

Abstract
Mesencephalic astrocyte-derived neurotrophic factor (MANF) is an endoplasmic reticulum (ER)–resident protein that plays a crucial role in attenuating ER stress responses. Although MANF is indispensable for the survival and function of mouse β-cells, its precise role in human β-cell development and function is unknown. In this study, we show that lack of MANF in humans results in diabetes due to increased ER stress, leading to impaired β-cell function. We identified two patients from different families with childhood diabetes and a neurodevelopmental disorder associated with homozygous loss-of-function mutations in the MANF gene. To study the role of MANF in human β-cell development and function, we knocked out the MANF gene in human embryonic stem cells and differentiated them into pancreatic endocrine cells. Loss of MANF induced mild ER stress and impaired insulin-processing capacity of β-cells in vitro. Upon implantation to immunocompromised mice, the MANF knockout grafts presented elevated ER stress and functional failure, particularly in recipients with diabetes. By describing a new form of monogenic neurodevelopmental diabetes syndrome caused by disturbed ER function, we highlight the importance of adequate ER stress regulation for proper human β-cell function and demonstrate the crucial role of MANF in this process.
Introduction
Autosomal-recessive causes of childhood-onset monogenic diabetes commonly present with syndromic diabetes (diabetes and additional nonautoimmune features). For instance, individuals carrying homozygous loss-of-function variants in WFS1, SLC19A2, and SLC29A3 develop diabetes in childhood with multiple nonautoimmune additional features (1–3). Autosomal-recessive genetic diseases are enriched in populations with high rates of consanguinity (4). Genetic studies of children with syndromic diabetes who are born to consanguineous parents facilitates the identification of novel recessive causes of syndromic diabetes.
Recent studies highlight the important role of mesencephalic astrocyte-derived neurotrophic factor (MANF) in maintaining normal endoplasmic reticulum (ER) homeostasis (5). The major phenotype in juvenile MANF knockout (KO) mice is insulin-deficient diabetes, indicating the important role of MANF in the β-cell (6). β-Cells are under high demand for insulin synthesis, folding, and secretion. ER is the intracellular compartment where proinsulin molecules fold to their native conformation before transport to Golgi for further granule packaging and secretion (7). Accumulation of unfolded and misfolded proteins in the ER leads to ER stress and activation of the unfolded protein response (UPR) (8). If prolonged, unresolved ER stress leads to cell death, which is mainly mediated by DNA damage-inducible transcript 3 (DDIT3/CHOP) (9). Therefore, any alterations in the mechanisms controlling protein folding may have a deleterious effect on β-cell function and survival (10).
In this study, we undertook whole-genome sequencing for children with syndromic diabetes, aiming to identify novel autosomal-recessive loss-of-function mutations (human KO), causing monogenic diabetes. We show that homozygous loss-of-function variants in MANF cause a novel multisystem disorder characterized by childhood-onset diabetes, short stature, deafness, developmental delay, and microcephaly. To study the role of MANF in human β-cell development and to create an experimental model for human diabetes caused by loss of MANF, we generated MANF KO human embryonic stem cells (hESC) and differentiated them into pancreatic islet endocrine cells. Our results show that lack of MANF does not affect the development of human β-cells but increases their susceptibility to ER stress–induced functional failure. Our experimental findings provide a pathogenic explanation for the phenotype of the patients.
Research Design and Methods
Patient Cohort
We investigated 32 children with syndromic diabetes with low likelihood of type 1 diabetes from 8 pediatric diabetes clinics across Turkey, a country with high rates of consanguinity (11). Syndromic diabetes was defined as the presence of any nonautoimmune features in addition to diabetes. All of the children were negative when tested for islet autoantibodies, and their type 1 diabetes genetic risk score was <50th centile of the population with type 1 diabetes (12). Ethical approval for the study was from University of Kocaeli and Dokuz Eylül University ethics committees (Kocaeli and Izmir, Turkey, respectively) and Wales Research Ethics Committee (Wales, U.K.). The study was conducted in accordance with the Declaration of Helsinki, and all subjects or their parents gave informed consent for genetic testing.
Molecular Genetic Analysis
The molecular genetic analysis plan to identify novel homozygous predicted loss-of-function variant causing syndromic diabetes is described in Supplementary Fig. 1.
We first performed targeted next-generation sequencing for all known monogenic diabetes genes as previously described (13). Individuals with likely pathogenic and pathogenic variants according to American College of Medical Genetics and Genomics guidelines were excluded (14). We calculated homozygosity from the off-target reads of a targeted next-generation sequencing gene panel using a SavvyHomozygosity tool (https://github.com/rdemolgen/SavvySuite) (15). We only took forward patients with calculated homozygosity >6.5% to increase the likelihood of identifying a novel homozygous pathogenic variant.
We performed gene discovery in the remaining eight individuals by whole-genome sequencing. Genome sequencing was performed using BGISEQ-500 on DNA extracted from peripheral blood leukocytes. The mean depth across these 8 genomes was 35. The sequencing data were analyzed using an approach based on the GenomeAnalysisToolkit (GATK) best practice guidelines. GATK HaplotypeCaller was used to identify variants that were annotated using Alamut batch version 1.11 (SOPHiA Genetics, Rouen, France), and variants that failed the QD2 VCF filter or had less than five reads supporting the variant allele were excluded. Copy number variants were called by SavvyCNV, which uses read depth to judge copy number states (https://github.com/rdemolgen/SavvySuite) (15).
We prioritized homozygous protein–truncating variants (frameshift, nonsense, and splice acceptor/donor excluding last exon) in these genomes. These variants are predicted to cause loss of function by nonsense-mediated mRNA decay (16). We investigated variants in the genes in which there are no individuals with homozygous protein–truncating variant in population and in which homozygous protein–truncating variants are not known to cause monogenic disease. We used multiple genome/exome population databases. These included gnomAD v2.1 (n = 135,743), gnomAD v3 (n = 71,702) (https://gnomad.broadinstitute.org) (17), TOPMed Freeze 5 (n = 62,784) (https://bravo.sph.umich.edu/freeze5/hg38/), and two populations with higher rates of consanguinity (The Greater Middle East [GME] Variome Project; n = 2,497) (http://igm.ucsd.edu/gme/index.php) (18) and Genes & Health (n = 8,921) (September 2019 download) (19).
Variant confirmation and cosegregation in family members were performed by Sanger sequencing (primer sequences available in Supplementary Table 1).
To identify additional patients with a variant in the putative candidate gene, we used literature review, internet search (20), and Sanger sequencing of the candidate gene in patients selected based on homozygosity over the candidate gene and/or similar clinical features as a proband. The patients were referred for diagnostic genetic monogenic diabetes testing performed at the Department of Molecular Genetics, Royal Devon and Exeter NHS Foundation Trust, and did not have a pathogenic variant in the known monogenic diabetes genes.
