

Abstract
Purpose of review:
Numerous studies have begun to unravel the genetic basis of not only aortic disease but also other forms of commonly encountered vascular diseases. The goal of this review is to provide clinicians a reference to help identify and diagnose different types of vascular disease with a genetic underpinning.
Recent findings:
Ongoing studies have identified numerous genes involved in the TGF-β signaling pathway that are also associated with thoracic aortic aneurysm and dissection, and it is possible to test for pathogenic variants in these genes in the clinical setting using commercially available genetic testing panels. Additional studies have begun to identify genetic variants associated with an increased risk of bicuspid aortic valve, abdominal aortic aneurysm, and fibromuscular dysplasia.
Summary:
With increased availability of low-cost genetic testing, clinicians are now able to not only definitively diagnose some vascular syndromes but also provide information on the risk of disease in other family members, as well as provide guidance in terms of family planning. As the cost of genetic testing continues to drop with the benefit of increasing insurance coverage, genetic data will increasingly become part of clinical care for many patients with vascular disease.
Keywords: vascular genetics, aneurysm, dissection, aortic disease
Introduction
Clinicians have long recognized that many vascular diseases exist as part of well-defined syndromes or cluster within families, thus suggesting a genetic contribution to disease pathogenesis. Indeed, early research efforts focusing on specific diseases, such as Marfan syndrome, successfully identified mutations responsible for these syndromes. With the recent advent of next-generation sequencing technology, low-cost genetic testing is increasingly available to both patients and providers, and greater access has dramatically expanded our understanding of the genetic basis of vascular disease. Historically, much attention has focused on thoracic aortic aneurysm and dissection (TAAD), and numerous genes in the TGF-β signaling pathway have been implicated in many cases of TAAD (Table 1).[1] However, improved genotyping as well as more extensive phenotyping has led to greater appreciation of the involvement of additional vascular beds in TAAD and other vascular syndromes as well as the involvement of other organs and biologic processes. In this review, we describe several vascular syndromes that affect both the thoracic aorta and other vascular beds and discuss their associated genes and mutations, many of which can be identified through commercially-available genetic testing panels.
Table 1.
Examples of Genetic Causes of Thoracic Aortic Aneurysm and Dissection
| Disease | Gene | Cardiovascular Manifestations | Other Manifestations |
|---|---|---|---|
| Marfan syndrome | FBN1 | Aortic root aneurysm, aortic dissection, mitral and tricuspid valve prolapse, pulmonary artery dilatation. | Ectopia lentis, myopia, retinal detachment, glaucoma, premature cataracts, scoliosis, joint laxity, elongated limbs relative to trunk length, arachnodactyly, pectus excavatum, pectus carinatum, dental crowding, and a high arched palate. |
| Loeys-Dietz syndrome | TGFB2, TGFB3, TGFBR1, TGFBR2, SMAD3 | Diffuse aortic and arterial aneurysm and dissection, arterial tortuosity, bicuspid aortic valve, atrial septal defect, patent ductus arteriosus, mitral valve prolapse, left ventricular hypertrophy. | Bifid uvula, cleft palate, hypertelorism, pectus carinatum, pectus excavatum, scoliosis, pes planus, joint contractures, neurocognitive abnormalities, early-onset osteoarthritis (SMAD3) |
| Familial thoracic aortic aneurysm and dissection | MYH11, ACTA2, TGFBR1, TGFBR2 | Ascending and descending aortic aneurysm and dissection, patent ductus arteriosus (MYH11), livedo reticularis (ACTA2), patent ductus arteriosus (ACTA2), bicuspid aortic valve (ACTA2), Moyamoya disease (ACTA2), premature stroke and coronary artery disease (ACTA2), cerebral, carotid, and popliteal artery aneurysms (TGFBR2) | |
| Vascular Ehlers-Danlos syndrome | COL3A1 | Aortic, visceral, extracranial, or limb artery aneurysm and dissection, spontaneous carotid-cavernous sinus fistulas, premature varicose veins | Smooth, velvety skin, easy skin tearing and scarring, delayed wound healing, striae, easy bruising, thin, delicate, and pinched nose, hollow cheeks, prominent eyes, hypermobile joints, intestinal rupture, uterine rupture, muscle or tendon rupture, clubfoot |
| Congenital contractural arachnodactyly | FBN2 | Aortic root dilatation, mitral valve prolapse | Arachnodactyly, scoliosis, joint contractures, muscle hypoplasia, pectus carinatum, crumpled-appearing ears |
| Shprintzen-Goldberg Syndrome | SKI | Ascending aortic aneurysm | Pectus carinatum, pectus excavatum, arachnodactyly, clef palate |
| Arterial tortuosity syndrome | SLC2A10 | Diffuse tortuosity, elongation, stenosis, and aneurysm of the aorta and large and medium arteries with risk of dissection, pulmonary artery stenosis | Soft, velvety, and hyperextensible skin, facial dysmorphisms, abdominal hernias, joint hypermobility |
| Turner Syndrome | 45, X | Ascending aortic aneurysm, aortic dissection, bicuspid aortic valve, aortic coarctation | Short stature, congenital lymphedema, shield chest, webbed neck, low-set ears, gonadal dysgenesis, learning disabilities, renal malformations, hypothyroidism |
Genetic analysis in vascular disease
Family history: Family history of vascular disease remains one of the strongest predictors of TAAD and is likely a more important component of the clinical evaluation than genetic testing in many patients.[2] Thus, the cornerstone of genetic analysis remains a thorough family history, which should include at least three generations and include the medical history of not only first-degree relatives but also grandparents, uncles, aunts, nephews, nieces, and cousins in each generation. Questions should focus on known cardiovascular diseases and syndromes but also include queries about sudden death, musculoskeletal complaints, height, and major surgeries that may have involved the aorta or other vessels. Utilizing a pedigree is helpful, and there are numerous web resources to facilitate creating pedigrees.[3]
- Understanding the spectrum of genetic disorders: The majority of vascular conditions for which clinicians consider genetic testing are examples of Mendelian disorders, in which variation in a gene is responsible for rare disease in either an individual (sporadic) or several related family members (inherited). Although all disease-causing variants may be located on a single gene, there are also conditions in which variants in several different genes are associated with the same syndrome.
- In other common, non-Mendelian disorders, there may be evidence of heritability but no causal variants associated with the disease. Instead, there may be dozens or even hundreds of variants, each of which may lend a small degree of genetic predisposition, associated with the disorder. Tools such as a genome-wide association study (GWAS) allow investigators to examine the association between numerous common alleles and a common disease state. This is not performed in the clinical setting and is instead utilized in large cohorts of patients with the disease of interest.
