

Abstract
Release of the neuromodulator noradrenaline signals salience during wakefulness, flagging novel or important experiences to reconfigure information processing and memory representations in the hippocampus. Noradrenaline is therefore expected to enhance hippocampal responses to synaptic input; however, noradrenergic agonists have been found to have mixed and sometimes contradictory effects on Schaffer collateral synapses and the resulting CA1 output. Here, we examine the effects of endogenous, optogenetically driven noradrenaline release on synaptic transmission and spike output in mouse hippocampal CA1 pyramidal neurons. We show that endogenous noradrenaline release enhances the probability of CA1 pyramidal neuron spiking without altering feedforward excitatory or inhibitory synaptic inputs in the Schaffer collateral pathway. β-adrenoceptors mediate this enhancement of excitation-spike coupling by reducing the charge required to initiate action potentials, consistent with noradrenergic modulation of voltage-gated potassium channels. Furthermore, we find the likely effective concentration of endogenously released noradrenaline is sub-micromolar. Surprisingly, although comparable concentrations of exogenous noradrenaline cause robust depression of slow afterhyperpolarization currents, endogenous release of noradrenaline does not, indicating that endogenous noradrenaline release is targeted to specific cellular locations. These findings provide a mechanism by which targeted endogenous release of noradrenaline can enhance information transfer in the hippocampus in response to salient events.
Keywords: CA1, hippocampus, locus coeruleus, noradrenaline, synaptic transmission
Introduction
The locus coeruleus (LC) is the major source of noradrenergic innervation projecting widely and diffusely throughout the brain (Berridge and Waterhouse 2003; Schwarz et al. 2015; Kebschull et al. 2016). LC neurons respond to novel or salient events, and the release of noradrenaline (NA) is proposed to signal that new schemas need to be formed (Aston-Jones and Cohen 2005; Bouret and Sara 2005; Yu and Dayan 2005; Sales et al. 2019). To achieve this, NA must activate brain areas associated with memory formation and reconfigure information processing to prioritize new information over internal representations. In addition, despite exhibiting very little activity during sleep, LC activity is critical in sleep–wake transitions in response to sensory input (Hayat et al. 2020) and exhibits tonic firing during exploration (Vankov et al. 1995). Therefore, LC activity plays a role in integrating real-time sensory input into cortical representations and decisions.
The hippocampus is important for memory formation, consolidation, and retrieval (Bird and Burgess 2008), and all subfields receive dense noradrenergic innervation (Walling et al. 2012; Schwarz et al. 2015; Wagatsuma et al. 2018). Rodents with disrupted LC input (Compton et al. 1995; Takeuchi et al. 2016), blockade of hippocampal adrenoceptors (Ji et al. 2003; Lemon et al. 2009) or deletion of NA (Murchison et al. 2004) all have impairments in memory and learning, whereas stimulation of LC fibers in the hippocampus reorganizes place cell representations and enhances memory (Takeuchi et al. 2016; Kaufman et al. 2020), leading to the idea that NA is an integral regulator of memory processing. However, the key targets for NA release and the mechanisms by which NA reconfigures neuronal networks in the hippocampus are not well understood.
NA signals via 3 principle G-protein-coupled receptor subtypes—α1-, α2-, and β-adrenoceptors (ARs) (Ramos and Arnsten 2007). β-ARs can be further sub-divided into β1-, β2-, and β3-ARs, which, despite all signaling via the Gs protein, have distinct downstream targets (Madison and Nicoll 1982, 1986; Hagena and Manahan-Vaughan 2017; Liu et al. 2017). Within the hippocampus, the major effects of NA on cellular excitability are mediated via β-ARs where the most robust effect is inhibition of the slow afterhyperpolarization (sAHP) (Madison and Nicoll 1982; Haas and Konnerth 1983; Sah and Isaacson 1995), which has been correlated with increased spiking and enhanced learning (Oh et al. 2003; Disterhoft and Oh 2006). In addition, NA also enhances intrinsic excitability via inhibition or downregulation of voltage-gated potassium channels located on the dendrites of CA1 pyramidal neurons (Yuan et al. 2002; Liu et al. 2017). In contrast, activation of α2-ARs hyperpolarizes neurons thereby reducing excitability (Madison and Nicoll 1986; Ul Haq et al. 2012). It is not clear which of these possible mechanisms is engaged by endogenous release of NA.
NA also modulates synaptic communication within hippocampal networks where it facilitates long-term potentiation (LTP) via β-AR enhancement of GluA1 surface expression (Hu et al. 2007; Hagena and Manahan-Vaughan 2012; O'Dell et al. 2015; Hansen and Manahan-Vaughan 2015a; Liu et al. 2017; Palacios-Filardo and Mellor 2019). However, NA has mixed acute effects on basal synaptic transmission. NA has either negligible or small depressant effects on excitatory inputs in the hippocampus (Heginbotham and Dunwiddie 1991; Katsuki et al. 1997), yet despite this can significantly augment population spikes (Dunwiddie et al. 1992; Raman et al. 1996; Lin et al. 2003; O'Dell et al. 2010; Murchison et al. 2011; Ul Haq et al. 2012; Liu et al. 2017). Within inhibitory networks, α1-ARs not only depolarize interneurons and enhance inhibitory drive onto CA1 pyramidal neurons but also depress evoked inhibitory synaptic transmission (Madison and Nicoll 1988; Doze et al. 1991; Bergles et al. 1996). The overall effect of NA on excitatory:inhibitory ratio is unknown, and it is unclear whether acute effects on synaptic transmission contribute to the output from CA1 pyramidal neurons.
Previous studies have primarily utilized pharmacological activation of ARs with supra-micromolar NA concentrations or specific AR agonists. However, estimates suggest that endogenous NA concentrations are sub-micromolar (Harley et al. 1996; Muller et al. 2014; Feng et al. 2019). Therefore, we sought to examine the acute effects of endogenous noradrenaline release from LC terminals on synaptic and cellular properties within the CA1 region of the hippocampus to test whether endogenous NA enhances CA1 output. We found that β-AR activation via NA released from LC terminals enhances the spike output of CA1 pyramidal neurons without altering feedforward excitatory or inhibitory synaptic input. Our data confirm estimates of effective endogenous NA in the sub-micromolar (600 nM) concentration range but found that it did not inhibit sAHP current and is therefore targeted to specific cellular domains. Instead, endogenous NA reduced the charge required to trigger action potentials, which enhanced excitation-spike coupling.
Methods
All experiments were performed in accordance with UK Home Office legislation after approval by the University of Bristol Animal Welfare and Ethics Review Board.
Stereotaxic Viral Vector Injections
Male mice aged 28–35 days were anesthetized for surgery with an intraperitoneal (IP) injection of a ketamine (70 mg/kg, Vetalar, Pharmacia, UK) and medetomidine (0.5 mg/kg, Domitor, Pfizer, UK) mixture. After positioning in a stereotaxic frame (Neurostar drill and injection robot), a burr hole (∅ 1.0 mm) was made over the right LC at coordinates from lambda, AP: 4.12 mm (corrected using variation from the average bregma-lambda distance of 4.2 mm, Franklin and Paxinos Mouse Atlas, 3rd edn), ML: 1 mm. A glass micropipette with a tip diameter of 20–30 μm pulled on a vertical puller (Harvard) containing the viral vector was advanced with a 20°-rostral angulation to a depth of 3.9 mm from brain surface. Three injections (250 nL each, at a speed of 125 nL/min) of the CAV2-PRS-ChR2-mCherry viral vector that expresses mCherry tagged ChR2 selectively in noradrenergic neurons (Li et al. 2016) were made at 3.5, 3.7, and 3.9 mm depths.
Ex Vivo Slice Preparation
Transverse dorsal hippocampal slices were prepared from P21 to P70 C57BL/6J male mice. Following cervical dislocation, brains were immediately removed and immersed in ice-cold sucrose-based cutting solution containing the following (in mM): 205 sucrose, 10 glucose, 26 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, and 5 MgSO4. Individual hippocampi were dissected and mounted on agar and 400-μm slices were cut using a VT1200 vibratome (Leica). Following dissection, slices were transferred to aCSF containing the following (in mM): 119 NaCl, 10 glucose, 26 NaHCO3, 2.5 KCl, 1 NaH2PO4, 1.3 MgSO4, and 2.5 CaCl2, maintained at ~35 °C for 30 min, and then stored at room temperature. Slices were left for a minimum of 1 h after dissection before recordings were made. All solutions were saturated with 95% O2 and 5% CO2.
Electrophysiological Recordings
Slices were submerged in a recording chamber (~3 mL) perfused at 2–4 mL/min with aCSF (as above) at 31–32 °C. CA1 pyramidal cells were visualized using infrared-differential interference contrast (DIC) optics on an Olympus BX-50WI microscope. Patch electrodes with a resistance of 2–8 MΩ were pulled from borosilicate filamented glass capillaries (1.5 OD × 0.86 ID × 100 L mm, Harvard Apparatus) using a vertical puller (PC-10; Narishige) or horizontal puller (P-87, Sutter Instruments). For voltage-clamp synaptic recordings, pipettes were filled with intracellular solution containing the following (mM): 130 CsMeSO3, 4 NaCl, 10 HEPES, 0.5 EGTA, 10 TEA, 2 Mg-ATP, 0.5 Na-GTP, 1 QX-314, at pH 7.3, 280–290 mOsm. For voltage-clamp sAHP current recordings pipettes were filled with intracellular solution (mM): 117 KMeSO3, 8 NaCl, 10 HEPES, 4 Mg-ATP, 0.3 Na-GTP, 0.2 EGTA, and 1 MgCl, pH 7.4, 280–290 mOsm. For current-clamp recordings, pipettes were filled with intracellular solution (mM): 125 K-gluconate, 5 NaCl, 1 MgCl2, 10 HEPES, 0.2 EGTA, 4 Mg-ATP, 0.3 Na-GTP, pH 7.4, 280–290 mOsm. Whole-cell patch-clamp recordings from CA1 pyramidal neurons were made with an AxoPatch 200B amplifier (Molecular Devices), and the current signals were low-pass filtered at 2 or 5 kHz (synaptic currents) or 1 kHz (sAHP currents), and digitized at 10 kHz (synaptic recordings) or 50 kHz (current clamp recordings) using a CED Power 1401 data acquisition board and Signal acquisition software (CED). Junction potentials were not corrected for.
