

Abstract
We present Augur, a method to prioritize the cell types most responsive to biological perturbations in single-cell data. Augur employs machine-learning framework to quantify the separability of perturbed and unperturbed cells within a high-dimensional space. We validate our method on single-cell RNA-seq, chromatin accessibility, and imaging transcriptomics datasets, and show that Augur outperforms existing methods based on differential gene expression. Augur identified the neural circuits restoring locomotion in mice following spinal cord neurostimulation.
Within a decade, single-cell technologies have scaled from individual cells to entire organisms1,2. Investigators are now able to quantify RNA and protein expression, resolve their spatial organization in complex tissues, and dissect their regulation in hundreds of thousands of cells. This exponential increase in scale is enabling a transition from atlasing of healthy tissues to delineating the cell type-specific responses to disease and experimental perturbation3,4. This shift requires a parallel analytical transition, from cataloguing the marked molecular differences between cell types to resolving more subtle phenotypic alterations within cell types. Existing tools focus on identifying individual genes or proteins with statistically significant differences between conditions5. However, inferences at the level of individual analytes are ill-suited to address the broader question of which cell types are most responsive to a perturbation in the multidimensional space of single-cell data. Such prioritizations could clarify the contribution of each cell type to organismal phenotypes such as disease state, or identify cellular subpopulations that mediate the response to external stimuli such as drug treatment. Cell type prioritization could also guide downstream investigation, including the selection of experimental systems such as Cre lines or FACS gates to support causal experiments. However, investigators currently lack bespoke tools to identify cell types affected by perturbation.
Here, we introduce Augur, a versatile method to prioritize cell types based on their molecular response to a biological perturbation (Fig. 1a). We reasoned that cell types most responsive to a perturbation should be more separable, within the multidimensional space of single-cell measurements, than less affected ones, and that the relative difficulty of this separation would provide a quantitative basis for cell type prioritization. We formalized this difficulty as a classification task, asking how accurately disease or perturbation state could be predicted from highly multidimensional single-cell measurements. For each cell type, Augur withholds a proportion of sample labels, and trains a classifier on the labeled subset. The classifier predictions are compared with the experimental labels, and cell types are prioritized based on the area under the receiver operating characteristic curve (AUC) of these predictions in cross-validation.
Fig. 1. Augur correctly prioritizes cell types in synthetic and experimental single-cell datasets.

a, Schematic overview of Augur. b, AUCs of Augur and a naive random forest classifier without subsampling in simulated scRNA-seq datasets containing increasing numbers of cells. Cell type prioritizations are confounded by training dataset size for the naive classifier, but Augur abolishes this confounding factor. The mean and standard deviation of n = 10 independent simulations are shown. Dotted lines show linear regression; shaded areas show 95% confidence intervals. c, Pearson correlations between the AUC of each cell type, and the number of cells of that type sequenced, across a compendium of 22 scRNA-seq datasets, for Augur and a naive random forest classifier without subsampling. d, Augur AUCs scale monotonically with both the proportion of DE genes and the magnitude of DE in simulated cell populations of n = 200 cells. e, Relationship between number of DE genes detected by a representative test for single-cell differential gene expression (Wilcoxon rank-sum test), and the proportion of DE genes simulated between the two populations, for simulated populations of between n = 100 and n = 1,000 cells. f, Cell type prioritization in simulated scRNA-seq data from a tissue with 5,000 cells, eight cell types and increasingly unequal numbers of cells per type, as quantified by the Gini coefficient. The Pearson correlation to the simulation ground truth (proportion of DE genes) is shown for Augur and a representative test for single-cell DE (Wilcoxon rank-sum test). The mean and standard deviation of n = 10 independent simulations are shown. Dashed line shows mean cell type Gini coefficient across n = 22 published scRNA-seq datasets (0.52). **, p < 0.01; ***, p < 0.001, two-sided paired t-test. g, Pearson correlation between cell type prioritizations (AUC/number of DE genes) and simulation ground truth for Augur and six tests for single-cell DE in simulated tissues containing eight cell types (n = 5,000 cells) with a cell type Gini coefficient of 0.50, approximately equal to the mean of 0.52 in 22 published scRNA-seq datasets. h, Cell type prioritizations of bone marrow-derived mononuclear phagocytes from four species stimulated with LPS for between two and six hours for Augur and a representative test for single-cell DE (Wilcoxon rank-sum test). i, Pearson correlation between cell type prioritizations and duration of LPS exposure for Augur and six tests for single-cell DE (in the same order as in g). Pearson correlations with a two-sided p-value less than 0.05 are shown in orange. j, Left, Augur cell type prioritizations mirror the number of DE genes in a microarray dataset of FACS-purified cells (two-sided Pearson correlation, n = 6 cell types matching between bulk and single-cell datasets). Right, the number of DE genes detected in the scRNA-seq dataset by a Wilcoxon rank-sum test is uncorrelated with the FACS gold standard. k, Pearson correlation between cell type prioritizations and the FACS gold standard for Augur and six tests for single-cell DE (n = 6 cell types matching between bulk and single-cell datasets). Pearson correlations with a two-sided p-value less than 0.05 are shown in orange. l, Reproducibility of cell type prioritization in two independent studies of Alzheimer’s disease (n = 6 cell types matching between single-cell datasets, two-sided Pearson correlation). m, Augur cell type prioritizations in a scATAC-seq dataset track with the number of DE genes in an RNA-seq dataset of FACS-purified cells (n = 3 cell types matching between bulk and single-cell data).
Because the amount of available training data typically has a strong effect on classifier performance, we anticipated that the uneven relative abundances of cell types in single-cell datasets could confound cell type prioritization. In both simulated data and a compendium of 22 published scRNA-seq datasets, we found that the AUC scaled with the number of cells, as opposed to the perturbation intensity (Extended Data Figs. 1a-b and 2a-b). To overcome this confound, Augur repeatedly draws small samples from the dataset, and reports the mean AUC across samples. We found this procedure abolished the dependence on the total number of cells (Fig. 1b-c, Extended Data Figs. 1c-d and 2b). Moreover, we established that Augur correctly prioritized cell types subjected to simulated perturbations of known intensities, finding the AUC increased monotonically with both the amount and magnitude of simulated differential expression (Fig. 1d and Extended Data Fig. 1e-f).
We compared Augur to previously described approaches that have attempted to prioritize cell types based on the relative number of genes passing a statistical threshold for differential expression (DE)3,6–9. In both simulated and experimental datasets, we found that the number of DE genes was strongly correlated with the number of cells per type (Extended Data Figs. 1g-i and 2c), causing abundant cell types with modest transcriptional perturbations to be prioritized over rare but more strongly perturbed cell types (Fig. 1e and Extended Data Fig. 1j). Consequently, we found that in simulated datasets with increasingly uneven cell type proportions, the performance of DE methods declined rapidly (Fig. 1f-g and Extended Data Fig. 3a-b). Importantly, this decline occurred even for cell type distributions that were substantially less uneven than those observed in real scRNA-seq data (Extended Data Fig. 3c). In contrast, Augur prioritized cell types with high accuracy regardless of cell type distributions (r ≥ 0.95; Fig. 1f). In published datasets, we confirmed this bias led DE methods to prioritize highly abundant cell types, even when such prioritizations were biologically unlikely (Extended Data Fig. 2d-f). For instance, DE methods identified oligodendrocytes as most strongly perturbed in Alzheimer’s disease10, likely due to their relative abundance in the brain (Extended Data Fig. 2g-j).
We next applied Augur to published scRNA-seq datasets with a quantitative measure of ground truth in order to evaluate its ability to prioritize cell types exposed to stimuli of known intensity. In homogenous cell populations, Augur detected the expected dose-response relationships in mononuclear phagocytes from four species stimulated with LPS ranging from two to six hours11, whereas DE methods yielded biologically incongruent results (Fig. 1h-i and Supplementary Fig. 1a). To evaluate the performance of Augur in a complex tissue, we analyzed a scRNA-seq dataset of PBMCs stimulated with interferon4, comparing cell type prioritizations to an independent microarray experiment on FACS-purified cells12. We observed an almost perfect correspondence between Augur and the number of DE genes in this FACS gold standard, whereas single-cell DE methods were uncorrelated with the gold standard (Fig. 1j-k and Supplementary Fig. 1b).
To evaluate the reproducibility of cell type prioritization, we applied Augur to two independent scRNA-seq studies comparing individuals with Alzheimer’s disease and healthy controls3,10. Augur produced nearly identical prioritizations, identifying the most profound transcriptional perturbations in neurons and endothelial cells (Fig. 1l). Similarly, we asked whether Augur could prioritize cell types from identical experimental perturbations, but obtained with orthogonal single-cell technologies. We applied Augur to scRNA-seq7 and single-cell imaging transcriptomics (STARmap)13 datasets from the visual cortex after exposure to light. Despite technical differences between the datasets, Augur consistently prioritized excitatory neurons, and even ranked subpopulations of excitatory neurons from specific cortical layers in identical order (Extended Data Fig. 3d). Finally, we applied Augur to single-cell ATAC-seq data from bone marrow-derived cells stimulated with LPS14, and found that Augur cell type prioritizations mirrored a gold standard from bulk RNA-seq of FACS-sorted cells (Fig. 1m)15.
Augur can flexibly incorporate continuous or multi-class sample labels in addition to conventional treatment versus control designs. We applied Augur to prioritize cell types of the prefrontal cortex based on quantitative measures of amyloid burden, neuritic plaques, and neurofibrillary tangles in individuals with Alzheimer’s disease3. Cell type prioritizations were strongly correlated to those based on clinical diagnosis, reflecting the pathogenesis of the disease (Supplementary Fig. 2). Likewise, Augur can readily be applied to prioritize cell types in datasets with more than two perturbations (Supplementary Fig. 3).
To apply Augur to single-cell datasets with more complex experimental designs, we devised a test for differential cell type prioritization (Extended Data Fig. 4a). Applying differential prioritization to a single-cell imaging transcriptomics (MERFISH) dataset16, Augur identified multiple neuron subtypes preferentially activated during parenting in either male or female mice (Extended Data Fig. 4b-c). Similarly, in a scRNA-seq dataset17, Augur prioritized several neuron subtypes with differential responses to whisker lesioning in Cx3cr1+/− and Cx3cr1−/− mice (Extended Data Fig. 4d).
