

Abstract
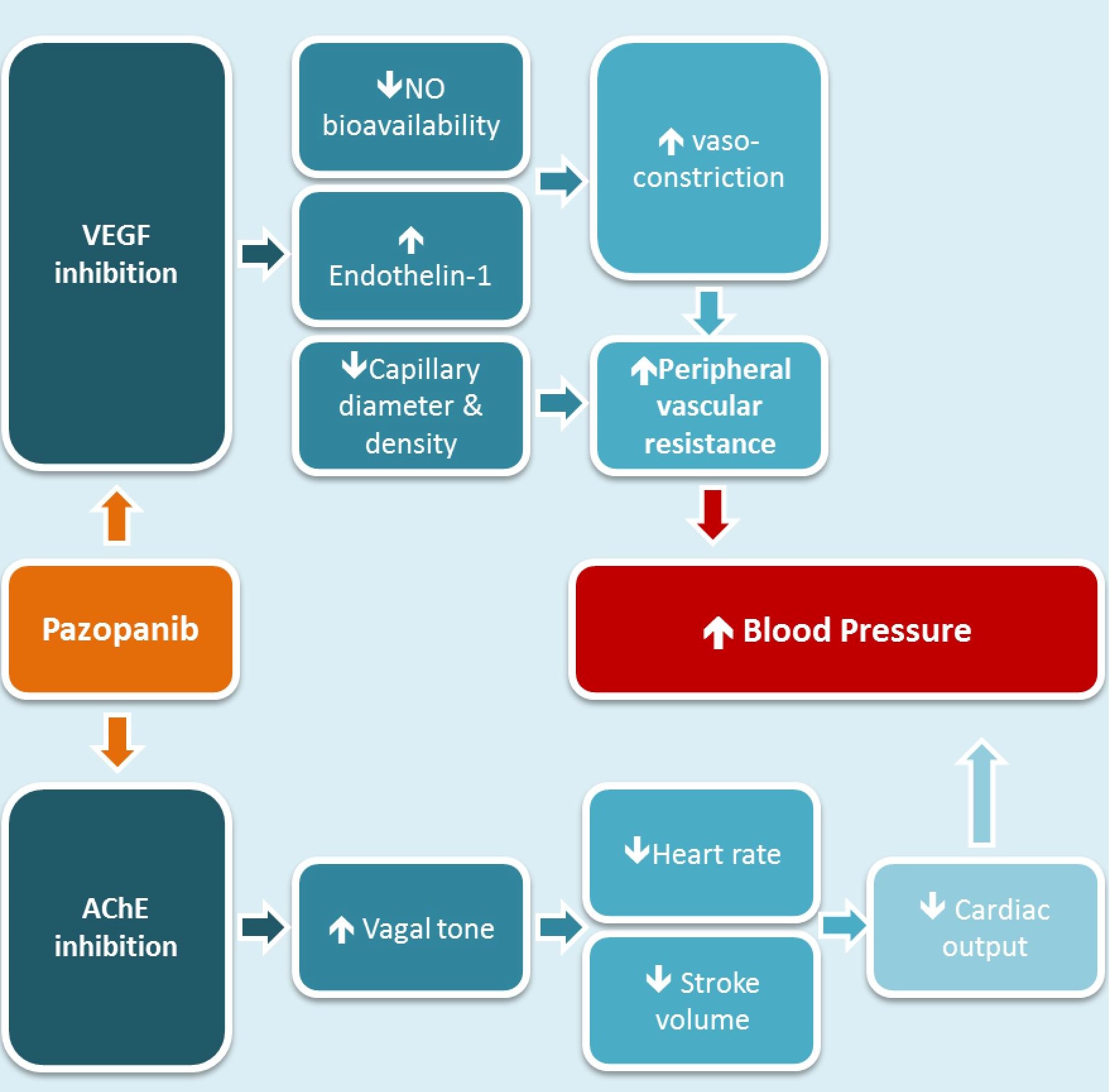
Drugs targeting the vascular endothelial growth factor (VEGF) signalling pathway are approved for several malignancies. Unfortunately, VEGF inhibitors lead to hypertension in 30-80% patients. Reduced nitric oxide synthase activity, microvascular rarefaction and increased vascular resistance have been proposed as potential mechanisms. We aimed to assess these mechanisms in patients receiving the VEGF inhibitor, pazopanib, for cancer.
27 normotensive patients with advanced solid malignancies received pazopanib 800mg od. Endothelial function was assessed using forearm plethysmography with intra-arterial infusions of acetylcholine (ACh). Detailed haemodynamic measurements were taken. Density and diameter of the conjunctival and episcleral microvasculature were evaluated using haemoglobin video imaging. Measurements were taken at baseline, 2 and 12 weeks after initiation of pazopanib, or earlier if patients became hypertensive.
By the end of the trial, systolic blood pressure increased by 12(95% CI:4, 19) mmHg; P=0.003, diastolic by 10(95% CI:5, 15) mmHg; P<0.001 and peripheral vascular resistance (PVR) by 888(95% CI:616, 1168) dynes*s/cm5; P<0.001. Forearm blood flow (FBF) improved: Ratio of ACh response at end of trial/baseline was 2.8 (95% CI:1.84, 4.25); P<0.001. Microvascular density in the sclera was reduced by -15.5(95% CI:-25.7, -5.3)%; P=0.003 and diameter by -2.09(95% CI:-3.95 to -0.19)μm; P=0.03. A post-hoc colorimetric assay revealed that pazopanib inhibited acetylcholinesterase activity by -56(95% CI:-62, -52)%; P<0.001.
