

Abstract
Ubiquitylation, a wide-spread post-translational protein modification in eukaryotes, marks cytosol‐invading bacteria as cargo for anti-bacterial autophagy.1–3 The identity of the ubiquitylated substrate on bacteria has remained unknown. Here we show that the ubiquitin coat on cytosol-invading Salmonella is formed through the unprecedented ubiquitylation of a non-proteinaceous substrate, the lipid A moiety of bacterial lipopolysaccharide (LPS), by the E3 ubiquitin ligase RNF213. RNF213 is a risk factor for Moyamoya disease (MMD)4,5, a progressive stenosis of the supraclinoid internal carotid artery that causes stroke, especially in children.6,7 RNF213 restricts the proliferation of cytosolic Salmonella and is essential for the generation of the bacterial ubiquitin coat, both directly, through ubiquitylation of LPS, and indirectly, through recruitment of LUBAC, a downstream E3 ligase that adds M1-linked ubiquitin chains onto pre-existing ubiquitin coats.8 In cells lacking RNF213 bacteria do not attract ubiquitin-dependent autophagy cargo receptors and fail to induce anti-bacterial autophagy. The ubiquitylation of LPS on cytosol-invading Salmonella requires the dynein-like core of RNF213 but not its RING domain. Instead, LPS ubiquitylation relies on an RZ finger in the E3 shell. We conclude that ubiquitylation extends beyond protein substrates, that LPS ubiquitylation triggers cell-autonomous immunity and we postulate that non- proteinaceous substances other than LPS may also become ubiquitylated.
The lifestyle of intracellular bacteria shields them from many canonical immune mechanisms while exposing them to a variety of cell-autonomous defences that are unique to individual subcellular compartments.9 Bacteria attempting to colonize the cytosol are restricted through selective macroautophagy (‘xenophagy’) in their ability to establish infections.3,10,11 Xenophagy is directed towards two types of ‘eat-me’ signals associated with cytosol-invading bacteria, galectin-8 and poly-ubiquitin.2,10,12–14 Galectin-8, a cytosolic lectin, binds glycans exposed on damaged host membranes, and accumulates near cytosol-invading bacteria during their escape from phagosomes.12,15 The bacterial ubiquitin coat is deposited by several host E3 ubiquitin ligases including LRSAM116, Parkin17, LUBAC8,18 and SMURF119, whose specific contributions to cell-autonomous immunity remain largely unknown. Of these, LUBAC, requires pre-existing ubiquitin for its own recruitment, indicating that cascades of E3 enzymes generate and shape the ubiquitin coat.8 The ubiquitinome of infected cells includes bacterial outer membrane proteins but whether the formation of the bacterial ubiquitin coat requires the ubiquitylation of specific bacterial substrates remains to be established.20,21
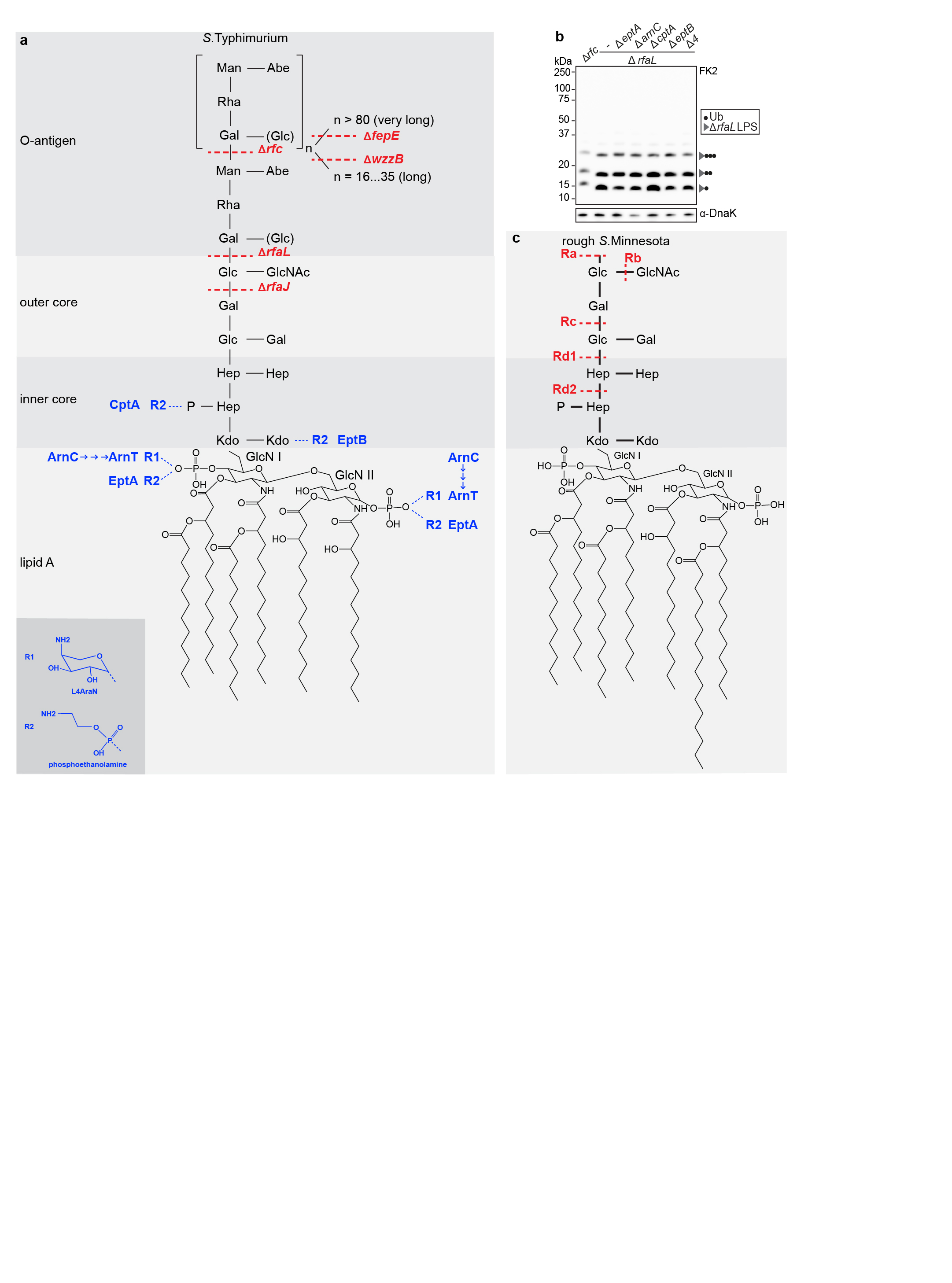
We deployed structured illumination microscopy (SIM), a superresolution technique, to study the ubiquitin coat of Salmonella enterica serovar Typhimurium (S.Typhimurium), a Gram‐negative bacterium that causes gastroenteritis in humans. Upon host cell invasion S.Typhimurium resides in Salmonella-containing vacuoles, from which a fraction of bacteria escapes into the cytosol. We observed that at 1h post infection (p.i.) poly-ubiquitin colocalized with galectin-8+ve remnants of Salmonella-containing vacuoles while at 4h p.i., when galectin- 8+ve membranes are no longer detected, poly-ubiquitin was associated with the bacterial surface (Fig.1a). Bacteria therefore become sequentially associated with poly-ubiquitin that resides either on damaged host membranes or the bacterium itself. To characterize the ubiquitin on the bacterial surface, we isolated bacteria from infected cells at 4h p.i. (Fig.1b). Immunoblotting with FK2, an antibody specific for conjugated ubiquitin, revealed a ubiquitin smear above 50kDa in wild type bacteria while S.Typhimurium Drfc, lacking the O-antigen polymerase required to synthesize smooth lipopolysaccharide (LPS)22 (Extended Data Fig.1a), carried distinct ubiquitylated products of low molecular weight (LMW), in addition to a faint ubiquitin smear above 100kDa. The distinct banding pattern in S.Typhimurium Drfc revealed the existence of a prominent oligo-ubiquitylated substrate in the bacterial ubiquitin coat, which, judged by the size of the smallest band (15kDa), has an apparent molecular weight of circa 7kDa. Ubiquitylation of LPS rather than a protein substrate explains the observed phenotypes: semi-rough LPS in S.Typhimurium Drfc has a matching molecular weight and the variable length of O-antigen chains in wild type bacteria converts the ubiquitylated LMW bands into a higher molecular weight smear. To differentiate protein from non-protein ubiquitylation we boiled bacterial lysates prepared in Bugbuster, a lysis reagent that supports native protein conformation, which eliminated the highest molecular weight ubiquitin smear both from wild type and mutant bacteria, indicative of protein ubiquitylation, while the distinct ubiquitin bands in S.Typhimurium Drfc and the majority of the ubiquitin smear in wild type bacteria were heat resistant, consistent with LPS ubiquitylation (Fig.1b). To further verify the existence of ubiquitylated LPS (Ub-LPS) we generated additional Salmonella mutants deficient in distinct steps of LPS biosynthesis, predicting that alterations in the LPS structure will cause corresponding changes in the ubiquitylation pattern (Fig.1c). S.Typhimurium DrfaL and Drfc, deficient in O-antigen ligase and polymerase, respectively, produce uniquely sized LPS lacking all O-antigen or displaying one O-antigen subunit only.22,23 When isolated from cells, S.Typhimurium DrfaL and Drfc carried ubiquitylated products that accurately matched the size difference of their respective LPS. Wild type bacteria produce O- antigens of three different sizes, namely long and very long chains, whose synthesis requires the O-antigen length regulators WzzB and FepE, respectively, and shorter chains synthesized by the O-antigen polymerase Rfc when not engaged by WzzB.22,23 In wild type bacteria, LPS with very long O-antigen chains was not ubiquitylated. Thus as predicted, ubiquitylation of S.Typhimurium DfepE was indistinguishable from wild type bacteria. In contrast, S.Typhimurium DwzzB, lacking LPS with long O-antigen chains but still producing shorter chains through apo-Rfc, carried ubiquitylated products of correspondingly reduced size. Finally, to confirm the specificity of FK2 for ubiquitin, we i) incubated S.Typhimurium Drfc extracted from cells with the deubiquitylating enzyme USP2, which depleted the characteristic Ub-LPS pattern (Fig.1d) and ii) isolated S.Typhimurium Drfc from cells expressing HA-tagged ubiquitin, which reproduced the characteristic LMW band pattern when probed with anti-HA antibody (Fig.1e). Ubiquitin lacking the C-terminal di-Gly motif was not conjugated. Taken together, our results demonstrate the ubiquitylation of LPS on cytosol-invading bacteria, a finding that extends the scope of ubiquitylation beyond a post-translational protein modification, to include an entirely novel class of biomolecules.