We also considered whether Augur could be applied directly to single-cell measures of transcriptome dynamics, such as the RNA velocity18, in order to specifically prioritize cell types undergoing an acute response to a perturbation on the timescale of transcription. We found that both experimental measurements19 and computational inference18 of transcriptional activity consistently captured more information than total RNA abundance in perturbations ranging from 45 min to 4 h in duration (Extended Data Fig. 5a-g). Conversely, we confirmed that transcriptome dynamics did not confer an appreciable information gain to cell type prioritization when the perturbation is chronic (Extended Data Fig. 5h-i).
We finally aimed to demonstrate the relevance of Augur to discover new biological mechanisms. We recently showed that targeted epidural spinal stimulation of lumbar segments (TESS), augmented by monoaminergic stimulation20, restores walking after spinal cord injury in individuals with paralysis21. However, the neural circuits engaged by this treatment remain enigmatic. We devised an experiment to expose the neuron subtypes recruited by TESS using single-cell transcriptomics (Fig. 2a). Mice received a severe contusion of the thoracic spinal cord that led to permanent paralysis of both legs. In the presence of serotonergic and D1 agonists, TESS immediately enabled walking in paralyzed mice (Fig. 2b-c). We performed single-nucleus RNA-seq of 18,514 nuclei from mice walking for 30 min with TESS and control mice, identifying all the major cell types of the lumbar spinal cord (Fig. 2d and Supplementary Fig. 4). We then subjected the 6,035 identified neurons to an additional round of clustering. This analysis identified 39 neuron subtypes expressing classical marker genes that were detected across experimental conditions (Fig. 2e and Extended Data Fig. 6).
Fig. 2. Augur identifies neuron subtypes that enable walking after paralysis.

a, Top, single-nucleus RNA-sequencing experimental design to prioritize neuron subtypes recruited by TESS. Middle, chronophotography of mice in the presence or absence of TESS and monoaminergic agonists. Bottom, stick diagram decompositions of right leg movements; leg endpoint trajectory with acceleration at toe-off; activity of extensor and flexor muscles of the ankle. b, Principal component analysis (n = 3 mice) of gait parameters for each condition (small circles). Large circles show the average per group. c, Bar plot shows the average scores on principal component 1 (PC1), which quantify the locomotor performance of paralyzed mice (n = 3) and mice walking with TESS (n = 3). d, Uniform manifold approximation and projection (UMAP) visualization of 18,514 nuclei, revealing the six major cell types of the mouse lumbar spinal cord. e, UMAP visualization of 6,035 neurons subjected to an additional round of sub-clustering and the 39 identified neuron subtypes. f, UMAP visualization of 6,035 neurons, colored by Augur cell type prioritization (AUC). The seven prioritized neuron subtypes with the highest AUCs are highlighted. g, Monosynaptically restricted anterograde tracing in Vsx2-Cre mice reveals V2a interneurons densely innervating motor neurons (ChAT). Similar results were obtained from three independent experiments. h, Dot plot showing expression of the immediate early gene Fos in neuron subtypes prioritized by Augur. i, Confirmation of colocalization of V2a, V1/V2b, and Spp1 marker genes (Vsx2, Slc6a5, and Spp1 respectively) and Fos by RNAscope in situ hybridization. Schematic indicates location of imaging for each marker within the spinal cord to aid specificity. Similar results were obtained from two independent experiments.
We reasoned that applying Augur directly to the RNA velocity of these neurons could prioritize subtypes that are immediately engaged by the therapy. Previous studies suggested that TESS generates an electrical field that depolarises proprioceptive afferent fibers22. Consistent with this prediction, Augur robustly prioritized interneurons with the molecular profiles of V2a and V1/V2b neurons, which are known to receive synapses from proprioceptive afferents (Fig. 2f and Extended Data Fig. 7). V2a interneurons have been implicated in left-right alternation23, whereas V2b interneurons are critical for flexor-extensor alternation24. Augur also prioritized Spp1-positive neurons, typically associated with motoneurons (Fig. 2f). Virus-mediated anatomical tracing in transgenic mice revealed dense synaptic projections from the prioritized interneurons onto motoneurons (Fig. 2g). The induction of immediate early genes in V2a and V1/V2b interneurons (Fig. 2h) confirmed their activation in response to TESS-enabled walking, a finding we verified by in situ hybridization (Fig. 2i and Extended Data Fig. 8). In contrast, interneurons not prioritized by Augur showed minimal amounts of Fos mRNA (Extended Data Fig. 8). These results illustrate the value of Augur to expose neural circuits underlying complex behaviors.
Augur is computationally efficient, requiring a median of 49.7 min and 2.3 GB of RAM to analyze our compendium of 22 scRNA-seq datasets (Supplementary Fig. 5a-b). Inherent to the design of Augur is the ability to scale to datasets containing hundreds of thousands or even millions of cells on a laptop (Supplementary Fig. 5c-d). Moreover, Augur is robust to sequencing depth and classifier hyperparameters (Supplementary Figs. 6-7), and is compatible with both 3’ and full-length sequencing protocols (Supplementary Fig. 8). Contrary to single-cell DE methods, Augur incorporates information from both highly and lowly expressed genes (Extended Data Fig. 9). Augur is robust to several forms of batch effect, and computational batch correction can rescue accurate cell type prioritization under highly confounded experimental designs (Extended Data Fig. 10). Conversely, a limitation of Augur is that inferences at the level of cell types aggregate continuous underlying gradients of response intensity within cell populations, in order to achieve accurate cell type prioritization. As an efficient and principled method for cell type prioritization, we envision that Augur will facilitate the interpretation of a growing resource of single-cell data spanning multiple experimental conditions, and help single-cell technologies realize their potential to pinpoint cell types underlying organism-level phenotypes.
Online Methods
Design and implementation of Augur
Single-cell technologies increasingly allow investigators to collect datasets that span multiple experimental conditions: for instance, patients with a particular disease compared to healthy controls, animals exposed to a specific behavioral stimulus compared to unstimulated animals, or organisms subject to a particular genetic manipulation and wild-type controls. A number of tools have been developed to identify individual analytes (for instance, genes, proteins, or accessible chromatin regions) that exhibit statistically significant differences between experimental conditions5,25. However, for many biological questions, the analytical level of interest is not individual differentially abundant features, but rather the specific cell types that are most strongly affected by a stimulus. For instance, investigators may design a single-cell transcriptomics experiment to identify particular cell types in a complex tissue that undergo the most marked transcriptional changes in response to treatment with a drug, in order to clarify its mechanism of action. We refer to the process of ranking cell types based on their molecular response to a biological perturbation as cell type prioritization.
We designed Augur as a method to prioritize cell types based on their molecular response to a perturbation in highly multidimensional single-cell data. We reasoned that cells undergoing a profound response to a given experimental stimulus should become more separable, in the space of molecular measurements, than cells that remain unaffected by the stimulus. We sought to design a quantitative metric of this separability that would be robust to heteroscedasticity between cell types, and account for the specific biological and technical variability within each cellular subpopulation. Accordingly, Augur quantifies this separability by asking how readily the experimental sample labels associated with each cell (e.g., treatment vs. control) can be predicted from molecular measurements alone. In practice, this is achieved by training a machine-learning model specific to each cell type, to predict the experimental condition from which each individual cell originated. The accuracy of each cell type-specific classifier is evaluated in cross-validation, providing a quantitative basis for cell type prioritization.
We reasoned that an ideal method for cell type prioritization would make no assumptions about the distributions of features provided as input, and more broadly, would be agnostic to the particular molecular features provided as input: that is, it would readily incorporate single-cell RNA-seq, proteomics, epigenomics, and imaging transcriptomics datasets, among other modalities. Accordingly, Augur uses a random forest classifier to predict sample labels for each cell type. Random forests have the advantage that they do not make any parametric assumptions about the distribution of the input features, and consequently are robust to both the nature of the molecular measurements themselves, as well as to the specific pre-processing and normalization steps applied to obtain the input features-by-cells matrix.
When training machine-learning models, model performance generally improves as the size of the training dataset increases. We anticipated that this well-known phenomenon could present a critical confound to cell type prioritization, because cell types are unevenly represented in most single-cell datasets for both biological and technical reasons. To account for this confound, Augur repeatedly draws small samples of fixed size from each cell-type specific gene expression matrix, and performs cross-validation on these subsampled matrices (by default, 50 subsamples of 20 cells per condition are drawn). Augur then reports the mean cross-validation AUC across many small subsamples. We confirmed that this procedure abolishes the relationship between the number of cells of a particular type and the cross-validation AUC, in both real and simulated datasets (Fig. 1b-c and Extended Data Figs. 1-2).
To further improve computational efficiency, Augur incorporates two feature selection steps to minimize the number of analytes provided to the classifier as input. First, for each cell type in turn, Augur removes genes with little cell-to-cell variation within that cell type. This procedure, commonly referred to as highly variable gene identification in the context of single-cell RNA-seq26, also has the effect of removing noise. To flexibly account for the mean-variance relationship without making assumptions about the form of this relationship, Augur fits a local polynomial regression between the mean and coefficient of variation27,28 using the ‘loess’ function, and ranks genes based on their residuals in this model. A fixed quantile of the most highly variable genes are retained for each cell type (specified using the ‘var_quantile’ parameter, which defaults to 50% in order to remove only features that show less-than-expected variation based on their mean abundance). Second, for each iteration, a random proportion of features are randomly removed to improve speed and memory usage (specified using the ‘feature_perc’ parameter, which also defaults to 50%). In combination, these steps significantly reduce the size of the matrix that must be taken out of a sparse representation for input to the classifier, from ~20,000 genes to ~5,000 genes in a typical scRNA-seq dataset. To avoid discarding information in datasets where fewer analytes are measured, feature selection is only performed for datasets exceeding a certain minimum number of features (with this cutoff set, by default, to 1,000).