To examine changes in spike probability in response to Schaffer collateral synaptic input, 10 square voltage stimuli (0.1 ms) at 10 Hz were delivered to axons in the stratum radiatum using a bipolar tungsten microelectrode. The stimulation intensity was adjusted such that approximately half of the resulting excitatory post-synaptic potential (EPSP) responses yielded action potentials (i.e., spike probability (Pspike) = 0.5). Following bath application of NA, current injection to the soma was used to adjust the membrane potential (Vm) back to its baseline value to determine whether any effects of NA were mediated by the change in Vm. Throughout experiments, input resistance (Rin) was monitored with hyperpolarizing current injections of 25 pA.
To induce endogenous NA release from LC terminals, hippocampal slices were stimulated with 5-ms duration pulses from a 473-nm LED (2.3 mW/mm2, Thor Labs) delivered through the objective lens. Tonic NA release was mimicked by 1 Hz repetitive stimulation for 10 min whereas phasic NA release was induced by 10 light pulses every 20 s at 25 Hz for 10 min. Both protocols finished 10 ms prior to the start of Schaffer collateral stimulation in each recording sweep. In all slices used for endogenous NA release, expression of ChR2-mCherry was confirmed post hoc by immunohistochemistry. Control experiments for light stimulation were conducted in naïve slices from animals that did not receive virus injection.
To measure feedforward excitatory and inhibitory synaptic input, CA1 pyramidal neurons were clamped at the GABAA receptor reversal potential (EGABA, ~−65 mV, measured separately in experiments using 10 μM NBQX) or the AMPA receptor reversal potential (EAMPA, ~+5 mV, measured separately in experiments using 50 μM of picrotoxin and 1 μM CGP-55845) to isolate excitatory and inhibitory post-synaptic currents (EPSCs and IPSCs) in response to stimulation of Schaffer collateral axons, respectively (5 pulses at 10 Hz, every 20 s). Stimulation intensity was adjusted to obtain EPSC or IPSC amplitudes of 50–150 pA. Following IPSC recordings, the AMPA/kainate receptor antagonist NBQX (10 μM) was applied to confirm IPSCs were generated by feedforward inhibition resulting from Schaffer collateral stimulation and not direct stimulation of the interneurons. Responses not inhibited by ≥75% within 10 min of NBQX application were discarded from subsequent analysis. Series resistance was monitored throughout voltage-clamp experiments, and synaptic recordings were rejected from analysis if the series resistance increased by more than 30% or greater than 30 MΩ. sAHP currents were elicited by depolarizing pyramidal cells by 70 mV from a holding potential of −50 mV for 120 ms every 20 s. Assessment of excitation-spike coupling was performed by 1-s depolarizing current injections with the current amplitude adjusted to be just supra-threshold for action potential initiation (rheobase).
Immunohistochemistry
For perfuse fixation, mice were given a terminal anesthetic (Pentobarbital, Euthatal) and then perfused with 4% paraformaldehyde. The cerebrum and brain stem were separated and post-fixed in 4% PFA for >24 h before cryoprotection in 30% sucrose in 0.1 M phosphate buffer (PB) at least 24 h before slice preparation. Brainstem or hippocampi were then mounted on a cryotome (Leica) using Cryomatrix (ThermoFisher Scientific) and 40 μm (pons) and 50 μm (hippocampus) slices cut then stored in 0.1 M PB + 0.02% NaN3. Free-floating slices were washed in 0.1 M PB (3×) before transfer to 50% alcohol:dH2O for 30 min and washing again in 0.1 M PB (3×). Slices were then incubated overnight in primary antibodies: rabbit anti-mCherry (1:4000, BioVision) and/or sheep anti-DβH (1:1000, Merck Millipore) in 0.1 M PB + 0.3% Triton X-100 containing 5% donkey or goat serum (Sigma-Aldrich). The following day slices were washed in 0.1 M PB (×3) and then incubated for >3 h with appropriate secondary antibodies: goat anti-rabbit Alexa-Fluor 594 (1:1000, Invitrogen) and/or donkey anti-sheep IgG Alexa-Fluor 488 (1:400, Jackson ImmunoResearch) in 0.1 M PB + 0.3% Triton X-100 containing 2% donkey or goat serum. Slices were then transferred to a final solution (0.1 M PB + 1:10 000 DAPI, Invitrogen) before mounting on slides. Slides were imaged using a Leica fluorescent microscope and LASX imaging software (Leica) and fluorescent neurons were manually counted to examine mCherry and DβH expression.
Data Analysis
Data were analyzed using Signal software (CED) and custom MATLAB scripts. In spike output experiments, spikes were detected on-line if the recorded voltage passed a threshold (0 mV). Spike probability was calculated as the average number of spikes per train of stimuli over a minute period divided by the number of EPSPs in a train (10). Spike latency was determined off-line using a custom MATLAB script that calculated the time between the stimulation artifact or initiation of current injection and the peak of the action potential. Jitter in spike latency was calculated as the standard deviation of spike latencies for the first spike response to stimulation. Spike threshold was calculated from the first derivative of the voltage trace at the point at which dV/dT exceeded 30 V/s. The charge transfer required to elicit an action potential was calculated as the area under the voltage trace between the start of current injection and the spike threshold. EPSP amplitudes were measured from average traces pooled from consecutive responses within each minute bin and calculated as the peak voltage reached 5–50 ms after stimulation. Only the first EPSP within a train was analyzed and only if no spike was detected on that EPSP. Input resistance was measured as the steady-state voltage deflection in response to current injections at the start of each sweep. IsAHP amplitude was measured as the peak current amplitude 100–500 ms after termination of the depolarizing pulse to ensure clear distinction from the medium afterhyperpolarization and compared to the current value 30 ms before the depolarizing pulse. A mean was then taken of the values 10 ms before and 10 ms after this peak amplitude. EPSC and IPSC amplitudes were measured at the detected peak amplitude 5–20 ms after the stimulation artifact and subtracted from the baseline current immediately before the stimulation artifact. To average across experiments for responses that varied substantially in baseline amplitude, responses were normalized to the average during the 10 min prior to NA application. The paired-pulse ratio (PPR) for PSCs was calculated as the second peak divided by the first peak (P2/P1).
Experimental unit was defined as cell, and only one cell was recorded per slice. On average ~2 cells were recorded per animal (range 1–4). Data were analyzed blind to the expression of ChR2. Data are plotted as the mean ± standard error of the mean (SEM). Data points on bar graphs used in analysis are the average of 5 min periods from each cell representing the experimental condition. Paired Student’s t-tests were used to compare normalized changes from baseline, whereas unpaired Student’s t-tests were used when comparing changes between two experimental groups (unless data were not normally distributed, wherein a Mann-Whitney test was used instead). Repeated-measures ANOVA with Tukey’s multiple comparison tests were used to analyze data involving multiple experimental conditions (i.e., baseline, drug/optostimulation, Vm reset). A one-way ANOVA with a Dunnett’s multiple comparison test was used to compare the different experimental conditions to a WT control group. *, **, ***, and **** denote P < 0.05, 0.01, 0.001, and 0.0001, respectively. Data were aggregated in Microsoft Excel, and statistical testing was performed using GraphPad 7. Traces and graphs were generated in GraphPad Prism 7 or MATLAB 2017a.
Drugs and Reagents
DL-noradrenaline hydrochloride and DL-propranolol hydrochloride were purchased from Sigma-Aldrich, fresh stock solutions (50 mM) were made up in dH2O each day and stored on ice. Picrotoxin (Sigma-Aldrich), CGP-55845 (HelloBio) and NBQX di-sodium salt (HelloBio) stock solutions (5 mM, 1 mM, and 20 μM, respectively) were made up in dH2O and stored at −20 °C.
Results
Optoactivation of LC Neurons
To measure the effects of endogenous NA release from LC fibers in the hippocampus, channelrhodopsin (ChR2) was expressed in noradrenergic neurons in LC by injection of CAV-PRS-ChR2-mCherry viral vector in mice (Li et al. 2016; Hirschberg et al. 2017) (Fig. 1A). 3 weeks after injection, strong ChR2-mCherry expression was found in cell bodies and fibers within the LC that co-localized almost exclusively with dopamine-β-hydroxylase (Fig. 1B,C; 94.5 ± 2.3% mCherry-expressing cells also stained positive for DβH, n = 5). Axonal fibers expressing ChR2-mCherry were also found in a dense network throughout all regions of the hippocampus (Fig. 1D). ChR2-mCherry expression was confirmed in all animals and slices used for subsequent electrophysiological experiments measuring the impact of endogenous NA release. Whole-cell current-clamp recordings from ChR2-mCherry-expressing LC neurons showed that brief 5-ms light pulses reliably elicited action potentials up to 25-Hz stimulation frequency but above this frequency action potentials became attenuated in amplitude and fidelity (average for each stimulation across 3 trials shown in Fig. 1E,F).
Figure 1.
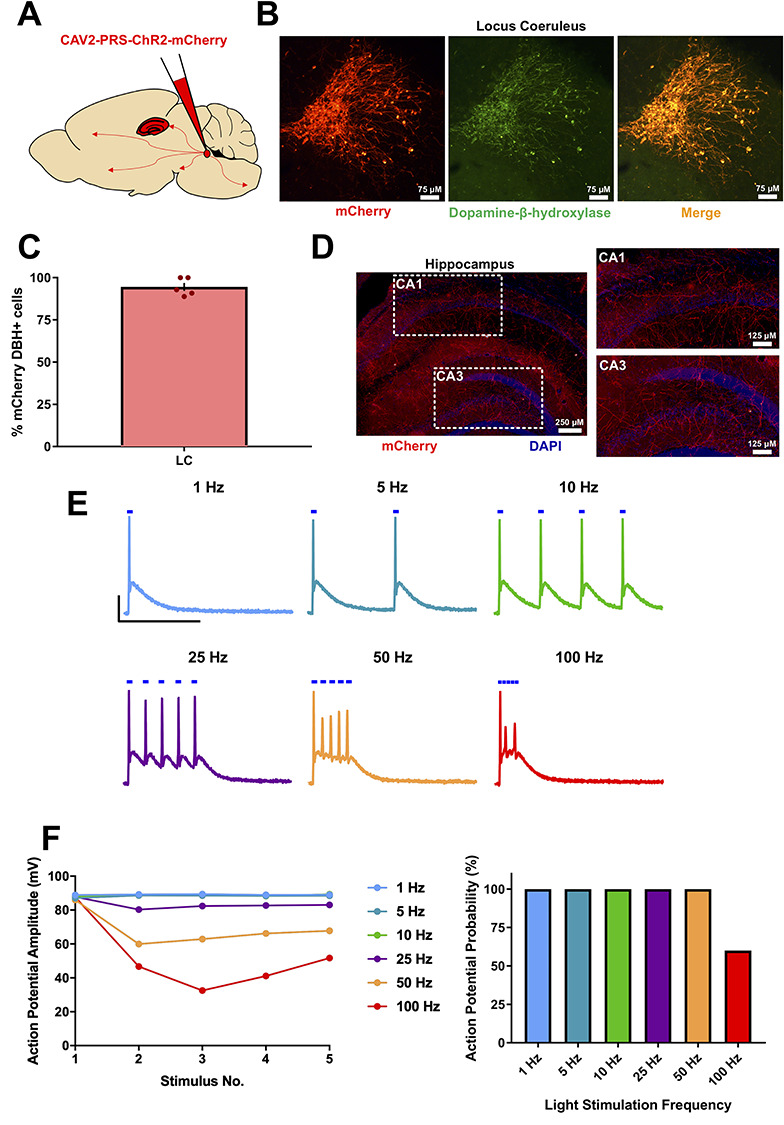
Characterization of the CAV2-PRS-mCherry-ChR2 viral vector expression in mice. (A) Schematic for viral vector injections into the Locus Coeruleus (LC). (B) mCherry fluorescence co-localized with dopamine-β-hydroxylase fluorescence, confirming successful noradrenergic neuronal targeting of the viral vector. (C) % of mCherry+ neurons also showing dopamine-β-hydroxylase expression. (D) mCherry fluorescence in LC fibers within the hippocampus confirming expression of ChR2 in axons. (E and F) Light stimulation of ChR2-mCherry-expressing LC neurons recorded in current-clamp configuration reliably elicited action potentials with stimulation frequencies up to 25 Hz after which amplitude and fidelity began to deteriorate. Scale bars = 25 mV, 200 ms.