Quantitative RT-PCR Analysis of MANF Expression in Human Blood
Peripheral blood was collected in PAXgene Blood RNA tubes (PreAnalytiX GmbH, Hombrechtikon, Switzerland) and RNA extracted using the PAXgene Blood RNA kit (Qiagen, Hilden, Germany). Total RNA was DNase-treated using the TURBO DNA-free kit (Ambion) and reverse transcribed using SuperScript IV Reverse Transcriptase (Invitrogen). Triplicate quantitative PCR (qPCR) reactions (5 µL) using TaqMan Gene Expression assays (Applied Biosystems) were carried out on a QuantStudio 12K Flex (Thermo Fisher Scientific). TaqMan Gene Expression assay identification numbers used are as follows: Hs00180640_m1 (MANF), Hs01060665_g1 (ACTB), Hs00831112_s1 (RPL11), Hs00984230_m1 (B2M), Hs00939627_m1 (GUSB), and Hs02786624_g1 (GAPDH). Expression of MANF was normalized using the geometric mean of five housekeeping genes (ACTB, RPL11, B2M, GUSB, and GAPDH).
Cell Culture and In Vitro Differentiation of hESC
hESC (WA01/H1 line; WiCell) were cultured on Matrigel-coated plates (BD Biosciences) in Essential 8 medium (A1517001; Life Technologies). Differentiation was carried out following a seven-stage protocol as previously described (21–23) with further modifications. The glucose concentration of each stage was as follows: S1–S4 cells were cultured in 10.5 mmol/L glucose; S5–S6 cells were cultured in 20.5 mmol/L glucose; and S7 cells were cultured in 5.5 mmol/L glucose.
Genome Editing
To recapitulate the gene mutations found in the patients, guide RNAs targeting the first exon of MANF gene for deletion were designed using Benchling (https://benchling.com). Guide RNAs with the highest quality score and lowest off-target score were selected and purchased, alongside the RNP components (HiFi Cas9 protein, crRNA, and tracrRNA), from Integrated DNA Technologies and used according to the manufacturer’s recommended protocol. Two million undifferentiated stem cells were electroporated with the RNP complex using the Neon Transfection system (1,100 V, 20 ms, 2 pulses) (Thermo Fisher Scientific) and plated on Matrigel-coated plates in Essential 8 medium containing 10 μmol/L ROCK inhibitor overnight. Afterward, cells were single-cell sorted, expanded, and screened for the desired deletion using PCR. Positive clones were validated by Sanger sequencing and characterized for pluripotency and chromosomal integrity.
Western Blot Analysis
To extract whole-cell protein from hESC-derived cells, cells were washed with ice-cold PBS and lysed with 300 μL Laemmli buffer (Bio-Rad Laboratories) for 10 min on ice. The cells were then sonicated (three times for 15 s each) and the homogenate centrifuged at 1,000 relative centrifugal force for 10 min at 4°C and heated at 95°C for 5 min. Equal amounts of supernatant were run on Any kD Mini-PROTEAN TGX gel (Bio-Rad Laboratories) at 100 V for 1 h in SDS running buffer (Bio-Rad Laboratories). Proteins were then transferred to a nitrocellulose membrane (Invitrogen) using the iBlot system (Invitrogen) for 5 min. The membrane was blocked in Tris-buffered saline containing 0.05% Tween and 5% low-fat dried milk for 90 min at 4°C. The membrane was then probed with the primary antibody overnight at 4°C, washed twice with Tris-buffered saline containing 0.05% Tween, and incubated with the corresponding secondary antibody for 1 h at room temperature. Chemiluminescence detection was performed following Amersham ECL (RPN2235; Cytiva) exposure using the ChemiDoc XRS+ System and Image Lab Software (Bio-Rad Laboratories). Details on the antibodies used are given in Supplementary Table 3.
Flow Cytometry Analysis
hESC-derived cells were dissociated into single cells by incubation with TrypLE for 7–10 min at 37°C and resuspended in cold FBS/PBS (5% v/v). For surface marker staining of CXCR4, 1 million cells were incubated with the directly conjugated antibody CD184/CXCR4 allophycocyanin at a final dilution of 1:10 for 30 min at room temperature. For intracellular markers, 1 million cells were first fixed in 250 μL of Cytofix/Cytoperm Buffer (554722; BD Biosciences) for 20 min at 4°C and then washed twice with BD Perm/Wash Buffer Solution (554723; BD Biosciences). Cell pellet was resuspended in 80 μL FBS/BD Perm/Wash buffer (4% v/v) and incubated with the corresponding directly conjugated antibody at a final dilution of 1:80 overnight at 4°C. After incubation with the antibody, cells were washed twice and analyzed using an FACSCalibur cytometer (BD Biosciences) and FlowJo software (Tree Star, Inc.). Gating was defined using isotype antibodies. Details on the antibodies used are given in Supplementary Table 3.
Quantitative RT-PCR
Gene expression was assessed at different stages of differentiation. Total RNA was extracted using the NucleoSpin RNA Plus kit (Macherey-Nagel). Quantification and quality of the RNA preparations were measured using a SimpliNano (GE Healthcare Life Sciences) spectrophotometer. For cDNA synthesis, 1.5 μg RNA was first denatured for 1 min at 65°C and then reverse transcribed using the GoScript Reverse Transcriptase kit (Promega). cDNA (50 ng) was amplified by quantitative RT-PCR using 5 times HOT FIREPol EvaGreen qPCR Mix Plus and 5 μL of forward and reverse primer mix at 2 μmol/L each in a final volume of 20 μL. The reagents were pipetted by the QIAgility (Qiagen) liquid-handling system into 100-well discs and run in Rotor-Gene Q (Qiagen) thermocycler programmed as follows: 95°C for 15 min, followed by 40 cycles of 95°C for 25 s, 57°C for 25 s, 72°C for 25 s, and finished by a melting step. Rotor-Gene Q software (Qiagen) was used to calculate cycle threshold (Ct) values. Cyclophilin G (PPIG) was used as an endogenous housekeeping gene to calculate ΔCt and normalized to undifferentiated hESC to calculate ΔΔCt values and relative gene expression. Exogenous positive control was used as a calibrator, and reverse-transcription reaction without template was used as a negative control. Details on the primers used are provided in Supplementary Table 2.
Induction of ER Stress
Fifty isletlike aggregates were manually picked and incubated in full S7 media supplemented with the corresponding concentration of ER stress inducers. After the treatment, aggregates were collected, fixed with 4% paraformaldehyde (PFA) for 3 h at room temperature, embedded in paraffin, and then processed for immunohistochemistry. A total of 1 μmol/L of thapsigargin (TG) (T9033; Sigma-Aldrich) and 5 µg/mL of tunicamycin (TM) (T7765; Sigma-Aldrich) were used for 48 h, while 1 µg/mL of brefeldin A (B5936; Sigma-Aldrich) was used for 24 h. DMSO was used as a vehicle control at 5 µL/mL.
Immunocytochemistry and Histology
For adherent cultures, cells were fixed in 4% PFA for 15 min at room temperature, permeabilized with 0.5% Triton X-100 in PBS, then blocked with Ultra V block (Thermo Fisher Scientific) for 10 min, and incubated with primary antibodies diluted in 0.1% Tween in PBS overnight at 4°C. After incubation, cells were washed twice with PBS and incubated with corresponding secondary antibodies diluted in 0.1% Tween in PBS for 1 h at room temperature. For isletlike aggregates paraffin embedding, aggregates were fixed in 4% PFA for 3 h at room temperature, briefly stained with eosin, then embedded in 2% low-melting agarose (Sigma-Aldrich), and transferred to paraffin blocks. For grafts, graft-containing kidney was retrieved, fixed in 4% PFA for 24 h at room temperature, and processed for paraffin embedding. Paraffin sections of 5-μm thickness were deparaffinized, and antigen retrieval was carried out using the Decloaking Chamber (Biocare Medical) in citrate buffer at 95°C for 12 min. Blocking and incubation with antibodies were performed as described above. For TUNEL assay, In Situ Cell Death Detection Kit, Fluorescein (#11684795910; Roche) was used according to the manufacturer’s instruction. Details on the antibodies used are provided in Supplementary Table 3.
