

Abstract
Obesity is a critical risk factor causing the development of metabolic diseases and cancers. Its increasing prevalence worldwide has aroused great concerns of the researchers on adipose development and metabolic function. During adipose expansion, adipogenesis is a way to store lipids as well as to avoid lipotoxicity in other tissues, and may be an approach to offset the negative metabolic effects of obesity. In this Review, the transcriptional regulation of adipogenesis is outlined to characterize numerous biological processes in research on the determination of adipocyte fate and regulation of adipogenic differentiation. Notably, one of the post‐transcriptional modifications of mRNA, namely, N6‐methyladenosine (m6A), has been recently found to play a role in adipogenesis. Here, the roles of m6A‐related enzymes and proteins in adipogenesis, with a particular focus on how these m6A‐related proteins function at different stages of adipogenesis, are mainly discussed. The Review also highlights the coordination role of the transcriptional and post‐transcriptional (RNA m6A methylation) regulation in adipogenesis and related biological processes. In this context, a better understanding of adipogenesis at both the transcriptional and post‐transcriptional levels may facilitate the development of novel strategies to improve metabolic health in obesity.
Keywords: adipogenesis, obesity, RNA m6A modification, transcription factors
This work provides an overview on the roles of m6A‐related enzymes and proteins in adipogenesis at different stages, and highlights the coordinating effect of transcriptional and post‐transcriptional (RNA m6A methylation) regulation on adipogenesis. A better understanding of adipogenesis at both the transcriptional and the post‐transcriptional levels may facilitate the development of novel strategies to fight against obesity.
1. Introduction
The global epidemic of obesity has always been increasing for the past 30 years. As judged by the body mass index, nearly 40% of the adult population are overweight and about 13% are deemed as obese.[ 1 ] It has been well established that obesity is correlated with a wide range of comorbidities such as insulin resistance and type 2 diabetes mellitus, nonalcoholic fatty liver, and dyslipidemia.[ 2 ] Hence, the epidemic of obesity highlights the importance of elucidating the mechanism of adipose development, particularly adipogenesis.
Over the past three decades, the mechanisms of adipogenesis have been extensively studied, and adipogenesis has been found to be regulated by a complex transcriptional and epigenetic cascade.[ 3 ] The “‘master regulator”’ of adipogenesis is peroxisome proliferator‐activated receptor (PPARγ), which is both necessary and sufficient for fat cell formation.[ 4 ] PPARγ and CCAAT‐enhancer‐binding proteins (C/EBPα) work together with other transcription factors to activate the target genes required for terminal adipogenic differentiation. More and more key transcription factors have been identified to regulate adipogenesis both in vivo and in vitro.[ 5 ] A series of excellent Reviews have provided details on the transcriptional regulation of adipogenesis.[ 6 , 7 , 8 ] Notably, it has been shown that mRNA N6‐methyladenosine (m6A) modification functions as a novel and critical post‐transcriptional modulator of gene expression, which may be a new perspective to further and better understand adipogenesis.[ 9 , 10 , 11 ]
This Review aims to outline the mechanisms of adipogenesis under transcriptional and RNA N6‐methyladenosine‐mediated post‐transcriptional regulation. Special emphasis will be placed on the novel role of mRNA m6A modification in adipogenesis and important proteins related to post‐transcriptional regulation based on recent advances in the understanding of m6A. Finally, the Review will also discuss the potential of therapeutically targeting the activity of m6A‐related enzymes for the reduction of obesity.
2. Physiology of Adipose Tissues
Adipose tissues are crucial for systemic metabolic homeostasis. Originated from different subcompartments of the mesoderm, adipose tissues have various distributions mainly in subcutaneous and visceral depots in the body,[ 12 , 13 ] and exert distinct functions due to different compositions of fat cell types, namely, white, brown, and beige adipocytes. White adipocytes, which make up the bulk of white adipose tissue (WAT), are capable of storing chemical energy formatted as unilocular lipid droplets.[ 13 ] By contrast, brown and beige adipocytes are characterized by the capability of dissipating energy to generate heat.[ 14 , 15 ] Characteristically, these adipocytes have abundant mitochondria, small lipid droplets and high uncoupling protein 1expression in human and mouse, while they can also be discriminated with high confidence by specific lineage as well as different depots. The detailed information about brown and beige adipogenesis has been well summarized in other excellent Reviews.[ 14 , 15 , 16 , 17 ] In this Review, we will focus on the adipogenesis of white adipocytes.
3. Molecular Mechanisms of Adipogenesis
3.1. Adipogenesis
Studies of WAT development have been mostly focused on the cellular mechanism of adipocyte differentiation, which is termed as adipogenesis and refers to the process that fibroblast‐like progenitor cells (mesenchymal stem cells) differentiate into new adipocytes (Figure 1 ).[ 3 , 6 , 8 ] Adipogenesis is required by normal adipose tissues for mechanical protection and thermal insulation, and is characterized as a way to support WAT to store excess calories and adapt to increased energy expenditure. Hence, the newly formed fat cells are used to effectively sequester lipids to prevent lipotoxicity in other nonadipose tissues such as liver, muscle, heart, and placenta.[ 8 ] For energy balance, WAT undergoes hypertrophy (size) and hyperplasia (number) of mature adipocytes in long‐term energy storage.[ 13 ] From the point of view of tissue and cell biology, WAT is a complex organ composed by heterogeneous cells, primarily adipocytes, preadipocytes, vascular endothelial cells, and immune cells. Through a process called adipogenesis, adipocytes are derived from mesenchymal stem cells (MSCs) that have the capacity to develop into several cell types (adipocytes, osteocytes, chondrocytes, myocytes).[ 3 ] At cellular level, with a process of commitment, the precursor cells become adipocyte lineages, which are called preadipocytes. Next, preadipocytes undergo several rounds of mitotic clonal expansion (MCE) and finally differentiate into adipocytes. Thus, the adipogenesis process comprises two main phases: commitment from MSCs to preadipocytes and terminal differentiation into mature adipocytes[ 6 ] (Figure 1).
Figure 1.
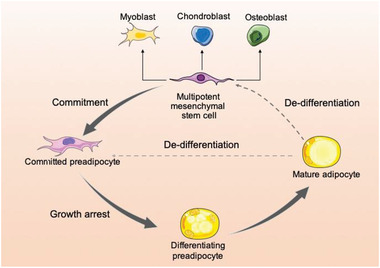
Overview of adipogenesis. Mesenchymal stem cells are also capable of forming adipocytes, myoblasts, osteoblasts, and chondroblasts. Adipogenesis is the process by which fibroblast‐like progenitor cells restrict their fate to adipogenic lineage (preadipocytes, also called adipogenic progenitor cells or adipogenic stem cells), finally differentiating to form adipocytes. There is now evidence showing that adipocytes can dedifferentiate back to fibroblast‐like progenitor cells in response to metabolic environments. The dedifferentiated cells can proliferate and redifferentiate to become adipocytes.
Clarification of the detailed mechanism of adipogenesis is still given top priority in current studies of adipose development and related metabolic diseases. A large number of transcription factors, coactivators/corepressors, epigenomic factors, and signaling pathways are involved in adipogenesis. The transcriptional cascade that promotes adipogenesis has also been studied in depth, and again, the most detailed information is focused on the transcription factors and signaling pathways that promote or repress adipogenesis (Figure 2 ).[ 13 , 18 ] The specific roles of epigenetic factors implicated in histone modification and chromatin remodeling in adipogenesis have also been well documented by some excellent Reviews.[ 7 , 19 , 20 , 21 ] Here, we will focus on the main transcription factors and signaling pathways involved in the two phases of adipogenesis.
Figure 2.
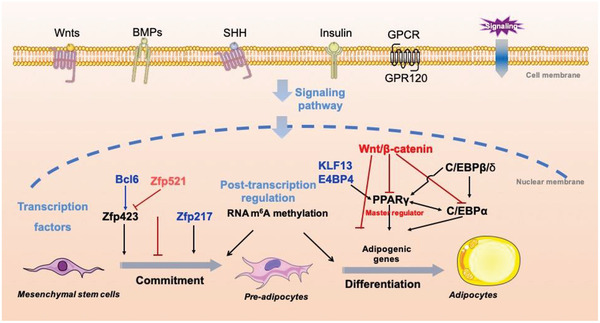
Regulation of adipogenesis. Adipogenesis is orchestrated by a complex of regulation cascades. When receiving a number of extracellular signals such as Wnts and BMPs, transcription factors, epigenetic and post‐transcriptional regulators are activated, promoting or inhibiting adipogenesis.
