

Abstract
Cross-coupling of two alkyl fragments is an efficient method to produce organic molecules rich in sp3-hybridized carbon centers, which are attractive candidate compounds in drug discovery. Enantioselective C(sp3)–C(sp3) coupling is challenging, especially of alkyl electrophiles without an activating group (aryl, vinyl, carbonyl). Here we report a strategy based on nickel hydride addition to internal olefins followed by nickel-catalyzed alkyl-alkyl coupling. This strategy enables enantioselective cross-coupling of non-activated alkyl halides with alkenyl boronates to produce chiral alkyl boronates. Employing readily available and stable olefins as pro-chiral nucleophiles, the coupling proceeds under mild conditions and exhibits broad scope and high functional group tolerance. Applications for the functionalization of natural products and drug molecules, as well as the synthesis of chiral building blocks and a key intermediate to (S)-(+)-Pregabalin, are demonstrated.
In drug discovery, it has been recognized that organic compounds with a greater 3-dimensional (3D) shape than flat aromatics have higher chances to succeed as drug candidates1,2. The fraction of sp3 carbons in a molecule is suggested as a descriptor for its 3D shape1. Because high-throughput synthesis has become a standard practice in the pharmaceutical industry, methods introducing sp3 carbons in a parallel manner, such as cross-coupling of alkyl electrophiles3–6, are highly valuable for drug development. However, the enantioselective cross-coupling of alkyl electrophiles, especially alkyl-alkyl coupling, remains challenging3,4,7,8.
One strategy for enantioselective C(sp3)–C(sp3) coupling consists of enantioselective metal hydride addition to an internal olefin to form a chiral metal-alkyl intermediate, followed by enantiospecific alkyl-alkyl coupling (Fig. 1a). This approach creates a stereogenic center at a carbon center of the olefin. The approach complements enantioconvergent alkyl-alkyl coupling of racemic alkyl electrophiles9–16, which is still limited in scope. While reports of stereoconvergent coupling of racemic α-zincated N-Boc-pyrrolidines with alkyl halides suggested the feasibility of this mode of alkyl-alkyl coupling17,18, the challenge rests on the ability of a metal hydride catalyst to perform both enantioselective addition to an internal olefin and coupling with non-activated alkyl electrophiles. Cu–H catalysed enantioselective functionalization of internal olefins is advancing rapidly in recent years19–25, but the coupling of the in-situ generated chiral organocopper intermediates with non-activated alkyl electrophiles remains elusive (Fig. 1b). Ni–H catalysis is more suited than Cu–H catalysis for coupling with alkyl electrophiles15,16,26,27, however, Ni–H insertion into an internal olefin typically leads to chain-walking to form a terminal, primary-alkyl nickel intermediate28–33, ablating the chirality generated in the initial insertion (Fig. 1c).
Figure 1. Strategies for enantioselective C(sp3)-C(sp3) cross-coupling.

a, Enantioselective metal hydride insertion to internal olefins followed by enantiospecific alkyl-alkyl coupling. b, Challenge for Cu–H chemistry: no precedent of cross coupling of non-activated alkyl electrophiles with a transient chiral alkyl–Cu intermediate. c, Challenge for Ni–H chemistry: an unwanted chain walking event results in an ablation of the newly generated stereocenter (initially a new stereocenter is created upon metal hydride insertion into an olefin, but the stereocenter is lost due to chain walking generating a primary alkyl–Ni species). d, This work: Ni-catalysed enantioselective cross-coupling of non-activated alkyl halides with internal olefins. e, Comparison to other catalytic methods for the synthesis of chiral alkyl boronates: asymmetric hydrogenation of specialized and hard-to-access substrates by Ir-catalysis, asymmetric hydroboration of olefins bearing a specific directing group by Cu-catalysis, Pd-catalysed asymmetric 1,2-metallate rearrangement followed by cross coupling which employs highly reactive and difficult to handle organolithium reagents (RLi), and Ni-catalysed enantioconvergent cross coupling of α-halo boronates with highly reactive organozinc reagents (RZnBr). These methods provide limited substrate scope and were less practical or general than the method described in the current work. DG = directing group
A directing α-aryl or α-boryl group can stabilize a branched organonickel intermediate27,34–36. Enantioselective coupling of such an intermediate with an alkyl electrophile remained elusive, although a single example of analogous coupling with PhI with low enantioselectivity (62% e.e.) has been reported36. Here we describe Ni–H catalysed enantioselective C(sp3)–C(sp3) cross-coupling of non-activated alkyl halides with alkenyl boronates (Fig. 1d). This coupling yields a diverse range of chiral alkyl boronic acid pinacol esters (Bpins), which are both versatile intermediates and important endpoints to bio-active molecules37–41. Chiral alkyl boronates might be prepared by hydroboration42, Matteson reaction43, or using enantioenriched α-lithiated benzoates44. However, these strategies either suffer from regioselectivity issues or require stoichiometric chiral reagents. Recently new approaches based on asymmetric catalysis such as hydrogenation45, directed hydroboration46, 1,2-metallate rearrangement47, and enantioconvergent Negishi coupling48 have been developed (Fig. 1e). Nevertheless, significant limitations still exist. For example, specialized and hard-to-access substrates were required for hydrogenation, and substrates with a specific directing group were necessary for hydroboration. On the other hand, methods based on 1,2-metallate rearrangement and Negishi coupling employ reactive organometallic reagents, which compromise functional group compatibility. By using readily available and stable olefins as nucleophiles and unactivated alkyl halides as electrophiles under mild reaction conditions, our method provides notable advantages in reaction efficiency, substrate availability and scope, as well as functional group tolerance. In particular, applications for the post-product functionalization of many drug molecules and natural products are demonstrated.