Principles of genetic testing
Clinical scenarios for genetic testing: Few professional guidelines exist to guide clinicians in pursuing genetic testing for TAAD. Nonetheless, testing may be useful in the following scenarios: individuals with additional phenotypic features suggestive of a known TAAD syndrome; individuals presenting younger than age 60, particularly without known risk factors; individuals with additional affected family members; and extreme or unusual phenotypes. Additionally, all at-risk relatives of a person with a confirmed genetic TAAD syndrome should undergo counseling and genetic testing with aortic imaging if the genetic diagnosis is confirmed.[4]
- Sequencing strategies: In the clinical setting, genetic testing is typically achieved through next-generation sequencing of one or a panel of genes commonly implicated in TAAD. Numerous companies offer such testing, which may be covered by insurers depending on the clinical setting and the policies of the specific payer.
- When confronted with a suspected TAAD syndrome, many providers choose to sequence the entire panel of TAAD genes, and this is often the only option in cases of ill-defined phenotypes that span several syndromes. In cases in which one specific syndrome is suspected, however, it is also reasonable to sequence only genes associated with that syndrome. Importantly, although a genetic diagnosis may influence decision-making in terms of aortic interventions, it is not required for the diagnosis and management of most TAAD syndromes. Furthermore, such testing should only be pursued by providers comfortable interpreting the test results and after a detailed discussion with patients and family members about the potential medical, legal, and ethical ramifications of such testing.
- Interpreting test results: In some instances, a clinician may strongly suspect a genetic cause for a disease despite inconclusive panel testing results. This occurs frequently, as detection rates are low in many cases of TAAD, particularly in non-syndromic cases. In these situations, sequencing of the entire coding portion of the genome (whole exome sequencing) or both the coding and non-coding portions of the genome (whole genome sequencing) can be considered, although these are typically pursued in research settings and are not commonly performed or covered by insurers in clinical practice.
- Genetic testing results are not binary. Variants detected by genetic testing are classified not only as pathogenic or benign but also as variants of unknown significance (VUS) that lack examples from other affected individuals or functional data to support pathogenicity. Such variants must be interpreted with caution.[5, 6]
- Another important consideration in genetic testing is the variability in detection rate of pathogenic variants. Due to considerable overlap in the clinical features of many TAAD syndromes, detection rate with a single-gene sequencing approach may be low, although this is somewhat improved by utilizing panel testing.[7–9] Detection rate also varies depending on the suspected clinical syndrome.[8, 9]
Role of genetic specialists: When available, referral to a genetic counselor may be helpful, particularly when screening multiple family members or clinically unaffected individuals. Additionally, collaborating with genetic counselors is encouraged to facilitate testing given the numerous issues regarding privacy, insurer and employer discrimination, interpretation of inconclusive results, and logistics. Counselors are also helpful in collecting and mapping additional family history, recognizing and discussing inheritance patterns and risk, and helping individuals cope with the psychosocial issues surrounding testing. Finally, all providers who lack expertise or comfort in interpreting genetic testing results should consider referral to a genetic specialist before initiating testing.
TAAD Syndromes
Marfan syndrome
Epidemiology: Marfan syndrome (MFS) is the most common heritable connective tissue disorder associated with TAAD with a prevalence of ~1:15,000.[10]
- Clinical presentation: The most important vascular manifestation of MFS is thoracic aortic aneurysm that predominantly affects the aortic root, often with concomitant effacement of the sinotubular junction (STJ) on aortic imaging. To avoid variability in aortic root measurements, the diameter is measured at the STJ and normalized for sex, age, and body surface area to obtain a Z-score.[11]
- Patients are at risk for type A aortic dissection even in the absence of aortic root dilatation. More recent data suggest individuals are also at risk for distal aortic aneurysm and dissection.[12] Additional cardiac manifestations include mitral and tricuspid valve prolapse and pulmonary artery dilatation, although pulmonary artery dissection is exceedingly rare.[13, 14]
- Clinical features are highly variable, but ocular and musculoskeletal features are additional hallmarks of MFS.
- Genetics: MFS is inherited in an autosomal dominant manner due to mutations in FBN1, which encodes the extracellular matrix protein fibrillin-1, an extracellular matrix glycoprotein that assembles into microfibrils to provide both elasticity and architectural support to the vessel wall.[17, 18]
- FBN1 mutations may lead to aortic pathology in MFS in part through elastic fiber fragmentation and degeneration of the tunica media.[19] However, it is increasingly apparent that the pathogenesis is also due to excessive signaling in the TGF-β pathway, which plays an essential role in many TAAD syndromes.[20, 21]
- Diagnosis: The diagnosis of MFS is based on the Revised Ghent criteria.[22] MFS should be suspected in individuals with aortic root enlargement (Z-score ≥2.0), ectopia lentis, a systemic score ≥7 (Table 2), and a family history of MFS. In such individuals, we recommend genetic testing for confirmatory purposes.
- MFS is confirmed in an individual with a pathogenic variant in FBN1 along with either ectopia lentis or aortic root enlargement (Z-score ≥2.0).
Clinical course: In general, with early detection, surveillance, and timely aortic intervention if necessary, the lifespan of individuals with MFS approaches that of the general population.[23] However, certain FBN1 mutations, in particular those lying in exons 24–32, are associated with more severe phenotypes and greater mortality.[24] Similarly, individuals with haploinsufficient FBN1 mutations, compared to those with dominant negative mutations, are less likely to exhibit progression of aortic root dilatation while on losartan, a known inhibitor of TGF-β signaling.[25]
Table 2.
Systemic Score for the Diagnosis of Marfan Syndrome
| Feature | Value |
|---|---|
| Wrist AND thumb sign | 3 |
| Wrist OR thumb sign | 1 |
| Pectus carcinatum | 2 |
| Pectus excavatum or chest asymmetry | 1 |
| Hindfoot deformity | 2 |
| Pes planus (flat feet) | 1 |
| Pneumothorax | 2 |
| Dural ectasia | 2 |
| Protrusio acetabulae | 2 |
| Reduced upper segment:lower segment AND increased arm span:height ratios | 1 |
| Scoliosis or thoracolumbar kyphosis | 1 |
| Reduced elbow extension | 1 |
| 3 of 5 facial features | 1 |
| Skin striae | 1 |
| Myopia | 1 |
| Mitral valve prolapse | 1 |
A score of ≥7 is considered a positive finding.
An interactive score calculator with photos and descriptions of each finding is available at https://www.marfan.org/dx/score
Adapted from Dietz H. Marfan Syndrome. 2017 ed. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 2001.
Loeys-Dietz syndrome
- Clinical presentation: Loeys-Dietz syndrome (LDS) is an autosomal dominant connective tissue disorder associated with diffuse aortic and arterial aneurysm and dissection as well as arterial tortuosity.[26] Other cardiac features of LDS include bicuspid aortic valve (BAV), atrial septal defect, patent ductus arteriosus (PDA), mitral valve prolapse, and left ventricular hypertrophy.[26]
- Common physical exam characteristics include a bifid uvula, cleft palate, and hypertelorism.[27] Skeletal abnormalities overlap with those of MFS and include pectus deformities, scoliosis, and pes planus, although tall stature is less common in LDS. Joint contractures are also frequently seen in LDS.[26]
- In contrast with MFS, some individuals with LDS also exhibit neurocognitive abnormalities.[27]
- Genetics: Mutations in genes encoding TGF-β ligands (TGFB2 and TGFB3) as well as TGF-β receptors (TGFBR1 and TGFBR2) have been associated with LDS.[1, 28] Similar to MFS, these mutations lead not only to weakened integrity of the extracellular matrix and medial degeneration but also to disruptions in TGF-β signaling. Commercial TAAD panels commonly test for these genes.