3.2. Roles of Transcription Factors in Adipogenesis
Numerous studies based on the prevalent cell model in vitro C3H10T1/2 and 3T3‐L1 have facilitated the faithful mimicking of the functional characteristics of adipogenesis. C3H10T1/2 line, which was established in 1973, has been well identified as a pluripotent line for studying the first phase of adipogenesis (commitment),[ 22 ] while another cell line, 3T3‐L1 preadipocyte cell line, has become the “gold standard” for investigating the second phase of adipogenesis (terminal differentiation).[ 6 , 23 ] Likewise, the primary stromal vascular fractions (SVFs) isolated from human and other animal fat tissues have become an ex vivo way to mimic adipose development.[ 24 ] Meanwhile, in vivo models involving mice with knockout or conditional knockout of certain genes are employed to overcome the drawbacks of in vitro studies and define the phenotypes observed from cell lines. In our group, by using piglets as a model, SVFs isolated from porcine adipose tissue and muscle treated with an adipogenic cocktail medium for adipogenesis were used to screen differential expression genes.[ 25 ] Furthermore, some important transcription factors have been well characterized. Currently, some Reviews have summarized the progress in the research on adipose tissues and lipid metabolism,[ 8 , 13 ] and clarified the transcriptional cascade of adipocyte differentiation.[ 7 , 26 ] Here, we will highlight the main regulators during commitment and terminal differentiation that are known to date.[ 27 , 28 ]
3.2.1. Transcription Factors Regulating Commitment
To dissect the mechanism of adipogenic commitment, in vivo and in vitro models have been employed for adipogenic research. However, due to the complexity of the regulation cascade in commitment, some original and pendent questions remain to be answered, such as how MSCs are restricted into preadipocytes.[ 29 ] Gupta et al. (2010) established adipogenic and nonadipogenic Swiss 3T3 fibroblast cell lines for screening vital factors of adipogenic commitment. It was observed that zinc finger protein 423 (Zfp423) was preferentially enriched in adipogenic fibroblasts (preadipocytes) in comparison with in nonadipogenic cell lines,[ 30 , 31 ] indicating that Zfp423 plays an important role in the commitment stage of adipogenesis. Consistent with the in vitro studies of function loss and gain, whole body knockout of Zfp423 resulted in smaller adipose tissues at embryo day 18.5.[ 30 ] These findings may help to mark Zfp423 as a critical transcription factor for commitment.[ 31 ] Notably, to find out the upstream regulators of Zfp423, a novel transcription factor, B cell lymphoma 6 (BCL6), has been reported and characterized by our group.[ 32 ] Knockdown of BCL6 was shown to reduce the expression of Zfp423. By RNA sequencing and promoter analysis, BCL6 was found to enhance the promoter activity of signal transducer and activator of transcription 1, and was identified as an upregulator of Zfp423, early B cell factor 1 (Ebf1), and PPARγ.[ 32 ] However, in vivo model of conditional knockout of BCL6 in adipose tissues showed that BCL6 deletion would lead to WAT expansion,[ 33 ] implying that BCL6 possibly plays different roles in regulating early adipogenesis and late lipogenesis. From some RNA‐seq data used to identify BCL6, we have also identified a novel transcription factor Zfp217 that regulates early adipogenesis of C3H10T1/2 cell line[ 25 , 34 ] (Figure 2), implying that the transcriptional atlas of commitment can be further improved. In addition, another group identified a repressor of adipogenesis from zinc finger protein family, namely, Zfp521,[ 35 ] which acts as a paralog of Zfp423 and inhibits commitment by binding to Ebf1 to block the expression of Zfp423 (Figure 2). Some other groups have also identified some new negative regulators of commitment, such as transcription factor 7‐like 1.[ 36 ]
Although a number of transcription factors have been documented, determination of how progenitor cells acquire a preadipocyte commitment remains quite challenging.[ 37 ] Much of our knowledge regarding the molecular mechanisms of adipogenic commitment is based on cell culture models involving fibroblast‐like cells (both primary cells and cell lines) that differentiate to form adipocytes in vitro. However, there is no single consensus marker or well defined markers of preadipocytes in vivo, reflecting the high heterogeneity of adipose progenitor cells.[ 8 , 38 , 39 ] Mice lineage‐tracking system and single‐cell sequencing have revealed heterogeneity among preadipocytes, providing novel insights into the mechanism of adipogenesis[ 40 , 41 , 42 ] (reviewed extensively elsewhere[ 38 , 39 ]).
3.2.2. Transcription Factors Regulating Terminal Differentiation
By taking advantages of cell lines and traditional adipogenic cocktail medium, terminal differentiation has been better clarified than commitment. As mentioned above, PPARγ is characterized as the master regulator of adipogenesis,[ 4 , 43 ] because of its indispensable role in adipocyte differentiation in vitro[ 4 ] and in vivo.[ 44 ] Even though a large number of regulators have been identified during commitment and terminal differentiation, they are finally dependent on the expression and/or activity of PPARγ during adipogenesis (Figure 2). It has also been found that mouse PPARγ has two truncated isoforms. Notably, PPARγ1 is widely expressed in several tissues and cell types, including adipose tissue, skeletal muscle, liver, and bone, whereas PPARγ2 expression is limited almost exclusively to adipocytes.[ 45 ] In fact, PPARγ2 has more adipogenic potential than PPARγ1, and is essential for effective adipogenesis in vitro.[ 4 , 43 ] Expression of mouse PPARγ2 is induced during the differentiation of cultured adipocyte cell lines and is strikingly adipose‐specific in vivo, which was identified as the first adipocyte‐specific transcription factor in mouse.[ 43 ] We have previously reported that eicosapentaenoic acid upregulates the expression of PPARγ1 by activating RXRa, and then PPARγ1 binds to the functional peroxisome proliferator responsive element in the promoter of adipocyte‐specific PPARγ2 to continuously activate the expression of PPARγ2 throughout the transdifferentiation process.[ 46 ] PPARγ1 enhances the expression of PPARγ2 during transdifferentiation of mouse myoblast cell line C2C12 into adiponectin‐expressed cells, namely, adipocytes.[ 46 ] To fully activate the adipogenic differentiation, PPARγ and its most important downstream effector C/EBPα functionally synergize[ 47 ] and bind to more than 90% of the same DNA binding sites to activate the transcription of genes expressed in mature adipocytes, such as fatty acid binding protein 4 (FABP4, also called aP2) and adiponectin.[ 48 ]
However, the expression levels of C/EBPα and PPARγ are low in the early process of adipogenesis, indicating that there are earlier modulators which regulate these transcription factors. C/EBPβ is another member of C/EBP family acting as an activator of PPARγ and C/EBPα transcription, which has transient high expression during the early stage of adipogenesis. It is evident that the regulatory factors targeting C/EBPβ are essential for the development of adipose tissues, and play a pivotal role in multiple rounds of MCE (Figure 2).[ 49 ] Another class of factors, namely, the kruppel‐like transcription factor (Klf) family, were shown to have high expression in early adipogenesis. The members including Klf4, Klf5, and Klf15 promote adipocyte differentiation, which is on the contrary inhibited by Klf2, Klf3, and Klf7.[ 50 ] Klf4 can bind to the promoter of C/EBPβ to induce the expression of PPARγ and Klf15. Likewise, another member of KLF family, Klf13, directly binds to the promoter of PPARγ to activate its expression.[ 51 ] In addition, Klf5 is regulated by the glucocorticoid receptor (GR).[ 52 ] Our group has identified E4 promoter‐binding protein 4 (E4BP4, also called NFIL3), which was significantly induced by glucocorticoid (Dexamethasone) via GR,[ 53 ] indicating that E4BP4 and Klf15 might be the targets of the cocktail medium to promote adipogenesis.[ 54 ] These results indicate that some hormone‐related signals such as E4BP4‐GR axis might help to regulate PPARγ from another perspective. Targeting these key transcription factors may facilitate a better understanding of the regulation of adipogenesis by transcriptional cascades (Figure 2).
3.3. Main Pathways and Receptors in Adipogenesis
In addition to the key regulatory factors, several signaling hormones and ligands have also been shown to be involved in adipogenesis (Figure 2).
3.3.1. Wingless and INT‐1 (Wnt) Signaling
Wnt signaling, for example, is perhaps the first characterized suppressor of adipogenesis.[ 55 , 56 ] During the commitment of MSCs, it was observed that Wnt signaling plays a role in commitment from C3H10T1/2 to A33 preadipocytes. R‐spondins‐2 and ‐3, which are activators of Wnt signaling, showed higher expression in committed A33 preadipocyte cell line. These findings indicate that Wnt signaling functions in the early commitment of adipogenesis.[ 6 ] Canonical Wnt signaling results in the stabilization of β‐catenin. In preadipocytes, this stabilization leads to a failure of PPARγ and C/EBPα expression and, in some cases, a shift toward an osteoblastic or other cell phenotypes.[ 55 , 57 , 58 , 59 ] In late adipogenic program, ablation of Wnt signaling promotes the expression of PPARγ, causing transdifferentiation of myoblasts into adipocytes in vitro.[ 55 ] Wnt may also redirect adipocyte dedifferentiation, implying that it is an intrinsic suppressor for both the induction and maintenance of adipose phenotype.[ 60 ]
3.3.2. Transforming Growth Factor‐β (TGF‐β)/Bone Morphogenetic Protein (BMP) Signaling
TGF‐β signaling also acts as a potent inhibitor of adipogenesis.[ 61 ] TGF‐β1 is a secreted protein that binds to TGF receptors, leading to the activation of SMAD proteins that induce the expression of extracellular matrix related genes such as collagen proteins to restrict adipogenesis by inhibiting PPARγ.[ 61 , 62 ] However, other molecules in the TGF‐β super family called BMPs, which were originally defined for their functions in cartilage and bone formation, have recently been shown to play different roles from TGF‐β in adipogenesis.[ 63 , 64 , 65 ] Interestingly, members of BMPs have distinct functions to promote adipogenesis of white and brown adipocytes.[ 63 , 64 , 65 , 66 ] BMP2 and BMP4, for example, have been identified as potent proadipogenic factors that drive adipogenesis at the early stage in vitro, whereas BMP7 plays a crucial role in promoting the adipogenesis and thermogenesis of brown adipocytes.[ 63 , 65 , 66 ] Notably, both Wnt and BMP signaling are involved in maintaining the development of various organs, implying that their functions at different stages may be spatiotemporally regulated.
3.3.3. Insulin Signaling
As a pancreatic peptide hormone, insulin plays a predominant physiological role in glucose metabolism, and functions to bind the insulin receptor or the related insulin‐like growth factor 1 (IGF1) receptor. The insulin signaling cascade activates insulin receptor substrates and then PI3K/AKT kinases, leading to the activation of downstream CREB, mTOR, and the family of forkhead proteins (FOXOs).[ 67 , 68 ] It is widely acknowledged that these intracellular effectors in insulin signaling are inevitably associated with nutritional physiology and metabolism, and elimination of them blocks adipogenesis.[ 69 , 70 , 71 , 72 , 73 , 74 , 75 , 76 , 77 ] Thus, insulin is an essential component of adipogenic differentiation medium in vitro and promotes adipogenesis in highly adipogenic progenitor cells by FACS.[ 40 ] Even though glucocorticoid signaling has been reported to sensitize preadipocytes to insulin signaling and consequently enhance adipogenic action of the insulin pathway,[ 78 ] it still needs to be clarified how other signals facilitate or suppress insulin signaling.[ 8 ]
3.3.4. G‐Protein‐Coupled Receptors (GPCRs)
Many eicosanoids signal via cell surface GPCRs to regulate adipogenesis, which has also been well clarified. Long chain fatty acids (LCFAs) function as the ligands of PPARγ, which is famous for its role as a nuclear receptor, as well as the ligands of specific GPCRs.[ 27 , 79 , 80 ] Treatment with LCFAs may not only activate PPARγ, but also activate the membrane fatty acid receptor GPR120 and subsequently enhance the expression of PPARγ by the ERK1/2‐Ca2+ pathway.[ 27 , 81 , 82 ]
Some other pathways such as Notch[ 83 , 84 , 85 , 86 ] and hedgehog[ 87 , 88 ] signaling may also play critical roles in adipogenesis, which have not been well characterized.[ 60 ] These pathways may help to expand our sights into the combating against obesity and related metabolic diseases. In short, the regulatory mechanism of adipogenesis by transcriptional cascades has been well manifested as illustrated in Figure 2.