Results and discussion
Reaction development
We recently developed Ni–H catalysed hydrocarbonation of alkenyl Bpins27. To achieve enantioselective C(sp3)–C(sp3) coupling based on this racemic reaction, we screened various chiral ligands and fine-tuned other reaction parameters. Our model reaction was the coupling of trans-1-hexenylboronic acid pinacol ester (1a) with 3-phenylpropyl iodide (2a) to give (S)-4,4,5,5-tetramethyl-2-(1-phenylnonan-4-yl)-1,3,2-dioxaborolane (3a) (Table 1). The optimized reaction conditions were established as the following: NiCl2 (15 mol%) as the Ni source, Bi-Ox L6 (20 mol%) as the ligand, diethoxy-methylsilane (DEMS, 2.5 equiv.) as the hydride source, KF (2.5 equiv.) as the base, a (3:2) mixture of DCE/DMF as the solvent, room temperature, and 40 hour reaction time. Under these conditions, 3a was obtained as a single regioisomer in 72% GC yield (69% isolated yield) and 92% e.e. (entry 1, Table 1). The main side products were an alkane originated from protodeiodination of 2a and an alkyl chloride originated from transhalogenation of 2a with NiCl2. Products that would form due to Suzuki-type cross-coupling or hydrosilylation of alkenyl Bpin or HI elimination from alkyl iodide were not detected.
Table 1. Summary of the effects of reaction parameters on the reaction efficiencya .

|
| Entry | Variants | Yield (%) | e.e. (%)b |
|---|---|---|---|
| 1 | none | 72 (69)c | 92 |
| 2 | L1 | 73 | 52 |
| 3 | L2 | 76 | 42 |
| 4 | L3 | 31 | 36d |
| 5 | L4 | 47 | 60 |
| 6 | L5 | 37 | 58 |
| 7 | NiBr2 | 52 | 80 |
| 8 | NiBr2.diglyme | 51 | 84 |
| 9 | Ni(COD)2 | 31 | 80 |
| 10e | CsF | 70 | 68 |
| 11 | PMHS | 57 | 91 |
| 12 | DMF as solvent | 73 | 66 |
| 13 | DCE as solvent | n.d. | n.d. |
See the Supporting Information for section 2 experimental details; all reactions were carried out in a 0.1 mmol scale with respect to 2a; corrected GC yields using n-dodecane as an internal standard were reported.
The enantiomeric excesses (e.e.s) were determined using HPLC analysis of the corresponding alcohol after stereospecific oxidation of the boronic ester (see Supplementary Information section 2 for full details).
Isolated yield is shown in the parenthesis.
The opposite enantiomer was enriched.
Reaction time = 12h. DMF = Dimethylformamide; DCE = 1,2-Dichloroethane; RT = room temperature; h = hour. n.d. = not detected; PMHS = Polymethylhydrosiloxane; DEMS = Diethoxy-methylsilane.
The influence of different reaction parameters in the outcome of the reaction is described in Supplementary Tables 1-6. A concise summary of key observations is shown in Table 1. Structurally related Pyr-Ox (L1 and L2) and Box (L3) ligands were inefficient in this transformation (entries 2-4, Table 1). Bi-Ox ligands with alkyl substituents gave lower yields or e.e.s (entries 5 and 6, Table 1). The reactions were sensitive to the substituents on the aryl units of the Bi-Ox ligands (see Supplementary Table 3). Other nickel(II) sources such as NiBr2 and NiBr2.diglyme afforded lower yields and enantioselectivities (entries 7 and 8, Table 1). The use of Ni(COD)2 as precatalyst led to lower yield and e.e. (entry 9). Among alkali metal fluoride bases, those containing a large cation (i.e., Rb+ and Cs+) gave good yields while those with a small cation (i.e., Li+ and Na+) shut down the hydroalkylation (see Supplementary Table 6)49. Compared to KF, CsF and RbF decreased the enantioselectivity (entry 10, Table 1 and see Supplementary Table 6)50. We suspect a noncovalent interaction network between the alkali metal cation, π-system of the phenyl unit of L6 and oxygen atom of the boronic ester was important for the enantioselectivity, which is then sensitive to the nature of the cation51,52. Reactions were sensitive to the substituents of hydrosilanes (see Supplementary Table 5). Whereas a variety of hydrosiloxanes could be used, less electrophilic hydrosilanes such as Et3SiH, PhSiH3, Ph3SiH and PhMe2SiH were inefficient hydride donors probably because they could not be activated by KF to form a reactive penta-coordinate hydrosilicate species53. Reactions using DEMS, PMHS (polymethylhydrosiloxane), and (EtO)3SiH gave similar enantioselectivity, but the reaction using DEMS had the highest yield (entry 11, Table 1 and see Supplementary Table 5). A lower amount of DEMS (1.5 equiv.) diminished the yield (53%) but not enantioselectivity, suggesting that a larger amount of DEMS was necessary to promote the formation and insertion of Ni–H against side reactions54. The mixed solvent (DCE/DMF) turned out as the best solvent to achieve high enantioselectivity. DCE alone was not a suitable solvent (entry 13, Table 1) likely because it cannot dissolve a sufficient amount of NiCl2. The reaction in DMF alone had a similar yield but lower e.e. than that in the mixed solvent (entry 12, Table 1), demonstrating a sensitivity of the enantioselectivity on the solvent properties. Using a pre-formed NiCl2-L6 complex as catalyst gave the product in 61% yield with 92% e.e. (see Supporting Information section 12.7), suggesting the presence of this species in the catalytic cycle.