Clinical course: The vascular phenotype in LDS is often more severe than MFS with individuals suffering aortic and arterial dissections or ruptures at comparatively smaller vessel calibers.[26] Among early described cases, the mean age of death was ~26 years, and aortic dissection or intracerebral hemorrhage can occur even in infancy or childhood.[27, 31, 32]
Familial thoracic aortic aneurysm and dissection
Clinical presentation: Cases of thoracic aortic aneurysm and dissection with a family history of aortic disease but an absence of syndromic features are often classified as familial TAAD. Familial TAAD can be inherited in an autosomal dominant manner, and several genetic mutations have been identified in such cases. Of note, however, up to 70% of families with familial TAAD will have no detectable pathogenic variants upon sequencing, so negative genetic testing does not rule out this condition.[33]
- Genetics: Mutations involving MYH11, which encodes a smooth muscle cell-specific myosin heavy chain, have been implicated in several families with both TAAD and PDA.[34, 35] However, among 93 families with familial TAAD but without PDA, investigators found no MYH11 mutations.[35] Overall, data suggest only ~1% of familial TAAD cases are attributable to MYH11 mutations.[33]
- In contrast, mutations in the gene encoding another smooth muscle cell protein, alpha-actin (ACTA2), are responsible for up to 14% of familial TAAD cases.[36] Some mutations in ACTA2 are also associated with livedo reticularis, PDA, and BAV.[37] Affected individuals are at risk for both type A and type B dissections as well as Moyamoya disease.[37, 38]
- Additional studies have shown that ACTA2 mutation carriers are also at heightened risk for premature stroke and coronary artery disease.[38]
- Penetrance is low for ACTA2 mutations, and only about 50% of carriers will exhibit aortic manifestations.
- Fewer than 5% of familial TAAD cases are attributed to mutations in TGFBR2.[39] Notably, these individuals lack the characteristic craniofacial findings of LDS. Beyond ascending aortic disease, TGFBR2 mutations are also associated with descending aortic disease as well as cerebral, carotid, and popliteal artery aneurysms.[37]
- Mutations in TGFBR1 without features of LDS are also associated with familial TAAD.[40]
Ehlers-Danlos syndrome
- Clinical presentation: Ehlers-Danlos syndrome (EDS) encompasses several connective tissue disorders with multi-system manifestations.
- Classic EDS, also known as types I and II, is characterized by smooth, velvety skin that scars and tears easily, delayed wound healing, wide striae, easy bruising, and hypermobile joints. Although rare, up to 6% of individuals with classic EDS develop aortic root dilatation.[41] However, frank aortic aneurysm and dissection are rare.[42] Mutations in COL5A1 and COL5A2 are frequently identified in classic EDS.[43]
- Hypermobile EDS, or type III, features widespread joint laxity along with varying degrees of musculoskeletal pain.[44] Skin features are similar to that of classic EDS, although typically less pronounced. Mild aortic root dilatation is rarely seen as is mitral valve prolapse.[44, 42] No clear pathologic variants have been identified in hypermobile EDS.
- In contrast, vascular, or type IV, EDS is characterized by a severe vascular phenotype potentially involving aneurysm or dissection of the aorta (Figure 1), visceral, extracranial, or limb arteries and spontaneous carotid-cavernous sinus fistulas.[45–47] Other adverse outcomes include spontaneous intestinal or uterine rupture leading to emergency surgery or death.[45]
- In addition to the characteristics of classic EDS, additional features include a thin, pinched nose, hollow cheeks, prominent eyes, spontaneous tendon or muscle rupture, premature varicose veins, and clubfoot.[45]
- Genetics: Mutations in the gene encoding type III collagen, COL3A1, have been identified as the cause of vascular EDS.[48] Type III collagen is a major component of the extracellular matrix within the tunica media and tunica adventitia, and these mutations lead to disruption of the extracellular matrix and weakening of the vessel wall.
- Interestingly, mutations leading to minimal production of type III collagen, in contrast to mutations leading to haploinsufficiency, are associated with a lower risk of aortic disease but a greater risk of visceral artery manifestations.[49] Haploinsufficiency mutations are also associated with a younger age of first presentation. COL3A1 null mutations, as opposed to splice site or missense mutations, are also associated with longer survival.[50]
Clinical course: Nearly 25% of individuals with vascular EDS suffer a major complication by age 20 with more than 80% affected by age 40.[51]
Figure 1:
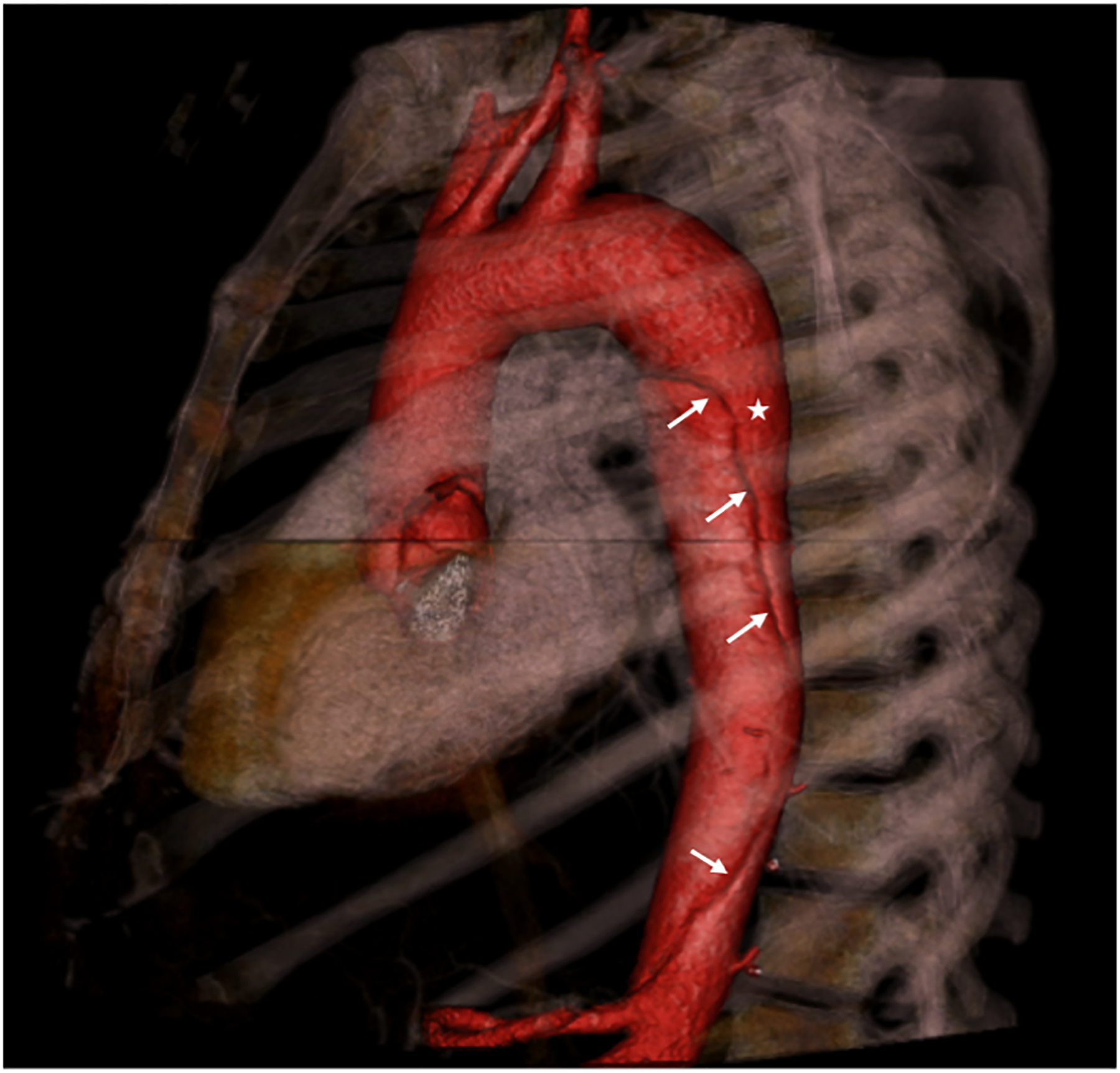
Sagittal 3-dimensional computer tomographic (CT) image of a type B aortic dissection in a patient with type IV Ehlers-Danlos syndrome. The aorta and its branches are colored red. The dissection flap is clearly visualized (arrows) with the true lumen marked by an asterisk.
