

Abstract
Following the discovery of cryptochrome-DASH (CRYD) as a new type of blue-light receptor cryptochrome, theoretical and experimental findings on CRYD have been reported. Early studies identified CRYD as highly homologous to the DNA repair enzyme photolyases (PLs), suggesting the involvement of CRYD in DNA repair. However, an experimental study reported that CRYD does not exhibit DNA repair activity in vivo. Successful PL-mediated DNA repair requires: (i) the recognition of UV-induced DNA lesions and (ii) an electron transfer reaction. If either of them is inefficient, the DNA repair activity will be low.
To elucidate the functional differences between CRYD and PL, we theoretically investigated the electron transfer reactivity and DNA binding affinity of CRYD and also performed supplementary experiments. The average electronic coupling matrix elements value for Arabidopsis thaliana CRYD (AtCRYD) was estimated to be 5.3 meV, comparable to that of Anacystis nidulans cyclobutane pyrimidine dimer PLs (AnPL) at 4.5 meV, indicating similar electron transfer reactivities. We also confirmed the DNA repair activity of AtCRYD for UV-damaged single-stranded DNA by the experimental analysis. In addition, we investigated the dynamic behavior of AtCRYD and AnPL in complex with double-stranded DNA using molecular dynamics simulations and observed the formation of a transient salt bridge between protein and DNA in AtCRYD, in contrast to AnPL in which it was formed stably. We suggested that the instability of the salt bridge between protein and DNA will lead to reduced DNA binding affinity for AtCRYD.
Keywords: electron transfer, electronic coupling matrix elements, salt bridge, DNA binding
Significance
Although cryptochrome-DASH (CRYD) is high sequence homology with photolyases (PLs), CRYD cannot repair UV-damaged DNA. We investigated the electron transfer reactivity in terms of the electronic coupling matrix elements TDA, using theoretical methods, indicated that TDA value of CRYD is comparable with PLs. Hence CRYD will exhibit the comparable electron transfer reactivity with PLs. We furthermore explored DNA binding activity, elucidated that CRYD does not form the stable salt bridges among the specific amino acid residues and DNA, whereas PLs form their stable salt bridges. Therefore, we indicated that the stable salt bridge is important in order to DNA repair.
Introduction
DNA damage by UV radiation (290–400 nm) results in the formation of two major types of UV-induced DNA lesions at adjacent pyrimidine bases: cis-syn cyclobutane pyrimidine dimers (CPDs) and pyrimidine-pyrimidone (6-4) photoproducts ((6-4)PPs) [1–3]. These lesions block cellular replication and transcription, leading to growth inhibition in plants and skin cancer in mammals. Photolyases (PLs) have been identified as enzymes responsible for the repair of these lesions [1,4,5]. Cryptochromes (CRYs) [6–8], which are blue light receptors, play key roles in various biological functions, such as entrainment of circadian rhythms in plants and magnetic receptors in birds [8–14]. Both PLs and CRYs belong to the PLs/CRYs superfamily, which includes proteins with diverse functions. PLs and CRYs share homologies in amino acid sequence and tertiary structure [1], besides sharing a flavin adenine dinucleotide (FAD) as a common cofactor that absorbs blue light in sunlight to exert their functions [2,4]. However, CRYs do not exhibit DNA repair activity in response to UV-induced DNA damage because the FAD in CRYs is hardly converted to the fully reduced form of FAD (FADH–) [15,16], which is the active redox state of FAD in PLs. In 2000, Brudler et al. identified a new type cryptochrome, cryptochrome-DASH (CRYD), in bacteria and plants [17]. The structure and the function of CRYD were investigated experimentally [17–21], and it was reported that Arabidopsis thaliana CRYD (AtCRYD) can selectively repair CPD on single-stranded DNA (ssDNA) but not on double-stranded DNA (dsDNA) [20]. The reason why CRYD does not exhibit this distinct DNA repair activity remains obscure.
The DNA repair process of PLs consists of the following steps: (1) recognition of UV-induced DNA lesions by PLs, accommodating the lesions in the active site; (2) light-excitation of the fully reduced state of FAD (FADH–*); (3) forward electron transfer (FET) from FADH–* to the UV-induced DNA lesions, forming a neutral semiquinoid FAD (FADH•) and an anion radical of the lesions; (4) rearrangement of the UV-induced DNA lesions in the anion radical state; and (5) a back electron transfer from the repaired pyrimidine bases to FADH• [1,22–26]. The FET reaction, a pivotal process for the light-driven DNA repair mechanism by PLs, has been explored in detail by theoretical and experimental analyses. Currently, FET is considered to occur using the superexchange mechanism via the adenine moiety of FAD. Stuchebrukhov et al. reported pioneering results regarding the superexchange mechanism via the adenine moiety by investigating the electron transfer (ET) pathway of CPD photolyases (CPD-PLs), using a tunneling current approach [27–30]. Liu et al. investigated the ET reaction of CPD-PLs using femtosecond spectroscopy and demonstrated that the adenine-mediated superexchange mechanism was dominant [25]. Recently, Miyazawa et al. and Sato et al. suggested the possibility of amino acid residue-mediated superexchange mechanism via a methionine and/or an asparagine residue in the active site [31,32] and Prytkova et al. suggested the direct ET from FADH–* to CPD [33]. Although the outline of the DNA repair mechanism for CPD-PLs has been revealed during the last two decades by theoretical [29–39] and experimental studies [23–26,40–45], there is room for further research on the DNA repair mechanism because the primary process in the DNA repair reaction, i.e., the recognition of UV-induced DNA lesions by PLs, is not clearly understood. An understanding of the whole DNA repair process would shed light on the origin of the functional differences between PLs and CRYD.
Here, we investigated the FET reaction and DNA binding of Anacystis nidulans CPD-PL (AnPL) and AtCRYD to identify the differences in the DNA repair activities of PLs and CRYs. In this study, the electronic coupling matrix elements TDA was estimated for AtCRYD using molecular dynamics (MD) simulations and quantum chemical calculations. In addition, the TDA value of an AtCRYD mutant, in which Gln395 was replaced with methionine residue (AtCRYDQ395M), was also estimated. Previous studies reported that the methionine (Met353) and the asparagine (Asn349) residues contributed to FET in AnPL [31,32]. The amino acid residue which is same position as Met353 of AnPL in AtCRYD replaced the glutamine residue (Gln395), whereas the amino acid residue which is same position of Asn349 of AnPL in AtCRYD was not replaced (Asn391) (the Supplementary Fig. S1 and S2). Therefore, comparing the TDA values of wild-type AtCRYD and mutant AtCRYD, we considered that the differences in the amino acid residues might affect the FET reactivity. The result indicated that the TDA value of AtCRYD was comparable to that of AtCRYDQ395M and AnPL, which were estimated in the previous study [32]. Our experimental analysis confirmed that AtCRYD exhibited DNA repair activity for ssDNA. Then, we measured the DNA repair activity for wild-type AtCRYD and mutant AtCRYD by mutating the three amino acid residues in AtCRYD (Arg443Lys/Glu444Pro/Asp445Leu) and found that both proteins cannot repair dsDNA. To elucidate the underlying reason, we compared the behaviors of AnPL and AtCRYD in complex with dsDNA containing the CPD, using MD simulations. The MD simulations indicated that the stable salt bridges between the two positively charged amino acid residues (Lys401 and Arg404 for AnPL, Arg443 and Arg446 for AtCRYD) and DNA were stable in AnPL, in contrast to those in AtCRYD, which were transient. We proposed that this instability of the salt bridges might be one factor that leads to lower DNA binding activity of AtCRYD for dsDNA. In addition, our theoretical and experimental analyses further indicated that the two negatively charged residues, Glu444 and Asp445, are not be the cause of the transient salt bridge.
Methods
In this study, we investigated two major issues: (1) the electron transfer (ET) reactivity of AtCRYD in complex with UV-damaged single-stranded DNA (ssDNA) containing a CPD (AtCRYD-ssDNA) and (2) the dynamical behavior of AnPL in complex with UV-damaged dsDNA containing a CPD (AnPL-dsDNA) and of AtCRYD in complex with UV-damaged dsDNA containing a CPD (AtCRYD-dsDNA) complex.