Unexpectedly, pazopanib led to an increase in ACh-mediated FBF response; likely due to the inhibition of acetylcholinesterase activity. Pazopanib increased PVR and reduced microvascular density and diameter, suggesting that microvascular rarefaction could be one of the key mechanisms behind VEGF inhibition-induced hypertension.
Clinical Trial Registration: The trial was registered publicly on https://clinicaltrials.gov (NCT01392352).
Keywords: Endothelial dysfunction, vascular endothelial growth factor inhibition, hypertension, rarefaction, peripheral resistance
Introduction
A recent large observational study in over 3 million patients has highlighted the increased risk of cardiovascular disease (CVD) in cancer survivors in comparison to the general population1. Interestingly, CVD mortality risk is highest within the first year after cancer diagnosis, suggesting that the high risk of CVD could be explained by aggressive therapy shortly after disease discovery. One of the treatments associated with known CV adverse events are tyrosine kinase inhibitors (TKI) targeting vascular endothelial growth factor (VEGF) receptors.
Pazopanib is an orally bio-available, small molecule multi-targeted TKI of VEGF receptors 1, 2, and 3, platelet-derived growth factor receptor α and β, and tyrosine-protein kinase CD117 (c-KIT)2. It is an established treatment for a number of solid tumours and has been licensed for use in metastatic renal cell carcinoma and soft tissue sarcoma3. It works by inhibiting angiogenesis, thereby leading to tumour hypoxia and shrinkage. Unfortunately, treatment with VEGF inhibitors is complicated by cardiovascular side effects, most commonly hypertension4. Hypertension is a class-effect of VEGF inhibitors and the incidence of hypertension varies depending on the agent used, occurring in approximately 40% of patients taking pazopanib5. An increase in blood pressure is seen in most patients on these therapies and 15% of patients develop severe hypertension. Although the incidence of hypertension is well documented with VEGF inhibitors, the underlying pathophysiology remains unclear.
Two main mechanisms for hypertension have been suggested: endothelial dysfunction and microvascular rarefaction6. In health, binding of VEGF to its receptors leads to activation of multiple pathways including the endothelial nitric oxide synthase (eNOS) pathway, resulting in increased nitric oxide (NO) production and vasodilatation. Thus, when VEGF signalling pathway is inhibited, the NO pathway is suppressed and the endothelin-1 pathway stimulated, promoting vasoconstriction and consequent hypertension7. An alternative mechanism is microvascular rarefaction. In health, VEGF maintains the integrity of the capillary and arteriole network. Experimental data suggest that when this pathway is inhibited, both microvascular density and diameter are reduced7. The subsequent diminution of the microvascular surface area leads to an increase in peripheral vascular resistance, and resultant increase in blood pressure8. However, these pathways have mainly been studied in animals, and the existing data in humans are conflicting with various vascular parameters measured independently, and/or biomarkers being assessed in separate small studies, using a variety of drugs (both TKIs and monoclonal antibodies) to inhibit VEGF.
Therefore, we designed a mechanistic clinical trial (HYPAZ, an open label investigation into HYpertension induced by PAZopanib therapy) where our primary objective was to determine whether reduced NO bioavailability was the causative mechanism underlying a rise in blood pressure induced by the TKI, pazopanib. Secondary and exploratory end points included determining the effect of pazopanib on capillary rarefaction, peripheral vascular resistance, cardiac output and arterial stiffness in order to draw a comprehensive picture of the possible mechanisms behind VEGF-induced hypertension.
Methods
Patients
Patients with advanced solid malignancies for whom pazopanib was considered a reasonable option by their oncologist were recruited. Patients were eligible if they were aged ≥18 years old, had an Eastern Cooperative Oncology Group (ECOG) performance status9 of 0-1, measurable disease by RECIST 1.110 (at least one lesion measuring 20mm) and had adequate baseline organ function. Patients were excluded if they were hypertensive (BP>150/100mmHg), had any history of acute cardiovascular events within 6 months, or were considered to be at an increased risk from treatment with anti-angiogenic therapy. A total of 31 eligible patients were recruited into the study from June 2011 to July 2014. Approval for this clinical trial was obtained from the Medicines Healthcare Regulatory Agency (MHRA) and the Research Ethics Committee (10/H0304/72). Written informed consent was received from each participant prior to performing any study related procedures. The studies were carried out in accordance with institutional guidelines and ICH Good Clinical Practice. The trial was registered publicly on clinicaltrials.gov (NCT01392352) and was sponsored by Cambridge University Hospitals NHS Foundation Trust and the University of Cambridge. The data that support the findings of this study are available from the corresponding author upon reasonable request.
Study Protocol
This was an open-label, non-randomized, exploratory, targeted safety Phase IIB study to investigate the mechanisms of hypertension caused by pazopanib therapy. The primary endpoint was change in forearm blood flow (FBF) ratio in response to intra-arterial acetylcholine (ACh) infusion (endothelium-dependent dilatation) using the technique of venous occlusion plethysmography. Secondary endpoints included FBF ratio responses to intra-arterial sodium nitroprusside (SNP) infusion (endothelium-independent dilatation) and L-N(γ)-mono-methyl-arginine (L-NMMA) infusion (basal NO production), as well as changes in blood pressure (BP), augmentation index (AIx), aortic pulse wave velocity (aPWV), cardiac output (CO), peripheral vascular resistance (PVR), minimum forearm vascular resistance (MFVR), and conjunctival and episcleral microvascular density and diameter.
