

Abstract
One of the biochemical abnormalities found in diabetic tissues is a decrease in the cytosolic oxidized to reduced forms of the nicotinamide adenine dinucleotide ratio (NAD+/NADH also known as pseudohypoxia) caused by oxidation of excessive substrates (glucose through the polyol pathway, free fatty acids and lactate). Subsequently, a decline in NAD+ levels as a result of the activation of poly adenine nucleotide diphosphate‐ribose polymerase (mainly in type 1 diabetes) or the inhibition of adenine nucleotide monophosphate‐activated protein kinase (in type 2 diabetes). Thus, replenishment of NAD+ levels by nicotinamide‐related compounds could be beneficial. However, these compounds also increase nicotinamide catabolites that cause oxidative stress. This is particularly troublesome for patients with diabetes, because they have impaired nicotinamide salvage pathway reactions at the level of nicotinamide phosphoribosyl transferase and phosphoribosyl pyrophosphate, which occurs by the following mechanisms. First, phosphoribosyl pyrophosphate synthesis from pentose phosphate pathway is compromised by a decrease in plasma thiamine and transketolase activity. Second, nicotinamide phosphoribosyl transferase expression is decreased because of reduced adenosine monophosphate‐activated protein kinase activity, which occurs in type 2 diabetes. The adenosine monophosphate‐activated protein kinase inhibition is caused by an activation of protein kinase C and D1 as a result of enhanced diacylglycerol synthesis caused by pseudohypoxia and increased fatty acids levels. In this regard, nicotinamide‐related compounds should be given with caution to treat diabetes. To minimize the risk and maximize the benefit, nicotinamide‐related compounds should be taken with insulin sensitizers (for type 2 diabetes), polyphenols, benfotiamine, acetyl‐L‐carnitine and aldose reductase inhibitors. The efficacy of these regimens can be monitored by measuring serum NAD+ and urinary nicotinamide catabolites.
Keywords: Adenosine monophosphate‐activated protein kinase, Nicotinamide, Nicotinamide adenine dinucleotide
Diabetes causes nicotinamide adenine dinucleotide metabolism dysregulation characterized by increasing consumption of nicotinamide adenine dinucleotide to produce nicotinamide and decreasing conversion of nicotinamide to nicotinamide adenine dinucleotide (salvage cascade) by biochemical abnormalities, which have been postulated for the etiology of diabetic complications. This results in increasing production of nicotinamide catabolites, which might cause oxidative stress. For diabetes patients, nicotinamide‐related compounds, such as nicotinamide riboside and nicotinamide mononucleotide might be useful after correcting these abnormalities.
Introduction
Both nicotinic acid and nicotinamide are known as vitamin B3 (niacin). Its deficiency causes pellagra, with symptoms including inflamed skin, diarrhea and dementia. In developed countries, it is mainly used as a supplement for various conditions 1 , 2 , 3 although its efficacy in humans is still not clear. Both forms of niacin are utilized to synthesize nicotinamide adenine dinucleotide (NAD+) 1 , 4 , 5 . However, as nicotinic acid causes flush and other symptoms, nicotinamide is mainly used at a high dose 5 . NAD+ metabolism is currently one of the hottest research topics in the field of aging following the recent increase in resveratrol research. It has been shown that human skin NAD+ content sharply declines as people age 6 . Similar findings were observed in plasma NAD+ levels 7 . Whether this decline relates to the aging process, and whether replenishment of NAD+ slows aging are hot issues. Nevertheless, nicotinamide and related compounds have been and are currently being tested to treat conditions, such as cardiovascular disease 8 and diabetes, in humans.
Our research focuses on diabetic complications 9 , NAD+ redox abnormalities in diabetes (pseudohypoxia) 9 , 10 , 11 , 12 , 13 , sirtuin‐1 (SIRT1) 14 , 15 , 16 , resveratrol 15 , 17 , 18 and adenosine monophosphate (AMP)‐activated protein kinase (AMPK) 16 , 18 , 19 , 20 , 21 , all of which control and at the same time are controlled by nicotinamide and NAD+. In both types of diabetes (type 1 and 2), the metabolism of NAD+ appears to be dysregulated. This is evident in animal models of diabetes and is highly suggested in human diabetes. The etiology of metabolic dysregulation in NAD+ is likely to coincide with the etiology of diabetic vascular and neural complications, which has been elucidated in the past five decades of research. However, our research interest has been more than diabetic complications studied in various tissues. As explained in this article, the main causative abnormality – a decrease in cytosolic NAD+/NADH – has been observed not only in the kidney, retina and nerves, typical tissues in the field of diabetic complications, but also found in the liver, heart, skeletal muscle and even in pancreatic β‐cells of individuals with diabetes, prediabetes and obesity 9 . In these tissues, the effects of this abnormality are still not well explored. For example, this redox change is the main force that increases diacylglycerol (DAG) and triacylglyerol synthesis. Consistent with this idea, expression of aldose reductase in the mouse liver causes the same redox change in the absence of diabetes, which leads to fatty liver ,as shown recently 22 . Thus, the data and research we present in this article are intended for researchers studying diabetes in general. We review the relationship between NAD+, diabetes and its complications. After an introduction to nicotinamide, AMPK, SIRT1 and resveratrol, we discuss how NAD+ dysregulation might be corrected, and how nicotinamide related compounds, such as nicotinamide riboside (NR) and nicotinamide mononucleotide (NMN), might be used as a treatment for diabetes, but with several caveats.
Pseudohypoxia and NAD+ metabolism abnormalities in diabetic tissues
In this section, we start to define the concept of pseudohypoxia. This term was first used in a paper by Professor Williamson 10 , who was a mentor of one of the authors of the present article (YI). Together we wrote a number of papers explaining the concept 10 , 11 , 13 , 21 , 23 , 24 , 25 , 26 , 27 , 28 . Pseudohypoxia is a state of lower than normal cytosolic NAD+/NADH ratio (the normal free ratio is ~600). It is known from studies carried out in the 1970s that various tissues (first observed in the liver) from diabetic animals have abnormal values in total NAD, and its ratio in reduced (NADH) and oxidized (NAD+) forms 29 , 30 , 31 . For example, the lens from rats post 6 weeks of streptozotocin‐induced diabetes has a three‐ to fourfold lower NAD+/NADH ratio compared with non‐diabetic rats due to increased NADH and decreased NAD+. Total NAD was decreased by 15%. Such a high decline was also observed with NADPH 29 . By using metabolite indicator methods 30 in the retina and sciatic nerve to calculate the free ratios of NAD+ and NADH in the cytosolic and mitochondrial compartments (i.e., by measuring lactate, pyruvate for cytosolic, and 2‐oxoglutarate, ammonium and glutamate for mitochondria), we and others found that a low NAD+/NADH ratio is only observed in the cytosol and not in mitochondria 10 , 11 , 13 , 23 , 26 , suggesting that the diabetic effect is, at least in early stages, solely in the cytosol 10 . Similar changes can be observed by incubating rat retina with high glucose for 2 h 12 , 32 . In this case, change occurred only in the ratio, not in total NAD. Clear involvement of the polyol pathway (oxidation of sorbitol to fructose) to cause a low NAD+/NADH ratio was observed in early diabetic rats 9 , 13 , and by incubating human red blood cells with high glucose, in which aldose reductase inhibitors prevented NAD+/NADH ratio change (Figure 1). Thus, acute hyperglycemia, such as post‐prandial hyperglycemia, likely changes the cellular NAD+/NADH ratio without decreasing the total NAD amount. However, this acute change of NAD+/NADH is enough to increase triose phosphate levels by glyceraldehyde phosphate dehydrogenase (GAPDH), the synthesis of diacylglycerol and production of superoxide 9 , 33 , as follows. Increased NADH produces glycerol‐3‐phospate (a precursor for DAG), which fuels electrons at mitochondria complex II through the glycerol phosphate shuttle by electron transfer from mitochondrial glycerol‐3‐phosphate dehydrogenase 34 , 35 . This leads to reverse electron flow to complex I, leading to superoxide production 36 . In addition, cytosolic NADH might directly produce oxidative stress (Figure 2).
Figure 1.
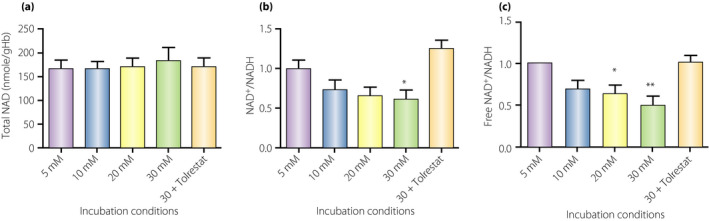
Total and free nicotinamide adenine dinucleotide (NAD+/NADH) ratio changes in human red blood cells incubated in the indicated conditions for 1 h. One hour incubation of red blood cells with high glucose decreased the ratios of oxidized to the reduced form of NAD+/NADH, which was completely prevented by 70 µmol/L aldose reductase inhibitor, tolrestat. See Data S1 for details of the experiment. * P < 0.05 vs 5 mM, ** P < 0.01 vs 5 mM
Figure 2.
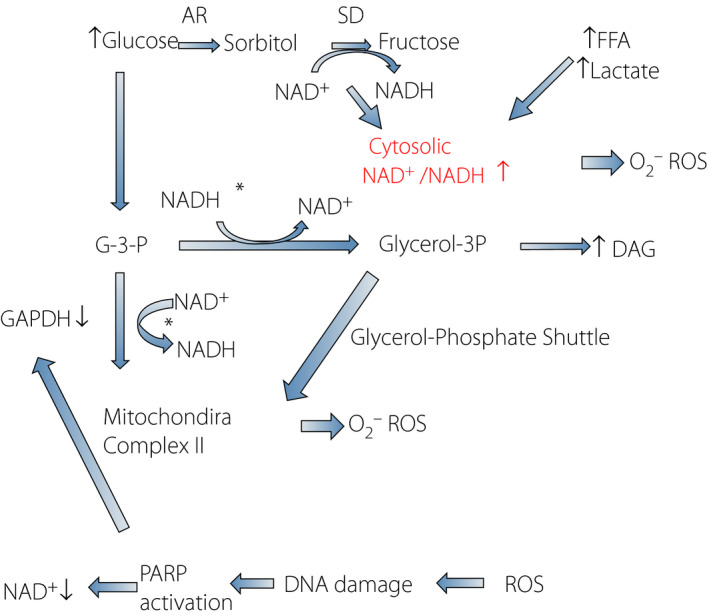
Biochemical cascades postulated to cause diabetic complications. Intracellular hyperglycemia increases in polyol pathway flux, free fatty acids (FFA) and lactate that cause a cytosolic nicotinamide adenine dinucleotide (NAD+/NADH) ratio increase (pseudohypoxia). This redox change induces: (i) production of reactive oxygen species (ROS) from cytosol and mitochondria; and (ii) increased flux to glycerol‐3‐phosphate and diacylglycerol (DAG). An excess of long‐chain acetyl coenzyme A also augments ROS and production of DAG. Increased NADH produces glycerol‐3‐phosphate, which fuels electrons at mitochondria complex II through the glycerol phosphate shuttle by electron transfer from mitochondrial glycerol‐3‐phosphate dehydrogenase. Chronically increased ROS production causes deoxyribonucleic acid damage and activates poly‐adenosine diphosphate ribosyl‐polymerase (PARP), which utilizes NAD+ as a substrate. PARP activation might cause post‐translational modification of glyceraldehyde phosphate dehydrogenase (GAPDH), resulting in inhibition and translocation. AR, aldose reductase; G‐3‐P, glyceraldehyde‐3‐phosphate; SD, sorbitol dehydrogenase.