Endogenous NA Enhances CA1 Spike Output Via β-AR Activation
We next sought to test whether endogenously released NA from LC fibers altered the spike output of CA1 pyramidal neurons in response to synaptic input (Fig. 2A). A series of 10 stimuli at 10 Hz were given to the Schaffer collateral fibers with the stimulation intensity adjusted such that approximately five of the 10 elicited EPSPs generated a spike (i.e., spike probability (Pspike) = 0.5). This gave us the opportunity to identify both positive and negative modulation of spike output. Optogenetic stimulation (1 Hz for 10 min) enhanced spike probability of CA1 neurons in slices taken from animals expressing ChR2 in LC by 42.3 ± 8.6% but not in slices from naïve animals and therefore not expressing ChR2 (Fig. 2B–D; 1 Hz ChR2, Pspike = 0.69 ± 0.05, n = 12, P = 0.0003 compared to baseline; 1 Hz non-ChR2, Pspike = 0.49 ± 0.05, n = 8, P = 0.835 compared to baseline). This enhancement of spike output by endogenous NA release was completely blocked by application of the β-AR-selective antagonist, propranolol (500 nM) applied 5 min prior and during light stimulation (Fig. 2E,F; 1 Hz propranolol, Pspike = 0.51 ± 0.08, n = 6, P = 0.903 compared to baseline). This indicates that the enhancement of spike output is mediated by activation of β-ARs and also that there is little endogenous NA tone within the slices.
Figure 2.
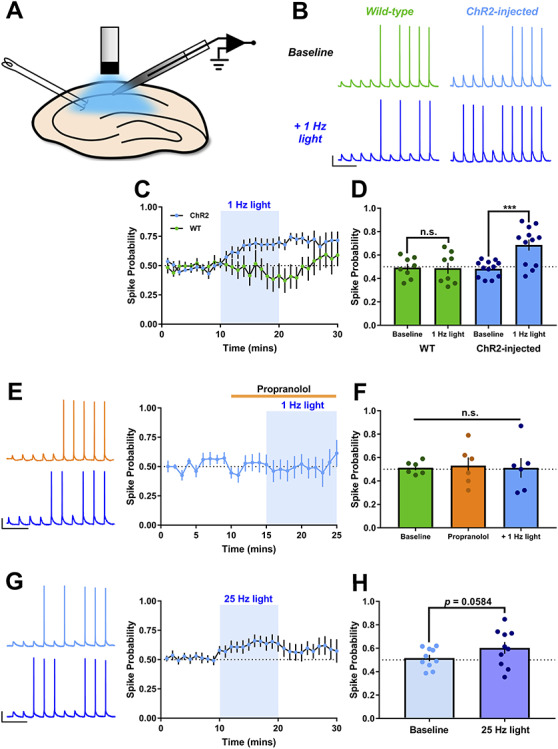
Endogenous NA release enhances CA1 spike output via β-AR activation. (A) Schematic showing ex vivo slice recording and optogenetic stimulation. (B–D) Example traces (B), experiment timecourse (C) and grouped data (D) show 1 Hz tonic light stimulation (10 min, LED) to release endogenous NA enhances the spike probability in CA1 pyramidal neurons in response to Schaffer collateral synaptic stimulation (10 stimuli at 10 Hz) in slices from ChR2-injected but not WT mice. Scale bars = 25 mV, 250 ms. (E and F) Pre-incubation of slices with the β-AR antagonist, propranolol (500 nM) for 5 min prior to light stimulation blocked the increase in spike probability. Scale bars = 25 mV, 250 ms. (G and H) Phasic light stimulation (10 pulses at 25 Hz, immediately before synaptic stimuli) also increased spike probability but the increase was more variable. Scale bars = 25 mV, 250 ms.
LC noradrenergic neurons exhibit two distinct firing modes often classed as tonic and phasic equating to prolonged firing at ~1 Hz (tonic) or burst firing at ~25 Hz (phasic) (Berridge and Waterhouse 2003; Aston-Jones and Cohen 2005; Bouret and Sara 2005; Vazey et al. 2018). These different modes will likely lead to different NA concentration profiles, which may change the effect of NA in a similar manner to that seen for dopamine release in the hippocampus (Rosen et al. 2015). We therefore next mimicked phasic release of NA using bursts of 10 light pulses at 25 Hz repeated every 20 s in slices from ChR2 expressing animals. Phasic NA release caused a similar but slightly smaller enhancement of spike output (Fig. 2G,H; Pspike = 0.59 ± 0.048, n = 11, P = 0.058 compared to baseline) compared to tonic (1 Hz) release. We therefore used tonic 1 Hz stimulation as the more effective stimulation frequency for subsequent experiments testing the effects of endogenous NA release.
Dose-Dependent Enhancement of CA1 Spike Output by Exogenous NA
The presence of ARs, potentially at some distance from NA terminals and release sites, makes it difficult to determine the precise effective concentration of endogenous NA at receptors. Reports suggest that NA release from LC terminals is likely in the sub-micromolar to low micromolar range (Harley et al. 1996; Muller et al. 2014; Feng et al. 2019) but this may vary depending on the brain region, the neuron type, and the target receptors. To estimate the concentration of endogenous NA release in our experiments, we explored the dose-response relationship for exogenous bath-applied NA concentrations ranging from 200 nM to 20 μM assayed on CA1 spike output.
200 nM NA had no effect on spike probability (Fig. 3A,B; Pspike = 0.57 ± 0.08, n = 8, P = 0.702 compared to baseline). However, 600 nM NA produced a robust increase in the spike output of CA1 pyramidal neurons by 37.6 ± 10.9% (Fig. 3C,D; Pspike = 0.72 ± 0.05, n = 7, P = 0.029 compared to baseline). This increase was accompanied by a small and variable depolarization of the resting membrane potential (ΔVm = +2.85 ± 1.55 mV, P = 0.114). In recordings where neurons depolarized, Vm was reset after 10 min to test whether the enhanced spike probability was due to depolarization but resetting Vm did not reverse the increase in spike probability (Pspike = 0.64 ± 0.10, n = 7, P = 0.481 compared to 600 nM NA). Surprisingly, when the concentration of exogenous NA was increased to 2 μM the enhancement of spike probability disappeared and this was not affected by resetting Vm (Fig. 3E,F; Pspike = 0.43 ± 0.08, n = 8, P = 0.934 compared to 2 μM NA). Furthermore, when the concentration of exogenous NA was further increased to 20 μM spike probability was dramatically reduced to 10.7 ± 10.1% (Fig. 3G,H; Pspike = 0.05 ± 0.04, P = 0.003 compared to baseline, n = 5). The reduction in spike probability was accompanied by a variable hyperpolarization of CA1 pyramidal neurons (ΔVm = −2.06 ± 1.31 mV, n = 5, P = 0.187 compared to baseline), but resetting Vm did not reverse the reduction in spike probability (0.08 ± 0.06, P = 0.877 compared to 20 μM NA).
Figure 3.
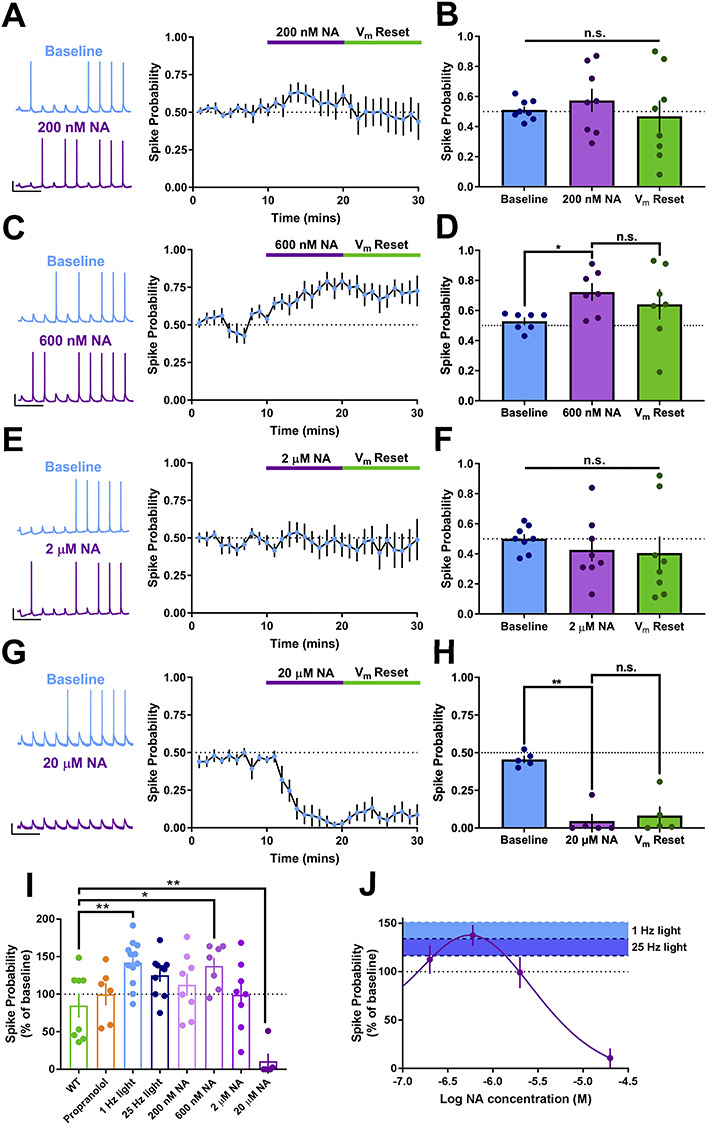
Biphasic dose-response relationship for exogenous NA on CA1 spike output. (A–H) Example traces, averaged timecourse and grouped data for spike probability experiments using exogenous application of 200 nM (A and B), 600 nM (C and D), 2 μM (E and F) and 20 μM NA (G and H). (I) Percentage change in spike probability for all conditions. (J) Dose-response curve for change in spike probability with concentration of NA. Overlaid blue shading indicates the mean and SEM change in spike probability produced by 1 or 25 Hz light stimulation of endogenous NA release. Overlap between blue shading and dose-response curve indicates likely concentration of endogenous NA release. Scale bars = 20 mV, 250 ms.
