

Abstract
At the end of mitosis, eukaryotic cells must segregate both copies of their replicated genome into two new nuclear compartments1. They do this either by first dismantling and later reassembling the nuclear envelope in a so called “open mitosis”, or by reshaping an intact nucleus and then dividing into two in a “closed mitosis”.2,3 While mitosis has been studied in a wide variety of eukaryotes for over a century4, it is not known how the double membrane of the nuclear envelope is split into two at the end of a closed mitosis without compromising the impermeability of the nuclear compartment5. In studying this problem in the fission yeast Schizosaccharomyces pombe, a classical model for closed mitosis5, we use genetics, live cell imaging and electron tomography to show that nuclear fission is achieved via local disassembly of the nuclear envelope (NE) within the narrow bridge that links segregating daughter nuclei. In doing so, we identify a novel inner NE-localised protein Les1 that restricts the process of local NE breakdown (local NEB) to the bridge midzone to prevent the leakage of material from daughter nuclei. The mechanics of local NEB in a closed mitosis closely mirror those of NEB in open mitosis3, revealing an unexpectedly deep conservation of nuclear remodelling mechanisms across diverse eukaryotes.
A key event in the process of cell division in eukaryotes is the partitioning of the nuclear genome into two nuclear compartments. To achieve this, replicated sister chromosomes detach from the inner nuclear envelope (NE)3, enabling them to be separated from one another through the work of a microtubule-based spindle6, before being sorted into two new, physically separate nuclei at mitotic exit. Eukaryotic cells have adopted a wide spectrum of strategies to coordinate nuclear remodelling with chromosome segregation7. At one extreme, in a so called “open mitosis”, cells first disassemble the nuclear lamina and the continuous nuclear envelope (NE) at mitotic entry, and then reverse this process by reassembling the structure around separated chromosomes at mitotic exit. At the other extreme, in a “closed mitosis”, because the nuclear/cytoplasmic compartment barrier remains intact throughout the division process, spindle poles must be inserted into the nuclear envelope8 to form an intranuclear spindle that can drive chromosome segregation. This spindle is then disassembled as the NE is divided into two9. While these different modes of nuclear division share key features, and despite there being a range of intermediate states10,11, the resolution of a closed mitosis presents a unique topological challenge. Currently it is not understood how this is overcome to enable organisms undergoing a closed mitosis to divide the double NE without compromising nuclear integrity.
To shed light on this process, we chose to study nuclear division in the fission yeast, Schizosaccharomyces pombe (S. pombe), which serves as an experimentally tractable example of an organism that undergoes a classic closed mitosis. Previous studies have shown that the S. pombe nucleus does not tear at mitotic exit12, as it does in the related yeast S. japonicus. Instead the nucleus constricts to form a dumbbell shape with a thin nuclear bridge around the anaphase spindle5. While the organisation and dynamics of the anaphase spindle have been studied in some detail13–15, it is not known how the nuclear envelope is then remodelled to generate two new nuclei at the end of this process without compromising the nuclear/cytoplasmic compartment barrier.
To explore this question, we used a synthetic nuclear-localised GFP construct to characterise the dynamics of nuclear fission, and to determine the extent to which the nuclear permeability barrier is maintained throughout the process (Fig. 1a, 1b). Although nuclear GFP levels remained constant throughout the division process (as expected for a closed mitosis), we observed a gradual loss of GFP from the nuclear bridge prior to nuclear division (Fig 1b, using an automated analysis pipeline described in Extended Data Fig. 2a and Methods). Importantly, this occurred without the visible leakage of GFP from daughter nuclei (Figure 1a). Imaging the nuclear envelope remodelling over the same time period is complicated by the fact that in fission yeast, as in other eukaryotes, the outer face of the nuclear envelope is continuous with the 3D mesh of tubules and sheets constituting the endoplasmic reticulum (ER; Fig. 1c and Extended Data Fig. 1a)16. In searching for a better marker of the inner nuclear envelope to image this process live, we homed in on a hitherto-uncharacterised candidate SPAC23C4.05c, owing to its homology to the stress-responsive NE protein Ish1 (Extended Data Fig. 1c-e) and its strong negative genetic interactions with ESCRTIII proteins (Extended Data Fig. 1b)17. SPAC23C4.05c localises exclusively to the nucleoplasmic surface of the inner NE throughout the cell cycle (Fig. 1c, Extended Data Fig. 2d-e) without labelling ER tubules or cortex. Strikingly, SPAC23C4.05c was also seen concentrated at the stalk of each daughter nucleus during anaphase - a phenotype for which we named the protein Les1, or LEM-like Enriched in Stalks (See Methods for further details).
Figure 1. Les1 domains define the site of nuclear division.

a. S. pombe cell (dotted outline) expressing a synthetic NLS-GFP construct undergoing mitosis; panels are maximum intensity projections of confocal images acquired every 30 seconds. Representative of >50 cells across 5 biological repeats. Dotted vertical lines represent maximum spindle length (Spindle max, t=0). b. Mean (darker lines) and standard deviations (lighter bands) of averaged single-cell traces (from 11 cells at t=300 to 19 cells at t=0, from 2 strains) aligned at spindle max (t=0) and normalised to daughter nuclear intensity at t=0. c. Single-plane Airyscan reconstructions of live cells at various stages of the cell cycle expressing a synthetic ER-localised mCherry construct (AHDL) and Les1 (Les1; SPAC23C4.05c) tagged with mNeonGreen at the endogenous locus. Green arrows mark the boundaries of the Les1 stalks, visible in late anaphase. Representative of >10 cells across 2 biological and 8 technical repeats. d. Reconstructions using SRRF at 28 second intervals on single-plane confocal slices, of a cell undergoing anaphase, expressing Atb2-mCherry (Tubulin) and Les1-mNeonGreen tagged at the endogenous loci. Green arrows mark Les1 stalk boundaries, which first become visible in mid-anaphase. Representative of >10 cells across 2 biological and 8 technical repeats. e. Les1 intensity in stalks (1.5 μm from nuclear periphery) over time: mean (darker line) and standard deviations (lighter band) of between 11 (t=300) and 36 (t=0) single-cell traces, aligned at spindle max (t=0) and normalised to maximum bridge intensity. All scale bars = 2 μm.
Using Les1 as a marker, we were then able to follow in detail the dynamic changes in nuclear shape that accompany spindle elongation – as a single nucleus divides into two via a characteristic dumbbell-shaped intermediate (Fig. 1d). The kinetics of spindle elongation are highly reproducible18, allowing us to align single-cell trajectories to the time point at which the spindle reaches its maximum length (see also Methods). At early stages of bridge formation, Les1 was found to concentrate in stalks originating at the neck of each daughter nucleus (Fig. 1d-e). At maximum spindle elongation, Les1 was visibly depleted from the midzone of the bridge (Fig. 1d); a process that was followed, within a few seconds, by the breakage of the spindle (Fig. 1d and Extended Data Fig. 2b).
Since these observations pointed to the midzone of the bridge as the site where nuclear fission likely occurs, we used correlative light microscopy and electron tomography of Les1-mNeonGreen/mCherry-Atb2 dual-labeled cells (Fig. 2a) to characterise early and late nuclear bridges (Fig. 2b-c and Extended Data Fig. 3a). In early bridges, the nuclear envelope was seen to envelop the spindle (narrowing at the base of the stalks and widening towards the midzone) and was studded with nuclear pores (Fig. 2b). At an intermediate stage, the nuclear pores were completely excluded from the stalk and clustered in a central bulge (Extended Data Fig. 3b). By contrast, in late stage bridges, while the NE still enveloped the spindle within stalks, there was no evidence of a continuous nuclear envelope within the midzone region of the bridge. Instead, spindle microtubules were seen projecting out of the two newly formed daughter nuclei, through stalk membranes that lacked nuclear pores into the cytoplasm (Fig. 2c). All that was left of the central part of the nuclear bridge at this stage were fragments of membrane - an observation that explains the loss of nuclear GFP from the midzone of the late anaphase bridge (Fig. 1).
Figure 2. Nuclear division occurs by local nuclear envelope breakdown.

a. Representative SRRF reconstructions from single-plane confocal slices of cells in early (lefthand panels) and late (right-hand panels) anaphase, expressing Les1-mNeonGreen and Atb2-mCherry (tubulin) tagged at the endogenous loci. Note that intact spindle microtubules still persist in the late anaphase bridge despite midzone clearance of Les1. Representative of >10 cells across 2 biological and 8 technical repeats. Scale bar = 2μm. b. Virtual slice through electron tomogram (left) and 3D segmentation model (right) of early anaphase bridge (nuclear envelope in green, microtubules in magenta). White region indicates magnified region in left panel. Blue arrows indicate nuclear pores. 1 technical repeat. Scale bars = 100 nm. c. Right to left: Full and clipped views of 3D segmentation models of late anaphase bridges (nuclear envelope in green, microtubules in magenta), superimposed on virtual slices through electron tomograms, shown without model in second from left. Magnified regions are different virtual slices of the region indicated by white insets. Magenta arrows indicate microtubules, green arrows indicate stalk tip and limit of intact nuclear envelope. Blue arrows indicate nuclear pores. Representative of 5 technical repeats. Scale bars = 100 nm.
If the nuclear envelope in the central region of the bridge is disassembled to induce nuclear division, as suggested by this unexpected observation, how is this distinct midzone region specified? A clue to this came from two observations made using EM. First, nuclear pores within early-stage bridges were found to be physically too closely apposed to spindle microtubules to have a full nuclear basket, based on steric considerations (Extended Data Fig. 3c, Extended Data Fig. 2d-e). Second, nuclear pores are completely absent from stalks in late-stage bridges (Figure 2c). Building on these data, when we tracked various components of the nuclear pore complex (NPC; individual proteins referred to as nucleoporins or “Nups”; Extended Data Fig. 2d) through nuclear division by light microscopy we discovered that the pores that enter the bridge are completely devoid of nuclear basket components Alm1 and Nup211 (Fig. 3a, Extended Data Fig. 4a-c). NPCs were also visibly depleted from stalk regions of the bridge where Les1 is concentrated (Fig. 3b-c, Extended Data Fig. 5a). Instead, as previously reported19,20, NPCs were found concentrated within the central bulging section of the bridge, where Les1 levels are low (Fig. 3b-c). Even here, the movement of NPC clusters within the midzone appeared constrained by the stalks on either side (Fig. 3c).