4. Post‐Transcriptional Regulation: RNA m6A Modification in Adipogenesis
Although DNA sequence conveys the majority of heritable information to subsequent generations, accumulating evidence demonstrates that epigenetics plays an important role in regulating cell‐type‐specific gene expression during cell differentiation.[ 89 , 90 ] Epigenetics includes several different mechanisms, such as DNA methylation, histone modification, and chromatin remodeling.[ 19 , 91 , 92 ] However, RNA can be also modified, which can be a key biomarker for several biological events.[ 11 ] For instance, N6‐methyladenosine (m6A), a common modification in mRNA, has been detected in 1970s and its roles in regulating gene expression are being uncovered.[ 11 ]
4.1. Overview of RNA m6A Modification
More than 150 distinct chemical modifications of RNAs have been found to date, and m6A has been identified as the most abundant modification on eukaryotic messenger RNA (mRNA).[ 93 ] m6A modification is widely distributed in viruses, bacteria, yeast, plants, and vertebrates.[ 94 , 95 , 96 , 97 ] In 2011, Jia et al. discovered the first demethylase, the fat mass, and obesity‐associated protein (FTO), confirming that m6A modification is dynamic and reversible.[ 98 ] Subsequently, m6A RNA IP sequence (MeRIP‐seq) was established based on specific m6A antibody immunoprecipitation,[ 99 , 100 ] which leads to numerous studies of RNA epigenomics and makes it one of the research hotspots. Further work showed that m6A modification is highly conserved in a sequence context as RRm6ACH (R represents G and A; H represents A, C, and U),[ 101 ] and is preferentially distributed around stop codons and enriched at 3 ‘UTR, 5‘UTR, and the inner exons.
Among the effectors of m6A, three types of key proteins are known to be involved in balancing m6A modification: “writer”‐methyltransferases, “eraser”‐demethylases, and “reader”‐m6A binding proteins. m6A modification is deposited to RNAs by a core methyltransferase complex composed of METTL3 and METTL14 heterodimer, in which METTL3 acts as the catalytic subunit and METTL14 facilitates RNA binding. Another key protein WTAP, which binds to METTL3/14, is required for optimal substrate recruitment and METTL3/14 localization.[ 93 ] The “eraser”‐demethylases comprise demethylases FTO and alkB homolog 5 (ALKBH5) to remove the methylation. The “reader”‐m6A binding proteins include YTHs, HNRNPs, and IGF2BPs, which recognize m6A and help to define biological events by post‐transcriptional regulation of mRNA[ 11 , 102 ] (Figure 3 ).
Figure 3.
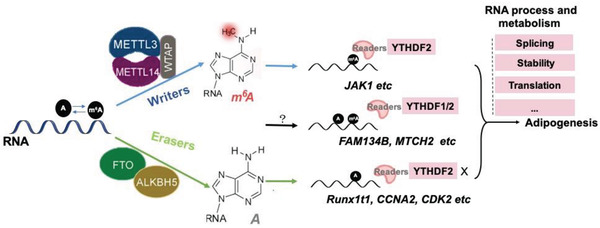
Impact of m6A on mRNA abundance in adipogenesis. The “writer”‐methyltransferase complexes METTL3, METTL14, and WTAP; “eraser”‐demethylase FTO and ALKBH5; “reader”‐m6A binding proteins YTH family and IGF2BPs affect m6A methylation and RNA metabolism of key genes (such as Runx1t1) to regulate adipogenesis.
m6A modification has been shown to regulate nearly every stage of mRNA processing, including mRNA processing and metabolism (export, splicing, decay, translation)[ 103 ] (Figure 3). Based on the understanding of its dynamic reversibility and widespread roles in mRNA metabolism, m6A modification was reported to play important roles in numerous physiological processes, such as animal development, gametogenesis, T cell homeostasis, stress responses, circadian rhythm, cell differentiation, and reprogramming, and is regarded as a post‐transcriptional regulatory mechanism.[ 10 , 103 , 104 , 105 ]
Researchers have also focused on the function of m6A modification in adipogenesis[ 9 , 28 , 106 ] and obesity‐related events. m6A was shown to be responsive to high‐fat treatment in mice.[ 107 ] Another group set up an obese porcine model (Landrace pigs as the lean‐type breed and Jinhua pigs as the obese‐type breed) to identify the unique peaks of m6A in mRNA using m6A sequencing.[ 108 ] These discrepant m6A distributions in vivo revealed that the variation of m6A modification may be related to adipose function and adipogenesis, and m6A‐related proteins play a specific and important role in modulating adipogenesis. Hence, we summarize recent studies to present the new molecular regulatory mechanisms of m6A on adipogenesis.
4.2. m6A “eraser” FTO Promotes Adipogenesis
A genome‐wide association analysis has identified the roles of FTO in obesity and related diseases.[ 109 , 110 ] A number of studies have reported the positive function of FTO in fat mass, body weight, and adipogenesis.[ 111 , 112 ] The m6A‐dependent role of FTO was first reported by Zhao et al. (2014), as the global m6A level was increased during adipogenesis of mouse 3T3‐L1 cells. Knockdown of FTO impaired adipogenesis, and overexpression of catalytically inactive FTO did not restore adipogenic differentiation.[ 106 ]
4.2.1. FTO Functions as a Cell Fate Regulator for Bone Marrow Stromal Cells (BMSCs) to Promote Adipogenic Commitment
FTO participates in GDF11‐FTO‐PPARγ axis to control the shift of osteoporotic BMSCs fate to the formation of more adipocytes instead of osteoblasts.[ 113 ] These results suggest that demethylase FTO and FTO‐mediated low m6A modification act as a switch to transform osteogenesis to adipogenesis.
4.2.2. FTO Enhances Adipogenesis by MCE Progress
It has also been shown that FTO regulates m6A modification in the mRNA of cyclin A2 (CCNA2) and cyclin‐dependent kinase 2 (CDK2), which are crucial cell cycle regulators, leading to delayed entry of MDI‐induced 3T3L1 cells into G2 phase.[ 114 ] It was found that FTO negatively regulates m6A levels and positively regulates adipogenesis in porcine preadipocytes.[ 115 ] Furthermore, it has been found that FTO deficiency suppresses Janus kinase 2 (JAK2) expression in an m6A‐dependent manner of porcine and mouse preadipocytes, leading to increases in the phosphorylation of STAT3 and attenuating the transcription of C/EBPβ, which inhibits adipocyte differentiation at the early MCE stage.[ 116 ] Furthermore, it was obeserved that FTO functions as a regulator of alternative splicing regulatory protein Srsf2, and the identified runt‐related transcription factor 1 (Runx1t1) is the core target gene of FTO.[ 106 ] Subsequent research revealed that FTO might exert its promoting effects on adipogenesis through enhancing the expression of the short isoform of RUNX1T1 at the early MCE stage.[ 112 ]
4.2.3. Recently, FTO Was Found to Regulate Autophagy during Adipogenesis
m6A modification also plays a critical role in regulating autophagy and adipogenesis of mouse 3T3‐L1 and porcine primary preadipocytes through targeting Atg5 and Atg7. Knockdown of FTO decreased the expression of ATG5 and ATG7, leading to attenuation of autophagosome formation, thereby inhibiting autophagy and adipogenesis.[ 117 ] These results provide a novel insight that m6A methylation regulates adipogenesis by targeting autophagy.
Collectively, increasing evidence supports that FTO‐dependent m6A demethylation plays a pivotal role in adipogenesis. FTO promotes adipogenesis partly by influencing MCE stage and autophagy, while other mechanisms for its adipogenesis‐promoting effect still need further elucidation. Even though a lot of studies have shown the role of demethylase FTO in adipogenesis, it still remains confusing how FTO protein itself is regulated. In the research on the upstream mechanism of FTO, our group has identified zinc finger protein (Zfp217) as a transcriptional regulator of FTO to enhance adipogenesis in an m6A‐dependent way, which provides a new perspective to understand the modulation of m6A modification during adipogenesis.[ 28 ]
4.3. m6A “Writer” METTL3 Represses Adipogenesis
Compared with FTO, METTL3 has an opposite function in adipogenesis. A recent study has reported that METTL3 deficiency appears to promote adipogenic differentiation in mouse 3T3L1 cells.[ 106 ]
METTL3 suppresses the commitment and MCE of adipogenesis in vitro. Interestingly, it was reported that the deletion of METTL3 causes inhibited bone formation and marrow fat accumulation. The loss of METTL3 compromised osteogenic potential and increased adipogenic differentiation through m6A RNA methylation of the critical regulator of lineage allocation in MSCs and osteoblast precursors.[ 118 , 119 ] Mechanically, knockdown of METTL3 decreased the m6A level of JAK1 and increased its expression in porcine BMSCs, resulting in upregulated STAT5 phosphorylation to bind to the promoter of C/EBPβ.[ 120 ] These findings uncover consistent molecular mechanisms of METTL3‐dependent m6A modification in JAK‐STAT‐C/EBPβ pathway, and provide novel insights into the regulation of the early MCE stage of adipogenesis by m6A‐related proteins.
However, other research groups have identified that METTL3, METTL14, and WTAP positively control adipogenesis by promoting cell cycle transition in MCE of 3T3‐L1 cells.[ 121 ] Consistent with the in vitro results, WTAP+/− mice did not show diet‐induced obesity with smaller size and number of adipocytes, resulting in increased energy expenditure, attenuated hepatic steatosis, and macrophage infiltration and better insulin sensitivity.[ 121 ] The observed complex and obscure regulatory effects of m6A modification on adipogenesis may be due to the efficiency of gene interference and complexity of in vivo study. Another view of point is that all these homozygous gene knockout mice are embryonic lethal, indicating the critical role of m6A writer in animal development.[ 121 ] Thus, further research is needed to reveal the role of methyltransferase complex in adipogenesis.
