, TGIF1, ZNF532, CPEB1, KCNA1, SPHK2, CBFA2T3, COL1A2, TBL1XR1, AIFM1, PTPRD, KLF6, RAB3C, KCNK10, PAPOLA, NAV3, FOSL2, ATL3, SH3D19, SCRG1, WWC1, SNX2 |
| CDS | FOS, DAB2IP, HIPK1, MYLIP, GRAMD1B, MAP2K4, MTF1, PTAR1, RRBP1, NOL4L, TSC1, HIVEP1, ATXN1, IKZF2, TRIP12, SSFA2, VPS4B, TECPR2, GOLGA8A, GOLGA8B, NSF, XYLT2, ELOA, ADAM10, GAA, RBL2, ARFGEF2, SYNJ1, BCL2L11, MCL1, SH3PXD2A, PDZD8, GOLGA3, IPO5, PHLPP2, ITGA5, ANKIB1, FNDC3B, MPP1, RAD21, ZNF17, ZNF776, MYH9, GPBP1L1, CD69, SCAF11, ATP2B4, DSTYK, TULP4, CUX1, CREB3L2, KIAA1109, RBM27, SETD5, TRIM36, TMF1, VPS52, ZBTB34, MBD2, UBR1, NPTN, CNOT2, TTLL7, XPR1, FKBP14, TMCC3, TCHP, PDCD6IP, LCOR, USP7, FRMD3, GNL1, RAB30, WNK4, SS18L1, CELSR2, MYT1L, COX4I1, TSFM, CACNA2D1, PVALB, NFIA, ADGRA2, PPM1B, FBXW7, B4GALT7, RASA1, ZBTB20, SLC35G1, KIAA1024, USF2, TAGAP, STX17, SEL1L3, TBC1D19, ANGPTL2, ADAM23, GIGYF2, LRP1B, SLC4A10, MAP3K20, ZNF385B, ARHGEF17, EPG5, HERC2, RYR3, EPHA8, CSMD1, ATP6V1B2, CHMP7, CPEB1, NRK, FRMPD3, DOK5, TACC2, SBNO1, ITM2B, SPTBN4, HNRNPU, HAS3, WWP2, SCN8A, GDF11, DTX2, PPP1R9A, PIK3CB, PEX5L, VWA5B2, TENM1, CSMD3, EFR3A, PTPRD, TTC28, NOTCH1, P3H3, EPS8, PDZD2, NIPBL, RAB3C, DCAF6, SNX13, PLEKHG3, BSN, ZC2HC1A, RBM47, REST, WDFY3, PTPRK, TCF21, GAS2L3, ADCY3, SRPK2, CTTNBP2, NRF1, NFIX, RHPN2, NPNT, OTUD4, ADAM19, CNTN4, OXSR1, ASB7, CLEC16A, FOXP1, ROBO2, BTLA, FSTL1, MYO18A, C2CD4C, PHF3, COL19A1 |
| Target genes of upregulated miRNAs | |
| 3′UTR | RECK, NCAPG, PAN3, KLHL42, GID4, CCL1, CD59, SLC20A1, PPP1R3B, NEGR1, THRB, PCDH17, FIGN, HTR2C, FAM126B, ETNK1 |
| 5′UTR | KAT6A, ZBTB26, TNRC6B |
| CDS | EPHA4, ADNP, TNRC6B, ATF7IP, FBXO11, NR2C2, PTPN14, SPRY4, KLF3, CASKIN1, ROBO2 |
Gene Ontology (GO) analysis was performed using DAVID for three categories: biological process (BP), cellular component (CC) and molecular function (MF). An overview of the top 10 terms significantly enriched with target genes for each of the above GO categories is presented in Fig. 6. The enriched GO BP terms mainly included regulation of transcription from RNA polymerase II promoter and protein ubiquitination involved in ubiquitin-dependent protein catabolic process; the CC terms were related to the cytoplasm, nucleus and nucleoplasm, while the MF terms focused on transcription factor activity and sequence-specific DNA binding. KEGG pathway analysis was performed on candidate targets of DE-miRNAs. Figure 7 shows the top 10 significantly enriched KEGG pathways, with PI3K-Akt signalling pathway, small cell lung cancer, viral carcinogenesis and microRNAs in cancer at the top of the list.
Figure 6.

Gene Ontology (GO) enrichment analysis of terms potentially regulated by DE-miRNAs. The target genes were annotated by DAVID in three categories: biological process, cellular component and molecular function. The top 10 significantly enriched terms are shown.
Figure 7.