A decline of NAD+ levels is clearly observed in chronic type 1 insulinopenic diabetic animal models, such as streptozotocin diabetic rats. A measurable decrease might take weeks to months to occur, depending on the tissues. The aorta from 1‐month‐old diabetic mice showed 60% decline in NAD+ levels 37 . This decrease is apparently caused by activation of poly adenosine diphosphate (ADP)‐D‐ribosyl polymerase (PARP), likely caused by oxidative stress‐mediated deoxyribonucleic acid (DNA) damage, as its inhibitor, BJ‐34, prevented it. Declining NAD+ levels is a signature of an imbalance in energy homeostasis, which is manifested as decreased adenosine triphosphate (ATP) levels 37 or a decreased ratio of ATP/(ADP or AMP). Declining ATP levels are reported in type 1 diabetic animal models 37 , as well as in the liver of human type 1 diabetic patients 38 .
Compared with type 1 diabetes, the declining NAD+ levels in type 2 diabetes (particularly in the hyperinsulinemic stage) and its animal models seem to be mild. A high‐fat diet in mice causes insulin resistance and impaired glucose tolerance similar to early type 2 diabetes. In this model, a decline of NAD+ was reported in the liver and white adipose tissue 39 . In addition, despite a modest increase of plasma glucose, activation of the polyol pathway and PARP are observed in the sciatic nerve 40 . Energy levels are also compromised, albeit slightly. Skeletal muscle ATP and high energy intermediate phosphocreatine levels in humans with type 2 diabetes were decreased by 12%, but ADP was not changed significantly. In the liver, ATP synthesis was decreased 41 . The activation of PARP in both types of diabetes, which appears to be a consequence of oxidative damage to DNA, accelerates the decreasing NAD+/NADH ratio further by inactivating GAPDH (Figure 2) 42 .
In addition to high glucose, non‐esterified fatty acids are increased in type 1 diabetes by insulinopenia, and type 2 diabetes by increased ectopic fat deposition. Fatty acid toxicity in the development of diabetes was firstly proposed by McGarry 43 , 44 . This theory was preceded by the seminal work of Randle 45 , who described fatty acid‐induced insulin resistance observed by tissue incubation. Recent findings of selective insulin resistance in the liver observed in type 2 diabetes concurs with these fatty acid toxicity theories 46 . In the development of diabetic complications and insulin resistance, free fatty acids (FFA) are utilized by the mitochondria, resulting in increased NADH that slows shuttling activities to mitochondria, which causes pseudohypoxia more easily. The second effect of increased FFA and FFA‐CoA in cytosol is to increase the synthesis of acyl‐glycerols including DAG (Figure 2).
A decreasing cytosolic NAD+/NADH ratio affects the cytosolic lactate/pyruvate ratio through lactate dehydrogenase, which induces higher lactate production. Thus, plasma lactate levels are increased in obesity 47 and diabetes 48 . This high level of plasma lactate, in turn, causes the NAD+/NADH ratio change in other cells through the lactate dehydrogenase reaction 9 . Thus, the uniqueness of lactate is that it can transmit the redox change from one organ to another. It was shown that a lactate infusion causes insulin resistance in rats 49 . Similarly, high plasma lactate was associated with a high risk of developing diabetes 50 .
Taken together, the redox change characterized by pseudohypoxia, which is caused by excessive substrate oxidation, leads to oxidative stress and DNA damage, occurs in various tissues, and is found in obesity, prediabetes and diabetes.
SIRT1 and AMPK
SIRT1 belongs to a family of sirtuins that use NAD+ as a substrate catalyzing the reaction of deacetylation or mono‐ADP‐ribosylation of proteins 19 . AMPK is known as a master regulator of energy metabolism, as it is activated by lowering the ratio of ATP to AMP or ADP 19 . Both SIRT1 and AMPK are known to activate genes involved in longevity, and work together as the master regulators of various metabolic pathways 14 , 18 , 19 , 51 , 52 . Well‐known targets of both are peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha (PGC1‐α), which activates mitochondrial biogenesis 53 , 54 , 55 , 56 , and forkhead box O3, which determines the fate of the cells (life or death signaling pathways) 14 , 57 , 58 . They also suppress pro‐inflammatory nuclear factor‐kappa B signaling 59 , 60 , 61 and upregulate autophagy by suppressing mammalian target of rapamycin signaling (Figure 3) 62 , 63 , 64 .
Figure 3.
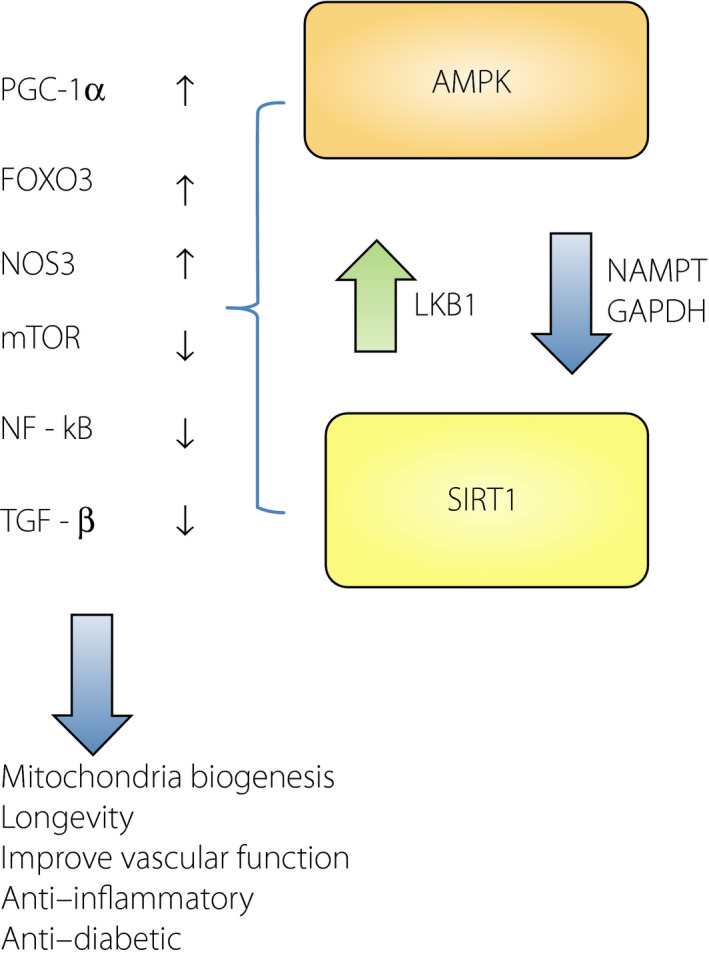
Adenosine monophosphate (AMP)‐activated protein kinase (AMPK) and sirtuin‐1 (SIRT1) interaction. AMPK and SIRT1 activate each other through liver kinas B1 (LKB1) and nicotinamide phosphoribosyltransferase (NAMPT), and regulate target molecules, such as peroxisome proliferator‐activated receptor gamma coactivator 1‐ alpha (PGC1‐α), forkhead box O3 (FOXO3), mammalian target of rapamycin (mTOR) and so on. Polyphenol, such as resveratrol, activates both AMPK and SIRT1 in multiple ways. NF‐κB, nuclear factor‐κB; TGF‐β, transforming growth factor‐β.
There is a positive reciprocal relationship between AMPK and SIRT1. Activation of AMPK has been shown to increase NAD+ levels, which results in SIRT1 activation 65 . This effect is mediated by upregulation of nicotinamide phosphoribosyltransferase (NAMPT) messenger ribonucleic acid and protein expression. Our results showed an absolute requirement of AMPK for the upregulation of NAD+. The enzyme is thought of as a rate‐limiting step to convert nicotinamide to NAD+ 66 . This increase in NAD+ level might cause SIRT1 (and other sirtuins) activation by two closely related mechanisms. One mechanism is to increase NAD+ levels and another is to decrease nicotinamide levels. As nicotinamide is a competitive inhibitor of sirtuin family enzymes 67 , 68 , the second effect affects activities of SIRT1 and other members of the family. For example, an increase in nuclear and cytoplasmic NAD+, and a decrease in nicotinamide might activate SIRT1 and 6.
We showed that SIRT1 deacetylates liver kinase B1 (LKB1), which is an upstream kinase of AMPK and causes its activation, thus controlling AMPK activity indirectly (Figure 3) 16 , 19 . LKB1 activity appears to play crucial roles in the context of diabetic complications. The Schwann cell‐specific LKB1 knockout mouse showed biochemical and morphological changes predominantly in peripheral nerves characterized by energy depletion, decreased NAD, NAD+/NADH ratio and axonal atrophy morphologically similar to diabetic neuropathy 69 .
Recently, there is an additional mechanism to activate SIRT1 by AMPK involving GAPDH phosphorylation 64 . In this mechanism, GAPDH Ser122 phosphorylation by AMPK causes GAPDH nuclear translocation where it binds SIRT1 directly and activates it. Thus, there is reciprocal or positive feedback relationship between SIRT1 and AMPK (Figure 3) 14 , 19 .
Relationship between cellular NAD+ and ATP
The main function of NAD+ is as the electron acceptor for catabolic pathways 70 . Under strong reduction of NAD+ (production of NADH), even glycolysis is inhibited at the step of GAPDH. NAD+ synthesis using nicotinamide requires at least three ATPs (Figure 4) 70 . In addition, the last step of NAD+ synthesis catalyzed by NMN adenylyltransferase (NMNAT) is completely reversible 71 , meaning that there might be equilibration between cytosolic NAD+ and ATP levels (Figure 4). Such a relationship was reported in hepatocytes. A titration experiment that changed cellular ATP levels clearly showed that cytosolic NAD+ positively correlates with ATP levels 72 . This indicates that if the NAD+ level becomes low enough, it affects ATP levels.
Figure 4.
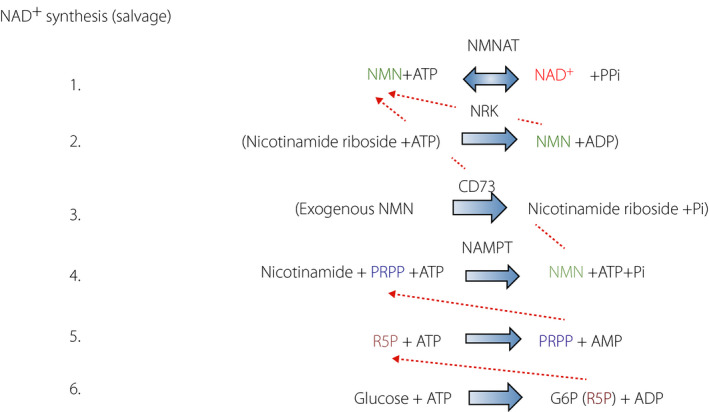
Nicotinamide adenine dinucleotide (NAD+) synthesis cascade from nicotinamide (salvage pathway). NAD+ catabolite, nicotinamide, can be salvaged to re‐synthesize NAD+ by the reaction of nicotinamide mononucleotide adenylyltransferase (NMNAT) from nicotinamide mononucleotide (NMN) (1). NMN is produced by nicotinamide phosphoribosyltransferase (NAMPT) (4) from phosphoribosyl diphosphate (PRPP), which originates from pentose phosphate pathway product, ribose‐5‐phosphate (5, 6). In addition to this endogenous cascade, nicotinamide riboside (NR), a milk ingredient, can produce NMN (2). Recently, NMN has been shown to boost NAD+. The majority of exogenously administered NMN is believed to be decomposed to nicotinamide riboside (3). One of the peculiarities of the salvage pathway is its dependency of adenosine triphosphates (ATPs). In addition, because of the reversibility of NMNAT reaction (1) there is a positive correlation between cytosolic NAD+ and ATP. NRK, nicotinamide riboside kinase (ribosylnicotineamide kinase); RSP, ribose‐5‐phosphate.