Setup for the initial structures
To estimate the electronic coupling matrix element TDA for evaluation of ET reactivity, theoretical calculations were performed using either the coordinates after geometry optimization of the X-ray crystal structure (PDB ID: 2VTB [21]) by quantum mechanics/molecular mechanics (QM/MM) calculation or the equilibrated structures of MD simulations, depending on the purpose. We used either i) each optimized structure of AtCRYD in complex with ssDNA containing a CPD, and AtCRYD mutant in which Gln395 of 2VTB was replaced with methionine (AtCRYDQ395M) by QM/MM calculation, or ii) many snapshots during 500 ns MD simulations for AtCRYD-ssDNA and AtCRYDQ395M-ssDNA to evaluate the contribution of the protein environment on the TDA value. In both cases, the artificial CPD-analog in 2VTB was converted to the CPD with the natural phosphate by applying a force field upon the tLeap procedure described below. These model structures were used as the initial structures for the QM/MM calculation and the 500 ns MD simulations.
For the observation of the dynamical behavior of AnPL-dsDNA and AtCRYD-dsDNA complexes, a complex structure of AtCRYD with dsDNA containing CPD is required, but it has not been solved to date. We, therefore, prepared a model structure of the AtCRYD-dsDNA complex. The structures of AnPL in complex with dsDNA containing a CPD-analog with a formacetal linkage in its internucleoside moiety (PDB ID: 1TEZ [46]) and AtCRYD (PDB ID: 2VTB) were aligned by TM-align [47], and the AnPL was replaced with AtCRYD (Supplementary Fig. S2). We used these model structures as the initial structures for the 1000ns MD simulations. Note that the CPD-analog in AnPL-dsDNA and AtCRYD-dsDNA was also converted to the natural CPD in the manner described above.
Amber force fields for each cofactor and cyclobutane pyrimidine dimer
In order to obtain the force fields of CPD, FADH–, 5,10-methenyltetrahydrofolate (MTHF), and 8-hydroxy-7,8-didemethyl-5-deazariboflavin (8-HDF), we performed a geometry optimization and electron structure calculation for each molecule with a B3LYP/6-31G(d) and MP2/6-31+G(d,p) level of theory using Gaussian 16 [48]. Here, the geometric coordinate of CPD was obtained from PDB ID: 1SNH [49], and FADH–, MTHF, and 8-HDF were obtained from PDB ID: 2VTB and 1TEZ, respectively. Each cofactor and CPD was parameterized using a general amber force field [50], and the partial charges were calculated using the restrained electrostatic potential method using the antechamber module of the AMBER16 program package [51].
Setup for quantum mechanics/molecular mechanics calculations and molecular dynamics simulations
For the QM/MM calculation and MD simulations to estimate the TDA value, we prepared initial structures using the tLeap module of the AMBER program package by assigning the force fields for AtCRYD-ssDNA and AtCRYDQ395M-ssDNA. Here, we adopted the AMBER ff14SB for protein [52], the AMBER OL15 for DNA [53], and the created force fields for CPD, FADH–, and MTHF. Since it has been supposed by theoretical and experimental studies that the protonation of Glu283 in AnPL affects the DNA repair reaction [35,46,54], Glu325 was protonated in AtCRYD-ssDNA and AtCRYDQ395M-ssDNA, and other amino acid residues maintained the standard state in physiological pH. We adopted a TIP3P model for water molecules [55] and constructed square simulation boxes with a margin of at least 12 Å from proteins to the box boundaries. Finally, to neutralize each system, counterions (Na+) were added.
To investigate the dsDNA binding mechanism for CPD-PL and CRYD, we used the modified X-ray crystal structure of AnPL-dsDNA and the modeled structure for AtCRYD-dsDNA. Each solvated system was created according to the procedures described above, and we adopted the created force field for 8-HDF in AnPL-dsDNA. Simulation boxes with a margin of at least 30 Å from proteins to the box boundaries were used.
The procedures of molecular dynamics simulations
In this study, we carried out the MD simulations as follows: (1) the obtained AMBER topology file was converted to a GROMACS topology file for each system using the ACPYPE tool [56], and then energy minimization was performed for 5000 steps. (2) The temperature of each system was heated from 0 K to 298 K during 100 ps. First, the temperature was heated from 0 K to 5 K for 10 ps under the NVT ensemble (T=5 K). Then, the temperature was gradually heated for nine steps under the NPT ensemble (P=1 bar; T=5 K, 50 K, 100 K, 150 K, 200 K, 225 K, 250 K, 275 K, and 298 K), where each step was carried out for 10 ps. (3) The MD simulations were carried out for 500 ns in the TDA values estimation or 1000 ns in the DNA binding study under the NPT ensemble (P=1 bar and T=298 K). Here a restriction for the 5'- and 3'-edge nucleobases of dsDNA (i.e., a force constant of 4184 kJ/(mol·nm2)) was applied during temperature heating and production run procedures. In all MD simulations, the linear constraint solver algorithm was used for constraints [57], the Nose-Hoover thermostat for temperature regulation [58–60], and the Berendsen barostat for pressure regulation [61]. The periodic boundary conditions were applied with the particle mesh Ewald method [62], and the simulation time step was 2 fs. A snapshot was recorded every 10 ps. In this study, all MD simulations were performed by the GROMACS version 5.0.4 program package [63].
Quantum mechanics/molecular mechanics calculation
We carried out the geometry optimization by the QM/MM calculation for the energy minimized structures of AtCRYD-ssDNA and AtCRYDQ395M-ssDNA by MM level using GROMACS. In the QM/MM calculation of AtCRYD-ssDNA (AtCRYDQ395M-ssDNA), the QM region included CPD, FADH–, Glu325, Asn391, and Gln395 (Met395), whereas the other molecules were in the MM region (Supplementary Fig. S3). We adopted our own N-layered integrated molecular orbital and molecular mechanics (ONIOM) method [64] and performed the geometry optimization with B3LYP/6-31G(d):AMBER94 levels at Gaussian 09 [65], where the electronic embedding is adopted to include the MM region charge distribution in the QM evaluations [66].
Electronic coupling matrix elements
To investigate the ET reaction in this study, the electronic coupling matrix elements TDA were estimated. The TDA value is usually used to evaluate the ET rate constants KET. KET is expressed as the following equation by the Marcus theory [67]:
| (1) |
where ΔG and λ are the driving force and the reorganization energy, respectively; ħ, KB and T are the Planck constant divided by 2π, Boltzmann constant, and temperature, respectively. To estimate the TDA value, we adopted the generalized Mulliken–Hush (GMH) method [68–70] with the configuration interaction singles (CIS) method [71]. The GMH method is expressed as follows:
| (2) |
where μ1 and μ2 are the corresponding state dipole moment vectors of initial state 1 and final state 2, respectively, and μ12 is the transition dipole moment from the CIS wave functions ψ1 to ψ2. ΔE12 is the difference in the excited state energies between ψ1 and ψ2. Here, we performed the CIS calculation using the GAMESS program [72].
We applied this method for the QM/MM optimized structure of each system (i.e., AtCRYD-ssDNA and AtCRYDQ395M-ssDNA), where the isoalloxazine ring of FADH–, the adenine moiety of FADH–, CPD, Glu325, Asn391, and Glu395 or Met395 for AtCRYD-ssDNA and AtCRYDQ395M-ssDNA were included in the QM region to calculate CIS, whereas the other atoms were treated as the point charge to simulate the surrounding environment of the QM region. To simplify the system further, the ribitol part of FADH– and the main chain of Glu, Asn, Gln, and Met were replaced with methyl group. Also, the internucleoside linkages and its neighboring oxygen atom of the 5'-side and the oxygen atom adjacent the internucleoside linkages of 3'-side in CPD replaced with H atom and the ribose part of the adenine moiety of FADH– were replaced with H atom (Supplementary Fig. S4). We also applied the same procedure for 200 MD snapshots of AtCRYD-ssDNA and AtCRYDQ395M-ssDNA.