The study design is depicted and detailed in supplemental files (Figure S1) Please see http://hyper.ahajournals.org. Patients made up to 8 visits; baseline research assessments were taken at visit 2, repeated 2 weeks later on visit 3, and the last research measurements were taken 12 weeks after the initiation of treatment (visit 8) or earlier if the patient became hypertensive, thus called visit HYP.
Haemodynamic measurements
All studies were conducted in a quiet, temperature-controlled room by trained members of the study team. Further details about the methods can be found in the supplemental files. Please see http://hyper.ahajournals.org
Venous Occlusion Plethysmography
Forearm blood flow (FBF) was measured using venous occlusion plethysmography (Hokanson Inc, Bellevue, USA) with calibrated mercury-in-silastic strain gauges as previously described11. Endothelium-dependent dilatation was assessed by incremental infusions of ACh (Novartis Pharmaceuticals, Basel, Switzerland) at doses of 7.5 and 15 μg/min, endothelium-independent dilatation by SNP (Nitroprussiat Fides, Madrid, Spain) at doses of 3 and 10 μg/min and basal NO production / the effect of non-selective NOS inhibition was assessed by an infusion of L-NMMA (Bachem Distribution Services GmBH, Weil am Rhein, Germany) at doses of 2 and 4 μmol/min.
Blood pressure and Arterial stiffness
Measurements were made in seated and supine position following at least five minutes of rest. Brachial BP was recorded using a validated oscillometric technique (HEM-705CP; Omron Corp., Japan). Arterial stiffness (Aix and aPWV) was measured using the SphygmoCor system as described previously12.
Minimum Forearm Vascular Resistance
Venous occlusion plethysmography was set up as described for FBF, but without arterial cannulation. After at least 10 minutes of supine rest, the baseline FBF was assessed. This was followed by inflation of a brachial cuff to supra-systolic pressure for 13 minutes in the non-dominant arm. Immediately after deflation of the cuff, FBF was measured for 3 minutes. The average of the three highest FBF rates (flowmax) during the recording was used for the subsequent calculation of MFVR by dividing MAP by flowmax. Vascular resistance is given in resistance unit (R unit); 1mmHg/ml/min/100ml forearm tissue13,14.
Cardiac output
Cardiac output was assessed using a non-invasive, inert gas rebreathing technique15.
Haemoglobin video imaging of conjunctiva and episclera
Haemoglobin video imaging of the conjunctiva and episclera is a technique by which blood flow through the entire ocular surface microcirculation can be imaged16, using a slit-lamp (Zeiss SL130) modified for haemoglobin imaging as described previously17. We aimed to image these circulations during visits 2, 3, and 8. However, by Visit 8, many patients found the technique too demanding and only 4 complete sets of good-quality video sequences could be obtained. Our formal analysis was therefore restricted to Visits 2 and 3.
Video Analysis
Microvascular density (percentage coverage) and diameter distribution were obtained by applying the pre-trained U-Net convolutional neural network18,19,20 on processed images using multiple open-source toolboxes21,22,23.
Acetylcholinesterase (AChE) inhibition assay
Whole blood samples were collected from 10 healthy volunteers and spiked with a range of concentrations (0.1-183 μM) of pazopanib (GW-786034, BioVision) and AChE activity was measured colorimetrically using a commercially available assay kit (#MAK119, SigmaAldrich/Merck).
Safety assessments
Safety data were collected continuously from patients until 28 days after the discontinuation of pazopanib. Adverse events were graded according to the National Cancer Institute’s Common Terminology Criteria for Adverse Events (CTCAE) version 3.0.
Statistical Analysis
A hierarchical mixed effects model was used to analyse the effect of pazopanib on FBF with fixed effects for dose, Visit, and their interaction, and unstructured random dose effects at the patient and patient-Visit level. The effect of pazopanib on the other haemodynamic measurements (BP, AIX, aPWV, CO, PVR, MFVR) were analysed using a simple ANCOVA model, with fixed effects for Visit, adjusting for baseline value. All parameters are reported with estimates and 95% confidence intervals, and/or means and standard deviations. A detailed description of the statistical analysis used in the trial can be found in the supplement file. Please see http://hyper.ahajournals.org
Results
Ninety-five patients were given study information, of whom 53 consented to screening. Thirty-one patients were initially considered eligible, but safety concerns were identified at the baseline visit in 4 patients (tumour adjacent to major vessels in 3 and new bleeding in 1) and a clinical decision was made not to commence pazopanib. Therefore, 27 patients, with a mean age of 60 years, were initiated on treatment with pazopanib at the standard dose of 800mg daily. See Table 1 for baseline characteristics and demographics at the screening Visit. Patients had a wide range of malignant disease. Patients were normotensive with an average blood pressure of 122/76 ± 15/8mmHg at the screening Visit. The flow of the patients though the study can be seen in Figure S2 in the supplemental files. Please see http://hyper.ahajournals.org
Table 1. Subject demographics and characteristics.