The Substrate scope
The scope of this enantioselective coupling method is broad (Table 2). In addition to an aryl group (3a, 3b, and 3j), primary alkyl iodides containing a pendant ether (3c), ketone (3d), ester (3e, 3f, 3k, and 3l), carbamate (3h), phthalimide (3i), and amine (3m and 3n) groups all reacted well. These data rule out the possibility of a specific group that directs the enantioselectivity of the coupling. A high level of functional group tolerance, unusual for cross-coupling of organometallic reagents, was achieved. For example, an unprotected OH group was compatible (3j). Despite the ability of Ni to activate aryl iodides, bromides and chlorides, our method tolerated these potentially reactive groups (3e-3g). Substrates containing medicinally relevant heterocycles such as furan (3k), thiophene (3l), indole (3m) and piperidine (3n) were also viable.
Table 2. Scope of Ni–H catalysed enantioselective C(sp3)–C(sp3) couplinga .
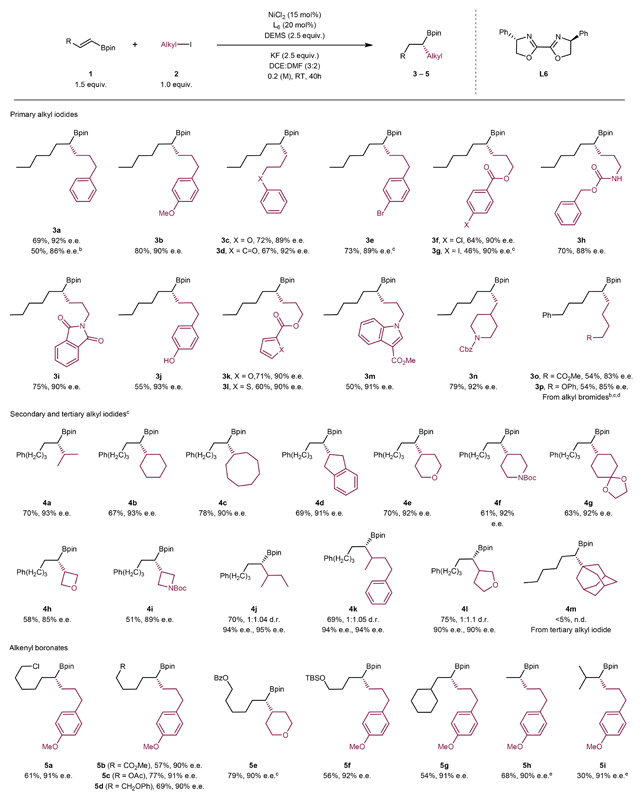
|
Conditions: All reactions were carried out with NiCl2 (15 mol%), ligand L12 (20 mol%), 1 (0.15 mmol), 2 (0.10 mmol), DEMS (0.25 mmol), KF (0.25 mmol) and DCE:DMF (0.5 mL) at room temperature for 40 hours. The enantiomeric excesses (e.e.s) were determined using HPLC analysis (see Supplementary Information section 6 for full details). b Alkyl bromide with 40 mol% KI was used. c 1 (0.1 mmol) and 2 (0.15 mmol) were used. d Reactions were conducted in a 0.2 mmol scale with respect to 1. e Reactions were conducted in a 0.2 mmol scale with respect to 2.
The coupling of alkyl bromides required an in-situ Br/I exchange and was slightly less efficient than the corresponding coupling of alkyl iodides. For example, the coupling of trans-1-hexenylboronic acid pinacol ester (1a) with 3-phenylpropyl bromide gave 3a in 50% yield and 86% e.e. in the presence of 40 mol% KI. By comparison, an analogous coupling using the corresponding alkyl iodide 2a gave 69% yield and 92% e.e.. Similar yields and e.e.s were obtained for two other alkyl bromides (3o and 3p). More inert alkyl electrophiles such as alkyl chloride or alkyl triflate were unsuitable coupling partners. Coupling of an alkyl triflate in the presence of 40 mol% KI gave only a trace amount of the desired product indicating the inefficiency of triflate/I exchange.
Enantioselective cross-coupling of two secondary alkyl fragments is challenging55. Thus, it is noteworthy that the present method also works for the coupling of secondary alkyl iodides, including both acyclic and cyclic substrates, delivering the corresponding alkyl Bpins with good yields and high enantioselectivity (4a-4i). Medicinally interesting cyclic groups such as indane, oxetane and azeditine were tolerated (4d-4i). No isomerization was observed in the alkyl fragments. Coupling of unsymmetrical secondary alkyl iodides gave good yields, but no diastereoselectivity (4j-4l). The enantioselectivities for both diastereomers were high (90-95% e.e.). The coupling of unactivated tertiary alkyl iodides such as tert-butyl iodide and 1-iodoadamentane was unsuccessful (e.g., 4m), likely due to their steric hindrance against oxidative addition to Ni.
A wide range of alkenyl Bpins could be used as nucleophiles to deliver the corresponding enantiomerically enriched alkyl Bpins (5a-5i) (Table 2). The coupling was regioselective at the carbon α-to the Bpin group. The alkenes can contain functional groups such as alkyl chloride (5a), ester (5b and 5c), ether (5d, 5e and 5f). Vinylboronate, a synthetically useful substrate posing a challenge in regioselectivity, was coupled in high enantioselectivity (90% e.e.) and regioselectivity (12:1 b/l). Coupling of a sterically demanding β,β’-disubstituted alkenyl Bpin was less efficient, giving the product in 30% yield. Coupling of a trisubstituted vinyl boronate 2-(cyclohex-1-en-1-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane was unsuccessful, probably due to difficulty in the addition of Ni–H to the olefin compared to protodeiodination of the alkyl iodide. Among organoboron reagents, Bpin derivatives seem unique for this transformation. The coupling of alkenyl 9-BBN derivative of boronic acid was unsuccessful, as these reagents seemed to decompose under the reaction conditions. In these cases, the majority of alkyl iodides remained intact.