Results and Discussion
Electron transfer reactivity for cryptochrome-DASH
To investigate the ET reaction of AtCRYD-ssDNA and AtCRYDQ395M-ssDNA, we evaluated the electronic coupling matrix elements TDA with the molecular configurations shown in Figure 1a. As shown in Figure 2, the relevant molecular orbitals (MOs) of each system were found to be similar. In all cases, MOs for the ground state S0 were populated in the distal side of the isoalloxazine ring toward the CPD, whereas those for the lowest singlet excited state S1 and the second singlet excited state S2 were populated in the proximal side of the isoalloxazine ring. MOs for the third singlet excited state S3 were populated in the 3'-side of CPD. These results indicated that S0→S1 and S0→S2 transitions were assigned as π→π* excitations of flavin (locally excited (LE) state), and S0→S3 indicated the charge transfer (CT) from FADH– to CPD. The excitation energies to S1, S2, and S3 for AtCRYD-ssDNA were 4.4643, 4.8752, and 5.2237 eV, respectively, while those for AtCRYDQ395M-ssDNA were 4.4432, 4.8812, and 4.9730 eV, respectively. And the oscillator strengths to S1, S2, and S3 for AtCRYD-ssDNA were 0.00, 0.25, and 0.00, respectively, while those for AtCRYDQ395M-ssDNA were 0.00, 0.25, and 0.00, respectively, (Table 1). These results indicate that the displacement of the electron amplitude from the distal side to the proximal side of the isoalloxazine ring occurs upon light-excitation (S0→S2) and subsequently being relaxes to the S1 state by internal conversion. The internal conversion rate of flavin determined by time-resolved absorption and fluorescence experiments showed that the relaxation S2→S1 would occur on an ultrafast time scale (<1 ps) [73,74]. These results indicate that ET occurs from FADH–* to CPD by the thermal excitation from the S1 state to the S3 state after relaxation.
Figure 1.
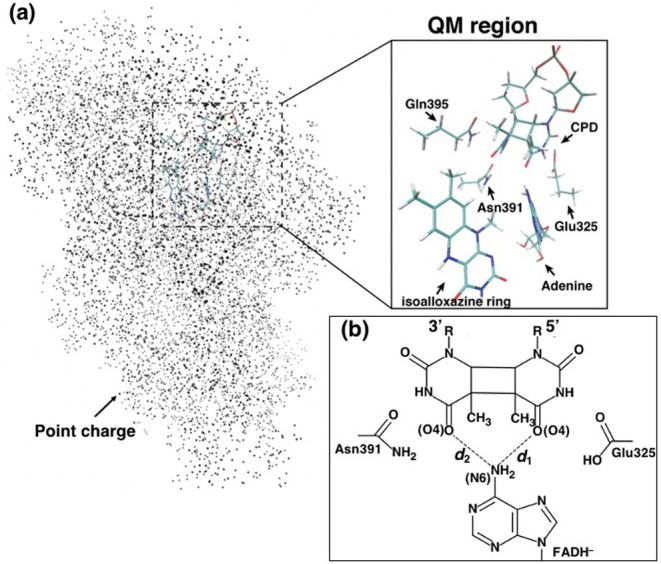
A calculation system of CIS for QM/MM optimized structures and MD snapshots. (a) The molecules in the QM region; isoalloxazine ring of FADH–, adenine moiety of FADH–, CPD, Glu325, Asn391, and Gln395 or Met395 for AtCRYD or AtCRYDQ395M. The other atoms are treated as point charges, which are shown as black dots. Notably, water molecules were taken into account as point charges, although they are not shown in this figure. (b) Schematic view of the relative position of the adenine moiety and the CPD. d1 and d2 are the distances between the adenine moiety (N6 atom) of FADH– and CPD (two O atoms).
Figure 2.
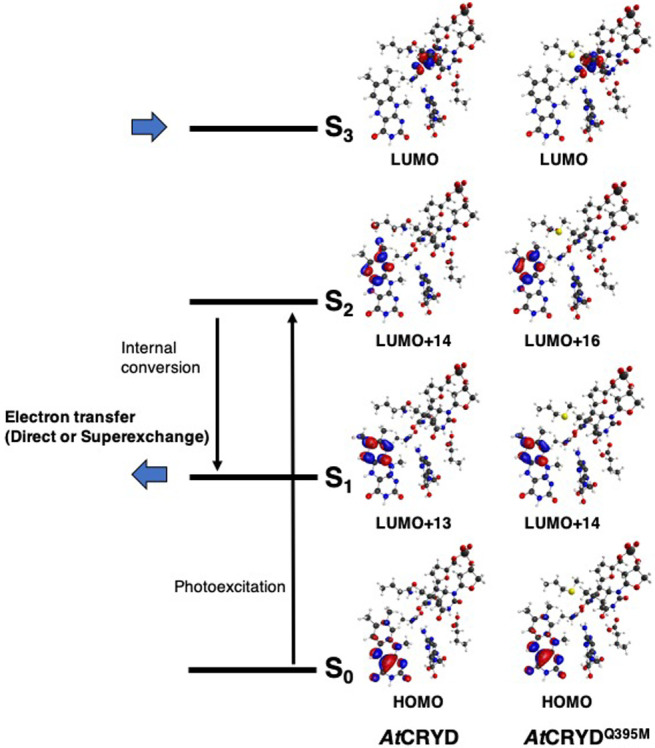
Molecular orbitals for each excited state calculated for the QM/MM optimized structures. S0 is the ground state, and Sn is nth singlet excited state. Here, the molecular orbitals of the largest CI coefficient of each excited state for QM region (Fig. 1a) were indicated.
Table 1.
Excited state, CI vector, and excitation energies [eV] for QM/MM optimized structure
| AtCRYD Excited statea | CI vector (top CI coefficient and transition are shown)b | Excitation energy (oscillator strength)b |
|---|---|---|
| S1 (LE state (I)) | –0.64 (HOMO→LUMO+13) | 4.4632 (0.00) |
| S2 (LE state (I)) | –0.56 (HOMO→LUMO+14) | 4.8752 (0.25) |
| S3 (CT state (C)) | –0.99 (HOMO→LUMO) | 5.2237 (0.00) |
| S5 (CT state (A)) | –0.86 (HOMO→LUMO+5) | 5.8203 (0.02) |
| AtCRYDQ395M Excited statea | CI vector (top CI coefficient and transition are shown)b | Excitation energy (oscillator strength)b |
| S1 (LE state (I)) | –0.64 (HOMO→LUMO+14) | 4.4432 (0.00) |
| S2 (LE state (I)) | –0.51 (HOMO→LUMO+16) | 4.8812 (0.25) |
| S3 (CT state (C)) | –0.99 (HOMO→LUMO) | 4.9730 (0.00) |
| S5 (CT state (A)) | 0.70 (HOMO→LUMO+4) | 5.8830 (0.05) |
a I, A, and C represent isoalloxazine ring, adenine moiety, CPD, respectively. b The calculation level of CIS/6-31G(d).
We then estimated the TDA value for each system using the obtained MOs. The TDA value was 6.5 meV for AtCRYD-ssDNA and was 3.1 meV for AtCRYDQ395M-ssDNA. To verify the structures obtained by QM/MM optimization, the distances between CPD and the adenine moiety of FADH– in AtCRD-ssDNA and AtCRDQ395M-ssDNA were evaluated. The measured distances d1 and d2 represent the relative position of adenine toward the CPD (i.e., the distances between the N6 of the adenine moiety and the two oxygens O4 of CPD; 5'-side of CPD for d1 and 3'-side for d2; Fig. 1b). The obtained distances are summarized in the Supplementary Table S1. In the comparison between d1 and d2, the relative position of the adenine moiety for AtCRYD-ssDNA was more biased to the 5'-side of the CPD than that of AnCRYDQ395M-ssDNA, with a difference of 0.48 Å compared to AtCRYDQ395M-ssDNA. Thus, it is suggested that the position of the adenine moiety gave a major contribution to the TDA value. Notably, ET reaction is generally reported to be sensitive to the protein environment [75]; hence, the contribution of the protein environments should be taken into account. To investigate the influence of the protein environments, we performed CIS calculation for the QM/MM optimized structure of AtCRYD-ssDNA, excluding the all point charges; however, we could not observe the CT state from FADH– to CPD within ten excitation states (S1–S10). Then we performed a CIS calculation for the system, excluding the point charge of ssDNA. As a result, the estimated excitation energy of the CT state (S3) from FADH– to CPD was 4.87 eV, which was smaller than that of the full system (5.22 eV). In contrast, the excitation energies of the LE states of FADH– (S1 and S2) were almost unchanged (<0.05 eV). The TDA value (7.0 meV) changed slightly as compared to that of the full system (6.5 meV). Thus, it is shown that the presence of the surrounding amino acid residues of active site and nucleotides have a vast influence on the electronic state of the active site.