Figure 3. Les1-restricted stepwise NPC disassembly drives local NEB.

a. Maximum intensity projections of confocal images acquired every 60 seconds of S. pombe expressing Alm1-mNeonGreen and Cut11-mCherry. Scale bar = 2 μm. Representative of >30 cells across 3 biological repeats. b. iSIM images of cells expressing Cut11-mCherry and Les1-mNeonGreen. Scale bar = 2 μm. Representative of >10 cells across 2 biological and 10 technical repeats. c. Kymograph of intensities averaged across a single confocal slice of a dividing cell (white rectangular box in inset) expressing Cut11-mCherry and Les1-mNeonGreen and imaged at 3 frames per second followed by denoising. Magenta arrows indicate a mobile but corralled NPC cluster. Representative of >10 cells across 2 technical repeats. d. Maximum intensity projections of confocal images of cells expressing Cut11-GFP and Atb2-mCherry and acquired at 10 second intervals. Scale bars = 2 μm. Representative of >30 cells across 3 biological repeats. e. Averaged normalised intensity traces for NLS-GFP (between 22 cells at t=-300 to 37 cells at t=0), Nup60-mNeonGreen (between 11 cells at t=-300 to 34 cells at t=0), and Cut11-mCherry (between 11 cells at t=-300 to 34 at t=0), aligned by spindle max (t=0). Dotted orange line indicates single exponential fit to NLS-GFP average. Data from two strains were combined based on the analysis described in Extended Data Fig. 6b. f. Confocal maximum intensity projections of imp1Δ cells expressing Les1-mNeonGreen and Cut11-mCherry in the presence (lower panel) or absence (upper panel) of 5 μM Latrunculin A (lower panel). Magenta arrows indicate persistent midzone Cut11 clusters. Scale bar = 2 μm. Representative of >20 cells across 2 biological and 4 technical repeats. g. Confocal maximum intensity projections of imp1Δ cells expressing Alm1-mNeonGreen and Cut11-mCherry. Magenta arrows indicate persistent midzone Cut11 clusters. Representative of >20 cells across 2 biological repeats. Scale bar = 5 μm.
Several lines of evidence indicate that Les1 accumulation - and the concomitant corralling of NPCs - requires a close association of the nuclear envelope with spindle microtubules but is not contingent upon the formation of a normal bridge midzone. First, occasional ‘stray’ NPC clusters located distal to the midzone always correspond to areas of local Les1 depletion and local bridge dilation (Extended Data Fig. 5c-f). Second, in cells with excess NE that generate thicker, floppy bridges, Les1 cannot form stalks and NPCs are no longer restricted to the midzone (Extended Data Fig. 6a). Finally, aberrant tubular projections containing spindle microtubules, induced either by reducing NE surface area or by acutely inhibiting Aurora kinase activity before cells enter mitosis (See Methods), accumulate Les1 and lack NPCs (Extended Data Fig. 6b-c). Taken together, these results indicate that the stalks, defined by Les1 accumulation at sites of microtubule-membrane contact within the bridge, function to restrict a population of basketless NPCs to the bridge midzone.
In an open mitosis, the stepwise removal of NPCs leads to NE fenestration and loss of structural integrity during nuclear envelope breakdown (NEB)11. We observed precisely this sequence of events in the S. pombe bridge midzone, in a process we term “local NEB”. Thus, in the few minutes following dumbbell collapse, as the nuclear NLS-GFP signal was seen disappearing from the bridge (Fig. 3e), NPCs were gradually lost from the midzone. This process began with the loss of nuclear ring Nups (Nup60) and was completed with the loss of transmembrane Nups (Cut11) (Fig. 3d-e, Extended Data Fig. 5g-i). While fast imaging revealed distinct clusters of NPCs disappearing at different times, this order of events was preserved within any single bridge (Extended Data Fig. 5i). Strikingly, while this was independent of Les1 itself (Extended Data Fig. 7d-e), both NPC disassembly and local NEB could be completely arrested through the deletion of the importin Imp120 (Fig. 3f-g, Extended Data Fig. 6d). Since local NEB precedes spindle disassembly (Fig. 3d-e, Fig. 2c), this also enables us to reinterpret the previously described Imp1 - mutant spindle phenotype20. As our new data make clear, Imp1-dependent removal of bridge NPCs and local nuclear envelope breakdown expose spindle microtubules to the cytoplasm, where cytoplasmic factors trigger their timely disassembly.
What is the function of Les1 in this context? In support of the idea that the site of NEB is restricted to the bridge midzone by Les1-defined stalks, the spatial organization of NPCs in the bridge was completely lost in the les1Δ strain. In these cells, NPC components were found to be uniformly distributed in both early and late stage bridges of les1Δ cells by light microscopy (Fig. 4a, Extended Data Fig. 7f) and EM (Fig. 4c and Extended Data Fig. 7a-b), leading to the onset of local NEB at a random location along the bridge (Extended Data Fig. 7f). At the same time, daughter nuclei in the les1Δ strain suffered transient leakages at the time of maximum spindle elongation, as measured by loss of nuclear NLS-GFP (Fig. 4a-b), which typically occurred in only one of the two daughter cell nuclei (Fig. 4a-b). These leaks were rapidly repaired in a manner that does not depend on Les1 (Fig. 4a). This likely explains the lack of a growth defect in the les1Δ strain, since the repair process is associated with recruitment of the ESCRTIII protein Cmp7 (Fig. 4d)21 to sites of local NEB (Fig. 4d and Extended Data Fig. 8b-c). In line with this, les1Δ is synthetically near-lethal when combined with deletions in cmp7 or lem2 - Cmp7’s binding partner (Extended Data Fig. 8d-e) – observations that are also consistent with the deeply conserved role for ESCRTIII proteins in NE sealing22–25 and repair26,27 across metazoan and fungal mitoses.
Figure 4. Les1 isolates nuclei from local NEB.

a. Maximum intensity projections of spinning disk confocal images of cells expressing Nup60-mCherry tagged at the endogenous locus and NLS-GFP, either wildtype (top panels) or carrying a Les1 deletion (lower panels), imaged at 30 second intervals. Orange arrows indicate sites of NLS-GFP leakage from one or both daughter nuclei. Representative of >50 cells across 4 biological repeats. Scale bars = 2 μm. b. Single cell intensity traces for wildtype (left 2 panels) and les1Δ cells (right 2 panels), indicating intensity in each daughter nucleus, aligned at spindle max (dotted line, t =0) and normalised individually to maximum intensity. Orange bar and text indicate the number of leaky nuclei. c. Virtual slices through electron tomograms of a dividing les1Δ cell. The approximate overlap in field of view of the tomograms is indicated by dashed lines. NPCs are indicated by blue arrowheads. Scale bars = 100 nm. Representative of 2 technical repeats. d. Confocal maximum intensity projections of les1Δ cells expressing Cmp7-mNeonGreen and Nup60-mCherry. Representative of >20 cells across 3 biological repeats. Scale bar = 2 μm. e. les1Δ cells expressing NLS-GFP and Nup60-mCherry, in the presence (lower panel) or absence (upper panel) of 10 μM Cerulenin. Orange arrows indicate sites of NLS-GFP leakage. Representative of >40 cells across 3 biological repeats. Scale bar = 5 μm. f. Schematic indicating relative intensity levels of key readouts of the nuclear division process, aligned relative to spindle max. g. Schematic illustrating the process of local NEB and the role of Les1 in structuring the bridge.
Taken together, these data suggest that Les1 stalks functionally isolate daughter nuclei from the process of Imp1-dependent local NEB at the centre of the bridge (Extended Data Fig. 8g). Consistent with this hypothesized role for Les1, deleting Les1 does not strongly impact the kinetics of NPC disassembly (Extended Data Fig. 7d-e) or average inter-NPC spacing (Extended Data Fig. 7b). Instead, Les1 likely acts to create a seal by cinching the inner nuclear envelope tightly around the spindle, leading to the segregation of excess membrane and bulky NPCs into the characteristic bulge at the centre of the bridge in wild-type cells, a structure conspicuously absent from les1Δ tomograms (Fig. 4c). In line with this, treating les1Δ cells with Cerulenin to increase membrane tension in the bridge (by inhibiting fatty acid biosynthesis in the ER28) forces NPCs back into the centre to rescue the nuclear leakage phenotype in the proportion of cells able to form a bridge (Fig. 4e and Extended Data Fig. 8f). Therefore, Les1 performs a critical role in nuclear division in S. pombe by ensuring that, while nuclei are topologically open to the cytoplasm at this stage of mitosis, the compartment boundary itself remains effectively closed (Fig. 4f-g).
In summary, in this study we identify a protein Les1 that positions the site of nuclear fission during S. pombe nuclear division. Through the study of Les1 localisation and its deletion mutant, we describe a novel process of local nuclear envelope breakdown that reveals an unexpectedly close similarity between the remodelling of the nuclear envelope in open and closed mitosis (Fig. 4g). In both cases, the new nuclear compartment is remodelled as the result of NPC disassembly. Thus, the key difference between mitotic strategies across the eukaryotic tree29 may only be one of degree, depending on the timing and localisation of NPC disassembly.