Figure 1. Ubiquitylation of lipopolysaccharide (LPS).

a, Structured illumination micrograph of HeLa cells at 1 h and 4 h p.i. with S. Typhimurium and immunostained for LPS, FK2 and Galectin-8. Scale bar, 3 pm. Lines indicate the plot profiles adjacent. b-f, Immunoblot analysis of the indicated S. Typhimurium strains, extracted from HeLa cells (b,c,d,f) or MEFs stably expressing HA-Ubiquitin or HA-Ubiquitin AGG (e). GroEL, loading controls for bacterial lysates. b, Bacterial lysates left at room temperature (RT) or incubated at 90°C for 15 minutes. c, right panel, Emerald300 stain of LPS extracted from indicated S. Typhimurium strains grown in Luria Broth (LB). d, f, S. Typhimurium Arfc extracted from HeLa cells 4 h p.i. and treated with 0 nM, 10 nM or 2 pM recombinant USP2 catalytic domain for 30 min (d) or lysed and incubated with 100 or 200 mM NaOH for 20 min (f). K48 Ub2 provides an amide-linked control. a-f, Representative of 3 biological repeats. For gel source data, see Supplementary Fig. 1.
Protein ubiquitylation targets primary amino and, occasionally, also hydroxy groups, resulting in amide‐ or ester-linked conjugates, respectively. The constitutive LPS core does not contain amino groups suitable for ubiquitylation. However, substoichiometric modifications triggered by environmental cues can introduce such moieties in the form of 4-amino-L-arabinose or phosphoethanolamine. To test whether LPS ubiquitylation requires substoichiometric amino groups in the LPS core, we deleted arnC, the undecaprenyl-phosphate 4-deoxy-4-formamido- L-arabinose transferase required to generate 4-amino-L-arabinose, and the phosphoethanolamine transferases eptA, eptB and cptA from S. Typhimurium DrfaL. LPS ubiquitylation proceeded unimpaired in single mutants as well as the pentuple knockout (S.Typhimurium ∆rfaL, ∆arnC, ∆eptA, ∆eptB, ∆cptA) (Extended Data Fig.1a,b). We conclude that none of the known amino functions in LPS is required for its ubiquitylation. Ester-linked ubiquitin, in contrast to amide-linked conjugates, is sensitive to mild alkaline hydrolysis. We found that LPS-ubiquitin, but not amide-linked ubiquitin used as a control, was hydrolyzed under alkaline conditions (Fig.1f), further confirming that LPS ubiquitylation does not proceed through amide linkage.
To reconstitute LPS ubiquitylation in vitro we incubated S.Typhimurium isolated from infected cells with recombinant E1 and E2 enzymes (UBE1 and UBCH5C, respectively), HeLa lysates as the source of E3 activity and FLAG-ubiquitin. We deployed three S.Typhimurium strains (Drfc, DrfaL and DrfaJ) that produce LPS of distinct sizes to unequivocally identify reaction products as Ub-LPS (Extended Data Fig.1a). Incubating the reaction for 30min or longer produced mono- and oligo-ubiquitylated LPS (Fig.2a). Mono-Ub-LPS appeared as a singlet in anti-FLAG and a doublet in FK2 blots due to the size difference between FLAG-Ub-LPS synthesized in vitro and wildtype Ub-LPS already present on cell-extracted bacteria. The generation of mono-FLAG-Ub-LPS indicates de novo LPS ubiquitylation in vitro.
Figure 2. RNF213 is required for LPS ubiquitylation.

a, In vitro ubiquitylation (IVU) of indicated S. Typhimurium strains extracted from infected HeLa cells. The IVU reaction was comprised of FLAG-ubiquitin, E1 enzyme (UBE1), E2 enzyme (UBCH5C), and HeLa lysate as E3 ligase donor, and was terminated at the indicated time points. b, Purification scheme for the LPS ubiquitylating activity. c, IVU of Hela-extracted S. Typhimurium Arfc with fractions eluted from MonoS column as E3 donor. Graph indicates the exclusive unique peptide count for RNF213 detected by mass spectrometry. d, Immunoblot analysis of indicated S. Typhimurium strains extracted from WT and RNF213KO HeLa cells. a,c,d, Blots were probed with the indicated antibodies. GroEL, loading control for bacterial lysates. S.T. Salmonella Typhimurium. a-c, n=1. d, Representative of 3 biological repeats. For gel and graph source data, see Supplementary Fig. 1 and source data Fig. 2.
To identify the enzyme generating mono-Ub-LPS, we fractionated HeLa lysates by sequential ammonium sulphate precipitation, hydrophobic interaction chromatography, gel filtration and ion exchange chromatography, followed by mass spectrometry (Fig.2b, Extended Data Fig.2a). RNF213 (ring finger protein 213) was the sole protein containing a domain characteristic for eukaryotic E3 ubiquitin ligases, i.e. a RING, HECT or RBR domain, whose peptide counts matched the LPS ubiquitylating activity eluting from the final column (Fig.2c). To test whether RNF213 is required for the ubiquitylation of LPS, we used siRNAs to deplete and CRISPR technology to knock-out RNF213. Cells lacking RNF213 failed to ubiquitylate LPS upon infection with wild type and Drfc S.Typhimurium (Fig.2d, Extended Data Fig.2b-d), revealing an essential role for human and murine RNF213 in ubiquitylating LPS on cytosol‐invading bacteria.
RNF213 is the largest recognized human E3 ligase with a mass of almost 600kDa. It features a large N-terminal disordered region followed by a stalk, a linker domain, a dynein-like core comprised of two catalytically active and four inactive AAA+ domains, and a C-terminal E3 module surrounding the RING domain (Fig.3a, Extended Data Fig.3a).24 RNF213 is the major susceptibility gene for Moyamoya disease (MMD), a cerebrovascular disorder characterized by bilateral stenosis of the supraclinoid internal carotid artery and abnormal formation of collateral vessels.4–7 MMD is caused by missense mutations in RNF213, localized mainly to domains surrounding the RING finger, the so-called E3 module. The (patho-) physiological role of RNF213 is not well understood; involvement in lipotoxicity, lipid droplet formation, cell death and NF-kB signalling have been reported.25–28
Figure 3. LPS ubiquitylation by RNF213 is a RING-independent, RZ finger-mediated reaction.

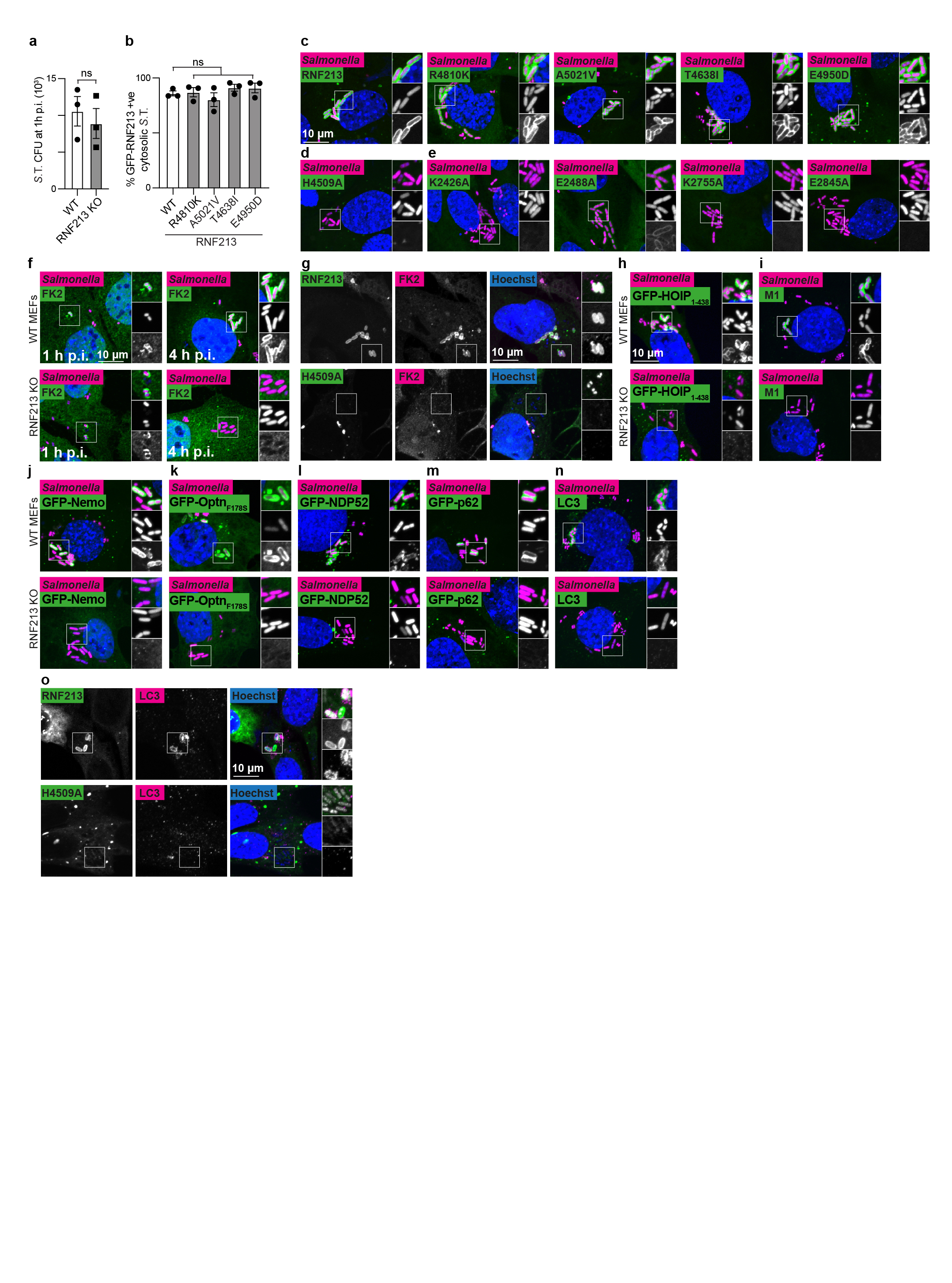
a, RNF213 domain structure. Critical residues in Walker A (K2426 and K2775) and Walker B motifs (E2488 and E2845) are indicated with red and yellow lines, respectively. RZ finger RNF213-ZNFX1 finger, CTD C-terminal domain. b, h, immunoblot analysis of S. Typhimurium Arfc extracted from WT or RNF213KO MEFs complemented with GFP or GFP-RNF213 alleles as indicated. GFP blots present the upper and lower part of continuous blots from which the middle parts have been removed as indicated by the dashed line. c-e, Immunoblot analysis of S.Typhimurium Arfc extracted from HeLa cells (c,d), or purified LPS from indicated S.Minnesota strains (e) and subjected to in vitro ubiquitylation using HeLa, Sf9 or Sf9 expressing huRNF213 lysates (c) or purified RNF213 (d,e). f, Immunoblot analysis of an IVU reaction using GST-tagged RNF213 fragments expressed in E. coli to assess autoubiquitylating activity. Representative of 3 experiments. b-f, h, Blots were probed with the indicated antibodies. Actin and GroEL, loading controls for mammalian and bacterial lysates respectively. g, RZ finger consensus amongst RNF213 and ZNFX1 proteins, human RNF213 protein sequence is depicted below. b, *, non-specific band. b,d-f,h, Representative of at least 3 biological repeats. c, n=1 For gel source data, see Supplementary Fig. 1.