Based on the above results, we estimated the TDA value to gauge the structural fluctuation using MD snapshots. First, we performed 500 ns MD simulations for AtCRYD-ssDNA and AtCRYDQ395M-ssDNA and estimated the root-mean-square deviation (RMSD) value (Supplementary Fig. S5). Each RMSD value was stable after 300 ns, and the average RMSD values for the last 200 ns were 2.3±0.2 and 2.4±0.1 Å in AtCRYD-ssDNA and AtCRYDQ395M-ssDNA, respectively. We, therefore, randomly chose 200 snapshots from the last 200 ns MD simulations and calculated the MOs for each snapshot in the same way as for the QM/MM optimized structures. A representative set of MOs selected from these 200 calculations is depicted in Figure 3, and the excitation energies and the oscillator strengths are summarized in Table 2. The S1 and S2 excited states were found to be in the same states (LE state of the isoalloxazine ring) as those for the QM/MM optimized structure, with excitation energies of ~4.0 eV and ~4.6 eV for AtCRYD-ssDNA and AtCRYDQ395M-ssDNA, respectively. In contrast, the S3 excited state was in the CT state from the isoalloxazine ring to the adenine moiety (CT-A), which was distinct from the result for the QM/MM optimized structures (Table 1). Finally, the S4 excited state was found to be the CT state from the isoalloxazine ring to CPD (CT-C), indicating that the CT-A state had decreased in energy due to the protein environment. In some of the MD snapshots, we also observed the S3 excited state as CT-C, but the rate of its occurrence was rather low (<10%). Indeed, such behavior was previously reported for AnCPD-PL by Lee et al. [38], who calculated the excited states for 20 structures containing FADH– and CPD with an ADC(2)/def2-SVP level of theory and reported that the orders of the CT-A and CT-C states depending on the structure. Although a hopping mechanism through the adenine moiety was considered, this mechanism was ruled out by a later experimental study [25]. Therefore, we assumed that the ET reaction from the isoalloxazine ring to CPD always occurs. Taking into account the similarity between the energy levels, we concluded that the S4–S6 excited states were dominant in the ET reaction for AtCRYD-ssDNA and AtCRYDQ395M-ssDNA (53% and 59%, respectively). Notably, the orders of the excited states higher than S4 for the MD snapshots were diverse, with similar excitation energies within a range of ~0.2 eV (Supplementary Table S2).
Figure 3.
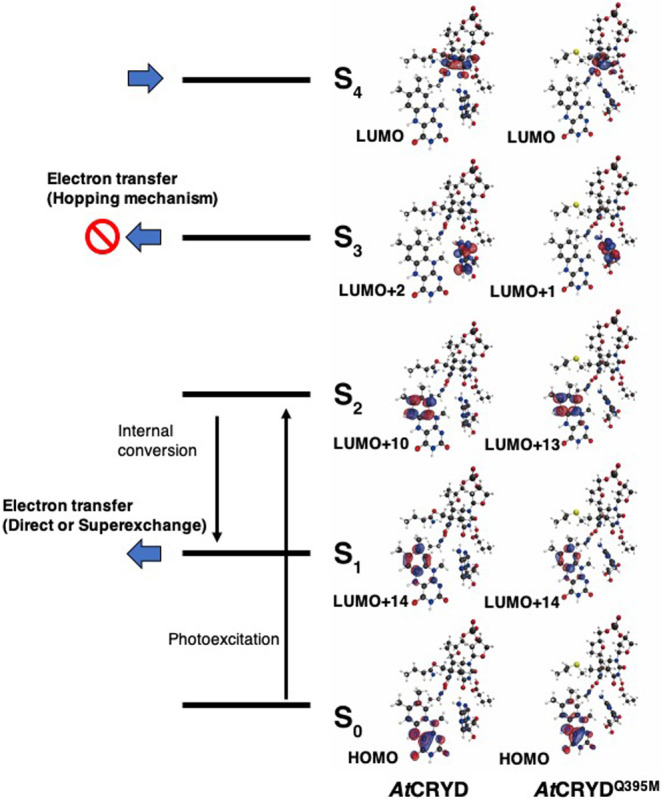
Representative molecular orbitals calculated for MD snapshot. S0 is the ground state, and Sn is nth singlet excited state. Here, the molecular orbitals of the largest CI coefficient of each excited state for QM region (Fig. 1a) were indicated.
Table 2.
Excited state, CI vector, and excitation energies [eV] for representative MD snapshot
| AtCRYD Excited statea | CI vector (top CI coefficient and transition are shown)b | Excitation energy (oscillator strength)b |
|---|---|---|
| S1 (LE state (I)) | 0.66 (HOMO→LUMO+14) | 4.1277 (0.03) |
| S2 (LE state (I)) | 0.80 (HOMO→LUMO+10) | 4.6373 (0.20) |
| S3 (CT state (A)) | 0.86 (HOMO→LUMO+2) | 5.2078 (0.02) |
| S4 (CT state (C)) | –0.92 (HOMO→LUMO) | 5.3534 (0.00) |
| AtCRYDQ395M Excited statea | CI vector (top CI coefficient and transition are shown)b | Excitation energy (oscillator strength)b |
| S1 (LE state (I)) | –0.75 (HOMO→LUMO+14) | 4.0096 (0.03) |
| S2 (LE state (I)) | 0.58 (HOMO→LUMO+13) | 4.6743 (0.17) |
| S3 (CT state (A)) | 0.91 (HOMO→LUMO+1) | 5.2596 (0.00) |
| S4 (CT state (C)) | 0.93 (HOMO→LUMO) | 5.4777 (0.00) |
a I, A and C represent isoalloxazine ring, adenine moiety and CPD, respectively. b The calculation level of CIS/6-31G(d).
The average TDA values for the MD snapshots were 5.3±4.6 meV for AtCRYD-ssDNA and 5.1±4.8 meV for AtCRYDQ395M-ssDNA, whereas these values for the QM/MM optimized structures were ~6.0 and ~3.0 meV, respectively. These changes indicated that the TDA value was remarkably affected by the protein environment. In the previous study using the same method, the TDA value of AnPL-dsDNA was estimated to be 4.5±3.7 meV [32], which is in a good agreement with the estimated values for AtCRYD-ssDNA and AtCRYDQ395M-ssDNA. In the previous experimental study, the ET rate constant of Escherichia coli CPD-PL was identified as 250 ps, with –0.44 eV for the driving force ΔG and 1.21 eV for the reorientation energy λ, corresponding to 3.0 meV of the estimated electronic coupling constant [25]. This result indicates that the ET rate constant of AtCRYD may be comparable to that of AnPL. As mentioned above, a difference in the lesion-binding site could be responsible for the reduction in the DNA repair activity in AtCRYD; however, the TDA value of AtCRYDQ395M, which a binding site resembling that of AnPL, was found to be identical to that of AtCRYD. This fact excludes the possibility that the methionine in the active site critically affects the ET rate constant, although the methionine is important for the stabilization of the active site [54] and the CPD-PL activity [46,76].