Implementation
Augur is implemented as an R package, available from https://github.com/neurorestore/Augur (Supplementary Fig. 9). Augur takes as input a features-by-cells (e.g., genes-by-cells for scRNA-seq) matrix, and a data frame containing metadata associated with each cell, minimally including the cell type annotations and sample labels to be predicted. Alternatively, a Seurat29, monocle330, or SingleCellExperiment31 object can be provided as input. To optimize both speed and memory usage, all computations are implemented for sparse matrices, up to the classification procedure itself. Because the feature selection, classification, and cross-validation procedures are independent for each cell type, Augur can readily be parallelized over the cell types in the input dataset, using the ‘mclapply’ package for parallelization, and runs on four cores by default.
Multiclass classification and regression
Augur quantifies the accuracy by which cell type labels can be predicted from molecular measurements using the area under the receiver operating characteristic curve (AUC), or the macro-averaged AUC in the case of multiclass classification. For experiments in which the perturbation can be interpreted as a continuous or ordinal variable, the classification objective is replaced with a regression task, and the accuracy of the corresponding random forest regression models is quantified using the concordance correlation coefficient (CCC)32, a measure of both the precision and accuracy of the relationship between predicted and experimental sample labels. By default, Augur returns the mean AUC (or CCC) for each cell type as a summary of cell type classification, but also calculates a larger suite of metrics for each fold of each subsampling iteration, including accuracy, precision, recall, sensitivity, specificity, negative predictive value, and positive predictive value, for users interested in investigating predictions in more detail. Augur also returns the feature importance associated with each input gene (Supplementary Fig. 11).
Differential prioritization
To compare cell type prioritizations between related conditions, we devised a permutation-based test for differential prioritization. In order to obtain a null distribution of AUCs for each cell type that reflected variability associated with number of cells sequenced, read depth, and other technical factors, we permuted sample labels within each cell type, and ran Augur on the permuted dataset. We repeated this permutation procedure 1,000 times. We then compared the observed difference between condition-specific AUCs, ΔAUCobs, for each cell type to the difference under permuted sample labels, ΔAUCrnd, and calculated permutation p-values33.
Simulations
We initially tested Augur on simulated scRNA-seq data, using the ‘Splatter’ R package34. Initial simulation parameters were estimated from the Kang et al. dataset4 using the ‘splatEstimate’ function, and populations of 100–1,000 cells from two experimental conditions were generated, in increments of 100. We then simulated differential expression in varying proportions of genes (using the ‘de.prob’ parameter), and with varying magnitudes (using the ‘de.facLoc’ parameter). To specifically evaluate the ability of Augur to abolish the relationship between the number of cells in a particular population and the AUC of sample label classification, we compared Augur to cell type prioritization using an identical feature selection and classification procedure, but without drawing small subsamples from the dataset, by setting the ‘n_subsamples’ argument to 0. We additionally implemented a cell type prioritization scheme based on the number of differentially expressed genes between conditions, as previously described3,6. Cell types were ranked based on the number of differentially expressed genes using six different tests for differential expression in single-cell transcriptomics datasets (t-test, Wilcoxon rank-sum test, likelihood ratio test35, logistic regression36, MAST37, and a negative binomial generalized linear model), implemented through the Seurat ‘FindMarkers’ function.
To evaluate the impact of different scRNA-seq protocols on cell type prioritization, we repeated these simulations with parameters estimated from libraries prepared by Ziegenhain et al.38, using six prominent scRNA-seq methods (CEL-seq2, Drop-seq, MARS-seq, SCRB-seq, Smart-seq, and Smart-seq2; Supplementary Fig. 8).
To evaluate the impact of mean expression levels on cell type prioritization, we binned genes based on their mean expression into quintiles, then repeated these simulation experiments with either Augur or a representative single-cell test for differential expression, the Wilcoxon rank-sum test, run separately on genes from each quintile (Extended Data Fig. 9). To ensure these two methods were provided with the same genes as input, filtering of lowly variable genes was performed for the entire gene expression matrix, then Augur was subsequently run with no additional feature selection. We additionally confirmed these trends were not an artefact of our simulated datasets by performing an identical differential expression analysis in the Kang et al. dataset4, again finding that the vast majority of differentially expressed genes were detected within the top 20% of most highly expressed genes..
To evaluate the impact of the distribution of cell type proportions on cell type prioritization in complex tissues, we simulated scRNA-seq experiments with eight cell types. These simulated cell types displayed a graded response to perturbation, having between 10% and 80% of their genes differentially expressed in response to the stimulus. The unevenness of the distribution of cell type frequencies, as quantified by the Gini coefficient, was systematically varied. A total of 5,000 cells were simulated, with the number of cells of each type drawn randomly from a gamma distribution such that the distribution of cell type frequencies achieved a prespecified Gini coefficient in the range from 0 to 0.7, in increments of 0.05. The accuracy of cell type prioritization was quantified as the correlation between the AUC, for Augur, or the number of differentially expressed genes, for the six single-cell tests for differential expression described above, and the proportion of differentially expressed genes under the simulation ground truth. To compare these simulations to real scRNA-seq datasets, we calculated the Gini coefficient of cell type frequencies using the ‘reldist’ R package across 22 published studies, as described below, obtaining a mean Gini coefficient of 0.52.
Finally, because separability within cell types can arise not only from the cell-intrinsic response to perturbation but also from a number of technical factors, we evaluated the impact of batch effects on cell type prioritization (Extended Data Fig. 10). In simulated populations of 200 cells from two experimental conditions, sequenced in two batches, we simultaneously varied both the proportion of differentially expressed genes and the location parameter for the batch effect factor log-normal distribution (‘batch.facLoc’), fixing the location parameter of the differential expression factor log-normal distribution (‘de.facLoc’) at 0.5, as above. Under the default model in Splatter, technical batch effects are orthogonal to both the magnitude of perturbation-dependent differential expression, and the likelihood that a given cell is observed in either the stimulated or unstimulated condition. Because the separability between conditions is effectively unchanged in this scenario (“scenario #1”), we extended the Splatter package to incorporate confounding between batch and differential expression (“scenario #2”), and between batch and experimental condition (“scenarios #3-5”). Confounding between batch and differential expression is achieved by adjusting the order of operations in Splatter such that differential expression is simulated prior to the application of a batch effect, with the result that the batch effect amplifies the perturbation in one of the two batches. Confounding between batch and condition is achieved by adjusting the proportion of cells from each experimental condition within each batch, such that one batch is more likely to contain cells from the stimulated population. The fork of the Splatter repository implementing confounded batch effects is available from https://github.com/jordansquair/splatter_batch. Last, we asked whether computational methods for batch effect correction could restore the expected gradient of perturbation response in confounded datasets. Using an exemplary approach, the mutual nearest neighbors method39 as implemented in the ‘batchelor’ R package, we found that computational correction of batch effects restored accurate cell type prioritization. We suggest exploration and, if necessary, computational correction of any batch effects prior to cell type prioritization with Augur.
RNA velocity analysis
To generate intronic and exonic read count matrices for each dataset, data were downloaded from the SRA and converted to FASTQ format using the SRA toolkit. In the case of inDrops data, annotated BAM files were obtained using dropTag40 with flags -s -S -c. Reads were then aligned to the latest Ensembl release (GRCm38.93), using STAR (v.2.5.3a)41. For Drop-seq data, files were first converted from FASTQ to BAM format using the Picard function ‘FastqtoSam’. Reads were then aligned to the latest Ensembl release using the Drop-seq toolkit (https://github.com/broadinstitute/Drop-seq). Next, count matrices of exonic and intronic reads were obtained using dropEst with flags -m -V -L eEBA -F. Barcodes were filtered to match those present in the processed datasets uploaded to the Gene Expression Omnibus (GEO) for each dataset. RNA velocity was subsequently calculated using the ‘velocyto’ R package18. Features were first chosen by filtering for genes with a minimum expression value per cell type using the function ‘filter.genes.by.cluster.expression’, with filters adjusted based on the read count distributions for each dataset (GSE102827: exon filter, 0.5, intron filter, 0.1; GSE130597: exon filter, 0.03, intron filter, 0.02; GSE103976, exon filter, 0.05, intron filter, 0.03). We then calculated gene-relative velocity using kNN pooling with k=10 (default) and fit.quantile = 0.01. By default, the function ‘gene.relative.velocity.estimates’ in velocyto.R returns a matrix containing only those features for which accurate estimates of γ and velocity could be obtained. Consequently, we ran Augur without either variable gene or random gene filters, as feature selection had already been performed during the creation of the RNA velocity matrix used as input. To compare AUCs for cell type prioritization on matrices of exonic or total counts, we retained only those genes for which velocity estimates could be calculated, and likewise disabled the variable gene and random gene filters. All other parameters were left as default.
Computational benchmarking
To quantify the computational resources required for cell type prioritization (Supplementary Fig. 5), we ran Augur with default settings on our compendium of 22 scRNA-seq datasets. The R package ‘peakRAM’ was used to monitor peak memory usage, and the base R function ‘system.time’ was used to monitor wall time.
Hyperparameter analysis
To characterize the robustness of Augur prioritizations to hyperparameters associated with its subsampling or feature selection procedures, the random forest classifier, and the choice of classifier itself, we evaluated the impact of systematically varying each of these parameters (Supplementary Fig. 6). We first investigated the impact of the number and size of subsamples from each cell-type-specific gene expression matrix on cell type prioritization, finding the ranks of each cell type stabilized around 50 subsamples. While larger subsample sizes generally yielded more robust ranks, these thresholds also precluded analysis of several cell types represented by fewer cells in existing datasets, and consequently we opted for an inclusive subsample size of 20 cells per experimental condition. Similarly, we ran Augur on gene expression matrices consisting of the top 10–100% of highly variable genes, followed by selection of a random subset of 10–100% of these, but found Augur was generally robust to the features provided as input. (We used the default thresholds of 50% on the variable gene and random selection filters throughout, unless otherwise specified). To assess the robustness of Augur prioritizations to random forest hyperparameters, we varied the number of trees in the forest between 10–1,000, the minimum number of cells required to split an internal node between 2–10, and the number of features sampled per split between 2–500. Finally, to assess the impact of the classifier itself, we implemented L1-penalized logistic regression in Augur using the R package ‘glmnet’, with the optimal value of the regularization parameter λ determined for each iteration using the function ‘cv.glmnet’.