| Variable | Mean ± SD |
|---|---|
| n | 27 |
| Type of malignant disease | metastatic renal cell carcinoma, soft tissue sarcoma, melanoma, cervical and ovarian cancer, thymic carcinoma, gastrointestinal and hepatobiliary system malignancies |
| Age, years (range) | 60±13 (25-82) |
| Gender | Women= 17, Men= 10 |
| Ethnicity, n | 26 White, 1 Chinese |
| Smoking status, n | 5 current smokers, 7 past smokers, 15 non-smokers |
| Height, cm | 168±9 |
| Weight, kg | 70±17 |
| Total Cholesterol, mmol/l | 4.8±1.0 |
| Glucose, mmol/l | 5.6±0.9 |
| Blood pressure, mmHg | 122/76 ± 15/8 |
| ECOG status, n | 0= 7, 1= 20 |
| Target lesion size, mm | 102±51 |
ECOG denotes The Eastern Cooperative Oncology Group performance status
Haemodynamic measurements
Blood pressure, cardiac output and arterial stiffness
Changes in haemodynamic parameters following pazopanib are listed in Table 2. After two weeks of treatment (Visit 3) systolic BP increased by 12 (95% CI: 6 to 18) mmHg; P<0.001 and diastolic BP by 8 (95% CI: 5 to 12) mmHg; P<0.001 and remained at the similar level at Visit 8/HYP. Similarly, after two weeks of treatment, we saw 24 hour ambulatory systolic BP increase by 6 (95% CI: -16, 25) mmHg; P=0.02 and diastolic BP by 6 (-6, 18) mmHg; P<0.001, and BP continued to stay elevated at Visit 8/HYP. A large increase in PVR of +504 (95% CI: 288 to 728) dynes*s/cm5; P<0.001 was observed at Visit 3 and +888 (95% CI: 616 to1168) dynes*s/cm5; P<0.001 at Visit 8/HYP. Aortic PWV increased by 1.3 (95% CI: 0.3 to 2.2) m/s; P=0.01 at Visit 8/HYP, importantly this change remained significant after adjusting for the change in MAP. MFVR increased after the initiation of pazopanib from 3.27 (95% CI: 2.82 to 3.72) to 4.38 (95% CI: 3.88 to 4.88) mmHg/ml/min/100ml; P= 0.01 at Visit 3 and remained high at Visit 8/HYP, 4.93 (85% CI: 3.57 to 6.29) mmHg/ml/min/100ml. The increase in MFVR was more pronounced in those patients who developed clinical hypertension with pazopanib compared with those who did not (P=0.023).
Table 2. Baseline haemodynamics and the change at week 2 and week 12.
| Variable | Baseline (Visit 2) Mean ±SD | Week 2 (Visit 3) Change(95% CI) | Week 12/HYP (Visit 8/HYP) Change(95% CI) |
|---|---|---|---|
| Systolic BP (mmHg) | 117±13 | +12 (6,18); P<0.001 | +12 (4,19); P<0.001 |
| Diastolic BP (mmHg) | 73±9 | +8 (5,12); P<0.001 | +10 (5,15); P<0.001 |
| MAP (mmHg) | 85±10 | +9 (4,13); P<0.001 | +10 (5,16); P<0.001 |
| Heart Rate (bpm) | 89±14 | -11(-16,-7); P<0.001 | -9(-15,-3); P<0.001 |
| Cardiac output (L/min) | 4.88±1.42 | -0.95 (-1.25, -0.65); P<0.001 | -1.35 (-1.73, -0.98); P<0.001 |
| Cardiac Index (L/min/m3) | 2.70±0.72 | -0.51 (-0.76, -0.27); P<0.001 | -0.86 (-1.18, -0.53); P<0.001 |
| PVR (dynes*s/cm5) * | 1552±576 | +504 (288, 728); P<0.001 | +888 (616, 1168); P<0.001 |
| AIx (%) † | 16±10 | +7 (4,11); P<0.001 | +6 (1,11); P<0.001 |
| Aortic PWV (m/s) ‡ | 8.0±2.1 | +0.4 (-0.3,1.2); P=0.23 | +1.3 (0.3,2.2); P<0.01 |
| 24h Ambulatory SBP (mmHg) | 118±12 | +6 (-16, 25); P=0.02 | +6 (-12, 24); P=0.08 |
| 24h Ambulatory DBP (mmHg) | 75±8 | +6 (-6, 18); P<0.001 | +5 (-5, 19); P=0.01 |
derived from PVR= (MAP/CO)*80
adjusted for HR
adjusted for MAP and HR. The significance was determined using a simple ANCOVA model, with fixed effects for Visit, adjusting for baseline value. AIx denotes augmentation index; BP, blood pressure; DBP, diastolic blood pressure; MAP, mean arterial pressure; PVR, peripheral vascular resistance; PWV, pulse wave velocity and SBP, systolic blood pressure.
Forearm Blood Flow
The primary endpoint of the trial was the change in forearm blood flow (FBF) ratio in response to intra-arterial ACh infusion. We saw a significant interaction between dose and Visit in ACh-mediated (endothelium-dependent) dilatation following pazopanib treatment (P=0.0006). This was apparent at Visit 3 and at Visit 8/HYP). See Figure 1 for the representation of the FBF results (Panel A: ACh). There was no change in endothelium-independent dilatation (P=0.75), as assessed by the change in FBF ratio during SNP infusion nor in basal NO production, as assessed by change in FBF during L-NMMA infusion (P=0.9) (Figure 1- panel B: SNP and panel C: L-NMMA).
Figure 1. Change in the forearm blood flow ratio (infused/ control arm).

FBF responses to Acetylcholine (panel A), Sodium nitroprusside (panel B), and NG-monomethyl-L-arginine (panel C) at baseline (blue line), at 2 weeks after initiation of pazopanib (red line) and at 12 weeks (green line) or when subject became hypertensive (Visit HYP). * denotes P<0.001 (a hierarchical mixed effects model). Data are expressed as a geometric mean FBF ratio (infused to control arm) ± SEM.