In the MD simulation for AtCRYD-ssDNA, the average distances d1 and d2 were 2.9±0.2 Å and 3.3±0.4 Å, respectively, whereas those for AtCRYDQ395M-ssDNA were 3.1±0.2 Å and 3.1±0.3 Å, respectively. To investigate the relationship between the relative position of the adenine–CPD pair and the TDA value, the TDA value for each snapshot was evaluated and mapped as functions of the distances d1 and d2 (Fig. 4). The distances d1 and d2 were distributed in the range of 2.6–4.0 Å; however, large TDA values (>15 meV) were mostly observed only when either d1 or d2 was shorter than 3.0 Å. This result indicates that the TDA value might decrease when the adenine moiety is located away from CPD. Rousseau et al. also reported that the contribution of the superexchange mechanism decreased when the adenine moiety was separated from CPD [39]. Therefore, the adenine moiety is very important for the ET reaction.
Figure 4.
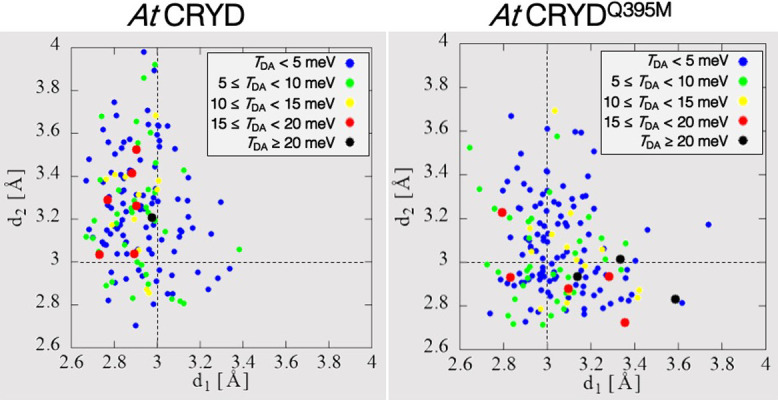
Correlation between distances, d1 and d2, and the TDA value for AtCRYD and AtCRYDQ395. Here blue, green, yellow, red and black dots express TDA<5.0 meV, 5.0≤TDA<10.0 meV, 10.0≤TDA<15.0 meV, 15.0≤TDA<20.0 meV, and TDA≥20.0 meV, respectively.
Molecular dynamics simulations for dsDNA binding state
Because the electron transfer efficiency of AtCRYD was estimated to be similar to that of AnPL, the functional differences between CRYD and PL have been speculated to originate from an upstream DNA repair process. Indeed, a previous experimental study demonstrated that AtCRYD is able to bind specifically to the CPD located in the bubble of dsDNA, indicating that the CPD binds weakly to the dsDNA [21]. A previous theoretical study suggested that the two negatively charged amino acid residues, Glu444 and Asp445, in AtCRYD would give rise to the repulsion of dsDNA [77]. The same sequence positions in AnPL are occupied by nonpolar amino acid residues (Pro402 and Leu403) (Supplementary Fig. S1). Therefore, we first considered the possibility that these negatively charged residues affect the DNA repair activity of AtCRYD. To confirm this, we prepared mutant AtCRYD (Supplementary Texts S1, S2 and Figs. S6 and S7) by mutating the three amino acid residues in AtCRYD to those of AnPL, namely Arg443Lys/Glu444Pro/Asp445Leu, and examining their DNA repair activities in vitro. Although the ssDNA containing the CPD was successfully repaired in both proteins, as expected from our results on ET reactivity, the repair of the CPD in the dsDNA was unsuccessful even for the triple mutant (Supplementary Text S3 and Fig. S8). This result indicated that the repulsion among two negatively charged residues and DNA will not be a dominant factor for the inhibition of DNA repair activity in AtCRYD.
To elucidate the other factors inhibiting DNA repair activity, we compared the behavior of the AnPL-dsDNA complex with that of the AtCRYD-dsDNA complex using MD simulations. We focused the formation of the salt bridge between Lys401, Arg404, and DNA in AnPL because it was reported that the salt bridge between protein and DNA relates to the DNA repair yield of PLs [78]. Although no salt bridge between the positively charged residues (Lys401 and Arg404) and the phosphate backbone of the DNA was found in the AnPL crystal structure [46], the stable salt bridges were observed in our MD simulations. The average value of the distances between Lys401 and the phosphate backbone of 5'-side of CPD (dNZ-O1 and dNZ-O2: the distances between the nitrogen atom NZ of Lys401 and the two oxygen atoms O1 or O2 of the phosphate) were close enough to form the salt bridge, with 4.2±1.0 Å for dNZ-O1 and 3.0±0.5 Å for dNZ-O2. The same held true for the Arg404 and the internucleoside phosphate of the CPD pair, and dNH1-O11, dNH1-O12, dNH2-O11, and dNH2-O12 (i.e., the distances between the two nitrogen atoms NH1 and NH2 of Arg404 and two oxygen atoms O11 and O12 of the internucleoside phosphate of CPD) were 3.0±0.6, 4.1±0.5, 4.4±0.9, and 4.5±0.7 Å, respectively, during the last 500 ns of 1000 ns MD simulations (Figs. 5a–c). The above conformational difference between the crystal structure and the MD simulation would be attributable to the presence of the natural phosphate at the internucleoside linkage in the CPD, instead of the nonpolar formacetal moiety. In the structural analysis of class II CPD-PL from Methanosarcina mazei, the replacement of the formacetal by the phosphate induced a rearrangement at the active site that allowed the CPD lesion to be accommodated more tightly than with the formacetal linkage [79]. Based on these results, formation of the salt bridge between DNA and amino acid residues is important for DNA binding, hence implying necessity to flip the CPD out of the DNA duplex at the beginning of the DNA repair process.
Figure 5.

The distance between an amino acid residue and CPD that form a salt bridge during 1000 ns MD simulations. For AnPL, dNZ-O1 and dNZ-O2 are the distances between nitrogen atom NZ of Lys401 and oxygen atoms O1 and O2 of CPD, respectively. dNH1-O11, dNH1-O12, dNH2-O11, and dNH2-O12 are the distance between nitrogen atoms NH1 and NH2 of Arg404 and oxygen atoms O11 and O12 of CPD, as indicated by the subscripts. For AtCRYD, d1NH1-O11, d1NH1-O12, d1NH2-O11, and d1NH2-O12 are the distances between nitrogen atoms NH1 and NH2 of Arg443 and oxygen atoms O11 and O12 of CPD, as indicated by the subscripts, while d2NH1-O11, d2NH1-O12, d2NH2-O11, and d2NH2-O12 are the distances between nitrogen atoms NH1 and NH2 of Arg446 and oxygen atoms O11 and O12 of CPD, as indicated by the subscripts. (a), (b) and (c) are each distance of AnPL. (a) the distances dNZ-O1 and dNZ-O2 are displayed, (b) the distances dNH1-O11 and dNH1-O12 are displayed, and (c) the distances dNH2-O11 and dNH2-O12 are displayed, respectively. On the other hand, (d), (e), (f), and (g) are each distance of AtCRYD. (d) the distances d1NH1-O11 and d1NH1-O12 are displayed, (e) the distances d1NH2-O11 and d1NH2-O12 are displayed, (f) the distances d2NH1-O11 and d2NH1-O12 are displayed, and (g) the distances d2NH2-O11 and d2NH2-O12 are displayed, respectively.
In contrast with AnPL-dsDNA, the two positively charged residues in AtCRYD (Arg443 and Arg446) did not participate in the electrostatic interactions during the MD simulations (Fig. 5), despite their location at the same position as AnPL (Supplementary Fig. S1). The Arg443 residue, corresponding to Lys401 in AnPL, was located away from the phosphate backbone of 5'-side of CPD and distal to the internucleoside phosphate moiety during the 900 ns MD simulations (10.9±3.1 Å for d1NH1-O11, 11.9±3.4 Å for d1NH1-O12, 10.4±3.5 Å for d1NH2-O11, and 11.3±3.8 Å for d1NH2-O12). However, we observed that Arg443 swung closer to the internucleoside phosphate at a distance of <4 Å, forming a salt bridge during the last 100 ns of MD simulations (Figs. 5d, e). The electrostatic interaction between Arg446, corresponding to Arg404 in AnPL, and the internucleoside phosphate of CPD seemed to be weak, with slightly longer distances compared to those observed for AnPL (5.9±2.3 Å for d2NH1-O11, 7.4±2.2 Å for d2NH1-O12, 5.4±1.7 Å for d2NH2-O11, and 6.5±1.8 Å for d2NH2-O12; Figs. 5f, g). Therefore, the salt bridge would be transiently formed upon DNA binding in the case of AtCRYD. These results indicate that the stability of the salt bridge in AtCRYD is much weaker than that of AnPL. The transient salt bridge formation observed here would inhibit proper DNA binding of AtCRYD. In addition, we attempted the MD simulations for the model structure of triple mutant AtCRYD, but the salt bridges were not improved. In particular, the salt bridge of Arg446, which was considered more important for DNA repair activity [78], was not definitely formed (Supplementary Fig. S9).