To sum up, these results comprehensively reveal that demethylase FTO and FTO‐mediated low m6A modification act as a switch, whereas methyltransferase METTL3 has the opposite effect in transforming osteogenesis to adipogenesis. After methylation or demethylation, the alteration of m6A methylation by FTO or METTL3 is ultimately performed by recruiting specific “reader” proteins to m6A sites, which impact the fate of target mRNAs by influencing RNA metabolism.[ 122 ]
4.4. m6A “Readers” in Adipogenesis
4.4.1. YTHs Mediate RNA Metabolism of Key Genes to Inhibit Adipogenesis
YT521‐B homology (YTH) domain family including YTHDF1–3 and YTHDC1–2 has been well identified as m6A readers. YTHDF1/3 were reported to promote the translation of target transcripts and YTHDF2 promotes the degradation of mRNA.[ 123 ] Previous studies have revealed that the degradation of key mRNA mediated by YTHDF2 may be the most significant impact of m6A methylation on adipogenesis. After m6A modification is erased or deposited by FTO or METTL3, YTHDF2 increases or decreases the recognition and degradation of methylated mRNAs of CCNA2, CDK2, JAK1/2, Atg5 or Atg7, which consequently alters the expression of target proteins and adipogenic differentiation.[ 114 , 116 , 117 , 120 ] Moreover, mRNA modification on single genes was also explored. Loss of m6A in the mRNA of FAM134B was found to facilitate adipogenesis, and YTHDF2 may recognize and bind to its m6A sites, resulting in reduced mRNA stability and protein expression.[ 124 ] It is worth noting that YTHDF2 maintains m6A methylation by limiting the activity of demethylase FTO under heat shock stress,[ 125 ] providing more potential ways for the “reader” proteins to participate in adipogenesis. Consistently, we also found that Zfp217 interacts with YTHDF2 to maintain the m6A‐demethylation activity of FTO to promote adipogenesis.[ 28 ]
In addition to the degradation of mRNA by YTHDF2, YTHDF1 also regulates adipogenesis through increasing protein expression. It was found that m6A positively mediates patatin‐like phospholipase domain containing 2 expression and functions by promoting translation most likely through YTHDF1.[ 108 ] Meanwhile, the research group has also revealed the role of m6A methylation of key genes in adipogenesis of intramuscular preadipocytes. They identified a unique methylated gene in Jinhua pigs, mitochondrial carrier 2, which promotes intramuscular adipogenesis in an m6A‐YTHDF1‐dependent manner.[ 126 ]
Besides YTHDF1/2, other YTH domain family proteins may also have functions in adipogenesis. Nuclear m6A reader YTHDC1 functions as a recruiter of mRNA splicing factors, and ZNF638 pre‐mRNA is a key target.[ 127 ] It is known that ZNF638 physically interacts and transcriptionally cooperates with C/EBPβ/δ and splicing regulators to promote adipogenesis.[ 128 , 129 ] These findings imply that YTHDC1 may affect adipogenesis partly through ZNF638, which is worthy of further study.
Overall, YTH family functions as a downstream regulator of mRNA methylation and metabolism to mediate adipogenesis.
4.4.2. Other m6A “Readers” in Adipogenesis
In addition to YTH family, some RNA binding proteins may also be m6A “readers” in adipogenesis. IGF2BPs, a distinct family of m6A readers, could maintain the stability and storage of mRNAs depending on m6A modification.[ 130 ] It was shown that overexpression of IGF2BP1 promoted adipocyte differentiation and lipid droplet accumulation of chicken preadipocytes, whereas inhibition of IGF2BP1 showed the opposite effect.[ 131 ] These results suggest that IGF2BPs promote adipogenesis in an m6A‐dependent manner and the target mRNAs of IGF2BPs deserve further research.
Human antigen R (HuR, also called ELAVL1), a well‐established RNA‐binding protein, binds to the U‐rich regions at the 3′‐UTR of thousands of transcripts to increase the mRNA stability and translation efficiency of the target genes.[ 54 ] HuR also serves as a potential m6A reader as identified by an RNA affinity chromatography approach.[ 99 ] Knockdown of METTL3 or METTL14 enhanced the binding of HuR to demethylated mRNA, which subsequently increased RNA stability in embryonic stem cells.[ 132 ] It has been reported that HuR binds to the mRNA of C/EBPβ, which is required for the onset of adipogenesis,[ 133 , 134 ] suggesting that it is a new way for HuR to act as an m6A “reader” to regulate adipogenesis.
Therefore, the functional outcome of m6A modification in regulating adipogenesis depends on the reading of specific m6A sites of transcripts by m6A “readers” to influence their RNA metabolism. These studies highlight the potential regulatory mechanisms by which m6A “readers” regulate adipogenesis in an m6A‐dependent manner.
5. Orchestration of Transcriptional and Post‐Transcriptional Regulation of Adipogenesis
In central dogma of molecular biology, mRNA plays a key role of connecting link between the preceding and the following. DNA is transcribed into mRNA, which is then translated into protein. Around mRNA, transcription factors recognize specific DNA sequences as “master regulators,” performing the first step of decoding genetic information.[ 135 ] A series of transcription factors along with DNA epigenetic regulators constitute a complex and orchestrated transcriptional cascade to regulate gene expression. In addition, post‐transcriptional regulators, especially m6A modification, affect almost every stage of mRNA processing to alter gene expression at the post‐transcriptional level, which controls adipogenesis and many other processes.[ 136 ]
To regulate gene expression, both transcrptional and post‐transcrptional regulation show spatiotemporal continuity. 1) The occurrence of post‐transcriptional modification depends on the initiation of transcription. 2) Then, the post‐transcriptional modification influences the translation of transcripts through participating in RNA processing and metabolism. 3) Finally, post‐transcriptional regulation could facilitate rapid remodeling of the transcriptome relative to transcriptional regulation.[ 137 ]
Thus, to clarify the coordination of transcriptional and post‐transcriptional regulation may be a critical and profound topic in current studies of adipogenesis.
5.1. m6A‐Related Proteins Modulate Adipogenesis through Transcriptional Regulation
5.1.1. The Transcription of Key Regulators Is Regulated by m6A Modification to Mediate Adipogenesis
In many pivotal biological events, m6A modification occurs in the mRNAs of some key transcription factors. The dynamic changes of methylated or demethylated mRNAs may alter the cellular levels of transcripts and proteins to affect cell fate[ 137 ] (Figure 4 ). In adipose tissues, researchers have revealed a potential relationship between transcriptional regulation and post‐transcriptional modification using MeRIP‐Seq.[ 138 ] These results reveal the potential mechanism by which m6A‐related proteins regulate the mRNA methylation of vital transcription factors.
Figure 4.
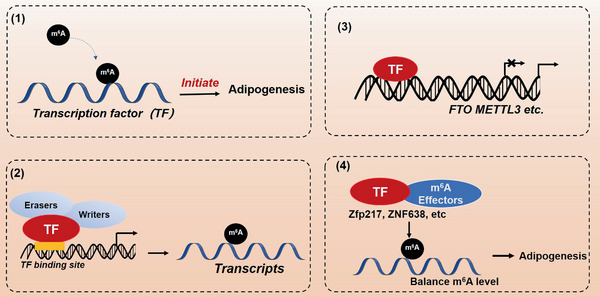
Complex regulation at transcriptional and post‐transcriptional levels by m6A modification in adipogenesis. (1) m6A‐related proteins regulate the transcription of key regulators to mediate adipogenesis in an m6A modification‐dependent manner. (2) m6A‐related proteins play a direct role in transcriptional regulation. (3) m6A‐related proteins may be transcriptionally regulated. (4) Besides transcriptional regulation, transcription factors interact with m6A‐related proteins to regulate m6A methylation.
During adipogenesis, the methylation of the master regulator, PPARγ, has been reported recently.[ 113 ] It was observed that FTO facilitates the differentiation of BMSCs into adipocytes rather than osteoblasts.[ 113 ] Furthermore, FTO binds to and demethylates the mRNA of PPARγ, and consequently enhances its expression. In FTO‐knockdown BMSCs, high m6A modification in the mRNA of PPARγ inhibited the expression of PPARγ, suggesting a negative role of m6A in adipogenesis.[ 113 ] These results suggest that the regulation of PPARγ by m6A modification may be one way besides transcription and activation of PPARγ to regulate adipogenesis.
Another key regulator in adipogenesis, C/EBPβ, has also been found to be regulated by m6A modification.[ 116 ] FTO deficiency inhibited the expression of JAK2 and phosphorylation of STAT3, leading to decreased expression of C/EBPβ,[ 116 ] while knockdown of METTL3 decreased the mRNA m6A level of JAK1, resulting in enhanced mRNA stability of JAK1, and activated phosphorylation of STAT5 to bind to the promoter of C/EBPβ in the adipogenic differentiation of pig BMSCs.[ 120 ] Thus, C/EBPβ may not be the direct target of RNA m6A modification in BMSCs, which has also been demonstrated in 3T3‐L1 cell line by some groups.[ 139 ] In our previous study, RNA m6A modification was also not detected in the mRNA of C/EBPβ in 3T3‐L1 cell line with or without adipogenic induction.[ 28 ] Thus, m6A might modulate the upstream regulators of key transcription factors to inhibit adipogenesis.
C/EBPα has been identified to be regulated by FTO in other biological events but not in adipogenesis, implying that C/EBPα might also be a target of m6A modification in adipogenesis.[ 140 ] Thus, it may be speculated that m6A modification in adipogenesis has multiple functions to directly modify some key transcription factors or modulate their upstream key regulators. Although some potential mechanisms have been identified to be related to m6A modification, further studies are needed to more comprehensively reveal the functions of RNA m6A methylation in the regulation of adipogenesis.
5.1.2. m6A‐Related Proteins Bind the DNA Sequence of Target Genes to Regulate Their Transcription
The nuclear localization of m6A effectors indicates that they may play different roles in different cell types and/or in response to different stimuli and stresses. Demethylase FTO has been well known for its role in obesity and related diseases and can act as a transcription cofactor. FTO is abundant in both the nucleus and cytoplasm of certain cell lines, but mostly in nucleus.[ 141 ] Interestingly, FTO is recruited to both unmethylated and methylated promoters and enhances the transactivation potential of the C/EBPs to modulate the transcriptional regulation of adipogenesis.[ 142 ] Furthermore, FTO was also reported to demethylate DNA N6‐methyldeoxyadenosine (6mA), and affect the transcriptional activation of C/EBPδ by demethylating 6mA at the promoter of C/EBPδ in adipogenesis.[ 139 ]
For methyltransferases, METTL3/14‐WTAP is distributed in nucleus to bind the DNA sequence of target genes to regulate their transcription. It was observed that METTL3 and/or METTL14 are located in the relevant chromatin sites, which co‐occurs with accumulated m6A methylation.[ 143 , 144 ] METTL3/14‐WTAP is recruited at desired chromatin loci, and cotranscriptionally regulates m6A installation most likely with transcription factors and histone marks.[ 93 ] Besides METTL3/14‐WTAP, PCIF1, a cap‐specific m6A methyltransferase, interacts with the C‐terminal domain of RNA polymerase II, suggesting that PCIF1 may perform its function in a cotranscriptional manner.[ 145 ] Likewise, it was found that transcription factors SMAD2 and SMAD3 recruit the methyltransferase complex (METTL3/14‐WTAP) and promote its binding to a subset of transcripts (i.e., NANOG) involved in early cell fate, which reveals the connection between transcriptional and post‐transcriptional regulation.[ 146 ] These findings suggest that methyltransferases can also function as transcriptional regulators during biological events. It would be interesting to further illustrate the function of methyltransferases in transcriptional regulation of adipogenesis.
Collectively, these findings unravel that FTO and methyltransferase complex may be implicated not only in m6A modification, but also in transcriptional regulation directly (Figure 4).