Methods
Phylogenetics and protein bioinformatics
Secondary structure models for Les1 were generated using the homology modelling and threading software tool I-TASSER30,31 (https://zhanglab.ccmb.med.umich.edu/I-TASSER/). All sequence homology searches were carried out using a local installation of the HMMER suite of profile-HMM search tools32 (http://hmmer.org/). Uniprot IDs for sequences analysed in Fig S1: O94559 (Les1_Sp), S9RL50 (Les1_So), S9VS84 (Les1_Scr), B6JWA6 (Les1_Sj), Q9Y7X6 (Ish1_Sp), S9PR74 (Ish1_So), S9X3G6 (Ish1_Scr), B6K568 (Ish1_Sj) and Q03104 (MSC1_S.cerevisiae). MAFFT33,34 (https://mafft.cbrc.jp/alignment/software/) was used to generate sequence alignment with the following command-line options:
mafft --maxiterate 1000 --localpair <infile.fasta> > <outfile.align>
Both trimmed and untrimmed alignments were used to generate phylogenetic trees, though in the case of Les1 this did not significantly affect the resulting trees. Alignments were trimmed, using TrimAl35 (http://trimal.cgenomics.org/trimal) with these command-line options:
trimAl/source/trimal -in <infile.align> -gt 0.6 -cons 40 -phylip -out <outfile.trim>
Maximum-likelihood trees were inferred using IQTREE36,37 1.6 (http://www.iqtree.org/), run using the model test function (for Extended Data Fig. 1d, LG+G4) and 1000 bootstraps:
iqtree-omp-1.5.4-MacOSX/bin/iqtree-omp -nt 4 -s <infile.trim> -m TEST -bb 1000 -redo
Conserved motifs were detected in alignments through comparisons with the PFAM database38,39 (https://pfam.xfam.org/). Selected regions of alignments were displayed using ESPript340 (http://espript.ibcp.fr) and default options. Although Ish1 and Les1 are annotated to be Type I LEA domain proteins (Uniprot; https://www.uniprot.org/uniprot/Q9Y7X6), no homology or profile HMM similarity was detected to any LEA domain families. Instead, the easily detected but poorly characterised Ish1 motif (PFAM PF10281), present in two copies in both SPAC23C4.05c and Ish1 (as well as S. cerevisiae MSC1; Extended Data Fig. S1c-e), bears similarity to the widely conserved HeH/LEM (PFAM PF12949) and SAP (PFAM PF02037) domains. To reflect this finding, we settled on the name LEM-like Enriched in Stalks (Les1) for the S. pombe protein SPAC23C4.05c and its best-match orthologs in Schizosaccharomyces species.
S. pombe culture
S. pombe cells were cultured using standard methods41,42 on solid (YES-agar) and liquid (YES) rich growth media (ForMedium), at a growth temperature of 32°C unless stated otherwise. All experiments were performed in exponential growth at 32°C with at least 48 hours of growth (>20 generations) in liquid YES before plating for live imaging. For live imaging, uncoated 35-mm dishes with polymer coverslips (No. 1.5 coverslip, 180 μm thick, Ibidi) were first coated with 1 mg/mL soybean lectin (in water, aliquots stored at -80°C, Sigma-Aldrich) for 15 minutes. After washing away the excess lectin with fresh YES, cells drawn from exponentially growing liquid cultures were allowed to settle for 30 minutes in a minimum volume of 500 μL of YES. The entire plating volume was replaced with 1 mL of fresh YES prewarmed to 32°C prior to transfer to the microscope. For drug treatment experiments, Latrunculin A (Sigma-Aldrich), Cerulenin (Sigma-Aldrich), and 1NM-PP1 (Calbiochem) were added to YES at 5 μM (10 mM stock in DMSO), 10 μM (10 mM stock in DMSO), and 5 nM (1 mM stock in DMSO) respectively. The maximal amount of DMSO (1/1000 in YES) added to cells across conditions had no detectable effect on the kinetics of nuclear division – thus, to ease direct kinetic comparisons across all strains and experiments in the paper, drug-treated cells were compared to untreated controls. For cell culture for electron tomography experiments, see section on correlative fluorescence microscopy and electron tomography.
Plasmids and S. pombe strain construction
The full genotypes of all strains used in this study are described in Extended Data Table 1, at the end of this document. Strains generated specifically for this study were constructed using standard methods41,42 for gene editing and crossing. Gene deletions and tagging were performed as previously described43 for PCR-based gene targeting, using standard primers designed with the Bahler lab web-interface scripts (http://bahlerlab.info/resources/), pFA6a vector templates carrying Hygromycin (Hph) or Kanamycin (Kan) resistance cassettes, and transformation using the Lithium Acetate method44. Antibiotic-resistant clones generated by this method were verified by PCR of the gene locus being targeted as well as fluorescence microscopy, if applicable. The exception to the standard workflow was for the pFA6a-mNeonGreen vectors used in this study, which carry a non-standard linker upstream of the mNeonGreen coding sequence. Instead of the standard 20-mer (CGGATCCCCGGGTTAATTAA) forward linker, these require a 21-mer forward linker (GATTCTGCTGGATCAGCTGGC). The reverse linker remains unchanged. One new pFA6a vector derivative was generated for this study, replacing the mCherry coding sequence in pFA6a-mCherry:Hph with the coding sequence for the photo-switchable fluorescent protein mEOS3.245 (Addgene) by standard restriction-digestion cloning (using restriction enzymes BamHI and AscI, NEB). This vector is available upon request. Crosses were performed by random spore analysis41,42 followed by marker selection (Hygromycin/Kanamycin resistance, ura/leu auxotrophy, or sensitivity to 5 μM 1NM-PP1 (Calbiochem) for strains carrying the Ark1-as3 allele, as appropriate) followed by additional screening for fluorescence, if applicable. The crosses shown in Extended Data Fig. 8d-e were carried out using tetrad dissection46, with each colony grown from a single spore. Briefly, cells were mated on low nitrogen media (EMM-Nitrogen) to produce tetrads of four haploid spores. After 2 days, cells were streaked onto nutrient-rich media (YES) for 2-3 h to degrade the protective membrane surrounding each tetrad. Individual spores were isolated using a Singer MSM300 Tetrad Micro dissecting unit. C-terminal truncation constructs with C-terminal mNeonGreen tags (Extended Data Fig. 8a) were generated at endogenous loci using the standard PCR-based method as described above, but with the following left-hand/forward primers:
-
Les1 (1 -94)
5’-ATTCTTGGCCTCAACGAAAGCTTGATGACTTTCTCCAAAATCATGGGG TAAAGTCACTGGACGTTCCTCCTATCGAGACTGATTCTGCTGGATCAGCTGGC-3’;
-
Les1 (1 -204)
5’-CCACCAATGATGAGTTGGAATCCTGGTCAAATAATCTACTCCTTTCTA TGTTGGATCAGAAAAACATTACAGTACCAATTGATTCTGCTGGATCAGCTGGC-3’;
-
Les1 (1 -291)
5’-TTTCTGTTCTTTCACCTCGGGAAACTCTTTTGAAAGAAGCATACGCTA ACCGCTTCACACCGCGTGTAATGATTGCCTCCGATTCTGCTGGATCAGCTGGC-3’.
These truncation sites were selected in order to delete either both Ish1 motifs and the C2H2 Zn finger, or the second Ish1 motif and the Zn finger, or just the Zn finger, with care taken to avoid truncating the protein in the middle of predicted secondary structural elements such as alphahelices. The first two constructs, Les1(1-94) and Les1 (1-204), did not generate any detectable expression by fluorescence microscopy. The localisation pattern of the third construct Les1 (1-291) is shown in Extended Data Fig. 8a.
Live-cell fluorescence microscopy
All strains were imaged live in regular growth medium (YES) in glass-bottom dishes (see S. pombe culture) within stage-top incubation chambers held at 32°C. No single dish was used for experiments lasting longer than 3 hours from the time of plating. Four microscopes were used for this study: 2 spinning disk confocal systems, a Nikon TiE widefield system with a VT-iSIM module and a Zeiss LSM880 with an Airyscan module. The first spinning disk microscope consists of a Nikon TiE inverted stand attached to a Yokogawa CSU-X1 spinning disk scan head and a Hamamatsu C9100-13 EMCCD camera. The entire system was controlled using Volocity software. Cells were imaged using a 100X oil-immersion CFI Plan Apochromat VC objective (1.4NA, working distance 0.13 mm) with an optional 1.5x additional magnification. The second spinning disk microscope consists of a Zeiss Axio Observer Z1 inverted stand attached to a Yokogawa CSU-W1 spinning disk scan head and a Photometrics Prime BSI Scientific CMOS detector. Cells were imaged using 63X oil-immersion Plan Apochromat (1.4NA, working distance 0.19 mm) and 100X oil-immersion Plan Apochromat (1.4NA, working distance 0.17 mm) objectives combined with an optional 1.5x additional magnification. The LSM880 is an inverted laser-scanning confocal microscope with an Axio Observer stand. Cells were imaged using a 63x oil-immersion Plan Apochromat objective (1.4NA, working distance 0.19 mm) and the Airyscan47 detector. Acquisition on the latter systems is controlled via the Zen software (Zeiss). In all cases, samples were illuminated with 488 nm (mNeonGreen or GFP) and 561 nm (mCherry) lasers and appropriate fluorescence filter sets for these fluorophores. Photoconversion of mEOS3.2 was carried out using a 405 nm laser, with the non-converted state imaged using a 488nm laser and and the same filter set as for mNeonGreen/GFP, and the converted state imaged using a 561nm laser and the same filter set as for mCherry. For regular live imaging on all systems, asynchronous cells were usually imaged using a 4.3 μm Z-stack with 16 slices at 270 nm vertical intervals, and time intervals ranging from 5 seconds to 120 seconds, never exceeding 30 minutes of continuous imaging. For Airyscan imaging, cells were imaged using a larger Z-stack at single timepoints. For SRRF and Hough fitting, cells were imaged with the system held at a single Z-plane and the imaging rate was increased to yield final reconstructed SRRF images at >3 frames per second. iSIM (“instant SIM”) images were acquired with a Visitech VT-iSIM module and Hamamatsu Flash4.0v3 scientific CMOS camera, attached to an inverted Nikon TiE microscope stand with Perfect Focus System and motorised stage (100x oil immersion 1.45 NA Plan Apo λ objective).
Image processing
All basic image processing (cropping, viewing stacks, scaling for visual presentation, producing maximum intensity projections) was carried out in Fiji48 ((ImageJ49). All time-lapse images subjected to Super-Resolution Radial Fluctuations (SRRF) analysis were processed with NanoJ-LiveSRRF, the newest implementation of NanoJ-SRRF50. NanoJ-LiveSRRF is available on request, expected to be available for download soon. NanoJ-SRRF is already released and available as open-source software for Fiji/ImageJ. Airyscan processing and iSIM 3D deconvolution were carried out using proprietary Zen (Zeiss) and Elements (Nikon) software respectively.