As we suggested above, the salt bridge formation would play a key role in lesion flipping. The formation of a transient salt bridge would be incompatible with accommodation of the CPD lesion in the binding site, presumably preventing the repair of dsDNA for AtCRYD. Therefore, we suggested that the formation of a transient salt bridge is one factor determining the DNA binding inhibition of AtCRYD by generating attraction between the positively charged residues (Arg443 and Arg446) and DNA rather than the repulsion by the negatively charged residues (Glu444 and Asp445). In addition, it was also indicated that the presence of the negatively charged residues is not responsible for the instability of the salt bridge.
Conclusion
In this study, we searched for the intrinsic origin of the functional differences between AtCRYD and AnPL by using theoretical calculations with supplementary experiments. Regarding their ET reactivities, we found that i) the energy levels of the molecular orbitals in excited states S3 or above varied depending on the protein environment, ii) the ET reactivity, described by electronic coupling matrix element TDA, for AtCRYD was in good agreement with that for AnPL, and iii) an introduction of a minor mutation, such as replacement of methionine with glutamine in the active site did not influence the ET reactivity. Comprehensively, these findings indicated that AtCRYD should be able to repair UV-damaged DNA if the lesion is properly accommodated in the active site. Therefore, it is suggested that the differences between AtCRYD and AnPL in biological function would be ascribed to the DNA binding properties. The MD simulations of AnPL-dsDNA and AtCRYD-dsDNA complexes indicated that DNA binding would be inhibited by transient salt bridge formation in AtCRYD and the repulsive interaction with DNA due to negatively charged residues (Glu444 and Asp445 in AtCRYD) would not be the cause of this instability. The proper formation of the salt bridge will be a key factor that leads the DNA repair function in AtCRYD. This finding of the transient salt bridge formation in AtCRYD will give further insights into the functional differences between AtCRYD and AnPL in future studies.
Acknowledgements
This work was supported by RIKEN’s special postdoctoral researcher program. The calculations were performed using Research Center for Computational Science (RCCS), Okazaki, Japan and RIKEN HOKUSAI System.
Conflicts of Interest
All the authors declare that they have no conflict of interest.
Author Contributions
R.S. and M.T. directed this project. R.S. and Y.M. performed the theoretical calculations and data analysis. R.M. and J.Y. performed the AtCRYD production and in vitro assays. R.S., N.O., J.Y. and M.T. wrote the manuscript.
Supplementary Material
References
- [1].Sancar A. Structure and function of DNA photolyase and cryptochrome blue-light photoreceptors. Chem. Rev. 103, 2203–2237 (2003). DOI: 10.1021/cr0204348 [DOI] [PubMed] [Google Scholar]
- [2].Jagger, J. Solar-UV actions on living cells. (Praeger, New York, 1985). [Google Scholar]
- [3].Mitchell D. L. & Nairn R. S.. The biology of the (6-4) photoproduct. Photochem. Photobiol. 49, 805–819 (1989). DOI: 10.1111/j.1751-1097.1989.tb05578.x [DOI] [PubMed] [Google Scholar]
- [4].Sancar A. Structure and function of DNA photolyase. Biochemistry 33, 2–9 (1994). DOI: 10.1021/bi00167a001 [DOI] [PubMed] [Google Scholar]
- [5].Brettel K. & Byrdin M.. Reaction mechanism of DNA photolyase. Curr. Opin. Struct. Biol. 20, 693–701 (2010). DOI: 10.1016/j.sbj.2010.07.003 [DOI] [PubMed] [Google Scholar]
- [6].Cashmore A. R. The cryptochrome family of blue/UV-A photoreceptors. J. Plant Res. 111, 267–270 (1998). [Google Scholar]
- [7].Cashmore A. R., Jarillo J. A., Wu Y.-J. & Liu D.. Cryptochromes: blue light receptors for plants and animals. Science 284, 760–765 (1999). DOI: 10.1126/science.284.5415.760 [DOI] [PubMed] [Google Scholar]
- [8].Partch C. L., Clarkson M. W., Özgür S., Lee A. L. & Sancar A.. Role of structural plasticity in signal transduction by the cryptochrome blue-light photoreceptor. Biochemistry 44, 3795–3805 (2005). DOI: 10.1021/bi047545g [DOI] [PubMed] [Google Scholar]
- [9].Más P., Devlin P. F., Panda S. & Kay S. A.. Functional interaction of phytochrome B and cryptochrome 2. Nature 408, 207–211 (2000). DOI: 10.1038/35041583 [DOI] [PubMed] [Google Scholar]
- [10].Mouritsen H., Janssen-Bienhold U., Liedvogel M., Feenders G., Stalleicken J., Dirks P., et al. Cryptochromes and neuronal-activity markers colocalize in the retina of migratory birds during magnetic orientation. Proc. Natl. Acad. Sci. USA 101, 14294–14299 (2004). DOI: 10.1073/pnas.0405968101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Li Q.-H. & Yang H.-Q.. Cryptochrome signaling in plants. Photochem. Photobiol. 83, 94–101 (2007). DOI: 10.1562/ 2006-02-28-IR-826 [DOI] [PubMed] [Google Scholar]
- [12].Ahmad M., Galland P., Ritz T., Wiltschko R. & Wiltschko W.. Magnetic intensity affects cryptochrome-dependent responses in arabidopsis thaliana. Planta 225, 615–624 (2007). DOI: 10.1007/s00425-006-0383-0 [DOI] [PubMed] [Google Scholar]
- [13].Ritz T., Wiltschko R., Hore P. J., Rodgers C. T., Stapput K., Thalau P., et al. Magnetic compass of birds is based on a molecule with optimal directional sensitivity. Biophys. J. 96, 3451–3457 (2009). DOI: 10.1016/j.bpj.2008.11.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Czarna A., Berndt A., Singh H. R., Grudziecki A., Ladurner A. G., Timinszky G., et al. Structures of drosophila cryptochrome and mouse cryptochrome1 provide insight into circadian function. Cell 153, 1394–1405 (2013). DOI: 10.1016/j.cell.2013.05.011 [DOI] [PubMed] [Google Scholar]
- [15].Song S.-H., Öztürk N., Denaro T. R., Arat N. Ö., Kao Y.-T., Zhu H., et al. Formation and function of flavin anion radical in cryptochrome 1 blue-light photoreceptor of monarch butterfly. J. Biol. Chem. 282, 17608–17612 (2007). DOI: 10.1074/jbc.M702874200 [DOI] [PubMed] [Google Scholar]
- [16].Kao Y.-T., Tan C., Song S.-H., Öztürk N., Li J., Wang L., et al. Ultrafast dynamics and anionic active states of the flavin cofactor in cryptochrome and photolyase. J. Am. Chem. Soc. 130, 7695–7701 (2008). DOI: 10.1021/ja801152h [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Brudler R., Hitomi K., Daiyasu H., Toh H., Kucho K., Ishimura M., et al. Identification of a new cryptochrome class: structure, function, and evolution. Mol. Cell 11, 59–67 (2003). DOI: 10.1016/s1097-2765(03)00008-x [DOI] [PubMed] [Google Scholar]
- [18].Daiyasu H., Ishikawa T., Kuma K., Iwai S., Todo T. & Toh H.. Identification of cryptochrome DASH from vertebrates. Genes Cells 9, 479–495 (2004). DOI: 10.1111/j.1356-9597.2004.00738 [DOI] [PubMed] [Google Scholar]