To compare changes in spike probability and construct a dose-response relationship we normalized changes in spike probability for each cell and averaged for each condition (Fig. 3I). This revealed a biphasic dose-response relationship for exogenous NA (Fig. 3J), from which it was possible to estimate the effective concentration of endogenous optogenetically-evoked NA. Endogenous NA evoked with 1 Hz optostimulation (light blue bar is the ± SEM range of this result set) yielded a change in spike probability comparable to the peak of the biphasic dose-response curve corresponding to 600 nM exogenous NA, providing a best estimate for the concentration of endogenous NA.
Effects of NA on Feedforward Excitatory and Inhibitory Synaptic Inputs to CA1
An enhancement of spike probability and excitation-spike coupling could result from changes in synaptic input to CA1 neurons or from changes to CA1 neuronal intrinsic excitability and synaptic integration. We assessed which of these factors is the key mediator of enhanced spike probability, focusing first on synaptic inputs.
Within the spike probability experiments it was possible to extract and quantify the strength of the synaptic input by measuring the amplitude of the first EPSP in the stimulus train, since the first EPSP rarely generated an action potential. Analysis of these data revealed that EPSP amplitude was not altered by endogenous NA release or by exogenous NA applied at concentrations of 200 nM, 600 nM or 2 μM, but was reduced by 20 μM NA (Fig. 4A–C,I; endogenous NA = 93.6 ± 2.3% of baseline, n = 11, P = 0.408 compared to WT; 600 nM NA = 95.13 ± 13.7% of baseline, n = 7, P = 0.750 compared to baseline; 20 μM NA, 77.16 ± 3.79% of baseline, n = 5, P = 0.049 compared to baseline), and this EPSP depression was not rescued by resetting Vm. These data suggest one cause of the reduced spike output in response to 20 μM NA is an attenuation of excitatory synaptic input. The data also indicate that enhancement of spike probability by endogenous NA release or 600 nM exogenous NA does not result from increases in excitatory synaptic input.
Figure 4.
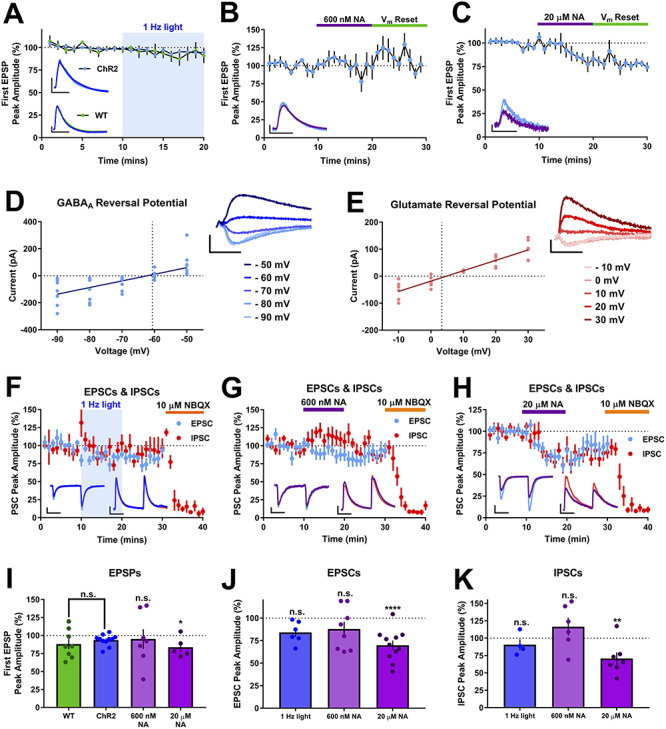
Endogenous NA release has no effect on Schaffer collateral feedforward excitatory and inhibitory synaptic transmission. (A–C) EPSPs recorded during spike probability experiments were unaffected by endogenous NA release (A) or 600 nM NA (B) but depressed by 20 μM NA (C). Plots in A show EPSPs recorded in slices from virus injected and WT animals. Insets in panels show example EPSPs. Scale bars = 2 mV, 20 ms. (D and E) Experimentally determined reversal potentials for GABA and glutamate receptors, respectively. Current-voltage plots and example traces for IPSCs (D) and EPSCs (E). Scale bars = 200 pA and 100 ms (GABAA reversal traces), 50 pA and 100 ms (glutamate reversal traces). (F–H) Time-course plots showing EPSC and IPSC amplitudes and the effect of endogenous NA release (F), 600 nM (G) and 20 μM NA (H). Scale bars = 100 pA, 50 ms (both traces in H and IPSCs in F and G), 25 pA, 50 ms (EPSCs, F and G). (I–K) Grouped data for EPSPs (I), EPSCs (J) and IPSCs (K) for each NA application.
Using the first EPSP from the spike probability experiments is a useful measure of Schaffer collateral excitatory synaptic input to the CA1, but does not provide information on inhibitory synaptic strength or the overall excitation:inhibition ratio, which may be modulated by NA (Madison and Nicoll 1988; Doze et al. 1991; Bergles et al. 1996). We examined this local microcircuit in more detail using whole-cell voltage-clamp recordings to isolate feedforward excitatory and inhibitory synaptic currents by holding CA1 pyramidal neurons at the experimentally determined reversal potentials for AMPA (~+5 mV) and GABAA (~−65 mV) receptors (Fig. 4D,E). Inhibitory inputs were confirmed as feedforward disynaptic inputs by application of the AMPA/Kainate receptor antagonist NBQX (10 μM) at the end of each experiment and resulting blockade of IPSCs. Neither endogenous NA release by 1 Hz light stimulation nor 600 nM exogenous NA produced any effect on feedforward excitatory or inhibitory synaptic responses (Fig. 4F,G,J,K; 1 Hz light stimulation, EPSCs = 84 ± 5% and IPSCs = 86 ± 8% of baseline, n = 6 and 5, respectively, P = 0.116 and P = 0.310, respectively; 600 nM NA, EPSCs = 89 ± 8% and IPSCs = 110.9 ± 10.6% of baseline, n = 8 and 6, respectively, P = 0.460 and P = 0.231, respectively). However, 20 μM exogenous NA depressed both feedforward EPSCs and IPSCs (Fig. 4H,J,K; EPSCs = 69.8 ± 5.3% and IPSCs = 70.7 ± 8.9% of baseline, n = 11 and 7, respectively, P = 0.00009 and 0.009, respectively). The reduction in EPSC amplitude with 20 μM NA is comparable to the depression of EPSPs in spike probability experiments. Paired-pulse ratio for EPSCs or IPSCs was not affected by either endogenous or 600 nM or 20 μM exogenous NA (light stimulation = 1.50 ± 0.12 versus baseline = 1.45 ± 0.15, n = 6, P = 0.557 compared to baseline; 600 nM NA = 1.46 ± 0.12 versus baseline = 1.50 ± 0.01, n = 8, P = 0.768 compared to baseline; 20 μM NA = 1.48 ± 0.11 versus baseline = 1.48 ± 0.06, n = 11, P = 0.970 compared to baseline). These data confirm the reduced excitatory synaptic input caused by 20 μM NA and indicate this is a contributor to the reduction in spike output. However, the data also show that endogenous NA release or exogenous NA concentrations that mimic those found endogenously have no acute effect on either excitatory or inhibitory feedforward synaptic transmission. This rules out a role for synaptic input changes underlying the enhancement of spike probability by NA and therefore suggests that changes to intrinsic cellular excitability are the most likely mediators.
Enhancement of Intrinsic Cellular Excitability by Endogenous NA Release
The best characterized target for β-ARs is inhibition of the sAHP, a Ca2+-activated K+ current with a slow decay and an important regulator of neuronal excitability (Oh et al. 2003; Disterhoft and Oh 2006). The sAHP can be triggered by repetitive trains of synaptic input or a strong depolarizing current delivered directly to the soma and sAHP inhibition produces a reliable increase in spike output (Madison and Nicoll 1982; Haas and Konnerth 1983; Sah and Isaacson 1995), making it a good candidate to mediate the observed enhancement of spike probability. sAHP currents (IsAHP) with amplitudes of 10–40 pA were induced with a depolarizing voltage step from −50 to +20 mV. These sAHP currents reduced gradually in amplitude to 84.8 ± 2.2% over 20 min in time matched controls. We next tested the most effective optostimulation protocol for NA release (1 Hz) to determine if endogenous NA could inhibit the sAHP. Surprisingly, NA release elicited by 1 Hz light stimulation had no effect on IsAHP amplitude when compared to light stimulation in non-ChR2 expressing controls (Fig. 5A,D; WT = 83 ± 6% of baseline, ChR2 = 85 ± 10% of baseline, n = 6 and 9, respectively, P = 0.859). In contrast, exogenous application of 600 nM or 20 μM NA rapidly and substantially inhibited IsAHP in comparison to time matched controls (Fig. 5B–D; 600 nM, 21 ± 9% of baseline, n = 5, P = 0.0003 compared to time matched control; 20 μM, 10 ± 8% of baseline, n = 6, P = 0.00009 compared to time matched control). These data indicate that the release of endogenous NA is not sufficient to activate the β-ARs linked with Ca2+-activated K+ channels underlying the sAHP and that inhibition of sAHP current does not underlie the enhancement of spike probability caused by endogenous NA.