The Bpin groups in products 3e and 4g were stereospecifically oxidized to give alcohols (3e’ and 4g’; see Supporting Information section 14). The X-ray crystal structures of 3e’ and 4g’ revealed the absolute configuration of the chiral carbon centers. By analogy, we assigned the corresponding absolute configurations to all products.
The reaction of a cis-alkenyl Bpin (1l) gave a product (3b) identical to the reaction of its trans-analogue (see Supporting Information section 12.1). However, under reaction conditions and even without a Ni catalyst, cis-alkenyl Bpin (1l) would be first converted to its trans-analogue (1a) prior to hydroalkylation (see Supporting Information section 12.2).
Synthetic applications & diversification of chiral products
Functionalization of drug molecules and natural products typically require mild reaction conditions and high functional group tolerance. As such, the present method is well suited for this purpose, and could be used to synthesize an array of chiral alkyl Bpins bearing complex or bio-active alkyl fragments derived from drugs and natural products (Table 3). Alkyl iodides bearing multiple stereocenters derived from nopol, a chiral terpinol (6a), naproxen (6b), a nonsteroidal anti-inflammatory drug, and a lithocholic acid derivative (6d) were all viable electrophiles, yielding potentially valuable products in synthetically useful yields and high diastereoselectivity. In addition, alkyl iodides derived from drugs such as gemfibrozil (6c), isoxepac (6f), indomethacin (6g), probenecid (6h) and adapalene (6i) as well as from a herbicide 2,4-D (6e) were transformed into the corresponding chiral alkyl Bpins with ease.
Table 3. Functionalization of natural product and drug derivatives.a .
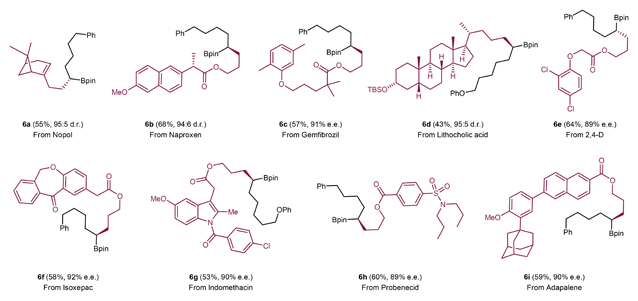
|
Conditions: All reactions were carried out with NiCl2 (15 mol%), ligand L12 (20 mol%), 1 (0.15 mmol), 2 (0.10 mmol), DEMS (0.25 mmol), KF (0.25 mmol) and DCE:DMF (0.5 mL) at room temperature for 40 hours. The enantiomeric excesses (e.e.s) and diastereomeric ratios (d.r.s) were determined using HPLC analysis (see Supplementary Information section 6 for full details).
Chiral alkyl Bpins are powerful intermediates in asymmetric organic synthesis because the C–B moiety can be easily transformed into a C–X moiety (X = C or heteroatom) with the conservation of chirality at the α-C center39,40. We provide several illustrative examples in Figure 2a for the transformation of one coupling product 4e. C–C, C–O, and C–Br bond formation reactions proceeded cleanly, affording chiral organic compounds (7-10) without erosion in enantiomeric excess. We also applied our method for the enantioselective synthesis of a key chiral amino alcohol intermediate to the drug (S)-(+)-Pregabalin (Fig. 2b)56. Coupling of trans-3-methyl-1-butenyl boronic acid pinacol ester (1m) with tert-butyl(2-iodoethoxy)diphenylsilane (2m’) provided 11 in 42% yield with 90% e.e. Stereospecific homologation of 11, amination and silyl ether-deprotection provided the amino alcohol intermediate 12 in 43% overall yield from 11. Conversion of 12 to pregabalin has been previously reported57. A gram-scale reaction between trans-5-phenyl-1-pentenyl boronic acid pinacol ester (1b) and 4-iodotetrahydro-2H-pyran (2v) using a reduced catalyst loading (10 mol%) afforded 4e in 62% yield (1.116 g) and 93% e.e. (Fig. 2c). A one-pot reaction sequence consisted of hydroboration of 1-hexyne to give alkenyl Bpin 1a in-situ, followed by cross-coupling with 2b without isolating 1a, yielded 3b in 74% yield and 90% e.e. (Fig. 2d). These results further showcase the preparative utility of the coupling.
Figure 2. Synthetic applications.

a, Conversion of chiral alkyl Bpins to a diverse array of valuable chiral products such as C–heteroarene coupling product 7, homologation followed by oxidation to primay alcohol bearing a β-tertiary carbon stereocenter 8, alkyl bromide 9 via C–Br bond formation and alcohol 10 through oxidation (see Supplementary Information section 7 for full details). b, Synthesis of 12, a key intermediate of (S)-(+)-Pregabalin (see Supplementary Information section 8 for full details). c, A gram-scale reaction using 10 mol% catalyst (see Supplementary Information section 10 for full details). d, One-pot asymmetric hydroalkylation without isolation of alkenyl Bpin (see Supplementary Information section 11 for full details). NBS = N-Bromosuccinimide; TBAF = tetrabutylammoniumfluoride, PMP = paramethoxyphenyl.