- [19].Lin C. & Todo T.. The cryptochrome. Genome Biol. 6, 220–228 (2005). DOI: 10.1186/gb-2005-6-5-220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Selby C. P. & Sancar A.. A cryptochrome/photolyase class of enzymes with single-stranded DNA-specific photolyase activity. Proc. Natl. Acad. Sci. USA 103, 17696–17700 (2006). DOI: 10.1073/pnas.0607993103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pokorny R., Klar T., Hennecke U., Carell T., Batschauer A. & Essen L.-O.. Recognition and repair of UV lesions in loop structures of duplex DNA by DASH-type cryptochrome. Proc. Natl. Acad. Sci. USA 105, 21023–21027 (2008). DOI: 10.1073/pnas.0805830106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Weber S. Light-driven enzymatic catalysis of DNA repair: a review of recent biophysical studies on photolyase. Biochim. Biophys. Acta 1707, 1–23 (2005). DOI: 10.1016/j.bbabio.2004.02.010 [DOI] [PubMed] [Google Scholar]
- [23].Kao Y.-T., Saxena C., Wang L., Sancar A. & Zhong D.. Direct observation of thymine dimer repair in DNA by photolyase. Proc. Natl. Acid. Sci. USA 102, 16128–16132 (2005). DOI: 10.1073/pnas.0506586102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Essen L.-O. & Klar T.. Light-driven DNA repair by photolyase. Cell. Mol. Life Sci. 63, 1266–1277 (2006). DOI: 10.1007/s00018-005-5447-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liu Z., Guo X., Tan C., Li J., Kao Y.-T., Wang L., et al. Electron tunneling pathways and role of adenine in repair of cyclobutane pyrimidine dimer by DNA photolyase. J. Am. Chem. Soc. 134, 8104–8114 (2012). DOI: 10.1021/ja2105009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kneuttinger A. C., Kashiwazaki G., Prill S., Hell K., Muller M. & Carell T.. Formation and direct repair of UV-induced dimeric DNA pyrimidine lesions. Photochem. Photobiol. 90, 1–14 (2014). DOI: 10.1111/php.12197 [DOI] [PubMed] [Google Scholar]
- [27].Stuchebrukhov A. A. Tunneling currents in electron transfer reactions in proteins. J. Chem. Phys. 104, 8424–8432 (1996). DOI: 10.1063/1.471592 [Google Scholar]
- [28].Stuchebrukhov A. A. Tunneling currents in electron transfer reaction in proteins. II. calculation of electronic superexchange matrix element and tunneling currents using nonorthogonal basis sets. J. Chem. Phys. 105, 10819–10829 (1996). DOI: 10.1063/1.472890 [Google Scholar]
- [29].Antony J., Medvedev M. & Stuchebrukhov A. A.. Theoretical study of electron transfer between the photolyase catalytic cofactor FADH– and DNA thymine dimer. J. Am. Chem. Soc. 122, 1057–1065 (2000). DOI: 10.1021/ja993784t [Google Scholar]
- [30].Medvedev M. & Stuchebrukhov A. A.. DNA repair mechanism by photolyase: electron transfer path from the photolyase catalytic cofactor FADH– to DNA thymine dimer. J. Theor. Biol. 210, 237–248 (2001). DOI: 10.1006/jtbi.2001.2291 [DOI] [PubMed] [Google Scholar]
- [31].Miyazawa Y., Nishioka H., Yura K. & Yamato T.. Discrimination of class I cyclobutane pyrimidine dimer photolyase from blue light photoreceptors by single methionine residue. Biophys. J. 94, 2194–2203 (2008). DOI: 10.1529/biophysj/107.119248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sato R., Kitoh-Nishioka H., Ando K. & Yamato T.. Electron transfer pathways of cyclobutane pyrimidine dimer photolyase revisited. J. Phys. Chem. B 122, 6912–6921 (2018). DOI: 10.1021/acs.jpcb.8b04333 [DOI] [PubMed] [Google Scholar]
- [33].Prytkova T. R., Beratan D. N. & Skourtis S. S.. Photoselected electron transfer pathways in DNA photolyase. Proc. Natl. Acid. Sci. USA 104, 802–807 (2007). DOI: 10.1073/pnas.0605319104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Harrison C. B., O’Neil L. L. & Wiest O.. Computational studies of DNA photolyase. J. Phys. Chem. A 109, 7001–7012 (2005). DOI: 10.1021/jp051075y [DOI] [PubMed] [Google Scholar]
- [35].Masson F., Laino T., Rothlisberger U. & Hutter J.. A QM/MM investigation of thymine dimer radical anion splitting catalyzed by DNA photolyase. Chemphyschem. 10, 400–410 (2009). DOI: 10.1002/cphc.200800624 [DOI] [PubMed] [Google Scholar]
- [36].Hassanali A. A., Zhong D. & Singer S. J.. An AIMD study of the CPD repair mechanism in water: reaction free energy surface and mechanistic implications. J. Phys. Chem. B 115, 3848–3859 (2011). DOI: 10.1021/jp107722z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wang H., Chen X. & Fang W.. Excited-state proton coupled electron transfer between photolyase and the damaged DNA through water wire: a photo-repair mechanism. Phys. Chem. Chem. Phys. 16, 25432–25441 (2014). DOI: 10.1039/c4cp04130e [DOI] [PubMed] [Google Scholar]
- [38].Lee W., Kodali G., Stanley R. & Matsika S.. Coexistence of different electron-transfer mechanism in the DNA repair process by photolyase. Chemistry 22, 11371–11381 (2016). DOI: 10.1002/chem.201600656 [DOI] [PubMed] [Google Scholar]
- [39].Rousseau B. J. G., Shafei S., Migliore A., Stanley R. J. & Beratan D. N.. Determinants of photolyase’s DNA repair mechanism in mesophiles and extermophiles. J. Am. Chem. Soc. 140, 2853–2861 (2018). DOI: 10.1021/jacs.7b11926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Essen L.-O. Photolyases and cryptochromes: common mechanisms of DNA repair and light-driven signaling? Curr. Opin. Struct. Biol. 16, 51–59 (2006). DOI: 10.1016/j.sbi.2006.01.004 [DOI] [PubMed] [Google Scholar]
- [41].Kao Y.-T., Saxena C., Wang L., Sancar A. & Zhong D.. Femtochemistry in enzyme catalysis: DNA photolyase. Cell Biochem. Biophys. 48, 32–44 (2007). DOI: 10.1007/s12013-007-0034-5 [DOI] [PubMed] [Google Scholar]
- [42].Tan C., Liu Z., Li J., Guo X., Wang L., Sancar A., et al. The molecular origin of high DNA-repair efficiency by photolyase. Nat. Commun. 6, 7302 (2015). DOI: 10.1038/ncomms8302 [DOI] [PubMed] [Google Scholar]
- [43].Liu Z., Wang L. & Zhong D.. Dynamics and mechanisms of DNA repair by photolyase. Phys. Chem. Chem. Phys. 17, 11933–11949 (2015). DOI: 10.1039/c4cp05286b [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zhang M., Wang L. & Zhong D.. Photolyase : dynamics and mechanisms of repair of sun-induced DNA damage. Photochem. Photobiol. 93, 78–92 (2017). DOI: 10.1111/php.12695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Zhang M. & Zhong D.. Photolyase: dynamics and electron-transfer mechanisms of DNA repair. Arch. Biochem. Biophys. 632, 158–174 (2017). DOI: 10.1016/j.abb.2017.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mees A., Klar T., Gnau P., Hennecke U., Eker A. P. M., Carell T., et al. Crystal structure of a photolyase bound to a CPD-like DNA lesion after in situ repair. Science 306, 1789–1793 (2004). DOI: 10.1126/science.1101598 [DOI] [PubMed] [Google Scholar]
- [47].Zhang Y. & Skolnick J.. TM-Align: a protein structure alignment algorithm based on the TM-score. Nucleic. Acids. Res. 33, 2302–2309 (2005). DOI: 10.1093/nar/gki524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. Gaussian16, revision B.01 (Gaussian Inc., Wallingford, CT, 2016). [Google Scholar]