Figure 5.
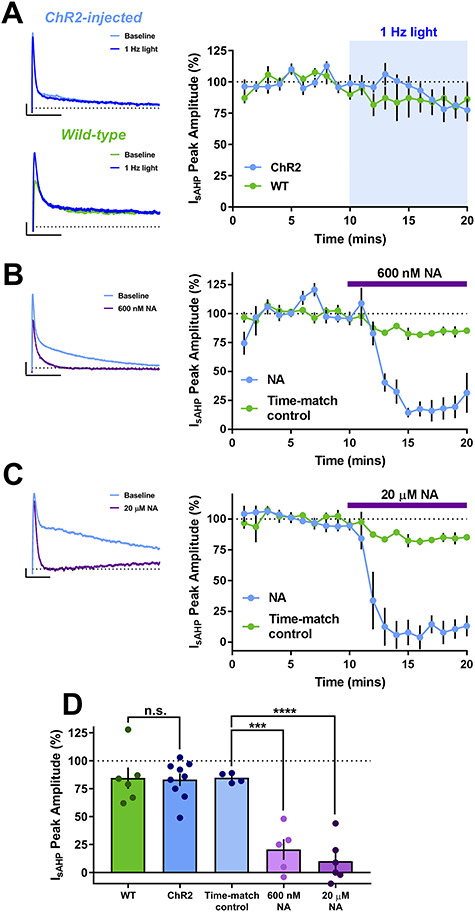
I sAHP currents are inhibited by exogenous 20 μM or 600 nM NA, but not endogenous NA release. (A–C) Timecourse and example traces showing IsAHP inhibition with 600 nM NA (B) and 20 μM NA (C) but not endogenous NA release in slices from virus injected mice or by light stimulation in slices from WT mice (A). Scale bars = 10 pA, 500 ms (WT), 25 pA, 500 ms (ChR2-injected), 50 pA, 500 ms (600 nM NA), 10 pA, 250 ms (20 μM NA). (D) Grouped data for virus injected and WT mice show no effect of endogenous NA release whereas both 600 nM and 20 μM NA depressed IsAHP when compared to a time-matched control.
Alternative mechanisms that could cause enhancement of spike probability include modulation of conductances active at rest or those active in response to depolarizing input. To monitor conductances active at rest we analyzed membrane potential and input resistance during the spike probability experiments. Average resting membrane potential was −65.1 ± 0.7 mV and input resistance 90.5 ± 2.6 MΩ and neither endogenous NA release or exogenous application of 600 nM or 20 μM NA produced consistent changes in membrane potential (Fig. 6A–D; ChR2 = −0.19 ± 0.69 mV, n = 12, P = 0.891 compared to baseline; WT = −0.01 ± 1.00 mV, n = 8, P = 0.897 compared to baseline; 600 nM NA = 2.85 ± 1.55 mV, n = 7, P = 0.114 compared to baseline; 20 μM NA = −2.06 ± 1.31, n = 5, P = 0.187 compared to baseline) or input resistance (Fig. 6E–H; ChR2 = 2.01 ± 5.38 MΩ, n = 12, P = 0.413 compared to baseline; WT = −4.49 ± 3.35 MΩ, n = 8, P = 0.383 compared to baseline; 600 nM NA = −0.80 ± 3.91, n = 7, P = 0.688 compared to baseline; 20 μM NA = −3.53 ± 3.82, n = 5, P = 0.278 compared to baseline) indicating no effect of NA on resting excitability.
Figure 6.
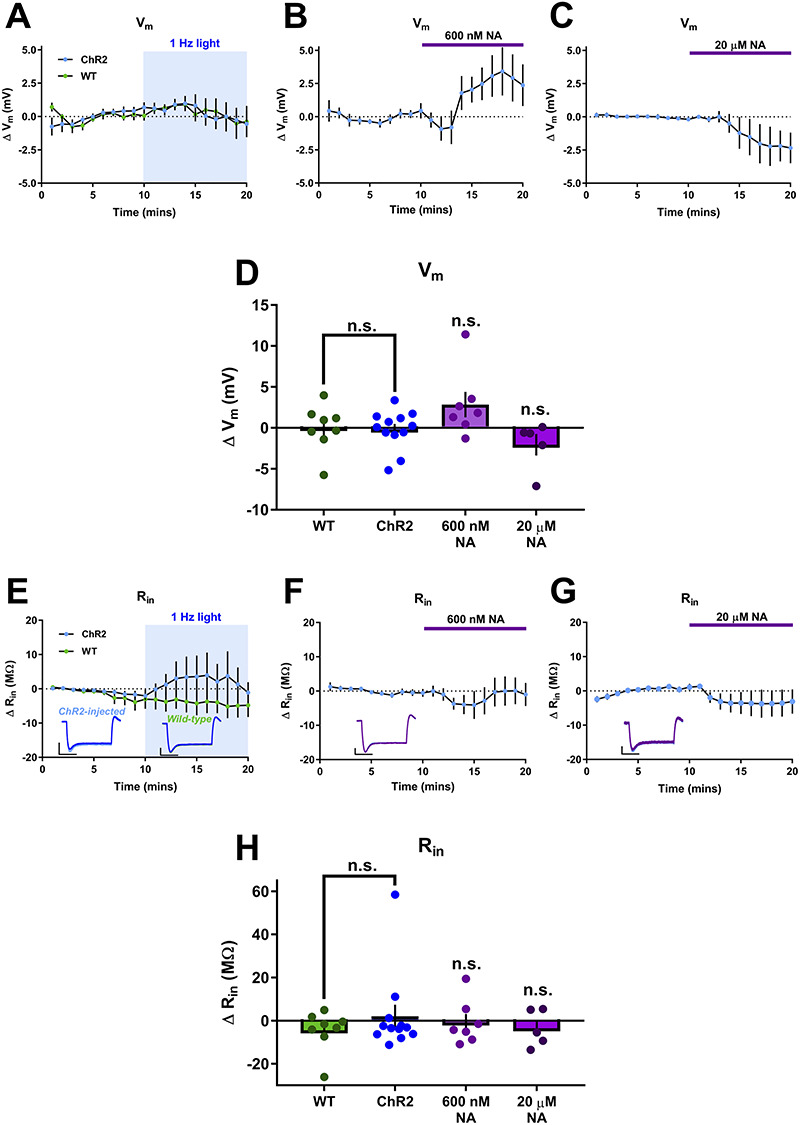
Endogenous NA release does not modulate resting intrinsic cellular properties. (A–C) Timecourse of changes in resting membrane potential (Vm) from baseline showing no effect of endogenous NA release (A) or 600 nM NA (B) but a hyperpolarization caused by 20 μM NA (C). (D) Group data for effects of all conditions on Vm. (E–G) Timecourse of changes in input resistance (Rin) from baseline showing no effect of endogenous NA release (E), 600 nM NA (F) and 20 μM NA (G). Example traces are inset. Scales bars = 5 mV, 200 ms. (H) Group data for effects of all conditions on Rin.
A final possible mechanism for the enhancement of spike probability by NA is a strengthening of the coupling between excitatory input and action potential generation. Therefore, we analyzed action potential initiation measures including the spike threshold and the spike latency, which is a measure of the amount of charge required to initiate an action potential. The action potential threshold was measured for the first spike in each train of synaptic stimuli before and during release of endogenous NA or application of exogenous 600 nM NA but not 20 μM NA since spiking was almost entirely prevented by application of this dose (Fig. 2). Interestingly, endogenous NA release, but not 600 nM exogenous NA, lowered the threshold for action potential initiation (Fig. 7A–C; endogenous NA = −1.92 ± 0.47 mV, n = 12, P = 0.028 compared to wild-type (WT); 600 nM NA = +1.73 ± 1.11 mV, n = 7, P = 0.155 compared to baseline). Furthermore, spike latency was reduced by both endogenous NA release and exogenous 600 nM NA (Fig. 7D–F; endogenous NA = 5.68 ± 0.39 to 5.18 ± 0.40, n = 12, P = 0.0095; 600 nM NA = 6.60 ± 0.75 to 5.62 ± 0.74, n = 7, P = 0.053 compared to baseline). Taken together, these data indicate a potential modulation of active conductances by NA enhancing the coupling between excitatory input and action potential initiation.
Figure 7.
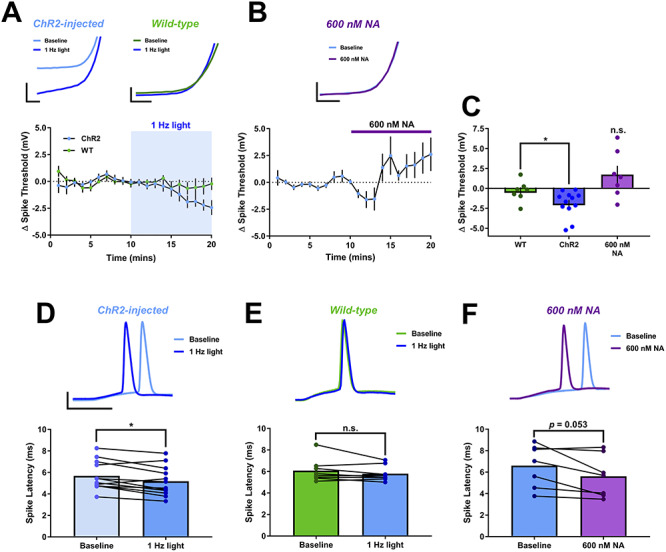
Endogenous NA release reduces spike latency. (A and B) Spike threshold in response to evoked EPSPs was lowered by endogenous NA release (A) but not 600 nM NA (B). Spike threshold is reduced in slices from virus injected animals but not in WT animals. Example traces are inset. Scale bars = 5 mV, 0.5 ms for expanded traces and 25 mV, 5 ms otherwise. (C) Group data for spike threshold. (D–F) Group data and example traces showing that spike latency in response to evoked EPSPs is reduced by endogenous NA release in slices from virus injected mice (D) or 600 nM NA (F) but not by light stimulation in slices from WT mice (E). Scale bars = 25 mV, 5 ms.
To confirm these findings, we assessed excitation-spike coupling using somatic current injections to initiate action potentials with current injection amplitude defined by the rheobase (the minimum somatic current injection to evoke a single action potential). In CA1 pyramidal neurons, K+ channels (predominantly the Kv1 family) expressed in the somatodendritic region are activated near the action potential threshold and cause a delay in action potential spiking, resulting in a longer action potential latency and increased jitter or variance in latency.