Static Glucose-Stimulated Insulin Secretion
Fifty isletlike aggregates were manually picked and preincubated in 3.3 mmol/L glucose-containing Krebs buffer in a 12-well plate placed on a rotating platform for 90 min at 37°C. Aggregates were then washed with Krebs buffer and sequentially incubated in Krebs buffer containing 3.3 mmol/L glucose, 16.6 mmol/L glucose, and 3.3 mmol/L glucose plus 30 mmol/L KCL, for a period of 30 min. Samples of 300 μL were collected from the supernatant of each treatment and stored at −80°C for insulin ELISA measurements. After the last incubation, aggregates were collected and lysed in acid ethanol for determination of total insulin content and DNA content. Stimulated insulin secretion results are presented as fractional release of total human insulin content after cell mass normalization using total DNA content.
Implantation of SC-Islets
All experiments were approved by the Animal Welfare Committee of Southern Finland (ESAVI/14852/2018). NOD-SCID-γ (0055577; The Jackson Laboratory) were maintained at Biomedicum Helsinki animal facility on a 12-h light/dark cycle with ad libitum food. Mice were anesthesized with isoflurane and subcutaneously injected with carprofen (Rimadyl, 5 mg/kg; Pfizer, Helsinki, Finland) and buprenorphine (Temgesic, 0.05–0.1 mg/kg; Reckitt Benckiser Pharmaceuticals Ltd, Berkshire, U.K.) as analgesics. Approximately 500 SC-islets were loaded on a polyethylene-50 tubing and transplanted under the kidney capsule as previously described (24). Mouse blood samples were collected monthly from the saphenous vein using heparin-coated capillary tubes. Blood plasma was separated by centrifugation (5,000 relative centrifugal force, 5 min, room temperature) and stored at −80°C for human C-peptide analysis.
Streptozotocin Treatment
After 3 months of implantation, mice were fasted for 4 h and then intraperitoneally injected with 130–150 mg/kg streptozotocin (STZ) (S0130; Sigma-Aldrich). STZ was solubilized in cold 0.1 mol/L citrate buffer composed of 0.1 mol/L citric acid monohydrate and 0.1 mol/L trisodium citrate dehydrate. Drinking water was replaced with 10% sucrose-containing tap water for 48 h to avoid hypoglycemia caused by unregulated insulin release from dying β-cells.
Intraperitoneal Glucose Tolerance Test
After 6 h of fasting, mice were intraperitoneally injected with 2 g glucose/kg in the form of 30% glucose in saline. Blood glucose levels were measured using a glucometer (OneTouch Ultra; LifeScan, Milpitas, CA), and blood samples were collected for measuring human C-peptide levels at 0, 15, 30, 60, and 90 min after glucose injection.
ELISA
Insulin levels were measured from the in vitro samples with the Insulin ELISA kit (Mercodia, Uppsala, Sweden), and human-specific C-peptide was measured from plasma samples with the Ultrasensitive C-peptide ELISA kit.
Image Acquisition and Analysis
For adherent cultures, an EVOS inverted microscope (Life Technologies) was used to image the cells. For paraffin sections, Zeiss Axio Observer Z1 with Apotome was used to image the cells, and then images were processed with ZEN 2 software. All paraffin sections were equally treated and imaged with the same microscope parameters. Image quantification was performed using CellProfiler software (25) and Fiji software (26).
Statistical Analysis
All data are presented as individual points with their mean ± SEM. Statistical significance evaluation was conducted by unpaired, two-tailed Student t test with Welch correction using Prism 8 software (GraphPad Software, La Jolla, CA). P values <0.05 were considered statistically significant (*P < 0.05; **P < 0.001; ***P < 0.0001).
Data and Resource Availability
The data generated and analyzed during this study are included in the published article and its Supplementary Material. Additional data and resources are available from the corresponding author upon reasonable request.
Results
Identification of Individuals With Homozygous Protein–Truncating Variants in MANF
We performed gene discovery by whole-genome sequencing on eight subjects with nonautoimmune diabetes and additional extrapancreatic features to identify novel homozygous loss-of-function causes of syndromic diabetes. The subjects did not have mutation in any of the known monogenic diabetes genes and had high genome-wide homozygosity (>6.5%) (Supplementary Fig. 1A). Analysis of genome sequencing data identified one individual (case 1) with a novel homozygous frameshift variant in MANF (Chr3[GRCh37]:g.51422846_51422858del, NM_006010.5:c.82_94del, p.Leu28Thrfs*33) (Fig. 1 and Supplementary Fig. 1B). The variant was not present in five large independent population exome/genome databases (combined total n = 281,647) (17). None of these population databases had any homozygous protein–truncating or rare homozygous missense (minor allele frequency <0.01) variants, suggesting rare homozygous variants in MANF are highly intolerant to humans.
Figure 1.

Identification of homozygous MANF mutation in two patients with childhood diabetes. A: Pedigrees of the two case subjects with homozygous MANF variants. Square symbolizes male, and circle symbolizes female. Black-shaded area indicates the presence of diabetes; blue and red areas indicate the presence of short stature and deafness, respectively, whereas the gray-shaded area indicates the presence of microcephaly and developmental delay. B: Schematic presentation of the MANF gene KO strategy using CRISPR/Cas9. The positions of patient mutations are marked with red arrows, and the guides used to delete exon 1 are presented as blue arrowheads. C: Representative immunoblot showing the absence of the MANF protein in the generated clone. α-Tubulin was used as a loading control.
Replication analysis identified a second independent individual (case 2) with a different homozygous protein–truncating variant in MANF (NM_006010.5:c.103+1G>T, essential donor splice site). This case was partially described by Yavarna et al. (27). Case 2 was part of the exome study of individuals with clinically suspected Mendelian disease (27). Similar to case 1, this variant is not seen in any population databases. The parents of both case subjects were heterozygous for MANF variants. Sanger sequencing of MANF from additional 187 patients presenting with diabetes and microcephaly, developmental delay, deafness, or short stature and/or are homozygous for the region incorporating MANF did not identify additional case subjects.
Both protein-truncating variants are in exon 1. They generate premature termination codons and are predicted to cause loss of MANF due to classical nonsense-mediated mRNA decay of the mutant mRNA pathway (28). In line with this, MANF mRNA in peripheral blood of case 1 was fivefold lower compared with control individuals without damaging MANF variants (P = 0.02) (Supplementary Fig. 1). These data suggest that both of these case subjects are lacking MANF and likely to represent a human KO of MANF.