For a structure-function analysis of RNF213 we complemented RNF213KO MEFs with RNF213 alleles (Fig.3b). RNF213 remained active upon deletion of the N-terminal disordered region (RNF213ΔN586). Further N-terminal shortening prevented efficient LPS ubiquitylation (RNF213ΔN1009), as did a C-terminal truncation (RNF2131-5186). To test the importance of the dynein-like core, we mutated the two catalytically active AAA+ domains in their Walker A and B motifs to disable ATP binding (RNF213K2426A, RNF213K2775A) and hydrolysis (RNF213E2488A, RNF213E2845A), respectively (Fig.3b).24,28 ATP binding by both domains was essential for efficient LPS ubiquitylation, as was ATP hydrolysis by the fourth but not the third AAA+ domain. We next tested RNF213 alleles from MMD patients. None of the chosen alleles (RNF213R4810K, RNF213A5021V, RNF213T4638I, RNF213E4950D) affected LPS ubiquitylation (Fig.3b). Finally, to test whether the RING domain mediates LPS ubiquitylation, we introduced inactivating point mutations (RNF213H4014N, RNF213W4024R). Neither mutation affected catalytic activity, nor did a deletion of the entire RING domain (RNFΔ3997-4036) (Fig3b). We conclude that RNF213-mediated LPS ubiquitylation requires a catalytically active AAA+ module but is independent of the RING domain, consistent with a recent report of RING-independent autoubiquitylation by RNF213.24
To fully reconstitute LPS ubiquitylation in vitro, we purified recombinant RNF213 from Sf9 cells (Extended Data Fig.3b), a cell type with no endogenous LPS ubiquitylating activity (Fig.3c, Extended Data Fig.3c). Purified RNF213 required the presence of ATP, as well as E1 and E2 enzymes, to ubiquitylate LPS on bacteria extracted from host cells, thus confirming a canonical reaction path (Fig.3d, Extended Data Fig.3d). Recombinant RNF213 also ubiquitylated purified rough LPS, including lipid A, which confirms RNF213 as a bona fide LPS E3 ubiquitin ligase and reveals lipid A as its minimal substrate (Fig.3e, Extended Data Fig.1c). The site(s) of ubiquitylation in lipid A remain unknown. Consistent with hydrolysis of ubiquitylated LPS under mild alkaline conditions (Fig.1f), RNF213-mediated ubiquitylation of lipid A may target hydroxy groups on either the sugars or fatty acids of lipid A, although ubiquitylation of the phosphate groups of lipid A is also conceivable. Taken together, we conclude that LPS ubiquitylation targeting the lipid A moiety is mediated by RING-independent E3 ligase activity in RNF213.
To identify the RNF213 domain(s) required for RING-independent activity, we screened RNF213 fragments expressed in E.coli for auto-ubiquitylating activity in vitro. RNF2134307-4702 became mono- and oligo-ubiquitylated upon incubation with E1 (UBE1) and UBCH7, a transthiolating E2 enzyme and reportedly24 the most active E2 for RNF213 (Fig.3f). The activity of this fragment tolerated further N- but not C-terminal truncation, revealing RNF2134429-4702 as the minimal active fragment. Neither fragment contained the RING domain. When mapped onto the structure of murine RNF213, RNF2134307-4702 comprised the middle and C-terminal lobe (C-lobe) of the E3 shell, RNF2134429-4702 the C-lobe only (Extended Data Fig.3a). While the C-lobe is not homologous to other proteins, inserted into the C-lobe resides a unique 27 amino acid peptide with sequence similarity to the NFX1-type zinc finger containing protein (ZNFX1), an interferon-induced RNA helicase with anti-viral function (Fig.3g, Extended Data Fig.3a,e).29 We therefore name this peptide the RNF213-ZNFX1- finger (RZ finger). The RZ finger is not resolved in the RNF213 structure24; it contains two conserved histidine and four conserved cysteine residues, suggesting it may form a Zn2+ complex. We tested the functional importance of the CHC3H-motif by creating RNF2134307-4702 H4509A and full length RNF213H4509A, which had lost the ability to auto-ubiquitylate in vitro and to ubiquitylate LPS in infected cells, respectively (Fig.3f,h). We conclude that the RZ finger presented by the C-lobe of the E3 shell promotes autoubiquitylation and is essential for RNF213-mediated LPS ubiquitylation, functionally similar to RING, HECT or RBR domains in other eukaryotic E3 ubiquitin ligases.
To test the functional importance of RNF213 for cell-autonomous immunity, we infected cells with S.Typhimurium. Lack of RNF213 had no effect on bacterial entry but cells deficient in RNF213 failed to restrict bacterial proliferation (Fig.4a, Extended Data Fig.4a). In wild type cells RNF213 was recruited to the surface of cytosolic S.Typhimurium, where it colocalized with ubiquitin (Fig.4b). Instant structured illumination microscopy (iSIM) revealed that RNF213 recruitment started focally and subsequently spread around the bacterium, indicating cooperative behaviour (Video1, Fig.4c). Recruitment of RNF213 to bacteria was not affected by MMD alleles (RNF213R4810K, RNF213A5021V, RNF213T4638I, RNF213E4950D) but required E3 ligase activity (RNF213H4509A) and a dynein-like core able to bind and hydrolyze ATP (RNF213K2426A, RNF213E2488A, RNF213K2775A, RNF213E2845A) (Fig.4d–e, Extended Data Fig.4b–e). Interestingly, RNF213E2488A, deficient in ATP hydrolysis due to a mutation in the Walker B motif of the third AAA+ domain, was impaired in forming a stable RNF213 coat (Fig.4e, Extended Data Fig.4e) despite efficiently ubiquitylating LPS on cytosolic bacteria (Fig.3b). Such phenotype revealed a specific requirement for the dynein-like AAA+ module in generating the RNF213 coat, possibly through effects on RNF213 oligomerization.28
Figure 4. RNF213 provides cell-autonomous immunity.

a, Fold replication of intracellular S. Typhimurium in WT and RNF213KO MEFs normalised to 1 h p.i. time point. Bacteria were counted by serial dilution of cell lysate on LB agar plates. b, Structured illumination micrograph of HeLa cells at 4 h p.i. with S. Typhimurium and immunostained for LPS, ubiquitin (FK2) and RNF213. Scale bar, 3 pm. Line indicates the plot profile adjacent. c, Still images from Video 1: Instant structured illumination microscopy (iSIM) of MEFs expressing FLAG-GFP-RNF213 infected with mCherry-expressing S. Typhimurium. Time p.i. as indicated. Scale bar, 1 pm d-e, Percentage of cytosolic S. Typhimurium positive for FLAG-GFP-RNF213 at 3 h p.i. in WT or RNF213KO MEFs stably expressing the indicated GFP-RNF213 alleles. f-o, Percentage of S. Typhimurium positive at 3h p.i. for FK2 (ubiquitin) (f,g), GFP- HOIP1-438 WT or T360A (h), M1-linked linear ubiquitin chains (i), GFP-Nemo (j), GFP-OptineurinF178S (k), GFP-NDP52 (l), GFP-p62 (m) and LC3 (n,o) in WT and RNF213KO MEFs (f,h-n) or RNF213KO MEFs stably expressing the indicated GFP-RNF213 alleles (g,o). Statistical significance was assessed by two-tailed unpaired Student’s t-test (a,d,f,h,i-o) or one-way anova (e,h). *P < 0.05, **P < 0.01. Data are expressed as the mean ± s.e.m. of 3 (a- m),4 (o) or 5 (n) independent biological repeats (see source data Fig. 4).