Similar to action potentials initiated by synaptic stimulation, the latency of action potentials initiated by somatic current injection was reduced by endogenous NA release (Fig. 8A,D,E; 594 ± 71 to 376 ± 104 ms, n = 7, P = 0.031 compared to baseline), recapitulated by 600 nM exogenous NA (Fig. 8B,D,E; 553 ± 104 to 271 ± 55 ms, n = 6, P = 0.006 compared to baseline) and blocked by the β-AR antagonist propranolol (500 nM) (Fig. 8C,D,E; 744 ± 75 to 628 ± 93 ms, n = 5, P = 0.106 compared to baseline). These effects on spike latency were mirrored by the spike jitter (Fig. 8A–C,F; endogenous NA, 229 ± 30 to 125 ± 33 ms, P = 0.004 compared to baseline; 600 nM NA, 190 ± 16 to 95 ± 33 ms, P = 0.033 compared to baseline; propranolol +600 nM NA, 189 ± 18 to 202 ± 28, P = 0.772 compared to baseline). The reduction in spike latency caused by NA could be due to a reduction in action potential threshold or a reduction in the amount of current injection required to reach threshold. In these experiments, the action potential threshold was not altered by NA (Fig. 8H; endogenous NA: −1.0 ± 2.5 mV, P = 0.697; 600 nM NA = −2.8 ± 1.5 mV, P = 0.131; propranolol +600 nM NA = −0.11 ± 0.7 mV, P = 0.885) but the amount of charge transfer was reduced (Fig. 8G; endogenous NA: 5.3 ± 1.2 to 2.8 ± 1.3 mV/S, P = 0.016; 600 nM NA = 6.1 ± 1.7 to 2.1 ± 1.0 mV/S, P = 0.008; propranolol +600 nM NA = 9.0 ± 1.8 to 6.64 ± 1.1 mV/S, P = 0.087). These results are entirely consistent with NA causing a down-regulation of voltage-dependent K+ channels, which reduces the amount of charge required to bring the membrane potential to the action potential threshold (Yuan et al. 2002; Liu et al. 2017).
Figure 8.
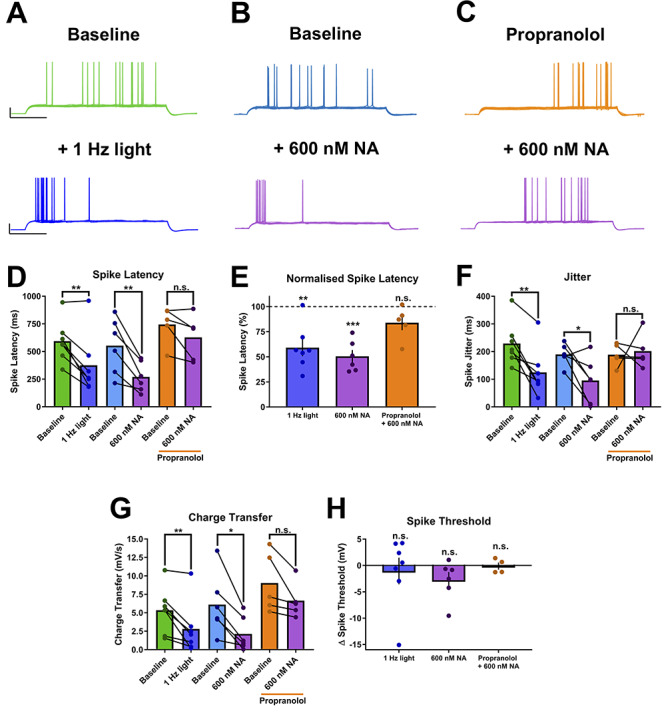
Excitation-spike coupling is enhanced by endogenous NA release. (A–C) Example traces showing a reduction in rheobase spike latency and jitter in response to endogenous NA release (A) or 600 nM NA (B) that is blocked with propranolol (C). Scale bars = 25 mV, 250 ms. (D–H) Group data showing the effects of endogenous NA release and 600 nM NA and the block with propranolol on spike latency (D and E), jitter (F), charge transfer required to elicit an action potential (G) and spike threshold (H).
Discussion
We demonstrate that endogenous NA release, elicited by optogenetic stimulation of noradrenergic fibers in the hippocampus, enhances CA1 pyramidal neuron spike output in response to Schaffer collateral synaptic input. Like many of the excitatory actions of NA, this effect is mediated via β-ARs (Dunwiddie et al. 1992; Raman et al. 1996; Lin et al. 2003; O'Dell et al. 2010; Murchison et al. 2011; Ul Haq et al. 2012; Liu et al. 2017). The enhancement of spike output was mimicked by application of relatively low concentrations of exogenous NA (600 nM) and but not by higher concentrations (2–20 μM) suggesting that the effective concentration of endogenously released NA is around 600 nM. This agrees very closely with recent estimates of NA release from LC terminals using genetically-encoded sensors (Feng et al. 2019). Surprisingly, this profile of endogenous NA release did not inhibit sAHP currents, even though exogenous NA consistently produces inhibition (Madison and Nicoll 1982; Haas and Konnerth 1983; Sah and Isaacson 1995) and neither did NA have an effect on synaptic input to CA1 pyramidal neurons. Instead the increased spike output resulted from enhancement of excitation-spike coupling consistent with a down-regulation of voltage-dependent K+ channels (Liu et al. 2017).
The use of optogenetic techniques to selectively stimulate neuromodulator release has highlighted differences between exogenous bath-applied and endogenous release of dopamine and serotonin in ex vivo brain slice experiments (Rosen et al. 2015; Teixeira et al. 2018). We also show that this is the case for NA and propose that such distinct effects are not only concentration-dependent, but also depend on the spatio-temporal release dynamics of neuromodulators. Complete inhibition of the sAHP with saturating concentrations (≥10 μM) of NA have been reported previously (Madison and Nicoll 1982; Haas and Konnerth 1983; Dunwiddie et al. 1992; Sah and Isaacson 1995), and we show much lower concentrations (600 nM) also inhibit IsAHP. However, the lack of IsAHP modulation by endogenously released NA suggests that NA is not released near the β-ARs, which modulate IsAHP. The precise location of noradrenergic terminals and NA release in CA1 is unknown, but the LC is conventionally thought to modulate this network via bulk “volume transmission” (Fuxe et al. 2010). In contrast, some evidence suggests that CA3 pyramidal neurons are synaptically targeted by the LC for more precise regulation (Walling et al. 2012). However, the dichotomy between exogenous and endogenous NA actions on IsAHP suggests there is some degree of compartmental targeting by LC terminals in CA1. This highlights the importance of NA transporters in shaping the spatiotemporal profile of NA by removing NA from the extracellular space. Indeed, application of uptake inhibitors increases the effective concentration (i.e., the concentration experienced by ARs) of endogenous NA released by optogenetic stimulation of LC terminals (Feng et al. 2019). NA transporters are less likely to alter the effective concentration of bath applied exogenous NA since the reserve pool of NA is much larger but this may potentially lead to an overestimation of the effective concentration of bath applied NA. It is also reported that LC terminals in the hippocampus may release dopamine as well as NA and indeed that dopamine mediates the majority of the effects of LC activation (Kempadoo et al. 2016; Takeuchi et al. 2016; Wagatsuma et al. 2018). We find no evidence for dopamine release from LC terminals since all of the effects documented here were blocked by the β-adrenergic antagonist propranolol.
In acute and cultured hippocampal CA1 neurons, Kv1.1, Kv1.2 and Kv1.6 channels are expressed in the somatodendritic and axonal compartments (Grosse et al. 2000), where they regulate spike generation at the axon initial segment (Guan et al. 2007; Liu et al. 2017). Such expression overlaps with that observed for β-ARs (Guo and Li 2007), so allowing rapid modulation of action potential initiation. Since the molecular identity (and therefore subcellular location) of the K+ channels underlying the sAHP have yet to be unequivocally defined (Andrade et al. 2012; King et al. 2015; Higham et al. 2019) it is plausible that these channels are located in the more distal dendrites only accessible to exogenous NA acting on β-ARs. In support of this, the sAHP in CA1 pyramidal neurons can be activated by subthreshold EPSPs, suggesting a dendritic location distant from the soma (Lancaster et al. 2001) and estimated to be 100–200 μm from the soma in CA1 pyramidal and rat basolateral amygdala neurons (Sah and Bekkers 1996; Power et al. 2011). Moreover, sAHP inhibition with exogenous NA concentrations as low as 300 nM has been demonstrated (Madison and Nicoll 1982, 1986). These studies support the notion that the present results are not due to optogenetically triggered NA being released at a concentration insufficient to modulate the sAHP, but rather that release is targeted to specific domains of the CA1 cells. Indeed a more distal location is likely given that the sAHP acts not only to regulate neuronal excitability by counteracting repetitive synaptic barrages in the dendrites (Andrade et al. 2012), but also shapes the temporal summation of EPSPs (Lancaster et al. 2001). In addition, the sAHP is inhibited by β1-ARs (Madison and Nicoll 1982, 1986) whereas Kv1.1 channels are inhibited by β2-ARs (Liu et al. 2017) and therefore the location and affinity of β1 and β2 ARs for endogenous NA may also contribute to the differential effect of endogenous NA on sAHP currents and Kv channels. It is also possible that the temporal profile of NA release resulting from tonic versus phasic LC activity could differentially activate receptor populations.
The absence of noradrenergic modulation of IsAHP or conductances active at resting membrane potentials indicates that the modulation of spike output is via regulation of excitation-spike coupling, which was supported by the finding that noradrenaline reduces the charge required to drive CA1 pyramidal neurons to fire action potentials. We found this was not due to a consistent reduction in action potential threshold and therefore that noradrenaline is likely modulating voltage-dependent conductances activated by subthreshold depolarization that normally regulate the coupling between sub-threshold excitation (either by synaptic input or somatic current injection) and action potential initiation. Since noradrenaline inhibits dendritic voltage-dependent potassium conductances including Kv4.2 and Kv1.1 (Hoffman and Johnston 1999; Yuan et al. 2002; Liu et al. 2017) that control the coupling between sub-threshold depolarization and action potential initiation (Hoffman et al. 1997) we propose that this is the mechanism by which endogenous NA release modulates excitation-spike coupling but our data do not distinguish which of these two potassium conductances mediate the effects of NA.