Lack of MANF Causes Diabetes, Deafness, Developmental Delay, Microcephaly, and Short Stature
Both case 1 and case 2 present a similar clinical phenotype of childhood onset diabetes, short stature (< −3 SD), bilateral sensorineural deafness, microcephaly, and developmental delay (Fig. 1 and Supplementary Table 2). The diabetes was diagnosed at 10 and 17 years of age, respectively. Case 1 had brief treatment (10 days) with insulin followed by diet to control diabetes for 1.5 years. Both patients are currently on insulin. They did not have autoantibodies associated with type 1 diabetes and have persistence of endogenous insulin secretion for ≥4 years postdiagnosis, with C-peptide levels of 240 pmol/L and 330 pmol/L, respectively. Case 2 also has partial hypopituitarism with deficiency in growth hormone, thyroid-stimulating hormone, and gonadotropins. Hypopituitarism has not been detected in case 1 by 14 years of age. Father of case 1 has a BMI of 29.4 kg/m2 and was diagnosed with type 2 diabetes at 33 years of age. He is currently 41 years old and being treated with metformin. He does not present any other features seen in case 1.
Generation of a MANF KO Stem Cell Model
The homozygous mutations in both case subjects were found in exon 1 of the MANF gene (Fig. 1A and B). Therefore, we created an experimental model for dissecting the role of MANF in human β-cell development and function using CRISPR-Cas9–mediated genome editing to generate a deletion encompassing the first exon of MANF gene in H1 hESC. The genomic DNA sequencing of the generated clone showed the successful deletion of the targeted sequence, which resulted in complete absence of MANF protein as shown by Western blot analysis (Fig. 1C and Supplementary Fig. 2A). The generated clone was karyotypically normal and expressed markers of pluripotency (Supplementary Fig. 2B–D). The KO clone, alongside its wild-type (WT) counterpart, was differentiated into pancreatic islets for further studies.
MANF KO Stem Cells Differentiate Normally to β-Cells
To study the role of MANF in pancreatic development, we differentiated MANF-KO H1 (H1-MANF-KO) and WT H1 hESC into pancreatic islets following an optimized stepwise differentiation protocol (Fig. 2A). Both cell lines differentiated with similar efficiencies into definitive endoderm lineage, in which ∼95% of the cells expressed the definitive endoderm cell surface marker CXCR4 (29) (Fig. 2B and Supplementary Fig. 3A). After 12 days of differentiation (at S4, pancreatic progenitor stage), both cell lines were similarly coexpressing the pancreatic progenitor markers PDX1 and NKX6-1 in ∼60% of the cells (Fig. 2B and Supplementary Fig. 3A). This was further confirmed by their similar levels of PDX1 and NKX6-1 mRNA expression (Fig. 2C). Immunocytochemistry analysis at this stage showed similar expression pattern of pancreatic progenitor and endocrine markers in both cell lines (Fig. 2D). At the end stage (S7 SC-islets and islet endocrine cells), both cell lines generated ∼35% insulin-producing cells (Fig. 2B and Supplementary Fig. 3A) and similar mRNA transcript levels of INS, GCG, and MAFA, a marker of mature β-cells (22) (Fig. 2C). Quantitative immunocytochemistry of these stem cell–derived islets (SC-islets) showed a similar ratio of monohormonal cells (INS+ and GCG+) (Fig. 2D and E). Taken together, these data indicate that MANF deficiency does not alter the differentiation of human pluripotent stem cells into pancreatic β-cells in vitro.
Figure 2.

MANF KO hESC differentiate normally to β-cells. A: Schematic presentation of the seven-stage differentiation protocol (for further details, see research design and methods). B: Graphical presentation of flow cytometry analysis of the differentiating hESC for specific relevant markers of each stage in H1 (WT) and H1-MANF-KO (N = 8 for S1 and S7 cells; N = 7 for S4 cells). For the definitive endoderm stage, CXCR4 was used as a surface marker, PDX1 and NKX6-1 double-positive staining was used as a marker of the pancreatic progenitor population (S4), while NKX6-1 and insulin (INS) double-positive staining was used as a marker of end-stage β-like cells (S7). C: Relative gene expression levels of pancreatic genes analyzed by RT-qPCR in H1 and H1-MANF-KO (N = 4–9). D: Immunocytochemistry analysis of S4 monolayer cells for PDX1, NKX6-1, chromogranin A (CHGA), and insulin (INS) (scale bars = 100 μm) and S7 aggregates for INS, glucagon (GCG), and the nuclear stain Hoechst (N = 6) (scale bars = 100 μm). E: Quantification of the percentage of INS-positive and GCG-positive cells in H1 and H1-MANF-KO SC-islets. H1 is presented as blue circles, and H1-MANF-KO is presented as red circles. Data are presented as mean ± SEM.
MANF KO Sensitizes β-Cells to ER Stress
To assess the effect of MANF-KO on the expression levels of ER stress markers, we used immunocytochemistry on S7 SC-islets. MANF-KO β-like cells showed marginally higher levels of BiP when compared with WT (Fig. 3A and B). This might indicate a higher level of ER stress in the MANF-KO β-like cells, which could be associated with the higher accumulation of proinsulin in these cells (Fig. 3A and B). Interestingly, MANF-KO cells displayed a significantly higher number of KI67+ proliferative β-like cells when compared with the WT (Fig. 3A and B). Furthermore, we confirmed our findings by quantifying the mRNA levels of ER stress markers using RT-qPCR, which showed an increasing trend in the expression levels of BiP (HSPA5), GRP170 (HYOU1), and TXNIP in MANF-KO cells (Fig. 3C and Supplementary Fig. 3B). However, both genotypes showed similar very low basal mRNA transcript levels of the inflammatory markers tumor necrosis factor-α, CXCL-10, interleukin-6, and interferon-α (data not shown).
Figure 3.

MANF KO β-cells tend to have induced ER stress and significantly reduced insulin-processing capacity. A: Immunocytochemistry analysis of H1 and H1-MANF-KO S7 aggregates for β-like cells (INS+), which express ER stress marker BiP (N = 5), proinsulin (PROINS) (N = 6), and proliferation marker (Ki67) (N = 8) (scale bars = 20 μmol/L). B: Quantified relative amounts of BiP, proinsulin, and Ki67 positive β-cells from A. C: Relative gene expression levels of ER stress genes analyzed by RT-qPCR in H1 and H1-MANF-KO (N = 4–9). D: ER stress–induced apoptosis of S7 aggregates. Quantification of the percentage of insulin-positive cells, which are labeled by TUNEL assay after treating the aggregates with the following chemical ER stress inducers: TM, TG (N = 3), and brefeldin A (BREF) (N = 5). DMSO was used as a vehicle control. E: Static glucose-stimulated insulin secretion of S7 SC-islets. Values are presented as fractional secretion of human insulin (hINS) over total insulin content. (N = 6). F: hINS content of S7 SC-islets. Cell mass is normalized by average DNA content of each aggregate (N = 6). G: Ratio of human proinsulin (hPROINS) content to hINS content per aggregate (N = 4 for H1 and 3 for H1-MANF-KO). H1 is presented as blue circles, and H1-MANF-KO is presented as red circles. Data are presented as mean ± SEM. *P < 0.05; ***P < 0.0001.