Congenital contractural arachnodactyly
- Clinical presentation: Congenital contractural arachnodactyly (CCA), or Beals-Hecht syndrome, is an autosomal dominant connective tissue disorder characterized by arachnodactyly, scoliosis, upper and lower extremity joint contractures, muscle hypoplasia, pectus carinatum, and crumpled-appearing ears.[52] Classically, although it shares many skeletal features with MFS, patients with CCA typically lacked cardiovascular abnormalities.
Genetics: Presumed pathogenic mutations in FBN2 have been identified in several individuals diagnosed with CCA. [52–54] However, sequencing of FBN2 in other probands has been unrevealing, suggesting other genes may be involved in this disease.
Shprintzen-Goldberg Syndrome
Clinical presentation: Shprintzen-Goldberg Syndrome is a connective tissue disorder with features that overlap with both MFS and LDS. This disorder is characterized by both ascending aortic aneurysm as well as skeletal abnormalities, including chest wall deformities, arachnodactyly, and cleft palate.[1]
- Genetics: The mutation responsible for this disorder has been mapped to SKI, a known repressor of TGF-β signaling.[55]
- Cultured fibroblasts from patients with Shprintzen-Goldberg Syndrome show markers of upregulated TGF-β signaling, and experimental knockdown of SKI paralogs in zebrafish leads to a similar craniofacial phenotype along with severe cardiac developmental abnormalities, thus further supporting the role of SKI in normal TGF-β signaling.[55]
Arterial tortuosity syndrome
- Clinical presentation: Arterial tortuosity syndrome is an autosomal recessive, non-inflammatory disease that is characterized by diffuse tortuosity, elongation, stenosis, and aneurysm of large and medium arteries, including the aorta, with an associated risk of dissection.[56, 57]
- Children with arterial tortuosity syndrome often present with dyspnea and cyanosis due to involvement of the pulmonary arteries.[58] Pulmonary artery stenosis occurs in approximately 60% of affected individuals with pulmonary hypertension, right ventricular hypertrophy, and right ventricular dysfunction representing significant causes of morbidity and mortality.[56] Individuals may also present with acute stroke as a consequence of arterial dissection.[56] Although individuals may present in adulthood, some of the most severe complications occur in childhood.
- Additional clinical features include soft, velvety, and hyperextensible skin, facial dysmorphisms, abdominal hernias, and joint hypermobility.[58]
Genetics: Mutations in SLC2A10, which encodes the glucose transporter GLUT10, have been implicated in arterial tortuosity syndrome.[57] Disruption of SLC2A10 leads to upregulation of TGF-β signaling within the arterial wall, similar to that seen in LDS. Of note, glucose metabolism is unaffected in these individuals.[56]
Turner Syndrome
Epidemiology: Turner Syndrome is the most common chromosomal disorder in women with a prevalence of 1:2,5000–3,000.[59, 60]
- Clinical presentation: Typical physical exam findings include short stature, congenital lymphedema, shield chest, webbed neck, and low-set ears. Amenorrhea, infertility, and delayed or absent breast development are common due to abnormal gonadal development. Other features of the syndrome include learning disabilities, renal malformations, and hypothyroidism.[60, 59]
Genetics: Turner syndrome is caused by an imbalance of X chromosomes. The bulk of cases are due to X chromosome monosomy, or complete absence of an X chromosome (45,X), with other cases attributed to duplication of the long arm of one X chromosome or mosaicism of 45,X.[60] In adults, it is typically diagnosed by karyotype.
Additional Vascular Disorders with a Genetic Contribution
Bicuspid aortic valve disease
Epidemiology: Sporadic BAV disease is the most common congenital heart disease, affecting up to 2% of the general population.[64, 65]
- Clinical presentation: In addition to aortic valvular stenosis and insufficiency, up to 75% of individuals with BAV will develop ascending aorta dilatation or aneurysm. In contrast to MFS, aneurysms in BAV typically spare the aortic root and STJ and tend to involve the proximal ascending aorta. These individuals are also at high risk of type A aortic dissection, and the risk of aneurysm is independent of valvular dysfunction.[66, 67]
- Family studies have shown a high heritability for BAV.[68] Additionally, individuals with a first-degree family member who has BAV have a ten-folder greater risk of also having BAV compared to the general population.[64] However, individual genes involved in non-syndromic BAV pathogenesis have remained elusive.
- Genetics: Mutations in NOTCH1 are associated with BAV and severe calcific valvular disease, although individuals in this study lacked aortic involvement.[69] A GWAS of 1,792 individuals with BAV identified a variant in a non-coding region near GATA4.[70] FBN1 mutations were also identified in two individuals with BAV and features of MFS.[71]
- Nonetheless, routine genetic testing in individuals with BAV is not warranted, and the heritability of BAV disease should instead be addressed by recommending screening echocardiograms in first-degree relatives of affected individuals [Class I, Level of Evidence C].[4]
Abdominal Aortic Aneurysm
- Epidemiology: Abdominal aortic aneurysm (AAA) is defined as a 50% increase in caliber relative to the adjacent normal aortic segment.