Endogenous NA release did not modulate synaptic inputs to CA1 in our experiments and only at higher exogenous concentrations of NA was synaptic transmission depressed. This broadly agrees with previous reports where the effect of NA on synaptic transmission is weak but generally depressing at higher doses (Madison and Nicoll 1988; Doze et al. 1991; Heginbotham and Dunwiddie 1991; Bergles et al. 1996; Katsuki et al. 1997). Interestingly, presynaptic α2-ARs can depress synaptic transmission in LC (Starke and Montel 1973) and are expressed on the terminals of CA3 axons (Milner et al. 1998). However, we found no evidence for a presynaptic depression of synaptic transmission since we saw no change in paired-pulse ratio on addition of 20 μM NA. Therefore, the major role for endogenous NA release on synaptic transmission is likely to be via regulation of long-term synaptic plasticity. Induction of LTP can be facilitated by enhancing postsynaptic excitability via downregulation of Kv1.1 (Liu et al. 2017) or inhibition of sAHP (Fuenzalida et al. 2007), which enhance back-propagating action potentials and widen the temporal window for associative plasticity (Lin et al. 2003; Liu et al. 2017). In addition, induction of LTP may also be facilitated by NA by enhancing Ca2+ influx through NMDARs via PKA-mediated phosphorylation (Raman et al. 1996) and expression of LTP can be facilitated by increased trafficking of AMPARs to the postsynaptic membrane as a result of AMPAR phosphorylation (Hu et al. 2007; Makino et al. 2011), DNA methylation and post-translational histone modifications (Maity et al. 2015). Coincident timing of LC activity and Schaffer collateral input drives the induction and regulates the persistency of NMDA receptor-dependent LTP in the CA1 (Lemon et al. 2009; Hansen and Manahan-Vaughan 2015b; Liu et al. 2017). Such intrinsic synaptic activity is absent in the present study, but this would not prevent the modulation of the Kv channels and altered spike probability we observed. Indeed, it is plausible that the coincident timing of LC and SC inputs may augment the effects of one another and lead to a greater degree of spike probability enhancement and downstream plasticity.
The LC typically shows spontaneous tonic spiking activity during wakefulness in the range 0.1–5 Hz (Aston-Jones and Bloom 1981; Vankov et al. 1995; Takeuchi et al. 2016; Xiang et al. 2019). During sleep states, particularly non-REM sleep, the LC is essentially silent, however an increase in LC activity precedes and gates arousal from sleep (Aston-Jones and Bloom 1981; Carter et al. 2010) and a slightly higher tonic level of firing (1–2 Hz) is commonly observed during exploratory behavior (Vankov et al. 1995; Takeuchi et al. 2016). Further, the selective optogenetic addition of a low level of noradrenergic activity during sleep alters hippocampal memory consolidation (Swift et al. 2018). Where phasic burst firing is thought to signal saliency, possibly via a network reset mechanism to allow the learning of a new rule or contingency that modifies place cell representations (Bouret and Sara 2005; Kaufman et al. 2020), within this experiment we found an increase in excitation-spike coupling with a modest stimulation of LC terminals (1 Hz) corresponding to a low level of NA (~600 nM). However, the level of NA in the hippocampal slice under basal conditions was biologically negligible as the application of propranolol was without effect on the excitation-spike coupling in the absence of optogenetic drive. The low frequency optogenetic activation of LC terminals may therefore promote memory consolidation (i.e., of a given environment) into an existing schema without resetting the network. In this context the β-AR mechanism acts to fundamentally alter the input-output relationship of the CA1 gateway for hippocampal information flow to the cortex without altering the synaptic communication within the local microcircuits. As such this represents a novel noradrenergic gating mechanism that can globally boost the output from the hippocampus to the cortex dependent on behavioral context driven by neuromodulator state.
Author Contributions
T.J.B. designed and performed research, analyzed data, and wrote the paper. A.E.P. and J.R.M. designed and supervised research and wrote the paper.
Funding
Biotechnology and Biological Sciences Research Council (BBSRC BB/M009122/1); Wellcome Trust (AEP-gr088373, JRM-101029).
Notes
We thank all members of the Mellor and Pickering groups for constructive discussion and Eric J. Kremer for provision of CAV vectors. Conflict of Interest: The authors declare no competing financial interests.
References
- Andrade R, Foehring RC, Tzingounis AV. 2012. The calcium-activated slow AHP: cutting through the Gordian knot. Front Cell Neurosci. 6:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aston-Jones G, Bloom FE. 1981. Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J Neurosci. 1:876–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aston-Jones G, Cohen JD. 2005. Adaptive gain and the role of the locus coeruleus-norepinephrine system in optimal performance. J Comp Neurol. 493:99–110. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Doze VA, Madison DV, Smith SJ. 1996. Excitatory actions of norepinephrine on multiple classes of hippocampal CA1 interneurons. J Neurosci. 16:572–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge CW, Waterhouse BD. 2003. The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Brain Res Rev. 42:33–84. [DOI] [PubMed] [Google Scholar]
- Bird CM, Burgess N. 2008. The hippocampus and memory: insights from spatial processing. Nat Rev Neurosci. 9:182–194. [DOI] [PubMed] [Google Scholar]
- Bouret S, Sara SJ. 2005. Network reset: a simplified overarching theory of locus coeruleus noradrenaline function. Trends Neurosci. 28:574–582. [DOI] [PubMed] [Google Scholar]
- Carter ME, Yizhar O, Chikahisa S, Nguyen H, Adamantidis A, Nishino S, Deisseroth K, de Lecea L. 2010. Tuning arousal with optogenetic modulation of locus coeruleus neurons. Nat Neurosci. 13:1526–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton DM, Dietrich KL, Smith JS, Davis BK. 1995. Spatial and non-spatial learning in the rat following lesions to the nucleus locus coeruleus. Neuroreport. 7:177–182. [PubMed] [Google Scholar]
- Disterhoft JF, Oh MM. 2006. Learning, aging and intrinsic neuronal plasticity. Trends Neurosci. 29:587–599. [DOI] [PubMed] [Google Scholar]
- Doze VA, Cohen GA, Madison DV. 1991. Synaptic localization of adrenergic disinhibition in the rat hippocampus. Neuron. 6:889–900. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Taylor M, Heginbotham LR, Proctor WR. 1992. Long-term increases in excitability in the CA1 region of rat hippocampus induced by beta-adrenergic stimulation: possible mediation by cAMP. J Neurosci. 12:506–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Zhang C, Lischinsky JE, Jing M, Zhou J, Wang H, Zhang Y, Dong A, Wu Z, Wu H et al. 2019. A genetically encoded fluorescent sensor for rapid and specific in vivo detection of norepinephrine. Neuron. 102:745, e748–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuenzalida M, Fernandez de Sevilla D, Buno W. 2007. Changes of the EPSP waveform regulate the temporal window for spike-timing-dependent plasticity. J Neurosci. 27:11940–11948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuxe K, Dahlstrom AB, Jonsson G, Marcellino D, Guescini M, Dam M, Manger P, Agnati L. 2010. The discovery of central monoamine neurons gave volume transmission to the wired brain. Prog Neurobiol. 90:82–100. [DOI] [PubMed] [Google Scholar]
- Grosse G, Draguhn A, Hohne L, Tapp R, Veh RW, Ahnert-Hilger G. 2000. Expression of Kv1 potassium channels in mouse hippocampal primary cultures: development and activity-dependent regulation. J Neurosci. 20:1869–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan D, Lee JC, Higgs MH, Spain WJ, Foehring RC. 2007. Functional roles of Kv1 channels in neocortical pyramidal neurons. J Neurophysiol. 97:1931–1940. [DOI] [PubMed] [Google Scholar]
- Guo NN, Li BM. 2007. Cellular and subcellular distributions of beta1- and beta2-adrenoceptors in the CA1 and CA3 regions of the rat hippocampus. Neuroscience. 146:298–305. [DOI] [PubMed] [Google Scholar]
- Haas HL, Konnerth A. 1983. Histamine and noradrenaline decrease calcium-activated potassium conductance in hippocampal pyramidal cells. Nature. 302:432–434. [DOI] [PubMed] [Google Scholar]
- Hagena H, Manahan-Vaughan D. 2012. Learning-facilitated long-term depression and long-term potentiation at mossy fiber-CA3 synapses requires activation of beta-adrenergic receptors. Front Integr Neurosci. 6:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagena H, Manahan-Vaughan D. 2017. The serotonergic 5-HT4 receptor: a unique modulator of hippocampal synaptic information processing and cognition. Neurobiol Learn Mem. 138:145–153. [DOI] [PubMed] [Google Scholar]
- Hansen N, Manahan-Vaughan D. 2015a. Locus coeruleus stimulation facilitates long-term depression in the dentate gyrus that requires activation of beta-adrenergic receptors. Cereb Cortex. 25:1889–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen N, Manahan-Vaughan D. 2015b. Hippocampal long-term potentiation that is elicited by perforant path stimulation or that occurs in conjunction with spatial learning is tightly controlled by beta-adrenoreceptors and the locus coeruleus. Hippocampus. 25:1285–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley CW, Lalies MD, Nutt DJ. 1996. Estimating the synaptic concentration of norepinephrine in dentate gyrus which produces beta-receptor mediated long-lasting potentiation in vivo using microdialysis and intracerebroventricular norepinephrine. Brain Res. 710:293–298. [DOI] [PubMed] [Google Scholar]
- Hayat H, Regev N, Matosevich N, Sales A, Paredes-Rodriguez E, Krom AJ, Bergman L, Li Y, Lavigne M, Kremer EJ et al. 2020. Locus-coeruleus norepinephrine activity gates sensory-evoked awakenings from sleep. Sci. Adv. 6:eaaz4232. doi: 10.1126/sciadv.aaz4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heginbotham LR, Dunwiddie TV. 1991. Long-term increases in the evoked population spike in the CA1 region of rat hippocampus induced by beta-adrenergic receptor activation. J Neurosci. 11:2519–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higham J, Sahu G, Wazen RM, Colarusso P, Gregorie A, Harvey BSJ, Goudswaard L, Varley G, Sheppard DN, Turner RW et al. 2019. Preferred formation of heteromeric channels between coexpressed SK1 and IKCa channel subunits provides a unique pharmacological profile of Ca(2+)-activated potassium channels. Mol Pharmacol. 96:115–126. [DOI] [PubMed] [Google Scholar]