To evaluate the role of MANF in β-cell survival, we quantified apoptotic cells using the TUNEL assay on S7 SC-islets treated with different ER stressors. In the basal condition, ∼1.5% of the insulin-positive cells were TUNEL+ in both MANF-KO and WT (Fig. 3D). Upon the induction of experimental ER stress using TM, an inhibitor of N-linked glycosylation, or TG, which depletes ER calcium (30), the percentage of apoptotic β-like cells increased similarly in both genotypes (∼8% with TM, and 5% with TG). However, upon the treatment with brefeldin A, an inhibitor of ER-to-Golgi cargo transfer (31), the percentage of MANF-KO apoptotic β-like cells was significantly higher than WT (29% vs. 18%) (Fig. 3D and Supplementary Fig. 3C). These findings suggest that depletion of MANF results in mildly increased levels of ER stress in stem cell–derived β-cells in vitro, making them more sensitive to additional ER stress.
Impaired Insulin Processing in MANF KO β-Cells
Next, we examined the functionality of the SC-islets by performing static insulin secretion tests. Both genotypes responded to high glucose with a twofold increase in human insulin secretion (Fig. 3E and Supplementary Fig. 3D). Additionally, KCl induced a fourfold increase of insulin release in both genotypes, indicating normal insulin secretion response to depolarization (Fig. 3E). Although both genotypes showed a similar pattern of insulin secretion, the absolute amount of insulin secreted by the MANF-KO SC-islets was lower, and their insulin content was significantly lower than in WT (Fig. 3F). Additionally, MANF-KO cells showed a significantly higher ratio of proinsulin to insulin content (Fig. 3G), even though proinsulin content was similar (Supplementary Fig. 3E). Thus, MANF-KO had a minor effect on β-cell function but a significant effect on the insulin- processing capacity of the β-cells in vitro.
Impaired Function of MANF KO β-Cells after Implantation
To evaluate the functionality of SC-islets in vivo, we implanted MANF-KO and WT SC-islets under the kidney capsule of immunocompromised (NOD-SCID-γ) mice. We followed the functionality of the grafted cells for 6 months by measuring the circulating glucose and human C-peptide levels (Fig. 4A). Blood glucose levels of the mice implanted with both MANF-KO and WT cells decreased over time from usual normoglycemic levels for mice (8 mmol/L) to human levels (4 mmol/L) by 3 months, suggesting that the grafted cells were able to efficiently control the blood glucose of transplanted mice (Fig. 4B). This was consistent with a gradual increase in the levels of circulating human C-peptide in randomly fed mice, reaching up to 2,800 pmol/L after 3 months (Fig. 4C). At 4 months postimplantation, the levels of C-peptide in fasted mice were significantly higher in mice implanted with WT cells compared with MANF-KO (Fig. 4C).
Figure 4.
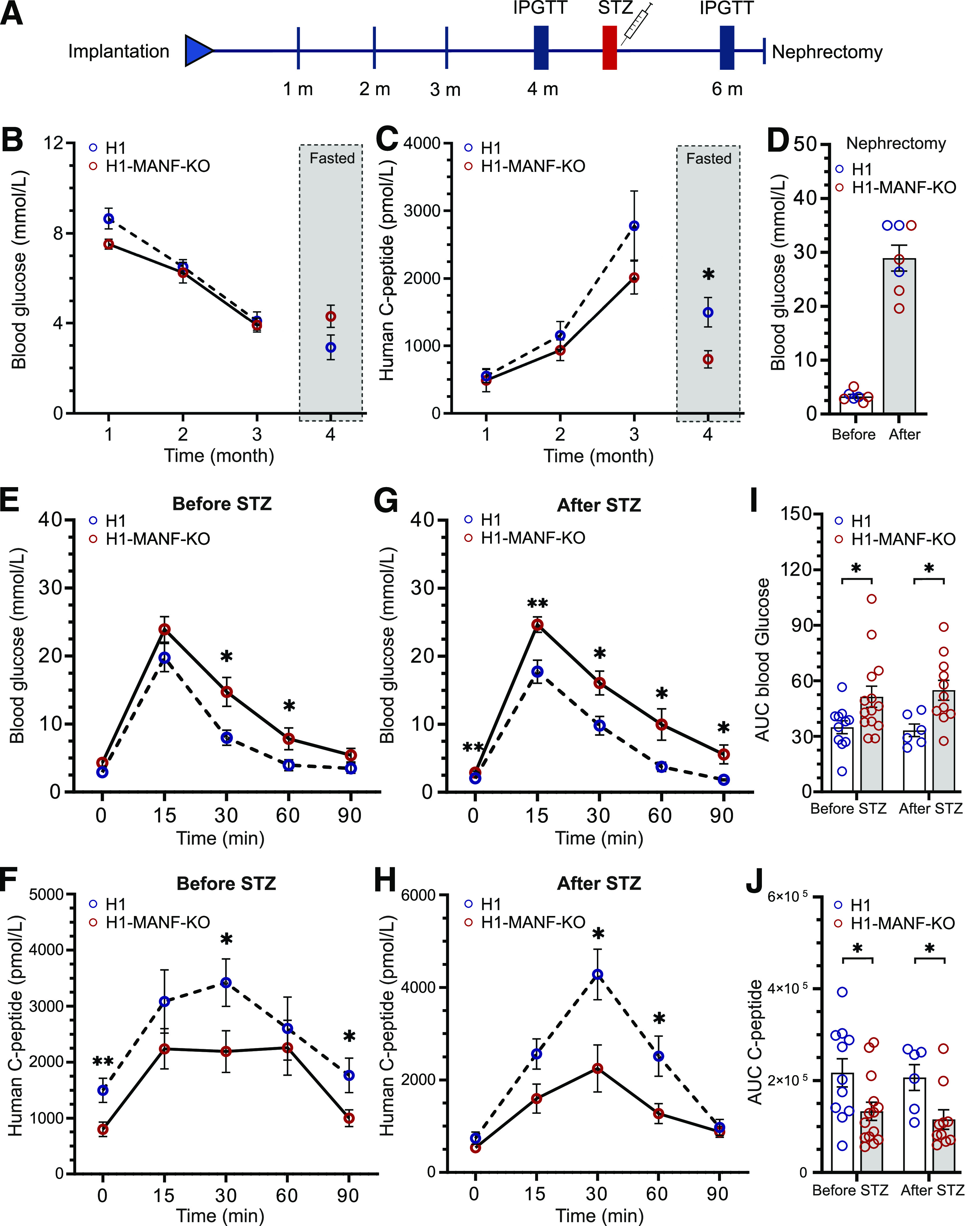
Impaired response to glucose of MANF KO cells in vivo. A: Schematic presentation of the in vivo workflow. B: Monthly follow-up of H1 and H1-MANF-KO in vivo functionality by measuring the blood glucose levels of implanted mice. Measurements of the first 3 months were done on randomly fed mice, while for the last month, mice were fasted (N = 11 for H1 and 14 for H1-MANF-KO). C: Monthly follow-up of H1 and H1-MANF-KO in vivo functionality by measuring the circulating human C-peptide levels of implanted mice. Measurements were done on randomly fed mice for the first 3 months, while for the last month, mice were fasted (N = 11 for H1 and 14 for H1-MANF-KO). D: Blood glucose measurements before and after removing the implanted kidney of STZ-induced diabetic mice by surgical unilateral nephrectomy (N = 7). E: Blood glucose measurements during an IPGTT of fasted H1- and H1-MANF-KO–implanted mice before STZ treatment (N = 11 for H1 and 14 for H1-MANF-KO). F: Levels of circulating human C-peptide during an IPGTT of fasted H1- and H1-MANF-KO–implanted mice before STZ treatment (N = 11 for H1 and 14 for H1-MANF-KO). G: Blood glucose measurements during an IPGTT of fasted H1- and H1-MANF-KO–implanted mice after STZ treatment (N = 6 for H1 and 11 for H1-MANF-KO). H: Levels of circulating human C-peptide during an IPGTT of fasted H1- and H1-MANF-KO–implanted mice after STZ treatment (N = 6 for H1 and 11 for H1-MANF-KO). I: Quantification of the efficiency of the implanted grafts to regulate the mice blood glucose during IPGTT by measuring the area under the curve (AUC) of E and G. J: Quantification of the ability of the implanted grafts to secrete human C-peptide by measuring the AUC of F and H. H1 is presented as blue circles, and H1-MANF-KO is presented as red circles. Data are presented as mean ± SEM. *P < 0.05; **P < 0.001. m, months.