5.2. m6A‐Related Proteins May Be Transcriptionally Regulated
Although numerous studies have reported that m6A‐related proteins such as METTL3, FTO, and YTHDF2 play important roles in regulating gene expression and adipogenesis, little is known about the upstream mechanism by which these m6A‐related proteins themselves are regulated (Figure 4). We previously reported an interesting mode of transcription and post‐transcription of gene expression in adipogenesis.[ 28 ] It was found that deficiency of transcription factor Zfp217 inhibited adipogenesis in 3T3L1 cells and led to a global increase in m6A mRNA methylation. However, Zfp217 is neither a methyltransferase nor a demethylase. It remains unclear how Zfp217 inhibits the mRNA methylation of m6A. We subsequently demonstrated that Zfp217 acts as a transcription factor and binds to the promoter of FTO gene to regulate its expression and promte adipogenesis.[ 28 ] Thus, these results reveal a pivotal mechanism to combine gene transcription with m6A mRNA modification, which may be a novel way to modulate m6A modification during adipogenesis and other vital events.
Notably, it was demonstrated that there is a feedback regulatory mechanism between transcriptional and post‐transcriptional level in R‐2‐hydroxyglutarate (R‐2HG) mediated anti‐leukemic activity.[ 140 ] R‐2HG exhibited a repressing effect on FTO demethylation activity, thereby increasing m6A methylation of the mRNA of key transcription factor C/EBPα, which would inhibit the expression of C/EBPα and leukemia cell proliferation/viability. Interestingly, low protein levels of C/EBPα in turn decreased mRNA level of FTO due to transcriptional regulation to enhance m6A modification. The feedback regulation between transcriptional and post‐transcriptional level provides some implications for dissecting the molecular mechanism of adipogenesis, and ultimately combating obesity and its associated risks of metabolic syndromes.
In addition, it was reported that METTL3 may be regulated by post‐transcriptional and post‐translational regulations. It has been shown that methyltransferase METTL3 could be influenced by miRNAs and SUMOylation,[ 147 , 148 ] and METTL3/14 complex is phosphorylated at several sites which might be functionally important.[ 149 ]
5.3. Besides Transcriptional Regulation, Transcription Factors Interact with m6A‐Related Proteins to Regulate m6A Methylation
Once a protein is identified as a transcription factor, the DNA binding domain and transcriptional activation domain will be well characterized. However, other key protein domains might be often ignored. It is recognized that transcription factors exert their functions through interacting with other proteins, suggesting their multifunctional roles in biological events (Figure 4). Notably, Zfp217 is known as a core component of protein complexes with histone deacetylase (HDAC), CoREST, and CtBPs,[ 150 , 151 , 152 ] and interacts with m6A methyltransferase METTL3 in embryonic stem cells (ESCs).[ 105 ] Zfp217 suppresses METTL3 activity, preventing the deposition of m6A on core transcripts to regulate pluripotency and reprogramming of ESCs. In regulation of adipogenesis, it was also validated that there is an actual interaction between Zfp217 and m6A “reader” protein YTHDF2. Zfp217 could act as a regulator of YTHDF2‐FTO competition, which sequesters YTHDF2 from limiting the accessibility of FTO to specific m6A sites.[ 28 ]
Interestingly, it was found that transcriptional coregulator ZNF638 interacts with another m6A “reader” HNRNPA2B1 in adipogenesis,[ 129 ] suggesting that ZNF638 may function in an m6A‐dependent manner. Therefore, these studies shed light on the novel role of transcription factors to orchestrate gene transcription and m6A RNA modification, which offers a new approach for dissecting the molecular mechanism of adipogenesis.
6. Therapeutic Implications for Obesity‐Related Metabolic Disorders
Given that m6A‐related proteins play critical roles in adipogenesis, they may be potential therapeutic targets of adipogenesis and obesity‐related metabolic disorders. For example, the classical methylation inhibitor, i.e., cycloleucine, can significantly decrease the mRNA m6A level in a dose‐dependent manner in porcine adipocytes and increase intracellular triglyceride, whereas the methyl donor betaine shows the opposite effect.[ 115 ] Epigallocatechin gallate (EGCG), a component in green tea, can effectively prevent obesity and the prevalence of type 2 diabetes mellitus.[ 153 ] Wu et al. found that EGCG reduces the expression of FTO, leading to an overall increase in levels of RNA m6A methylation and ultimately inhibiting adipogenesis by blocking the MCE at the early stage.[ 154 ] Although the above substances have been found to affect adipogenesis through RNA m6A methylation, their functions and effects remain to be validated in vivo.
Importantly, small‐molecule inhibitors have been developed to inhibit the catalytic activity of m6A effectors to combat obesity and cancer. Recently, virtual screening approach was employed to find FTO inhibitors from FDA‐approved drugs; as a result, entacapone was identified as a chemical inhibitor of FTO.[ 155 ] Entacapone inhibits FTO demethylation activity, which thereby increases the energy expenditure and improves glucose tolerance of mice. FTO deficiency regulates FOXO1 expression by demethylating m6A sites, and entacapone was confirmed to regulate gluconeogenesis and thermogenesis through FTO–FOXO1 axis in vivo (Figure 5 ). FB23 and FB23‐2 were reported to directly bind to FTO and selectively inhibit the m6A demethylase activity of FTO. FB23‐2 significantly affects the proliferation, differentiation, and apoptosis of human acute myeloid leukemia cells in vitro and in vivo.[ 156 ] These results suggest that small‐molecule inhibitors specifically targeting FTO could be developed for fighting against obesity‐related metabolic disorders and other events (Figure 5). The findings broaden our horizon that numerous regulatory molecules affecting m6A modification can serve as effective treatment strategies of obesity‐related metabolic disorders (summarized in Table 1 ).[ 157 , 158 , 159 , 160 , 161 , 162 , 163 , 164 ]
Figure 5.
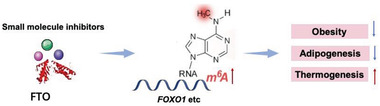
Potential inhibitors of FTO to regulate obesity‐related homeostasis.
Table 1.
Functions of compounds targeting m6A effectors in homeostasis
| Regulator | Target protein | Effect | Primary functions | Refs. |
|---|---|---|---|---|
| Rhein et al. | FTO | Inhibit | Reversibly bind FTO enzyme and competitively prevent the recognition of m6A substrates | [158] |
| MO‐I‐500 | FTO | Inhibit | Inhibit survival and/or colony formation of SUM149‐MA cells | [158, 160] |
| Meclofenamic acid (MA) and MA2, the ethyl ester form of MA | FTO | Inhibit | Inhibit glioblastoma stem cells tumorigenesis | [161, 162] |
| R‐2‐hydroxyglutarate (R‐2HG) | FTO | Inhibit | Antileukemic activity | [140] |
| Epigallocatechin gallate | FTO | Inhibit | Reduce the expression of FTO and adipogenesis | [154] |
| IOX3 | ALKBH5 | Inhibit | Inhibitor of ALKHB5 | [163] |
| Citrate | ALKBH5 | Inhibit | Modest inhibitor of ALKHB5 | [164] |
With regard to methyltransferase, there have been few reports about targeting METTL3 to regulate adipogenesis and combat obesity. It is known that METTL3 may also have other functions besides its catalytic activity in cancers, and cytoplasmic‐localized METTL3 was proposed as a potential m6A reader.[ 93 , 165 ] Thus, development of inhibitors to target special RNA m6A methyltransferase has good application prospects in future studies.
Overall, the relationship between transcriptional and post‐transcriptional regulation by m6A‐related proteins may provide some novel therapeutic targets for combating obesity.
7. Conclusion and Perspective
In summary, from the above Review, the transcriptional mechanisms of adipogenesis have been well studied in the past three decades, while there have been relatively fewer studies of the post‐transcription of adipocyte differentiation, particularly RNA modifications. In this context, we summarized the progress in the studies of m6A modification in adipogenesis and identified Zfp217, a pivotal modulator of transcriptional and post‐transcriptional regulation in adipogenesis, which may help to better explore the orchestrated regulation at different levels in adipocyte differentiation. Besides, the regulation model for gene expression that coordinates RNA m6A methylation and gene transcription may provide a new approach to better understand other biological events.
Despite of the rapid progress in the studies of transcriptional and post‐transcriptional regulation of adipogenesis, many questions remain to be answered.
First, although both in vitro cell lines and in vivo models have provided reliable methods to manifest the key regulators in adipogenesis, the detailed mechanism for commitment phase is still unclear. Recently, a group led by Patrick Seale applied scRNA‐seq and FACS to identify new surface markers for progenitor cells and committed preadipocytes in WAT, defining a mesenchymal cell hierarchy involved in adipocyte formation.[ 8 , 38 , 40 ]
Second, the role of m6A in adipogenesis needs to be better characterized. The roles of RNA processing and metabolism modulated by m6A need to be further demonstrated in adipogenesis, and more importantly in lipid metabolism, which has not been reported yet. To find the “classical” drugs from FDA system for targeting novel m6A‐related enzymes such as FTO[ 10 , 155 ] may facilitate a better application of these potential drugs to treat obesity. Answering these questions may promote our understanding of m6A‐dependent adipogenesis, provide new insights into adipogenesis, and develop new approaches to treat obesity, cancer, and other metabolic diseases.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgements
T.S. and Y.Y. contributed equally to this work. This work was supported by the National Natural Science Foundation of China (No. 31472075), The modern agricultural industry technology system (CARS‐35), and Fundamental Research Funds for the Central Universities of China (No. 2662017PY017).
Biographies
Tongxing Song is an associate professor from the Department of Animal Nutrition and Feed Science at Huazhong Agricultural University (HZAU). His research mainly focuses on transcriptional and post‐transcriptional regulation of adipogenesis. He received his Ph.D. of Molecular Nutrition from HZAU at Wuhan, China in 2018. After his work as a postdoctoral fellow at Purdue University, USA until 2019, he is currently working in HZAU.

Jian Peng is a professor (Grade II) and doctoral supervisor from the Department of Animal Nutrition and Feed Science at Huazhong Agricultural University (HZAU). She received her Ph.D. of Animal Genetics, Breeding, and Reproduction in 1999 from HZAU, China. After her work as a visiting scholar fellow at University of Manitoba, Canada until 1998, she came back to HZAU and worked as an associate professor. In 2000, she was appointed as a professor at the Department of Animal Sciences at HZAU. Her work focuses mainly on adipose tissue development and maternal lipid metabolism.

Song T., Yang Y., Jiang S., Peng J., Novel Insights into Adipogenesis from the Perspective of Transcriptional and RNA N6‐Methyladenosine‐Mediated Post‐Transcriptional Regulation. Adv. Sci. 2020, 7, 2001563 10.1002/advs.202001563
Contributor Information
Siwen Jiang, Email: jiangsiwen@mail.hzau.edu.cn.