Image denoising for fast-acquired data
Denoising (Fig. 3c, Extended Data Fig. 4b) was performed using the Noise2Noise image restoration technique51 as implemented using the CSBDeep/CARE framework52. Three separate Noise2Noise models were trained independently for Alm1-mNeonGreen images, Les1-mNeonGreen images and Cut11-mCherry images. Training data comprised 2000 pairs of intensity-normalised 64x64 pixel patches in two adjacent frames, randomly selected from across all acquired datasets for each model. Validation data comprised an additional 200 pairs of 64x64 pixel patches randomly selected and normalised via the same method. Each model was trained with the following network parameters: training loss = mean-squared error, UNet kernel size = 3x3, training steps per epoch = 200, training epochs = 100. Jupyter notebooks for network training and prediction and trained Noise2Noise models are available at https://github.com/superresolusian/local-NEB.
Statistics and reproducibility
Through the entire paper, the basic independent biological unit of comparison is the single cell (undergoing nuclear division). For experiments in the paper using light microscopy, biological repeats refer to cells drawn from two different cultures and plated separately; technical repeats refer to cells drawn from the same culture but plated separately, usually imaged on the same day. For high-resolution (in time or space) imaging of strains already analysed by conventional confocal imaging, we focused on collecting data from additional technical repeats. The number of cells indicated in the legends that accompany representative images is a conservative estimate of the number of cells, across biological and technical repeats, at the same cell cycle stage. Special considerations apply for electron microscopy (see “Correlative fluorescence microscopy and electron tomography”). Since cells for all experiments were cultured at 32°C in rich YES growth media, we were able to pool cells across technical and biological repeats, as well as different clones carrying the same deletion or fluorescent protein tag, for population-level analyses (See “Analysis framework for single-cell trajectories” and “Population-level analyses”).
Analysis framework for single-cell trajectories
Analysis of single-cell trajectories was carried out using custom software written for the opensource platform Fiji48. This is available from https://github.com/superresolusian/local-NEB.
ROI selection
Regions of Interest (ROIs) containing dividing cells were manually selected in time series data. Only division events that completed were selected for analysis (Extended Data Fig. 2a, ‘Manually select ROIs’, ‘Extracted ROI’). A ROI devoid of nuclei over the whole timelapse was also selected for background subtraction of measured intensities.
Detecting divisions
Each ROI was maximum-intensity projected and this projection was then blurred, binarised, hole-filled and skeletonised using ImageJ1 ‘Binary’ functions in Fiji48. The longest path of the skeleton structure was assumed to correspond to the dividing nucleus, and the angle formed by this line-like path measured to be the division angle, 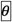 (Extended Data Fig. 2a, ‘Determine division angle’).
(Extended Data Fig. 2a, ‘Determine division angle’).
Circle detection
Within ROIs, nuclei were identified for each frame and their radii determined using a custom-written Fiji plugin implementing the circular Hough transform53 (Extended Data Fig. 2a, ‘Circle detection’; each coloured circle denotes a separate detected nucleus). For two-colour images, the mNeonGreen channel was used to identify nuclei due to their superior signal-to-noise ratio, and these nuclei centroid coordinates were assumed to be the same across both channels. For NLS images, a Sobel filter was applied to extract the perimeters of the nuclei prior to performing the circular Hough transform.
Identification of dividing nucleus pairs
For all possible pairs of detected circles in the ROI, the angle between the circle centres was calculated. Circle pairs oriented at angles different from the division angle by more than 30° were rejected. For proteins distributed along the whole length of the bridge between the two daughter nuclei (e.g. Les1 as shown in Fig. 1d), all candidate bridge paths in each ROI frame were identified by blurring, binarising, hole-filling and skeletonising the images (Skeleton paths shown in Extended Data Fig. 2a, ‘Path segmentation’). Paths of length < 3 pixels were rejected as these corresponded to isolated nuclei (e.g. Extended Data Fig. 2a, ‘Path segmentation’, blue paths). The endpoints of the remaining paths were then checked against the coordinates of the remaining circle pairs. Paths without anchoring circle detections were rejected (e.g. Extended Data Fig. 2a, ‘Path segmentation’, pink paths), as were any detected circles lacking an associated path (e.g. Extended Data Fig. 2a, ‘Circle detection’, red circle). The final result for each frame was either that no division events were successfully detected, or two nuclei and joining path were detected (Extended Data Fig. 2a, ‘Filter circles and paths satisfying selection criteria’). Following breakage of the bridge, there is no longer a continuous path between the daughter nuclei. In this case, for a detected circle pair at an appropriate angle we joined two paths, each with one endpoint anchored at one of the circles, with a straight line between the ‘free’ endpoints (Extended Data Fig. 2a, ‘Filter circles and paths satisfying selection criteria’, yellow dotted line). For proteins not continuously present along the whole length of the bridge (e.g. NLS as shown in Fig. 1a) the path corresponding to the bridge location was defined as a straight line between the two daughter nucleus circle centres.
Definition of bridge and timepoints
The bridge was defined as the connection between the two daughter nuclei excluding any pixels within the nuclei perimeters. The initiation of division was defined as the first frame in which two distinct nuclei were successfully detected, and dumbbell appearance was defined as the first frame where a bridge could be discerned (i.e. the first timepoint where all path pixels were not contained by daughter nuclei). Nucleus separation distance was defined as the Euclidean distance between the centroids of the daughter nuclei.
Measurement of nuclear intensities
Nuclear intensities were the background-subtracted average intensities within the detected circles (Fig.1b ‘Nucleus’, Fig. 4b).
Measurement of bridge intensities
Total bridge intensity was the background-subtracted average intensity along the bridge path (Fig. 1b ‘Bridge’, Fig. 3e, Extended Data Fig. 4a, Extended Data Fig. 5h, Extended Data Fig. 7d, Extended Data Fig. 8f). In all cases, the path linewidth was set to 5 (i.e. 2 pixels perpendicularly either side of the path) to account for the full thickness of the bridges.
Measurement of vertical displacement of NE proteins
Circle detection and analysis was again performed using the circular Hough transform, but this time on whole frames (i.e. no manually selected ROIs) so that all nuclei within a single time frame were detected. All data analysed had Cut11-mCherry as a reference protein in one channel and another NE protein of interest in the second channel. For each nucleus, the difference in radius between the two channels was calculated (Extended Data Fig. 2c,e). SRRF processing was performed on images prior to radius measurement to increase resolution. As a control, Nup60 was labelled with a green-to-red photoconvertable fluorescent protein (mEOS3.2) and images taken before and after photoconversion. These two channels were then analysed using the same pipeline as for the Cut11 two-colour strains to check that there were no systematic errors in radius measurement between red and green channels. The values obtained for various Nups provide a good match with a recent electron microscopy analysis of the S. pombe NPC54, which also provided the estimate of the width of the lumen between inner and outer nuclear envelopes.
Spindle detection and breakpoint measurement
Spindles were segmented from ROIs in images containing fluorescently labelled tubulin (mCherry-Atb2) by blurring, binarisation, hole-filling and skeletonisation. The longest path of the skeleton was determined for each frame; the frame in which spindle breakage occurred was determined as the first frame where the maximum skeleton path length decreased by ≥ 25% compared to the maximum skeleton path length measured across all previous frames.
Quantifying early bridge intensities
Normalised bridge intensity for early bridges (Extended Data Fig. 7c) was calculated as
where:
f: the first frame in a nuclear division sequence in which a bridge appears that has 1 μm ≤ bridge length ≤ 2μm
I bridge(f): the average intensity along the bridge length in frame f (with a linewidth of 3 pixels for averaging adjacent to the bridge axis)
I bg(f): the average intensity of the image background in frame f
I nuc(f): the average intensity of interphase nuclei in frame f.
Bridge numbers were as follows: Cut11 n=19, Cut11-les1Δ n=27 (p=0.0179, unpaired two-tailed t-test); Nup60 n=44, Nup60-les1Δ n=102 (p=0.0722, unpaired two-tailed t-test).
Data curation
Every detected nucleus division was manually checked to ensure that the correct nuclei had been identified, and ROIs containing false detections were excluded from analysis.
Manual quantification
The breakpoint analysis in Extended Data Fig. 7f was scored manually due to the high error rates of identifying the precise site of breakage from automatically extracted time series data. The timings in Extended Data Fig. 6e-g (with reference to the first appearance of the nuclear bridge) were manually quantified. This was due to the presence of a second green label (Cdc15-mNeonGreen) in addition to Cut11-GFP: while this allowed the visualisation of the cytokinetic ring, it also prevented the automated analysis of maximum intensity projections.
Population-level analyses
Unless otherwise specified, single-cell trajectories (see section above for specific measurements) were aligned to the time of maximal spindle elongation (if a tubulin label not present, measured indirectly using the maximal separation between daughter nuclei as a proxy for spindle length) – making use of the fact that spindle elongation kinetics are highly reproducible from cell to cell. When combining trajectories from different strains (e.g. Fig. 3e), we made use of the observation that mitotic timing tends to be consistent even when spindles reach different maximum lengths (Extended Data Fig. 5b). This is probably due to a Klp9-dependent adjustment in spindle elongation rates between strains (strains with longer spindles elongate their spindles faster)55. Statistical analyses were carried out using GraphPad (mean and standard deviation of averaged traces), MATLAB (ANOVA; mean and standard deviation of averaged traces) and the Estimation Stats platform56 (two-sided Mann-Whitney). Graphs and heatmaps were generated using either GraphPad or MATLAB.