Pathway enrichment analysis for genes potentially regulated by DE-miRNAs. Genes regulated by DE-miRNAs were retrieved and analysed for enrichment in KEGG pathways using DAVID. The P value was − log10 transformed. The top 10 enriched KEGG pathways are reported.
Discussion
The role of miRNAs in canine MCT has not yet been elucidated. The current study aimed to characterize the miRNA profile of canine MCTs using FFPE samples. A multi-step approach was adopted: the pilot part of the study profiled miRNAs in MCTs and healthy adjacent margins via next-generation sequencing. In the second step, the DE-miRNAs were validated by performing RT-qPCR on the samples selected for sequencing and on a separate independent group of FFPE samples. Our results showed that the expression of 63 miRNAs, of which 18 were upregulated and 45 were downregulated, was significantly affected in MCTs. Nine DE-miRNAs were then validated in a larger group by RT-qPCR, demonstrating that five of these DE-miRNAs, namely, miR-21-5p, miR-92a-3p, miR-338, miR-379 and miR-885, were effectively modulated. The diagnostic accuracy of three DE-miRNAs—miR-21, miR-379 and miR-885—was excellent, and the AUC of their combination increased to 0.9854 with 100% sensitivity and 94.4% specificity. Due to their limited nucleotide length, miRNAs have shown higher stability than DNA and mRNA in sample types such as FFPE tissues16. However, the preparation of miRNA NGS libraries from FFPE samples is particularly challenging because of the intersample heterogeneity of the RNA quality17. In the present investigation, the library preparations were performed in parallel to avoid the batch effect, but mappable miRNA reads were produced from only 2 of 10 tumours. Moreover, mast cells, which release their granules into the tumour mass9, may also affect library preparation.
Mast cells are crucial players in allergies, immune responses, angiogenesis and the maintenance of tissue function and integrity18, also promoting tissue repair19. Furthermore, mast cells modulate the tumour microenvironment by performing a two-pronged role: they perform a pro-neoplastic role by releasing mitogenic and pro-angiogenic factors such as histamine, IL-10, TNF, FGF2, VEGF, IL-18 and MMP20, that promote immune suppression, proliferation and angiogenesis; and they perform an anti-neoplastic role by inhibiting cell growth and motility and promoting antitumour inflammatory reactions and apoptosis21. Few studies have investigated the dysregulation of miRNAs in MCTs in dogs. Using real-time PCR-based TaqMan Low-Density miRNA Arrays, Fenger and colleagues22 demonstrated that the expression level of miR-9 was increased in high-grade canine MCTs, promoting an invasive phenotype. Furthermore, circulating miRNA-126 resulted in exacerbation of non-epithelial neoplasms, including MCT23.
The DE-miRNAs identified herein have been related to neoplasms in humans and, in some cases, in dogs. Of the five miRNAs that were found to be differentially regulated, two, namely, miR-21 and miR-379 were upregulated, whereas three, namely, miR-92a-3p, miR-338, and miR-885, were downregulated. MiR-21, which was found to be upregulated in the present study, has been widely investigated in cancer, and its upregulation has been associated with cell proliferation, invasion, apoptosis and drug resistance24,25. MiR-21 is frequently overexpressed in human cancers, including breast cancer, lung cancer, pancreatic cancer, ovarian cancer, glioma, liver neoplasms, gastric cancer, colorectal cancer and kidney cancer26, and in canine oral melanoma27, hepatocellular carcinoma28 and malignant mammary tumours29. In humans, overexpression of miR-21 has been related to downregulation of tumour suppressor genes, including programmed cell death 4 (PDCD4), matrix metalloproteinases (MMPs), phosphatase and tensin homolog (PTEN), reversion inducing cysteine-rich protein (RECK), and phosphoinositide 3-kinase (PI3K)26. MiR-379, which was also upregulated, is an onco-suppressor miRNA. MiR-379 negatively regulates cell proliferation, migration and invasion in several human cancers, including nasopharyngeal carcinoma30, cervical carcinoma31, gastric cancer32 and bladder cancer33, by targeting tumor protein D52 (TPD52), V-crk avian sarcoma virus CT10 oncogene homolog-like (CRKL), focal adhesion kinase (FAK) and mouse double minute 2 (MDM2).