Haemoglobin video imaging of conjunctiva and episclera
Representative images from the conjunctival haemoglobin video imaging can be seen in Figure 2; Panel A). Following 2 weeks of treatment (at Visit 3), there was a significant change in both the density -6.49 (95% CI: -10.31 to -2.66) %; P=0.003) and diameter -2.09 (95% CI: -3.95 to -0.19) μm; P=0.03, of vessels in the ocular surface microcirculations (Figure 2; Panel B).
Figure 2. Haemoglobin imaging.

Panel A) Representative images from the conjunctival and episcleral haemoglobin video imaging (1 pixel equals 10μm2). One-minute videos were recorded using a modified slit lamp. The videos were aligned and overlaid to yield a single image, which was segmented and stratified according the vessel diameter using a pre-trained U-Net convolutional neural network. The stratification is represented by the different colours in the images shown. Panel B) Microvascular density (blue) and diameter (orange) at baseline and at two weeks after initiation of treatment. Data are expressed as a mean ± SEM. * denotes P<0.05.
Pazopanib plasma concentrations
Trough plasma pazopanib concentrations ranged from 4.3μM to 210μM. Geometric mean pazopanib concentration was 75 ±2.0μM at Visit 3 and 75±1.7μM at Visit 8/HYP.
Acetylcholinesterase activity
In whole blood samples from healthy subjects, pazopanib led to a dose-dependent decrease in AChE activity (Figure 3). At the highest tested dose of pazopanib (183 μM) there was a 55.9 (95% CI: -61.8 to -50.1) %; P<0.001 inhibition of AChE activity. At the pazopanib concentration (57.9μM) that most closely represented the observed mean trough concentration in the trial, there was 40 (95% CI; -43.3 to -36.7) %; P< 0.001 inhibition of AChE activity.
Figure 3. Percentage inhibition of acetylcholine esterase (AChE) activity.

AChE activity was assessed in whole blood samples taken from 10 healthy volunteers and spiked in a range of concentration of pazopanib. ANOVA P<0.001. Data are expressed as a mean ± SEM.
Adverse Events
Adverse events were collected for all subjects for the duration of the study and are listed in Table S1 (related adverse events) and S2 (serious adverse events) in the supplemental files. Further details can be found in the supplemental files. Please see http://hyper.ahajournals.org
Related adverse events were in line with the adverse events reported for other, similar populations of patients treated with pazopanib.
Discussion
Hypertension is a common and serious side effect associated with VEGF-inhibitors. The HYPAZ trial aimed to gain a better understanding of the underlying mechanisms in otherwise normotensive patients with cancer.
As expected, trough concentrations of pazopanib reached steady state after two weeks of dosing. Both systolic and diastolic blood pressure increased after two weeks of treatment and this increase was sustained throughout the study. Nearly all patients experienced a rise in BP from their baseline values, and 4 out of 13 patients who finished the trial reached the pre-determined static threshold definition of hypertension (defined as SBP of >160 mmHg and/or DBP of >100mmHg, as per national treatment guidelines at the time of the study protocol submission) and were then initiated on anti-hypertensive therapy. Our data are in keeping with the published literature which demonstrates that approximately 90% of patients taking VEGF inhibitors experience a rise in blood pressure, with 15% of patients developing a severe form of clinically diagnosed hypertension5.
Our study saw an increase in ACh-mediated dilatation following pazopanib. These unexpected results contradict previous data from animal and human studies that have repeatedly demonstrated a key role of the NO pathway in VEGF inhibitor-induced hypertension. Facemire et al. demonstrated in a murine model, that VEGFR2 blockade leads to reduced eNOS expression, thus clearly showing the deleterious effect of VEGF blockade on eNOS24. Subsequently, in human studies, plasma nitrate, a biomarker of NO bioavailability, is reduced following treatments inhibiting VEGF25. Using Doppler flowmetry and iontophoresis of pilocarpine, Mourad et al. demonstrated reduced vasodilation following bevacizumab in patients with metastatic colorectal cancer26. Finally, using the same technique as used in the present study, Thijs et al. demonstrated that acute bevacizumab infusion reduced the vasodilatory effect to ACh in healthy volunteers27.
The unpredicted results of our FBF study led us to consider, post hoc, that pazopanib may act as an inhibitor of AChE, leading to the observed improvement in ACh-mediated dilatation, without improving endothelial function per se. AChE is the enzyme responsible for the hydrolysis of ACh, and hence its inhibition would increase the half-life of ACh, leading to increased local concentrations of ACh in the forearm. This would potentially confound our efforts to assess endothelial function with this particular challenge agent. In subsequent ex vivo study in healthy volunteers, we demonstrated that in the presence of 210μM pazopanib, there was a 55% inhibition of AChE activity. Importantly, a 40% inhibition was seen at the pazopanib concentration (57.9μM), which most closely represented the observed trough concentration in the trial, therefore suggesting that the inhibition seen in ex vivo studies is likely to be clinically relevant. We believe this is the first study to demonstrate that pazopanib has an inhibitory effect on AChE in humans. Following the completion of our study, a computational analysis reported that pazopanib shares structural and functional properties with donepezil, a marketed AChE inhibitor used for the treatment of dementia28. This unexpected pharmacological profile of pazopanib would also explain the discrepancy between the findings of Thijs et al.27 and our data, since bevacizumab is a monoclonal antibody against VEGF with a different molecular structure to pazopanib, thus would not lead to AChE inhibition. Our data relating to AChE inhibition could explain the observed vagal adverse events including bradycardia and reduced stroke volume, resulting in decreased cardiac output and GI toxicity.