Mechanistic considerations
When radical clock 5-hexenyliodide (2n’) was used as a substate, cyclization occurred. The product originated from cyclization of the 5-hexenyl radical (13) was obtained in 11% yield and 91% e.e. (Fig. 3a). The coupling product originated from uncyclized 5-hexenyl radical was also detected but it was difficult to purify and isolate. This data supports the intermediacy of alkyl radicals. When 1.0 equiv. of radical scavenger (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) was added to the reaction, no desired product was detected (Fig. 3b).
Figure 3. Mechanistic studies of the catalytic enantioselective C(sp3)–C(sp3) cross-coupling.

a, Radical clock experiment with an alkene-tethered alkyl iodide 2n’ afforded the formation of 13 under standard conditions, suggesting the intermediacy of an alkyl radical and its subsequent cyclization and C(sp3)–C(sp3) coupling (see Supplementary Information section 12.4 for full details). b, Reaction was completely shut down by the addition of a radical scavanger TEMPO (see Supplementary Information section 12.5 for full details). c, Probe for origin of regioselectivity by using a tridentate Py-box ligand (L20) instead of L6 (see Supplementary Information section 12.6 for full details). d, Reactions with substrates where the alkenyl group is distal to the Bpin group: chain-walking products were formed with high enantioselectivity under standard conditions (see Supplementary Information section 13 for full details). e, Outline of a possible reaction pathway for Ni-catalysed enantioselective C(sp3)–C(sp3) cross coupling. r.r = Regioisomeric ratio; PMP = paramethoxyphenyl.
The regioselectivity of the reactions might be due to two factors: i) Stabilization of the α-boryl Ni-alkyl intermediate by the vacant p-orbital of the Bpin unit. Similar regioselectivity was observed in the addition of Cp2ZrHCl (Schwartz’s reagent) to alkenyl BBN58. ii) Coordination of the oxygen atom of the Bpin unit to the Ni center. We tested the second possibility using a tridentate Py-box ligand (L20), which was expected to hamper the coordination of the oxygen atom of the Bpin to the Ni center. While the coupling yield was modest (31%), the reaction is highly α-selective (r.r. = 13:1) (Fig. 3c). This result suggests that the regioselectivity is not likely due to oxygen coordination.
We tested several substrates (1n, 1o, and 1p) where the alkenyl group is distal to the Bpin group (Fig. 3d). Products originated from hydroalkylation at α-C to Bpin were obtained in low to modest yields but high enantioselectivity (90% and above). Other regioisomers were also formed. These data indicate chain-walking of the distal alkenyl group mediated by Ni–H until forming the stable α-boryl Ni-alkyl species.
The mechanism of this Ni–H catalysed enantioselective C(sp3)–C(sp3) cross-coupling is proposed in Figure 3e, analogous to a previous proposal on Ni-catalysed hydroalkylation15. Under the reaction conditions, a chiral L*Ni(I)–Cl species (A) is formed as the actual catalyst, which undergoes single electron transfer (SET) with an alkyl iodide to generate an alkyl radical and L*ClNi(II)–I (B). The reaction of B with a hydrosilane generates a Ni–H species L*ClNi(II)–H (C) that inserts to alkenyl Bpin. The insertion is regioselective at the α-C to the boryl group, generating a chiral alkyl intermediate (D). The resulting Ni-alkyl intermediate (D) then recombines with the alkyl radical to give a high-valent Ni(III) complex (E), which undergoes reductive elimination to give the product. We propose the stereoselective step is the insertion of a chiral Ni–H into the olefin. Alternatively, the reaction proceeds by reversible Ni-alkyl homolysis followed by stereoselective reductive elimination59. The reaction profile excludes a kinetic resolution process (see Supporting Information section 12.3).
Conclusion
In summary, we have developed Ni–H catalysed enantioselective C(sp3)–C(sp3) coupling of non-activated alkyl iodides with alkenyl Bpins. By employing readily available and stable olefin as nucleophiles, this coupling enables the streamlined synthesis of chiral alkyl Bpins under mild conditions, with a previously unattained scope and functional group tolerance. Examples in post-product functionalization and chiral syntheses demonstrate the potential utility of this method in drug discovery.
Methods
General Procedure for probing the scope of enantioselective C(sp3)–C(sp3) cross-coupling of non-activated alkyl electrophiles
To an oven-dried 10 mL Teflon-screw capped test tube were added NiCl2 (1.9 mg, 15 μmol, 0.15 equiv.) and L6 (5.8 mg, 0.02 mmol, 0.20 equiv.). The vial was introduced in a nitrogen-filled glovebox. A magnetic stir bar (6x15 mm), anhydrous DCE (0.30 mL) and DMF (0.20 mL) were added, and the mixture was stirred for 40 minutes at room temperature. Then anhydrous KF (14.5 mg, 0.25 mmol, 2.50 equiv.) was added to it and the stirring was continued for additional 2–3 minutes, at which point alkenyl boronic acid pinacol ester 1 (0.15 mmol, 1.00 equiv.) was added and the mixture was stirred for additional 1 minute. Then alkyl iodide 2 (0.10 mmol, 1.50 equiv.) was added to the resulting mixture [for cross coupling with secondary alkyl iodides: alkenyl boronic acid pinacol ester 1 (0.10 mmol, 1.00 equiv.) and secondary alkyl iodide (0.15 mmol, 1.50 equiv.) were used]. Stirring was further continued for 5 minutes, then DEMS (43.0 μL, 0.25 mmol, 2.50 equiv.) was added dropwise to it. The test tube was then sealed with airtight electrical tapes and removed from the glove box and stirred at RT for 40 hours maintaining 460 rpm. The crude reaction mixture was directly subjected to flash column chromatography by using a mixture of hexane and EtOAc to obtain 3a – 6i.