Our results demonstrated that miR-92a, miR-338 and miR-885 were downregulated in canine MCTs. MiR-92a belongs to the miR-17-92a cluster, which is dysregulated in many different types of human tumours34. The mechanisms by which miR-92a promotes tumorigenesis include augmenting tumour proliferation, inhibiting tumour apoptosis, and enhancing tumour invasion and metastasis35 by targeting PTEN in oesophageal squamous cell cancer36 and Dickkopf-related protein 3 (DKK3) in osteosarcoma37. These features identify miR-92a as an onco-miRNA. However, onco-suppressor activities of the miR-17-92a cluster have also been reported, including anti-proliferative and senescence effects in bladder cancer cells38 and in prostate39 and gastric40 cancers by targeting, among other pathways, the NOTCH/EP4 pathway. Similarly, the role of miR-338 is controversial, as it has been associated with both pro- and antitumour roles. MiR-338 targets oncogenes such as RAB32 and EYA2, and its downregulation in cancer is also linked to overexpression of epidermal growth factor receptor (EGFR)41,42 and MET transcriptional regulator (MACC1)43. MiR-338 plays a tumour-promoting role in melanoma that is linked to tumour growth and metastasis44. MiR-338 is also involved in hypoxia-induced epithelial-to-mesenchymal transition by targeting HIF-1α45. MiR-885 is a tumour suppressor miRNA that interferes with cell proliferation and migration by targeting SOCS in colorectal cancer46 and the Wnt/β-catenin pathway in hepatocellular carcinoma47.
Gene Ontology and KEGG pathway analysis suggested that DE-miRNAs have an impact on transcription activities, cell cycle progression and cell survival and, in general, on several pathways involved in cancer development. This hypothesis is supported by gene expression analysis of canine cutaneous MCTs10,48. Gene expression profiling of metastatic and non-metastatic MCTs using an array approach identified differentially expressed genes involved in apoptosis, cell cycle arrest and loss of cell polarity and adhesion48. Comparison between these genes and the genes potentially modulated by DE-miRNAs identified in the present study showed that seven genes (Fos Proto-Oncogene or FOS, Histone Deacetylase or HDAC, Striatin or STRN, Neurofibromin 2 or NF2, Phosphoinositide-3-Kinase Regulatory Subunit 3 or PIK3R3, Rho Guanine Nucleotide Exchange Factor or ARHGEF, C-Type Lectin Domain Containing or CLEC) are potentially modulated by downregulation of miR-92a, miR-338 and miR-885. Conversely, Sprouty RTK Signaling Antagonist or SPRY can be modulated by upregulation of miR-21 and miR-379. Transcriptome analysis comparing low- and high-risk canine MCTs using next-generation RNA sequencing identified 71 differentially expressed genes associated with cell proliferation and the extracellular matrix10. Comparison between these genes and the candidate target genes identified in the present study showed that eight genes belonging to the kelch-like (KLHL), collagen (COL), matrix metallopeptidase (MMP), multi-domain (WW, PDZ, FERM) containing protein (FRMPD), C-type lectin domain (CLEC) and suppressor of cytokine signaling (SOCS) families may be downregulated. Conversely, genes belonging to the KLHL and 5-Hydroxytryptamine Receptor (HTR) families may be upregulated DE-miRNAs. Previous results obtained via two different strategies, an array-based approach and RNA-seq, support our hypothesis that the dysregulation of miRNAs identified in this study may influence the expression of genes involved in cell proliferation, survival and tumour spread10.
Although the prognostic role of both the Patnaik5 and Kiupel4 grading systems in canine MCTs is widely accepted, histological grading alone cannot accurately predict the risk of local and distant metastases3–5,9,49. Nodal metastases have been reported in 20–49% of cutaneous MCTs at first presentation, and identification of lymph node involvement is crucial for accurate tumour staging and prognosis3,49,50. Recently, a novel classification system for the evaluation of nodal metastasis in canine MCTs has been proposed and correlated with the clinical outcome, providing evidence that dogs diagnosed with early metastatic/overt metastatic (HN2-HN3) nodes have a shorter life expectancy7. In our study, a three-miRNA signature (miR-379-miR-21-miR-885) accurately discriminated between healthy adjacent tissue and MCT tissue (AUC = 0.9854) and identified patients with early nodal metastases (AUC = 0.8923). Since the number of enrolled patients did not allow us to perform discriminant analysis of parameters such as survival time and progression-free interval, the present results provide a background to investigate new biomarkers of MCT outcome in different matrices, including blood, to support clinical decision making.
In conclusion, the present study demonstrated that the expression levels of miR-21, miR-379, miR-92a, miR-885 and miR-338 in the tumour microenvironment are changed compared to those in healthy adjacent tissues and differ in dogs with early nodal metastases compared to those without nodal involvement, suggesting that these miRNAs may epigenetically modulate genes involved in MCT progression and metastasis. Our study provides insights into the emerging roles of miRNAs in veterinary oncology, although more efforts are required to establish the role and molecular targets of the investigated DE-miRNAs. Since the sample size influences the clinical sensitivity and specificity of the test, further studies are necessary to confirm the diagnostic value of miRNAs by increasing the number of patients.