- Hirschberg S, Li Y, Randall A, Kremer EJ, Pickering AE. 2017. Functional dichotomy in spinal- vs prefrontal-projecting locus coeruleus modules splits descending noradrenergic analgesia from ascending aversion and anxiety in rats. Elife. 6:e29808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DA, Johnston D. 1999. Neuromodulation of dendritic action potentials. J Neurophysiol. 81:408–411. [DOI] [PubMed] [Google Scholar]
- Hoffman DA, Magee JC, Colbert CM, Johnston D. 1997. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature. 387(6636):869– 6675. [DOI] [PubMed] [Google Scholar]
- Hu H, Real E, Takamiya K, Kang MG, Ledoux J, Huganir RL, Malinow R. 2007. Emotion enhances learning via norepinephrine regulation of AMPA-receptor trafficking. Cell. 131:160–173. [DOI] [PubMed] [Google Scholar]
- Ji JZ, Zhang XH, Li BM. 2003. Deficient spatial memory induced by blockade of beta-adrenoceptors in the hippocampal CA1 region. Behav Neurosci. 117:1378–1384. [DOI] [PubMed] [Google Scholar]
- Katsuki H, Izumi Y, Zorumski CF. 1997. Noradrenergic regulation of synaptic plasticity in the hippocampal CA1 region. J Neurophysiol. 77:3013–3020. [DOI] [PubMed] [Google Scholar]
- Kaufman AM, Geiller T, Losonczy A. 2020. A role for the locus coeruleus in hippocampal CA1 place cell reorganization during spatial reward learning. Neuron. 105:1018–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebschull JM, Garcia da Silva P, Reid AP, Peikon ID, Albeanu DF, Zador AM. 2016. High-throughput mapping of single-neuron projections by sequencing of barcoded RNA. Neuron. 91:975–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempadoo KA, Mosharov EV, Choi SJ, Sulzer D, Kandel ER. 2016. Dopamine release from the locus coeruleus to the dorsal hippocampus promotes spatial learning and memory. Proc Natl Acad Sci U S A. 113:14835–14840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King B, Rizwan AP, Asmara H, Heath NC, Engbers JD, Dykstra S, Bartoletti TM, Hameed S, Zamponi GW, Turner RW. 2015. IKCa channels are a critical determinant of the slow AHP in CA1 pyramidal neurons. Cell Rep. 11:175–182. [DOI] [PubMed] [Google Scholar]
- Lancaster B, Hu H, Ramakers GM, Storm JF. 2001. Interaction between synaptic excitation and slow afterhyperpolarization current in rat hippocampal pyramidal cells. J Physiol. 536:809–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemon N, Aydin-Abidin S, Funke K, Manahan-Vaughan D. 2009. Locus coeruleus activation facilitates memory encoding and induces hippocampal LTD that depends on beta-adrenergic receptor activation. Cereb Cortex. 19:2827–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Hickey L, Perrins R, Werlen E, Patel AA, Hirschberg S, Jones MW, Salinas S, Kremer EJ, Pickering AE. 2016. Retrograde optogenetic characterization of the pontospinal module of the locus coeruleus with a canine adenoviral vector. Brain Res. 1641:274–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YW, Min MY, Chiu TH, Yang HW. 2003. Enhancement of associative long-term potentiation by activation of beta-adrenergic receptors at CA1 synapses in rat hippocampal slices. J Neurosci. 23:4173–4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Cui L, Schwarz MK, Dong Y, Schluter OM. 2017. Adrenergic gate release for spike timing-dependent synaptic potentiation. Neuron. 93:394–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA. 1982. Noradrenaline blocks accommodation of pyramidal cell discharge in the hippocampus. Nature. 299:636–638. [DOI] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA. 1986. Actions of noradrenaline recorded intracellularly in rat hippocampal CA1 pyramidal neurones, in vitro. J Physiol. 372:221–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA. 1988. Norepinephrine decreases synaptic inhibition in the rat hippocampus. Brain Res. 442:131–138. [DOI] [PubMed] [Google Scholar]
- Maity S, Rah S, Sonenberg N, Gkogkas CG, Nguyen PV. 2015. Norepinephrine triggers metaplasticity of LTP by increasing translation of specific mRNAs. Learn Mem. 22:499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino Y, Johnson RC, Yu Y, Takamiya K, Huganir RL. 2011. Enhanced synaptic plasticity in mice with phosphomimetic mutation of the GluA1 AMPA receptor. Proc Natl Acad Sci USA. 108:8450–8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner TA, Lee A, Aicher SA, Rosin DL. 1998. Hippocampal alpha2a-adrenergic receptors are located predominantly presynaptically but are also found postsynaptically and in selective astrocytes. J Comp Neurol. 395:310–327. [PubMed] [Google Scholar]
- Muller A, Joseph V, Slesinger PA, Kleinfeld D. 2014. Cell-based reporters reveal in vivo dynamics of dopamine and norepinephrine release in murine cortex. Nat Methods. 11:1245–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murchison CF, Schutsky K, Jin SH, Thomas SA. 2011. Norepinephrine and ss(1)-adrenergic signaling facilitate activation of hippocampal CA1 pyramidal neurons during contextual memory retrieval. Neuroscience. 181:109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murchison CF, Zhang XY, Zhang WP, Ouyang M, Lee A, Thomas SA. 2004. A distinct role for norepinephrine in memory retrieval. Cell. 117:131–143. [DOI] [PubMed] [Google Scholar]
- O'Dell TJ, Connor SA, Gelinas JN, Nguyen PV. 2010. Viagra for your synapses: enhancement of hippocampal long-term potentiation by activation of beta-adrenergic receptors. Cell Signal. 22:728–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Dell TJ, Connor SA, Guglietta R, Nguyen PV. 2015. Beta-adrenergic receptor signaling and modulation of long-term potentiation in the mammalian hippocampus. Learn Mem. 22:461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh MM, Kuo AG, Wu WW, Sametsky EA, Disterhoft JF. 2003. Watermaze learning enhances excitability of CA1 pyramidal neurons. J Neurophysiol. 90:2171–2179. [DOI] [PubMed] [Google Scholar]
- Palacios-Filardo J, Mellor JR. 2019. Neuromodulation of hippocampal long-term synaptic plasticity. Curr Opin Neurobiol. 54:37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power JM, Bocklisch C, Curby P, Sah P. 2011. Location and function of the slow afterhyperpolarization channels in the basolateral amygdala. J Neurosci. 31:526–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Tong G, Jahr CE. 1996. Beta-adrenergic regulation of synaptic NMDA receptors by cAMP-dependent protein kinase. Neuron. 16:415–421. [DOI] [PubMed] [Google Scholar]
- Ramos BP, Arnsten AF. 2007. Adrenergic pharmacology and cognition: focus on the prefrontal cortex. Pharmacol Ther. 113:523–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen ZB, Cheung S, Siegelbaum SA. 2015. Midbrain dopamine neurons bidirectionally regulate CA3-CA1 synaptic drive. Nat Neurosci. 18:1763–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P, Isaacson JS. 1995. Channels underlying the slow afterhyperpolarization in hippocampal pyramidal neurons: neurotransmitters modulate the open probability. Neuron. 15:435–441. [DOI] [PubMed] [Google Scholar]
- Sah P, Bekkers JM. 1996. Apical dendritic location of slow afterhyperpolarization current in hippocampal pyramidal neurons: implications for the integration of long-term potentiation. J Neurosci. 16:4537–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sales AC, Friston KJ, Jones MW, Pickering AE, Moran RJ. 2019. Locus coeruleus tracking of prediction errors optimises cognitive flexibility: an active inference model. PLoS Comput Biol. 15:e1006267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz LA, Miyamichi K, Gao XJ, Beier KT, Weissbourd B, DeLoach KE, Ren J, Ibanes S, Malenka RC, Kremer EJ et al. 2015. Viral-genetic tracing of the input-output organization of a central noradrenaline circuit. Nature. 524:88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starke K, Montel H. 1973. Involvement of alpha-receptors in clonidine-induced inhibition of transmitter release from central monoamine neurones. Neuropharmacology. 12:1073–1080. [DOI] [PubMed] [Google Scholar]
- Swift KM, Gross BA, Frazer MA, Bauer DS, Clark KJD, Vazey EM, Aston-Jones G, Li Y, Pickering AE, Sara SJ et al. 2018. Abnormal locus coeruleus sleep activity alters sleep signatures of memory consolidation and impairs place cell stability and spatial memory. Curr Biol. 28:3599, e3594–3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi T, Duszkiewicz AJ, Sonneborn A, Spooner PA, Yamasaki M, Watanabe M, Smith CC, Fernandez G, Deisseroth K, Greene RW et al. 2016. Locus coeruleus and dopaminergic consolidation of everyday memory. Nature. 537:357–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira CM, Rosen ZB, Suri D, Sun Q, Hersh M, Sargin D, Dincheva I, Morgan AA, Spivack S, Krok AC et al. 2018. Hippocampal 5-HT input regulates memory formation and Schaffer collateral excitation. Neuron. 98:992, e1004–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ul Haq R, Liotta A, Kovacs R, Rosler A, Jarosch MJ, Heinemann U, Behrens CJ. 2012. Adrenergic modulation of sharp wave-ripple activity in rat hippocampal slices. Hippocampus. 22:516–533. [DOI] [PubMed] [Google Scholar]
- Vankov A, Herve-Minvielle A, Sara SJ. 1995. Response to novelty and its rapid habituation in locus coeruleus neurons of the freely exploring rat. Eur J Neurosci. 7:1180–1187. [DOI] [PubMed] [Google Scholar]
- Vazey EM, Moorman DE, Aston-Jones G. 2018. Phasic locus coeruleus activity regulates cortical encoding of salience information. Proc Natl Acad Sci U S A. 115:E9439–E9448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagatsuma A, Okuyama T, Sun C, Smith LM, Abe K, Tonegawa S. 2018. Locus coeruleus input to hippocampal CA3 drives single-trial learning of a novel context. Proc Natl Acad Sci USA. 115:E310–E316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walling SG, Brown RA, Miyasaka N, Yoshihara Y, Harley CW. 2012. Selective wheat germ agglutinin (WGA) uptake in the hippocampus from the locus coeruleus of dopamine-beta-hydroxylase-WGA transgenic mice. Front Behav Neurosci. 6:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang L, Harel A, Gao H, Pickering AE, Sara SJ, Wiener SI. 2019. Behavioral correlates of activity of optogenetically identified locus coeruleus noradrenergic neurons in rats performing T-maze tasks. Sci Rep. 9:1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu AJ, Dayan P. 2005. Uncertainty, neuromodulation, and attention. Neuron. 46:681–692. [DOI] [PubMed] [Google Scholar]
- Yuan LL, Adams JP, Swank M, Sweatt JD, Johnston D. 2002. Protein kinase modulation of dendritic K+ channels in hippocampus involves a mitogen-activated protein kinase pathway. J Neurosci. 22:4860–4868. [DOI] [PMC free article] [PubMed] [Google Scholar]