The AUC of cell type prioritization ranges from 0 to 1, where an AUC of 0.5 corresponds to the accuracy of a random classifier, and an AUC of 1 represents perfect classification. Cell type prioritization is most effective when the distribution of AUCs spans a wide range, distinguishing cell types that are unaffected by the perturbation from those that are profoundly affected. However, in situations where all cell types are undergoing a profound perturbation response, or when datasets are sequenced very deeply (and thus more information is available to the classifier), many cell types may have an AUC of 1, representing perfect separability. In this case, Augur hyperparameters may be modified in order to purposefully degrade the performance of the classifier, and thereby achieve a broader range in the distribution of AUCs across cell types. Conceptually, this can be thought of as effectively the opposite of the hyperparameter tuning step that would typically be performed during the development of a machine-learning classifier. Importantly, this intervention is feasible because Augur hyperparameters have remarkably little effect on the rank of different cell types: that is, the cell type prioritizations as such (Supplementary Fig. 6 and Extended Data Fig. 7d). However, although the cell type prioritizations remain consistent, a subset of parameters have a marked effect on the magnitude of the AUCs (Supplementary Fig. 10). Empirically, we suggest decreasing the number of trees in the random forest classifier in scenarios where perfect classification can be achieved for many cell types (Supplementary Fig. 10g). Alternatively, the number of trees may be increased in scenarios where all AUCs are close to 0.5 (for instance, cells undergoing an exceptionally subtle perturbation, or very sparsely sequenced datasets).
Downsampling analysis
Motivated by the observation that only a fraction of reads at conventional depths are required to detect transcriptional programs and assign cell types42, we also evaluated the impact of sequencing depth on Augur cell type prioritizations by downsampling published scRNA-seq datasets to between 5–95% of their original depths (Supplementary Fig. 7). Reads were downsampled from the processed count matrices using the ‘downsampleMatrix’ function from the ‘DropletUtils’ package43, with the argument ‘bycol = FALSE’ in order to sample without replacement from all reads in the entire dataset rather than from each cell individually.
Preprocessing and analysis of published single-cell datasets
Data from a total of 28 published single-cell studies was processed and analyzed with Augur as described in detail in Supplementary Note 1. Unless otherwise noted, expression matrices and metadata were stored as Seurat objects, and genes detected in less than three cells were removed.
Application of Augur to TESS
To experimentally validate the ability of Augur to uncover new biological mechanisms and identify neuron subtypes involved in complex behaviors, we applied Augur to investigate the neural circuits underlying the functional response to targeted epidural electrical stimulation (TESS) following a field-standard contusion spinal cord injury44,45 using single-nucleus transcriptomics. Details on the animal model, surgical procedures, post-surgical care, electrochemical stimulation, and kinematic analysis are provided in Supplementary Note 2.
Single-nucleus RNA sequencing
Single nucleus dissociation was completed with a modified protocol based on our previous work46. Briefly, animals were euthanized by isoflurane inhalation and cervical dislocation. The thoracic SCI site was rapidly dissected and frozen on dry ice. Spinal cords were dounced in 500 μl sucrose buffer (0.32 M sucrose, 10 mM HEPES [pH 8.0], 5 mM CaCl2, 3 mM Mg-acetate, 0.1 mM EDTA, 1 mM DTT) and 0.1% Triton X-100 with the Kontes Dounce Tissue Grinder. 2 mL of sucrose buffer was added and filtered through a μm cell strainer. The lysate was subsequently centrifuged at 3200 g for 10 min at 4°C. The supernatant was decanted, and 3 mL of sucrose buffer added to the pellet and incubated for 1 min. The pellet was homogenized using an Ultra-Turrax and 12.5 mL of density buffer (1 M sucrose, 10 mM HEPES [pH 8.0], 3 mM Mg-acetate, 1 mM DTT) was added below the nuclei layer. The tube was centrifuged at 3200 g at 4°C and supernatant immediately poured off. The nuclei on the bottom half of the tube wall were collected with 100 μl PBS with 0.04% BSA and 0.2 U/μl RNase inhibitor. Resuspended nuclei were filtered through a 30 μm strainer. The nuclei suspension was finally adjusted to 1000 nuclei/μl.
Library preparation
Library preparation was carried out with 10x Genomics Chromium Single Cell Kit Version 2. The nuclei suspension was added to the Chromium RT mix to achieve loading numbers of 5,000. For downstream cDNA synthesis (13 PCR cycles), library preparation and sequencing, the manufacturer’s instructions were followed.
Read alignment
Reads were aligned to the latest Ensembl release (GRCm38.93), and a matrix of unique molecular identifier (UMI) counts was obtained using CellRanger count. Velocyto18 was subsequently used to obtain count matrices of exonic and intronic reads. Seurat29 was used to calculate quality control metrics, including the number of genes detected, number of UMIs per cell, and % mitochondrial genes in order to filter low-quality cells appropriately (cells with number of genes expressed < 200; cells with % mitochondrial reads > 5%; genes expressed in < 3 cells). The matrix used for downstream analysis consisted of 19,954 genes and 18,514 cells.
Clustering and integration
To integrate datasets across different experimental conditions, we took advantage of recently developed bioinformatic tools that align datasets from multiple conditions into a unified space29. Gene expression data was first normalized using regularized negative binomial models47, then integrated across batches using Seurat29. Batch effects were regressed out using the ‘latent.vars’ argument. Normalized and integrated gene expression matrices were clustered using Seurat29 to identify cell types in the integrated dataset using a standard workflow, including highly variable gene identification, principal component analysis, nearest-neighbor graph construction, and graph-based community detection. Following the identification of coarse-grained cell types (e.g., ‘neuron’), we identified fine-grained neuron subtypes by sub-clustering major cell types. We used clustering trees48 to guide the decision of the optimal resolution (Extended Data Fig. 7a). Cell types were manually annotated by using differential expression analysis to identify marker genes5,29. Putative cell types were assigned on the basis of marker gene expression, guided by previous work46,49–51.
RNA velocity
RNA velocity was calculated using the ‘velocyto’ R package18. Velocyto estimates cell velocities from their spliced and unspliced mRNA content. We generated the annotated spliced and unspliced reads using the ‘run10x’ function of the Velocyto command line tool, as described above. We then calculated gene-relative velocity using kNN pooling with k=10 (default).
Viral tract tracing
All surgeries on mice were performed at EPFL under general anaesthesia with isoflurane in oxygen-enriched air using an operating microscope, and rodent stereotaxic apparatus (David Kopf). To trace the efferent connections of Vsx2 (V2a) neurons AAV-DJ-hSyn Flex mGFP 2 A synaptophysin mRuby (Stanford Vector Core Facility, reference AAV DJ GVVC-AAV-100, titer 1.15E12 genome copies per ml52) was injected on each side of the cord of Vsx2-Cre mice at the L2 spinal level, 0.25 μl 0.6 mm below the surface at 0.1 μl per minute using glass micropipettes (ground to 50 to 100 μm tips) connected via high-pressure tubing (Kopf) to 10-μl syringes under the control of microinfusion pumps.
Immunohistochemistry
After terminal anaesthesia by barbiturate overdose, mice were perfused transcardially with 4% paraformaldehyde and spinal cords processed for immunofluorescence as previously described53,54. Primary antibodies were: goat anti-choline acetyltransferase (ChAT, 1:50, Millipore, AB144P). Secondary antibodies were: Alexa Fluor 647 Donkey Anti Goat (1:200; Life Technologies, AB32849). Immunofluorescence was imaged digitally using a slide scanner [Olympus VS-120 Slide scanner] or confocal microscope [Zeiss LSM880 + Airy fast module with ZEN 2 Black software (Zeiss, Oberkochen, Germany)]. Images were digitally processed using ImageJ (NIH) or Imaris (Bitplane, v.9.0.0).
RNAscope
We confirmed the in situ localization of cell type markers and the expression of the immediate early gene Fos using RNAscope. Briefly, 16 μm cryosections were obtained from fixed-frozen spinal cords of animals undergoing identical experimental procedures. We used these sections to confirm the localization of Spp1 (cat. no. 435191), Slc6a5 (cat. no. 409741-C3) and Vsx2 (cat. no. 438341). We additionally included an analysis of negative controls that were not prioritized by Augur including Cck (cat. no. 402271-C3), Npy (cat. no. 313321), Rorb (cat. no. 444271-C3), Pnoc (cat. no. 437881), Gal (cat. no. 400961-C3), and Trh (cat. no. 436811 neurons). All these cell types have also been validated elsewhere46,50,51. We combined gene markers with Fos (cat. no. 316921-C2) to confirm the presence of immediate early gene activation in these cell types46. To detect the transcripts we used the RNAscope assay for fixed frozen tissue (Advanced Cell Diagnostics)55. Probes were designed and provided by Advanced Cell Diagnostics, Inc. Staining was performed according to standard procedures, using the RNAscope Fluorescent Multiplex Reagent Kit (cat. no. 323133).
Visualization
Throughout the manuscript, box plots show the median (horizontal line), interquartile range (hinges) and smallest and largest values no more than 1.5 times the interquartile range (whiskers), and error bars show the standard deviation.
Extended Data
Extended Data Fig. 1. Augur overcomes confounding factors to cell type prioritization in simulated cell populations.