Correlative fluorescence microscopy and electron tomography
Correlative microscopy was done as described before for resin-embedded yeast cells57,58, with minor modifications. In brief, Les1-mNeonGreen/mCherry-Atb2 expressing cells and Cut11-GFP/mCherry-AHDL expressing les1Δ cells were grown in YES at 32°C to mid-log phase, pelleted by vacuum-filtration and high-pressure frozen in the 100 μm recess of aluminium platelets (Wohlwend) using an HPM100 (Leica Microsystems). Samples were freeze-substituted and embedded in Lowicryl HM20 (Polysciences) according to the published protocol57, except with 0.03% uranyl acetate in the freeze-substitution solution, and for the les1Δ experiment, samples were shaken on dry ice for 2 h during freeze-substitution. Resin blocks were sectioned at 320 nm nominal thickness, picked up onto carbon-coated copper grids (AGS160, Agar Scientific) and imaged on the same day on a Nikon TE2000 or Ti2 microscope using a 100x TIRF objective, a NEO sCMOS DC-152Q-C00-FI camera (Andor), and a Niji LED light source. Based on the fluorescence images, cells with profiles in which an elongated bridge was visible within the section plane were selected for electron tomography. 15 nm protein A-coated gold beads (EMS) were adhered to the grids prior to Reynolds’ lead citrate staining. Dual-axis electron tomographic tilt series were acquired approximately from +60° to -60° on a TF20 electron microscope (FEI) operated in STEM mode, using a 50 μm C2 aperture, at 1° increment and 1.1 nm pixel size on an axial bright field detector59, using SerialEM60. Both wild type and les1Δ data are each from one high-pressure freezing and freeze-substitution experiment. Tomograms were reconstructed using IMOD61. Segmentation models were generated using Amira (Thermo Fisher Scientific) by manual tracing of membranes and microtubules, followed by extensive simplification and smoothening of the generated surfaces. Therefore, segmentation models are for purely illustrative purposes. Segmentation models are mirrored relative to the original tomograms, thus the corresponding electron tomographic slices in figure panels Fig. 2b, Fig. 2c, and Extended Data Fig. 3c are shown flipped relative to the original tomograms. Some of the electron tomographic slices shown in figures have been mildly gauss-filtered to improve visibility. NPC nearest neighbour distances were measured in IMOD61. An imod model file was manually generated of the centres of all visible nuclear pores, and the programme imod-dist was used to obtain distances of each pore to all other pores. For each nuclear pore, the shortest distance was determined to be the distance to its nearest neighbour.
Extended Data
Extended Data Figure 1. ER topology; Les1 structure and phylogeny.

a. Airyscan reconstructions of cells expressing the mCherry-AHDL synthetic construct. Orange arrows indicate tubules linking the outer nuclear envelope to the cortical ER. Purple arrows indicate the displacement of the cortical ER by the division ring (not shown). The blue bar indicates the central ER plate formed during late anaphase. Representative of >20 cells across 3 technical repeats. Scale bar = 2 μm. b. Genome-wide genetic interaction scores (E-MAP) with Les1/SPAC23C4.05c. Data from Frost et al. 2012. c. Schematic representation of Les1 sequence with positions of conserved motifs indicated. d. Phylogenetic tree of Les1 homologs across Schizosaccharomyces species with single S. cerevisiae homolog MSC1 grouping separately, indicating a duplication in the lineage leading up to Schizosaccharomyces. Bootstrap values indicated at nodes. See Methods for details of tree construction. e. Alignment of Les1 homologs with key conserved motifs highlighted. Color-coded by % similarity. See Methods for details.
Extended Data Figure 2. Image analysis workflow and its application to NE protein localisation and spindle breakdown timings.

a. Schematic demonstrating pipeline for detecting and measuring nuclei and bridges in timelapse data. Individual steps are described fully in the Methods. Representative images shown here are of Les1-mNeonGreen and Atb2-mCherry. b. The delay between reaching maximum spindle length (spindle max.) and spindle breakage, in seconds. Spindle breakage always follows maximal extension. Sample size(n)=39 individual cells drawn from 3 biological repeats of the entire experiment. Central line represents the mean and the error bars represent the standard deviation of the population. c. Images of two-colour strains used for relative localisation of NE and NPC components and nucleus radii (r) as measured using the circular Hough transform, representative of >130 individual cells each drawn from 2 biological repeats. e. The vertical displacement (relative to the plane of the nuclear envelope) of various nuclear pore complex components (schematised in d; Nup60, in the nuclear ring; Alm1 and Nup211 in the basket; transmembrane Nup Pom34) and NE membrane proteins (Cta4) relative to Cut11. These measurements correspond to the differences in r as shown in panel c. Nup60-mEOS was used as an internal control, with the values representing the displacement of photo-converted Nup60-mEOS (red channel) relative to un-converted Nup60-mEOS (green channel). The dotted lines represent an estimate of the thickness of the nuclear envelope (see Methods for details). n= 393 (Cta4), 455 (Pom34), 266 (Les1), 185 (Nup60), 280 (Nup211), 218 (Alm1), and 135 (Nup60-mEOS) individual cells, each drawn from across 2 biological repeats of the entire experiment. The measure of centre (central line) shows the mean of each population and the error bars represent the standard deviation.
Extended Data Figure 3. Electron tomography of mid- and early-stage bridges with correlative fluorescence images.

a. Fluorescence images of resin sections through cells expressing Les1-mNeonGreen (green) and Atb2-mCherry (magenta), corresponding to cells that were imaged by electron tomography and are shown in Figure 2b (left image), Figure 2c top panel (middle image) and Figure 2c lower panel (right image). Images have been rotated to match approximately the orientation of electron tomograms. Scale bars = 1 μm. Representative of 1 (Figure 2b) and 5 technical repeats (Figure 2c), respectively. b. Fluorescence image of resin section through a cell expressing Les1-mNeonGreen (green) and Atb2-mCherry (magenta), corresponding to cell imaged by electron tomography shown to the right, indicated by white region. Scale bar = 1 μm. The right two panels are virtual slices through electron tomograms of the cell shown to the left. The approximate overlap in field of view of the tomograms is indicated by dashed lines. Note that no NPCs are visible in the stalk part shown in top image. NPCs (indicated by blue arrowheads) are constrained to the midzone (bottom image). Scale bars = 100 nm. Representative of 2 technical repeats. c. Early stage bridge of a dividing cell; same electron tomogram and cell as shown in Figure 2b. Top panel is the segmentation model; white area corresponds to the panels below. Inset slice 1 is a virtual slice through the electron tomogram showing the centres of microtubules, indicated by white arrowheads. Inset slice 2 is 18 nm apart in z and shows a nuclear pore, indicated by blue arrowheads. The NPC thus projects less than 18 nm into the nucleoplasm before encountering the microtubules. 1 technical repeat. Scale bars = 50 nm.
Extended Data Figure 4. Dynamics of individual nucleoporins.

a. Averaged normalised intensity traces for Alm1-mNeonGreen (between 9 cells at t=-300 to 26 cells at t=0) aligned by spindle max (t=0). The central line represents the mean of the population; the shaded area depicts the standard deviation. Images on right are confocal maximum intensity projections of dividing cells expressing Alm1-mNeonGreen and Cut11-mCherry, representative of >20 cells across 3 biological repeats. Scale bar = 2 μm. b. Kymograph of intensities averaged across a single confocal plane of a dividing cell expressing Cut11-mCherry and Alm1-mNeonGreen and imaged at 3 frames per second followed by denoising (see Methods). Representative of >10 cells across 2 technical repeats. c. Confocal maximum intensity projections of dividing cells expressing Nup211-mNeonGreen and Cut11-mCherry. Representative of >30 cells across 3 biological repeats. Scale bar = 2 μm. d. Schematic indicating effect of deleting Nem1 on NE surface area and tension. e. Confocal maximum intensity projections of nem1Δ cells expressing Nup211-mNeonGreen and Cut11-mCherry. Representative of >30 cells across 2 biological repeats. Scale bar = 2 μm. Note that the basket Nup is able to enter the nuclear bridge in this condition. f. Confocal maximum intensity projections of dividing cells expressing various NPC subcomplex components tagged with mNeonGreen at their C termini, co-expressed in each case with Cut11-mCherry. Each set of panels is representative of >20 cells across 2 biological repeats. Scale bars = 2 μm.
Extended Data Figure 5. Relative localisation of Les1 and nucleoporins.

a. Averaged line traces (darker lines = mean, lighter bands = standard deviation) of Les1 and Cut11 intensities along the bridge for 16 cells aligned at bridge length 3 μm. b. Time from bridge formation to maximum spindle length measured for strains pooled to generate Figure 3g (GD173, GD250) and S4d (GD176, GD253). Numbers in brackets (n) indicate number of cells in each population, with bars representing mean and standard deviation. The ANOVA F statistic and p-value are listed above each plot. The line and pairwise p-value within each plot refer to the comparison between pooled strains. c. Single Airyscan reconstructions of cells expressing Cut11 tagged at the endogenous locus with mNeonGreen and a synthetic mCherry-AHDL construct. Magenta arrow highlights stray nuclear pore cluster accompanied by widening of the nuclear envelope. Representative of >10 cells across 2 technical replicates. Scale bar = 2 μm. d. Maximum intensity projections of confocal images acquired every 60 seconds of cells expressing Les1-mNeonGreen and Cut11-mCherry. Bridge formation is at t=0. Green arrows mark boundaries of Les1 stalks, magenta arrow indicates stray pore cluster. Representative of >50 cells across 6 biological repeats. Scale bar = 2 μm. e. SRRF reconstruction from single confocal planes of a cell expressing Les1-mNeonGreen and Cut11-mCherry. Magenta arrow indicates stray nuclear pore cluster, with examples marked upon the line scans along the bridges of 3 illustrative cells in f. Representative of >20 cells across 4 biological repeats. g. Confocal maximum intensity projections of cells expressing Cut11-GFP and Atb2-mCherry and acquired at 10 second intervals. Scale bars = 2 μm. Representative of >20 cells across 2 biological repeats. h. Relative NPC decay rates, calculated for a strain expressing both Nup60 and Cut11 (GD173; between 11 cells at t=-300 and 34 cells at t=0), showing similar relative rates to a cross-strain comparison, as in b. or Figure 3g. Line shows mean of the population. i. Kymograph generated using 10 fps single plane imaging of a strain expressing Nup60 and Cut11 tagged at the endogenous loci with mNeonGreen and mCherry respectively. Blue (Nup60) and magenta (Cut11) arrows represent the staggered decay of individual clusters of nuclear pores. Representative of >10 cells drawn from 2 technical replicates.