Jian Peng, Email: pengjian@mail.hzau.edu.cn.
References
- 1. WHO , Obesity, overweight Fact sheet, http://www.who.int/mediacentre/factsheets/fs311/en/ (accessed April 2019).
- 2. Ohgo S., Hasegawa S., Hasebe Y., Mizutani H., Nakata S., Akamatsu H., Exp. Dermatol. 2013, 22, 769. [DOI] [PubMed] [Google Scholar]
- 3. Farmer S. R., Cell Metab. 2006, 4, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tontonoz P., Hu E., Spiegelman B. M., Cell 1994, 79, 1147. [DOI] [PubMed] [Google Scholar]
- 5. Lowe C. E., O'rahilly S., Rochford J. J., J. Cell Sci. 2011, 124, 2681. [DOI] [PubMed] [Google Scholar]
- 6. Tang Q. Q., Lane M. D., Annu. Rev. Biochem. 2012, 81, 715. [DOI] [PubMed] [Google Scholar]
- 7. Lee J.‐E., Schmidt H., Lai B., Ge K., Mol. Cell. Biol. 2019, 39, e00601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ghaben A. L., Scherer P. E., Nat. Rev. Mol. Cell Biol. 2019, 20, 242 [DOI] [PubMed] [Google Scholar]
- 9. Zhao Y., Shi Y., Shen H., Xie W., J. Hematol. Oncol. 2020, 13, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wu J., Frazier K., Zhang J., Gan Z., Wang T., Zhong X., Obes. Rev. 2020, 21, e12942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhao B. S., Roundtree I. A., He C., Nat. Rev. Mol. Cell Biol. 2017, 18, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Olefsky J. M., Cell 2008, 134, 914. [DOI] [PubMed] [Google Scholar]
- 13. Rosen E. D., Spiegelman B. M., Cell 2014, 156, 20.24439368 [Google Scholar]
- 14. Inagaki T., Sakai J., Kajimura S., Nat. Rev. Mol. Cell Biol. 2016, 17, 480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kajimura S., Spiegelman B. M., Seale P., Cell Metab. 2015, 22, 546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harms M., Seale P., Nat. Med. 2013, 19, 1252. [DOI] [PubMed] [Google Scholar]
- 17. Wang W., Seale P., Nat. Rev. Mol. Cell Biol. 2016, 17, 691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rosen E. D., Macdougald O. A., Nat. Rev. Mol. Cell Biol. 2006, 7, 885. [DOI] [PubMed] [Google Scholar]
- 19. Zhou Y., Peng J., Jiang S., Eur. J. Cell Biol. 2014, 93, 170. [DOI] [PubMed] [Google Scholar]
- 20. Sun W., von Meyenn F., Peleg‐Raibstein D., Wolfrum C., Adv. Sci. 2019, 6, 1900275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ling C., Rönn T., Cell Metab. 2019, 29, 1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reznikoff C. A., Brankow D. W., Heidelberger C., Cancer Res. 1973, 33, 3231. [PubMed] [Google Scholar]
- 23. Green H., Kehinde O., Cell 1974, 1, 113. [Google Scholar]
- 24. Maumus M., Peyrafitte J.‐A., D'angelo R., Fournier‐Wirth C., Bouloumié A., Casteilla L., Sengenès C., Bourin P., Int. J. Obes. 2011, 35, 1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jiang S., Wei H., Song T., Yang Y., Peng J., Jiang S., PLoS One 2013, 8, e77094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. de Sá P. M., Richard A. J., Hang H., Stephens J. M., Comp. Physiol. 2017, 7, 635. [DOI] [PubMed] [Google Scholar]
- 27. Song T., Yang Y., Zhou Y., Wei H., Peng J., Cell. Mol. Life Sci. 2017, 74, 2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Song T., Yang Y., Wei H., Xie X., Lu J., Zeng Q., Peng J., Zhou Y., Jiang S., Peng J., Nucleic Acids Res. 2019, 47, 6130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cawthorn W. P., Scheller E. L., Macdougald O. A., J. Lipid Res. 2012, 53, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gupta R. K., Arany Z., Seale P., Mepani R. J., Ye L., Conroe H. M., Roby Y. A., Kulaga H., Reed R. R., Spiegelman B. M., Nature 2010, 464, 619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gupta R. K., Mepani R. J., Kleiner S., Lo J. C., Khandekar M. J., Cohen P., Frontini A., Bhowmick D. C., Ye L., Cinti S., Spiegelman B. M., Cell Metab. 2012, 15, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hu X., Zhou Y., Yang Y., Peng J., Song T., Xu T., Wei H., Jiang S., Peng J., Open Biol. 2016, 6, 160065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Senagolage M. D., Sommars M. A., Ramachandran K., Futtner C. R., Omura Y., Allred A. L., Wang J., Yang C., Procissi D., Evans R. M., Han X., Bederman I. R., Barish G. D., Cell Rep. 2018, 25, 3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xiang H., Zhong Z.‐X., Peng Y.‐D., Jiang S.‐W., Int. J. Mol. Sci. 2017, 18, 1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kang S., Akerblad P., Kiviranta R., Gupta R. K., Kajimura S., Griffin M. J., Min J., Baron R., Rosen E. D., PLoS Biol. 2012, 10, e1001433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cristancho A. G., Schupp M., Lefterova M. I., Cao S., Cohen D. M., Chen C. S., Steger D. J., Lazar M. A., Proc. Natl. Acad. Sci. USA 2011, 108, 16271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nic‐Can G. I., Rodas‐Junco B. A., Carrillo‐Cocom L. M., Zepeda‐Pedreguera A., Peñaloza‐Cuevas R., Aguilar‐Ayala F., Rojas‐Herrera R., Int. J. Mol. Sci. 2019, 20, 3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vishvanath L., Gupta R. K., J. Clin. Invest. 2019, 129, 4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kahn C. R., Wang G., Lee K. Y., J. Clin. Invest. 2019, 129, 3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Merrick D., Sakers A., Irgebay Z., Okada C., Calvert C., Morley M. P., Percec I., Seale P., Science 2019, 364, 2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang Q. A., Song A., Chen W., Schwalie P. C., Zhang F., Vishvanath L., Jiang L., Ye R., Shao M., Tao C., Gupta R. K., Deplancke B., Scherer P. E., Cell Metab. 2018, 28, 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rodeheffer M. S., Birsoy K., Friedman J. M., Cell 2008, 135, 240. [DOI] [PubMed] [Google Scholar]
- 43. Tontonoz P., Hu E., Graves R. A., Budavari A. I., Spiegelman B. M., Genes Dev. 1994, 8, 1224. [DOI] [PubMed] [Google Scholar]
- 44. Barak Y., Nelson M. C., Ong E. S., Jones Y. Z., Ruiz‐Lozano P., Chien K. R., Koder A., Evans R. M., Mol. Cell 1999, 4, 585. [DOI] [PubMed] [Google Scholar]
- 45. Vidal‐Puig A., Jimenez‐Liñan M., Lowell B. B., Hamann A., Hu E., Spiegelman B., Flier J. S., Moller D. E., J. Clin. Invest. 1996, 97, 2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Luo H., Zhou Y., Hu X., Peng X., Wei H., Peng J., Jiang S., Cell Cycle 2015, 14, 1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wu Z., Rosen E. D., Brun R., Hauser S., Adelmant G., Troy A. E., Mckeon C., Darlington G. J., Spiegelman B. M., Mol. Cell 1999, 3, 151. [DOI] [PubMed] [Google Scholar]
- 48. Lefterova M. I., Zhang Y., Steger D. J., Schupp M., Cristancho A., Feng D., Zhuo D., Stoeckert C., Liu X., Lazar M., Genes Dev. 2008, 22, 2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Qi‐Qun T., Otto T. C., M Daniel L., Proc. Natl. Acad. Sci. USA 2003, 100, 850.12525691 [Google Scholar]
- 50. Wu Z., Wang S., Dev. Biol. 2013, 373, 235. [DOI] [PubMed] [Google Scholar]
- 51. Jiang S., Wei H., Song T., Yang Y., Zhang F., Zhou Y., Peng J., Jiang S., Cell Biosci. 2015, 5, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mori T., Sakaue H., Iguchi H., Gomi H., Okada Y., Takashima Y., Nakamura K., Nakamura T., Yamauchi T., Kubota N., Kadowaki T., Matsuki Y., Ogawa W., Hiramatsu R., Kasuga M., J. Biol. Chem. 2005, 280, 12867. [DOI] [PubMed] [Google Scholar]
- 53. Yang Y., Wei H., Song T., Cai A., Zhou Y., Peng J., Jiang S., Peng J., Mol. Cell. Endocrinol. 2017, 450, 43. [DOI] [PubMed] [Google Scholar]
- 54. Sun S., Zhang X., Lyu L., Li X., Yao S., Zhang J., J. Biol. Chem. 2016, 291, 25823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ross S. E., Science 2000, 289, 950. [DOI] [PubMed] [Google Scholar]
- 56. Bennett C. N., Ross S. E., Longo K. A., Bajnok L., Hemati N., Johnson K. W., Harrison S. D., Macdougald O. A., J. Biol. Chem. 2002, 277, 30998. [DOI] [PubMed] [Google Scholar]
- 57. Bowers R. R., Lane M. D., Cell Cycle 2008, 7, 1191. [DOI] [PubMed] [Google Scholar]
- 58. Isakson P., Hammarstedt A., Gustafson B., Smith U., Diabetes 2009, 58, 1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cawthorn W. P., Bree A. J., Yao Y., Du B., Hemati N., Martinez‐Santibañez G., Macdougald O. A., Bone 2012, 50, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Song T., Kuang S., Clin. Sci. 2019, 133, 2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Choy L., Skillington J., Derynck R., J. Cell Biol. 2000, 149, 667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Choy L., Derynck R., J. Biol. Chem. 2003, 278, 9609. [DOI] [PubMed] [Google Scholar]
- 63. Wang E. A., Israel D. I., Kelly S., Luxenberg D. P., Groundwater 1993, 9, 57. [DOI] [PubMed] [Google Scholar]
- 64. Jin W., Takagi T., Kanesashi S.‐N., Kurahashi T., Nomura T., Harada J., Ishii S., Dev. Cell 2006, 10, 461. [DOI] [PubMed] [Google Scholar]