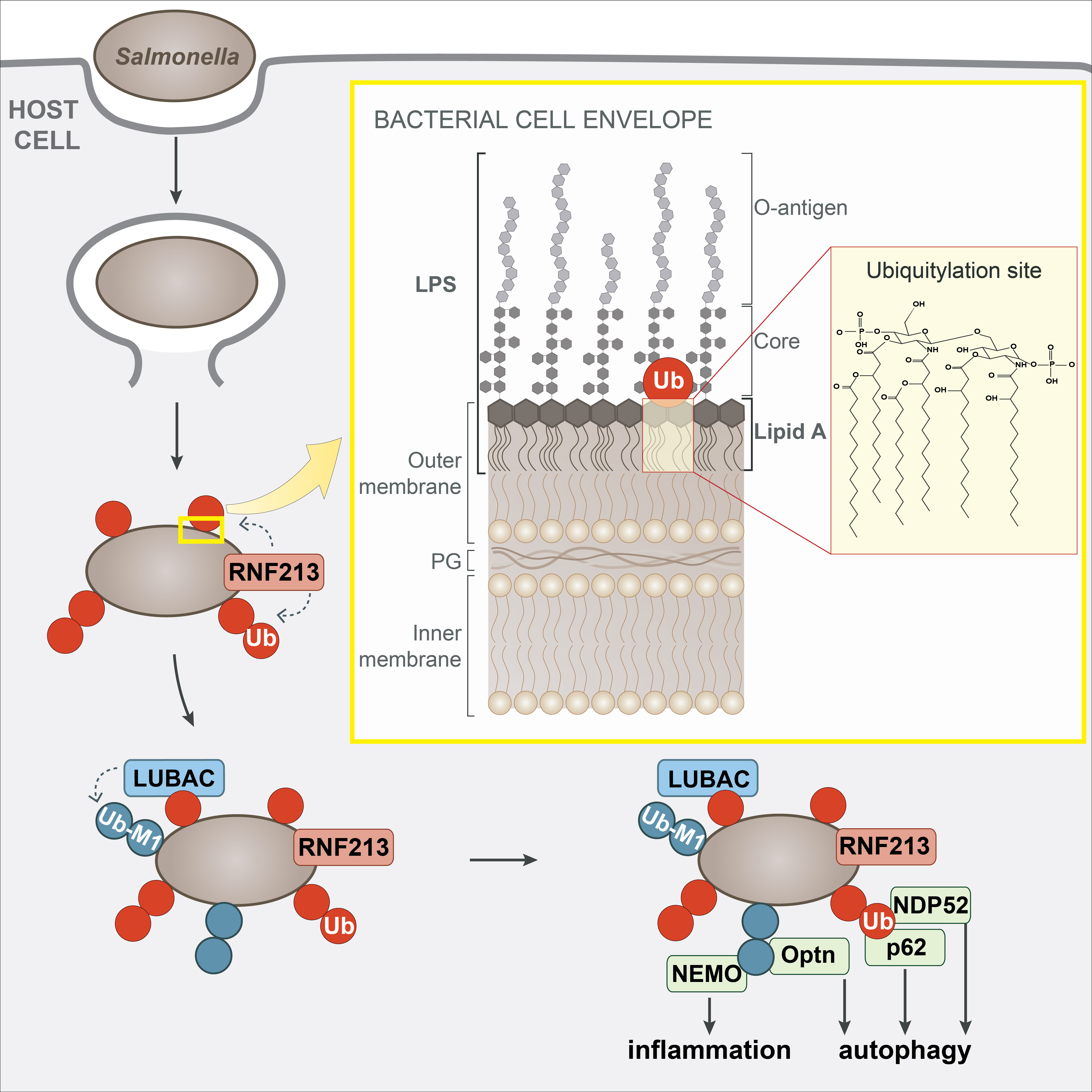
Cells deficient in RNF213 efficiently ubiquitylated remnants of Salmonella-containing vacuoles at 1h p.i. but failed to create a ubiquitin coat on the surface of S.Typhimurium at 4h p.i. (Fig.1a,4f, Extended Data Fig.4f), revealing that ubiquitylated LPS is an essential component of the bacterial ubiquitin coat. Complementation with wild type RNF213 but not with catalytically inactive RNF213H4509A restored the ubiquitin coat (Fig.4g, Extended Data Fig.4g). Deficiency in RNF213 curtailed recruitment of the LUBAC subunit HOIP (Fig.4h, Extended Data Fig.4h), as did inactivation of the ubiquitin binding site in HOIP1-438 T360A8, indicating that LUBAC recruitment requires RNF213-mediated LPS ubiquitylation. Consequently, in the absence of RNF213, LUBAC did not add M1-linked ubiquitin chains to the bacterial ubiquitin coat (Fig.4i, Extended Data Fig.4i), resulting in failure to recruit Nemo, the M1-specific adaptor subunit of the IKK complex (Fig.4j, Extended Data Fig.4j). The inability of RNF213KO cells to generate a bacterial ubiquitin coat at 4h p.i. abolished recruitment of ubiquitin-binding autophagy cargo receptors, including LUBAC-dependent Optineurin as well as LUBAC-independent NDP52 and p628 (Fig.4k–m, Extended Data Fig.4k–m), which resulted in failure of anti-bacterial autophagy, as indicated by the absence of LC3+ve S.Typhimurium (Fig.4n, Extended Data Fig.4n). Complementation with wild type RNF213 but not with catalytically inactive RNF213H4509A restored the recruitment of LC3 to S.Typhimurium (Fig.4o, Extended Data Fig.4o). We conclude that RNF213 is essential for the generation of the bacterial ubiquitin coat and that cells lacking RNF213 suffer defects in cell-autonomous immunity manifested as an inability to restrict the proliferation of S.Typhimurium (Extended Data Fig.5).
The identification of RNF213 as a novel immune sensor reveals an unexpected link between MMD and infection. Although MMD-predisposing RNF213 alleles were not impaired in their ability to ubiquitylate LPS, activation of mutant RNF213 by bacterial or other infections, may nevertheless contribute to the development of MMD in susceptible individuals. Activation during infection may explain the low penetrance of certain MMD risk alleles, such as RNF213R4810K, an allele encoded by 16 million Asians of which only 0.5% develop MMD.30 LPS ubiquitylation by RNF213 requires ATPase activity in its dynein-like core. Interestingly, to access LPS embedded in bacterial membranes, the cytosolic LPS receptor Caspase-431 also requires nucleotide hydrolysis, which is provided in trans by guanylate-binding proteins (GBPs)32,33, a family of interferon-induced GTPases. Nucleotide hydrolysis may thus represent a common requirement to sense LPS in cytosol-invading bacteria. Our discovery of LPS ubiquitylation expands the scope of ubiquitylation beyond a mere post-translational protein modification. Further non-proteinaceous ubiquitylation substrates may exist, both in cytosol‐invading pathogens and in host cells, which may be targeted by either eukaryotic RING, HECT or RBR E3 ligases, by prokaryotic NEL or Sde E3 ligases, or by unconventional E3 ligase activities that rely on novel domains, such as the RZ finger in the E3 shell of RNF213.
Methods
Plasmids, antibodies and chemicals
M6P plasmids were used to produce recombinant murine leukemia viruses (MLV) for the stable expression of proteins in mammalian cells.34 pETM30 plasmids were used for protein expression in E.coli. To express RNF213 in mammalian cells, an inducible PiggyBac transposon system was used.35 RNF213 cDNA was kindly provided by Dr Daisuke Morito. To facilitate the generation of RNF213 alleles, a codon optimized RNF213 cDNA was gene synthesized (Genewiz). Mutations and gene truncations were generated by PCR using NEB HiFi and verified by sequencing.
Primary antibodies used: FK2 (diluted 1:200, Enzo Life Science, BML-PW8810), ±-GroEL (1/1000, Enzo Life Science, ADI-SPS-875-F), α-DnaK (1:1000, Enzo Life Science, ADI-SPA- 880), α-HA.11 (1:1000, Convance, MMS-101R), α-actin (1:1000, Abcam, ab8227), α-RNF213 (1:1000, Merck, HPA003347), α-FLAG-M2-HRP (1:1000, Merck, A8592) and α-GFP (1:1000, JL8, Clontech, 632381), α-LPS (1:100, BioRad, 8210-0407), α-Galectin 8 (1:100, R&D Systems, AF1305), α-M1 (1:1000, 1F11/3F5/Y102L, GenenTech) and α-LC3 (1:100, Cosmo Bio, CTB-LC3-2-IC). Secondary antibodies used: Thermo Fisher Scientific (1:500, Alexa-conjugated anti-mouse, anti-goat, anti-human and anti-rabbit antisera) and Dabco (1:5000, HRP-conjugated reagents). Purified LPS used: Adipogen (wt LPS from S. Minnesota (IAX- 100-020), Ra (IAX-100-016), Rb (IAX-100-015), Rc (IAX-100-017), Rd1 (IAX-100-018), Rd2 (IAX-100-021), Re (IAX-100-021), Lipid A (IAX-100-001)).
Cell culture
HeLa cells were obtained from the European Collection of Authenticated Cell Cultures. Wild type mouse embryonic fibroblasts (MEFs) were kindly provided by Dr Chihiro Sasakawa.36 Cells were grown in IMDM supplemented with 10% FCS at 37°C in 5% CO2. All cell lines tested negative for Mycoplasma. Stable cell lines were generated by retroviral transduction or, for expression of different FLAG-GFP-RNF213 alleles, were generated using an inducible Piggybac transposon system.35 Briefly, RNF213KO MEFs seeded in 24 well plates were transfected with 1 μg of PiggyBac plasmids35 and 1 μg of pBase37 using Lipofectamine 2000 and 2 days later cells were selected with puromycin. Protein expression was induced with 1 μg/mL doxycycline for at least 15 h in the presence or absence of 20 μM ZVAD.