Materials and methods
Inclusion criteria and sample collection
Thirty-seven formalin-fixed, paraffin-embedded (FFPE) samples, including 21 MCT samples and 16 healthy adjacent tissue samples (dermal tissue at the excision margins), were selected from the archives of the Department of Veterinary Medicine of the Università degli Studi di Milano. Samples were collected from client-owned dogs that underwent veterinary consultation and surgery during routine oncological management of canine mast cell tumour. All experimental procedures were reviewed and approved by the Ethics Committee of the University of Milano (approval number 118/19). Patients were recruited after the owner provided written informed consent. All experiments were performed following the relevant guidelines and regulations. Samples were trimmed and processed according to currently recommended guidelines51 (Table 1). Cutaneous MCTs at first presentation without distant metastasis52 and sentinel/regional lymph nodes were surgically removed and histologically classified53 and graded4,5. In addition, neoplastic involvement of sentinel lymph nodes was categorized as previously described7,52.
For all samples, after bright field microscopy observation of the haematoxylin–eosin-stained slide, the corresponding paraffin block was penetrated using a biopsy punch with a plunger (Miltex) to collect a portion of the tumour (21 MCTs) or a portion of the healthy dermal connective tissue (16 margins); the latter samples were used as controls. For MCT samples, areas of necrosis, haemorrhage or inflammation were avoided, if present.
miRNA extraction and next-generation sequencing (NGS)
MiRNAs were extracted using an miRNeasy FFPE kit (Qiagen, Cat. No. 217504) following the manufacturer’s instructions. The RNA quality and quantity were verified according to MIQE guidelines54. The RNA concentration was determined in a Qubit 2.0 fluorometer with a Qubit microRNA Assay Kit (Invitrogen, Cat. No. Q32880). A pilot NGS study was performed on 10 MCTs and 7 healthy adjacent tissue samples (Table 1). Small RNA transcripts were converted into barcoded cDNA libraries. Library preparation was performed as previously reported55 using an NEBNext Multiplex Small RNA Library Prep Set (Cat. No. NEB#E7560) for Illumina, and sequencing was performed in a NextSeq 500 sequencer (Illumina Inc., USA).
Computational analysis
The output of the NextSeq500 Illumina sequencer was demultiplexed using bcl2fastq Illumina software embedded in the docker4seq package56. miRNA expression quantification was performed using the workflow and implementation previously described57. In brief, after adapter trimming with cutadapt58, sequences were mapped using SHRIMP59 to Canis familiaris precursor miRNAs available in miRBase 22.0-March 2018 (https://www.mirbase.org/). Using GenomicsRanges60, an R script, was used to identify the number of reads on precursor miRNAs mapping to the expected location on mature miRNAs. The detected counts were organized in a table including all analysed samples. For visualization purposes, only CPM (counts per million reads) values were used. Differential expression analysis was conducted using the DESeq2 Bioconductor package15 implemented in the docker4seq package. Differential expression analysis was performed using the abovementioned count table comparing the tumour and control groups using an adjusted P value of ≤ 0.05 and an absolute log2 fold change (log2FC) of ≥ 1 as the threshold criteria.
miRNA quantification by RT-qPCR
Small RNAs were extracted using an miRNeasy kit for FFPE samples (Qiagen, Cat. No. 217504). The Caenorhabditis elegans miRNA cel-miR-39 (25 fmol final concentration) (Qiagen, Cat. No. 219610) was used as a synthetic spike-in control due to its lack of sequence homology to canine miRNAs. RNA extraction was then carried out according to the manufacturer’s instructions.
To obtain cDNA, reverse transcription was performed using a TaqMan Advanced miRNA cDNA Synthesis Kit (Cat. No. A28007, Applied Biosystems) following the manufacturer’s instructions.
Quantitative real-time PCR (RT-qPCR) was performed to validate the sequencing results following the MIQE guidelines54. The selected DE-miRNAs included miR-370-3p (assay ID 478326_mir), miR-379-5p (assay ID 478077_mir), miR-92a-3p (assay ID 477827_mir), miR-21-5p (assay ID rno481342_mir), miR-26a-5p (assay ID mmu481013_mir), miR-342-3p (assay ID 478043_mir), miR-885-5p (assay ID 478207_mir), miR-375-3p (assay ID 481141_mir), and miR-338-3p (assay ID rno480884_mir). The endogenous controls were selected from sequencing data based on microRNA that did not show significant differences, with a log2 fold change equal to zero and the lowest standard error, and included miR-122-5p (assay ID rno480899_mir), miR-128-3p (assay ID mmu480912_mir) and miR-101 (custom probe SO_66039417_6871885).