This could be the case with excess DNA damage. In cells undergoing DNA damage, PARP is activated, which produces high levels of nicotinamide at the expense of NAD+. Although PARP enzyme activity is necessary to maintain genetic stability in the long term 73 , there are numerous reports showing that the inhibition of PARP is beneficial at least temporarily 73 , 74 . This is likely due to maintaining NAD+ and ATP levels. Streptozotocin, used to render rats diabetic, causes β‐cell DNA damage. This activates PARP, resulting in lowering NAD and ATP, and apoptosis. A high‐dose injection of nicotinamide (250 mg/kg) has been used to prevent streptozotocin‐treated rats from developing full diabetes. Similarly, nicotinamide prevents autoimmune diabetes in animal models, possibly through inhibition of PARP and prevention of β‐cell NAD+ depletion 75 . This was a main rationale for the European Nicotinamide Diabetes Trial (ENDIT), a clinical trial to prevent type 1 diabetes by nicotinamide. Nicotinamide can inhibit PARP activity by 90% at 1 mmol/L. Although nicotinamide levels were not measured in pancreatic β‐cells, such levels of nicotinamide have been shown to prevent PARP activation and also promote NAD+ synthesis 76 .
AMPK activity in type 1 and 2 diabetes
Energy homeostasis is believed to be regulated by AMP‐activated protein kinase, which is activated by lower ATP/(ADP or AMP) ratios 19 , 77 . AMPK activation upregulates catabolic processes and downregulates anabolic processes to restore ATP levels 19 , 77 . Because of the energy imbalance reported in type 1 diabetes, AMPK could be increased. We observed this in the retina from streptozotocin‐induced diabetic rats (Figure 5).
Figure 5.
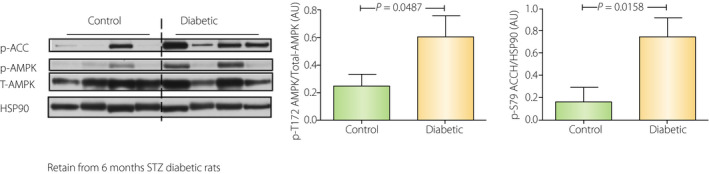
Activation of adenosine monophosphate‐activated protein kinase (AMPK) in the retina from 6 months streptozotocin (STZ) diabetic rats. Male Wistar rats with bodyweight of 150 g were obtained from Charles River (Boston, MA, USA). Diabetes was induced by a single injection of 50 mg/kg STZ. At 6 months’ duration of diabetes, rats were killed with a lethal dose of pentobarbital and their eyes were quickly removed. The retina was sampled and frozen in liquid nitrogen. The whole retina was ground with Western blot sample buffer, then subjected to sodium dodecyl sulphate‐polyacrylamide gel electrophoresis and Western blotting. Type 1 insulinopenic diabetes showed an increased phospho‐Thr172 AMPK and phospho‐Ser79 ACC (AMPK substrate) in the retina, which reflects decreasing energy levels.
In contrast to type 1 diabetes, AMPK activity is thought to be downregulated, not upregulated in the tissues of type 2 diabetes. The clinically approved drug for type 2 diabetes, metformin, is known to activate AMPK 78 , and its action is believed to promote beneficial effects in preventing fatty liver and cardiovascular events. Thus, although type 2 diabetes might have an energy production problem likely by mitochondrial dysfunction 79 , 80 , 81 , the energy levels are not low enough to activate AMPK. Rather, AMPK is downregulated. We observed this phenomenon in the retinas from db/db mice (Figure 6). High‐fat diet and type 2 diabetes are associated with insulin resistance and lipid metabolism abnormalities resulting in high DAG levels 10 , 82 . High DAG activates protein kinase C and D1, which phosphorylates AMPK alpha1 S487 83 and alpha2 S491 84 , respectively. The phosphorylation of these sites inhibits AMPK activity.
Figure 6.
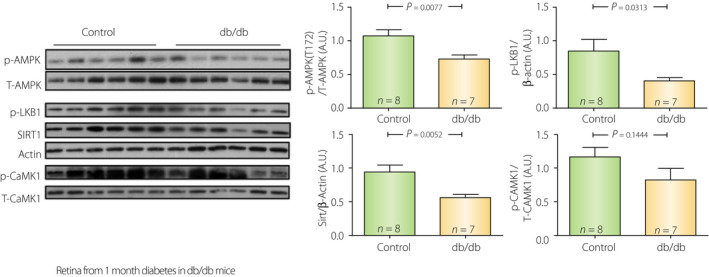
Suppression of sirtuin‐1 (SIRT1)–liver kinase B1 (LKB1)–adenosine monophosphate‐activated protein kinase (AMPK) cascade in db/db type 2 diabetic mice. In contrast to streptozotocin rats (Figure 5), retinas from 3 month‐old db/db mice with 1 month of confirmed diabetes (vs non‐diabetic db/w+ littermates) showed decreased pAMPK, pLKB1 and Sirt1 protein levels.
Nicotinamide is a substrate for NAD+ synthesis, and a product and inhibitor of sirtuins
The metabolic cascade of NAD+ synthesis from nicotinamide is shown in Figures 4 and 7. Nicotinamide is incorporated into the synthetic salvage cascade with phosphoribosyl diphosphate (PRPP), which is a product of the pentose phosphate pathway 66 , 70 . Production of PRPP requires ATPs 70 . The nicotinamide incorporation reaction by NAMPT itself does not require ATP, but this reaction is strongly activated when coupled with ATP hydrolysis 85 . The NMNAT reaction that converts NMN to NAD+ also uses ATP.
Figure 7.
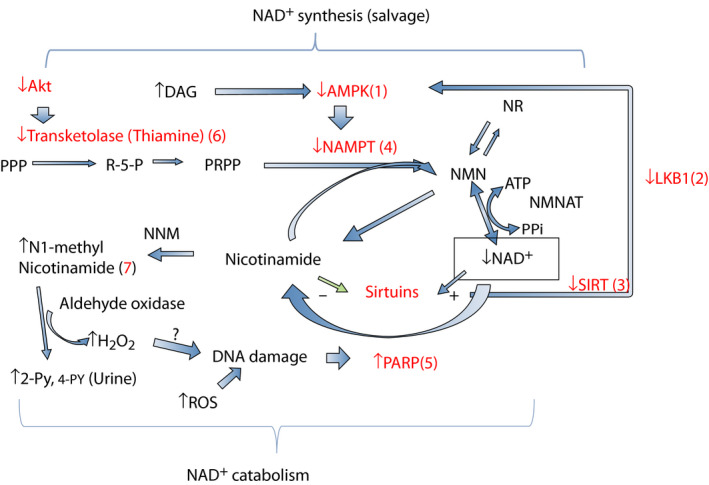
Effects of diabetes on nicotinamide adenine dinucleotide (NAD+) metabolisms. In type 2 diabetes, reduced sirtuin‐1 (SIRT1)–liver kinase B1 (LKB1)–adenosine monophosphate‐activated protein kinase (AMPK) cascade (1–3) decreases nicotinamide phosphoribosyltransferase (NAMPT) expression (4), which decreases NAD+ synthesis. Increased reactive oxygen species (ROS) activates poly‐adenosine diphosphate ribosyl‐polymerase (PARP) (5) and increases NAD+ consumption. This reaction occurs strongly in insulinopenic type 1 diabetes. In both diabetes, decreased plasma thiamine and reduced transketolase activity might cause impaired phosphoribosyl diphosphate (PRPP) production (6). Excessive nicotinamide increases nicotinamide catabolites N1‐nicotinamide, N‐methyl‐2‐pyridone‐5‐carboxamide (2‐PY), 4‐PY production (7) and produces more H2O2 by aldehyde oxidase.
Endogenous nicotinamide is mainly produced by sirtuin family enzymes and PARP (poly ADP‐D‐ribosyl polymerase or NAD+ ADP‐ribosyltransferase) 1 , 4 . Both enzymes mediate gene repair, DNA stability 73 and increase under stress conditions, such as fasting and aging. They use NAD+ as the substrate and produce nicotinamide as a product. The Km for NAD+ of sirtuin family members is 100–200 µmol/L, whereas that for PARP1 is 20–60 µmol/L 4 . The estimated nuclear (cytosol) NAD+ levels are approximately 70 µmol/L 4 . Therefore, increasing NAD+ levels would increase sirtuin family enzyme activity, but not PARP1. Both enzymes can be inhibited by nicotinamide. The IC50 for sirtuins is reported to be 30–70 µmol/L 86 , whereas PARP1 can be inhibited 90% by 1 mmol/L nicotinamide 87 . In the case of DNA damage, PARP is activated. Because of the higher IC50 for nicotinamide inhibition and low Km for NAD+, the PARP enzyme can consume cellular NAD+ to the levels low enough to cause ATP depletion and cell death.
Nicotinamide catabolites and the effects of diabetes
Unused nicotinamide for the salvage reaction is mainly methylated and oxidized, and excreted in urine (Figure 7). The major nicotinamide catabolites found in human urine are N‐methyl‐2 or 4‐pyridone‐5‐carboxamide (2 or 4‐YP) and N‐methylnicotinamide 88 . In both rodent models of type 2 diabetes (db/db mouse; obese Zucker rat) and human type 2 diabetes, urinary section of N‐methylnicotinamide and 2‐YP are increased 89 . This suggests downregulation of nicotinamide salvage activity. The conversion of N‐methylnicotinamide to 2‐ or 4‐PY is mediated by aldehyde oxidase 88 , which produces H2O2 as a by‐product. In humans, this enzyme is widely expressed in many tissues and cells 90 . In Caenorhabditis elegans, this enzyme is responsible for a transient increase of H2O2 production, and is required for the lifespan extension effects of Sir2.1 91 . Such hormesis action (short and mild stress, which is beneficial for health) could occur in higher organisms too. In humans, physical exercise promotes health benefits because of increasing reactive oxygen species (hormesis). Skeletal muscle produces and secretes interleukin‐6, which is known to activate AMPK 92 , 93 . Consistent with this scenario, anti‐oxidant treatment was shown to inhibit exercise’s beneficial effects 94 . However, sustained oxidative stress could be harmful. Thus, production of high nicotinamide and H2O2 could damage the cells (Figure 7). Indeed, some suggest that nicotinamide overload causes oxidative stress and insulin resistance 95 , 96 . There is a distinct possibility that oxidative stress produced by this reaction contributes to DNA damage and PARP activation, which could form a vicious cycle.
The enzyme that converts nicotinamide to N‐methylnicotinamide, nicotinamide n‐methyltransferase (NNM) utilizes the universal methyl donor S‐adenosyl‐L‐methionine as a co‐substrate, and because of this, it might control local S‐adenosyl‐L‐methionine concentration and methylation reactions, including phosphatase and epigenetics 97 .
Control of NAD+ synthesis by substrate PRPP and diabetes
Nicotinamide is not the only molecule determining NAD+ synthesis by NAMPT. The reaction also requires PRPP, which is another substrate, and is considered as rate limiting. PRPP comes from ribose‐5‐phosphate, which is the product of the pentose phosphate pathway (Figures 4,7) 70 . The production of ribose‐5‐phosphate, PRPP and NAD+ synthesis was shown to be regulated by the transketolase reaction. Phosphorylation of trasketolase at T382 by protein kinase B greatly activates the enzyme activity and NAD+ production 98 . Thus, insufficient protein kinase B activity in diabetes likely contributes to the reduction of transketolase activity. Transketolase activity requires thiamine as the co‐factor 70 . In both type 1 and 2 diabetes, plasma thiamine concentration is reported to be decreased by 75% as a result of high renal clearance 99 , 100 .