Four months after implantation, the mice were challenged with an intraperitoneal glucose tolerance test (IPGTT). Blood glucose excursions were significantly higher in mice transplanted with MANF-KO grafts (Fig. 4E and I). This was associated with lower secretion of human C-peptide (Fig. 4F and J). The glucose tolerance test was repeated after eradication of mouse β-cells using a single high dose of STZ. The mice carrying WT grafts maintained normal glucose tolerance and high human C-peptide secretion in this test. However, the mice transplanted with MANF-KO grafts showed reduced glucose clearance (Fig. 4G and I) and a substantially lower increase in human C-peptide secretion (area under the curve; P = 0.02) (Fig. 4H and J). To confirm the functionality of the grafts and the success of the STZ treatment in eradicating the mouse β-cells, grafts were retrieved after 6 months by unilateral nephrectomy, which resulted in immediate hyperglycemia in all mice (Fig. 4D). Taken together, these data show impaired functionality of implanted MANF-KO β-cells.
Elevated ER Stress and Proinsulin Accumulation in MANF KO Grafts
To further evaluate the effect of MANF-KO on the implanted SC-islets, grafts were retrieved at 1, 2, 4, and 6 months and examined by immunohistochemistry. Staining for INS and GCG in 6-month-old grafts showed no differences in the proportions of β- and α cells between the genotypes, further indicating that MANF depletion did not affect pancreatic endocrine differentiation (Fig. 5A and B). The percentage of INS+ cells in the grafts increased gradually with time (Supplementary Fig. 4E), while the number of proliferating β-cells gradually decreased (Supplementary Fig. 4C and D). Interestingly, MANF-KO β-cells showed significantly higher levels of proinsulin (50% PROINSHI+INS+/INS+) compared with their WT counterparts (35% PROINSHI+INS+/INS+) in the 6-month-old grafts (Fig. 5C and D). A similar pattern was observed in the grafts retrieved at 2, and 4 months, suggesting an important defect in proinsulin processing in the ER (Supplementary Fig. 4G). In addition, immunohistochemistry analysis for ER stress markers in 6-month grafts revealed significantly higher levels of the ER stress markers BiP (Fig. 5E and F and Supplementary Fig. 4H) and GRP170 (Supplementary Fig. 4A and B) in MANF-KO grafts compared with the WT grafts (23.3% vs. 15.4% BiPHI+INS+/INS+ cells and 27.9% vs. 10.2% GRP170HI+INS+/INS+ cells). Finally, TUNEL assay for apoptotic INS+ cells revealed an absence of β-cell death in the grafts of both genotypes (Supplementary Fig. 4F).
Figure 5.

Increased ER stress and accumulation of proinsulin (PROINS) in MANF KO grafts. A: Immunohistochemistry analysis of 6-month-old grafts for insulin (INS) and glucagon (GCG) (scale bars = 50 μmol/L). B: Quantification of the percentage of INS- and GCG-positive cells from A (N = 6 for H1 and 9 for H1-MANF-KO). C: Immunohistochemistry analysis of 6-month-old grafts for INS and PROINS (scale bars = 50 μmol/L). D: Quantification of the relative number of INS- and PROINS-positive cells from C (N = 6 for H1 and 9 for H1-MANF-KO). E: Immunohistochemistry analysis of 6-month-old grafts for INS cells, which are expressing the ER stress marker BiP (scale bar = 50 μmol/L). F: Quantification of the relative number of BiP-positive β-cells from E (N = 6 for H1 and 4 for H1-MANF-KO). H1 is presented as blue circles, and H1-MANF-KO is presented as red circles. Data are presented as mean ± SEM. **P < 0.001.
Discussion
In this study, we report MANF homozygous loss-of-function mutations as a novel genetic cause of childhood onset diabetes, deafness, developmental delay, microcephaly, and short stature. We used differentiation of hESC lacking MANF to the pancreatic islets to model the consequences of these genetic defects. This revealed the crucial role of MANF in mitigating β-cell ER stress and how its absence leads to impaired proinsulin processing and impaired β-cell function.
Strong genetic, clinical, and functional data suggest that lack of MANF is indeed the cause of diabetes in these patients. The childhood progressive diabetes is unlikely to be type 1 diabetes as they lacked islet autoantibodies, had low genetic risk of type 1 diabetes, and had significant residual C-peptide 4 years after diagnosis (32). Both patients with different homozygous mutations have strikingly similar extra pancreatic phenotypes with microcephaly, developmental delay, sensorineural deafness, and short stature. The brain is sensitive to ER stress–mediated damage and almost always affected along with pancreas in other ER stress–mediated monogenic diabetes forms (33). This strongly suggests that MANF deficiency is etiological also for these features.
Both global- and β-cell–specific loss of MANF results in severe early onset diabetes in transgenic mice (6,34). The two patients identified in this study were diagnosed with diabetes at 10 and 17 years of age, suggesting a slower progression of the β-cell failure in humans. In our MANF-KO hESC model, obvious signs of severe ER stress and functional deterioration were only evident in the in vivo model when the stem cell–derived β-cells became responsible for the insulin secretion of the host with diabetes, suggesting again that loss of MANF leads to progressively impaired β-cell function and diabetes.
MANF-KO mice present neurological phenotypes that resemble those in the human MANF-KO patients. MANF inactivation in the mouse inner ear leads to a progressive death of outer hair cells of the cochlea accompanied with hearing loss (35). Similarly to the patients, MANF-KO mice show a growth defect and short stature that is independent of diabetes (6).
The ER stress response is a crucial adaptive pathway for β-cell function and survival. ER stress can be triggered by the accumulation of misfolded protein that is unable to exit the ER, as in the case of the dominant INS mutations causing severe forms of diabetes in Akita mice and humans (23,36). Pathogenic mutations in genes involved in the UPR also lead to neonatal diabetes, as in the case of EIF2AK3 mutations in Wolcott-Rallison syndrome (37), IER3IP1 mutations (38), or YIPF5 mutations (39). Childhood-onset diabetes has been described in individuals carrying WFS1 and CISD2 mutations, leading to Wolfram syndrome (1), and recessive mutations in the DNAJC3 (40) and PPP1R15B genes (41). Indeed, the MANF syndrome clinical features of diabetes, microcephaly, and other neurological features are overlapping with those seen in patients with the above mutations (33,42). Thus, homozygous MANF mutations constitute a novel form of childhood-onset diabetes caused by dysregulated ER stress, and therefore, MANF should be included in the gene panels testing for monogenic diabetes.