a-b, Area under the receiver operating characteristic curve (AUC) of a random forest classifier trained in three-fold cross-validation to distinguish two simulated populations of cells1, with the total number of cells increasing from n = 100 to n = 1,000 and the proportion of differentially expressed genes between the two populations varying from 0% to 100%, a, or the location parameter of the differential expression factor log-normal distribution varying from 0.1 to 1.0, b.
c-d, As in a-b, but with the naive random forest classifier replaced with the subsampling procedure employed by Augur.
e-f, Relationship between Augur AUC and the proportion of differentially expressed genes, e, or the location parameter of the differential expression factor log-normal distribution, f, in distinguishing two simulated populations (n = 200 cells total). The mean and standard deviation of n = 10 independent simulations are shown. Inset, two-sided Pearson correlation.
g, Cell type prioritizations (AUC or number of differentially expressed genes) for a naive random forest classifier, Augur, and an exemplary single-cell differential expression test2, the Wilcoxon rank-sum test, for two simulated populations of cells with 50% of genes differentially expressed and a log-normal location parameter of 0.5, with the total number of cells increasing from n = 100 to n = 1,000 cells. Like a naive random forest strategy, the number of differentially expressed genes detected by the Wilcoxon rank-sum test scales linearly with the number of cells. The mean and standard deviation of n = 10 independent simulations are shown. Dotted lines show linear regression; shaded areas show 95% confidence intervals.
h-i, Number of differentially expressed genes detected by six tests for single-cell differential gene expression between two simulated populations of cells, with the total number of cells increasing from 100 to 1,000 and the proportion of differentially expressed genes between the two populations varying from 0% to 100%, h, or the location parameter of the differential expression factor log-normal distribution varying from 0.1 to 1.0, i.
j, Relationship between number of differentially expressed genes detected by five tests for single-cell differential gene expression and the proportion of differentially expressed genes simulated between the two populations, for simulated populations of between 100 and 1,000 cells (see also Fig. 1e). All single-cell differential expression tests detect a larger number of differentially expressed genes in a large population of cells with modest transcriptional perturbation (20% of genes differentially expressed) than in a smaller population of cells with more profound perturbation (70% of genes differentially expressed).
Extended Data Fig. 2. Augur overcomes confounding factors to cell type prioritization in a compendium of published single-cell RNA-seq datasets.

a, Overview of n = 22 published scRNA-seq datasets comparing two or more experimental conditions, used to verify the relationship between cell type prioritizations from a random forest classifier, Augur, or single-cell differential expression tests. Left, heatmap indicating the species of origin, the sequencing protocol, and whether cells or nuclei were sequenced. Right, properties of each dataset, including the total number of cell types identified in the original studies; the total number of cells sequenced; the number of cells per type (red bars indicate mean); and the mean number of reads for cells of each type.
b, Pearson correlations between the AUC of each cell type, and the number of cells of that type sequenced, across 22 datasets for Augur, bottom, and a naive random forest classifier without subsampling, top, as shown in Fig. 2c.
c, Pearson correlations between the number of differentially expressed genes per cell type, at 5% FDR, and the number of cells of that type sequenced, across 22 datasets for six statistical tests for single-cell differential expression.
d, Number of cells in the top-ranked cell type across 22 datasets for Augur, bottom, and a naive random forest classifier without subsampling, top.
e, Number of cells in the top-ranked cell type across 22 datasets for six statistical tests for single-cell differential expression.
f, Jaccard index between the top-ranked 1 to 5 cell types across 22 datasets, comparing Augur and six statistical tests for single-cell differential expression.
g, Cell type prioritizations in the Grubman et al., 20193 dataset by Augur and a representative test for single-cell differential expression, the Wilcoxon rank-sum test (“DE”).
h, Relationship between AUC and number of differentially expressed genes per cell type, at 5% FDR, in the Grubman et al., 2019 dataset. Dotted line shows linear regression.
i, Relationship between AUC and number of cells sequenced in the Grubman et al., 2019 dataset. Augur cell type prioritizations are uncorrelated with the number of cells per type. Dotted line shows linear regression; inset shows two-sided Pearson correlation.
j, Relationship between number of differentially expressed genes and number of cells sequenced in the Grubman et al., 2019 dataset. Cell type prioritizations based on the number of differentially expressed genes are strongly correlated with the number of cells per type. Dotted line shows linear regression; inset shows two-sided Pearson correlation.
Extended Data Fig. 3. Augur overcomes confounding factors to cell type prioritization in simulated tissues and across single-cell modalities.

a, Number of cells within each of eight cell types in a simulated tissue with increasingly unequal cell type proportions, as quantified by the Gini coefficient.
b, Cell type prioritization in simulated scRNA-seq data from a tissue with 5,000 cells distributed in eight cell types, with 10-80% of genes DE in response to perturbation, and increasingly unequal numbers of cells per type (as quantified by the Gini coefficient). The correlation to simulation ground truth (proportion of DE genes) is shown for five tests for single-cell differential gene expression. The mean and standard deviation of n = 10 independent simulations are shown. Dashed line shows mean Gini coefficient of cell type frequencies across 22 published scRNA-seq datasets. **, p < 0.01; ***, p < 0.001, two-sided paired t-test.
c, Inequality of cell type proportions in published scRNA-seq data. Top, Gini coefficient of cell type proportions across 22 published scRNA-seq datasets. Horizontal line and shaded area show the mean and standard deviation of the Gini coefficient across all datasets. Bottom, number of cells of each type across 22 published scRNA-seq datasets.
d, Comparison of cell type prioritization in independent scRNA-seq and single cell imaging transcriptomics (STARmap) studies of the mouse visual cortex after light exposure. Left, Augur cell type prioritization in the STARmap dataset4. Bottom, Augur cell type prioritization in the scRNA-seq dataset5. Center, correspondence between cell types defined in the scRNA-seq and STARmap datasets, quantified as the Spearman correlation coefficient between average profiles for each cell type across 139 genes present in both datasets.
Extended Data Fig. 4. Differential cell type prioritization in single-cell RNA-seq data.

a, Schematic overview of the permutation-based test for differential prioritization with Augur. First, cell type prioritization is performed within each of two conditions separately, yielding condition-specific AUCs for each cell type. Next, sample labels are randomly permuted within each cell type, and cell type prioritization is performed on shuffled data, yielding a null distribution of AUCs for each cell type and condition. AUCs for matching cell types are compared across conditions to calculate a ‘ΔAUC score’ for each cell type, and a null distribution of ΔAUC scores is calculated using the permuted data. Permutation p-values can then be calculated for each cell type, enabling the identification of statistically significant differences in cell type prioritization between conditions, as well as the condition in which the cell type is more transcriptionally separable.
b, Neuron subtypes with statistically significant differences in AUC between female and male mice during parenting, in a single-cell imaging transcriptomics experiment employing multiplexed error robust fluorescence in situ hybridization (MERFISH)6 (n = 79 subtypes). Eleven subtypes have significantly higher AUCs in female parents, whereas two have significantly higher AUCs in male parents.
c, Relationship between differential prioritization ΔAUC for parenting between male and female mice, and AUC for sex in naive mice. Several neuronal subtypes preferentially activated during parenting in female mice are also transcriptionally distinct in naive mice, such as the I-32 cluster, which is enriched for aromatase expression, and expresses multiple sex steroid hormone receptors6.
d, Neuron subtypes with statistically significant differences in AUC in response to whisker lesioning in Cx3cr1+/− as compared to Cx3cr1−/− mice, in a single-cell RNA-seq experiment7 (n = 28 subtypes). Four subtypes are have significantly higher AUCs in homozygous mice, whereas one subtype has a significantly higher AUC in heterozygous mice.
Extended Data Fig. 5. Cell type prioritization from transcriptional dynamics in acute experimental perturbations.

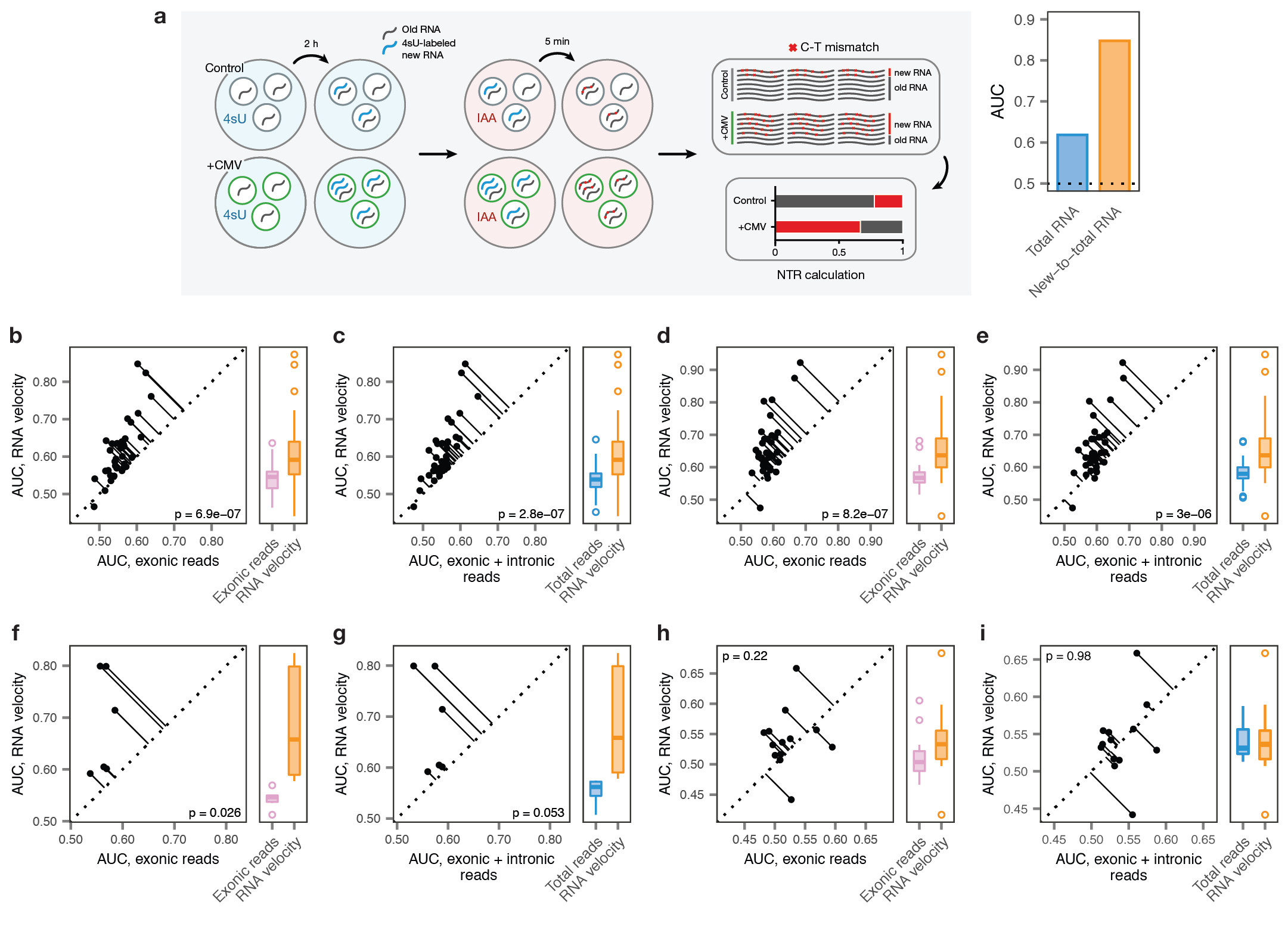
a, Left, schematic overview of the scSLAM-seq8 workflow. Cells are exposed to the nucleoside analogue 4-thiouridine (4sU), which is incorporated during transcription and converted to a cytosine analogue by iodoacetamide prior to RNA sequencing. This labeling permits in silico deconvolution of RNA molecules transcribed before and after 4sU exposure (‘old’ and ‘new’, respectively), and calculation of the ratio of new to total RNA (NTR), an experimental analogue to the computationally determined ‘RNA velocity’8,9. Right, AUCs for mouse fibroblasts exposed to lytic mouse cytomegalovirus (CMV) at 2 h post-infection, calculated by applying Augur to either total RNA or the NTR. The greater separability for the NTR reflects additional information specifically captured by the temporal dynamics of RNA expression in the context of this acute perturbation8.