RNA interference
HeLa cells were transfected with Silencer Select siRNAs against RNF213 (Thermo Fisher Scientific, s33570 and s33660) or the nontargeting Silencer Select Negative Control (Thermo Fisher Scientific, No.1) using Lipofectamine RNAiMAX (Thermo Fisher Scientific). Experiments were performed 3 days post transfection.
Generation of KO cells
To make RNF213KO cells, oligonucleotides for the gRNA (mRNF213: GAAGCGGTACATCATACGTG, hRNF213 : GCTGAAAGCGGGCGCACTGC) were phosphorylated with T4 PNK (New England Biolabs) and cloned into pSpCas9(BB)-2A- GFP (Addgene, PX458). Subsequently, cells were transfected and next day, GFP positive single cells were sorted into a 96 well plate. Resulting HeLa clones were screened for lack of RNF213 expression by immunoblotting. Disruption of RNF213 exons in HeLa and MEFs were verified by sequencing.
Bacteria
Escherichia coli strains MC1601 and NEB 10-beta were used for plasmid production and BL21 strain was used for protein purification.
Salmonella enterica serovar Typhimurium strain 12023 and isogenic ΔrfaL were kindly provided by Dr David Holden (Imperial College London).
To generate mutants in S.Typhimurium we deployed phage l Red recombinase38. Mutants in cptA, eptB, arnC, eptB were generated in S.Typhimurium lacking rfaL. The following primers were used:
wzzB fwd
(CATTAATCCTATGGCATATATTTGCTTTATGGCTACACTGTCTCCAGCTTCATCCTTTTT TTAGTTAGGGTATCTATGGTGTAGGCTGGAGCTGCTTC)
wzzB rev
(AAAAAACCGGGCAATGCCCGGTTTTTTAATGAGAAATTTTACCTGTCGTAGCCGACCAC CATCCGGCAAAGAAGCTTACATATGAATATCCTCCTTAG)
fepE fwd
(GATAATTCTGACTTGCTGTTAGAATCTCTGACAGGAATGTGTTCTTTCATTGGATAAATT TTTCAGGTCATACGGCATGGTGTAGGCTGGAGCTGCTTC)
fepE rev
(GGTACCGCTGGGGCGGCGATAGTCGCCCACACTGATGACAAAGCCGGATATCGCTAT CCGGCTTTTCGGGTAAATCACATATGAATATCCTCCTTAG)
rfc fwd
(TTTGCCTGATGGTAATATTTTTAATACTAAGCATTTTTTCTAAAGGCTCTATTGGTGTAG GCTGGAGCTGCTTC)
rfc rev
(ACAATTTTTACGCTTCAGAGCCAAATAAAACGGCGGCATTGCCGCCGTATAACTTACAT ATGAATATCCTCCTTAG)
cptA fwd
(TATCCGGACTACGGGAGAGAAGCACGCCAGGCCCGATAAGCGTTAGCGTTATCGGGC AACATACAGGATGGTGTAGGCTGGAGCTGCTTC)
cptA rev
(GTTTGCTTCAGATTCTTTTAAGGTTCAGGCGTTACGTTTGGCGCTACGATTAAAGACAG GCTCTCATTCTACATATGAATATCCTCCTTAG)
eptA fwd
(AAGTTCTTAAGGTTCACTTAATTTTACTTTGTCACGATTAGC
(AAGTTCTTAAGGTTCACTTAATTTTACTTTGTCACGATTAGCGTCACCGAATCGATGGA CGCATCAACATGGTGTAGGCTGGAGCTGCTTC)
eptA rev
(GCCTTCGGTTTGCGCGGCGAGTATTAACCCCTGTAATAATAGCGTGTCGTCTTCAACA ATCAGTATCTTCACATATGAATATCCTCCTTAG)
arnC fwd
(CCGGACATGACCGAGAGTGACTTTGATCGAGTCATTACCGCCCTTCATCAGATAGCAG GACAATAAGCATGGTGTAGGCTGGAGCTGCTTC)
arnC rev
(ATACCCGGCATCCAGTACGGCCTGCACCCCCTGACATCCCATATCGTGATAGGCAAAA ATAACGGCTTTCACATATGAATATCCTCCTTAG)
eptB fwd
(GCGTTAGCCGCATATTTACCTGTTTGATTAAAGAATCGTTGTACAGGTCGTTTTTATCC CGATTCCCAGGGTTTGTTTGCATGGTGTAGGCTGGAGCTGCTTC)
eptB rev
CATCTTACTGCTCAGGCGCACGCCCGTCAATCCCTTAGTCGCAAAATACAATTCAATCG
GCGAGAAAGTCGGCAGACAACCATATGAATATCCTCCTTAG
rfaJ fwd
(GAAGCATTATACTTCGGGTATAAATTATTATATAGCCTACTTTAAACGTAAACTTCTTGA ATAAAACCCATAGGTGATGTAATGGTGTAGGCTGGAGCTGCTTC)
rfaJ rev
(TTTTCACCGTCTTTTTTGACAAAGACGGTATAGTTTTTAATCTTTTTTTCAATAATCATAA TGGAGATTTAGGGAGGGGAACATATGAATATCCTCCTTAG)
Resistance cassettes were removed using Flp recombinase and genotypes were verified by sequencing.
Bacterial infections and extraction
S. Typhimurium was grown overnight in Luria broth (LB) and sub-cultured (1:33) in fresh LB for 3.5 h before infection.
To enumerate intracellular bacteria, MEFs in 24-well plates were infected with 20 μl of 1:15 diluted sub-culture for 8 min at 37°C. Following two washes with warm PBS and incubation in IMDM supplemented with 10% FCS and 100μg/ml gentamycin for 2 h, cells were cultured in IMDM supplemented with 10% FCS and 20 μg/ml gentamycin. To determine intracellular S.Typhimurium colony forming units (CFUs), cells from triplicate wells were lysed in 1 ml of cold PBS containing 0.1% Triton X-100. Serial dilutions were plated in duplicate on LB agar.
To extract bacteria from host cells for biochemical analysis, cells seeded in a 6-well plate, were infected with 100 μl of undiluted subculture for 20-30 min at 37°C. Cells were incubated in media containing 100 μg/ml gentamycin, washed once with PBS and lysed in 1 ml ice cold PBS containing 0.1% Triton X-100. Lysates were centrifugated at 300 g for 5 min at 4°C and supernatant containing bacteria was collected and centrifugated at 16,100 g for 10 min at 4°C. The bacterial pellet was washed once with PBS, followed by bacterial lysis in 50 μl BugBuster (Merck) including 2 mM iodoacetamide for 5 min at RT. The BugBuster lysate was centrifugated at 16,100 g for 10 min at RT, the supernatant was split up in 2 fractions: one was directly mixed with Laemmli buffer (Bio-Rad) containing 100 mM DTT and boiled for 5 min; the other fraction (used to further purify ubiquitylated LPS) was heated at 90°C for 15 min, centrifugated at 16,100 g for 10 min at RT and this heat-cleared supernatant was mixed with Laemmli buffer containing 100 mM DTT. For mild alkaline hydrolysis of LPS-ubiquitin, samples were further incubated with 100 or 200 mM NaOH at 37 °C for 20 min. K48-linked diubiquitin (Boston Biochem) was used as an amide-linked control. Samples were boiled for 5 min and subjected to immunoblot analysis.
Immunoblot analysis
Post-nuclear supernatants from HeLa or MEF cells were obtained following lysis with RIPA buffer (50 mM Tris pH7.4, 150 mM NaCl, 1% NP-40, 0.5% NaDOC, 0.1% SDS and 1x Halt protease inhibitor cocktail (Thermo Scientific)). Cleared supernatants were mixed with Laemmli buffer containing 100 mM DTT or 5% ²-mercaptoethanol and samples were boiled for 5 minutes. Samples were run on NuPAGE 4-12 % Bis-Tris gels (Thermo Fisher Scientific) in MES or MOPS SDS running buffer (Formedium) at 165 V for 45 minutes. An overnight wet transfer was used to transfer RNF213 protein onto methanol-activated PVDF membranes (Millipore). For other proteins, samples were transferred onto PVDF membranes using the Turbo transfer system (BioRad) for 7 min at 1.3 A. Membranes were blocked for 1 h in TBS-T (100 mM Tris pH 7.4, 150 mM NaCl, 0.1 % Tween 20) containing 5% milk (Marvel), followed by overnight incubation with primary antibodies in TBS-T containing 5% milk. Membranes were washed in TBS-T, incubated with secondary HRP-conjugated antibodies (Dabco,1/5000) in TBS-T containing 5% milk followed by 3 times 10 min TBS-T washes. In case of FK2 staining, milk was replaced by 2% BSA throughout the whole procedure. Heat cleared bacterial lysates were used to visualize LPS-ubiquitin with FLAG or FK2 antibody, non-heat cleared bacterial lysates were used to probe for GroEL as a loading control. Visualization following immunoblotting was performed using ECL detection reagents (Amersham Bioscience) and a ChemiDoc MP imaging system (BioRad).
LPS extraction and visualization
S. Typhimurium strains were grown overnight at 37°C in 2 mL of LB in a shaking incubator. Bacteria were centrifugated at 16,100 g for 5 min, resuspended in 100 μL of SDS buffer (2 % P-mercaptoethanol, 2 % SDS and 10 % glycerol, 50 mM Tris-HCl pH 6, 0.025 % Bromophenol Blue) and incubated at 95°C for 15 min. Ten μL of proteinase K (20 μg/mL, Qiagen) was added, and the samples were incubated for 3 h at 59°C. 200 μL of ice-cold water-saturated phenol was added to each tube. Samples were then thoroughly vortexed and incubated at 65°C for 15 min, vortexing an additional 2 to 3 times throughout. After cooling to RT, 1 mL of anhydrous diethyl ether was added to each tube. Samples were spun at 16,100 g for 10 min in a cold centrifuge. The bottom blue layer was carefully extracted and mixed with 200 μL of ice-cold water saturated phenol. Once again, samples were incubated at 65°C for 15 min before being mixed with 1 mL of diethyl ether. Tubes were centrifugated at 16,100 g for 10 min and the bottom blue layer was extracted and combined with 150 μL of 2X SDS buffer. Samples were separated by SDS PAGE and stained using the Pro-QTM Emerald 300 Stain Kit, according to the manufacturer’s instructions. Stained gels were visualized using a ChemiDoc MP imaging system (Biorad).