Similar to Wolfram syndrome, the MANF syndrome has childhood-onset diabetes with progressive β-cell dysfunction, developmental delay, and deafness. However, it lacks optic atrophy, diabetes insipidus, and urological defects that are common in Wolfram syndrome. Furthermore, the short stature and microcephaly seen in MANF syndrome are not reported in Wolfram syndrome. The difference in the clinical features indicates some differences in the pathogenesis. Further work with additional patients and animal models will be needed to fully understand these differences.
High expression of ER stress markers in human adult β-cells has been linked to higher tendency of proliferation (43). Additionally, induction of mild ER stress by hyperglycemia has been reported to induce β-cell proliferation (44,45). Conversely, subthreshold chronic ER stress has been shown to inhibit the proliferation of β-cells (46). We observed a similar pattern in our model in which the in vitro mild ER stress significantly induced the proliferation of MANF-KO β-cells, while after implantation and exposure to more severe ER stress, there was a trend of decreased proliferation (Fig. 3 and Supplementary Fig. 4D).
As a compensatory response to ER stress, UPR tends to attenuate protein translation to decrease the folding load on the ER (8). This may result in decreased insulin synthesis and processing capacity in ER-stressed β-cells. A previous study on Wolfram syndrome, which is caused by ER dysfunction (47), reported that although the low levels of UPR signaling decreased the insulin content of β-cells, it did not alter their glucose-stimulated insulin secretion in vitro (48). These results are well in line with the findings of our study, since we can detect a lower level of insulin content and secretion in the MANF-KO β-cells compared with WT cells, while the insulin response to glucose stimulation was preserved. Additionally, the significantly higher ratio of proinsulin to insulin content in the MANF-KO cells, observed both in vitro and in vivo, indicates impaired insulin-processing capacity, also reported in human prediabetes (49,50).
In conclusion, our findings indicate the essential role of MANF in protecting β-cells from ER stress–induced failure and apoptosis in humans. Our study also highlights the potential of the modeling platform, which enables detailed studies of isogenic stem cell–derived β-cells both in vitro and in vivo. The mild phenotype observed in vitro, representing an immature β-cell population, followed by an exacerbated phenotype in more actively functioning β-cells in vivo, is likely to recapitulate the pathogenic evolution of the diabetic phenotype in patients with MANF mutation.
Article Information
Acknowledgments. The authors thank the families for participating in the study. The authors also thank Solja Eurola, Jarkko Ustinov, Heli Grym, and Anni Laitinen (Stem Cells and Metabolism Research Program in the Faculty of Medicine of the University of Helsinki, Finland) for expert technical assistance and Rebecca Ward (Institute of Biomedical and Clinical Science, College of Medicine and Health, University of Exeter, UK) for help with Sanger sequencing.
Funding. The genetic studies were funded by Wellcome Trust awarded to K.A.P. (grant 110082/Z/15/Z). K.A.P. is a Wellcome Trust Fellow (grant 219606/Z/19/Z). A.T.H. is a Wellcome Trust Senior Investigator (WT098395/Z/12/Z) and is also supported by a National Institute for Health Research (NIHR) (Senior Investigator award). The NIHR Exeter Clinical Research Facility is a partnership among the University of Exeter Medical School, College of Medicine and Health, and Royal Devon and Exeter NHS Foundation Trust. The experimental studies were funded by the Academy of Finland (grant 297466 and MetaStem Center of Excellence grant 312437), the Sigrid Jusélius Foundation, the Novo Nordisk Foundation Center for Basic Metabolic Research, and JDRF (grant 2-SRA-2018-496-A-B, principal investigator Mart Saarma, co-principal investigator T.O.). H.M. is a member of the Doctoral Program in Integrative Life Science at University of Helsinki.
The views expressed are those of the authors and not necessarily those of the NIHR, Wellcome Trust, or the Department of Health and Social Care, U.K.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. H.M., K.A.P., D.B., A.T.H., J.S.-V., and T.O. conceived the project and planned the experiments. K.A.P., K.D., T.B.-O., K.C., J.M.L., and M.W. analyzed the genetic and clinical data. M.W. developed the genome sequencing and homozygosity mapping pipelines. D.B. performed genome editing and edited the manuscript. H.I., V.L., A.N., and J.S.-V. assisted in data acquisitions. M.L. and V.C. edited the manuscript. H.M. performed SC-islet differentiation, acquired, and analyzed the data and wrote the related first draft of the manuscript. K.A.P. identified the causative gene in the patients and wrote the related first draft of the manuscript. All authors reviewed and edited the manuscript. T.O. is the guarantor of this work and, as such, had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Prior Presentation. Preliminary results from this study were presented at the 55th Annual Meeting of the European Association for the Study of Diabetes, Barcelona, Spain, 16–20 September 2019.
Footnotes
This article contains supplementary material online at https://doi.org/10.2337/figshare.13622993.
H.M. and K.A.P. contributed equally to this work.
J.S.-V. and T.O. contributed equally to this work.
References
- 1.Vilan A, Faria O, Campos MM. [Wolfram syndrome]. Rev Neurol 2009;49:221–222 [in Spanish] [PubMed] [Google Scholar]
- 2.Labay V, Raz T, Baron D, et al. Mutations in SLC19A2 cause thiamine-responsive megaloblastic anaemia associated with diabetes mellitus and deafness. Nat Genet 1999;22:300–304 [DOI] [PubMed] [Google Scholar]
- 3.Noavar S, Behroozi S, Tatarcheh T, Parvini F, Foroutan M, Fahimi H. A novel homozygous frame-shift mutation in the SLC29A3 gene: a new case report and review of literature. BMC Med Genet 2019;20:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fareed M, Afzal M. Genetics of consanguinity and inbreeding in health and disease. Ann Hum Biol 2017;44:99–107 [DOI] [PubMed] [Google Scholar]
- 5.Lindahl M, Saarma M, Lindholm P. Unconventional neurotrophic factors CDNF and MANF: structure, physiological functions and therapeutic potential. Neurobiol Dis 2017;97:90–102 [DOI] [PubMed] [Google Scholar]
- 6.Lindahl M, Danilova T, Palm E, et al. MANF is indispensable for the proliferation and survival of pancreatic β cells. Cell Rep 2014;7:366–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fonseca SG, Gromada J, Urano F. Endoplasmic reticulum stress and pancreatic β-cell death. Trends Endocrinol Metab 2011;22:266–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007;8:519–529 [DOI] [PubMed] [Google Scholar]
- 9.Oyadomari S, Koizumi A, Takeda K, et al. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest 2002;109:525–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev 2008; 29:42–61 [DOI] [PubMed] [Google Scholar]
- 11.Koc I. Prevalence and sociodemographic correlates of consanguineous marriages in Turkey. J Biosoc Sci 2008;40:137–148 [DOI] [PubMed] [Google Scholar]