Nicotinamide, NAD+ synthesis and SIRT1‐AMPK cascade
AMPK activators have been shown to increase NAD+ levels, which results in SIRT1 activation (Figures 3,7) 65 . There are a few points we would like to highlight here based on our data. We found that AMPK activation in HepG2 cells incubated with 1 mmol/L metformin or 15 µmol/L resveratrol for 16 h increased NAD+ levels by approximately 30% (Figure 8a). Additionally, the messenger ribonucleic acid expression of the rate‐limiting enzyme, NAMPT, was increased in 2–5 h by incubation with 1 mmol/L 5‐aminoimidazole‐4‐carboxamide ribonucleotide, an AMPK activator, although the effect was transient (Figure 8b). Increased NAD+ levels with 1 mmol/L metformin were also observed in wild‐type mouse embryonic fibroblasts, whereas no increase was observed in AMPK α1&2 double knockout mouse embryonic fibroblasts (Figure 9a). These results clearly show that AMPK activators increase NAD+ in an AMPK‐dependent fashion. We also found that addition of 10 mmol/L nicotinamide in the culture media (5 mmol/L glucose minimal essential medium) was more effective at increasing NAD+ (Figure 8a). We assessed the Km values of NAMPT for nicotinamide using wild‐type mouse embryonic fibroblasts and human embryonic kidney 293 (HEK293) cells that were transfected with a human NAMPT1 plasmid. We found a 10‐fold higher Km for nicotinamide in human NAMPT (10 µmol/L) than in mouse NAMPT (0.9 µmol/L; Figure 9b). These values are in good agreement with some of the values previously reported 101 , 102 . This means that human cells do not efficiently utilize nicotinamide for synthesis of NAD+ if the concentration of nicotinamide is low.
Figure 8.
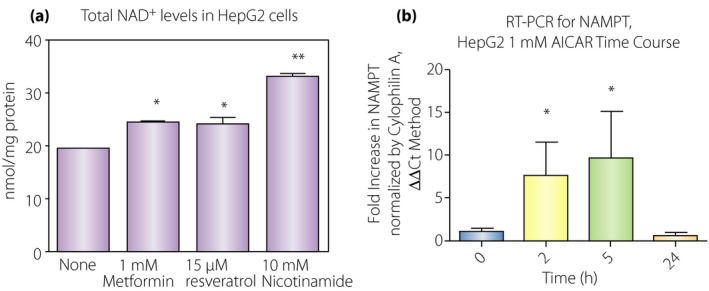
Adenosine monophosphate‐activated protein kinase (AMPK) activation, nicotinamide adenine dinucleotide (NAD+) levels and nicotinamide phosphoribosyltransferase (NAMPT) expression. (a) HepG2 cells cultured in minimal essential medium were incubated with the indicated concentration of reagents overnight. Total NAD+ was then assessed in these cells. AMPK activators, metformin and resveratrol increased NAD levels (n = 3, *P < 0.05 vs none). 10 mmol/L nicotinamide without AMPK activators could also increase NAD+ levels (n = 3, **P < 0.01 vs none). (b) NAMPT expression was stimulated by1 mmol/L 5‐aminoimidazole‐4‐carboxamide ribonucleotide (an AMPK activator). NAMPT messenger ribonucleic acid expression was increased seven‐ to tenfold by activation of AMPK (n = 3, *P < 0.05) in 2–5 h, but returned to baseline at 24 h.
Figure 9.
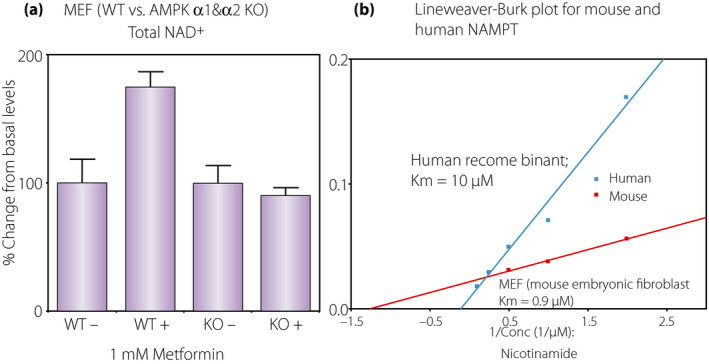
(a) Requirement of adenosine monophosphate‐activated protein kinase (AMPK) for increasing nicotinamide adenine dinucleotide (NAD+) levels by metformin. Mouse embryonic fibroblasts (MEF) were incubated with(+)/without(−) 1 mmol/L metformin overnight and NAD+ was assessed. Metformin was effective to increase NAD+ in wild‐type MEFs, but ineffective in AMPK α1&2 double knockout MEFs. (b) Km for nicotinamide of mouse versus human nicotinamide phosphoribosyltransferase (NAMPT). Mouse NAMPT Km was determined from cell protein homogenates of wild‐type MEFs. Human NAMPT1 was overexpressed in HEK293 cells and protein homogenates were used to determine the Km for nicotinamide. An average of two measurements were used to draw the plots. Estimated Km for nicotinamide was 0.9 µmol/L for mouse NAMPT and 10 µmol/L for human NAMPT.
Therefore, would incubation with high nicotinamide benefit human cells exposed to high glucose? We assessed this effect on cellular senescence in cultured human retinal pericytes. When human retinal pericytes were incubated with 25 mmol/L glucose for 2 weeks, a percentage of senescent cells (senescence‐associated galactosidase‐positive cells) was increased by fourfold (Figure 10). This increase was further augmented by an addition of 1 mmol/L nicotinamide. In contrast, the addition of 20 µmol/L resveratrol in 25 mmol/L glucose medium completely prevented the increase. The results are consistent with that NAD+ dysregulation might occur under high glucose, and under this condition, nicotinamide worsens the in vitro aging.
Figure 10.
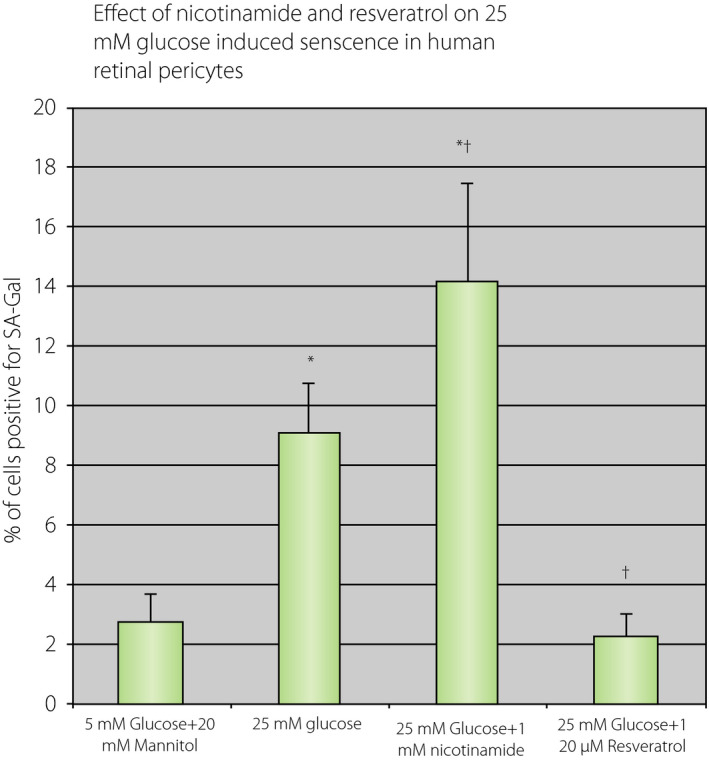
Effects of 25 mmol/L glucose and addition of 1 mmol/L nicotinamide or 25 µmol/L resveratrol on premature senescence in culture human retinal pericytes. Human retinal pericytes were cultured in 5% fetal bovine serum with EGM2 media for 10 days with the addition of indicated concentration of reagents. The cells were fixed and senescence associated galactosidase staining was carried out. Then, 25 mmol/L glucose increased a number of senescence cells by threefold (*P < 0.05 vs 5 mmol/L glucose, n = 4). The addition of 1 mmol/L nicotinamide in the medium further increased (*P < 0.05 vs 5 mmol/L glucose and † P < 0.05 vs 25 mmol/L glucose, n = 4); 20 µmol/L resveratrol treatment eliminated 25 mmol/L glucose effects († P < 0.05 vs 25 mmol/L glucose).
Summary of the plausible mechanisms of dysregulation of NAD+ metabolism in diabetes
As mentioned in the beginning, dysregulation of NAD+ metabolism occurs in various tissues in diabetes, which might be the etiology of diabetic complications and type 2 diabetes. Increased NAD+ catabolism for DNA repair appears to be a primary cause for the decrease in NAD levels, especially in insulinopenic type 1 diabetes models, such as the streptozotocin‐induced diabetic rat (Figure 11). Continuous low levels of tissue NAD+ and ATP in this model attest that NAMPT‐mediated compensatory mechanism somehow does not work properly, albeit even with activation of AMPK. The reason is still not clear. ATP levels might be too low to spare for NAD+ synthesis. Imai et al. 103 and others found that the conversion ratio of tryptophan to nicotinamide was lower in streptozotocin‐ and alloxan‐induced diabetes, suggesting that de novo synthesis of NAD+ from tryptophan appears to be impaired. This phenomenon was not observed in a type 2 diabetes model; Goto‐Kakizaki rats 104 .
Figure 11.
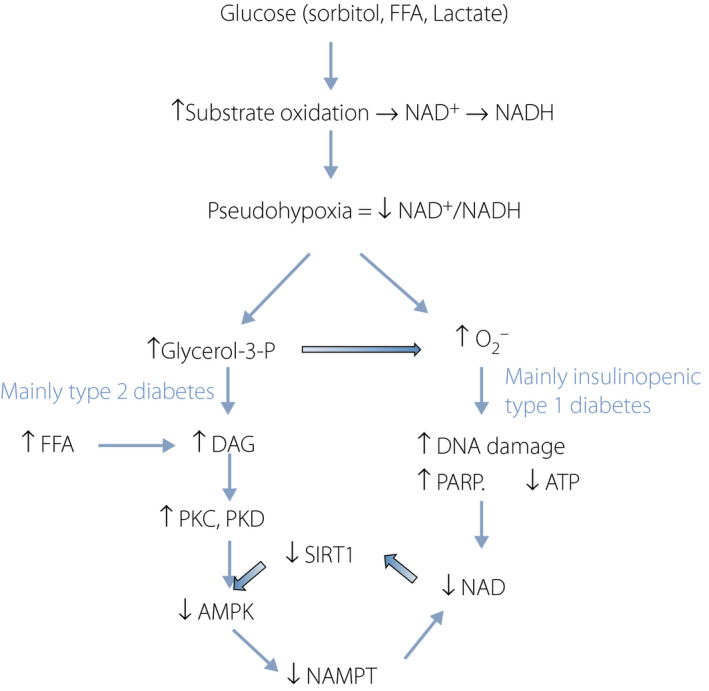
Summary of the cascade leading to lower nicotinamide adenine dinucleotide (NAD+) levels in diabetes. High glucose increases sorbitol levels by aldose reductase. Oxidation of sorbitol, free fatty acid (FFA) and lactate cause production of excess NADH. This leads to a low NAD+/NADH ratio (pseudohypoxia), which mainly causes activation of two pathways: diacylglycerol (DAG)–protein kinase C (PKC) and (DAG)–protein kinase D (PKD) cascade, which occur preferentially in type 2 diabetes and high levels of oxidative stress that occur in insulinopenic type 1 diabetes. Both cascades decrease NAD+ levels. ATP, adenosine triphosphate; DNA, deoxyribonucleic acid; PARP, poly adenosine diphosphate‐D‐ribosyl polymerase; SIRT1, sirtuin 1.
In type 2 diabetes animal models, in addition to an increase in NAD consumption by PARP, inactivation of AMPK is another factor (Figure 11). Impaired AMPK activity was reported in type 2 diabetes in skeletal muscle 105 , and fat tissue in obese individuals 106 . As aforementioned, activation of protein kinase C and protein kinase D by increased DAG is likely a part of the mechanism.