Protein production in insect cells
pOP806_pACEBac1 2xStrep-RNF213-3xFLAG plasmid was transformed into DH10EmBacY (DH10Bac with YFP reporter). Blue-white screening was used to isolate colonies containing recombinant baculoviral shuttle vectors (bacmids) and bacmid DNA was extracted combining cell lysis and neutralisation using buffer P1, P2 and N3 (Qiagen) followed by isopropanol precipitation. A 6-well of Sf9 cells (Oxford Expression Technologies Ltd) grown at 27°C in Insect-Xpress (Lonza) without shaking, were transfected with bacmid plasmid using PEI transfection reagent. After 7 days, virus P1 was collected and used 1/100 to transduce 100 mL (1× 10^6 cells / mL) Sf9 cells. After 7 days, virus P2 was harvested. To express protein, 1 L Sf9 cells was transduced with 1/100 dilution of P2 virus and cells were incubated at 27 °C with 140 rpm shaking for 3 or 4 days. Cell pellets were snap frozen and stored at -80°C. To lyse cells, they were thawed in 50 mM HEPES, 200 mM KCl, 1 mM TCEP, pH 7.2 containing 1x universal nuclease (Pierce) and protease inhibitor tablets (Roche). The lysate was centrifugated at 20,000 g for 30 min at 4°C and cleared lysate was applied to a 5 mL StrepTrap HP column (GE Healthcare) and eluted with 50 mM HEPES, 200 mM KCl, 1 mM TCEP, containing 2.5 mM desthiobiotin pH 8. PD10 columns were used to buffer exchange to 50 mM HEPES, 200 mM KCl and 0.25 mM TCEP, pH 7.2 and RNF213 protein (>1 mg/mL) was snap frozen in liquid nitrogen and stored at -80°C. All purification steps were carried out at 4°C, using an ÄKTA pure 25 (GE Healthcare).
Protein production in E.coli
GST-RNF213 protein fragments were purified from BL21 (DE3)pLac1 cells that were transformed with pETM30 plasmids and grown overnight on TYE plates containing 50 μg/mL kanamycin. An overnight starter culture was diluted 1/1000 in 1 L of LB containing 50 μg/mL kanamycin and grown at 37°C at 220 rpm until OD600 ~ 0.9 was reached. The flask was cooled to 16°C and protein expression was induced overnight with 100 μM IPTG at 220 rpm at 16°C. Cell pellets were resuspended in IVU reaction buffer containing 1.43 mM ²-mercaptoethanol, 2 mg/ml DNasel and EDTA free protease inhibitor tablets. The suspension was homogenised using an EmulsiFlex-C3 (Avestin) for two passes at ~15,000 psi and cleared by centrifugation at 30,000 rpm for 30 min at 4°C. The clarified lysate was applied to Amintra glutathione resin (Expedeon), washed with IVU reaction buffer containing 1.43 mM ²-mercaptoethanol and eluted with IVU reaction buffer containing 1.43 mM P-mercaptoethanol and 20 mM glutathione, pH 8. Protein was buffer exchanged into 1x IVU buffer with 1 mM TCEP, snap frozen and stored at -80°C.
For the purification of His-UBE1, an N-terminal GST-ubiquitin fusion protein was expressed and lysed in ²-mercaptoethanol-free lysis. The His-UBE1 ²-mercaptoethanol-free clarified lysate was supplemented with 10 mM ATP and 10 MgCh and incubated at RT for 30 min with Amintra glutathione resin (Expedeon), which were equilibrated with 50 mM Tris pH 8.5 and 2 mM ATP. The resin was then washed with DTT-free high salt buffer supplemented with 5 mM MgCl2. His-UBE1 was eluted in DTT-containing buffer and protein containing fractions were applied to anion-exchange 6 mL Resource Q column (GE Healthcare) with a 0-25% linear gradient from buffer A (25 mM Tris pH 8.5, 10 mM DTT, 50 mM NaCl) to buffer B (25 mM T ris pH 8.5, 10 mM DTT, 1000 mM NaCl) and size-exclusion chromatography (HiLoad 16/600 Superdex 75 pg, GE Healthcare) into buffer C (25 mM Tris pH 8.5, 10 mM DTT, 200 mM NaCl).
In vitro deubiquitylation (DUB) reaction
A 15 cm dish of HeLa cells was infected with 100 μL ∆rfc S.Typhimurium for 30 min. At 4 h p.i. bacteria were extracted as described above. Bacterial pellets were washed and resuspended in DUB reaction buffer (50 mM Tris pH 7.5, 50 mM NaCl and 5 mM DTT) and incubated with USP2 catalytic domain (Boston Biochem) for 30 min at 37°C.
In vitro ubiquitylation (IVU) reaction
First, E2 was pre-charged in a 5x reaction by incubating 10 μM of UBCH5C or UBCH7 (Boston Biochem) in IVU reaction buffer (30 mM Hepes pH7.4, 100 mM NaCl, 10 mM MgCI2) containing 50 mM ATP, 5 mM DTT, 200 μM FLAG-ubiquitin (Boston Biochem) and 0.2 μM UBE1, for 15 minutes at 37°C. Precharged E2 reaction was diluted to a 1× reaction with substrate (washed bacterial pellet obtained from a 10 cm dish of HeLa cells infected with S.Typhimurium for 4 h or purified LPS (Adipogen)) and E3 ligase (cell lysates or 350 nM recombinant RNF213). The reaction mix was incubated at 37°C for 1 h, after which the reaction was centrifugated at 16,100 g and the bacterial pellet or ubiquitylated LPS was washed twice in IVU reaction buffer containing 4 M urea. The bacterial pellet was lysed in BugBuster, the purified LPS solubilised in BugBuster and further processed for immunoblot analysis as described previously.
Purification of E3 ligase activity for LPS ubiquitylation
Suspension HeLa cells were used as a source for the unknown E3. HeLa lysate was dounce- homogenised in IVU reaction buffer containing 1 mM DTT and ultracentrifuged at 90,000 rpm in a TLA120.2 rotor. One mL of lysate was salted out using a 1 - 1.6 M (NH4)2SO4 cut, which was applied to a Butyl FF 1mL column (GE Healthcare) in IVU reaction buffer containing 1mM DTT and 1M (NH4)2SO4. The column was washed by reducing the salt concentration to 700 mM (NH4)2SO4, RNF213 activity was eluted by a further reduction to 0 mM (NH4)2SO4. The enzymatic activity was enriched in the last step elution, this fraction was concentrated to 500 μl volume (vivaspin 2, 10,000 MWCO PES) and applied to a Superdex 200 10/300 column (GE Healthcare), equilibrated in IVU reaction buffer containing 1 mM DTT and 10% glycerol. The fractions with enzymatic activity were applied to a MonoS column and protein was gradient eluted (100 to 500 mM NaCl) using IVU reaction buffer containing 1 mM DTT and 10% glycerol. The eluted fractions were analysed by mass spectrometry. To assess activity, IVU reactions were performed using proteins fractions dialysed in IVU reaction buffer containing 1 mM DTT (Slide-A-Lyzer, Thermo Scientific, 69570).
Mass spectrometry
Fractions eluted from the MonoS column were prepared for mass spectrometric analysis by in solution enzymatic digestion. Briefly, proteins were reduced in 10 mM DTT, and then alkylated with 55 mM iodoacetamide. After alkylation, proteins were digested with trypsin (Promega, UK) overnight at 37 °C at an enzyme to protein ratio of 1:20. The resulting peptides were analysed by nano-scale capillary LC-MS/MS using an Ultimate U3000 HPLC (ThermoScientific Dionex, San Jose, USA) to deliver a flow of approximately 300 nL/min. A C18 Acclaim PepMap100 5 μm, 100 μm × 20 mm nanoViper (ThermoScientific Dionex, San Jose, USA), trapped the peptides prior to separation on a 25 cm PicoCHIP nanospray column packed with Reprosil-PUR C18 AQ (New Objective Inc., Littleton, USA). Peptides were eluted with a 60 minute gradient of acetonitrile (2%v/v to 80%v/v). The analytical column outlet was directly interfaced via a nano-flow electrospray ionisation source, with a hybrid dual pressure linear ion trap mass spectrometer (Orbitrap Velos, ThermoScientific, San Jose, USA). Data dependent analysis was carried out, using a resolution of 30,000 for the full MS spectrum, followed by ten MS/MS spectra in the linear ion trap. MS spectra were collected over a m/z range of 300-2000. MS/MS scans were collected using a threshold energy of 35 for collision induced dissociation. LC-MS/MS data were then searched against a protein database (UniProt KB) using the Mascot search engine programme (Matrix Science, UK)39. Database search parameters were set with a precursor tolerance of 5 ppm and a fragment ion mass tolerance of 0.8 Da. Two missed enzyme cleavages were allowed and variable modifications for oxidized methionine, carbamidomethyl cysteine, pyroglutamic acid, phosphorylated serine, threonine and tyrosine were included. MS/MS data were validated using the Scaffold programme (Proteome Software Inc., USA).40 All data were additionally interrogated manually.