b-e, Cell type prioritization based on exonic reads, total RNA, or RNA velocity for cells of the mouse visual cortex after exposure to light for 1 h, b-c, or 4 h, d-e, in the Hrvatin et al., 2018 dataset5. The AUC is significantly higher for RNA velocity than for either exonic reads (1 h, n = 34 cell types, 4 h, n = 35 cell types; two-sided paired t-tests: b, 1 h, p = 6.9 × 10-7; d, 4 h, p = 8.2 × 10-7) or total RNA (c, 1 h, p = 2.8 × 10-7; e, 4 h, p = 3.0 × 10-6), reflecting additional information specifically captured by acute transcriptional dynamics.
f-g, Cell type prioritization based on exonic reads, total RNA, or RNA velocity in an Act-seq10 dataset, which minimizes transcriptional changes induced by single-cell dissociation. Cell types of the medial amygdala in mice subjected to 45 min of immobilization stress and control mice were profiled by Drop-seq11 after treatment with the transcription inhibitor actinomycin D. The AUC is higher for RNA velocity than for either exonic reads (f, p = 0.026, n = 6 cell types) or total RNA (g, p = 0.053), reflecting the additional information specifically captured by acute transcriptional dynamics, and indicating this is not an artefact related to the transcriptional perturbations known to be induced by conventional dissociation procedures12.
h-i, Cell type prioritization based on exonic reads, total RNA, or RNA velocity in a chronic perturbation. Cell types of the lateral hypothalamic area were profiled by Drop-seq11 in mice after 9-16 weeks of maintenance on either high-fat diet or control diet13. No significant difference in AUCs was observed for RNA velocity compared to either exonic reads (h, p = 0.22, n = 13 cell types) or total RNA (i, p = 0.98), consistent with the time scale of the experimental perturbation.
Extended Data Fig. 6. Subclustering of single-neuron transcriptomes identifies 39 neuron subtypes in the mouse lumbar spinal cord.

See also Extended Data Fig. 7a.
a, Dot plot showing expression of one marker gene per cell type for the 39 neuron subtypes of the mouse lumbar spinal cord.
b, Neuron subtype detection across experimental conditions (n = 6,035 neurons). TESS, targeted electrical epidural stimulation of the lumbar spinal cord.
c, Proportion of neurons of each subtype detected in each experimental condition.
d, Neuron subtype detection across experimental replicates (n = 3 mice per condition).
e, Proportion of neurons of each subtype detected in each experimental replicate.
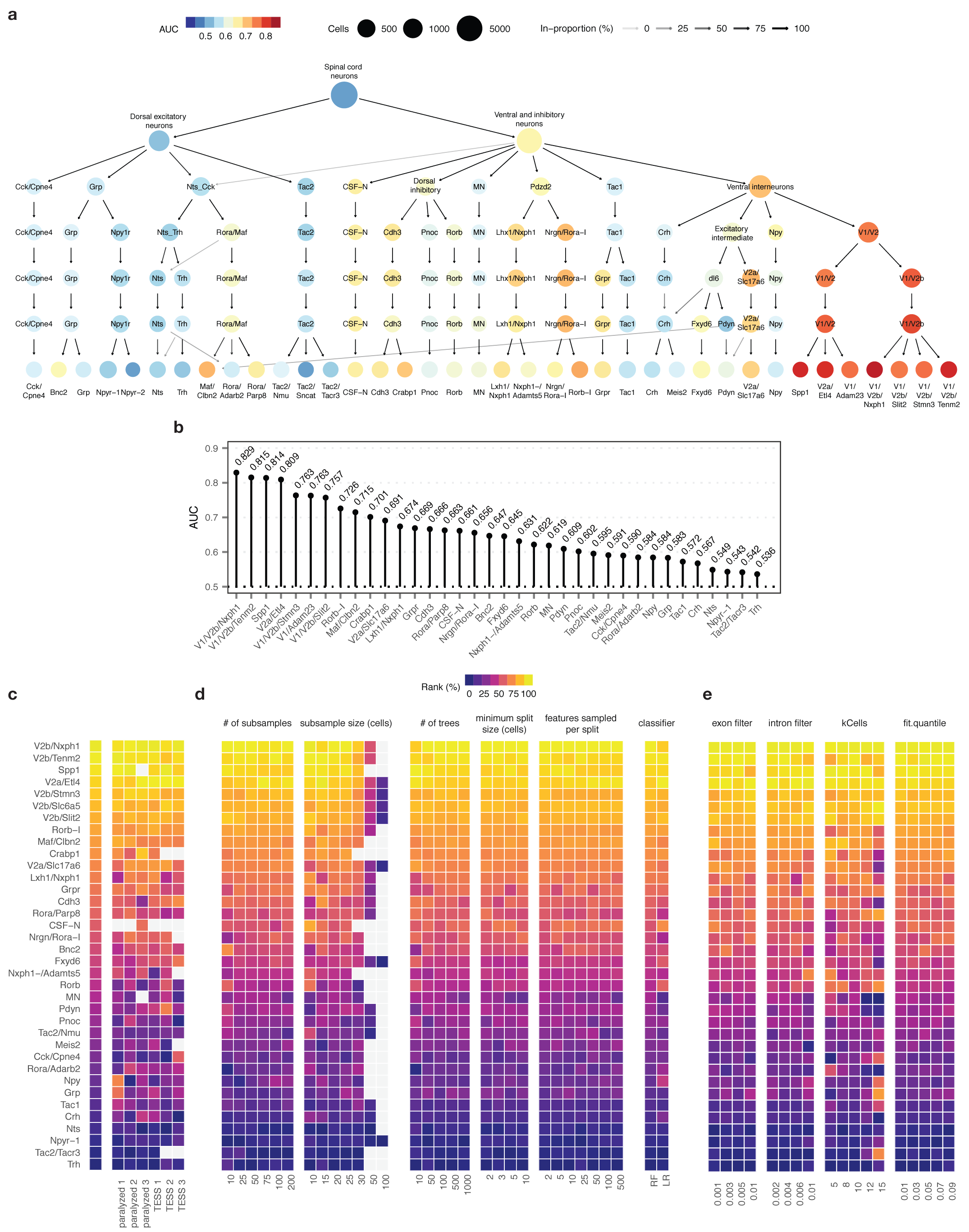
Extended Data Fig. 7. Robustness of Augur cell type prioritizations for mouse lumbar spinal cord neurons.

a, Clustering tree14 of mouse spinal cord neurons over seven clustering resolutions, revealing the hierarchical relationships between spinal cord neuron subtypes. Node color reflects AUCs for cell type prioritization in targeted electrical epidural stimulation.
b, AUCs for each of 37 neuron subtypes represented by at least 20 cells in both control and TESS-treated mice.
c-e, Robustness of cell type prioritization for neuron subtypes of the mouse lumbar spinal cord.
c, Impact of systematically withholding cells from each of six replicates (n = 3 per group) on cell type prioritization. Left, cell type prioritization with all six replicates, as in Fig. 2f. Grey tiles indicate neuron subtypes that were not represented by at least 20 cells in each condition after removal of cells from an experimental replicate.
d, Impact of varying Augur parameters, including the number of subsamples and the size of each subsample; random forest-specific hyperparameters (number of trees, minimum split size, number of features sampled per split); and the choice of classifier (random forest, RF; L1-penalized logistic regression, LR) on cell type prioritization. Grey tiles indicate sample sizes larger than the number of cells of that type in the dataset.
e, Impact of varying RNA velocity parameters, including exonic and intronic expression filters, the number of cells in the k-nearest neighbors pooling, and the extreme quantiles used to fit γ coefficients, on cell type prioritization.
Extended Data Fig. 8. Absence of colocalization of canonical marker genes for cell types not prioritized by Augur and Fos by RNAscope in situ hybridization.

Schematic indicates imaging location for each marker within the spinal cord. Bottom, proportion of cells expressing Fos from cell types prioritized by Augur (n = 3 cell types) or not prioritized by Augur (n = 6 cell types). Cell types prioritized by Augur are significantly more likely to express Fos after walking with TESS, compared to controls (p = 0.01, two-sided Fisher’s exact test), whereas cell types not prioritized by Augur do not display a statistically significant difference (p = 0.74). Error bars show standard deviation of the sample proportion.
Extended Data Fig. 9. Impact of mean gene expression level on cell type prioritization.