- Genetics: Despite the apparent role of genetics in AAA pathogenesis, no single gene has been implicated. GWAS and candidate gene analyses have found associations with AAA for genes involved in lipid pathways (LDLR and SORT1), vascular integrity (LRP1), and inflammation (IL6R).[78–81]
- However, most cases of AAA are not part of a cardiovascular syndrome, and routine genetic testing in such patients is not warranted.
Fibromuscular Dysplasia
- Epidemiology: Fibromuscular dysplasia (FMD) is a non-atherosclerotic, non-inflammatory vascular disease characterized by arterial tortuosity, stenosis, occlusion, aneurysm, or dissection.[82]
- FMD primarily affects women with a mean age of ~52, although the age range among 447 individuals in the U.S. registry is 5–83.[83]
- Clinical presentation: Commonly affected vessels include the carotid, vertebral, and renal arteries, although the coronary, visceral, and limb arteries may be less frequently involved.
- Depending on the vascular bed involved, patients may present with stroke, transient ischemia attack, pulsatile tinnitus, headache, myocardial infarction, resistant hypertension, or sudden onset flank or abdominal pain.
Genetics: 7.3% of individuals with FMD report a family member also diagnosed with FMD, and 23.5% of FMD patients report a family history of arterial aneurysm.[83] However, findings showing a specific genetic association are limited. Recently, a GWAS of 651 FMD patients found an association with an intronic allele in PHACTR1, which has previously been associated with coronary artery disease, migraine, and cervical artery dissection.[84] Additional studies are ongoing.
Spontaneous Visceral Artery Dissection
- Clinical presentation: Isolated spontaneous dissection of the celiac or superior mesenteric artery is a rare but potentially fatal vascular disease.
- Individuals often present with sudden onset abdominal or back pain, although this may also be detected incidentally on axial imaging studies.
Genetics: Familial cases are rare. Although it is reasonable to pursue genetic testing to rule out vascular EDS, panel testing is typically unrevealing in our experience, and to our knowledge, there are no published reports documenting a genetic predisposition.
Clinical course: Although the initial presentation can be complicated by bowel ischemia, hemorrhagic shock, or death, most individuals can be successfully managed without surgical intervention and without recurrence of symptoms.[85]
Conclusions
With the growing utilization of diagnostic imaging, we are increasingly detecting new cases of both syndromic and non-syndromic vascular diseases. Although researchers have made great strides in explaining the genetic and molecular basis for some of these diseases, our ability to act on this information in a meaningful and responsible way often lags, much to the disappointment of patients and providers alike. Greater access to genetic testing is only likely to worsen this frustration. We hope this review and others like it will provide clinicians a foundation for genetic testing in patients with vascular disease as well as a reference for important diseases and syndromes that will increasingly become part of the panoply of conditions we are expected to recognize and manage.
Sources of Funding
This work was supported by NIH T32 HL007575 and K12 HL133117 (AA).
Footnotes
Disclosures
No potential conflicts of interest relevant to this article were reported.
References
- 1.Lindsay ME, Dietz HC. The genetic basis of aortic aneurysm. Cold Spring Harbor perspectives in medicine. 2014;4(9):a015909. doi: 10.1101/cshperspect.a015909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ma WG, Chou AS, Mok SCM, Ziganshin BA, Charilaou P, Zafar MA et al. Positive family history of aortic dissection dramatically increases dissection risk in family members. International journal of cardiology. 2017;240:132–7. doi: 10.1016/j.ijcard.2017.04.080. [DOI] [PubMed] [Google Scholar]
- 3.Pyeritz RE. The family history: the first genetic test, and still useful after all those years? Genetics In Medicine. 2011;14:3. doi: 10.1038/gim.0b013e3182310bcf. [DOI] [PubMed] [Google Scholar]
- 4.Hiratzka LF, Bakris GL, Beckman JA, Bersin RM, Carr VF et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the Diagnosis and Management of Patients With Thoracic Aortic Disease: A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation. 2010;121(13):e266–e369. doi: 10.1161/CIR.0b013e3181d4739e. [DOI] [PubMed] [Google Scholar]
- 5.Rehm HL. Disease-targeted sequencing: a cornerstone in the clinic. Nature reviews Genetics. 2013;14(4):295–300. doi: 10.1038/nrg3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arndt A-K, MacRae CA. Genetic testing in cardiovascular diseases. Current opinion in cardiology. 2014;29(3):235–40. doi: 10.1097/HCO.0000000000000055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campens L, Callewaert B, Muiño Mosquera L, Renard M, Symoens S, De Paepe A et al. Gene panel sequencing in heritable thoracic aortic disorders and related entities – results of comprehensive testing in a cohort of 264 patients. Orphanet Journal of Rare Diseases. 2015;10:9. doi: 10.1186/s13023-014-0221-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ziganshin BA, Bailey AE, Coons C, Dykas D, Charilaou P, Tanriverdi LH et al. Routine Genetic Testing for Thoracic Aortic Aneurysm and Dissection in a Clinical Setting. Ann Thorac Surg. 2015;100(5):1604–11. doi: 10.1016/j.athoracsur.2015.04.106. [DOI] [PubMed] [Google Scholar]
- 9.Proost D, Vandeweyer G, Meester JA, Salemink S, Kempers M, Ingram C et al. Performant Mutation Identification Using Targeted Next-Generation Sequencing of 14 Thoracic Aortic Aneurysm Genes. Human mutation. 2015;36(8):808–14. doi: 10.1002/humu.22802. [DOI] [PubMed] [Google Scholar]
- 10.Groth KA, Hove H, Kyhl K, Folkestad L, Gaustadnes M, Vejlstrup N et al. Prevalence, incidence, and age at diagnosis in Marfan Syndrome. Orphanet Journal of Rare Diseases. 2015;10:153. doi: 10.1186/s13023-015-0369-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Devereux RB, de Simone G, Arnett DK, Best LG, Boerwinkle E, Howard BV et al. Normal limits in relation to age, body size and gender of two-dimensional echocardiographic aortic root dimensions in persons >/=15 years of age. Am J Cardiol. 2012;110(8):1189–94. doi: 10.1016/j.amjcard.2012.05.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.den Hartog AW, Franken R, Zwinderman AH, Timmermans J, Scholte AJ, van den Berg MP et al. The Risk for Type B Aortic Dissection in Marfan Syndrome. Journal of the American College of Cardiology. 2015;65(3):246–54. doi: 10.1016/j.jacc.2014.10.050. [DOI] [PubMed] [Google Scholar]; • Provides a longitudinal assessment of patients with Marfan syndrome and documents a high rate of type B dissection among these individuals, particularly in those with a previous history of prophylactic aortic surgery.
- 13.Lundby R, Rand-Hendriksen S, Hald JK, Pripp AH, Smith HJ. The pulmonary artery in patients with Marfan syndrome: a cross-sectional study. Genetics in medicine : official journal of the American College of Medical Genetics. 2012;14(11):922–7. doi: 10.1038/gim.2012.82. [DOI] [PubMed] [Google Scholar]
- 14.Pati PK, George PV, Jose JV. Giant Pulmonary Artery Aneurysm With Dissection in a Case of Marfan Syndrome. Journal of the American College of Cardiology. 2013;61:685. [DOI] [PubMed] [Google Scholar]
- 15.Pepe G, Giusti B, Sticchi E, Abbate R, Gensini GF, Nistri S. Marfan syndrome: current perspectives. The application of clinical genetics. 2016;9:55–65. doi: 10.2147/TACG.S96233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dietz H. Marfan Syndrome 2017 ed. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 2001. [Google Scholar]; •• This review provides a detailed description of the clinical manifestations of Marfan syndrome as well as its diagnosis and management.