Protein alignments
We identified animal homologs of human RNF213 (hRNF213) from major animal lineages with hRNF213 as a query for iterative JACKHMMER41 searches performed on combined UniProt and TrEMBL databases (with e-value cut-off = 0.001 and query length cut-off = 60%) and PSI- BLAST42 searches on non-redundant database (with e-value cut-off = 0.001 and query length cut-off = 60%). We collected animal homologs of hRNF213 (i.e. animal orthologs of hRNF213 and homologous RZ finger regions of hRNF213 and ZNFX1) and then we constructed multiple sequence alignments of orthologs/homologs using MSAPROBs43 which were refined using HMMER profiles. These alignments were further manually corrected based on Jalview44 conservation scores. Logo was constructed using WebLogo.45
Microscopy
Cells were grown on glass coverslips. After infection, cells were washed twice with PBS and fixed in 4% paraformaldehyde for 15 min. Cells were washed twice in PBS and then permeabilised in PBS with 0.1% Triton X-100 and blocked with PBS with 2% BSA. When staining for LC3, cells were permeabilised with 0.05% saponin in PBS with 2% BSA. Coverslips were incubated with primary antibodies followed by Alexa conjugated secondary antibodies in blocking solution for at least 1 h at room temperature. Coverslips were then mounted either in Prolong gold mounting medium or ProLong Gold Antifade Mountant (Invitrogen) for confocal imaging or super resolution microscopy, respectively.
Marker-positive bacteria were scored by eye on a Zeiss Axio Imager microscope using a 100x/1.4NA Oil immersion lens, amongst at least 200 bacteria per coverslip.
Super-resolution images were acquired using an Elyra S1 structured illumination microscope (SIM; Carl Zeiss). The system has four laser excitation sources (405 nm, 488 nm, 561 nm and 640 nm) with fluorescence emission filter sets matched to these wavelengths. SIM images were obtained using a ×63/1.4 NA oil immersion lens with grating projections at 3 rotations and 5 phases in accordance with the manufacturer’s instructions. The number of z planes varied with sample thickness. Super-resolution images were calculated from the raw data using Zeiss ZEN software and plot profiles were made with FiJi ImageJ.
For live-cell resolution imaging cells were seeded in 35 mm glass bottom MatTek microwell dishes. After infection, cells were washed 4 times in warm PBS and incubated in Leibovitz’s L-15 supplemented with 100μg/ml gentamycin for the duration of the experiment. Images were acquired using a 100X super resolution Apo TIRF oil objective on a Nikon Eclipse Ti2 with a VisiTech iSIM high speed super resolution system. Movies were analyzed using FiJi ImageJ.
Quantification and statistical analysis
All data were tested for statistical significance with Prism software (GraphPad Prism 8). The unpaired two-tailed Student’s t-test was used to test whether two samples originated from the same population. Differences between more than two samples were tested using a one-way analysis of variance (ANOVA). Unless otherwise stated, all experiments were performed at least three times and the data were combined for presentation as a mean±s.e.m. All differences not specifically indicated as significant were not significant (P>0.05). Significant values are indicated as *P<0.05 and **P<0.01. Statistical details, including sample size (n), are reported in the figures and figure legends.
Microscopy: for scoring marker-positive bacteria, at least three independent experiments with two technical replicates each were performed. Bacteria were scored by visual counting of n > 200 bacteria per replicate. Graphs show mean ± s.e.m.
Scoring intracellular CFUs: to score bacterial burdens, cells from triplicate wells were lysed and bacteria were plated in duplicate on LB agar. Each experiment was performed at least three times. Bacterial colonies were counted using the aCOLyte3 system (Synbiosis). Graphs show mean ± s.e.m.
Extended Data
Extended Data Figure 1. LPS structure in S. Typhimurium and S.Minnesota.

The composition of lipid A, inner core, outer core and O-antigen from S.Typhimurium (a) and rough variants of S.Minnesota (c). S.Typhimurium mutants deficient in specific steps of LPS biosynthesis and truncated LPS species from S.Minnesota are indicated in red. Substoichiometric modifications introducing amino groups and the enzymes responsible are indicated in blue. ArnC, the undecaprenyl-phosphate 4-deoxy-4-formamido-L-arabinose transferase, functions upstream of ArnT and is required for the ultimate incorporation of L4AraN into LPS. EptA, EptB and CptA incorporate phosphoethanolamine into LPS. b, Immunoblot analysis of the indicated S. Typhimurium strains, extracted from HeLa cells. Blots were probed with the indicated antibodies. DnaK, loading controls for bacterial lysates. A4 AeptA AarnC AcptA AeptB in ArfaL background. Representative of 3 biological repeats. For gel source data, see Supplementary Fig. 1.
Extended Data Figure 2. RNF213 is required for LPS ubiquitylation.

a, In vitro ubiquitylation (IVU) of S. Typhimurium Arfc extracted from infected HeLa cells. The reaction comprised FLAG-ubiquitin, E1 enzyme (UBE1), E2 enzyme (UBCH5C), and fractionated HeLa cell lysate as indicated. In the chromatograms depicted below each blot, light grey indicates fractions with little or no LPS-ubiquitylating activity, whereas blue indicates fractions with LPS-ubiquitylating activity used for further fractionation or mass spectrometry.
b,Immunoblot analysis of HeLa cells transfected with the indicated siRNAs and WT HeLa and RNF213KO HeLa cells c, d, Immunoblot analysis of S. Typhimurium Arfc (c,d left panel) or WT (d right panel) extracted from HeLa cells transfected with the indicated siRNAs (c) or extracted from WT or RNF213KO MEFs (d). Representative of 3 biological repeats. Blots were probed with the indicated antibodies. Actin and GroEL or DnaK, loading controls for mammalian and bacterial lysates, respectively. *, non-specific band. For gel source data, see Supplementary Fig. 1.
Extended Data Figure 3. LPS ubiquitylation by RNF213 is a RING-independent, RZ finger-mediated reaction.

a, RNF213 domain structure (pdb 6TAX) (left panel) with domain colours corresponding to Fig.3a and zoom in on the E3 module (middle and right panel) indicating N-terminal (N), middle (M) and C-terminal (C) lobes of the E3 shell domain. Red arrows point at the position of the unresolved RZ finger. b, Coomassie-stained gel of purified RNF213. c left panel, d left panel, Immunoblot analysis of S.Typhimurium Arfc extracted from HeLa cells and subjected to in vitro ubiquitylation using HeLa, Sf9 or Sf9 expressing huRNF213 lysates (c) or purified RNF213 (d) corresponding to Fig.3c,d. c right panel, Coomassie gel and immunoblot analysis of lysates used in Fig.3c. d right panel, Immunoblot analysis of IVU reaction supernatants, separated under non-reducing and reducing (+pme) conditions corresponding to Fig.3d. c,d, Blots were probed with indicated antibodies. e, Alignment and conservation scores of RZ fingers in RNF213 and ZNFX1 from the indicated species. Colour and height signify degree of conservation (from Jalview) (upper panel). RZ finger alignment (lower panel). b,d, Representative of 3 biological repeats. c, n=1. For gel source data, see Supplementary Fig. 1.
Extended Data Figure 4. RNF213 provides cell-autonomous immunity.

a, S. Typhimurium CFU 1 h p.i. extracted from WT and RNF213KO MEFs. Bacteria were counted by serial dilution of cell lysate on LB agar plates. Data are expressed as the mean ±s.e.m. of three experiments. b, Percentage of cytosolic S. Typhimurium positive for FLAG-GFP-RNF213 at 3 h p.i. in RNF213KO MEFs stably expressing the indicated GFP- RNF213 alleles. c-o, representative confocal micrographs for Fig.4d–o. c-e, WT or RNF213KO MEFs stably expressing the indicated GFP-RNF213 alleles infected with mCherry-expressing S. Typhimurium, fixed 3 h p.i. f-o, WT and RNF213KO MEFs (f,g,n), RNF213KO MEFs stably expressing GFP-RNF213 H4509A (g,o) or WT and RNF213KO MEFs stably expressing GFP- HOIP1-438 WT or T360A (h), GFP-Nemo (j), GFP-OptineurinF178S (k), GFP-NDP52 (l), GFP- p62 (m), infected with mCherry-expressing (c-f, h-n) or BFP-expressing (g,o) S. Typhimurium, fixed 3 h p.i. and stained for FK2 (ubiquitin) (f,g), M1-linked linear ubiquitin chains (i) or LC3 (n,o). g,o, For better visibility of bacteria, Hoechst/BFP channel has been depicted in white in the zoomed sections. c-n, Scale bar, 10 pm. Statistical significance was assessed by twotailed unpaired Student’s t-test (a) or one-way anova (b). ns, not significant. Data are expressed as the mean ± s.e.m. of 3 independent experiments (a,b, source data Extended Data Fig.4) and micrographs are representative of 3 biological repeats (c-o)
Extended Data Figure 5. Model of RNF213-mediated LPS ubiquitylation during bacterial infection.

Damage of Salmonella-containing vacuoles releases S.Typhimurium into the host cytosol, where RNF213 associates with the bacterial surface and ubiquitylates LPS, resulting in LUBAC recruitment and the deposition of M1-linked ubiquitin chains linked to an unidentified substrate. Recruitment of the autophagy cargo receptors NDP52 and p62 requires RNF213 but not LUBAC, while that of Optn and the IKK subunit NEMO relies on the activity of both RNF213 and LUBAC. Yellow insert: The structure of the Gram-negative cell envelope. PG peptidoglycan. Red insert: Lipid A, the minimal substrate for RNF213-mediated ubiquitylation of LPS. Ubiquitylation of lipid A is predicted to target its hydroxy or phosphate groups. Note that the C6’ OH function is not available as a potential ubiquitylation site when core is present.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank the LMB mass spectrometry facility for analysing samples (Farida Begum, Sew Peak Chew and Mark Skehel), Daisuke Morito for providing RNF213 cDNA and Christina Gladkova and Alexander von der Malsburg for advice. This work was supported by a PhD Fellowship from the Boehringer Ingelheim Trust to VD, and by grants from the MRC (U105170648) and the Wellcome Trust (WT104752MA) to FR.