NAD+ targeting treatments for patients with diabetes
Ideally, what we need are methods to increase NAD+ levels without increasing nicotinamide or its catabolites (Figure 12). Increasing expression of intracellular NAMPT is the primary goal to do so. Thus, AMPK activation is the key target for type 2 diabetes. It was shown in humans that aerobic, as well as resistance, exercise, which both activate skeletal muscle AMPK, increased the expression of NAMPT in skeletal muscle by 25–30% 107 . In contrast, it failed to do so in adipose tissue, suggesting that the modality of AMPK activation must be specific for the target tissue in which AMPK needs to be activated. Thus, starvation should activate AMPK and increase NAMPT activity in the liver. In diabetes patients, in addition to exercise and caloric restriction, the effect can be mimicked by taking metformin. Metformin activates AMPK in many tissues 78 . This activation was attributed to decreased fat accumulation and glucose production in the liver of obese and diabetic mice, although the latter appears to be AMPK independent, as shown in AMPK‐null mice 108 . In addition to metformin, we previously showed that glucagon‐like peptide‐1 can activate AMPK in endothelial cells 109 . Sodium–glucose cotransporter 2 inhibitor, canagliflozin, is also found to activate AMPK through inhibiting mitochondria 110 . As insulin potentially suppresses SIRT1‐AMPK activity, increasing insulin levels in type 2 diabetes might work negatively.
Figure 12.
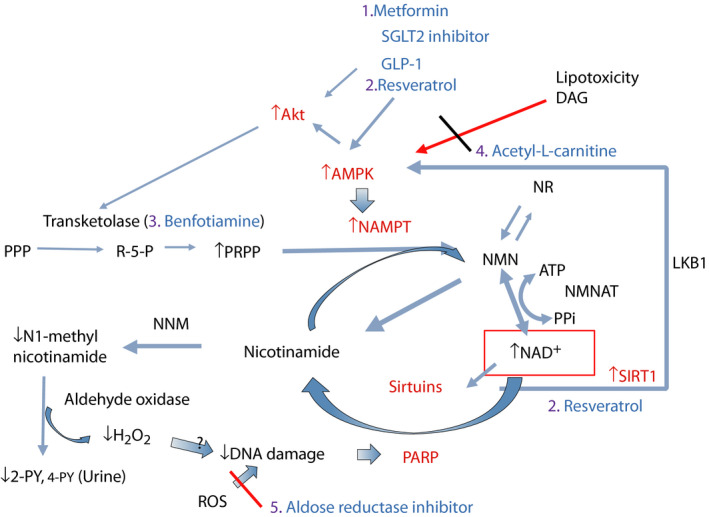
Proposed treatments to correct nicotinamide adenine dinucleotide (NAD+) levels in diabetes. Figure 7 showed abnormalities of NAD+ metabolism occurring in diabetes. To correct these abnormalities, proposed regimens are: (1) metformin, sodium–glucose cotransporter 2 (SGLT2) inhibitor, glucagon‐like peptide‐1 (GLP‐1); (ii) resveratrol, other polyphenols or sirtuin‐activating compoundssirtuin‐activating compounds (STACs) to activate sirtuin 1 (SIRT1)–adenosine monophosphate‐activated protein kinase (AMPK) for type 2 diabetes; (iii) benfotiamine to boost substrate (phosphoribosyl diphosphate [PRPP]) production; (iv) acetyl‐L‐carnitine to block diacylglycerol (DAG) production and lipotoxicity; and (v) aldose reductase inhibitor if glucose control is inadequate. 2‐PY, N‐methyl‐2‐pyridone‐5‐carboxamide; Akt, protein kinase B; AMPK, AMP‐activated protein kianse; ATP, adenosine triphosphate; DNA, deoxyribonucleic acid; LKB1, liver kinase B1; NMN, nicotinamide mononucleotide; NMNAT, nicotinamide mononucleotide adenylyltransferase; NNM, nicotinamide n‐methyltransferase; PPi, pyrophosphate; PPP, pentose phosphate pathway; R‐5‐P, ribose‐5‐P; ROS, reactive oxygen species.
Resveratrol activates both SIRT1 and AMPK 14 , 18 . SIRT1 activation occurs by: (i) direct binding at the N‐terminus, which activates it in a substrate‐specific manner 111 ; (ii) resveratrol's phosphodiesterase inhibitor activity 112 , 113 , which leads to increasing cyclic AMP‐dependent protein kinase activity, then phosphorylating SIRT1 at Ser434 114 ; and (iii) binding lamin A, which stabilizes SIRT1 nuclear localization 115 . AMPK activation is induced by: (i) activation of the SIRT1–LKB1–AMPK cascade 15 ; (ii) ATP synthesis inhibitor activity 116 ; and (iii) phosphodiesterase inhibitor action, which increases cyclic AMP, leading to increased intracellular Ca2+ levels through exchange protein directly activated by cyclic AMP‐Ca2+/calmodulin‐dependent protein kinase kinase‐β, which activates AMPK 112 . In humans, bioavailability of resveratrol is a major problem. Previously, now a defunct company, Sirtris developed SRT501 a proprietary formulation of resveratrol to increase bioavailability. In a phase Ib study, patients with type 2 diabetes treated for 28 days with SRT501 showed significantly lower fasting glucose levels (https://www.gsk‐studyregister.com/study/SRT‐501‐006?legacy=true). Similar studies have been carried out and recently systematically reviewed 117 , suggesting that resveratrol consistently lowered fasting blood glucose, insulin levels, homeostatic model assessment index and blood pressure. However, the effects on glycated hemoglobin were negligible. As compared to SRT501, the bioavailability of other currently available resveratrols is currently unknown. Quercetin might have similar properties 18 , but the efficacy has not been well studied.
For fatty acid toxicity, the acyl group in acetyl coenzyme A can be sequestrated by free carnitine. It was shown that both carnitine or acetyl‐L‐carnitine are effective in improving insulin‐mediated glucose disposal in humans 118 . Acetyl‐L‐carnitine and propyonyl‐L‐carnitine were tested on various complications in diabetic animal models, showing their efficacy. We previously reported that the levels of DAG can be decreased by acetyl‐L‐carnitine 23 , 24 , which also normalized suppression of energy utilization by Na+‐K+‐ATPase and redox change. Currently, acetyl‐L‐carnitine is prescribed for diabetic neuropathy in some countries. There is borderline efficacy for this condition 119 , 120 , 121 .
In case glycemic levels are not well controlled, polyol pathway activation is the main source of NAD+/NADH change. Activation of polyol pathway was postulated to be the main cause for PARP activation in diabetic rats 122 , 123 . This observation was not examined in human diabetes. However, aldose reductase inhibitor could be prescribed for the treatment.
It is not clear whether a flux from ribose‐5‐P of pentose phosphate pathway to PRPP is decreased in human diabetes. Based on decreased plasma thiamine levels observed, this might be occurring. At least supplementation with thiamine, or the more active compound, benfotimine, is not harmful. Benfotiamine was shown to prevent diabetic retinal and kidney complications in diabetic animals 124 , 125 . In human diabetes, benfotiamine was tested on diabetic neuropathy and found to have borderline efficacy 126 , 127 .
Nicotinamide and its related compounds for human diabetes
Enzymatic activities involving nicotinamide metabolism varies depending on species and even on strains in rodents. Thus, the effects of nicotinamide and its related compounds cannot be extrapolated from rodents to humans. Despite of effectiveness in rodent models, nicotinamide did not produce a positive result in human diabetes. The European Nicotinamide Diabetes Intervention Trial failed to show any preventative effect of nicotinamide for the onset of type 1 diabetes 75 . Rather, there is a report suggesting that nicotinamide administration causes insulin resistance in human 128 . Zhou et al. 95 reported that N‐methylnicotinamide by itself causes insulin resistance, and suggested that nicotinamide overload might cause type 2 diabetes. In agreement with this observation, human adipose tissue NNMT, the enzyme that produces N‐methylnicotinamide, activity correlates positively with adiposity and insulin resistance 129 , and serum N‐methylnicotinamide levels are associated with obesity and diabetes 130 .
Nicotinamide riboside, which is contained in cow milk 131 , has recently been tested in humans 132 , 133 , and showed increased blood NAD+ levels. It produces NMN by the action of nicotinamide ribose kinase 131 (Figure 4) or nicotinamide by purine nucleotide phosphorylase. If NR goes through the nicotinamide ribose kinase reaction, then PRPP is not required nor is it controlled by the NAMPT reaction.
In rodents, NR increases skeletal muscle NAD+ levels. However, this is not the case for humans. This might be related to the different Km for nicotinamide, as aforementioned. NR 1 g per day given to older people for 21 days 70 , 71 , 72 , 73 , 74 , 75 , 76 , 77 , 78 , 79 , 80 increased NAD+ levels in the blood, but not in skeletal muscle 134 . Blood and skeletal muscle nicotinamide levels were not increased, whereas methylnicotinamide and N‐methyl‐2‐pyridone‐5‐carboxamide (2‐PY) levels were increased in muscle, blood and urine. This study suggests that NNM activity might be high enough not to accumulate nicotinamide. Another study in which NR 2 g/day was given to obese and insulin‐resistant men for 12 weeks also showed no increase in NAD+ levels in skeletal muscle, nor in mitochondrial content and function 135 . Instead, it reduces NAMPT protein expression by 15%. This study also found skeletal muscle NAMPT protein levels were negatively correlated with plasma C‐peptide during oral glucose tolerance test and fasting insulin levels. Collectively, the study did not prove the efficacy of NR, rather it appears to suppress SIRT1‐AMPK. Neither study reported insulin levels nor glucose tolerance test.
More recently, NMN was tested in rodents 39 , 136 . Compared with NR, the metabolism of orally administered NMN is not fully understood. It is believed that unlike NR, NMN does not penetrate cell membranes because of phosphate group 1 , 4 . In vivo, CD73 (NADase) removes the phosphate group producing NR, which can be incorporated in cells (Figure 4) 1 , 4 , 5 . The compound has not been extensively tested in humans, but similar results of NR might be expected.
Collectively, in light of potential harmful effects of nicotinamide catabolites, nicotinamide‐related compound is not recommended for diabetes treatment until more studies are carried out.
Potential drugs in development
NNMT inhibitor has been developed and tested in mice 137 , 138 . Inhibition of the enzyme might divert nicotinamide from catabolism toward more NAD+ synthesis. NNMT overexpression decreases NAD+ levels and causes fatty‐liver in mice 139 . This enzyme inhibition should reduce a consumption of the universal methyl donor, S‐adenosyl‐L‐methionine, used for nicotinamide methylation, and reduces the production of S‐adenosyl‐L‐homocysteine. Potentially, it reduces atherogenic homocysteine levels and facilitates an induction of epigenetic change by methylation. A potential side‐effect is to increase nicotinamide levels that inhibit sirtuins.
A direct AMPK activator has been developed, and is in clinical trials for efficacy and safety 140 . A direct NAMPT activator was identified recently 141 , but is still in a very early stage of development. Imeglimin is a new antidiabetic drug that recently finished phase III clinical testing in Japan. In rodents, the drug increases mitochondrial numbers, and reduces oxidative stress and DAG production, and maintains high carnitine levels in high‐fat high sucrose mice 142 . If the similar effects are observed in humans, it should benefit NAD+ metabolism.