Quantitation was performed in a scaled down reaction volume (15 µl) in a CFX Connect Real-Time PCR Detection System (Bio-Rad) using 7.5 μl of 2X TaqMan Fast Advanced Master Mix (catalogue number 4444557), 0.75 μl of miRNA-specific TaqMan Advance assay reagent (20X), 1 μl of cDNA and water to make up the remaining volume. The thermal cycling profile was as follows: 50 °C for 2 min, 95 °C for 3 min and 40 cycles at 95 °C for 15 s and 60 °C for 40 s. No-RT controls and no-template controls were included. The geometric mean of the reference miRNA abundance was used for normalization. Relative quantification of target miRNAs was carried out after sample normalization using the geometric mean of the reference miRNA abundance in Bio-Rad CFX Maestro Software using the 2-ΔΔCq method.
miRNA target prioritization
The target genes of DE-miRNAs were retrieved using MiRWalk 3.061, which includes 3 miRNA target prediction programs (miRDB62, miRTarBase63 and TargetScan64). Analysis was performed on the entire gene sequence (including the 5′UTR, CDS, and 3′UTR). The list of target genes predicted by at least two of the three tools was included in further analysis, mRNA functional enrichment analysis was performed using the DAVID bioinformatic resource65,66, and biological pathways in the KEGG database67 were examined for enrichment.
Statistical analysis
Statistical analysis was performed using XLStat software for Windows (Addinsoft, New York, USA). Data were tested for normality using the Shapiro–Wilk test; as the data were not normally distributed, the nonparametric Wilcoxon signed-rank test for paired samples was applied. Quantitative (miRNA quantification and tumour size) and qualitative (lymph node HN classification7) variables were used for ordination analysis using the ‘Multiple Factor Analysis’ (MFA) function. Receiver operating characteristic (ROC) analysis was performed as previously reported to determine the diagnostic accuracyy68. Statistical significance was accepted at a P value of ≤ 0.05.
Supplementary information
Acknowledgements
The funding for this research was provided by Linea 2-2018, awarded by Università degli Studi di Milano, and by Fondo per il Finanziamento delle Attività Base di Ricerca—FFABR-Lecchi, awarded by Ministero dell’Istruzione, dell’Università e della Ricerca.
Author contributions
C.L. and V.Z. designed the study. F.C. and C.L. guided the performance of the experiments. D.S, R.F. and L.E.C. enrolled patients and surgically removed the tumours. V.G. performed histological classification and assessed the grading of tumours. V.Z. and M.A. performed the laboratory experiments. R.C. performed the bioinformatic data analysis. C.L., V.Z. and G.M. performed the statistical analysis. C.L., F.C. and D.S. provided the funding. C.L. and V.Z. wrote the main manuscript. All authors critically read and approved the manuscript.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-75877-x.
References
- 1.Welle MM, Bley CR, Howard J, Rüfenacht S. Canine mast cell tumours: A review of the pathogenesis, clinical features, pathology and treatment. Vet. Dermatol. 2008;19:321–339. doi: 10.1111/j.1365-3164.2008.00694.x. [DOI] [PubMed] [Google Scholar]
- 2.Mullins MN, et al. Evaluation of prognostic factors associated with outcome in dogs with multiple cutaneous mast cell tumors treated with surgery with and without adjuvant treatment: 54 cases (1998–2004) J. Am. Vet. Med. Assoc. 2006;228:91–95. doi: 10.2460/javma.228.1.91. [DOI] [PubMed] [Google Scholar]
- 3.Stefanello D, et al. Comparison of 2- and 3-category histologic grading systems for predicting the presence of metastasis at the time of initial evaluation in dogs with cutaneous mast cell tumors: 386 cases (2009–2014) J. Am. Vet. Med. Assoc. 2015;246:765–769. doi: 10.2460/javma.246.7.765. [DOI] [PubMed] [Google Scholar]
- 4.Kiupel M, et al. Proposal of a 2-tier histologic grading system for canine cutaneous mast cell tumors to more accurately predict biological behavior. Vet. Pathol. 2011;48:147–155. doi: 10.1177/0300985810386469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patnaik AK, Ehler WJ, MacEwen EG. Canine cutaneous mast cell tumor: Morphologic grading and survival time in 83 dogs. Vet. Pathol. 1984;21:469–474. doi: 10.1177/030098588402100503. [DOI] [PubMed] [Google Scholar]