Extended Data Figure 6. Response of nuclear division dynamics to acute perturbations.

a. Confocal maximum intensity projections of nem1Δ cells expressing Les1-mNeonGreen and Cut11-mCherry. Note the absence of Les1 stalk formation. Schematic indicates opposing effects of nem1Δ and Cerulenin treatment. Representative of >30 cells drawn from 2 biological repeats. Scale bar = 2 μm. b. Two examples of nuclei attempting to divide in the presence of 10 μM Cerulenin. Note Les1 stalk formation along aberrant bridge-like projections. Magenta arrows indicate NPC clusters. Representative of >30 cells drawn from 3 biological repeats. c. Dividing cells expressing the Ark1-as3 analog-sensitive allele, either co-expressing Les1-mNeonGreen and mCherry-Atb2 or Les1-mNeonGreen and Cut11-mCherry and treated with 5 nM 1NM-PP1 and 5 μM Latrunculin A. In both cases, representative of >30 cells across 2 biological repeats. Scale bars = 2 μm. d. iSIM image of imp1Δ cells expressing Les1-mNeonGreen and Cut11-mCherry. Arrows highlight isolated NPC clusters in Les1-depleted regions. Representative of >40 cells across 2 biological replicates. Scale bar = 2 μm. e. Time-lapse confocal images of cells expressing Cut11-GFP, Cdc15-mNeonGreen, and mCherry-AHDL acquired at 60 second intervals with frames displayed at 3-minute intervals. Representative of >50 cells across 3 biological replicates. Scale bars = 4 μm. Treatment with Latrunculin A depolymerises the actin ring (marked by Cdc15) but has a minimal impact on the time of nuclear division, as marked by the time from bridge formation to complete NPC signal loss (f). Numbers above and below the horizontal bar represent the difference in means with 95% confidence interval and the two-sided Mann-Whitney p-value. n=180 (untreated) and 146 (Lat A) individual cells in each population, in each case pooled from 3 biological replicates. Overlaid on individual data points, the upper and lower extent of the boxes span the inter-quartile range and the central bar denotes the median. g. Cytokinetic ring constriction only begins after nuclear division completes, and it takes almost 30 minutes for the ring to constrict and septation to complete. n=222 (NPC loss to constriction start), 103 (constriction start to end) and 111 (constriction end to septation) individual cells in each population, in each case pooled from 3 biological replicates. Overlaid on individual data points, the upper and lower extent of the boxes span the inter-quartile range and the central bar denotes the median.
Extended Data Figure 7. Nucleoporin localisation and dynamics in les1Δ cells.

a. Fluorescence image of resin section through a les1Δ cell expressing Cut11-GFP (green) and a synthetic mCherry-AHDL construct (magenta), corresponding to the cell that was imaged by electron tomography shown in Figure 4c. Representative of 2 technical repeats. Scale bar = 1 μm. b. Inter-pore distances for tomograms shown in Figures S3b (WT Mid-stage), 2b (WT Early stage) and tomogram not shown (les1Δ Mid-stage). See Methods for details on measurement. n=26 (WT mid-stage), 18 (WT early stage) and 18 (les1Δ mid-stage) individual nuclear pores in each dataset. The central lines represent the mean of the population, with the error bars representing standard deviation. c. Bridge intensity at bridge formation as an indirect readout of nucleoporin copy number in individual cells, for wild-type (WT) and les1Δ cells. n=19 (WT Cut11), 27 (les1Δ Cut11), 44 (WT Nup60) and 102 (les1Δ Nup60) individual cells pooled from a minimum of 2 biological replicates. Bars overlaid on top of individual data points represent the mean (central line) and standard deviation (error bars). p-values derive from a two-tailed unpaired t-test. See Methods for details. d. Decay curves for NLS-GFP (orange, from 28 cells at t=-300 to 39 cells at t=0), Nup60 (blue, from 10 cells at t=-300 to 15 cells at t=0) and Cut11 (magenta, from 10 cells at t=-300 to 15 cells at t=0) in a les1Δ background, drawn from a minimum of 2 biological replicates. Each trace was normalized by division by maximum bridge signal for that cell prior to averaging. The plot on the left represents the population averages for each marker in a les1Δ background, and the dotted orange line indicates exponential fit to the NLS-GFP average. The 3 subsequent plots show the averages (darker line) and standard deviation (shaded area) for each marker (NLS-GFP, orange; Nup60, blue; Cut11, magenta) in a les1Δ background, overlaid on the wild-type equivalents in gray. e. les1Δ cells expressing Alm1-mNeonGreen and Cut11-mCherry. Arrow indicates a small cluster of Alm1 that enters the bridge but then disappears. Representative of >20 cells from 2 biological repeats. Scale bar = 2 μm. f. SRRF reconstructions of confocal slices at 28 second intervals of les1Δ cells expressing Nup60-mNeonGreen and Cut11-mCherry tagged at the endogenous loci, aligned relative to spindle max (t=0). Representative of >10 cells across 2 technical replicates. Magenta arrow indicates breakpoint. Scale bar = 2 μm. On the right, the cumulative distribution (42 cells from 2 strains across 3 biological replicates) of breakpoint locations relative to the midzone in les1Δ cells. The shaded gray area represents the mean +/- standard deviation of breakpoint locations in wildtype cells (31 cells from 2 biological replicates), with the cumulative distribution as a gray line.
Extended Data Figure 8. Les1 truncations and genetic interactions.

a. Schematic indicating Les1 truncation constructs, with numbers representing amino acid positions starting from 1 at the N-terminus. Lower panels, cells expressing Les1 (1-291)-mNeonGreen, replacing Les1 at the endogenous locus, as well as Cut11-mCherry. See Methods for details on the truncation constructs (all at endogenous locus, replacing endogenous copy). Note the absence of detectable stalks and the delocalisation of Cut11 in the bridge. Representative of >10 cells across 2 biological repeats. Scale bar = 5 μm. b. Confocal maximum intensity projections of cells expressing Cmp7-mNeonGreen and Les1-mScarlet. Arrows indicate Cmp7 foci at the tips of retracting stalks. Representative of >20 cells across 4 biological repeats. Scale bar = 2 μm. c. Single iSIM slice of a dividing cell expressing Cmp7-mNeonGreen and Les1-mScarlet, representative of >5 cells drawn from 2 technical repeats. Scale bar = 2 μm. d. Tetrad dissection assay for les1Δ crossed with either lem2Δ or cmp7Δ, showing colonies grown from individual spores. See Methods for details. e. Single image of double deletion strains, derived from the tetrad assay clones shown in (d), expressing a synthetic NLS-GFP construct. Scale bar = 10 μm. Representative of >200 cells drawn from 2 biological repeats. f. Averaged line traces (darker lines = mean, lighter bands = standard deviation) of Nup60-mCherry intensities in les1Δ cells (gray: control; blue: treated with 10 μM Cerulenin) at bridge length 3 μm (n=45 cells for cerulenin-treated; n=27 cells for control). g. Confocal maximum intensity projections of les1Δ lem2Δ cells expressing Cut11-mCherry, either in the presence (lower panel) or absence (upper panel) of 5 μM Latrunculin A. Representative of >20 cells across 3 technical repeats. Scale bar = 2 μm.
Extended Data Table 1. Complete list of S. pombe strains used in this study.
List of all the Schizosaccharomyces pombe strains used in this study along with their full genotypes and any relevant strain construction notes.