Monitoring the ‘healthiness’ of NAD+ metabolism
How can we assess NAD+ metabolism and the effectiveness of interventions in humans? In healthy conditions, our hypothesis is that nicotinamide production by PARP or sirtuins is low, and produced nicotinamide should be preferentially used for NAD+ salvage processes and not for catabolic processes. Therefore, ‘healthiness’ might be monitored by measuring levels of tissue NAD+ and levels of urinary nicotinamide catabolites. In humans, measuring tissue NAD+ is very challenging in the clinical setting. Blood NAD+ could be used as a surrogate maker, which reflects red blood cell NAD+ content. Previously we found the RBC NAD+ content varies from 100 to 200 nmol/g Hb, and NADH levels are approximately 1/10th of NAD+. It can be assessed by various methods, including enzyme cycling assay 143 . Urinary nicotinamide catabolites (N‐methylnicotinamide, 2‐PY and 4‐PY) can be assessed by high‐performance liquid chromatography 144 and nuclear magnetic resonance 134 . Consistent with our hypothesis, nicotinamide catabolites are known to be increased by type 2 diabetes in both rodent models and humans 89 . In addition, a recent report showed that C57BL/6 mice fed with a high‐fat diet had a two‐ to threefold increase in 2‐PY and 4‐PY, as compared with mice fed a normal chow diet, and those levels were significantly reduced by treatment with metformin + vildagliptin (dipeptidyl peptidase inhibitor) 145 , also attesting our hypothesis. It is known that both NAD+ salvage activity 146 and nicotinamide catabolites 147 show diurnal variations as a result of circadian rhythm. Thus, timing for measuring these metabolites will be an issue. Previous studies did not take this into an account. After standardization of the timing, NAD metabolism ‘health index’ can be assessed as (blood NAD+) / (urinary nicotinamide catabolites), which are expected to be low with diabetes and become higher in healthy individuals. A previous study giving NR to humans 134 showed that blood NAD+ levels increased two‐ to threefold, but urine methylnicotinamide and 2‐Py levels increased eight‐ to tenfold. This suggests NR supplementation did not improve ‘healthiness.’ This interpretation agrees with another NR study that showed decreased NAMPT expression.
Concluding Remarks
In this review article, we pointed out that the etiology of NAD+ dysregulation remarkably coincides with the cascades postulated for the etiology of diabetic complications and other abnormalities induced by redox change. We also presented the new possibility that the nicotinamide catabolic cascade might play an important role, especially in oxidative stress production and epigenetic change in diabetes. For the treatment of type 2 diabetes and its complications, we recommend first to take AMPK‐activating medicines, such as metformin, glucagon‐like peptide‐1 agonist and sodium–glucose cotransporter 2 inhibitor. In particular, clinical data showing the ketogenic effect of sodium–glucose cotransporter 2 inhibitor seems to alleviate fatty acid toxicity and to reverse selective insulin resistance in the liver by mobilizing peripheral fat to the liver. In many countries, benfotiamine and acetyl‐L‐carnitine are available, which might help. If glucose control is not sufficient, aldose reductase inhibitors could be taken, although they are only available in certain countries. Taking a nicotinamide supplement, whether NR or NMN, at a high dose is not currently recommended until safety is established. However, with other regimens to improve ‘healthiness’ of NAD+ metabolism, it might be useful, as shown in a recent case report that acetyl‐L‐carnitine and nicotinamide treatment could prevent the development of type 1 diabetes 148 .
Disclosure
The authors declare no conflict interest.
Supporting information
Data S1 | Supplemental materials.
Acknowledgment
This research was funded by Chongqing Municipal Health and Health Committee (grant number #2017ZDXM033) and by District of Chongqing Liangjiang New Area (Public Document of the District [2019] #39) to FL, a Boston Area Diabetes Endocrinology Research Center grant to JMC, and Kilo Diabetes VASCULAR Foundation and National Science and Education (50814133) to YI.
J Diabetes Investig. 2020
References
- 1. Kirkland JB, Meyer‐Ficca ML. Niacin. Adv Food Nutr Res 2018; 83: 83–149. [DOI] [PubMed] [Google Scholar]
- 2. Peechakara BV, Gupta M. Vitamin B3. Treasure Island, FL: StatPearls, 2018. [Google Scholar]
- 3. Takahashi N, Li F, Fushima T, et al Vitamin B3 nicotinamide: a promising candidate for treating preeclampsia and improving fetal growth. Tohoku J Exp Med 2018; 244: 243–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Houtkooper RH, Canto C, Wanders RJ, et al The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev. 2010; 31: 194–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hwang ES, Song SB. Nicotinamide is an inhibitor of SIRT1 in vitro, but can be a stimulator in cells. Cell Mol Life Sci 2017; 74: 3347–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Massudi H, Grant R, Braidy N, et al Age‐associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS One 2012; 7: e42357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Clement J, Wong M, Poljak A, et al The Plasma NAD(+) Metabolome is dysregulated in "Normal" aging. Rejuvenation Res 2019; 22: 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lazzopina P, Mounsey A, Handler L. Clinical inquiries: does niacin decrease cardiovascular morbidity and mortality in CVD patients? J Fam Pract. 2018; 67: 314–316. [PubMed] [Google Scholar]
- 9. Ido Y. Pyridine nucleotide redox abnormalities in diabetes. Antioxid Redox Signal 2007; 9: 931–942. [DOI] [PubMed] [Google Scholar]
- 10. Williamson JR, Chang K, Frangos M, et al Hyperglycemic pseudohypoxia and diabetic complications. Diabetes 1993; 42: 801–813. [DOI] [PubMed] [Google Scholar]
- 11. Ido Y, Williamson JR. Hyperglycemic cytosolic reductive stress 'pseudohypoxia': implications for diabetic retinopathy. Invest Ophthalmol Vis Sci 1997; 38: 1467–1470. [PubMed] [Google Scholar]
- 12. Nyengaard JR, Ido Y, Kilo C, et al Interactions between hyperglycemia and hypoxia: implications for diabetic retinopathy. Diabetes 2004; 53: 2931–2938. [DOI] [PubMed] [Google Scholar]
- 13. Ido Y, Kilo C, Williamson JR. Cytosolic NADH/NAD+, free radicals, and vascular dysfunction in early diabetes mellitus. Diabetologia 1997; 40(Suppl 2): S115–S117. [DOI] [PubMed] [Google Scholar]
- 14. Ido Y. Diabetic complications within the context of aging: nicotinamide adenine dinucleotide redox, insulin C‐peptide, sirtuin 1‐liver kinase B1‐adenosine monophosphate‐activated protein kinase positive feedback and forkhead box O3. J Diabetes Investig 2016; 7: 448–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lan F, Cacicedo JM, Ido Y. Activation of AMPKK‐AMPK cascade by Silence Information Regulator 2 (Sir2). Diabetes 2005; 54(Supple 1): A383. [Google Scholar]
- 16. Lan F, Cacicedo JM, Ruderman N, et al SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP‐activated protein kinase activation. J Biol Chem 2008; 283: 27628–27635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ido Y, Duranton A, Lan F, et al Resveratrol prevents oxidative stress‐induced senescence and proliferative dysfunction by activating the AMPK‐FOXO3 cascade in cultured primary human keratinocytes. PLoS One 2015; 10: e0115341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lan F, Weikel KA, Cacicedo JM, Ido Y. Resveratrol‐induced AMP‐activated protein kinase activation is cell‐type dependent: lessons from basic research for clinical application. Nutrients 2017; 9: 751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ruderman NB, Xu XJ, Nelson L, et al AMPK and SIRT1: a long‐standing partnership? Am J Physiol Endocrinol Metab 2010; 298: E751–E760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Suchankova G, Nelson LE, Gerhart‐Hines Z, et al Concurrent regulation of AMP‐activated protein kinase and SIRT1 in mammalian cells. Biochem Biophys Res Commun. 2009; 378: 836–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ido Y, Carling D, Ruderman N. Hyperglycemia‐induced apoptosis in human umbilical vein endothelial cells: inhibition by the AMP‐activated protein kinase activation. Diabetes 2002; 51: 159–167. [DOI] [PubMed] [Google Scholar]
- 22. Sanchez‐Lozada LG, Andres‐Hernando A, Garcia‐Arroyo FE, et al Uric acid activates aldose reductase and the polyol pathway for endogenous fructose and fat production causing development of fatty liver in rats. J Biol Chem 2019; 294: 4272–4281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ido Y, McHowat J, Chang KC, et al Neural dysfunction and metabolic imbalances in diabetic rats: prevention by acetyl‐L‐carnitine. Diabetes 1994; 43: 1469–1477. [DOI] [PubMed] [Google Scholar]
- 24. Ido Y, Chang C, Williamson JR. Acetyl‐L‐carnitine normalizes diabetes‐induced changes in myocardial l‐carnitine, Na/K ATPase, and lactate/pyruvate ratio. Diabetes 1995; 44; (Supple 1): 36A. [Google Scholar]
- 25. Williamson JR, Ido Y. Understanding retinal cytosolic reductive stress. Invest Ophthalmol Vis Sci 1998; 39: 1295–1296. [PubMed] [Google Scholar]
- 26. Williamson JR, Kilo C, Ido Y. The role of cytosolic reductive stress in oxidant formation and diabetic complications. Diabetes Res Clin Pract 1999; 45: 81–82. [DOI] [PubMed] [Google Scholar]
- 27. Ido Y, Chang K, Woolsey TA, et al NADH: sensor of blood flow need in brain, muscle, and other tissues. FASEB J 2001; 15: 1419–1421. [DOI] [PubMed] [Google Scholar]
- 28. Ido Y, Chang K, Williamson JR. NADH augments blood flow in physiologically activated retina and visual cortex. Proc Natl Acad Sci USA 2004; 101: 653–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Varma SD, Kinoshita JH. Sorbitol pathway in diabetic and galactosemic rat lens. Biochim Biophys Acta 1974; 338: 632–640. [Google Scholar]
- 30. Gumaa KA, McLean P, Greenbaum AL. Compartmentation in relation to metabolic control in liver. Essays Biochem 1971; 7: 39–86. [PubMed] [Google Scholar]
- 31. Travis SF, Morrison AD, Clements RS Jr, et al Metabolic alterations in the human erythrocyte produced by increases in glucose concentration. The role of the polyol pathway. J Clin Investig 1971; 50: 2104–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Van den Enden MK, Nyengaard JR, Ostrow E, et al Elevated glucose levels increase retinal glycolysis and sorbitol pathway metabolism. Implications for diabetic retinopathy. Invest Ophthalmol Vis Sci 1995; 36: 1675–1685. [PubMed] [Google Scholar]
- 33. Vedantham S, Ananthakrishnan R, Schmidt AM, et al Aldose reductase, oxidative stress and diabetic cardiovascular complications. Cardiovasc Hematol Agents Med Chem 2012; 10: 234–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mracek T, Drahota Z, Houstek J. The function and the role of the mitochondrial glycerol‐3‐phosphate dehydrogenase in mammalian tissues. Biochim Biophys Acta 2013; 1827: 401–410. [DOI] [PubMed] [Google Scholar]
- 35. Orr AL, Quinlan CL, Perevoshchikova IV, et al A refined analysis of superoxide production by mitochondrial sn‐glycerol 3‐phosphate dehydrogenase. J Biol Chem 2012; 287: 42921–42935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nishikawa T, Edelstein D, Du XL, et al Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000; 404: 787–790. [DOI] [PubMed] [Google Scholar]
- 37. Soriano FG, Pacher P, Mabley J, et al Rapid reversal of the diabetic endothelial dysfunction by pharmacological inhibition of poly(ADP‐ribose) polymerase. Circ Res 2001; 89: 684–691. [DOI] [PubMed] [Google Scholar]