- 65. Huang H., Song T.‐J., Li X., Hu L., He Q., Liu M., Lane M. D., Tang Q.‐Q., Proc. Natl. Acad. Sci. USA 2009, 106, 12670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tseng Y.‐H., Kokkotou E., Schulz T. J., Huang T. L., Winnay J. N., Taniguchi C. M., Tran T. T., Suzuki R., Espinoza D. O., Yamamoto Y., Ahrens M. J., Dudley A. T., Norris A. W., Kulkarni R. N., Kahn C. R., Nature 2008, 454, 1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Accili D., Taylor S. I., Proc. Natl. Acad. Sci. USA 1991, 88, 4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Haeusler R. A., Mcgraw T. E., Accili D., Nat. Rev. Mol. Cell Biol. 2018, 19, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chaika O. V., Chaika N., Volle D. J., Wilden P. A., Pirrucello S. J., Lewis R. E., J. Biol. Chem. 1997, 272, 11968. [DOI] [PubMed] [Google Scholar]
- 70. Laustsen P. G., Michael M. D., Crute B. E., Cohen S. E., Ueki K., Kulkarni R., Keller S., Lienhard G., Kahn C., Genes Dev. 2002, 16, 3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tseng Y.‐H., Kriauciunas K. M., Kokkotou E., Kahn C. R., Mol. Cell. Biol. 2004, 24, 1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Solheim M. H., Winnay J. N., Batista T. M., Molven A., Njølstad P. R., Kahn C. R., Diabetes 2018, 67, 1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Park J.‐Y., Kim Y., Im J., You S., Lee H., J. Evidence‐Based Complementary Altern. Med. 2014, 2014, 1 [Google Scholar]
- 74. Xu J., Liao K., J. Biol. Chem. 2004, 279, 35914. [DOI] [PubMed] [Google Scholar]
- 75. George S., Science 2004, 304, 1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yeh W. C., Bierer B. E., Mcknight S. L., Proc. Natl. Acad. Sci. USA 1995, 92, 11086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Nakae J., Kitamura T., Kitamura Y., Biggs W. H., Arden K. C., Accili D., Dev. Cell 2003, 4, 119. [DOI] [PubMed] [Google Scholar]
- 78. Tomlinson J. J., Boudreau A., Wu D., Salem H. A., Carrigan A., Gagnon A., Mears A. J., Sorisky A., Atlas E., Haché R. J. G., Mol. Endocrinol. 2010, 24, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Talukdar S., Bae E. J., Imamura T., Morinaga H., Fan W., Li P., Lu W., Watkins S., Olefsky J., Cell 2010, 142, 687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ichimura A., Hirasawa A., Poulain‐Godefroy O., Bonnefond A., Hara T., Yengo L., Kimura I., Leloire A., Liu N., Iida K., Choquet H., Besnard P., Lecoeur C., Vivequin S., Ayukawa K., Takeuchi M., Ozawa K., Tauber M., Maffeis C., Morandi A., Buzzetti R., Elliott P., Pouta A., Jarvelin M.‐R., Körner A., Kiess W., Pigeyre M., Caiazzo R., Van Hul W., Van Gaal L., Horber F., Balkau B., Lévy‐Marchal C., Rouskas K., Kouvatsi A., Hebebrand J., Hinney A., Scherag A., Pattou F., Meyre D., Koshimizu T.‐A., Wolowczuk I., Tsujimoto G., Froguel P., Nature 2012, 483, 350. [DOI] [PubMed] [Google Scholar]
- 81. Song T., Zhou Y., Peng J., Tao Y.‐X., Yang Y., Xu T., Peng J., Ren J., Xiang Q., Wei H., Mol. Cell. Endocrinol. 2016, 434, 1. [DOI] [PubMed] [Google Scholar]
- 82. Song T., Peng J., Ren J., Wei H.‐k., Peng J., Biomed Res. Int. 2015, 2015, 1 [Google Scholar]
- 83. Bi P., Kuang S., Trends Endocrinol. Metab. 2015, 26, 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Bi P., Shan T., Liu W., Yue F., Yang X., Liang X.‐R., Wang J., Li J., Carlesso N., Liu X., Kuang S., Nat. Med. 2014, 20, 911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. James A. W., Leucht P., Levi B., Carre A. L., Xu Y., Helms J. A., Longaker M. T., Tissue Eng., Part A 2010, 16, 2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Varjosalo M., Taipale J., Genes Dev. 2008, 22, 2454. [DOI] [PubMed] [Google Scholar]
- 87. Fontaine C., Cousin W., Plaisant M., Dani C., Peraldi P., Stem Cells 2008, 26, 1037. [DOI] [PubMed] [Google Scholar]
- 88. Suh J. M., Gao X., Mckay J., Mckay R., Salo Z., Graff J. M., Cell Metab. 2006, 3, 25. [DOI] [PubMed] [Google Scholar]
- 89. Kouzarides T., Cell 2007, 128, 693. [DOI] [PubMed] [Google Scholar]
- 90. Reik W., Nature 2007, 447, 425. [DOI] [PubMed] [Google Scholar]
- 91. Allis C. D., Jenuwein T., Nat. Rev. Genet. 2016, 17, 487. [DOI] [PubMed] [Google Scholar]
- 92. Holoch D., Moazed D., Nat. Rev. Genet. 2015, 16, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Shi H., Wei J., He C., Mol. Cell 2019, 74, 640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Desrosiers R., Friderici K., Rottman F., Proc. Natl. Acad. Sci. USA 1974, 71, 3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Perry R. P., Kelley D. E., Cell 1974, 1, 37. [Google Scholar]
- 96. Krug R. M., Morgan M. A., Shatkin A. J., J. Virol. 1976, 20, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Zhong S., Li H., Bodi Z., Button J., Vespa L., Herzog M., Fray R. G., Plant Cell 2008, 20, 1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Jia G., Fu Y., Zhao X., Dai Q., Zheng G., Yang Y., Yi C., Lindahl T., Pan T., Yang Y.‐G., He C., Nat. Chem. Biol. 2011, 7, 885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Dominissini D., Moshitch‐Moshkovitz S., Schwartz S., Salmon‐Divon M., Ungar L., Osenberg S., Cesarkas K., Jacob‐Hirsch J., Amariglio N., Kupiec M., Sorek R., Rechavi G., Nature 2012, 485, 201. [DOI] [PubMed] [Google Scholar]
- 100. Meyer K. D., Saletore Y., Zumbo P., Elemento O., Mason C. E., Jaffrey S. R., Cell 2012, 149, 1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Schibler U., Kelley D. E., Perry R. P., J. Mol. Biol. 1977, 115, 695. [DOI] [PubMed] [Google Scholar]
- 102. Roundtree I. A., Evans M. E., Pan T., He C., Cell 2017, 169, 1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Liu J., Harada B. T., He C., Trends Cell Biol. 2019, 29, 487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Frye M., Harada B. T., Behm M., He C., Science 2018, 361, 1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Aguilo F., Zhang F., Sancho A., Fidalgo M., Di Cecilia S., Vashisht A., Lee D.‐F., Chen C.‐H., Rengasamy M., Andino B., Jahouh F., Roman A., Krig S. R., Wang R., Zhang W., Wohlschlegel J. A., Wang J., Walsh M. J., Cell Stem Cell 2015, 17, 689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Zhao X., Yang Y., Sun B.‐F., Shi Y., Yang X., Xiao W., Hao Y.‐J., Ping X.‐L., Chen Y.‐S., Wang W.‐J., Jin K.‐X., Wang X., Huang C.‐M., Fu Y., Ge X.‐M., Song S.‐H., Jeong H. S., Yanagisawa H., Niu Y., Jia G.‐F., Wu W., Tong W.‐M., Okamoto A., He C., Danielsen J. M. R., Wang X.‐J., Yang Y.‐G., Cell Res. 2014, 24, 1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Li X., Yang J., Zhu Y., Liu Y., Shi X., Yang G., Int. J. Mol. Sci. 2016, 17, 1336. [Google Scholar]
- 108. Wang X., Sun B., Jiang Q., Wu R., Cai M., Yao Y., Liu Q., Shi H., Feng J., Wang Y., Int. J. Obes. 2018, 42, 1912 [DOI] [PubMed] [Google Scholar]
- 109. Deng X., Su R., Stanford S., Chen J., Front. Endocrinol. 2018, 9, 396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Dina C., Meyre D., Gallina S., Durand E., Körner A., Jacobson P., Carlsson L. M. S., Kiess W., Vatin V., Lecoeur C., Delplanque J., Vaillant E., Pattou F., Ruiz J., Weill J., Levy‐Marchal C., Horber F., Potoczna N., Hercberg S., Le Stunff C., Bougnères P., Kovacs P., Marre M., Balkau B., Cauchi S., Chèvre J.‐C., Froguel P., Nat. Genet. 2007, 39, 724. [DOI] [PubMed] [Google Scholar]
- 111. Merkestein M., Sellayah D., Int. J. Endocrinol. 2015, 2015, 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Merkestein M., Laber S., Mcmurray F., Andrew D., Sachse G., Sanderson J., Li M., Usher S., Sellayah D., Ashcroft F. M., Cox R. D., Nat. Commun. 2015, 6, 6792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Shen G.‐S., Zhou H.‐B., Zhang H., Chen B., Liu Z.‐P., Yuan Y., Zhou X.‐Z., Xu Y.‐J., Biochim. Biophys. Acta, Mol. Basis Dis. 2018, 1864, 3644. [DOI] [PubMed] [Google Scholar]
- 114. Wu R., Liu Y., Yao Y., Zhao Y., Bi Z., Jiang Q., Liu Q., Cai M., Wang F., Wang Y., Wang X., Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2018, 1863, 1323. [DOI] [PubMed] [Google Scholar]
- 115. Wang X., Zhu L., Chen J., Wang Y., Biochem. Biophys. Res. Commun. 2015, 459, 201. [DOI] [PubMed] [Google Scholar]
- 116. Wu R., Guo G., Bi Z., Liu Y., Zhao N., Chen N., Wang F., Wang Y., Wang X., Biochim. Biophys. Acta, Gene Regul. Mech. 2019, 1862, 796 [DOI] [PubMed] [Google Scholar]
- 117. Wang X., Wu R., Liu Y., Zhao Y., Bi Z., Yao Y., Liu Q., Shi H., Wang F., Wang Y., Autophagy 2020, 16, 1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Wu Y., Xie L., Wang M., Xiong Q., Guo Y., Liang Y., Li J., Sheng R., Deng P., Wang Y., Zheng R., Jiang Y., Ye L., Chen Q., Zhou X., Lin S., Yuan Q., Nat. Commun. 2018, 9, 4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Tian C., Huang Y., Li Q., Feng Z., Xu Q., Int. J. Mol. Sci. 2019, 20, 551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Yao Y., Bi Z., Wu R., Zhao Y., Liu Y., Liu Q., Wang Y., Wang X., FASEB J. 2019, 33, 7529. [DOI] [PubMed] [Google Scholar]