- 6.Kiupel M, Camus M. Diagnosis and prognosis of canine cutaneous mast cell tumors. Vet. Clin. N. Am. Small. Anim. Pract. 2019;49:819–836. doi: 10.1016/j.cvsm.2019.04.002. [DOI] [PubMed] [Google Scholar]
- 7.Weishaar KM, Thamm DH, Worley DR, Kamstock DA. Correlation of nodal mast cells with clinical outcome in dogs with mast cell tumour and a proposed classification system for the evaluation of node metastasis. J. Comp. Pathol. 2014;151:329–338. doi: 10.1016/j.jcpa.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 8.Sledge DG, Webster J, Kiupel M. Canine cutaneous mast cell tumors: A combined clinical and pathologic approach to diagnosis, prognosis, and treatment selection. Vet. J. 2016;215:43–54. doi: 10.1016/j.tvjl.2016.06.003. [DOI] [PubMed] [Google Scholar]
- 9.Blackwood L, et al. European consensus document on mast cell tumours in dogs and cats. Vet. Comp. Oncol. 2012;10:e1–e29. doi: 10.1111/j.1476-5829.2012.00341.x. [DOI] [PubMed] [Google Scholar]
- 10.Pulz LH, et al. Identification of two molecular subtypes in canine mast cell tumours through gene expression profiling. PLoS One. 2019;14:e0217343. doi: 10.1371/journal.pone.0217343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bracken CP, Scott HS, Goodall GJ. A network-biology perspective of microRNA function and dysfunction in cancer. Nat. Rev. Genet. 2016;17:719–732. doi: 10.1038/nrg.2016.134. [DOI] [PubMed] [Google Scholar]
- 12.Di Leva G, Garofalo M, Croce CM. miRNA in cancer. Annu. Rev. Pathol. 2014;9:287–314. doi: 10.1146/annurev-pathol-012513-104715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rupaimoole R, Calin GA, Lopez-Berestein G, Sood AK. MiRNA deregulation in cancer cells and the tumor microenvironment. Cancer Discov. 2016;6:235–246. doi: 10.1158/2159-8290.CD-15-0893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu J, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 15.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peiró-Chova L, et al. High stability of microRNAs in tissue samples of compromised quality. Virch. Arch. 2013;463:765–774. doi: 10.1007/s00428-013-1485-2. [DOI] [PubMed] [Google Scholar]
- 17.Jones W, et al. Deleterious effects of formalin-fixation and delays to fixation on RNA and miRNA-Seq profiles. Sci. Rep. 2019;9:6980. doi: 10.1038/s41598-019-43282-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maurer M, et al. What is the physiological function of mast cells? Exp. Dermatol. 2003;12:886–910. doi: 10.1111/j.0906-6705.2003.0109a.x. [DOI] [PubMed] [Google Scholar]
- 19.Noli C, Miolo A. The mast cell in wound healing. Vet. Dermatol. 2001;12:303–313. doi: 10.1046/j.0959-4493.2001.00272.x. [DOI] [PubMed] [Google Scholar]
- 20.da Silva EZ, Jamur MC, Oliver C. Mast cell function: A new vision of an old cell. J. Histochem. Cytochem. 2014;62:698–738. doi: 10.1369/0022155414545334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dyduch G, Kaczmarczyk K, Okoń K. Mast cells and cancer: Enemies or allies? Pol. J. Pathol. 2012;63:1–7. [PubMed] [Google Scholar]
- 22.Fenger JM, et al. Overexpression of miR-9 in mast cells is associated with invasive behavior and spontaneous metastasis. BMC Cancer. 2014;14:84. doi: 10.1186/1471-2407-14-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heishima K, et al. Circulating microRNA-214 and -126 as potential biomarkers for canine neoplastic disease. Sci. Rep. 2017;7:2301. doi: 10.1038/s41598-017-02607-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feng YH, Tsao CJ. Emerging role of microRNA-21 in cancer. Biomed. Rep. 2016;5:395–402. doi: 10.3892/br.2016.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grimaldi A, et al. Non-coding RNAs as a new dawn in tumor diagnosis. Semin. Cell Dev. Biol. 2018;78:37–50. doi: 10.1016/j.semcdb.2017.07.035. [DOI] [PubMed] [Google Scholar]
- 26.Javanmardi S, Aghamaali MR, Abolmaali SS, Mohammadi S, Tamaddon AM. miR-21, an oncogenic target miRNA for cancer therapy: Molecular mechanisms and recent advancements in chemo and radio-resistance. Curr. Gene Ther. 2017;16:375–389. doi: 10.2174/1566523217666170102105119. [DOI] [PubMed] [Google Scholar]