- [49].Lee J.-H., Park C.-J., Shin J.-S., Ikegami T., Akutsu H. & Choi B.-S.. NMR structure of the DNA decamer duplex containing double T•G mismatches of cis-syn cyclobutane pyrimidine dimer: implications for DNA damage recognition by the XPC-hHR23B complex. Nucleic. Acids. Res. 32, 2474–2481 (2004). DOI: 10.1093/nar/gkh568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wang J. M., Wolf R. M., Caldwell J. W., Kollman P. A. & Case D. A.. Development and testing of a general AMBER force field. J. Comput. Chem. 25, 1157–1174 (2004). DOI: 10.1002/jcc.20035 [DOI] [PubMed] [Google Scholar]
- [51].Case, D. A., Betz, R. M., Botello-Smith, W., Cerutti, D. S., Cheatham, I., Darden, T. A., et al. AMBER 2016 (University of California, San Francisco, 2016). [Google Scholar]
- [52].Malter J. A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K. E. & Simmerling C.. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 (2015). DOI: 10.1021/acs.jctc.5b00255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Galindo-Murillo R., Robertson J. C., Zgarbova M., Sponer J., Otyepka M., Jurecka P., et al. Assessing the current state of amber force field modifications for DNA. J. Chem. Theory Comput. 12, 4114–4127 (2016). DOI: 10.1021/acs.jctc.6b00186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sato R., Kitoh-Nishioka H., Ando K. & Yamato T.. Computational study on the roles of amino acid residues in the active site formation mechanism of blue-light photoreceptors. Chem. Phys. Lett. 633, 247–251 (2015). DOI: 10.1016/j.cplett.2015.05.066 [Google Scholar]
- [55].Jorgensen W. L., Chandrasekhar J. & Nadura J. D.. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983). DOI: 10.1063/1.445869 [Google Scholar]
- [56].Sousa da Silva A. W. & Vranken W. F.. ACPYPE—antechamber python parser interface. BMC Res. Notes 5, 367 (2012). DOI: 10.1186/1756-0500-5-367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Hess B., Bekker H., Berendsen H. J. C. & Fraaije J. G. E. M.. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997). DOI: 10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H [Google Scholar]
- [58].Nose S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 52, 255–268 (1984). DOI: 10.1080/0026897840010201 [Google Scholar]
- [59].Hoover W. G. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A Gen. Phys. 31, 1695–1697 (1985). DOI: 10.1103/PhysRevA.31.1695 [DOI] [PubMed] [Google Scholar]
- [60].Evans D. J. & Holian B. L.. The Nose-Hoover thermostat. J. Chem. Phys. 83, 4069–4074 (1985). DOI: 10.1063/1.449071 [Google Scholar]
- [61].Berensen H. J. C., Postma J. P. M., van Gunsteren W. F., DiNola A. & Haak J. R.. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 (1984). DOI: 10.1063/1.448118 [Google Scholar]
- [62].Darden T., York D. & Pedersen L.. Particle mesh ewald: an N-log(N) method for ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993). DOI: 10.1063/1.464397 [Google Scholar]
- [63].Abraham, M. J., van der Spoel, D., Lindahl, E. & Hess, B. GROMACS user manual version 5.0.4. www.gromacs.org (2014). [Google Scholar]
- [64].Svensson M., Humbel S., Froese R. D. J., Matsubara T., Sieber S. & Morokuma K.. ONIOM: a multilayered integrated MO+MM method for geometry optimizations and single point energy predictions. A test for Diels-Alder reactions and Pt(P(t-Bu)3)2+H2 oxidative addition. J. Phys. Chem. 100, 19357–19363 (1996). DOI: 10.1021/jp962071j [Google Scholar]
- [65].Frish, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. Gaussian 09, Revision D.01 (Gaussian Inc., Wallingford, CT, 2009). [Google Scholar]
- [66].Bakowies D. & Thiel W.. Hybrid models for combined quantum mechanical and molecular mechanical approaches. J. Phys. Chem. 100, 10580–10594 (1996). DOI: 10.1021/jp9536514 [Google Scholar]
- [67].Marcus R. A. & Sutin N.. Electron Transfers in Chemistry and Biology. Biochim. Biophys. Acta Bioenerg. 811, 265–322 (1985). DOI: 10.1016/0304-4173(85)90014-X [Google Scholar]
- [68].Cave R. J. & Newton M. D.. Generalization of the Mulliken-Hush treatment for the calculation of electron transfer matrix elements. Chem. Phys. Lett. 249, 15–19 (1996). DOI: 10.1016/0009-2614(95)01310-5 [Google Scholar]
- [69].Cave R. J. & Newton M. D.. Calculation of electronic coupling matrix elements for ground and excited state electron transfer reactions: comparison of the generalized Mulliken-Hush and block diagonalization methods. J. Chem. Phys. 106, 9213–9226 (1997). DOI: 10.10163/1.474023 [Google Scholar]
- [70].Chen H.-C. & Hsu C.-P.. Ab initio characterization of electron transfer coupling in photoinduced systems: generalized Mulliken-Hush with configuration-interaction singles. J. Phys. Chem. A 109, 11989–11995 (2005). DOI: 10.1021/jp053712q [DOI] [PubMed] [Google Scholar]
- [71].Szabo, A. & Ostlund, N. S. Modern quantum chemistry: introduction to advanced electronic structure theory (Dover, New York, 2012). [Google Scholar]
- [72].Schmidt M. W., Baldrige K. K., Boatz J. A., Elbert S. T., Gordon M. S., Jensen J. H., et al. General atomic and molecular electronic structure system. J. Comput. Chem. 14, 1347–1363 (1993). DOI: 10.1002/jcc.540141112 [Google Scholar]
- [73].Enescu M., Lindqvist L. & Soep B.. Excited-state dynamics of fully reduced flavins and flavoenzymes studied at subpicosecond time resolution. Photochem. Photobiol. 68, 150–156 (1998). DOI: 10.1111/j.1751-1097.1998.tb02482.x [DOI] [PubMed] [Google Scholar]
- [74].Gauden, M., Yeremenko, S., Lann, W., van Stokkum, I. H. M., Ihalainen, J. A., van Grondelle R., et al. Photocycle of the flavin-binding photoreceptor AppA, a bacterial transcriptional antirepressor of photosynthesis genes. Biochemistry 44, 3653–3662 (2005) DOI: 10.1021/bi047359a [DOI] [PubMed] [Google Scholar]
- [75].Gray H. B. & Winkler J. R.. Electron transfer in proteins. Annu. Rev. Biochem. 65, 537–561 (1996). DOI: 10.1146/annurev.bi.65.070196.002541 [DOI] [PubMed] [Google Scholar]
- [76].Yamada D., Dokainish H. M., Iwata T., Yamamoto J., Ishikawa T., Todo T., et al. Functional conversion of CPD and (6-4) photolyases by mutation. Biochemistry 55, 4173–4183 (2016). DOI: 10.1021/acs.biochem.6b00361 [DOI] [PubMed] [Google Scholar]
- [77].Sato R., Harada R. & Shigeta Y.. The binding structure and affinity of photodamaged duplex DNA with members of the photolyase/cryptochrome family: a computational study. Biophys. Physcobiol. 15, 18–27 (2018). DOI: 10.2142/biophysico.15.0_18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Terai Y., Sato R., Yumiba T., Harada R., Shimizu K., Toga T., et al. Coulomb and CH-π interactions in (6-4) photolyase-DNA complex dominate DNA binding and repair abilities. Nucleic. Acids. Res. 46, 6761–6772 (2018). DOI: 10.1093/nar/gky364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Maestre-Reyna, M.,Yamamoto, J., Huang W.-C., Tsai M.-D., Essen L.-O. & Bessho Y.. Twist and turn: a revised structural view on the unpaired bubble of class II CPD photolyase in complex with damaged DNA. IUCrJ 5, 608–618 (2018). DOI: 10.1107/S205225251800996X [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.