Supplementary Material
Acknowledgement
This work is supported by the Swiss National Science Foundation. We thank Dr. Farzaneh Fadaei Tirani for the determination of the X-ray crystal structures of compounds 3e’ and 4g’ . Correspondence and requests for materials should be addressed to Xile Hu.
Footnotes
Author contributions
S.B. and X.H. conceived the project. S.B. designed and optimized the synthetic method. S.B. and R.M. studied the scope, application and mechanism. All authors analyzed the data and co-wrote the manuscript. X.H. directed the research. S.B. and R.M. contributed equally to this work.
Competing Interests Statement
The authors declare no competing interests.
Data Availability statement
Crystallographic data for 3e’ and 4g’ reported in this Article have been deposited at the Cambridge Crystallographic Data Centre, under deposition number CCDC 2011678 (3e’) and CCDC 1971802 (4g’). Copies of the data can be obtained free of charge via www.ccdc.cam.ac.uk. All other data supporting the findings of this study, including experimental procedures and compound characterization, NMR, HPLC and X-ray analysis are available within the Article and its Supplementary Information.
References
- 1.Lovering F, Bikker J, Humblet C. Escape from flatland: increasing saturation as an approach to improving clinical success. J Med Chem. 2009;52:6752–6756. doi: 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- 2.Ritchie TJ, Macdonald SJ. The impact of aromatic ring count on compound developability--are too many aromatic rings a liability in drug design? Drug Discov Today. 2009;14:1011–1020. doi: 10.1016/j.drudis.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 3.Choi J, Fu GC. Transition metal-catalyzed alkyl-alkyl bond formation: another dimension in cross-coupling chemistry. Science. 2017;356:eaaf7230. doi: 10.1126/science.aaf7230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fu GC. Transition-metal catalysis of nucleophilic substitution reactions: a radical alternative to SN1 and SN2 processes. ACS Cent Sci. 2017;3:692–700. doi: 10.1021/acscentsci.7b00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu X. Nickel-catalyzed cross coupling of non-activated alkyl halides: a mechanistic perspective. Chem Sci. 2011;2:1867–1886. [Google Scholar]
- 6.Rudolph A, Lautens M. Secondary alkyl halides in transition-metal-catalyzed cross-coupling reactions. Angew Chem Int Ed. 2009;48:2656–2670. doi: 10.1002/anie.200803611. [DOI] [PubMed] [Google Scholar]
- 7.Cherney AH, Kadunce NT, Reisman SE. Enantioselective and enantiospecific transition-metal-catalyzed cross-coupling reactions of organometallic reagents to construct C-C bonds. Chem Rev. 2015;115:9587–9652. doi: 10.1021/acs.chemrev.5b00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glasspoole BW, Crudden CM. Cross-coupling: the final frontier. Nat Chem. 2011;3:912–913. doi: 10.1038/nchem.1210. [DOI] [PubMed] [Google Scholar]
- 9.Owston NA, Fu GC. Asymmetric alkyl-alkyl cross-couplings of unactivated secondary alkyl electrophiles: stereoconvergent suzuki reactions of racemic acylated halohydrins. J Am Chem Soc. 2010;132:11908–11909. doi: 10.1021/ja105924f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilsily A, Tramutola F, Owston NA, Fu GC. New directing groups for metal-catalyzed asymmetric carbon-carbon bond-forming processes: stereoconvergent alkyl-alkyl Suzuki cross-couplings of unactivated electrophiles. J Am Chem Soc. 2012;134:5794–5797. doi: 10.1021/ja301612y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zultanski SL, Fu GC. Catalytic asymmetric γ-alkylation of carbonyl compounds via stereoconvergent Suzuki cross-couplings. J Am Chem Soc. 2011;133:15362–15364. doi: 10.1021/ja2079515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arp FO, Fu GC. Catalytic enantioselective Negishi reactions of racemic secondary benzylic halides. J Am Chem Soc. 2005;127:10482–10483. doi: 10.1021/ja053751f. [DOI] [PubMed] [Google Scholar]
- 13.Fischer C, Fu GC. Asymmetric nickel-catalyzed Negishi cross-couplings of secondary α-bromo amides with organozinc reagents. J Am Chem Soc. 2005;127:4594–4595. doi: 10.1021/ja0506509. [DOI] [PubMed] [Google Scholar]
- 14.Son S, Fu GC. Nickel-catalyzed asymmetric Negishi cross-couplings of secondary allylic chlorides with alkylzincs. J Am Chem Soc. 2008;130:2756–2757. doi: 10.1021/ja800103z. [DOI] [PubMed] [Google Scholar]
- 15.Wang Z, Yin H, Fu GC. Catalytic enantioconvergent coupling of secondary and tertiary electrophiles with olefins. Nature. 2018;563:379–383. doi: 10.1038/s41586-018-0669-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou F, Zhang Y, Xu X, Zhu S. NiH-Catalyzed remote asymmetric hydroalkylation of alkenes with racemic α-bromo amides. Angew Chem Int Ed. 2019;58:1754–1758. doi: 10.1002/anie.201813222. [DOI] [PubMed] [Google Scholar]