| Strain | Genotype | Reference | Strain construction details |
|---|---|---|---|
| SO4913 | cut11-GFP:ura4+ pBiP1-mCherry-AHDL:leu1+ h+ | Snezhana Oliferenko lab | For original AHDL 32 |
| SO6600 | pBiP1-NLS-GFP-NLS:leu1+_ade-_leu1-32_ura4D-18 h- | Snezhana Oliferenko lab | Integration at leu1 locus |
| MBY5861 | cut11-mCherry:ura4+ h- | Mohan Balasubramanian lab | |
| MBY6659 | pAct1 Lifeact-GFP:leu+ atb2-mCherry:Hph leu1-32 ura4-D18 h- | Mohan Balasubramanian lab | pCDL1484 integrated into MBY5856 |
| SI235 | Hph<<ark1-as3 h- | Silke Hauf lab | Hph integrated 390 bp upstream of Ark1 start codon; mutations are L166A (gatekeeper), S229A Q28R Q176R (suppressor) |
| SI236 | Hph<<ark1-as3 h+ | Silke Hauf lab | Hph integrated 390 bp upstream of Ark1 start codon; mutations are L166A (gatekeeper), S229A Q28R Q176R (suppressor) |
| PN1 | 972 h-(wild type) | Paul Nurse lab | |
| PN2 | 972 h+ (wild type) | Paul Nurse lab | |
| GD111 | Hph<<ark1-as3 atb2-mCherry:Hph h? | This study | MBY6659 X SI236 |
| GD121 | cut11-mNeonGreen:Kan Hph<<ark1-as3 atb2-mCherry:Hph h? | This study | cut11-mNeonGreen:Kan transformed into GD111 |
| GD130 | les1-mNeonGreen:Kan Hph<<ark1-as3 atb2-mCherry:Hph h? | This study | les1-mNeonGreen:Kan transformed into GD111 |
| GD138 | les1-mNeonGreen:Kan h+ | This study | les1-mNeonGreen:Kan transformed into 972 h+ |
| GD141 | les1-mNeonGreen:Kan atb2-mCherry:Hph h? | This study | GD138 X MBY6659 |
| GD155 | Hph<<ark1-as3 pBiP1-mCherry-AHDL::leu1+ h+ | This study | SO4913 X SI235 |
| GD171 | les1::Hph cut11-mCherry:ura4+ h- | This study | les1::Kan transformed into MBY5861 |
| GD172 | les1-mNeonGreen:Kan cut11-mCherry:ura4+ h+ | This study | GD138 X MBY5861 |
| GD173 | nup60-mNeonGreen:Kan cut11-mCherry:ura4+ h- | This study | nup60-mNeonGreen:Kan transformed into MBY5861 |
| GD175 | nup60-mNeonGreen:Kan Hph<<ark1-as3 atb2-mCherry:Hph h? | This study | nup60-mNeonGreen:Kan transformed into GD111 |
| GD176 | nup60-mNeonGreen:Kan les1::Hph cut11-mCherry:ura4+h- | This study | nup60-mNeonGreen:Kan transformed into GD171 |
| GD206 | les1::Hph pBiP1-NLS-GFP-NLS:leu1+_ade-_leu1-32_ura4D-18 h- | This study | les1::Hph transformed into SO6600 |
| GD220 | alm1-mNeonGreen:Kan cut11-mCherry:ura4+ h- | This study | alm1-mNeonGreen:Kan transformed into MBY5861 |
| GD224 | nup211-mNeonGreen:Kan cut11-mCherry:ura4+ h- | This study | nup211-mNeonGreen:Kan transformed into MBY5861 |
| GD225 | pom34-mNeonGreen:Kan cut11-mCherry:ura4+ h- | This study | pom34-mNeonGreen:Kan transformed into MBY5861 |
| GD227 | cdc15-mNeonGreen:Kan cut11-mCherry:ura4+ h- | This study | cdc15-mNeonGreen:Kan transformed into MBY5861 |
| GD229 | nup60-mEOS3.2:Kan h+ | This study | nup60-mEOS3.2:Kan transformed into 972 h+ |
| GD250 | nup60-mCherry:Kan h+ pBiP1-NLS-GFP-NLS:leu1+_ade-_leu1-32_ura4D-18 h- | This study | nup60-mCherry:Kan transformed into SO6600 |
| GD253 | les1::Hph nup60-mCherry:Kan h+ pBiP1-NLS-GFP-NLS:leu1+_ade-_leu1-32_ura4D-18 h- | This study | les1::Hph transformed into GD250 |
| GD255 | les1-mNeonGreen:Kan Hph<<ark1-as3 pBiP1-mCherry-AHDL::leu1+ h+ | This study | les1-mNeonGreen:Kan transformed into GD255 |
| GD257 | cta4-mNeonGreen:Kan cut11-mCherry:ura4+ h- | This study | cta4-mNeonGreen:Kan transformed into MBY5861 |
| GD259 | nup60-mCherry:Hph les1::NatR cmp7-mNeonGreen:Kan h- | This study | nup60-mCherry:Hph transformed into GD273 |
| GD261 | nup120-mNeonGreen:Kan cut11-mCherry:ura4+ h- | This study | nup120-mNeonGreen:Kan transformed into MBY5861 |
| GD263 | nup82-mNeonGreen:Kan cut11-mCherry:ura4+ h- | This study | nup82-mNeonGreen:Kan transformed into MBY5861 |
| GD264 | nup37-mNeonGreen:Kan cut11-mCherry:ura4+ h- | This study | nup37-mNeonGreen:Kan transformed into MBY5861 |
| GD265 | nup44-mNeonGreen:Kan cut11-mCherry:ura4+ h- | This study | nup44-mNeonGreen:Kan transformed into MBY5861 |
| GD266 | nup40-mNeonGreen:Kan cut11-mCherry:ura4+ h- | This study | nup40-mNeonGreen:Kan transformed into MBY5861 |
| GD270 | nem1::Hph nup211-mNeonGreen:Kan cut11-mCherry:ura4+ h- | This study | nem1::Hph transformed into GD224 |
| GD273 | les1::NatR cmp7-mNeonGreen:Kan h- | This study | les1::NatR transformed into KG18766 |
| KG18766 | cmp7-mNeonGreen:Kan h- | Kathy Gould lab | cmp7-mNeonGreen:Kan transformed into 972 h- |
| GD275 | nem1::Hph les1-mNG:kanR cut11-mCh:ura4+ h+ | This study | nem1::Hph transformed into GD172 |
| GD181 | imp1::Kan les1::Hph cut11-mCherry:ura4+ h- | This study | imp1::Kan transformed into GD171 |
| GD139 | les1:mScarlet:Hph h+ | This study | les1:mScarlet:Hph transformed into 972 h+ |
| GD140 | cmp7-mNeonGreen:Kan les1:mScarlet:Hph h+ | This study | KG18766 crossed with GD139 |
| GD283 | les1(1-204):mNeonGreen cut11-mCherry:ura4+ h- | This study | les1(1-204):mNeonGreen transformed into MBY5861 |
| GD284 | les1(1-291):mNeonGreen cut11-mCherry:ura4+ h- | This study | les1(1-291):mNeonGreen transformed into MBY5861 |
| GD285 | les1(1-94):mNeonGreen cut11-mCherry:ura4+ h- | This study | les1(1-94):mNeonGreen transformed into MBY5861 |
| GD290 | imp1::Hph alm1-mNeonGreen:Kan cut11-mCherry:ura4+ h- | This study | imp1::Hph transformed into GD220 |
| GD292 | imp1::Hph les1-mNeonGreen:Kan cut11-mCherry:ura4+ h+ | This study | imp1::Hph transformed into GD172 |
Acknowledgements
We would like to thank Mohan Balasubramanian, Snezhana Oliferenko, Silke Hauf, Kathy Gould, Jürg Bahler, Paul Nurse and their labs for sharing S. pombe strains, plasmids, expertise and S. pombe protocols; James O. Patterson for the kind gift of pFA6a-mNeonGreen plasmids; the LMB EM facility for EM support; Tim-Oliver Buchholz (MPI-CBG/CSBD, Dresden, Germany) for advice on the Noise2Noise implementation. We would like to acknowledge David Albrecht, Ishier Raote, Agathe Chaigne, Pedro Pereira, Caron Jacobs and members of the Baum lab, in particular Giulia Paci, Giulia Cazzagon, and Helen Matthews, as well as 3 anonymous reviewers, for their feedback on this manuscript. G.D. was funded by a European Union Marie Sklodowska-Curie Individual Fellowship (704281-CCDSA) and the Wellcome Trust (203276/Z/16/Z). Si.C. and R.H. were supported by the UK BBSRC (BB/R021805/1; BB/S507532/1), the UK Medical Research Council (MR/K015826/1), and the Wellcome Trust (203276/Z/16/Z). Sc.C was supported by the Francis Crick Institute which receives its core funding from Cancer Research UK (FC001121), the UK Medical Research Council (FC001121), and the Wellcome Trust (FC001121). W.K. was funded by the Medical Research Council (MC_UP_1201/8). B.B. was supported by UCL’s Institute for the Physics of Living Systems, the MRC-LMCB, by the Wellcome Trust (203276/Z/16/Z), and by Cancer Research UK (C1529/A28276).
Footnotes
Author Contributions
G.D. co-conceived the project, designed and implemented all the experiments, generated strains and reagents, acquired, analysed and interpreted the data (with the exception of the EM data shown in parts of Figs 2 and 4, and Extended Data Figs. 3, and 7), and led the drafting of the paper. Si.C. and U.S. created new software used in the work, and analysed and assisted in the interpretation of data. Sc.C. advised on experimental design, helped to generate strains and reagents, and provided protocols and training. R.H. advised on the creation of new software used for the project. W.K. performed all electron tomography and analysis of electron tomograms (parts of Figs 2 and 4, and Extended Data Figs. 3, and 7). B.B. co-conceived and supervised the project, provided advice on experimental design, implementation and analysis, and co-drafted the manuscript. All authors provided input during the manuscript drafting stage.
Competing Interests Statement
We declare that none of the authors have competing financial or non-financial interests as defined by Nature Research.
Data Availability Statement
All code used for the analyses in this paper is made freely and publicly available via a dedicated repository (https://github.com/superresolusian/local-NEB; see Code Availability Statement). Bulk microscopy time series data, comprising >50 files with an average size >1GB, are available upon request. The S. pombe strains generated for and used in this study (Extended Data Table 1) are available upon request. Source data required to reproduce all the graphs and conclusions of the manuscript, including those presented as Extended Data, are included in the paper and its supplementary information files.
Code Availability Statement
All custom software designed for and used in this study is freely available on GitHub in a public repository at https://github.com/superresolusian/local-NEB. The use of this code is governed by an MIT license.