Cell type prioritizations were performed using both Augur and a representative single-cell differential expression method, the Wilcoxon rank-sum test, using the entire transcriptome (left column) or genes divided into five quintiles based on mean expression (right columns). Insets show two-sided Pearson correlations throughout.
a, Relationship between Augur cell type prioritizations (AUC) and the proportion of differentially expressed genes between two simulated populations of cells (n = 200 cells total), as shown in Supplementary Fig. 1e. The mean and standard deviation of n = 10 independent simulations are shown.
b, As in a, but with Augur applied to each quintile of gene expression separately. The AUC remains strongly correlated with the ground-truth perturbation intensity, regardless of mean expression levels (r ≥ 0.92).
c, Relationship between Augur cell type prioritizations (AUC) and the location parameter of the differential expression factor log-normal distribution between two simulated populations of cells (n = 200 cells total), as shown in Supplementary Fig. 1f. The mean and standard deviation of n = 10 independent simulations are shown.
d, As in c, but with Augur applied to each quintile of gene expression separately. The AUC remains strongly correlated with the ground-truth perturbation intensity, regardless of mean expression levels (r ≥ 0.95).
e-f, As in a-b, but showing the number of differentially expressed genes detected by a Wilcoxon rank-sum test at 5% FDR, either across the entire transcriptome, e, or within each expression quintile, f. No differentially expressed genes are detected at 5% FDR outside of the top expression quintile.
g-h, As in c-d, but showing the number of differentially expressed genes detected by a Wilcoxon rank-sum test at 5% FDR, either across the entire transcriptome, g, or within each expression quintile, h. No differentially expressed genes are detected at 5% FDR outside of the top expression quintile.
i, Cell type prioritization in simulated scRNA-seq data from a tissue with 5,000 cells, distributed in eight cell types, with increasingly unequal numbers of cells per type, as quantified by the Gini coefficient and shown in Fig. 1f. The correlation to simulation ground truth (proportion of DE genes) is shown for Augur and a representative test for single-cell DE (Wilcoxon rank-sum test). The mean and standard deviation of n = 10 independent simulations are shown.
j, As in i, but with both Augur and the Wilcoxon rank-sum test applied to each quintile of gene expression separately.
k, Pearson correlation between Augur cell type prioritizations (AUC) and simulation ground truth (proportion of DE genes) in simulated scRNA-seq data from tissue with eight cell types, subjected to perturbations of varying intensity, as quantified by the the location parameter of the differential expression factor log-normal distribution. The mean of n = 10 independent simulations is shown for each perturbation intensity..
l, As in k, but with Augur applied to each quintile of gene expression separately. Augur incorporates information from lowly expressed genes even in subtle perturbations.
m, Number of differentially expressed genes detected by a Wilcoxon rank-sum test at 5% FDR for each cell type in the Kang et al. dataset15, within each expression quintile, confirming the simulations in a-l reflect trends in real data.
Extended Data Fig. 10. Impact of batch effects on cell type prioritization.

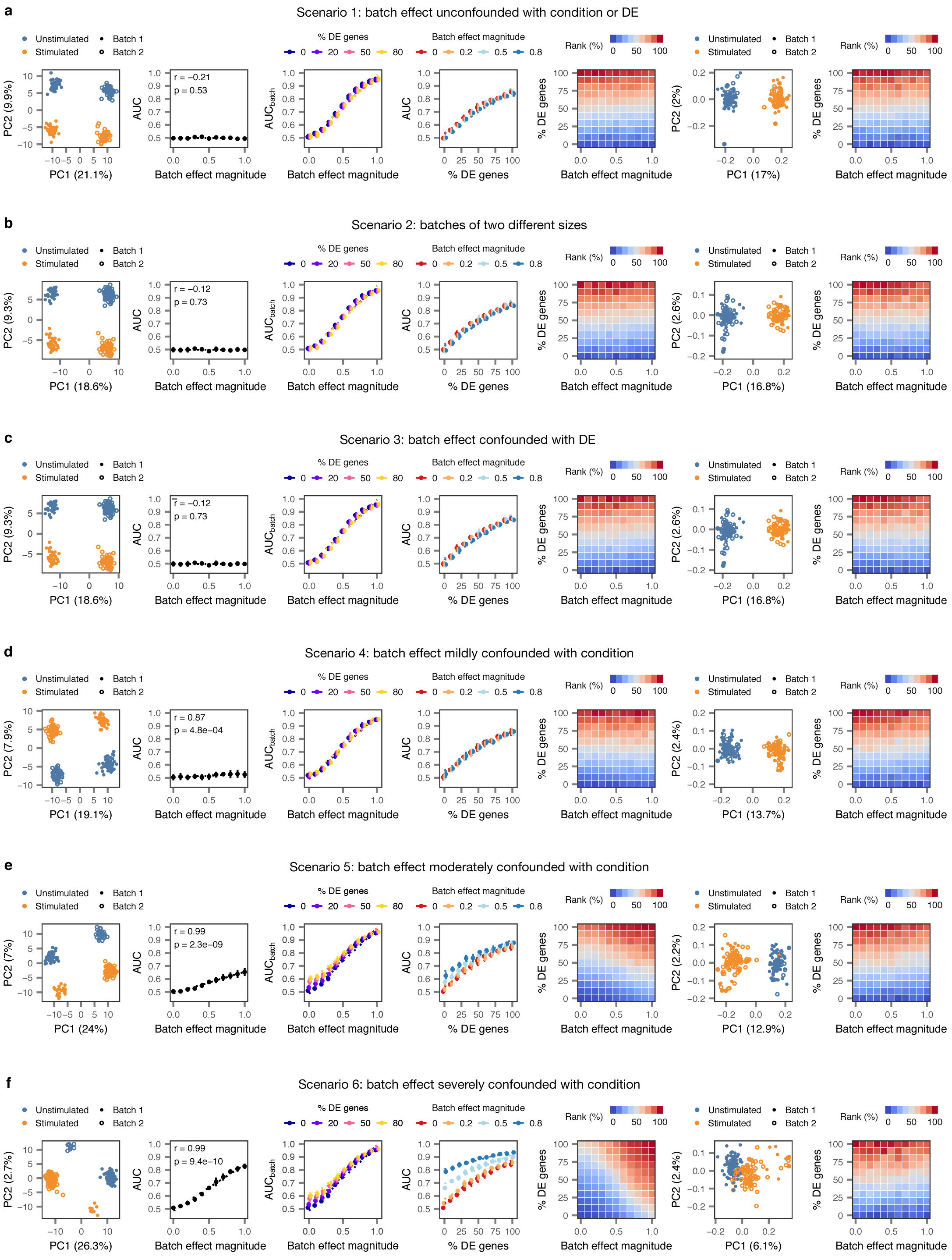
Two populations of cells (n = 200 cells total) were simulated, with each condition sequenced in two batches, and varying degrees of perturbation-dependent differential expression and/or technical batch effects were introduced according to five different batch effect scenarios. For each of the five scenarios, the following panels are shown from left to right:
i, Principal component analysis (PCA) of a representative simulation.
ii, Correlation between AUC and magnitude of simulated batch effect with 0% of genes differentially expressed in response to perturbation, reflecting the introduction of a spurious difference between conditions where none exists (inset, two-sided Pearson correlation).
iii, Correlation between AUC and magnitude of simulated batch effect when the random forest classifier is tasked with predicting batch rather than condition (AUCbatch), confirming the batch effect introduces the expected separability.
iv, Correlation between proportion of genes differentially expressed in response to perturbation and AUC for simulated populations of cells with no batch effect, and batch effects of three different magnitudes.
v, Cell type prioritizations in simulated populations of cells with varying perturbation intensity (% DE genes) and batch effect magnitudes.
vi, As in i, but after computational batch effect correction by alignment of mutual nearest neighbors16.
vii, As in v, but after computational batch effect correction by alignment of mutual nearest neighbors.
a, Impact of batch effects on cell type prioritization when technical batch is unconfounded with either condition or differential expression.
b, Impact of batch effects on cell type prioritization when batch #1 is twice as large as batch #2.
c, Impact of batch effects on cell type prioritization when perturbation-dependent differential expression is stronger in one of the two batches.
d, Impact of batch effects on cell type prioritization when technical batch is mildly confounded with condition (simulated cells are overrepresented in batch 1 by a factor of 20%).
e, Impact of batch effects on cell type prioritization when technical batch is moderately confounded with condition (simulated cells are overrepresented in batch 1 by a factor of 50%).
f, Impact of batch effects on cell type prioritization when technical batch is severely confounded with condition (simulated cells are overrepresented in batch 1 by a factor of 80%).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank D. Arneson, D. Avey, R. Mitra, A. Haber, O. Yilmaz, G. Chew, J. Polo, L. Adlung, I. Amit, D. Kim, D. Anderson, M. Basiri, R. Wirka, T. Quertermous, and F. Zhang for providing data and/or cell type annotations. This work was supported by a Consolidator Grant from the European Research Council [ERC-2015-CoG HOW2WALKAGAIN 682999] (to G.C.), the Swiss National Science Foundation (to G.C.; subside 310030_185214 and 310030_192558), Genome Canada and Genome British Columbia (to L.J.F.; project 214PRO), and Wings for Life (to M.A.S.). This work was also supported in part by the Intramural Research Program of the NIH, NINDS (to K.M. and A.L.). This work was enabled in part by the support provided by WestGrid and Compute Canada (to A.A.P. and L.J.F.), and through computational resources and services provided by Advanced Research Computing at the University of British Columbia (to L.J.F.). M.A.S. is supported by the Canadian Institutes of Health Research (CIHR) (Vanier Canada Graduate Scholarship, Michael Smith Foreign Study Supplement), an Izaak Walton Killam Memorial Pre-Doctoral Fellowship, a UBC Four Year Fellowship, a Vancouver Coastal Health–CIHR–UBC MD/PhD Studentship, a Brain Canada Hubert van Tol fellowship and a BCRegMed Collaborative Research Travel Grant. J.W.S. is supported by a CIHR Banting Postdoctoral fellowship.
Footnotes
Author contributions
M.A.S. and J.W.S. contributed equally to this work. M.A.S. and J.W.S. designed and implemented Augur, and performed all computational analyses. M.A.S., J.W.S., and M.G. processed published datasets. J.W.S., C.K., M.A.A., T.H.H., and M.M. performed experimental validation work, including viral tract tracing and RNAscope. C.K., K.J.E.M., and A.J.L. performed nucleus extraction and single-nucleus RNA-seq. M.G. and Q.B. analyzed experimental validation data. A.A.P., L.J.F., G.L.M., and G.C. supervised the work. M.A.S., J.W.S., and G.C. wrote the manuscript. All authors contributed to its editing.