Ocular surface haemoglobin video imaging was performed and a pre-trained machine learning code applied to interpret the data. Following only 2 weeks of treatment, a significant reduction in both microvascular density and diameter were observed, demonstrating clearly that pazopanib led to microvascular rarefaction. Our study is the first investigate the effect of VEGF inhibition on microvascular rarefaction using a technique in which microcirculations can be revisited, allowing a comparison of exactly the same arterioles, capillaries and venules over time. Our results confirm previous findings in humans, albeit using very different methodology, to look at microvascular effects of VEGF inhibition26,29.
Our data demonstrate that treatment with pazopanib led to a large increase in peripheral vascular resistance. Our findings support the results of a previous study demonstrating that treatment with sunitinib increased PVR30. Together with the fact that CO was reduced, as a result of the fall in heart rate and stroke volume, our results highlight the importance of increased PVR as the causative haemodynamic mechanism behind the blood pressure increment seen in this trial. Additionally, our study demonstrated an increase in minimum forearm vascular resistance following VEGF-inhibition. Although, there are no reference values for MFVR, a study by Svedsen et al. gives an indication of normative values by demonstrating that MFVR was 3.7 R units in normotensive and, 3.9 R units in hypertensive subjects14. This indicates that the MFVR in our study was normal at baseline, with 3.27 R units, and grossly elevated at the end of the trial at 4.93 R units. Together with the changes seen in the ocular microvasculature, the elevated MFVR suggests a structural remodelling of resistance vessels.
Our study demonstrated significant increases in arterial stiffness as measured by augmentation index and aortic PWV following pazopanib. Importantly, these changes remained significant after adjusting for confounding factors, such as MAP and HR. The increase observed in AIx could be a result of vasoconstriction at the sites of pressure wave reflection, as indicated by the increase in PVR and microvascular rarefaction. This would lead to an increase in the magnitude of the reflected pressure wave and subsequent increase in systolic BP. The observed pressure-independent increase in aortic PWV suggests that there may be direct structural changes in aortic wall properties, perhaps via changes in the composition of aortic extra cellular matrix or fragmentation of elastic fibres. Our data are supportive of the results of Alivon et al. who demonstrated that treatment with TKIs led to increased large artery stiffness, but in contrast to our study, they showed no change in AIx31.
The deleterious changes we observed in both the microvasculature and large arteries could contribute towards the increased risk of CVD in cancer patients. This was highlighted recently by a large study of cancer survivors in whom CVD mortality was highest within the first year of cancer diagnosis1. Together, our results and the observations in the cancer survivors, highlight the importance of the burgeoning area of cardio-oncology in attempt to minimise CVD complications in oncology patients. A detailed understanding of the underlying cardiovascular mechanisms of anti-cancer therapies could lead to better stratification and treatment of the haemodynamic burdens imposed on this population in order to minimise CVD complications and maximise survival rates.
Limitations
The complex study design and the underlying malignant disease made recruitment and retention of patients difficult, resulting in the study being relatively small and having a high dropout rate due to early disease progression in patients (n=14). Therefore, we were not able to compare the haemodynamic changes in hypertensive versus non-hypertensive patients, as originally planned. The small number of patients may have led to some of the secondary outcomes being underpowered, such as the L-NMMA response. No placebo group was enrolled as it was unethical to deny treatment from cancer patients who had already exhausted other treatment options. The unexpected AChE inhibition by pazopanib underlines the difficulty of working with drugs with a wide range of pharmacological activities. Thus, as it stands, the current study is unable to draw definitive conclusions about the role of endothelial dysfunction in VEGF-induced hypertension. Ideally, an alternative endothelium-dependent challenge agent, not hydrolysed by AChE, such as bradykinin, should be used to determine if there is a direct endothelium-dependent effect of pazopanib on NO bioavailability.
Perspective
We have demonstrated that the VEGF inhibitor pazopanib led to structural remodelling of the resistance vasculature, as evidenced by microvascular rarefaction and a significant increase in PVR, with a subsequent increase in BP. Our findings highlight the importance of altered microvascular structure and increased peripheral vascular resistance as the key mediators of hypertension seen with pazopanib and potentially other VEGF inhibitors. Our data supports the current expert opinion for the use of calcium-channel blockers and potassium sparing diuretics32 in the treatment of VEGF inhibition-induced hypertension due to their ability to reduce PVR. However further studies are needed, particularly as drugs which reduce resistance by relaxing pre-capillary resistance vessels may not be specific when rarefaction is the primary cause of increased PVR.
Supplementary Material
{kind=link}
Novelty and Significance.
What Is New?
The HYPAZ trial is the first to examine multiple concurrent potential mechanisms underlying hypertension in humans brought on by pazopanib.
Pazopanib unexpectedly led to an increase in ACh-mediated forearm blood flow (i.e. increased vasodilatation), but this is most likely due to inhibition of acetylcholinesterase activity by the drug, which has not been shown in humans previously.
Peripheral vascular resistance and minimum forearm resistance were increased and microvascular diameter and density were reduced by the treatment, resulting in an increase of blood pressure.
What is relevant?
Hypertension is a common adverse effect of VEGF inhibition, and it appears to be driven by an increase in PVR.
Hypertension associated with TKIs therefore should be treated with anti-hypertensives with vasodilatory effects, such as calcium channel antagonists.
The additional inhibitory effect of pazopanib on ACh esterase could explain other commonly seen side effects, such as bradycardia.
Summary
We have demonstrated that the VEGF inhibitor pazopanib led to structural remodelling of the resistance vasculature, as evidenced by microvascular rarefaction and a significant increase in PVR, with a subsequent increase in BP.