- 12.Patel KA, Oram RA, Flanagan SE, et al. Type 1 diabetes genetic risk score: a novel tool to discriminate monogenic and type 1 diabetes. Diabetes 2016;65:2094–2099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellard S, Lango Allen H, De Franco E, et al. Improved genetic testing for monogenic diabetes using targeted next-generation sequencing. Diabetologia 2013;56:1958–1963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Richards S, Aziz N, Bale S, et al.; ACMG Laboratory Quality Assurance Committee . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laver TW, De Franco E, Johnson MB, et al. SavvyCNV: genome-wide CNV calling from off-target reads. bioRxiv. 13 June 2019 [preprint]. DOI: 10.1101/617605 [Google Scholar]
- 16.Rivas MA, Pirinen M, Conrad DF, et al.; GTEx Consortium; Geuvadis Consortium . Human genomics. Effect of predicted protein-truncating genetic variants on the human transcriptome. Science 2015;348:666–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karczewski KJ, Francioli LC, Tiao G, et al.; Genome Aggregation Database Consortium . The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581:434–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scott EM, Halees A, Itan Y, et al. Characterization of greater middle eastern genetic variation for enhanced disease gene discovery. Nat Genet 2016;48:1071–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Narasimhan VM, Hunt KA, Mason D, et al. Health and population effects of rare gene knockouts in adult humans with related parents. Science 2016;352:474–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat 2015;36:928–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pagliuca FW, Millman JR, Gürtler M, et al. Generation of functional human pancreatic β cells in vitro. Cell 2014;159:428–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rezania A, Bruin JE, Arora P, et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol 2014;32:1121–1133 [DOI] [PubMed] [Google Scholar]
- 23.Balboa D, Saarimäki-Vire J, Borshagovski D, et al. Insulin mutations impair beta-cell development in a patient-derived iPSC model of neonatal diabetes. eLife 2018;7:e38519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saarimäki-Vire J, Balboa D, Russell MA, et al. An activating STAT3 mutation causes neonatal diabetes through premature induction of pancreatic differentiation. Cell Rep 2017;19:281–294 [DOI] [PubMed] [Google Scholar]
- 25.McQuin C, Goodman A, Chernyshev V, et al. CellProfiler 3.0: next-generation image processing for biology. PLoS Biol 2018;16:e2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rueden CT, Schindelin J, Hiner MC, et al. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 2017;18:529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yavarna T, Al-Dewik N, Al-Mureikhi M, et al. High diagnostic yield of clinical exome sequencing in Middle Eastern patients with Mendelian disorders. Hum Genet 2015;134:967–980 [DOI] [PubMed] [Google Scholar]
- 28.Khajavi M, Inoue K, Lupski JR. Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur J Hum Genet 2006;14:1074–1081 [DOI] [PubMed] [Google Scholar]
- 29.D’Amour KA, Agulnick AD, Eliazer S, Kelly OG, Kroon E, Baetge EE. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat Biotechnol 2005;23:1534–1541 [DOI] [PubMed] [Google Scholar]
- 30.Abdullahi A, Stanojcic M, Parousis A, Patsouris D, Jeschke MG. Modeling acute ER stress in vivo and in vitro. Shock 2017;47:506–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tran K, Li Y, Duan H, Arora D, Lim HY, Wang W. Identification of small molecules that protect pancreatic β cells against endoplasmic reticulum stress-induced cell death. ACS Chem Biol 2014;9:2796–2806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones AG, Hattersley AT. The clinical utility of C-peptide measurement in the care of patients with diabetes. Diabet Med 2013;30:803–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cnop M, Toivonen S, Igoillo-Esteve M, Salpea P. Endoplasmic reticulum stress and eIF2α phosphorylation: the Achilles heel of pancreatic β cells. Mol Metab 2017;6:1024–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Danilova T, Belevich I, Li H, et al. MANF is required for the postnatal expansion and maintenance of pancreatic β-cell mass in mice. Diabetes 2019;68:66–80 [DOI] [PubMed] [Google Scholar]
- 35.Herranen A, Ikäheimo K, Lankinen T, et al. Deficiency of the ER-stress-regulator MANF triggers progressive outer hair cell death and hearing loss. Cell Death Dis 2020;11:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mizobuchi N, Hoseki J, Kubota H, et al. ARMET is a soluble ER protein induced by the unfolded protein response via ERSE-II element. Cell Struct Funct 2007;32:41–50 [DOI] [PubMed] [Google Scholar]
- 37.Delépine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, Julier C. EIF2AK3, encoding translation initiation factor 2-α kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat Genet 2000;25:406–409 [DOI] [PubMed] [Google Scholar]
- 38.Shalev SA, Tenenbaum-Rakover Y, Horovitz Y, et al. Microcephaly, epilepsy, and neonatal diabetes due to compound heterozygous mutations in IER3IP1: insights into the natural history of a rare disorder. Pediatr Diabetes 2014;15:252–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Franco E, Lytrivi M, Ibrahim H, et al. YIPF5 mutations cause neonatal diabetes and microcephaly through endoplasmic reticulum stress. J Clin Invest 2020;130:6338–6353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Synofzik M, Haack TB, Kopajtich R, et al. Absence of BiP co-chaperone DNAJC3 causes diabetes mellitus and multisystemic neurodegeneration. Am J Hum Genet 2014;95:689–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abdulkarim B, Nicolino M, Igoillo-Esteve M, et al. A missense mutation in PPP1R15B causes a syndrome including diabetes, short stature, and microcephaly. Diabetes 2015;64:3951–3962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stone SI, Abreu D, McGill JB, Urano F. Monogenic and syndromic diabetes due to endoplasmic reticulum stress. J Diabetes Complications 2021;35:107618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xin Y, Dominguez Gutierrez G, Okamoto H, et al. Pseudotime ordering of single human β-cells reveals states of insulin production and unfolded protein response. Diabetes 2018;67:1783–1794 [DOI] [PubMed] [Google Scholar]
- 44.Porat S, Weinberg-Corem N, Tornovsky-Babaey S, et al. Control of pancreatic β cell regeneration by glucose metabolism. Cell Metab 2011;13:440–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sharma RB, O’Donnell AC, Stamateris RE, et al. Insulin demand regulates β cell number via the unfolded protein response. J Clin Invest 2015;125:3831–3846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Szabat M, Page MM, Panzhinskiy E, et al. Reduced insulin production relieves endoplasmic reticulum stress and induces β cell proliferation. Cell Metab 2016;23:179–193 [DOI] [PubMed] [Google Scholar]
- 47.Riggs AC, Bernal-Mizrachi E, Ohsugi M, et al. Mice conditionally lacking the Wolfram gene in pancreatic islet beta cells exhibit diabetes as a result of enhanced endoplasmic reticulum stress and apoptosis. Diabetologia 2005;48:2313–2321 [DOI] [PubMed] [Google Scholar]
- 48.Shang L, Hua H, Foo K, et al. β-cell dysfunction due to increased ER stress in a stem cell model of Wolfram syndrome. Diabetes 2014;63:923–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mykkänen L, Haffner SM, Kuusisto J, Pyörälä K, Hales CN, Laakso M. Serum proinsulin levels are disproportionately increased in elderly prediabetic subjects. Diabetologia 1995;38:1176–1182 [DOI] [PubMed] [Google Scholar]
- 50.Rodriguez-Calvo T, Zapardiel-Gonzalo J, Amirian N, et al. Increase in pancreatic proinsulin and preservation of β-cell mass in autoantibody-positive donors prior to type 1 diabetes onset. Diabetes 2017;66:1334–1345 [DOI] [PMC free article] [PubMed] [Google Scholar]