- 38. Gancheva S, Bierwagen A, Begovatz P, et al Newly diagnosed type 1 diabetes patients exhibit lower hepatic ATP concentrations despite normal hepatocellular lipid accumulation. Diabetologie und Stoffwechsel 2015; 10(S 01): P23. [Google Scholar]
- 39. Yoshino J, Mills KF, Yoon MJ, et al Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet‐ and age‐induced diabetes in mice. Cell Metab 2011; 14: 528–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Obrosova IG, Ilnytska O, Lyzogubov VV, et al High‐fat diet induced neuropathy of pre‐diabetes and obesity: effects of "healthy" diet and aldose reductase inhibition. Diabetes 2007; 56: 2598–2608. [DOI] [PubMed] [Google Scholar]
- 41. Schmid AI, Szendroedi J, Chmelik M, et al Liver ATP synthesis is lower and relates to insulin sensitivity in patients with type 2 diabetes. Diabetes Care 2011; 34: 448–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Du X, Matsumura T, Edelstein D, et al Inhibition of GAPDH activity by poly(ADP‐ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J Clin Investig 2003; 112: 1049–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McGarry JD. What if Minkowski had been ageusic? An alternative angle on diabetes. Science 1992; 258: 766–770. [DOI] [PubMed] [Google Scholar]
- 44. McGarry JD. Banting lecture 2001: dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes 2002; 51: 7–18. [DOI] [PubMed] [Google Scholar]
- 45. Randle PJ, Garland PB, Hales CN, et al The glucose fatty‐acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963; 1: 785–789. [DOI] [PubMed] [Google Scholar]
- 46. Brown MS, Goldstein JL. Selective versus total insulin resistance: a pathogenic paradox. Cell Metab 2008; 7: 95–96. [DOI] [PubMed] [Google Scholar]
- 47. Lovejoy J, Newby FD, Gebhart SS, et al Insulin resistance in obesity is associated with elevated basal lactate levels and diminished lactate appearance following intravenous glucose and insulin. Metabolism 1992; 41: 22–27. [DOI] [PubMed] [Google Scholar]
- 48. Crawford SO, Hoogeveen RC, Brancati FL, et al Association of blood lactate with type 2 diabetes: the Atherosclerosis Risk in Communities Carotid MRI Study. Int J Epidemiol 2010; 39: 1647–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vettor R, Lombardi AM, Fabris R, et al Lactate infusion in anesthetized rats produces insulin resistance in heart and skeletal muscles. Metabolism 1997; 46: 684–690. [DOI] [PubMed] [Google Scholar]
- 50. Juraschek SP, Selvin E, Miller ER, et al Plasma lactate and diabetes risk in 8045 participants of the atherosclerosis risk in communities study. Ann Epidemiol 2013; 23, 791–796.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen H, Liu X, Chen H, et al Role of SIRT1 and AMPK in mesenchymal stem cells differentiation. Ageing Res Rev 2014; 13: 55–64. [DOI] [PubMed] [Google Scholar]
- 52. Wang Y, Liang Y, Vanhoutte PM. SIRT1 and AMPK in regulating mammalian senescence: a critical review and a working model. FEBS Lett 2011; 585: 986–994. [DOI] [PubMed] [Google Scholar]
- 53. Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC‐1{alpha}. J Biol Chem 2005; 280: 16456–16460. [DOI] [PubMed] [Google Scholar]
- 54. Rodgers JT, Lerin C, Gerhart‐Hines Z, et al Metabolic adaptations through the PGC‐1 alpha and SIRT1 pathways. FEBS Lett 2008; 582: 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sriwijitkamol A, Ivy JL, Christ‐Roberts C, et al LKB1‐AMPK signaling in muscle from obese insulin‐resistant Zucker rats and effects of training. Am J Physiol Endocrinol Metab 2006; 290: E925–E932. [DOI] [PubMed] [Google Scholar]
- 56. Wan Z, Root‐McCaig J, Castellani L, et al Evidence for the role of AMPK in regulating PGC‐1 alpha expression and mitochondrial proteins in mouse epididymal adipose tissue. Obesity 2014; 22: 730–738. [DOI] [PubMed] [Google Scholar]
- 57. Li XN, Song J, Zhang L, et al Activation of the AMPK‐FOXO3 pathway reduces fatty acid‐induced increase in intracellular reactive oxygen species by upregulating thioredoxin. Diabetes 2009; 58: 2246–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Davila D, Connolly NM, Bonner H, et al Two‐step activation of FOXO3 by AMPK generates a coherent feed‐forward loop determining excitotoxic cell fate. Cell Death Differ 2012; 19: 1677–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Salminen A, Hyttinen JM, Kaarniranta K. AMP‐activated protein kinase inhibits NF‐kappaB signaling and inflammation: impact on healthspan and lifespan. J Mol Med 2011; 89: 667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yang Z, Kahn BB, Shi H, et al Macrophage alpha1 AMP‐activated protein kinase (alpha1AMPK) antagonizes fatty acid‐induced inflammation through SIRT1. J Biol Chem 2010; 285: 19051–19059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhang Z, Lowry SF, Guarente L, et al Roles of SIRT1 in the acute and restorative phases following induction of inflammation. J Biol Chem 2010; 285: 41391–41401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Giovannini L, Bianchi S. Role of nutraceutical SIRT1 modulators in AMPK and mTOR pathway: evidence of a synergistic effect. Nutrition 2017; 34: 82–96. [DOI] [PubMed] [Google Scholar]
- 63. Liu M, Wilk SA, Wang A, et al Resveratrol inhibits mTOR signaling by promoting the interaction between mTOR and DEPTOR. J Biol Chem 2010; 285: 36387–36394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chang C, Su H, Zhang D, et al AMPK‐dependent phosphorylation of GAPDH triggers Sirt1 activation and is necessary for autophagy upon glucose starvation. Mol Cell 2015; 60: 930–940. [DOI] [PubMed] [Google Scholar]
- 65. Canto C, Gerhart‐Hines Z, Feige JN, et al AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009; 458: 1056–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem 2004; 279: 50754–50763. [DOI] [PubMed] [Google Scholar]
- 67. Sauve AA, Schramm VL. Sir2 regulation by nicotinamide results from switching between base exchange and deacetylation chemistry. Biochemistry 2003; 42: 9249–9256. [DOI] [PubMed] [Google Scholar]
- 68. Bitterman KJ, Anderson RM, Cohen HY, et al Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J Biol Chem 2002; 277: 45099–45107. [DOI] [PubMed] [Google Scholar]
- 69. Beirowski B, Babetto E, Golden JP, et al Metabolic regulator LKB1 is crucial for Schwann cell‐mediated axon maintenance. Nat Neurosci 2014; 17: 1351–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Salway JG. Metabolism at a Glance. Oxford: Blackwell Publishers, 1999; 111 p. [Google Scholar]
- 71. Berger F, Lau C, Dahlmann M, et al Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem 2005; 280: 36334–36341. [DOI] [PubMed] [Google Scholar]
- 72. Devin A, Guerin B, Rigoulet M. Cytosolic NAD+ content strictly depends on ATP concentration in isolated liver cells. FEBS Lett 1997; 410: 329–332. [DOI] [PubMed] [Google Scholar]
- 73. Kirkland JB. Poly ADP‐ribose polymerase‐1 and health. Exp Biol Med 2010; 235: 561–568. [DOI] [PubMed] [Google Scholar]
- 74. Li F, Drel VR, Szabo C, et al Low‐dose poly(ADP‐ribose) polymerase inhibitor‐containing combination therapies reverse early peripheral diabetic neuropathy. Diabetes 2005; 54: 1514–1522. [DOI] [PubMed] [Google Scholar]
- 75. Gale EA, Bingley PJ, Emmett CL, et al European Nicotinamide Diabetes Intervention Trial (ENDIT): a randomised controlled trial of intervention before the onset of type 1 diabetes. Lancet 2004; 363: 925–931. [DOI] [PubMed] [Google Scholar]
- 76. Szkudelski T. Streptozotocin‐nicotinamide‐induced diabetes in the rat. Characteristics of the experimental model. Exp Biol Med 2012; 237: 481–490. [DOI] [PubMed] [Google Scholar]
- 77. Hardie DG. Biochemistry. Balancing cellular energy. Science 2007; 315: 1671–1672. [DOI] [PubMed] [Google Scholar]
- 78. Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia 2017; 60: 1577–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Apostolopoulou M, Strassburger K, Herder C, et al Metabolic flexibility and oxidative capacity independently associate with insulin sensitivity in individuals with newly diagnosed type 2 diabetes. Diabetologia 2016; 59: 2203–2207. [DOI] [PubMed] [Google Scholar]
- 80. Phielix E, Jelenik T, Nowotny P, et al Reduction of non‐esterified fatty acids improves insulin sensitivity and lowers oxidative stress, but fails to restore oxidative capacity in type 2 diabetes: a randomised clinical trial. Diabetologia 2014; 57: 572–581. [DOI] [PubMed] [Google Scholar]
- 81. Szendroedi J, Phielix E, Roden M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol 2011; 8: 92–103. [DOI] [PubMed] [Google Scholar]
- 82. Das Evcimen N, King GL. The role of protein kinase C activation and the vascular complications of diabetes. Pharmacol Res 2007; 55: 498–510. [DOI] [PubMed] [Google Scholar]
- 83. Heathcote HR, Mancini SJ, Strembitska A, et al Protein kinase C phosphorylates AMP‐activated protein kinase alpha1 Ser487. Biochem J 2016; 473: 4681–4697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Coughlan KA, Valentine RJ, Sudit BS, et al PKD1 Inhibits AMPKalpha2 through phosphorylation of serine 491 and impairs insulin signaling in skeletal muscle cells. J Biol Chem 2016; 291: 5664–5675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Burgos ES, Schramm VL. Weak coupling of ATP hydrolysis to the chemical equilibrium of human nicotinamide phosphoribosyltransferase. Biochemistry 2008; 47: 11086–11096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Guan X, Lin P, Knoll E, et al Mechanism of inhibition of the human sirtuin enzyme SIRT3 by nicotinamide: computational and experimental studies. PLoS One 2014; 9: e107729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ushiro H, Yokoyama Y, Shizuta Y. Purification and characterization of poly (ADP‐ribose) synthetase from human placenta. J Biol Chem 1987; 262: 2352–2357. [PubMed] [Google Scholar]
- 88. Stanulovic M, Chaykin S. Metabolic origins of the pyridones of N 1 ‐methylnicotinamide in man and rat. Arch Biochem Biophys 1971; 145: 35–42. [DOI] [PubMed] [Google Scholar]
- 89. Salek RM, Maguire ML, Bentley E, et al A metabolomic comparison of urinary changes in type 2 diabetes in mouse, rat, and human. Physiol Genom 2007; 29: 99–108. [DOI] [PubMed] [Google Scholar]
- 90. Moriwaki Y, Yamamoto T, Takahashi S, et al Widespread cellular distribution of aldehyde oxidase in human tissues found by immunohistochemistry staining. Histol Histopathol 2001; 16: 745–753. [DOI] [PubMed] [Google Scholar]
- 91. Schmeisser K, Mansfeld J, Kuhlow D, et al Role of sirtuins in lifespan regulation is linked to methylation of nicotinamide. Nat Chem Biol 2013; 9: 693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Pedersen BK. IL‐6 signalling in exercise and disease. Biochem Soc Trans 2007; 35(Pt 5): 1295–1297. [DOI] [PubMed] [Google Scholar]
- 93. Pedersen BK, Fischer CP. Beneficial health effects of exercise–the role of IL‐6 as a myokine. Trends Pharmacol Sci 2007; 28: 152–156. [DOI] [PubMed] [Google Scholar]
- 94. Ristow M, Zarse K, Oberbach A, et al Antioxidants prevent health‐promoting effects of physical exercise in humans. Proc Natl Acad Sci USA 2009; 106: 8665–8670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Zhou SS, Li D, Sun WP, et al Nicotinamide overload may play a role in the development of type 2 diabetes. World J Gastroenterol 2009; 15: 5674–5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Zhou Y, Chen N, Li D, et al Excess nicotinamide induces reactive oxygen species generation, insulin resistance, and epigenetic change in rats and humans. Diabetes 2018. 10.2337/db18-783-P [DOI] [Google Scholar]