Acknowledgements
None
Sources of Funding
This research was supported by the National Institute for Health Research/Wellcome Trust Clinical Research Facility, the Cambridge Clinical Trials Unit at Cambridge University Hospitals NHS Foundation Trust and the Cambridge Cancer Trials Centre (CCTC) Early Phase Team.
The study was funded as an investigator initiated trial by GSK who also provided free drug supply. The Cambridge Experimental Cancer Medicine Centre (ECMC) provided funding for scanning. KMMP, DJ, JC. IBW and CMM acknowledge funding from The NIHR Cambridge Biomedical Research Centre (BRC), which is a partnership between Cambridge University Hospitals NHS Foundation Trust and the University of Cambridge, funded by the National Institute for Health. Research (NIHR). This research was supported by the NIHR Cambridge Biomedical Research Centre (BRC-1215-20014). The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care. CBS acknowledges support from the EPSRC Centre for Mathematical Imaging in Healthcare No. EP/N014588/1. LY acknowledges funding from The Wellcome Trust Translational Medicine and Therapeutics Programme (100780/Z/12/Z) and the Raymond and Beverly Sackler fellowship. CB acknowledges support from the 4TU programme -Precision Medicine.
Footnotes
Disclosures
JC is a full time employee of Cambridge University Hospitals NHS Foundation Trust, but was seconded by the Trust for 50% of his NHS salaried time to work on GSK clinical trials until 2020. He received no employee benefits or shares/dividends or income from GSK. SSF is an employee of GSK and has received personal fees and other benefits from GSK outside the submitted work. Novartis Europharm Limited (Dublin, Ireland) is the current marketing authorisation holder of pazopanib (Votrient®) and has reviewed this manuscript. Other authors declare no conflicts.
References
- 1.Sturgeon KM, Deng L, Bluethmann SM, Zhou S, Trifiletti DM, Jiang C, Kelly SP, Zaorsky NG. A population-based study of cardiovascular disease mortality risk in US cancer patients. Eur Heart J. 2019;40:3889–3897. doi: 10.1093/eurheartj/ehz766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bukowski RM, Yasothan U, Kirkpatrick P. Pazopanib. Nat Rev Drug Discov. 2010;9:17–18. doi: 10.1038/nrd3073. [DOI] [PubMed] [Google Scholar]
- 3.Keisner SV, Shah SR. Pazopanib. Drugs. 2011;71:443–454. doi: 10.2165/11588960-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 4.Qi W-X, Lin F, Sun Y-J, Tang L-N, He A-N, Yao Y, Shen Z. Incidence and risk of hypertension with pazopanib in patients with cancer: a meta-analysis. Cancer Chemother Pharmacol. 2013;71:431–439. doi: 10.1007/s00280-012-2025-5. [DOI] [PubMed] [Google Scholar]
- 5.Agarwal M, Thareja N, Benjamin M, Akhondi A, Mitchell GD. Tyrosine Kinase Inhibitor-Induced Hypertension. Curr Oncol Rep. 2018;20:65. doi: 10.1007/s11912-018-0708-8. [DOI] [PubMed] [Google Scholar]
- 6.Robinson ES, Khankin EV, Karumanchi SA, Humphreys BD. Hypertension Induced by VEGF Signaling Pathway Inhibition: Mechanisms and Potential Use as a Biomarker. Semin Nephrol. 2010;30:591–601. doi: 10.1016/j.semnephrol.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Jesus-Gonzalez N, Robinson E, Moslehi J, Humphreys BD. Management of antiangiogenic therapy-induced hypertension. Hypertension. 2012;60:607–615. doi: 10.1161/HYPERTENSIONAHA.112.196774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levy BI, Ambrosio G, Pries AR, Struijker-Boudier HA. Microcirculation in hypertension: a new target for treatment? Circulation. 2001;104:735–740. doi: 10.1161/hc3101.091158. [DOI] [PubMed] [Google Scholar]
- 9.Oken MM, Creech RH, Tormey DC, Horton J, Davis TE, McFadden ET, Carbone PP. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol. 1982;5:649–655. [PubMed] [Google Scholar]
- 10.Schwartz LH, Litière S, de Vries E, Ford R, Gwyther S, Mandrekar S, Shankar L, Bogaerts J, Chen A, Dancey J, Hayes W, et al. RECIST 1.1 – Update and Clarification: From the RECIST Committee. Eur J Cancer. 2016;62:132–137. doi: 10.1016/j.ejca.2016.03.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilkinson IB, Webb DJ. Venous occlusion plethysmography in cardiovascular research: methodology and clinical applications. BrJClinPharmacol. 2001;52:631–646. doi: 10.1046/j.1365-2125.2001.01495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilkinson IB, Fuchs SA, Jansen IM, Spratt JC, Murray GD, Cockcroft JR, Webb DJ. Reproducibility of pulse wave velocity and augmentation index measured by pulse wave analysis. J Hypertens. 1998;16:2079–2084. doi: 10.1097/00004872-199816121-00033. [DOI] [PubMed] [Google Scholar]
- 13.Mathiassen ON, Buus NH, Olsen HW, Larsen ML, Mulvany MJ, Christensen KL. Forearm plethysmography in the assessment of vascular tone and resistance vasculature design: new methodological insights. Acta Physiologica. 2006;188:91–101. doi: 10.1111/j.1748-1716.2006.01611.x. [DOI] [PubMed] [Google Scholar]