Footnotes
Author Contributions
EW performed and analyzed experiments leading to the discovery of LPS ubiquitylation. EO identified and characterized RNF213 with contributions from ACC (superresolution microscopy, complementation and analysis of RNF213KO MEFs), KB (restriction of Salmonella by RNF213), CP (generation of Salmonella knockouts and analysis of RNF213KO MEFs), VD (validation of RNF213KO cells), BS (bioinformatic analysis). EO and FR designed the study and wrote the manuscript.
Competing interests
The authors declare no competing interests.
Data Availability
All data are included in the paper and its supplementary information files. For gel source images, see Supplementary Fig.1. Source data are provided with this paper. Materials can be obtained from the corresponding authors upon request.
References
- 1.Huang J, Brumell JH. Bacteria–autophagy interplay: a battle for survival. Nat Rev Microbiol. 2014;12:101–114. doi: 10.1038/nrmicro3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perrin AJ, Jiang X, Birmingham CL, So NSY, Brumell JH. Recognition of 666 Bacteria in the Cytosol of Mammalian Cells by the Ubiquitin System. Curr Biol. 2004;14:806–811. doi: 10.1016/j.cub.2004.04.033. [DOI] [PubMed] [Google Scholar]
- 3.Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722–737. doi: 10.1038/nri3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kamada F, et al. A genome-wide association study identifies RNF213 as the first Moyamoya disease gene. J Hum Genet. 2011;56:34–40. doi: 10.1038/jhg.2010.132. [DOI] [PubMed] [Google Scholar]
- 5.Liu W, et al. Identification of RNF213 as a Susceptibility Gene for Moyamoya Disease and Its Possible Role in Vascular Development. Plos One. 2011;6:e22542. doi: 10.1371/journal.pone.0022542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scott RM, Smith ER. Moyamoya Disease and Moyamoya Syndrome. New Engl J Med. 2009;360:1226–1237. doi: 10.1056/NEJMra0804622. [DOI] [PubMed] [Google Scholar]
- 7.Kuroda S, Houkin K. Moyamoya disease: current concepts and future perspectives. Lancet Neurology. 2008;7:1056–1066. doi: 10.1016/S1474-4422(08)70240-0. [DOI] [PubMed] [Google Scholar]
- 8.Noad J, et al. LUBAC-synthesized linear ubiquitin chains restrict cytosol-invading bacteria by activating autophagy and NF-κB. Nat Microbiol. 2017;2:17063. doi: 10.1038/nmicrobiol.2017.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schaible UE, Prof, Haas A., Prof . Intracellular Niches of Microbes. Wiley-Blackwell; 2009. [DOI] [Google Scholar]
- 10.Birmingham CL, Smith AC, Bakowski MA, Yoshimori T, Brumell JH. Autophagy 683 Controls Salmonella Infection in Response to Damage to the Salmonella -containing Vacuole. J Biol Chem. 2006;281:11374–11383. doi: 10.1074/jbc.M509157200. [DOI] [PubMed] [Google Scholar]
- 11.Nakagawa I, et al. Autophagy Defends Cells Against Invading Group A Streptococcus. Science. 2004;306:1037–1040. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- 12.Thurston TLM, Wandel MP, Muhlinen N, Foeglein Á, von Randow F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature. 2012;482:414–418. doi: 10.1038/nature10744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thurston TLM, Ryzhakov G, Bloor S, Muhlinen N, von Randow F. The TBK1 691 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol. 2009;10:1215–1221. doi: 10.1038/ni.1800. [DOI] [PubMed] [Google Scholar]
- 14.Wild P, et al. Phosphorylation of the Autophagy Receptor Optineurin Restricts Salmonella Growth. Science. 2011;333:228–233. doi: 10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dupont N, et al. Shigella Phagocytic Vacuolar Membrane Remnants Participate in the 696 Cellular Response to Pathogen Invasion and Are Regulated by Autophagy. Cell Host Microbe. 2009;6:137–149. doi: 10.1016/j.chom.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 16.Huett A, et al. The LRR and RING Domain Protein LRSAM1 Is an E3 Ligase Crucial for 699 Ubiquitin-Dependent Autophagy of Intracellular Salmonella Typhimurium. Cell Host Microbe. 2012;12:778–790. doi: 10.1016/j.chom.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manzanillo PS, et al. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature. 2013;501:512–516. doi: 10.1038/nature12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wijk SJL.vanet al. Linear ubiquitination of cytosolic Salmonella Typhimurium activates NF-κB and restricts bacterial proliferation. Nat Microbiol. 2017;2:17066. doi: 10.1038/nmicrobiol.2017.66. [DOI] [PubMed] [Google Scholar]
- 19.Franco LH, et al. The Ubiquitin Ligase Smurf1 Functions in Selective Autophagy of 706 Mycobacterium tuberculosis and Anti-tuberculous Host Defense. Cell Host Microbe. 2017;21:59–72. doi: 10.1016/j.chom.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fiskin E, Bionda T, Dikic I, Behrends C. Global Analysis of Host and Bacterial Ubiquitinome in Response to Salmonella Typhimurium Infection. Mol Cell. 2016;62:967–981. doi: 10.1016/j.molcel.2016.04.015. [DOI] [PubMed] [Google Scholar]
- 21.Engström P, et al. Evasion of autophagy mediated by Rickettsia surface protein OmpB is critical for virulence. Nat Microbiol. 2019;4:2538–2551. doi: 10.1038/s41564-019-0583-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raetz CRH, Whitfield C. Lipopolysaccharide Endotoxins. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whitfield C, Trent MS. Biosynthesis and Export of Bacterial Lipopolysaccharides. Annu Rev Biochem. 2014;83:99–128. doi: 10.1146/annurev-biochem-060713-035600. [DOI] [PubMed] [Google Scholar]
- 24.Ahel J, et al. Moyamoya disease factor RNF213 is a giant E3 ligase with a dynein-like core and a distinct ubiquitin-transfer mechanism. Elife. 2020;9:e56185. doi: 10.7554/eLife.56185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Piccolis M, et al. Probing the Global Cellular Responses to Lipotoxicity Caused by Saturated Fatty Acids. Mol Cell. 2019;74:32–44.:e8. doi: 10.1016/j.molcel.2019.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sugihara M, et al. The AAA+ ATPase/ubiquitin ligase mysterin stabilizes cytoplasmic lipid droplets. J Cell Biol. 2019;218:949–960. doi: 10.1083/jcb.201712120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banh RS, et al. PTP1B controls non-mitochondrial oxygen consumption by regulating RNF213 to promote tumour survival during hypoxia. Nat Cell Biol. 2016;18:803–813. doi: 10.1038/ncb3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morito D, et al. Moyamoya disease-associated protein mysterin/RNF213 is a novel AAA+ ATPase, which dynamically changes its oligomeric state. Sci Rep-uk. 2014;4:4442. doi: 10.1038/srep04442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y, et al. Mitochondria-localised ZNFX1 functions as a dsRNA sensor to initiate antiviral responses through MAVS. Nat Cell Biol. 2019;21:1346–1356. doi: 10.1038/s41556-019-0416-0. [DOI] [PubMed] [Google Scholar]
- 30.Hitomi T, LIU W, Kobayashi H, Harada KH, Koizumi A. Distribution of Moyamoya Disease Susceptibility Polymorphism p.R4810K in RNF213 in East and Southeast Asian Populations. Neurol Med-chir. 2012;52:299–303. doi: 10.2176/nmc.52.299. [DOI] [PubMed] [Google Scholar]
- 31.Shi J, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187–92. doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- 32.Wandel MP, et al. Guanylate-binding proteins convert cytosolic bacteria into caspase-4 signaling platforms. Nat Immunol. 2020:1–12. doi: 10.1038/s41590-020-0697-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Santos JC, et al. Human GBP1 binds LPS to initiate assembly of a caspase-4 activating platform on cytosolic bacteria. Nat Commun. 2020;11:3276. doi: 10.1038/s41467-020-16889-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Randow F, Sale JE. Retroviral transduction of DT40. Sub-cellular biochemistry. 2006;40:383–386. doi: 10.1007/978-1-4020-4896-8_30. [DOI] [PubMed] [Google Scholar]
- 35.Glover JD, et al. A novel piggyBac transposon inducible expression system identifies a role for AKT signalling in primordial germ cell migration. Plos One. 2013;8:e77222. doi: 10.1371/journal.pone.0077222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ogawa M, et al. A Tecpr1-Dependent Selective Autophagy Pathway Targets Bacterial Pathogens. Cell Host Microbe. 2011;9:376–389. doi: 10.1016/j.chom.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 37.Yusa K, Zhou L, Li MA, Bradley A, Craig NL. A hyperactive piggyBac transposase for mammalian applications. Proc National Acad Sci. 2011;108:1531–1536. doi: 10.1073/pnas.1008322108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc National Acad Sci. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perkins DN, Pappin DJC, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 40.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical Statistical Model To Estimate the Accuracy of Peptide Identifications Made by MS/MS and Database Search. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 41.Johnson LS, Eddy SR, Portugaly E. Hidden Markov model speed heuristic and iterative HMM search procedure. Bmc Bioinformatics. 2010;11:431. doi: 10.1186/1471-2105-11-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu Y, Schmidt B, Maskell DL. MSAProbs: multiple sequence alignment based on pair hidden Markov models and partition function posterior probabilities. Bioinformatics. 2010;26:1958–1964. doi: 10.1093/bioinformatics/btq338. [DOI] [PubMed] [Google Scholar]
- 44.Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crooks GE, Hon G, Chandonia J-M, Brenner SE. WebLogo: A Sequence Logo Generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are included in the paper and its supplementary information files. For gel source images, see Supplementary Fig.1. Source data are provided with this paper. Materials can be obtained from the corresponding authors upon request.