- 17.Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352(6333):337–9. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- 18.Pyeritz RE. Etiology and pathogenesis of the Marfan syndrome: current understanding. Ann Cardiothorac Surg. 2017;6(6):595–8. doi: 10.21037/acs.2017.10.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grewal N, Gittenberger-de Groot AC. Pathogenesis of aortic wall complications in Marfan syndrome. Cardiovascular Pathology. 2018;33:62–9. doi: 10.1016/j.carpath.2018.01.005. [DOI] [PubMed] [Google Scholar]
- 20.Holm TM, Habashi JP, Doyle JJ, Bedja D, Chen Y, Van Erp C et al. Noncanonical TGF Signaling Contributes to Aortic Aneurysm Progression in Marfan Syndrome Mice. Science (New York, NY). 2011;332(6027):358–61. doi: 10.1126/science.1192149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312(5770):117–21. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB et al. The revised Ghent nosology for the Marfan syndrome. Journal of medical genetics. 2010;47(7):476–85. doi: 10.1136/jmg.2009.072785. [DOI] [PubMed] [Google Scholar]
- 23.Silverman DI, Burton KJ, Gray J, Bosner MS, Kouchoukos NT, Roman MJ et al. Life expectancy in the Marfan syndrome. Am J Cardiol. 1995;75(2):157–60. [DOI] [PubMed] [Google Scholar]
- 24.Faivre L, Collod-Beroud G, Loeys, Child A, Binquet C, Gautier E et al. Effect of Mutation Type and Location on Clinical Outcome in 1,013 Probands with Marfan Syndrome or Related Phenotypes and FBN1 Mutations: An International Study. American Journal of Human Genetics. 2007;81(3):454–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Franken R, den Hartog AW, Radonic T, Micha D, Maugeri A, van Dijk FS et al. Beneficial Outcome of Losartan Therapy Depends on Type of FBN1 Mutation in Marfan Syndrome. Circulation Cardiovascular genetics. 2015;8(2):383–8. doi: 10.1161/circgenetics.114.000950. [DOI] [PubMed] [Google Scholar]
- 26.MacCarrick G, Black JH, Bowdin S, El-Hamamsy I, Frischmeyer-Guerrerio PA, Guerrerio AL et al. Loeys–Dietz syndrome: a primer for diagnosis and management. Genetics in Medicine. 2014;16(8):576–87. doi: 10.1038/gim.2014.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loeys BL, Schwarze U, Holm T, Callewaert BL, Thomas GH, Pannu H et al. Aneurysm Syndromes Caused by Mutations in the TGF-β Receptor. New England Journal of Medicine. 2006;355(8):788–98. doi: 10.1056/NEJMoa055695. [DOI] [PubMed] [Google Scholar]
- 28.Bertoli-Avella AM, Gillis E, Morisaki H, Verhagen JMA, de Graaf BM, van de Beek G et al. Mutations in a TGF-beta ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J Am Coll Cardiol. 2015;65(13):1324–36. doi: 10.1016/j.jacc.2015.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van de Laar IM, van der Linde D, Oei EH, Bos PK, Bessems JH, Bierma-Zeinstra SM et al. Phenotypic spectrum of the SMAD3-related aneurysms-osteoarthritis syndrome. Journal of medical genetics. 2012;49(1):47–57. doi: 10.1136/jmedgenet-2011-100382. [DOI] [PubMed] [Google Scholar]
- 30.van de Laar IM, Oldenburg RA, Pals G, Roos-Hesselink JW, de Graaf BM, Verhagen JM et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet. 2011;43(2):121–6. doi: 10.1038/ng.744. [DOI] [PubMed] [Google Scholar]
- 31.Malhotra A, Westesson PL. Loeys-Dietz syndrome. Pediatr Radiol. 2009;39(9):1015. doi: 10.1007/s00247-009-1252-3. [DOI] [PubMed] [Google Scholar]
- 32.Williams JA, Loeys BL, Nwakanma LU, Dietz HC, Spevak PJ, Patel ND et al. Early surgical experience with Loeys-Dietz: a new syndrome of aggressive thoracic aortic aneurysm disease. Ann Thorac Surg. 2007;83(2):S757–63; discussion S85–90. doi: 10.1016/j.athoracsur.2006.10.091. [DOI] [PubMed] [Google Scholar]
- 33.Milewicz DM, Regalado E. Heritable Thoracic Aortic Disease Overview GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 2003. [Google Scholar]
- 34.Khau Van Kien P, Mathieu F, Zhu L, Lalande A, Betard C, Lathrop M et al. Mapping of familial thoracic aortic aneurysm/dissection with patent ductus arteriosus to 16p12.2-p13.13. Circulation. 2005;112(2):200–6. doi: 10.1161/circulationaha.104.506345. [DOI] [PubMed] [Google Scholar]
- 35.Pannu H, Tran-Fadulu V, Papke CL, Scherer S, Liu Y, Presley C et al. MYH11 mutations result in a distinct vascular pathology driven by insulin-like growth factor 1 and angiotensin II. Hum Mol Genet. 2007;16(20):2453–62. doi: 10.1093/hmg/ddm201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guo DC, Pannu H, Tran-Fadulu V, Papke CL, Yu RK, Avidan N et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39(12):1488–93. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- 37.Milewicz DM, Carlson AA, Regalado ES. Genes Predisposing to Thoracic Aortic Aneurysms and Dissections: Associated Phenotypes, Gene-Specific Management, and Genetic Testing. Cardiology clinics. 2010;28(2):191–7. doi: 10.1016/j.ccl.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo DC, Papke CL, Tran-Fadulu V, Regalado ES, Avidan N, Johnson RJ et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am J Hum Genet. 2009;84(5):617–27. doi: 10.1016/j.ajhg.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pannu H, Fadulu VT, Chang J, Lafont A, Hasham SN, Sparks E et al. Mutations in transforming growth factor-beta receptor type II cause familial thoracic aortic aneurysms and dissections. Circulation. 2005;112(4):513–20. doi: 10.1161/circulationaha.105.537340. [DOI] [PubMed] [Google Scholar]
- 40.Tran-Fadulu V, Pannu H, Kim DH, Vick GW 3rd, Lonsford CM, Lafont AL et al. Analysis of multigenerational families with thoracic aortic aneurysms and dissections due to TGFBR1 or TGFBR2 mutations. Journal of medical genetics. 2009;46(9):607–13. doi: 10.1136/jmg.2008.062844. [DOI] [PubMed] [Google Scholar]
- 41.Wenstrup RJ, Meyer RA, Lyle JS, Hoechstetter L, Rose PS, Levy HP et al. Prevalence of aortic root dilation in the Ehlers-Danlos syndrome. Genetics in medicine : official journal of the American College of Medical Genetics. 2002;4(3):112–7. doi: 10.1097/00125817-200205000-00003. [DOI] [PubMed] [Google Scholar]