- 14.Svendsen MB, Khatir DS, Peters CD, Christensen KL, Buus NH. Differential effects of age on large artery stiffness and minimal vascular resistance in normotensive and mildly hypertensive individuals. Clinical Physiology and Functional Imaging. 2015;35:359–367. doi: 10.1111/cpf.12171. [DOI] [PubMed] [Google Scholar]
- 15.Clemensen P, Christensen P, Norsk P, Gronlund J. A modified photo- and magnetoacoustic multigas analyzer applied in gas exchange measurements. JApplPhysiol. 1994;76:2832–2839. doi: 10.1152/jappl.1994.76.6.2832. [DOI] [PubMed] [Google Scholar]
- 16.Meyer PA. The circulation of the human limbus. Eye. 1989;3(2):121–127. doi: 10.1038/eye.1989.19. [DOI] [PubMed] [Google Scholar]
- 17.Meyer PAR. Re-orchestration of blood flow by micro-circulations. Eye (Lond) 2018;32:222–229. doi: 10.1038/eye.2017.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ronneberger O, Fischer P, Brox T. U-Net: Convolutional Networks for Biomedical Image Segmentation. In: Navab N, Hornegger J, Wells WM, Frangi AF, editors. Medical Image Computing and Computer-Assisted Intervention – MICCAI 2015. Cham: Springer International Publishing; 2015. pp. 234–241. [Google Scholar]
- 19.Staal J, Abramoff MD, Niemeijer M, Viergever MA, van Ginneken B. Ridge-based vessel segmentation in color images of the retina. IEEE Transactions on Medical Imaging. 2004;23:501–509. doi: 10.1109/TMI.2004.825627. [DOI] [PubMed] [Google Scholar]
- 20.Dominique Zosso, Giang Tran, Stanley Osher. A unifying retinex model based on non-local differential operators. 2013 doi: 10.1117/12.2008839. [Internet] [DOI] [Google Scholar]
- 21.Evangelidis G. Image alignment toolbox: A Matlab toolbox for image alignment and registration. [cited 2019 Dec 17];2013 [Internet]. Available from: https://sites.google.com/site/imagealignment/home.
- 22.Zosso D. Non-local retinex. [cited 2019 Dec 17];2019 [Internet]. Available from: https://www.mathworks.com/matlabcentral/fileexchange/47562-non-local-retinex.
- 23.orobix/retina-unet. Orobix. [cited 2020 Sep 16];2020 [Internet]. Available from: https://github.com/orobix/retina-unet.
- 24.Facemire CS, Nixon AB, Griffiths R, Hurwitz H, Coffman TM. Vascular endothelial growth factor receptor 2 controls blood pressure by regulating nitric oxide synthase expression. Hypertension. 2009;54:652–658. doi: 10.1161/HYPERTENSIONAHA.109.129973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pandey AK, Singhi EK, Arroyo JP, Ikizler TA, Gould ER, Brown J, Beckman JA, Harrison DG, Moslehi J. Mechanisms of VEGF-Inhibitor Associated Hypertension and Vascular Disease. Hypertension. 2018;71:e1–e8. doi: 10.1161/HYPERTENSIONAHA.117.10271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mourad J-J, des Guetz G, Debbabi H, Levy BI. Blood pressure rise following angiogenesis inhibition by bevacizumab. A crucial role for microcirculation. Ann Oncol. 2008;19:927–934. doi: 10.1093/annonc/mdm550. [DOI] [PubMed] [Google Scholar]
- 27.Thijs AMJ, van Herpen CML, Sweep FCGJ, Geurts-Moespot A, Smits P, van der Graaf WTA, Rongen GA. Role of endogenous vascular endothelial growth factor in endothelium-dependent vasodilation in humans. Hypertension. 2013;61:1060–1065. doi: 10.1161/HYPERTENSIONAHA.111.00841. [DOI] [PubMed] [Google Scholar]
- 28.Yang Y, Li G, Zhao D, Yu H, Zheng X, Peng X, Zhang X, Fu T, Hu X, Niu M, Ji X, et al. Computational discovery and experimental verification of tyrosine kinase inhibitor pazopanib for the reversal of memory and cognitive deficits in rat model neurodegeneration. Chem Sci. 2015;6:2812–2821. doi: 10.1039/c4sc03416c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steeghs N, Rabelink TJ, op ’t Roodt J, Batman E, Cluitmans FHM, Weijl NI, de Koning E, Gelderblom H. Reversibility of capillary density after discontinuation of bevacizumab treatment. Ann Oncol. 2010;21:1100–1105. doi: 10.1093/annonc/mdp417. [DOI] [PubMed] [Google Scholar]
- 30.Catino AB, Hubbard RA, Chirinos JA, Townsend R, Keefe S, Haas NB, Puzanov I, Fang JC, Agarwal N, Hyman D, Smith AM, et al. Longitudinal Assessment of Vascular Function With Sunitinib in Patients With Metastatic Renal Cell Carcinoma. Circ Heart Fail. 2018;11:e004408. doi: 10.1161/CIRCHEARTFAILURE.117.004408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alivon M, Giroux J, Briet M, Goldwasser F, Laurent S, Boutouyrie P. Large artery stiffness and hypertension after antiangiogenic drugs: influence on cancer progression. J Hypertens. 2015;33:1310–1317. doi: 10.1097/HJH.0000000000000550. [DOI] [PubMed] [Google Scholar]
- 32.Waliany S, Sainani KL, Park LS, Zhang CA, Srinivas S, Witteles RM. Increase in Blood Pressure Associated With Tyrosine Kinase Inhibitors Targeting Vascular Endothelial Growth Factor. JACC CardioOncology. 2019;1:24–36. doi: 10.1016/j.jaccao.2019.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.