Competing interests
G.C. is a founder and shareholder of GTXmedical, a company with no direct relationships with the present work. M.A.S., J.W.S., and G.C. are named as co-inventors on a patent application related to this work.
Code availability
Augur is available from GitHub (https://github.com/neurorestore/Augur) and as Supplementary Software 1.
Data availability
Raw sequencing data and count matrices have been deposited to the Gene Expression Omnibus (GSE142245).
References
- 1.Tang F, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6:377–382. doi: 10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- 2.Cao J, et al. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science. 2017;357:661–667. doi: 10.1126/science.aam8940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mathys H, et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature. 2019;570:332–337. doi: 10.1038/s41586-019-1195-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kang HM, et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat Biotechnol. 2018;36:89–94. doi: 10.1038/nbt.4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soneson C, Robinson MD. Bias, robustness and scalability in single-cell differential expression analysis. Nat Methods. 2018;15:255–261. doi: 10.1038/nmeth.4612. [DOI] [PubMed] [Google Scholar]
- 6.Rossi MA, et al. Obesity remodels activity and transcriptional state of a lateral hypothalamic brake on feeding. Science. 2019;364:1271–1274. doi: 10.1126/science.aax1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hrvatin S, et al. Single-cell analysis of experience-dependent transcriptomic states in the mouse visual cortex. Nat Neurosci. 2018;21:120–129. doi: 10.1038/s41593-017-0029-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Avey D, et al. Single-Cell RNA-Seq Uncovers a Robust Transcriptional Response to Morphine by Glia. Cell Rep. 2018;24:3619–3629.e4. doi: 10.1016/j.celrep.2018.08.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen R, Wu X, Jiang L, Zhang Y. Single-Cell RNA-Seq Reveals Hypothalamic Cell Diversity. Cell Rep. 2017;18:3227–3241. doi: 10.1016/j.celrep.2017.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grubman A, et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat Neurosci. 2019;22:2087–2097. doi: 10.1038/s41593-019-0539-4. [DOI] [PubMed] [Google Scholar]
- 11.Hagai T, et al. Gene expression variability across cells and species shapes innate immunity. Nature. 2018;563:197–202. doi: 10.1038/s41586-018-0657-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mostafavi S, et al. Parsing the interferon transcriptional network and its disease associations. Cell. 2016;164:564–578. doi: 10.1016/j.cell.2015.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X, et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science. 2018;361 doi: 10.1126/science.aat5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lareau CA, et al. Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility. Nat Biotechnol. 2019;37:916–924. doi: 10.1038/s41587-019-0147-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reyes M, et al. Multiplexed enrichment and genomic profiling of peripheral blood cells reveal subset-specific immune signatures. Sci Adv. 2019;5:eaau9223. doi: 10.1126/sciadv.aau9223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moffitt JR, et al. Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science. 2018;362 doi: 10.1126/science.aau5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gunner G, et al. Sensory lesioning induces microglial synapse elimination via ADAM10 and fractalkine signaling. Nat Neurosci. 2019;22:1075–1088. doi: 10.1038/s41593-019-0419-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.La Manno G, et al. RNA velocity of single cells. Nature. 2018;560:494–498. doi: 10.1038/s41586-018-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Erhard F, et al. scSLAM-seq reveals core features of transcription dynamics in single cells. Nature. 2019;571:419–423. doi: 10.1038/s41586-019-1369-y. [DOI] [PubMed] [Google Scholar]
- 20.Courtine G, et al. Transformation of nonfunctional spinal circuits into functional states after the loss of brain input. Nat Neurosci. 2009;12:1333–1342. doi: 10.1038/nn.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wagner FB, et al. Targeted neurotechnology restores walking in humans with spinal cord injury. Nature. 2018;563:65–71. doi: 10.1038/s41586-018-0649-2. [DOI] [PubMed] [Google Scholar]
- 22.Formento E, et al. Electrical spinal cord stimulation must preserve proprioception to enable locomotion in humans with spinal cord injury. Nat Neurosci. 2018;21:1728–1741. doi: 10.1038/s41593-018-0262-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crone SA, et al. Genetic ablation of V2a ipsilateral interneurons disrupts left-right locomotor coordination in mammalian spinal cord. Neuron. 2008;60:70–83. doi: 10.1016/j.neuron.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 24.Zhang J, et al. V1 and v2b interneurons secure the alternating flexor-extensor motor activity mice require for limbed locomotion. Neuron. 2014;82:138–150. doi: 10.1016/j.neuron.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crowell HL, et al. On the discovery of population-specific state transitions from multi-sample multi-condition single-cell RNA sequencing data. BioRxiv. 2019 doi: 10.1101/713412. [DOI] [Google Scholar]
- 26.Yip SH, Sham PC, Wang J. Evaluation of tools for highly variable gene discovery from single-cell RNA-seq data. Brief Bioinformatics. 2019;20:1583–1589. doi: 10.1093/bib/bby011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brennecke P, et al. Accounting for technical noise in single-cell RNA-seq experiments. Nat Methods. 2013;10:1093–1095. doi: 10.1038/nmeth.2645. [DOI] [PubMed] [Google Scholar]
- 28.Grün D, Kester L, van Oudenaarden A. Validation of noise models for single-cell transcriptomics. Nat Methods. 2014;11:637–640. doi: 10.1038/nmeth.2930. [DOI] [PubMed] [Google Scholar]
- 29.Stuart T, et al. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao J, et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature. 2019;566:496–502. doi: 10.1038/s41586-019-0969-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amezquita RA, et al. Orchestrating single-cell analysis with Bioconductor. Nat Methods. 2020;17:137–145. doi: 10.1038/s41592-019-0654-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin LI. A concordance correlation coefficient to evaluate reproducibility. Biometrics. 1989;45:255–268. [PubMed] [Google Scholar]
- 33.Phipson B, Smyth GK. Permutation P-values should never be zero: calculating exact P-values when permutations are randomly drawn. Stat Appl Genet Mol Biol. 2010;9(39) doi: 10.2202/1544-6115.1585. [DOI] [PubMed] [Google Scholar]
- 34.Zappia L, Phipson B, Oshlack A. Splatter: simulation of single-cell RNA sequencing data. Genome Biol. 2017;18:174. doi: 10.1186/s13059-017-1305-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McDavid A, et al. Data exploration, quality control and testing in single-cell qPCR-based gene expression experiments. Bioinformatics. 2013;29:461–467. doi: 10.1093/bioinformatics/bts714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ntranos V, Yi L, Melsted P, Pachter L. A discriminative learning approach to differential expression analysis for single-cell RNA-seq. Nat Methods. 2019;16:163–166. doi: 10.1038/s41592-018-0303-9. [DOI] [PubMed] [Google Scholar]
- 37.Finak G, et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015;16:278. doi: 10.1186/s13059-015-0844-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ziegenhain C, et al. Comparative Analysis of Single-Cell RNA Sequencing Methods. Mol Cell. 2017;65:631–643.e4. doi: 10.1016/j.molcel.2017.01.023. [DOI] [PubMed] [Google Scholar]
- 39.Haghverdi L, Lun ATL, Morgan MD, Marioni JC. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat Biotechnol. 2018;36:421–427. doi: 10.1038/nbt.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petukhov V, et al. dropEst: pipeline for accurate estimation of molecular counts in droplet-based single-cell RNA-seq experiments. Genome Biol. 2018;19:78. doi: 10.1186/s13059-018-1449-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heimberg G, Bhatnagar R, El-Samad H, Thomson M. Low Dimensionality in Gene Expression Data Enables the Accurate Extraction of Transcriptional Programs from Shallow Sequencing. Cell Syst. 2016;2:239–250. doi: 10.1016/j.cels.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Griffiths JA, Richard AC, Bach K, Lun ATL, Marioni JC. Detection and removal of barcode swapping in single-cell RNA-seq data. Nat Commun. 2018;9:2667. doi: 10.1038/s41467-018-05083-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scheff SW, Rabchevsky AG, Fugaccia I, Main JA, Lumpp JE. Experimental modeling of spinal cord injury: characterization of a force-defined injury device. J Neurotrauma. 2003;20:179–193. doi: 10.1089/08977150360547099. [DOI] [PubMed] [Google Scholar]
- 45.Squair JW, et al. Integrated systems analysis reveals conserved gene networks underlying response to spinal cord injury. Elife. 2018;7 doi: 10.7554/eLife.39188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sathyamurthy A, et al. Massively Parallel Single Nucleus Transcriptional Profiling Defines Spinal Cord Neurons and Their Activity during Behavior. Cell Rep. 2018;22:2216–2225. doi: 10.1016/j.celrep.2018.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hafemeister C, Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. BioRxiv. 2019 doi: 10.1101/576827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zappia L, Oshlack A. Clustering trees: a visualization for evaluating clusterings at multiple resolutions. Gigascience. 2018;7 doi: 10.1093/gigascience/giy083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rosenberg AB, et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science. 2018;360:176–182. doi: 10.1126/science.aam8999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Häring M, et al. Neuronal atlas of the dorsal horn defines its architecture and links sensory input to transcriptional cell types. Nat Neurosci. 2018;21:869–880. doi: 10.1038/s41593-018-0141-1. [DOI] [PubMed] [Google Scholar]
- 51.Zeisel A, et al. Molecular architecture of the mouse nervous system. Cell. 2018;174:999–1014.e22. doi: 10.1016/j.cell.2018.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grimm D, et al. In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J Virol. 2008;82:5887–5911. doi: 10.1128/JVI.00254-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anderson MA, et al. Astrocyte scar formation aids central nervous system axon regeneration. Nature. 2016;532:195–200. doi: 10.1038/nature17623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Asboth L, et al. Cortico-reticulo-spinal circuit reorganization enables functional recovery after severe spinal cord contusion. Nat Neurosci. 2018;21:576–588. doi: 10.1038/s41593-018-0093-5. [DOI] [PubMed] [Google Scholar]
- 55.Wang F, et al. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J Mol Diagn. 2012;14:22–29. doi: 10.1016/j.jmoldx.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Augur is available from GitHub (https://github.com/neurorestore/Augur) and as Supplementary Software 1.
Raw sequencing data and count matrices have been deposited to the Gene Expression Omnibus (GSE142245).