References
- 1.LaJoie D, Ullman KS. Coordinated events of nuclear assembly. Curr Opin Cell Biol. 2017;46:39–45. doi: 10.1016/j.ceb.2016.12.008. [DOI] [PubMed] [Google Scholar]
- 2.Sazer S, Lynch M, Needleman D. Deciphering the evolutionary history of open and closed mitosis. Curr Biol. 2014;24:R1099–103. doi: 10.1016/j.cub.2014.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ungricht R, Kutay U. Mechanisms and functions of nuclear envelope remodelling. Nat Rev Mol Cell Biol. 2017;18:229–245. doi: 10.1038/nrm.2016.153. [DOI] [PubMed] [Google Scholar]
- 4.Yanagida M. The role of model organisms in the history of mitosis research. Cold Spring Harb Perspect Biol. 2014;6 doi: 10.1101/cshperspect.a015768. a015768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang D, Oliferenko S. Remodeling the nuclear membrane during closed mitosis. Current Opinion in Cell Biology. 2013;25:142–148. doi: 10.1016/j.ceb.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 6.Walczak CE, Cai S, Khodjakov A. Mechanisms of chromosome behaviour during mitosis. Nat Rev Mol Cell Biol. 2010;11:91–102. doi: 10.1038/nrm2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sazer S, Lynch M, Needleman D. Deciphering the evolutionary history of open and closed mitosis. Curr Biol. 2014;24:R1099–103. doi: 10.1016/j.cub.2014.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tamm T, et al. Brr6 drives the Schizosaccharomyces pombe spindle pole body nuclear envelope insertion/extrusion cycle. J Cell Biol. 2011;195:467–484. doi: 10.1083/jcb.201106076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boettcher B, Barral Y. The cell biology of open and closed mitosis. Nucleus. 2013;4:160–5. doi: 10.4161/nucl.24676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harel A, et al. Persistence of major nuclear envelope antigens in an envelope-like structure during mitosis in Drosophila melanogaster embryos. J Cell Sci. 1989;94(Pt 3):463–70. doi: 10.1242/jcs.94.3.463. [DOI] [PubMed] [Google Scholar]
- 11.Lénárt P, et al. Nuclear envelope breakdown in starfish oocytes proceeds by partial NPC disassembly followed by a rapidly spreading fenestration of nuclear membranes. J Cell Biol. 2003;160:1055–1068. doi: 10.1083/jcb.200211076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gu Y, Yam C, Oliferenko S. Divergence of mitotic strategies in fission yeasts. Nucleus-Austin. 2012;3:220–225. doi: 10.4161/nucl.19514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ding R, McDonald KL, McIntosh JR. Three-dimensional reconstruction and analysis of mitotic spindles from the yeast, Schizosaccharomyces pombe. J Cell Biol. 1993;120:141–151. doi: 10.1083/jcb.120.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ward JJ, Roque H, Antony C, Nédélec F. Mechanical design principles of a mitotic spindle. Elife. 2014;3:e03398. doi: 10.7554/eLife.03398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loiodice I, et al. Quantifying Tubulin Concentration and Microtubule Number Throughout the Fission Yeast Cell Cycle. Biomolecules. 2019;9:86. doi: 10.3390/biom9030086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang D, Vjestica A, Oliferenko S. The Cortical ER Network Limits the Permissive Zone for Actomyosin Ring Assembly. Curr Biol. 2010;20:1029–1034. doi: 10.1016/j.cub.2010.04.017. [DOI] [PubMed] [Google Scholar]
- 17.Frost A, et al. Functional Repurposing Revealed by Comparing S. pombe and S. cerevisiae Genetic Interactions. Cell. 2012;149:1339–1352. doi: 10.1016/j.cell.2012.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krüger LK, Sanchez J-L, Paoletti A, Tran PT. Kinesin-6 regulates cell-size-dependent spindle elongation velocity to keep mitosis duration constant in fission yeast. Elife. 2019;8 doi: 10.7554/eLife.42182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flor-Parra I, et al. Importin α and vNEBD Control Meiotic Spindle Disassembly in Fission Yeast. Cell Rep. 2018;23:933–941. doi: 10.1016/j.celrep.2018.03.073. [DOI] [PubMed] [Google Scholar]
- 20.Lucena R, et al. Nucleocytoplasmic transport in the midzone membrane domain controls yeast mitotic spindle disassembly. J Cell Biol. 2015;209:387–402. doi: 10.1083/jcb.201412144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gatta AT, Carlton JG. The ESCRT-machinery: closing holes and expanding roles. Curr Opin Cell Biol. 2019;59:121–132. doi: 10.1016/j.ceb.2019.04.005. [DOI] [PubMed] [Google Scholar]
- 22.Olmos Y, Hodgson L, Mantell J, Verkade P, Carlton JG. ESCRT-III controls nuclear envelope reformation. Nature. 2015;522:236–239. doi: 10.1038/nature14503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vietri M, et al. Spastin and ESCRT-III coordinate mitotic spindle disassembly and nuclear envelope sealing. Nature. 2015;522:231–235. doi: 10.1038/nature14408. [DOI] [PubMed] [Google Scholar]
- 24.Pieper GH, Sprenger S, Teis D, Oliferenko S. ESCRT-III/Vps4 Controls Heterochromatin-Nuclear Envelope Attachments. Dev Cell. 2020 doi: 10.1016/j.devcel.2020.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.von Appen A, et al. LEM2 phase separation promotes ESCRT-mediated nuclear envelope reformation. Nature. 2020;1-4 doi: 10.1038/s41586-020-2232-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gu M, et al. LEM2 recruits CHMP7 for ESCRT-mediated nuclear envelope closure in fission yeast and human cells. Proc Natl Acad Sci. 2017;114:E2166–E2175. doi: 10.1073/pnas.1613916114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thaller DJ, et al. An ESCRT-LEM protein surveillance system is poised to directly monitor the nuclear envelope and nuclear transport system. Elife. 2019;8 doi: 10.7554/eLife.45284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yam C, He Y, Zhang D, Chiam K-H, Oliferenko S. Divergent strategies for controlling the nuclear membrane satisfy geometric constraints during nuclear division. Curr Biol. 2011;21:1314–9. doi: 10.1016/j.cub.2011.06.052. [DOI] [PubMed] [Google Scholar]
- 29.McIntosh JR, Roos UP, Neighbors B, McDonald KL. Architecture of the microtubule component of mitotic spindles from Dictyostelium discoideum. J Cell Sci. 1985;75:93–129. doi: 10.1242/jcs.75.1.93. [DOI] [PubMed] [Google Scholar]
- 30.Yang J, et al. The I-TASSER suite: Protein structure and function prediction. Nature Methods. 2014;12:7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roy A, Kucukural A, Zhang Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol. 2011;7 doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Katoh K. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Katoh K, Rozewicki J, Yamada KD. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2019;20:1160–1166. doi: 10.1093/bib/bbx108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kalyaanamoorthy S, Minh BQ, Wong TKF, Von Haeseler A, Jermiin LS. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587–589. doi: 10.1038/nmeth.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Finn RD, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–30. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.El-Gebali S, et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019;47:D427–D432. doi: 10.1093/nar/gky995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42:W320–4. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Forsburg SL, Rhind N. Basic methods for fission yeast. Yeast. 2006;23:173–83. doi: 10.1002/yea.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moreno S, Klar A, Nurse P. [56] Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 1991;194:795–823. doi: 10.1016/0076-6879(91)94059-l. [DOI] [PubMed] [Google Scholar]
- 43.Bähler J, et al. Heterologous modules for efficient and versatile PCR-based gene targeting inSchizosaccharomyces pombe. Yeast. 1998;14:943–951. doi: 10.1002/(SICI)1097-0061(199807)14:10<943::AID-YEA292>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 44.Murray JM, Watson AT, Carr AM. Transformation of Schizosaccharomyces pombe: Lithium Acetate/ Dimethyl Sulfoxide Procedure. Cold Spring Harb Protoc. 2016 doi: 10.1101/pdb.prot090969. pdb.prot090969 2016. [DOI] [PubMed] [Google Scholar]
- 45.Zhang M, et al. Rational design of true monomeric and bright photoactivatable fluorescent proteins. Nat Methods. 2012;9:727–729. doi: 10.1038/nmeth.2021. [DOI] [PubMed] [Google Scholar]
- 46.Escorcia W, Forsburg SL. Methods in Molecular Biology. Vol. 1721. Humana Press Inc; 2018. Tetrad dissection in fission yeast; pp. 179–187. [DOI] [PubMed] [Google Scholar]
- 47.Huff J, et al. The new 2D Superresolution mode for ZEISS Airyscan. Nat Methods. 2017;14:1223–1223. [Google Scholar]
- 48.Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gustafsson N, et al. Fast live-cell conventional fluorophore nanoscopy with ImageJ through super-resolution radial fluctuations. Nat Commun. 2016;7 doi: 10.1038/ncomms12471. 12471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lehtinen J, et al. Noise2Noise: Learning Image Restoration without Clean Data. 35th Int Conf Mach Learn ICML 2018; 2018. pp. 4620–4631. [Google Scholar]
- 52.Weigert M, et al. Content-aware image restoration: pushing the limits of fluorescence microscopy. Nat Methods. 2018;15:1090–1097. doi: 10.1038/s41592-018-0216-7. [DOI] [PubMed] [Google Scholar]
- 53.Ballard DH. Generalizing the Hough transform to detect arbitrary shapes. Pattern Recognit. 1981;13:111–122. [Google Scholar]
- 54.Asakawa H, et al. Asymmetrical localization of Nup107-160 subcomplex components within the nuclear pore complex in fission yeast. PLOS Genet. 2019;15:e1008061. doi: 10.1371/journal.pgen.1008061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krüger LK, Sanchez J-L, Paoletti A, Tran PT. Kinesin-6 regulates cell-size-dependent spindle elongation velocity to keep mitosis duration constant in fission yeast. Elife. 2019;8 doi: 10.7554/eLife.42182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ho J, Tumkaya T, Aryal S, Choi H, Claridge-Chang A. Moving beyond P values: data analysis with estimation graphics. Nat Methods. 2019;16:565–566. doi: 10.1038/s41592-019-0470-3. [DOI] [PubMed] [Google Scholar]
- 57.Kukulski W, et al. Precise, Correlated Fluorescence Microscopy and Electron Tomography of Lowicryl Sections Using Fluorescent Fiducial Markers. Methods in cell biology. 2012;111:235–257. doi: 10.1016/B978-0-12-416026-2.00013-3. [DOI] [PubMed] [Google Scholar]
- 58.Bharat TAM, Hoffmann PC, Kukulski W. Correlative Microscopy of Vitreous Sections Provides Insights into BAR-Domain Organization In Situ. Structure. 2018;26:879–886. doi: 10.1016/j.str.2018.03.015. e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hohmann-Marriott MF, et al. Nanoscale 3D cellular imaging by axial scanning transmission electron tomography. Nat Methods. 2009;6:729–31. doi: 10.1038/nmeth.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mastronarde DN. Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol. 2005;152:36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 61.Kremer JR, Mastronarde DN, McIntosh JR. Computer Visualization of Three-Dimensional Image Data Using IMOD. J Struct Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All code used for the analyses in this paper is made freely and publicly available via a dedicated repository (https://github.com/superresolusian/local-NEB; see Code Availability Statement). Bulk microscopy time series data, comprising >50 files with an average size >1GB, are available upon request. The S. pombe strains generated for and used in this study (Extended Data Table 1) are available upon request. Source data required to reproduce all the graphs and conclusions of the manuscript, including those presented as Extended Data, are included in the paper and its supplementary information files.
All custom software designed for and used in this study is freely available on GitHub in a public repository at https://github.com/superresolusian/local-NEB. The use of this code is governed by an MIT license.