- 17.Cordier CJ, Lundgren RJ, Fu GC. Enantioconvergent cross-couplings of racemic alkylmetal reagents with unactivated secondary alkyl electrophiles: catalytic asymmetric Negishi α-alkylations of N-Boc-pyrrolidine. J Am Chem Soc. 2013;135:10946–10949. doi: 10.1021/ja4054114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mu X, Shibata Y, Makida Y, Fu GC. Control of vicinal stereocenters through nickel-catalyzed alkyl-alkyl cross-coupling. Angew Chem Int Ed. 2017;56:5821–5824. doi: 10.1002/anie.201702402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deutsch C, Krause N, Lipshutz BH. CuH-catalyzed reactions. Chem Rev. 2008;108:2916–2927. doi: 10.1021/cr0684321. [DOI] [PubMed] [Google Scholar]
- 20.Pirnot MT, Wang YM, Buchwald SL. Copper hydride catalyzed hydroamination of alkenes and alkynes. Angew Chem Int Ed. 2016;55:48–57. doi: 10.1002/anie.201507594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu S, Niljianskul N, Buchwald SL. Enantio- and regioselective CuH-catalyzed hydroamination of alkenes. J Am Chem Soc. 2013;135:15746–15749. doi: 10.1021/ja4092819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miki Y, Hirano K, Satoh T, Miura M. Copper-catalyzed intermolecular regioselective hydroamination of styrenes with polymethylhydrosiloxane and hydroxylamines. Angew Chem Int Ed. 2013;52:10830–10834. doi: 10.1002/anie.201304365. [DOI] [PubMed] [Google Scholar]
- 23.Wang YM, Bruno NC, Placeres AL, Zhu S, Buchwald SL. Enantioselective synthesis of carbo- and heterocycles through a CuH-catalyzed hydroalkylation approach. J Am Chem Soc. 2015;137:10524–10527. doi: 10.1021/jacs.5b07061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang Y, Perry IB, Buchwald SL. Copper-catalyzed enantioselective addition of styrene-derived nucleophiles to imines enabled by ligand-controlled chemoselective hydrocupration. J Am Chem Soc. 2016;138:9787–9790. doi: 10.1021/jacs.6b06299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bandar JS, Ascic E, Buchwald SL. Enantioselective CuH-catalyzed reductive coupling of aryl alkenes and activated carboxylic acids. J Am Chem Soc. 2016;138:5821–5824. doi: 10.1021/jacs.6b03086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu X, et al. Practical carbon-carbon bond formation from olefins through nickel-catalyzed reductive olefin hydrocarbonation. Nat Commun. 2016;7:11129. doi: 10.1038/ncomms11129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bera S, Hu X. Nickel-catalyzed regioselective hydroalkylation and hydroarylation of alkenyl boronic esters. Angew Chem Int Ed. 2019;58:13854–13859. doi: 10.1002/anie.201907045. [DOI] [PubMed] [Google Scholar]
- 28.Buslov I, Becouse J, Mazza S, Montandon-Clerc M, Hu X. Chemoselective alkene hydrosilylation catalyzed by nickel pincer complexes. Angew Chem Int Ed. 2015;54:14523–14526. doi: 10.1002/anie.201507829. [DOI] [PubMed] [Google Scholar]
- 29.Gaydou M, Moragas T, Julia-Hernandez F, Martin R. Site-selective catalytic carboxylation of unsaturated hydrocarbons with CO2 and water. J Am Chem Soc. 2017;139:12161–12164. doi: 10.1021/jacs.7b07637. [DOI] [PubMed] [Google Scholar]
- 30.Zhou F, Zhu J, Zhang Y, Zhu S. NiH-Catalyzed reductive relay hydroalkylation: a strategy for the remote C(sp3-H alkylation of alkenes. Angew Chem Int Ed. 2018;57:4058–4062. doi: 10.1002/anie.201712731. [DOI] [PubMed] [Google Scholar]
- 31.Sommer H, Julia-Hernandez F, Martin R, Marek I. Walking metals for remote functionalization. ACS Cent Sci. 2018;4:153–165. doi: 10.1021/acscentsci.8b00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun SZ, Borjesson M, Martin-Montero R, Martin R. Site-selective Ni-catalyzed reductive coupling of α-haloboranes with unactivated olefins. J Am Chem Soc. 2018;140:12765–12769. doi: 10.1021/jacs.8b09425. [DOI] [PubMed] [Google Scholar]
- 33.Sun SZ, Romano C, Martin R. Site-selective catalytic deaminative alkylation of unactivated olefins. J Am Chem Soc. 2019;141:16197–16201. doi: 10.1021/jacs.9b07489. [DOI] [PubMed] [Google Scholar]
- 34.He Y, Cai Y, Zhu S. Mild and regioselective benzylic C-H functionalization: Ni-catalyzed reductive arylation of remote and proximal olefins. J Am Chem Soc. 2017;139:1061–1064. doi: 10.1021/jacs.6b11962. [DOI] [PubMed] [Google Scholar]
- 35.Vasseur A, Bruffaerts J, Marek I. Remote functionalization through alkene isomerization. Nat Chem. 2016;8:209–219. doi: 10.1038/nchem.2445. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Y, Han B, Zhu S. Rapid access to highly functionalized alkyl boronates by NiH-catalyzed remote hydroarylation of boron-containing alkenes. Angew Chem Int Ed. 2019;58:13860–13864. doi: 10.1002/anie.201907185. [DOI] [PubMed] [Google Scholar]
- 37.Davidson M, Hughes AK, Marder TB, Wade K. Contemporary Boron Chemistry. The Royal Society of Chemistry; Cambridge: 2000. [Google Scholar]
- 38.Liu SY, Stephan DW. Contemporary research in boron chemistry. Chem Soc Rev. 2019;48:3434–3435. doi: 10.1039/c9cs90053e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leonori D, Aggarwal VK. Stereospecific couplings of secondary and tertiary boronic esters. Angew Chem Int Ed. 2015;54:1082–1096. doi: 10.1002/anie.201407701. [DOI] [PubMed] [Google Scholar]