- 97. Pissios P. Nicotinamide N‐methyltransferase: more than a vitamin B3 clearance enzyme. Trends Endocrinol Metab 2017; 28: 340–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Saha A, Connelly S, Jiang J, et al Akt phosphorylation and regulation of transketolase is a nodal point for amino acid control of purine synthesis. Mol Cell 2014; 55: 264–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Thornalley PJ, Babaei‐Jadidi R, Al Ali H, et al High prevalence of low plasma thiamine concentration in diabetes linked to a marker of vascular disease. Diabetologia 2007; 50: 2164–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Luong KV, Nguyen LT. The impact of thiamine treatment in the diabetes mellitus. J Clin Med Res 2012; 4: 153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Hara N, Yamada K, Shibata T, et al Nicotinamide phosphoribosyltransferase/visfatin does not catalyze nicotinamide mononucleotide formation in blood plasma. PLoS One 2011; 6: e22781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Rongvaux A, Shea RJ, Mulks MH, et al Pre‐B‐cell colony‐enhancing factor, whose expression is up‐regulated in activated lymphocytes, is a nicotinamide phosphoribosyltransferase, a cytosolic enzyme involved in NAD biosynthesis. Eur J Immunol 2002; 32: 3225–3134. [DOI] [PubMed] [Google Scholar]
- 103. Imai E, Sano M, Fukuwatari T, et al Tryptophan‐nicotinamide metabolism in rats with streptozotocin‐induced diabetes: association of dietrary vitamin C content. J Jpn Soc Nutr Food Sci 2011; 64: 313–321. [Google Scholar]
- 104. Imai E, Shibata K. Oral glucose tolerance and tryptophan metabolism in non‐obese and non‐insulin‐dependent diabetic goto‐kakizaki rats fed high‐tryptophan diets. J Nutr Sci Vitaminol 2018; 64: 48–55. [DOI] [PubMed] [Google Scholar]
- 105. Coughlan KA, Valentine RJ, Ruderman NB, et al Nutrient excess in AMPK downregulation and insulin resistance. J Endocrinol Diabetes Obes 2013; 1: 1008. [PMC free article] [PubMed] [Google Scholar]
- 106. Xu XJ, Gauthier MS, Hess DT, et al Insulin sensitive and resistant obesity in humans: AMPK activity, oxidative stress, and depot‐specific changes in gene expression in adipose tissue. J Lipid Res 2012; 53: 792–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. de Guia RM, Agerholm M, Nielsen TS, et al Aerobic and resistance exercise training reverses age‐dependent decline in NAD(+) salvage capacity in human skeletal muscle. Physiol Rep 2019; 7: e14139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Foretz M, Hebrard S, Leclerc J, et al Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Investig 2010; 120: 2355–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Krasner NM, Ido Y, Ruderman NB, et al Glucagon‐like peptide‐1 (GLP‐1) analog liraglutide inhibits endothelial cell inflammation through a calcium and AMPK dependent mechanism. PLoS One 2014; 9: e97554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Hawley SA, Ford RJ, Smith BK, et al The Na+/glucose cotransporter inhibitor canagliflozin activates AMPK by inhibiting mitochondrial function and increasing cellular AMP levels. Diabetes 2016; 65: 2784–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Hubbard BP, Sinclair DA. Small molecule SIRT1 activators for the treatment of aging and age‐related diseases. Trends Pharmacol Sci 2014; 35: 146–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Park SJ, Ahmad F, Philp A, et al Resveratrol ameliorates aging‐related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 2012; 148: 421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Wan D, Zhou Y, Wang K, et al Resveratrol provides neuroprotection by inhibiting phosphodiesterases and regulating the cAMP/AMPK/SIRT1 pathway after stroke in rats. Brain Res Bull 2016; 121: 255–262. [DOI] [PubMed] [Google Scholar]
- 114. Gerhart‐Hines Z, Dominy JE Jr, Blattler SM, et al The cAMP/PKA pathway rapidly activates SIRT1 to promote fatty acid oxidation independently of changes in NAD(+). Mol Cell 2011; 44: 851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Ghosh S, Liu B, Zhou Z. Resveratrol activates SIRT1 in a Lamin A‐dependent manner. Cell Cycle 2013; 12: 872–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Zheng J, Ramirez VD. Inhibition of mitochondrial proton F0F1‐ATPase/ATP synthase by polyphenolic phytochemicals. Br J Pharmacol 2000; 130: 1115–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Zhu X, Wu C, Qiu S, et al Effects of resveratrol on glucose control and insulin sensitivity in subjects with type 2 diabetes: systematic review and meta‐analysis. Nutr Metab 2017; 14: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Mingrone G. Carnitine in type 2 diabetes. Ann N Y Acad Sci 2004; 1033: 99–107. [DOI] [PubMed] [Google Scholar]
- 119. Sima AA, Calvani M, Mehra M, et al Acetyl‐L‐carnitine improves pain, nerve regeneration, and vibratory perception in patients with chronic diabetic neuropathy: an analysis of two randomized placebo‐controlled trials. Diabetes Care 2005; 28: 89–94. [DOI] [PubMed] [Google Scholar]
- 120. Bene J, Hadzsiev K, Melegh B. Role of carnitine and its derivatives in the development and management of type 2 diabetes. Nutr Diabetes 2018; 8: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Sergi G, Pizzato S, Piovesan F, et al Effects of acetyl‐L‐carnitine in diabetic neuropathy and other geriatric disorders. Aging Clin Exp Res 2018; 30: 133–138. [DOI] [PubMed] [Google Scholar]
- 122. Obrosova IG, Pacher P, Szabo C, et al Aldose reductase inhibition counteracts oxidative‐nitrosative stress and poly(ADP‐ribose) polymerase activation in tissue sites for diabetes complications. Diabetes 2005; 54: 234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Drel VR, Pacher P, Stevens MJ, et al Aldose reductase inhibition counteracts nitrosative stress and poly(ADP‐ribose) polymerase activation in diabetic rat kidney and high‐glucose‐exposed human mesangial cells. Free Radic Biol Med 2006; 40: 1454–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Babaei‐Jadidi R, Karachalias N, Ahmed N, et al Prevention of incipient diabetic nephropathy by high‐dose thiamine and benfotiamine. Diabetes 2003; 52: 2110–2120. [DOI] [PubMed] [Google Scholar]
- 125. Hammes HP, Du X, Edelstein D, et al Benfotiamine blocks three major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy. Nat Med 2003; 9: 294–299. [DOI] [PubMed] [Google Scholar]
- 126. Haupt E, Ledermann H, Kopcke W. Benfotiamine in the treatment of diabetic polyneuropathy–a three‐week randomized, controlled pilot study (BEDIP study). Int J Clin Pharmacol Ther 2005; 43: 71–77. [DOI] [PubMed] [Google Scholar]
- 127. Stracke H, Gaus W, Achenbach U, et al Benfotiamine in diabetic polyneuropathy (BENDIP): results of a randomised, double blind, placebo‐controlled clinical study. Exp Clin Endocrinol Diabetes 2008; 116: 600–605. [DOI] [PubMed] [Google Scholar]
- 128. Greenbaum CJ, Kahn SE, Palmer JP. Nicotinamide's effects on glucose metabolism in subjects at risk for IDDM. Diabetes 1996; 45: 1631–1634. [DOI] [PubMed] [Google Scholar]
- 129. Kannt A, Pfenninger A, Teichert L, et al Association of nicotinamide‐N‐methyltransferase mRNA expression in human adipose tissue and the plasma concentration of its product, 1‐methylnicotinamide, with insulin resistance. Diabetologia 2015; 58: 799–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Liu M, Li L, Chu J, et al Serum N(1)‐methylnicotinamide is associated with obesity and diabetes in Chinese. J Clin Endocrinol Metab 2015; 100: 3112–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Bieganowski P, Brenner C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss‐Handler independent route to NAD+ in fungi and humans. Cell 2004; 117: 495–502. [DOI] [PubMed] [Google Scholar]
- 132. Martens CR, Denman BA, Mazzo MR, et al Chronic nicotinamide riboside supplementation is well‐tolerated and elevates NAD(+) in healthy middle‐aged and older adults. Nat Commun 2018; 9: 1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Conze D, Brenner C, Kruger CL. Safety and metabolism of long‐term administration of NIAGEN (Nicotinamide Riboside Chloride) in a randomized, double‐blind, placebo‐controlled clinical trial of healthy overweight adults. Sci Rep 2019; 9: 9772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Elhassan YS, Kluckova K, Fletcher RS, et al Nicotinamide riboside augments the aged human skeletal muscle NAD(+) metabolome and induces transcriptomic and anti‐inflammatory signatures. Cell Rep 2019; 28, 1717–1728.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Dollerup OL, Chubanava S, Agerholm M, et al Nicotinamide riboside does not alter mitochondrial respiration, content or morphology in skeletal muscle from obese and insulin resistant men. J Physiol 2020; 598: 731–754. [DOI] [PubMed] [Google Scholar]
- 136. Poddar SK, Sifat AE, Haque S, et al Nicotinamide mononucleotide: exploration of diverse therapeutic applications of a potential molecule. Biomolecules 2019; 9: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Brachs S, Polack J, Brachs M, et al Genetic Nicotinamide N‐Methyltransferase (Nnmt) deficiency in male mice improves insulin sensitivity in diet‐induced obesity but does not affect glucose tolerance. Diabetes 2019; 68: 527–542. [DOI] [PubMed] [Google Scholar]
- 138. Kannt A, Rajagopal S, Kadnur SV, et al A small molecule inhibitor of Nicotinamide N‐methyltransferase for the treatment of metabolic disorders. Sci Rep 2018; 8: 3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Komatsu M, Kanda T, Urai H, et al NNMT activation can contribute to the development of fatty liver disease by modulating the NAD (+) metabolism. Sci Rep 2018; 8: 8637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Steinberg GR, Carling D. AMP‐activated protein kinase: the current landscape for drug development. Nat Rev Drug Discov 2019; 18: 527–551. [DOI] [PubMed] [Google Scholar]
- 141. Gardell SJ, Hopf M, Khan A, et al Boosting NAD(+) with a small molecule that activates NAMPT. Nat Commun 2019; 10: 3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Vial G, Chauvin MA, Bendridi N, et al Imeglimin normalizes glucose tolerance and insulin sensitivity and improves mitochondrial function in liver of a high‐fat, high‐sucrose diet mice model. Diabetes 2015; 64: 2254–2264. [DOI] [PubMed] [Google Scholar]
- 143. Lowry OH. Amplification by enzymatic cycling. Mol Cell Biochem 1980; 32: 135–146. [DOI] [PubMed] [Google Scholar]
- 144. Maeta A, Sano M, Fukuwatari T, et al Simultaneous measurement of nicotinamide and its catabolites, nicotinamide N‐oxide, N(1)‐methyl‐2‐pyridone‐5‐carboxamide, and N(1)‐methyl‐4‐pyridone‐3‐carboxamide, in mice urine. Biosci Biotechnol Biochem 2014; 78: 1306–1309. [DOI] [PubMed] [Google Scholar]
- 145. Pelantova H, Bartova S, Anyz J, et al Metabolomic profiling of urinary changes in mice with monosodium glutamate‐induced obesity. Anal Bioanal Chem 2016; 408: 567–578. [DOI] [PubMed] [Google Scholar]
- 146. Nakahata Y, Sahar S, Astarita G, et al Circadian control of the NAD+ salvage pathway by CLOCK‐SIRT1. Science 2009; 324: 654–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Okamoto H, Ishikawa A, Yoshitake Y, et al Diurnal variations in human urinary excretion of nicotinamide catabolites: effects of stress on the metabolism of nicotinamide. Am J Clin Nutr 2003; 77: 406–410. [DOI] [PubMed] [Google Scholar]
- 148. Fernandez IC, Del Carmen Camberos M, Passicot GA, et al Children at risk of diabetes type 1. Treatment with acetyl‐L‐carnitine plus nicotinamide ‐ case reports. J Pediatr Endocrinol Metab 2013; 26: 347–355. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 | Supplemental materials.