- 42.Atzinger CL, Meyer RA, Khoury PR, Gao Z, Tinkle BT. Cross-sectional and longitudinal assessment of aortic root dilation and valvular anomalies in hypermobile and classic Ehlers-Danlos syndrome. J Pediatr. 2011;158(5):826–30.e1. doi: 10.1016/j.jpeds.2010.11.023. [DOI] [PubMed] [Google Scholar]
- 43.Malfait F, Coucke P, Symoens S, Loeys B, Nuytinck L, De Paepe A. The molecular basis of classic Ehlers-Danlos syndrome: a comprehensive study of biochemical and molecular findings in 48 unrelated patients. Human mutation. 2005;25(1):28–37. doi: 10.1002/humu.20107. [DOI] [PubMed] [Google Scholar]
- 44.Tinkle B, Castori M, Berglund B, Cohen H, Grahame R, Kazkaz H et al. Hypermobile Ehlers-Danlos syndrome (a.k.a. Ehlers-Danlos syndrome Type III and Ehlers-Danlos syndrome hypermobility type): Clinical description and natural history. Am J Med Genet C Semin Med Genet. 2017;175(1):48–69. doi: 10.1002/ajmg.c.31538. [DOI] [PubMed] [Google Scholar]
- 45.Oderich GS, Panneton JM, Bower TC, Lindor NM, Cherry KJ, Noel AA et al. The spectrum, management and clinical outcome of Ehlers-Danlos syndrome type IV: a 30-year experience. Journal of Vascular Surgery. 2005;42(1):98–106. doi: 10.1016/j.jvs.2005.03.053. [DOI] [PubMed] [Google Scholar]
- 46.North KN, Whiteman DA, Pepin MG, Byers PH. Cerebrovascular complications in Ehlers-Danlos syndrome type IV. Annals of neurology. 1995;38(6):960–4. doi: 10.1002/ana.410380620. [DOI] [PubMed] [Google Scholar]
- 47.Parfitt J, Chalmers RT, Wolfe JH. Visceral aneurysms in Ehlers-Danlos syndrome: case report and review of the literature. Journal of Vascular Surgery. 2000;31(6):1248–51. doi: 10.1067/mva.2000.105667. [DOI] [PubMed] [Google Scholar]
- 48.Frank M, Albuisson J, Ranque B, Golmard L, Mazzella JM, Bal-Theoleyre L et al. The type of variants at the COL3A1 gene associates with the phenotype and severity of vascular Ehlers–Danlos syndrome. European Journal of Human Genetics. 2015;23(12):1657–64. doi: 10.1038/ejhg.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shalhub S, Black JH, Cecchi AC, Xu Z, Griswold BF, Safi HJ et al. Molecular diagnosis in vascular Ehlers-Danlos syndrome predicts pattern of arterial involvement and outcomes. Journal of Vascular Surgery. 2014;60(1):160–9. doi: 10.1016/j.jvs.2014.01.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pepin MG, Schwarze U, Rice KM, Liu M, Leistritz D, Byers PH. Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV). Genetics in Medicine. 2014;16(12):881–8. doi: 10.1038/gim.2014.72. [DOI] [PubMed] [Google Scholar]; • This study showed that among patients with vascular Ehlers-Danlos syndrome, COL3A1 null mutations are associated with longer survival compared to other mutations.
- 51.Pepin M, Schwarze U, Superti-Furga A, Byers PH. Clinical and Genetic Features of Ehlers–Danlos Syndrome Type IV, the Vascular Type. New England Journal of Medicine. 2000;342(10):673–80. doi: 10.1056/nejm200003093421001. [DOI] [PubMed] [Google Scholar]
- 52.Callewaert BL, Loeys BL, Ficcadenti A, Vermeer S, Landgren M, Kroes HY et al. Comprehensive clinical and molecular assessment of 32 probands with congenital contractural arachnodactyly: report of 14 novel mutations and review of the literature. Human mutation. 2009;30(3):334–41. doi: 10.1002/humu.20854. [DOI] [PubMed] [Google Scholar]
- 53.Gupta PA, Putnam EA, Carmical SG, Kaitila I, Steinmann B, Child A et al. Ten novel FBN2 mutations in congenital contractural arachnodactyly: delineation of the molecular pathogenesis and clinical phenotype. Human mutation. 2002;19(1):39–48. doi: 10.1002/humu.10017. [DOI] [PubMed] [Google Scholar]
- 54.Nishimura A, Sakai H, Ikegawa S, Kitoh H, Haga N, Ishikiriyama S et al. FBN2, FBN1, TGFBR1, and TGFBR2 analyses in congenital contractural arachnodactyly. American journal of medical genetics Part A. 2007;143a(7):694–8. doi: 10.1002/ajmg.a.31639. [DOI] [PubMed] [Google Scholar]
- 55.Doyle AJ, Doyle JJ, Bessling SL, Maragh S, Lindsay ME, Schepers D et al. Mutations in the TGF-beta repressor SKI cause Shprintzen-Goldberg syndrome with aortic aneurysm. Nat Genet. 2012;44(11):1249–54. doi: 10.1038/ng.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Callewaert BL, Willaert A, Kerstjens-Frederikse WS, De Backer J, Devriendt K, Albrecht B et al. Arterial tortuosity syndrome: clinical and molecular findings in 12 newly identified families. Human mutation. 2008;29(1):150–8. doi: 10.1002/humu.20623. [DOI] [PubMed] [Google Scholar]
- 57.Coucke PJ, Willaert A, Wessels MW, Callewaert B, Zoppi N, De Backer J et al. Mutations in the facilitative glucose transporter GLUT10 alter angiogenesis and cause arterial tortuosity syndrome. Nat Genet. 2006;38(4):452–7. doi: 10.1038/ng1764. [DOI] [PubMed] [Google Scholar]
- 58.Ritelli M, Chiarelli N, Dordoni C, Reffo E, Venturini M, Quinzani S et al. Arterial Tortuosity Syndrome: homozygosity for two novel and one recurrent SLC2A10 missense mutations in three families with severe cardiopulmonary complications in infancy and a literature review. BMC Med Genet. 2014;15:122. doi: 10.1186/s12881-014-0122-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Elsheikh M, Dunger DB, Conway GS, Wass JA. Turner’s syndrome in adulthood. Endocr Rev. 2002;23(1):120–40. doi: 10.1210/edrv.23.1.0457. [DOI] [PubMed] [Google Scholar]
- 60.Sybert VP, McCauley E. Turner’s Syndrome. New England Journal of Medicine. 2004;351(12):1227–38. doi: 10.1056/NEJMra030360. [DOI] [PubMed] [Google Scholar]
- 61.Gøtzsche CO, Krag-Olsen B, Nielsen J, Sørensen KE, Kristensen BO. Prevalence of cardiovascular malformations and association with karyotypes in Turner’s syndrome. Archives of Disease in Childhood. 1994;71:433–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bondy CA. Aortic Dissection in Turner Syndrome. Current opinion in cardiology. 2008;23(6):519–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Matura LA, Ho VB, Rosing DR, Bondy CA. Aortic dilatation and dissection in Turner syndrome. Circulation. 2007;116(15):1663–70. doi: 10.1161/circulationaha.106.685487. [DOI] [PubMed] [Google Scholar]