- 27.Ushio N, et al. Identification of dysregulated microRNAs in canine malignant melanoma. Oncol. Lett. 2019;17:1080–1088. doi: 10.3892/ol.2018.9692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lai YC, et al. Aberrant expression of microRNAs and the miR-1/MET pathway in canine hepatocellular carcinoma. Vet. Comp. Oncol. 2018;16:288–296. doi: 10.1111/vco.12379. [DOI] [PubMed] [Google Scholar]
- 29.Boggs RM, Wright ZM, Stickney MJ, Porter WW, Murphy KE. MicroRNA expression in canine mammary cancer. Mamm. Genome. 2008;19:561–569. doi: 10.1007/s00335-008-9128-7. [DOI] [PubMed] [Google Scholar]
- 30.Zhao X, Chu J. Microrna-379 suppresses cell proliferation, migration and invasion in nasopharyngeal carcinoma by targeting tumor protein D52. Exp. Ther. Med. 2018;16:1232–1240. doi: 10.3892/etm.2018.6302. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31.Shi X, et al. MicroRNA-379 suppresses cervical cancer cell proliferation and invasion by directly targeting V-crk avian sarcoma virus CT10 oncogene homolog-like (CRKL) Oncol. Res. 2018;26:987–996. doi: 10.3727/096504017X15140534417184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu M, Qin S, Cao F, Ding S, Li M. MicroRNA-379 inhibits metastasis and epithelial-mesenchymal transition via targeting FAK/AKT signaling in gastric cancer. Int. J. Oncol. 2017;51:867–876. doi: 10.3892/ijo.2017.4072. [DOI] [PubMed] [Google Scholar]
- 33.Wu D, et al. MicroRNA-379-5p plays a tumor-suppressive role in human bladder cancer growth and metastasis by directly targeting MDM2. Oncol. Rep. 2017;37:3502–3508. doi: 10.3892/or.2017.5607. [DOI] [PubMed] [Google Scholar]
- 34.Li M, et al. miR-92a family and their target genes in tumorigenesis and metastasis. Exp. Cell Res. 2014;323:1–6. doi: 10.1016/j.yexcr.2013.12.025. [DOI] [PubMed] [Google Scholar]
- 35.Zhang X, Li Y, Qi P, Ma Z. Biology of MiR-17-92 cluster and its progress in lung cancer. Int. J. Med. Sci. 2018;15:1443–1448. doi: 10.7150/ijms.27341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li X, Guo S, Min L, Guo Q, Zhang S. miR-92a-3p promotes the proliferation, migration and invasion of esophageal squamous cell cancer by regulating PTEN. Int. J. Mol. Med. 2019;44:973–981. doi: 10.3892/ijmm.2019.4258. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Yu H, et al. MiR-92a modulates proliferation, apoptosis, migration, and invasion of osteosarcoma cell lines by targeting Dickkopf-related protein 3. Biosci. Rep. 2019;39:20. doi: 10.1042/BSR20190410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tao J, et al. microRNA-18a, a member of the oncogenic miR-17-92 cluster, targets Dicer and suppresses cell proliferation in bladder cancer T24 cells. Mol. Med. Rep. 2012;5:167–172. doi: 10.3892/mmr.2011.591. [DOI] [PubMed] [Google Scholar]
- 39.Ottman R, Levy J, Grizzle WE, Chakrabarti R. The other face of miR-17-92a cluster, exhibiting tumor suppressor effects in prostate cancer. Oncotarget. 2016;7:73739–73753. doi: 10.18632/oncotarget.12061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shin VY, et al. MiR-92 suppresses proliferation and induces apoptosis by targeting EP4/Notch1 axis in gastric cancer. Oncotarget. 2018;9:24209–24220. doi: 10.18632/oncotarget.24819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Y, Qin H. miR-338-3p targets RAB23 and suppresses tumorigenicity of prostate cancer cells. Am. J. Cancer Res. 2018;8:2564–2574. [PMC free article] [PubMed] [Google Scholar]
- 42.Liang Y, et al. The EGFR/miR-338-3p/EYA2 axis controls breast tumor growth and lung metastasis. Cell Death Dis. 2017;8:e2928. doi: 10.1038/cddis.2017.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang C, Li H, Wang J, Zhang J, Hou X. MicroRNA-338-3p suppresses cell proliferation, migration and invasion in human malignant melanoma by targeting MACC1. Exp. Ther. Med. 2019;18:997–1004. doi: 10.3892/etm.2019.7644. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 44.Long J, Luo J, Yin X. MiR-338-5p promotes the growth and metastasis of malignant melanoma cells via targeting CD82. Biomed. Pharmacother. 2018;102:1195–1202. doi: 10.1016/j.biopha.2018.03.075. [DOI] [PubMed] [Google Scholar]
- 45.Shan Y, et al. MicroRNA-338 inhibits migration and proliferation by targeting hypoxia-induced factor 1α in nasopharyngeal carcinoma. Oncol. Rep. 2015;34:1943–1952. doi: 10.3892/or.2015.4195. [DOI] [PubMed] [Google Scholar]