- 40.Sandford C, Aggarwal VK. Stereospecific functionalizations and transformations of secondary and tertiary boronic esters. Chem Commun. 2017;53:5481–5494. doi: 10.1039/c7cc01254c. [DOI] [PubMed] [Google Scholar]
- 41.Collins BSL, Wilson CM, Myers EL, Aggarwal VK. Asymmetric Synthesis of Secondary and TertiaryBoronic Esters. Angew Chem Int Ed. 2017;56:11700–11733. doi: 10.1002/anie.201701963. [DOI] [PubMed] [Google Scholar]
- 42.Brown HC, Zweifel G. Hydroboration as a convenient procedure for the asymmetric synthesis of alcohols of high optical purity. J Am Chem Soc. 1961;83:486–487. [Google Scholar]
- 43.Hall DG, editor. Boronic Acids: Preparation, Applications in Organic Synthesis and Medicine. Wiley–VCH; Weinheim: 2006. [Google Scholar]
- 44.Burns M, et al. Assembly-line synthesis of organic molecules with tailored shapes. Nature. 2014;513:183–188. doi: 10.1038/nature13711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ganic A, Pfaltz A. Iridium-catalyzed enantioselective hydrogenation of alkenylboronic esters. Chem Eur J. 2012;18:6724–6728. doi: 10.1002/chem.201200246. [DOI] [PubMed] [Google Scholar]
- 46.Xi Y, Hartwig JF. Diverse asymmetric hydrofunctionalization of aliphatic internal alkenes through catalytic regioselective hydroboration. J Am Chem Soc. 2016;138:6703–6706. doi: 10.1021/jacs.6b02478. [DOI] [PubMed] [Google Scholar]
- 47.Zhang L, et al. Catalytic conjunctive cross-coupling enabled by metal-induced metallate rearrangement. Science. 2016;351:70–74. doi: 10.1126/science.aad6080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schmidt J, Choi J, Liu AT, Slusarczyk M, Fu GC. A general, modular method for the catalytic asymmetric synthesis of alkylboronate esters. Science. 2016;354:1265–1269. doi: 10.1126/science.aai8611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stoltz BM, et al. Potassium tert-butoxide-catalyzed dehydrogenative C–H silylation of heteroaromatics: a combined experimental and computational mechanistic study. J Am Chem Soc. 2017;139:6867–6879. doi: 10.1021/jacs.6b13031. [DOI] [PubMed] [Google Scholar]
- 50.Wynn DA, Roth MM, Pollard BD. The solubility of alkali-metal fluorides in non-aqueous solvents with and without crown ethers, as determined by flame emission spectrometry. Talanta. 1984;31:1036–1040. doi: 10.1016/0039-9140(84)80244-1. [DOI] [PubMed] [Google Scholar]
- 51.Chen Z-M, Hilton MJ, Sigman MS. Palladium-catalyzed enantioselective redox-relay Heck arylation of 1,1-disubstituted homoallylic alcohols. J Am Chem Soc. 2016;138:11461–11464. doi: 10.1021/jacs.6b06994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sawamura M, Ito Y. Catalytic asymmetric synthesis by means of secondary interaction between chiral ligands and substrates. Chem Rev. 1992;92:857–871. [Google Scholar]
- 53.Corriu RJP, Guerin C, Henner B, Wang Q. Pentacoordinate hydridosilicates: synthesis and some aspects of their reactivity. Organometallics. 1991;10:2297–2303. [Google Scholar]
- 54.Bandar JS, Pirnot MT, Buchwald SL. Mechanistic studies lead to dramatically improved reaction conditions for the Cu-catalyzed asymmetric hydroamination of olefins. J Am Chem Soc. 2015;137:14812–14818. doi: 10.1021/jacs.5b10219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huo H, Gorsline BJ, Fu GC. Catalyst-controlled doubly enantioconvergent coupling of racemic alkyl nucleophiles and electrophiles. Science. 2020;367:559–564. doi: 10.1126/science.aaz3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Frampton JE. Pregabalin: a review of its use in adults with generalized anxiety disorder. CNS Drugs. 2014;28:835–854. doi: 10.1007/s40263-014-0192-0. [DOI] [PubMed] [Google Scholar]
- 57.Mujahid M, Muthukrishnan M. A new enantioselective synthesis of the anticonvulsant drug pregabalin (lyrica) based on a hydrolytic kinetic resolution method. Chirality. 2013;25:965–969. doi: 10.1002/chir.22253. [DOI] [PubMed] [Google Scholar]
- 58.Zheng B, Srebnik M. Preparation and selective cleavage reactions of boron-zirconium 1,1-bimetalloalkanes. Tetrahedron Lett. 1993;34:4133–4136. [Google Scholar]
- 59.Gutierrez O, Tellis JC, Primer DN, Molander GA, Kozlowski MC. Nickel-catalyzed cross-coupling of photoredox-generated radicals: uncovering a general manifold for stereoconvergence in nickel-catalyzed cross-couplings. J Am Chem Soc. 2015;137:4896–4899. doi: 10.1021/ja513079r. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Crystallographic data for 3e’ and 4g’ reported in this Article have been deposited at the Cambridge Crystallographic Data Centre, under deposition number CCDC 2011678 (3e’) and CCDC 1971802 (4g’). Copies of the data can be obtained free of charge via www.ccdc.cam.ac.uk. All other data supporting the findings of this study, including experimental procedures and compound characterization, NMR, HPLC and X-ray analysis are available within the Article and its Supplementary Information.