- 64.Siu SC, Silversides CK. Bicuspid aortic valve disease. J Am Coll Cardiol. 2010;55(25):2789–800. doi: 10.1016/j.jacc.2009.12.068. [DOI] [PubMed] [Google Scholar]
- 65.Braverman AC, Guven H, Beardslee MA, Makan M, Kates AM, Moon MR. The bicuspid aortic valve. Current problems in cardiology. 2005;30(9):470–522. doi: 10.1016/j.cpcardiol.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 66.Tadros TM, Klein MD, Shapira OM. Ascending aortic dilatation associated with bicuspid aortic valve: pathophysiology, molecular biology, and clinical implications. Circulation. 2009;119(6):880–90. doi: 10.1161/circulationaha.108.795401. [DOI] [PubMed] [Google Scholar]
- 67.Freeze SL, Landis BJ, Ware SM, Helm BM. Bicuspid Aortic Valve: a Review with Recommendations for Genetic Counseling. Journal of genetic counseling. 2016;25(6):1171–8. doi: 10.1007/s10897-016-0002-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Calloway TJ, Martin LJ, Zhang X, Tandon A, Benson DW, Hinton RB. Risk factors for aortic valve disease in bicuspid aortic valve: a family-based study. American journal of medical genetics Part A. 2011;155a(5):1015–20. doi: 10.1002/ajmg.a.33974. [DOI] [PubMed] [Google Scholar]
- 69.Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN et al. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437(7056):270–4. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- 70.Yang B, Zhou W, Jiao J, Nielsen JB, Mathis MR, Heydarpour M et al. Protein-altering and regulatory genetic variants near GATA4 implicated in bicuspid aortic valve. Nature communications. 2017;8:15481. doi: 10.1038/ncomms15481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pepe G, Nistri S, Giusti B, Sticchi E, Attanasio M, Porciani C et al. Identification of fibrillin 1 gene mutations in patients with bicuspid aortic valve (BAV) without Marfan syndrome. BMC Med Genet. 2014;15:23. doi: 10.1186/1471-2350-15-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Darling RC, Messina CR, Brewster DC, Ottinger LW. Autopsy study of unoperated abdominal aortic aneurysms. The case for early resection. Circulation. 1977;56(3 Suppl):Ii161–4. [PubMed] [Google Scholar]
- 73.Lederle FA, Johnson GR, Wilson SE, Chute EP, Hye RJ, Makaroun MS et al. The aneurysm detection and management study screening program: validation cohort and final results. Aneurysm Detection and Management Veterans Affairs Cooperative Study Investigators. Arch Intern Med. 2000;160(10):1425–30. [DOI] [PubMed] [Google Scholar]
- 74.Kent KC. Abdominal Aortic Aneurysms. New England Journal of Medicine. 2014;371(22):2101–8. doi: 10.1056/NEJMcp1401430. [DOI] [PubMed] [Google Scholar]
- 75.Baird PA, Sadovnick AD, Yee IM, Cole CW, Cole L. Sibling risks of abdominal aortic aneurysm. Lancet. 1995;346(8975):601–4. [DOI] [PubMed] [Google Scholar]
- 76.Blanchard JF, Armenian HK, Friesen PP. Risk factors for abdominal aortic aneurysm: results of a case-control study. Am J Epidemiol. 2000;151(6):575–83. [DOI] [PubMed] [Google Scholar]
- 77.Verloes A, Sakalihasan N, Koulischer L, Limet R. Aneurysms of the abdominal aorta: familial and genetic aspects in three hundred thirteen pedigrees. J Vasc Surg. 1995;21(4):646–55. [DOI] [PubMed] [Google Scholar]
- 78.Bown MJ, Jones GT, Harrison SC, Wright BJ, Bumpstead S, Baas AF et al. Abdominal aortic aneurysm is associated with a variant in low-density lipoprotein receptor-related protein 1. Am J Hum Genet. 2011;89(5):619–27. doi: 10.1016/j.ajhg.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jones GT, Bown MJ, Gretarsdottir S, Romaine SP, Helgadottir A, Yu G et al. A sequence variant associated with sortilin-1 (SORT1) on 1p13.3 is independently associated with abdominal aortic aneurysm. Hum Mol Genet. 2013;22(14):2941–7. doi: 10.1093/hmg/ddt141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Boucher P, Gotthardt M, Li WP, Anderson RG, Herz J. LRP: role in vascular wall integrity and protection from atherosclerosis. Science. 2003;300(5617):329–32. doi: 10.1126/science.1082095. [DOI] [PubMed] [Google Scholar]
- 81.Harrison SC, Smith AJ, Jones GT, Swerdlow DI, Rampuri R, Bown MJ et al. Interleukin-6 receptor pathways in abdominal aortic aneurysm. Eur Heart J. 2013;34(48):3707–16. doi: 10.1093/eurheartj/ehs354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Olin JW, Gornik HL, Bacharach JM, Biller J, Fine LJ, Gray BH et al. Fibromuscular Dysplasia: State of the Science and Critical Unanswered Questions. Circulation. 2014;129:1048–78. [DOI] [PubMed] [Google Scholar]
- 83.Olin JW, Froehlich J, Gu X, Bacharach JM, Eagle K, Gray BH et al. The United States Registry for Fibromuscular Dysplasia: results in the first 447 patients. Circulation. 2012;125:3182–90. [DOI] [PubMed] [Google Scholar]
- 84.Kiando SR, Tucker NR, Castro-Vega L-J, Katz A, D’Escamard V, Tréard C et al. PHACTR1 Is a Genetic Susceptibility Locus for Fibromuscular Dysplasia Supporting Its Complex Genetic Pattern of Inheritance. PLOS Genetics. 2016;12(10):e1006367. doi: 10.1371/journal.pgen.1006367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.DeCarlo C, Ganguli S, Borges JC, Schainfeld RM, Mintz AJ, Mintz J et al. Presentation, treatment, and outcomes in patients with spontaneous isolated celiac and superior mesenteric artery dissection. Vascular Medicine. 2017;22(6):505–11. doi: 10.1177/1358863X17729770. [DOI] [PubMed] [Google Scholar]
- 86.Tanaka Y, Yoshimuta T, Kimura K, Iino K, Tamura Y, Sakata K et al. Clinical characteristics of spontaneous isolated visceral artery dissection. J Vasc Surg. 2018;67(4):1127–33. doi: 10.1016/j.jvs.2017.08.054. [DOI] [PubMed] [Google Scholar]