- 46.Su M, Qin B, Liu F, Chen Y, Zhang R. Mir-885-5p upregulation promotes colorectal cancer cell proliferation and migration by targeting suppressor of cytokine signaling. Oncol. Lett. 2018;16:65–72. doi: 10.3892/ol.2018.8645. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47.Zhang Z, et al. miR-885-5p suppresses hepatocellular carcinoma metastasis and inhibits Wnt/β-catenin signaling pathway. Oncotarget. 2016;7:75038–75051. doi: 10.18632/oncotarget.12602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blacklock KB, et al. Identification of molecular genetic contributants to canine cutaneous mast cell tumour metastasis by global gene expression analysis. PLoS One. 2018;13:e0208026. doi: 10.1371/journal.pone.0208026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferrari R, et al. The impact of extirpation of non-palpable/normal-sized regional lymph nodes on staging of canine cutaneous mast cell tumours: A multicentric retrospective study. Vet. Comp. Oncol. 2018;16:505–510. doi: 10.1111/vco.12408. [DOI] [PubMed] [Google Scholar]
- 50.Marconato L, et al. Therapeutic impact of regional lymphadenectomy in canine stage II cutaneous mast cell tumours. Vet. Comp. Oncol. 2018;16:580–589. doi: 10.1111/vco.12425. [DOI] [PubMed] [Google Scholar]
- 51.Kamstock DA, et al. Recommended guidelines for submission, trimming, margin evaluation, and reporting of tumor biopsy specimens in veterinary surgical pathology. Vet. Pathol. 2011;48:19–31. doi: 10.1177/0300985810389316. [DOI] [PubMed] [Google Scholar]
- 52.Stefanello D, et al. Ultrasound-guided cytology of spleen and liver: A prognostic tool in canine cutaneous mast cell tumor. J. Vet. Intern. Med. 2009;23:1051–1057. doi: 10.1111/j.1939-1676.2009.0354.x. [DOI] [PubMed] [Google Scholar]
- 53.Thompson JJ, et al. Canine subcutaneous mast cell tumors: Cellular proliferation and KIT expression as prognostic indices. Vet. Pathol. 2011;48:169–181. doi: 10.1177/0300985810390716. [DOI] [PubMed] [Google Scholar]
- 54.Bustin SA, et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009;55:611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 55.Pardini B, et al. microRNA profiles in urine by next-generation sequencing can stratify bladder cancer subtypes. Oncotarget. 2018;9:20658–20669. doi: 10.18632/oncotarget.25057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Beccuti M, et al. SeqBox: RNAseq/ChIPseq reproducible analysis on a consumer game computer. Bioinformatics. 2018;34:871–872. doi: 10.1093/bioinformatics/btx674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cordero F, Beccuti M, Arigoni M, Donatelli S, Calogero RA. Optimizing a massive parallel sequencing workflow for quantitative miRNA expression analysis. PLoS One. 2012;7:e31630. doi: 10.1371/journal.pone.0031630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011;17:10. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 59.Rumble SM, et al. SHRiMP: Accurate mapping of short color-space reads. PLoS Comput. Biol. 2009;5:e1000386. doi: 10.1371/journal.pcbi.1000386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lawrence M, et al. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013;9:e1003118. doi: 10.1371/journal.pcbi.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sticht C, De La Torre C, Parveen A, Gretz N. Mirwalk: An online resource for prediction of microRNA binding sites. PLoS One. 2018;13:e0206239. doi: 10.1371/journal.pone.0206239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wong N, Wang X. miRDB: An online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015;43:D146–D152. doi: 10.1093/nar/gku1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hsu SD, et al. MiRTarBase: A database curates experimentally validated microRNA-target interactions. Nucleic Acids Res. 2011;39:D163–169. doi: 10.1093/nar/gkq1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4:20. doi: 10.7554/eLife.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 67.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40:D109–114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lecchi C, et al. Characterization of circulating miRNA signature in water buffaloes (Bubalus bubalis) during Brucella abortus infection and evaluation as potential biomarkers for non-invasive diagnosis in vaginal fluid. Sci. Rep. 2019;9:1945. doi: 10.1038/s41598-018-38365-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.