- 121. Kobayashi M., Ohsugi M., Sasako T., Awazawa M., Umehara T., Iwane A., Kobayashi N., Okazaki Y., Kubota N., Suzuki R., Waki H., Horiuchi K., Hamakubo T., Kodama T., Aoe S., Tobe K., Kadowaki T., Ueki K., Mol. Cell Biol. 2018, 38, e00116‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Chen J., Fang X., Zhong P., Song Z., Hu X., RNA Biol. 2019, 16, 991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Roundtree I. A., He C., Curr. Opin. Chem. Biol. 2016, 30, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Cai M., Liu Q., Jiang Q., Wu R., Wang X., Wang Y., IUBMB Life 2019, 71, 580. [DOI] [PubMed] [Google Scholar]
- 125. Zhou J., Wan J., Gao X., Zhang X., Jaffrey S. R., Qian S.‐B., Nature 2015, 526, 591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Jiang Q., Sun B., Liu Q., Cai M., Wu R., Wang F., Yao Y., Wang Y., Wang X., FASEB J. 2018, 33, 2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Xiao W., Adhikari S., Dahal U., Chen Y.‐S., Hao Y.‐J., Sun B.‐F., Sun H.‐Y., Li A., Ping X.‐L., Lai W.‐Y., Wang X., Ma H.‐L., Huang C.‐M., Yang Y., Huang N., Jiang G.‐B., Wang H.‐L., Zhou Q., Wang X.‐J., Zhao Y.‐L., Yang Y.‐G., Mol. Cell 2016, 61, 507. [DOI] [PubMed] [Google Scholar]
- 128. Meruvu S., Hugendubler L., Mueller E., J. Biol. Chem. 2011, 286, 26516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Du C., Ma X., Meruvu S., Hugendubler L., Mueller E., J. Lipid Res. 2014, 55, 1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Huang H., Weng H., Sun W., Qin X., Shi H., Wu H., Zhao B. S., Mesquita A., Liu C., Yuan C. L., Hu Y.‐C., Hüttelmaier S., Skibbe J. R., Su R., Deng X., Dong L., Sun M., Li C., Nachtergaele S., Wang Y., Hu C., Ferchen K., Greis K. D., Jiang X., Wei M., Qu L., Guan J.‐L., He C., Yang J., Chen J., Nat. Cell Biol. 2018, 20, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Chen J., Ren X., Li L., Lu S., Chen T., Tan L., Liu M., Luo Q., Liang S., Nie Q., Zhang X., Luo W., Int. J. Mol. Sci. 2019, 20, 2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Wang Y., Li Y., Toth J. I., Petroski M. D., Zhang Z., Zhao J. C., Nat. Cell Biol. 2014, 16, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Gantt K., Cherry J., Tenney R., Karschner V., Pekala P. H., J. Biol. Chem. 2005, 280, 24768. [DOI] [PubMed] [Google Scholar]
- 134. Jones H., Carver M., Pekala P. H., Biochem. Biophys. Res. Commun. 2007, 355, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Lambert S. A., Jolma A., Campitelli L. F., Das P. K., Yin Y., Albu M., Chen X., Taipale J., Hughes T. R., Weirauch M. T., Cell 2018, 172, 650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Yue Y., Liu J., He C., Genes Dev. 2015, 29, 1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Heck A. M., Wilusz C. J., Biochim. Biophys. Acta, Gene Regul. Mech. 2019, 1862, 194402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Tao X., Chen J., Jiang Y., Wei Y., Chen Y., Xu H., Zhu L., Tang G., Li M., Jiang A., Shuai S., Bai L., Liu H., Ma J., Jin L., Wen A., Wang Q., Zhu G., Xie M., Wu J., He T., Huang C., Gao X., Li X., BMC Genomics 2017, 18, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Martin Carli J. F., Leduc C. A., Zhang Y., Stratigopoulos G., Leibel R. L., J. Lipid Res. 2018, 59, 1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Su R., Dong L., Li C., Nachtergaele S., Wunderlich M., Qing Y., Deng X., Wang Y., Weng X., Hu C., Yu M., Skibbe J., Dai Q., Zou D., Wu T., Yu K., Weng H., Huang H., Ferchen K., Qin X., Zhang B., Qi J., Sasaki A. T., Plas D. R., Bradner J. E., Wei M., Marcucci G., Jiang X., Mulloy J. C., Jin J., He C., Chen J., Cell 2018, 172, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Gulati P., Avezov E., Ma M., Antrobus R., Lehner P., O'rahilly S., Yeo G. S. H., Biosci. Rep. 2014, 34, e00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Wu Q., Saunders R. A., Szkudlarek‐Mikho M., De La Serna I., Chin K.‐V., Biochem. Biophys. Res. Commun. 2010, 401, 390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Xiang Y., Laurent B., Hsu C.‐H., Nachtergaele S., Lu Z., Sheng W., Xu C., Chen H., Ouyang J., Wang S., Ling D., Hsu P.‐H., Zou L., Jambhekar A., He C., Shi Y., Nature 2017, 543, 573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Knuckles P., Carl S. H., Musheev M., Niehrs C., Wenger C., Bühler M., Nat. Struct. Mol. Biol. 2017, 24, 561. [DOI] [PubMed] [Google Scholar]
- 145. Akichika S., Hirano S., Shichino Y., Suzuki T., Nishimasu H., Ishitani R., Sugita A., Hirose Y., Iwasaki S., Nureki O., Suzuki T., Science 2019, 363, 0080. [DOI] [PubMed] [Google Scholar]
- 146. Bertero A., Brown S., Madrigal P., Osnato A., Ortmann D., Yiangou L., Kadiwala J., Hubner N. C., De Los Mozos I. R., Sadée C., Lenaerts A.‐S., Nakanoh S., Grandy R., Farnell E., Ule J., Stunnenberg H. G., Mendjan S., Vallier L., Nature 2018, 555, 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Cai X., Wang X., Cao C., Gao Y., Zhang S., Yang Z., Liu Y., Zhang X., Zhang W., Ye L., Cancer Lett. 2018, 415, 11. [DOI] [PubMed] [Google Scholar]
- 148. Du Y., Hou G., Zhang H., Dou J., He J., Guo Y., Li L., Chen R., Wang Y., Deng R., Huang J., Jiang B., Xu M., Cheng J., Chen G.‐Q., Zhao X., Yu J., Nucleic Acids Res. 2018, 46, 5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Schöller E., Weichmann F., Treiber T., Ringle S., Treiber N., Flatley A., Feederle R., Bruckmann A., Meister G., RNA 2018, 24, 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Cowger J. J. M., Zhao Q., Isovic M., Torchia J., Oncogene 2007, 26, 3378. [DOI] [PubMed] [Google Scholar]
- 151. Lee D.‐F., Walsh M. J., Aguiló F., Trends Biochem. Sci. 2016, 41, 986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Kwak S., Kim T. W., Kang B.‐H., Kim J.‐H., Lee J.‐S., Lee H.‐T., Hwang I.‐Y., Shin J., Lee J.‐H., Cho E.‐J., Youn H.‐D., Nucleic Acids Res. 2018, 46, 6592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Wolfram S., Raederstorff D., Preller M., Wang Y., Teixeira S. R., Riegger C., Weber P., J. Nutr. 2006, 136, 2512. [DOI] [PubMed] [Google Scholar]
- 154. Wu R., Yao Y., Jiang Q., Cai M., Liu Q., Wang Y., Wang X., Int. J. Obes. 2018, 42, 1378. [DOI] [PubMed] [Google Scholar]
- 155. Peng S., Xiao W., Ju D., Sun B., Hou N., Liu Q., Wang Y., Zhao H., Gao C., Zhang S., Cao R., Li P., Huang H., Ma Y., Wang Y., Lai W., Ma Z., Zhang W., Huang S., Wang H., Zhang Z., Zhao L., Cai T., Zhao Y.‐L., Wang F., Nie Y., Zhi G., Yang Y.‐G., Zhang E. E., Huang N., Sci. Transl. Med. 2019, 11, 7116. [DOI] [PubMed] [Google Scholar]
- 156. Huang Y., Su R., Sheng Y., Dong L., Dong Z., Xu H., Ni T., Zhang Z. S., Zhang T., Li C., Han L., Zhu Z., Lian F., Wei J., Deng Q., Wang Y., Wunderlich M., Gao Z., Pan G., Zhong D., Zhou H., Zhang N., Gan J., Jiang H., Mulloy J. C., Qian Z., Chen J., Yang C.‐G., Cancer Cell 2019, 35, 677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Weng H., Huang H., Wu H., Qin X., Zhao B. S., Dong L., Shi H., Skibbe J., Shen C., Hu C., Sheng Y., Wang Y., Wunderlich M., Zhang B., Dore L. C., Su R., Deng X., Ferchen K., Li C., Sun M., Lu Z., Jiang X., Marcucci G., Mulloy J. C., Yang J., Qian Z., Wei M., He C., Chen J., Cell Stem Cell 2018, 22, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Chen B., Ye F., Yu L., Jia G., Huang X., Zhang X., Peng S., Chen K., Wang M., Gong S., Zhang R., Yin J., Li H., Yang Y., Liu H., Zhang J., Zhang H., Zhang A., Jiang H., Luo C., Yang C.‐G., J. Am. Chem. Soc. 2012, 134, 17963. [DOI] [PubMed] [Google Scholar]
- 159. Zheng G., Cox T., Tribbey L., Wang G. Z., Iacoban P., Booher M. E., Gabriel G. J., Zhou L., Bae N., Rowles J., He C., Olsen M. J., ACS Chem. Neurosci. 2014, 5, 658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Singh B., Kinne H. E., Milligan R. D., Washburn L. J., Olsen M., Lucci A., PLoS One 2016, 11, e0159072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Cui Q., Shi H., Ye P., Li L., Qu Q., Sun G., Sun G., Lu Z., Huang Y., Yang C.‐G., Riggs A. D., He C., Shi Y., Cell Rep. 2017, 18, 2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Huang Y., Yan J., Li Q., Li J., Gong S., Zhou H., Gan J., Jiang H., Jia G.‐F., Luo C., Yang C.‐G., Nucleic Acids Res. 2014, 43, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Aik W., Scotti J. S., Choi H., Gong L., Demetriades M., Schofield C. J., Mcdonough M. A., Nucleic Acids Res. 2014, 42, 4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Xu C., Liu K., Tempel W., Demetriades M., Aik W., Schofield C. J., Min J., J. Biol. Chem. 2014, 289, 17299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Deng X., Su R., Weng H., Huang H., Li Z., Chen J., Cell Res. 2018, 28, 507. [DOI] [PMC free article] [PubMed] [Google Scholar]